

SUMMARY
Improvements in tumor immunotherapies depend on better understanding of the anti-tumor T cell response. By studying human tumor-draining lymph nodes (TDLNs), we found that activated CD8+ T cells in TDLNs shared functional, transcriptional, and epigenetic traits with TCF1+ stem-like cells in the tumor. The phenotype and TCR overlap suggested that these TDLN cells were precursors to tumor-resident stem-like CD8+ T cells. Murine tumor models revealed that tumor-specific CD8+ T cells were activated in TDLNs but lacked an effector phenotype. These stem-like cells migrated into the tumor, where additional co-stimulation from antigen-presenting cells drove effector differentiation. This model of CD8+ T cell activation in response to cancer is different from that of canonical CD8+ T cell activation to acute viruses, and it proposes two stages of tumor-specific CD8+ T cell activation: initial activation in TDLNs and subsequent effector program acquisition within the tumor after additional co-stimulation.
In brief
Better understanding of the fundamental mechanisms controlling anti-tumor T cell responses is needed. Here, Prokhnevska et al. describe a two-step T cell activation response to cancer: (1) initial priming in tumor-draining lymph nodes resulting in a stem-like phenotype and (2) a co-stimulation-dependent phase in the tumor to acquire effector programming.
Graphical Abstract
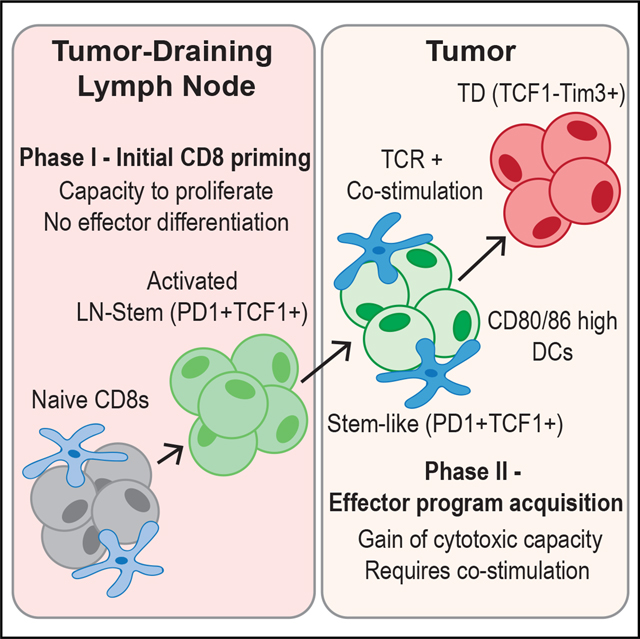
INTRODUCTION
CD8+ T cells are a crucial part of the adaptive immune response to cancer, and infiltration of these cells into tumors can predict patient survival and response to checkpoint therapy.1–7 Understanding why some patients have high infiltration of these cells and others have practically none requires a better understanding of the fundamental mechanisms that control the T cell response against cancer.
A productive T cell response is characterized by activation, expansion, and migration to the site of inflammation to kill target cells. The central paradigm describing T cell activation is that naive T cells are primed in the secondary lymphoid organs that drain infected tissue. This initial activation requires three main signals: signal 1, through MHC-peptide interaction with the T cell receptor (TCR); signal 2, through co-stimulation from a variety of different receptors; and signal 3, received from various cytokines. Upon receiving all three signals, T cells undergo many rounds of proliferation and express genes related to their effector function, like granzymes, perforin, and interferon (IFN), within the first 48 h after activation.8 These activating signals are only required for a short period of time as cells can be stimulated for 24 h and then placed in antigen-free conditions while still executing the effector program of many rounds of cell division and expression of cytotoxic molecules and cytokines over the following few days.9–11 Co-stimulation and cytokines are critical for the acquisition of effector function, as cells that receive a normal physiological amount of TCR signal, without these additional signals, undergo poor proliferation and express a lower amount of cytotoxic and effector molecules.12–14 This model of T cell activation implies that once CD8+ T cells receive these critical signals to activate, they are committed to proliferate, acquire cytotoxic potential, and now only need to encounter cells presenting their cognate antigen to kill.
