

Summary
CRISPR screens are used extensively to systematically interrogate the phenotype-to-genotype problem. In contrast to early CRISPR screens, which defined core cell fitness genes, most current efforts now aim to identify context-specific phenotypes that differentiate a cell line, genetic background or condition of interest, such as a drug treatment. While CRISPR-related technologies have shown great promise and a fast pace of innovation, a better understanding of standards and methods for quality assessment of CRISPR screen results is crucial to guide technology development and application. Specifically, many commonly used metrics for quantifying screen quality do not accurately measure the reproducibility of context-specific hits. We highlight the importance of reporting reproducibility statistics that directly relate to the purpose of the screen and suggest the use of metrics that are sensitive to context-specific signal. A record of this paper’s Transparent Peer Review process is included in the Supplemental Information.
Keywords: Reproducibility, CRISPR Screening, Genetic Interactions
Graphical Abstract
eTOC blurb (in brief)
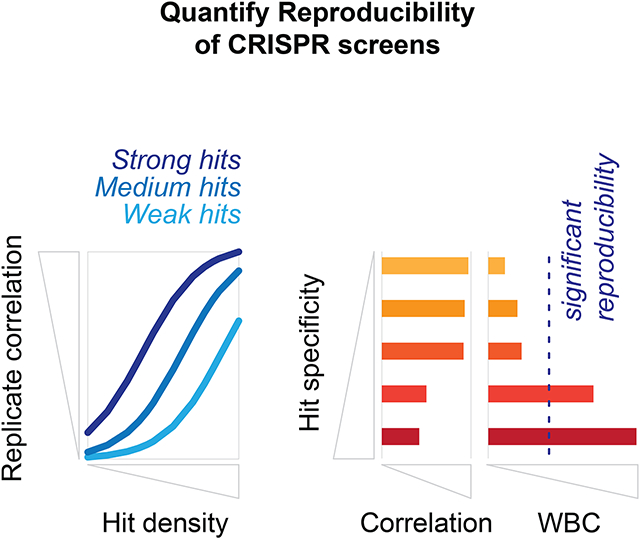
Reproducibility measurements are crucial for data interpretation. The signal of interest in context-specific CRISPR screens is often sparse. We show that sparsity complicates the interpretation of standard reproducibility measures like replicate correlation. We provide recommendations for reporting reproducibility of CRISPR screens and present the WBC score as an improved metric.
CRISPR screens see widespread use in the functional genomics community for interrogating gene function. Most prominently, loss-of-function CRISPR screens measure how the perturbation of each individual gene, across a library of targeted genes, affects cell fitness within a pool of cells. Each gene’s measurement in essence is composed of three elements: a fitness effect common to all cell types, a fitness effect specific to the biological context of the experiment (including cell type), and measurement error. Focusing on the first element, CRISPR screens completed across hundreds of different human cell types have now definitively identified the core genes that are essential across many cell types 1,2. Given that the core essential genes have been well-established, many CRISPR loss-of-function screens now focus on identifying genes that are essential in specific contexts, including different cell types, genetic backgrounds, or environmental conditions 1,3-8. Context-specific gene essentiality is important to explore because it can potentially help to guide functional annotation of the majority of genes in the human genome or elucidate disease mechanisms and therapy possibilities. However, the reproducibility of the context-specific effects discovered by CRISPR screens is inconsistently reported. Biological replicate screen reproducibility is typically reported using correlation measures on the level of normalized readcount data or fitness effects. At those processing steps, the data largely reflects covariation due to the gRNA representation in the library and/or the consensus (not context-specific) gene essentiality, respectively – neither provides an accurate estimate of the reproducibility of context-specific effects, which is often the main focus of the screen. Moreover, the interpretation of the commonly used metric, a correlation coefficient, is unclear due to the typical sparsity of effects in such screens.
To illustrate our point, we assess alternative reproducibility metrics across data processing levels of differential genome-wide CRISPR-Cas9 screens to identify genetic interactions (GI) with the fatty acid synthase (FASN) 9. First, we report the Pearson correlation coefficient (PCC) between independently replicated screens at the following points of data processing: starting gRNA abundance, end gRNA abundance, a fitness score reflecting the log-fold-change (LFC) between the end and starting gRNA abundance, the context-specific effect as measured by the differential LFC (dLFC; raw GI score), and finally, a fully normalized dLFC score (expressed as the qGI score, see 9 for details) (Figure 1a).
Figure 1: Reproducibility metrics for context-specific signal in CRISPR screens.

(a) Summary of the context-specific CRISPR-Cas9 screening and differential effect identification process. (b) Between-replicate Pearson correlation coefficients (PCC) for start and endpoint readcount data, the log2 foldchange (LFC) thereof and the differential LFC (dLFC) and qGI scores. Bars represent the mean of the three pairwise comparisons, dots represent the individual pairs. Screens were independently performed (starting from preparation and transfection of the gRNA library). (c) Within-FASN KO replicate to between FASN KO and non-FASN KO screen ratio of PCCs (WBC; see methods for details). Bars and dots represent the same as explained in (b). (d) Ranking of LUR1 (previous C12orf49) among the 17,804 genes screened in FASN KO cells at each data processing step as defined in 9. Ranks are means of the three biological replicates (e, f) Between-replicate PCC (e) and WBC (f) of LFC and dLFC data from DepMap genome-wide screens in 693 cell lines. (g, h) Comparison of between screen replicate PCCs on LFC and dLFC level for each of the 693 DepMap screens with the dLFC WBC. The four cell lines shown in (i) are highlighted. (i) Reproducibility of dLFC effects in four cell lines with different sets of replicate PCCs and WBCs. The consensus fitness is the per-gene mean LFC value across all replicates and 693 cell lines. The cell line-specific fitness is the per-gene LFC measured in each given cell line (SKBR3 = violet, HCC1187 = purple, MEL202 = cyan, A2780 = green). Circle size indicates each gene’s dLFC reproducibility and corresponds to the per-gene dLFC product between replicate screens. (j) PCC between simulated screening data with normally distributed noise at increasing numbers of hits with weak, medium and strong amplitude. Hit strength is defined as a multiple of the standard deviation of the noise distribution (σ).
Within-context (FASN KO) replicate correlations were highest for starting readcounts (r = 0.97 for gRNA, r = 0.97 for gene-level measures), reflecting that the gRNA library distribution is reproducible (Figure 1b). Removing this library effect from the endpoint readcounts to obtain LFC fitness values also results in high PCCs at the gene-level (r = 0.92) and slightly lower gRNA-level PCCs of 0.82 (Figure 1b). This shows that both unwanted technical features of the experiment and general fitness effects are highly reproducible. However, in this context, we aim to identify GIs with FASN, i.e. FASN-specific fitness effects, and thus, all of the measures above fail to measure the reproducibility associated with the focus of our screen. Once dLFC values are computed between the FASN KO query and wild-type reference screens, the PCC between replicate screens drops substantially to 0.3 (gRNA-level) and 0.5 (gene-level) and further to 0.21 (gRNA-level) and 0.42 (gene-level) when experimental artifacts are computationally normalized, which is reflected in the qGI score (Figure 1b). In summary, the replicate correlations decrease with more accurate quantification of the biological signal of interest, which is context-specific effects (in this case, GIs). Importantly, fitness score (LFC)-based replicate correlations cannot approximate context-specific effect reproducibility (Figure S1a-c), and such comparisons are particularly problematic for comparative evaluation of data from different sets of genes or different cell models.
To illustrate why focusing on the appropriate screen statistics is important for reporting reproducibility, we further analyzed the FASN KO, but compared it to five non-FASN KO screens. Under the simple assumption that biological replicates of the same genetic screen should exhibit more similarity than genetic screens with different query mutations, we computed a score that captures the similarity of two or more replicates of the same screen relative to the similarity of different screens, which we will subsequently refer to as the Within-vs-Between context replicate Correlation (WBC) score (see STAR Methods). Despite their high correlations, readcounts and LFC data did not distinguish within-context (same KO) replicates from between-context (different KO) pairs (Figure 1c), confirming that the high similarity does not indicate reproducibility of the main quantity of interest. In contrast, despite low within-context replicate PCCs, the dLFC measure exhibited strong WBC scores (z > 3), and these were further improved in the qGI score (Figure 1c). We note that only the context-specific scores (dLFC and qGI) capture the biologically relevant signal in this case, which are genetic interactions with the FASN query mutation. For example, only dLFC and qGI scores are able to identify the gene LUR1 as a top interacting partner (Figure 1d), which was recently characterized as playing a functional role in lipid metabolism with FASN 9. Using simple replicate PCC as a measure of reproducibility, one would conclude that these context-specific scores are of lower quality than the less biologically relevant scores from earlier stages of data processing, but a context-specific reproducibility score such as the WBC score suggests the opposite. Both the metric one chooses to quantify reproducibility and the stage of data processing at which this measurement is taken are important for making accurate conclusions about data quality.