This paradigm has been applied to cancer and is central to ideas like the cancer immunity cycle where T cells are primed in tumor-draining lymph nodes (TDLNs) and then migrate to tumors where they kill cancer cells.15–18 However, there are discrepancies with what this canonical T cell activation system predicts and what is observed in cancer. Description of a non-cytotoxic TCF1+ CD8+ T cell that resides within tumors does not fit this model.1,19–25 TCF1+ CD8+ T cells have encountered antigen, have undergone many rounds of proliferation, but have not expressed cytotoxic genes like perforin or granzyme B (GZMB). However, upon various stimulations, these stem-like cells give rise to cytotoxic daughter cells.26 Due to these properties, these cells have been termed “stem-like” or exhausted precursors. The critical role of stem-like CD8+ T cells in cancer is shown by checkpoint blockade and adoptive transfer trials.21,23,27–29 Because this cell does not fit with ideas predicted by the standard T cell activation model, this study aimed to investigate how this TCF1+ CD8+ T cell arises and generally how T cell activation in cancer occurs. We find that T cells responding to cancer proliferate in TDLNs but fail to express effector molecules, and only once in the tumor do these cancer-specific T cells undergo effector differentiation.
RESULTS
Activated PD1+ CD8+ T cells in human TDLNs are the precursor of stem-like cells in tumors
To understand how CD8 T cells are primed in cancer, we analyzed matching tumor and non-metastatic TDLNs from patients undergoing surgery for prostate, kidney, and bladder cancers. As we had previously found in over 100 patients, there was a wide range of CD8 T cell infiltration in these tumors,1 and the activated CD8 population always comprised both a TCF1+ stem-like and a TIM3+ terminally differentiated (TD) CD8 T cell (Figures 1A, S1A, and S1B). Stem-like CD8 T cells expressed high CD127 and CD28 and low TIM3, CD39, and GZMB (Figure 1C). By comparison, the TCF1−TIM3+ TD population expressed high CD39 and GZMB and lower CD28 and CD127 (Figures 1A and 1C). Due to their co-expression of TIM3 and CD39, we have used both markers interchangeably to denote the TD CD8+ tumor-infiltrating lymphocyte (TIL) population. When we analyzed TDLNs from prostate and kidney tumors, we identified a population of activated PD1+CD45RA− cells (Figure S1C). These cells expressed markers like CD69 and CXCR3 that distinguished them from naive CD8+ T cells (Figure S1D). All these activated cells in the TDLNs were phenotypically similar to the stem-like cells in the tumor, having high expression of TCF1, CD127, and CD28 and no expression of GZMB, CD39, and TIM3 (Figures 1B, 1C, and S1D).
Figure 1. Activated CD8+ T cells in human TDLNs have a similar phenotype to tumor-infiltrating stem-like CD8+ T cells.