To demonstrate the generality of our findings beyond genetic interaction screens, we performed a similar analysis on the reproducibility of cell line-specific effects in 693 screens within the Cancer Dependency Map 1,8. We made the assumption that screens performed in the same cell line (replicate screens in this case), contain context (cell line)-specific effects that distinguish a given cell line from other cell lines, and that those effects are quantified by dLFC rather than LFC values. We tested how the PCC and WBC quantify screen replication and how those metrics change when we focus on the cell line-specific (dLFC) signal. Again, we found that the within-context (same cell line) replicate correlation decreased substantially between the fitness effect (LFC; mean r = 0.81) and the cell line-specific deviation from the consensus fitness profile (dLFC; mean r = 0.47) (Figure 1e). In contrast, the WBC score indicates that the dLFC metric reflects context-specific signal with much higher quality (Figure 1f), which is the main goal in building such a cancer dependency map. Specifically, significant (z > 3) and highly significant (z > 5) reproducibility scores for cell line-specific effects are found for 99.9% and 91.3% of all cell lines by using the dLFC metric, respectively, while only 10.1% and 1.2% of cell lines reach the same significance level when using the LFC metric (Figure 1f). A metric like the WBC score provides additional resolution compared to simple correlation measures, emphasizing the reproducibility of context-specific effects (Figure 1g-i; Figure S1d). We note that the antagonistic relationship between replicate correlation and data normalization extends to non-CRISPR screens, including the most comprehensive GI data to date, recorded in yeast (Figure S2a, b) 10,11.
Perhaps one of the reasons why informative reproducibility measures are not consistently reported is that they tend to be relatively low, which may be viewed as evidence for poor quality data. How high should we expect replicate correlation to be for a high-quality CRISPR screen? Global metrics such as the PCC are highly impacted by the sparsity of the signal one is measuring, and in most cases, one would expect context-specific genetic effects to be rare 8-10. For instance, simulated genome-wide screening data (~18k genes) with normally distributed noise and 18 (0.1% density) or 180 (1% density) strong (12σ) true hits, densities typical of context-specific screens, would result in a PCC of 0.13 and 0.59, respectively. In contrast, a hit-density of 10%, which is more typical for pure fitness phenotypes in genome-wide screens, results in a PCC of 0.93 (Figure 1j). Thus, we should expect low PCC measures in genome-scale screens even where a small number of hits are highly reproducible due to the sparsity of context-specific fitness effects.
We note that there have been other complementary efforts to establish best practices for conducting CRISPR screens and analyzing the resulting data 2,12,13. In particular, Behan et al. recognized the challenges of computing correlation between replicate screens based on whole dependency profiles. Specifically, they noted that including the core essential genes in this calculation inflates the correlation such that replicates of the same screen are generally less distinguishable from replicates of different screens. Second, they noted that including guides targeting genes that never showed phenotypes led to pessimistic estimates of reproducibility due to the sparsity of signal across the dependency profile. Behan et al. addressed these issues by pre-processing the data to find the most variable signal (excluding both core essential genes and genes with no phenotypes) and to compute correlations on that subset of the data, which provides a more informative report of the data reproducibility. We address related issues here, but rather than pre-filtering of profiles, which may depend on the specific gRNA library used or a large collection of screens, we instead suggest that reproducibility analysis should be performed on scores that capture context-specific signal (e.g. dLFC). Furthermore, we propose a new metric, the WBC score, that is more directly interpretable than a correlation coefficient when applied to a sparse profile. Our suggested approach can be applied to a variety of CRISPR screening contexts.