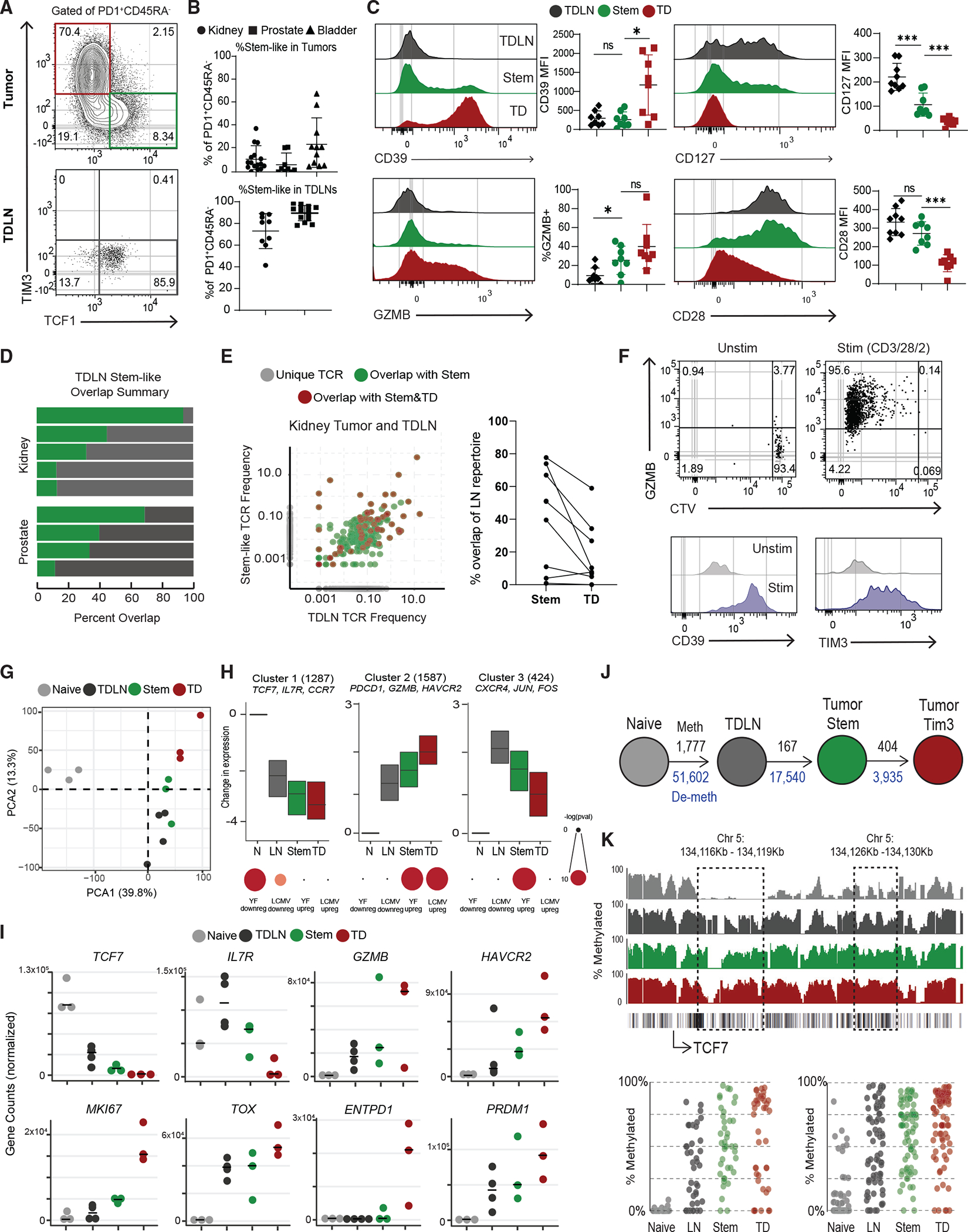
(A) Representative plots showing activated CD8+ T cells in human tumor and tumor-draining lymph nodes (TDLNs).
(B) Summaries of the proportion of stem-like CD8+ T cells in human kidney (n = 16), prostate (n = 32), and bladder (n = 12) tumors and kidney (n = 9) and prostate (n = 14) TDLNs.
(C) Phenotype of CD8+ T cell populations in TDLN and tumor.
(D) Summary of TCR repertoire overlap between activated CD8+ T cells from TDLNs and tumor-stem-like CD8+ T cells in human kidney and prostate cancer.
(E) TCR repertoire overlap in a representative patient between activated CD8+ T cells from TDLNs, stem-like CD8+ TILs, and TD CD8+ TILs. The proportion of the detected TCR clonotype in each patient that is unique or shared between the populations is shown. Summary showing the TCR overlap between stem-like and TD CD8+ TILs as a proportion of the LN CD8+ T cell repertoire.
(F) Sorted CTV-labeled activated CD8+ T cells from human TDLNs were cultured in vitro with anti-CD3/28/2 beads for 7 days.
(G) PCA of RNA-seq of naive CD8+ T cells, activated LN-stem CD8+, and stem-like CD8+ and TD CD8+ TILs.
(H) Gene expression patterns from K-means clustering of all CD8+ subsets. Gene set enrichment using genes from each cluster, compared with common effector CD8+ T cell signatures from yellow fever D14 and LCMV Arm.
(I) Normalized gene counts showing expression of selected genes in the sorted CD8+ T cell populations.
(J) Schematic of the number of methylated regions that are at least 25% different from naive in the TDLN and tumor CD8+ T cell populations. Black numbers show methylated regions, and blue numbers show demethylated regions.
(K) Specific methylation changes in TCF7. Traces show total methylation in regions. Boxed regions show significantly differentially methylated regions. Dot plots showed methylation of each CpG in boxed region. Median and 95% confidence intervals (CIs) are shown. *p < 0.05 determined by Mann-Whitney test.
Based on the phenotypic similarity of these activated T cells in TDLNs to stem-like cells in tumors, we were interested in whether these populations were clonally related. To test this, we performed TCR sequencing on activated CD8+ T cells from TDLNs (PD1+CD45RA−CD28+CD39−), as well as on stem-like (PD1+CD39−CD28+) and TD (PD1+CD39+) cells from matching kidney or prostate tumors. In every patient, we detected TCR overlap between the activated T cells from TDLNs and the stem-like subset in the tumor (Figure 1D). Additionally, several TCRs found in the TDLN population were detected in both stem-like and TD CD8+ TILs from matching tumors, implying a clear lineage relationship (Figure 1E). The PD1− CD8+ T cells sorted from the same TDLNs showed less overlap or similarity with tumor populations (Figure S1E). Together with the phenotype data above, these data suggest a lineage relationship between these 3 populations of cells, where the PD1+ CD45RA−TCF1+ cells in TDLNs, from here on referred to as “LN-stems,” appear to be the precursor to stem-like cells in the tumor, which then give rise to the TD CD8+ TILs.
We were next interested in if the LN-stem CD8s shared functional characteristics with stem-like cells in tumors. To determine whether the LN-stem CD8+ T cells also share these functional roles, we labeled LN-stems with CellTrace Violet (CTV) and stimulated them in vitro with anti-CD3/28/2 beads for 7 days. These cells all diluted CTV and increased expression of GZMB, CD39, and TIM3, as we had previously seen for stem-like CD8+ T cells isolated from tumors, suggesting a shared ability to proliferate and differentiate (Figure 1F).
Given the proposed relationship between LN-stem CD8+ T cells and stem-like and TD cells in tumors, we were interested in the transcriptional and epigenetic changes that occurred through these stages. We first performed RNA-seq on sorted naive (CD45RA+ CCR7+), LN-stem, tumor-stem-like, and TD CD8+ T cells from kidney tumors and TDLNs. Principal-component analysis (PCA) showed that all three subsets of activated CD8+ T cells clustered away from naive CD8+ T cells, and the LN-stem CD8+ T cells clustered more closely to the stem-like population from the tumor (Figure 1G). Unbiased K-means clustering was performed to identify common and differential gene expression patterns in the data. The first cluster identified genes that were highly expressed in naive CD8+ T cells and were progressively decreased from LN-stems to tumor subsets.
These genes were enriched with genes turned off in effector CD8+ T cells from both yellow fever (YF) and lymphocytic choriomeningitis virus (LCMV) Armstrong (Arm) (Figures 1H, 1I, and S1F). The second cluster contained genes that were progressively increased in LN-stems, which were further increased in tumor subsets. This second cluster was mostly enriched in genes found increased in effector CD8+ T cells from YF and LCMV Arm (Figures 1H and 1I). The last cluster identified genes that are increased in LN-stems but that progressively decrease as they differentiate, such as CXCR4 and CD28 (Figures 1H, 1I, S1F, and Table S1). We also performed gene set enrichment analysis (GSEA), comparing the LN-stem population with tumor-stem population, and found higher IFN-β signaling and many proliferative pathways active in the tumor-stem populations (Figure S1G). Finally, comparing LN-stem with TD cells in tumors indicated that energy production and cell cycle were increased in the TD cells (Figure 1H).