In conclusion, we highlight the importance of reporting appropriate reproducibility statistics for CRISPR screens. Biological replicate screens should be performed to establish the quality of data in any screening context, and importantly, the reported statistics should directly relate to the purpose of the screen. In addition to standard correlation measures, we suggest the use of additional metrics, such as the WBC, that are sensitive to context-specific signal.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Maximilian Billmann maximilian.billmann@gmail.com).
Materials availability
No materials have been generated for this study.
Data and code availability
All data had been publicly available prior to this study 8,9,11. The code implementing the WBC score is provided as Supplemental Method S1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
METHODS
Replicate correlation of genome-wide CRISPR-Cas9 screens in HAP1 FASN KO cells
HAP1 genome-wide CRISPR-Cas9 screening data was taken from 9. The three biological replicates were independently screened (including gRNA library preparation and transfection). The gRNA-level comparisons use values from 70,006 gRNAs that had an initial abundance at the start of the experiment of at least 40 readcounts. The gene-level comparisons use values from 17,804 genes, considering a gene whenever at least two gRNA sequences pass all QC thresholds. The fitness scores represent the log2-fold-change (LFC) between the start and endpoint gRNA abundance. The quantitative genetic interaction (qGI) score represents the differential fitness effect between a wildtype control and query gene (here FASN) knockout screen after correcting query gene-unspecific screening artifacts. LFC and qGI scores were generated as described in 9.
The Within-vs-Between context replicate Correlation (WBC) score
To define the Within-vs-Between context replicate Correlation (WBC) score for a given screen, its biological replicate correlation is scaled to its expected background correlation distribution: the mean and standard deviation of its correlation with screens performed in another context (e.g. query mutation). This converts the correlation coefficients into a metric with an unambiguous statistical interpretation that can be interpreted as a z-score. Notably, each context (e.g. a set of screens done in a cell line within a larger set of screens covering multiple cell lines) creates its own background correlation distribution. This is important, because even at the same data processing level, signal sparsity substantially differs between contexts, and the background correlation is dependent on the abundance of the within-context signal. An example for this trend is illustrated for 693 distinct cell lines taken from the DepMap (Figure S3a, b).
For the example shown in detail in this work, the FASN genetic interaction screens, genome-wide CRISPR-Cas9 screens completed in isogenic HAP1 cells were divided into those harboring a FASN loss-of-function mutation (n = 3) and those harboring a mutation different from FASN (n = 5), namely LDLR, SREBF1, SREBF2, ACACA and C12orf49/LUR1. At each data processing step, all pairwise Pearson correlation coefficients (PCC) within the FASN KO context and between FASN KO and each of the 5 remaining KO contexts were computed. From these comparisons, the mean PCC () and the WBC score were computed as follows:
Where:
Here, N refers to the 3 possible pairwise comparisons between FASN replicated screens (within-FASN KO), and M refers to the 15 possible pairwise combinations between FASN replicated screens and LDLR, SREBF1, SREBF2, ACACA and C12orf49/LUR1 screens.
While larger N and M provide more robust estimates of the WBC, we found that WBCs derived from any combination of 2, 3 or 4 of the LDLR, SREBF1, SREBF2, ACACA and C12orf49/LUR1 screens as well as only using 2 FASN KO screens provided stable measures of context-specific signal that distinguished scores derived from different stages of data processing (Figure S4a, b).
Cancer Dependency Map (DepMap) replicate screen comparison
LFC gRNA data (20Q3 release) was downloaded from the DepMap website [https://depmap.org/portal/download/]. gRNA values were mapped and mean-summarized per gene to obtain gene-level LFC data (shown in Figure 1e). Only cell lines with at least two replicates were considered in this analysis. To generate dLFC values, all screens were initially adjusted by quantilenormalizing gene-level LFC data using the R function normalizeQuantiles. Next, the scores for each gene across all screens were median centered at 0 so that if a gene was more essential in a cell line compared to its median fitness score, it had a negative score. The reproducibility of the cell line-specific signal was computed both on LFC and dLFC-level using the within-cell line PCC and the WBC comparing within-cell line PCCs to between-cell line PCCs.
Bin-wise replicate correlation analysis of LFC and qGI scores
To test how CRISPR screening fitness scores (LFC) affect the reproducibility of context-specific (qGI) scores, each gene was assigned to one of five bins with the intent to keep the range of qGI scores constant in each bin and having five incrementally increasing ranges of LFC scores in those bins. This was done by moving a window along the vector of qGI scores (representing the mean qGI score of both replicated screens). In each window, all genes had similar qGI but potentially different LFC scores. Genes with the most extreme LFC values were assigned to the first bin, genes with the next most extreme LFC values to the second bin and so on, thereby generating five bins with similar qGI ranges but different LFC ranges.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Critical commercial assays | ||
| Deposited data | ||
| CRISPR screening readcount, log2-foldchange and qGI data | Aregger et al.9 | GEO: GSE148627 |
| DepMap gRNA-level log-foldchange data | https://depmap.org/portal/download/ | 20Q3: Achilles_logfold_c hange.csv |
| SGA yeast fitness and genetic interaction data | https://boonelab.ccbr.utoronto.ca/condition_sga 11 | File S3 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| R version 4.2.1 | https://www.r-project.org/ | NA |
| Bowtie v0.12.8 | http://bowtie-bio.sourceforge.net/index.shtml | NA |
| code implementing the WBC score | This Paper | Supplemental Information Method S1 |
| Other | ||
Highlights.
Reproducibility metric interpretability is key for omics data analyses and integration
Context-specific CRISPR screens often have low hit density
Hit density determines the correlation coefficient range for CRISPR screen replicates
The WBC score provides a hit-specific interpretation for a replicate correlation
Acknowledgements
We thank all members from the Myers, Moffat, Boone and Andrews laboratory for fruitful discussions. This research was funded by grants from the National Science Foundation (MCB 1818293), the National Institutes of Health (R01HG005084, R01HG005853), the Ontario Research Fund, the Canada Research Chairs Program. and the CIHR (PJT-463531). JM holds the Glaxosmithkline Chair in Genetics and Genome Biology, The Hospital for Sick Children, Toronto, Ontario, Canada.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
Co-author Brenda Andrews is on the advisory board of Cell Systems.
References
- 1.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. (2019). Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568, 511–516. 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- 3.Han K, Jeng EE, Hess GT, Morgens DW, Li A, and Bassik MC (2017). Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol 35, 463–474. 10.1038/nbt.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Najm FJ, Strand C, Donovan KF, Hegde M, Sanson KR, Vaimberg EW, Sullender ME, Hartenian E, Kalani Z, Fusi N, et al. (2018). Orthologous CRISPR–Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol 36, 179–189. 10.1038/nbt.4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A, Licon K, Klepper K, Pekin D, Beckett AN, et al. (2017). Combinatorial CRISPR–Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 14, 573–576. 10.1038/nmeth.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeWeirdt PC, Sanson KR, Sangree AK, Hegde M, Hanna RE, Feeley MN, Griffith AL, Teng T, Borys SM, Strand C, et al. (2021). Optimization of AsCas12a for combinatorial genetic screens in human cells. Nat. Biotechnol 39, 94–104. 10.1038/s41587-020-0600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dempster JM, Rossen J, Kazachkova M, Pan J, Kugener G, Root DE, and Tsherniak A (2019). Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv. 10.1101/720243. [DOI] [Google Scholar]
- 8.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576.e16. 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aregger M, Lawson KA, Billmann M, Costanzo M, Tong AHY, Chan K, Rahman M, Brown KR, Ross C, Usaj M, et al. (2020). Systematic mapping of genetic interactions for de novo fatty acid synthesis identifies C12orf49 as a regulator of lipid metabolism. Nat. Metab 2, 499–513. 10.1038/s42255-020-0211-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. (2016). A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420–aaf1420. 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costanzo M, Hou J, Messier V, Nelson J, Rahman M, VanderSluis B, Wang W, Pons C, Ross C, Ušaj M, et al. (2021). Environmental robustness of the global yeast genetic interaction network. Science 372, eabf8424. 10.1126/science.abf8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doench JG (2018). Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet 19, 67–80. 10.1038/nrg.2017.97. [DOI] [PubMed] [Google Scholar]
- 13.Hanna RE, and Doench JG (2020). Design and analysis of CRISPR–Cas experiments. Nat. Biotechnol 38, 813–823. 10.1038/s41587-020-0490-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data had been publicly available prior to this study 8,9,11. The code implementing the WBC score is provided as Supplemental Method S1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.