Whole-genome DNA methylation analysis of these cell populations showed a similar trend. Most epigenetic changes occurred between naive and LN-stems, with 51,602 demethylated and 1,777 methylated regions compared with naive CD8s. Epigenetic changes continue in the tumor subsets with 17,540 new regions demethylated in tumor-stem-like TILs and 3,935 demethylated regions occurring specifically in tumor TD TILs (Figures 1J, S1I, S1J, and Table S2). These methylation changes clustered into similar patterns to those found in the transcriptional analysis. The first cluster showed genes that are progressively methylated as CD8+ T cells differentiate (Figures 1H, 1K, and S1H). For example, in Figure 1K, there are two regions in the TCF7 loci that get progressively methylated as LN-stems differentiate into stem-like and then TD CD8+ TILs. The second cluster included genes that were progressively demethylated and transcriptionally increased, such as CTLA4, EOMES, and PRDM1 (Figures 1H and S1H). The last cluster contained regions that were demethylated in the LN-stems and tumor stems but then re-methylated in tumor TDs (Figure S1H). Together, these data suggest that CD8+ T cells in TDLNs are generally more similar to the stem-like population in the tumor.
Tumor antigen-specific cells in TDLNs of mouse models are in a stem-like state
Although our data suggest a two-step differentiation model, several other models of CD8+ T cell activation are possible. One plausible explanation for the observed data in Figure 1 is that naive cells are initially primed in tumors, and then only stem-like cells migrate back to lymph nodes30 (Figure S2A). Another possibility is that effector CD8+ T cell activation occurs first, and those cells de-differentiate to a stem-like phenotype later. A third possibility is that these subsets both activate in the TDLNs and migrate to the tumor independently (Figure S2B).
To better identify which of these models explains the origin of CD8+ T cell phenotypes in cancer, we looked at studying these cells in mouse models. We first examined if mouse models of cancer had similar populations of CD8+ T cells to what we had found in patients. We examined TRAMPC1 tumors that expressed the LCMV glycoprotein (GP) (TRAMPC1-GP) and used both GP33 (LCMV GP epitope) and the endogenous SPAS1 antigen31 to analyze the tumor-specific CD8+ T cell response in TDLNs and tumors. Tumor-specific CD8+ T cells were present in both tumor and TDLNs and had an activated CD44+PD1+ phenotype (Figures 2A and S2D). In the tumor, both GP33+ and SPAS1+ CD8+ T cells had stem-like (Tcf1+ Tim3−) and TD (Tcf1−Tim3+) phenotypes (Figure 2B). Both tumor-specific and bulk activated TD CD8+ TILs had higher expressions of Gzmb, Ki67, and Blimp1, whereas stem-like CD8+ TILs expressed higher CD127 (Figure 2C). When we analyzed B16-GP, a melanoma tumor that grows in C57BL/6 mice, and RENCA-HA, a kidney tumor that grows in BALB/c mice, the tumor antigen-specific (GP33+ or PR8+) CD8+ TILs contained the same stem-like and TD populations. In the TDLN of each mouse model, all the tumor antigen-specific CD8+ T cells expressed high Tcf1 and did not express Tim3 (Figures 2B and S2E). The LN-stems increased expression of PD1, CD44, and Ki67, turned off CD127, and overall did not express the effector-associated molecules Gzmb and Blimp1 (Figures 2C and S2F). These data show that in three separate mouse models across different mouse strains, tumor-specific CD8+ T cells in TDLNs are activated, retain Tcf1 expression, and do not express effector molecules like Gzmb, all of which parallel the phenotype we observed in human TDLNs.
Figure 2. Tumor-specific CD8+ T cells activated in TDLNs to acquire a stem-like phenotype.
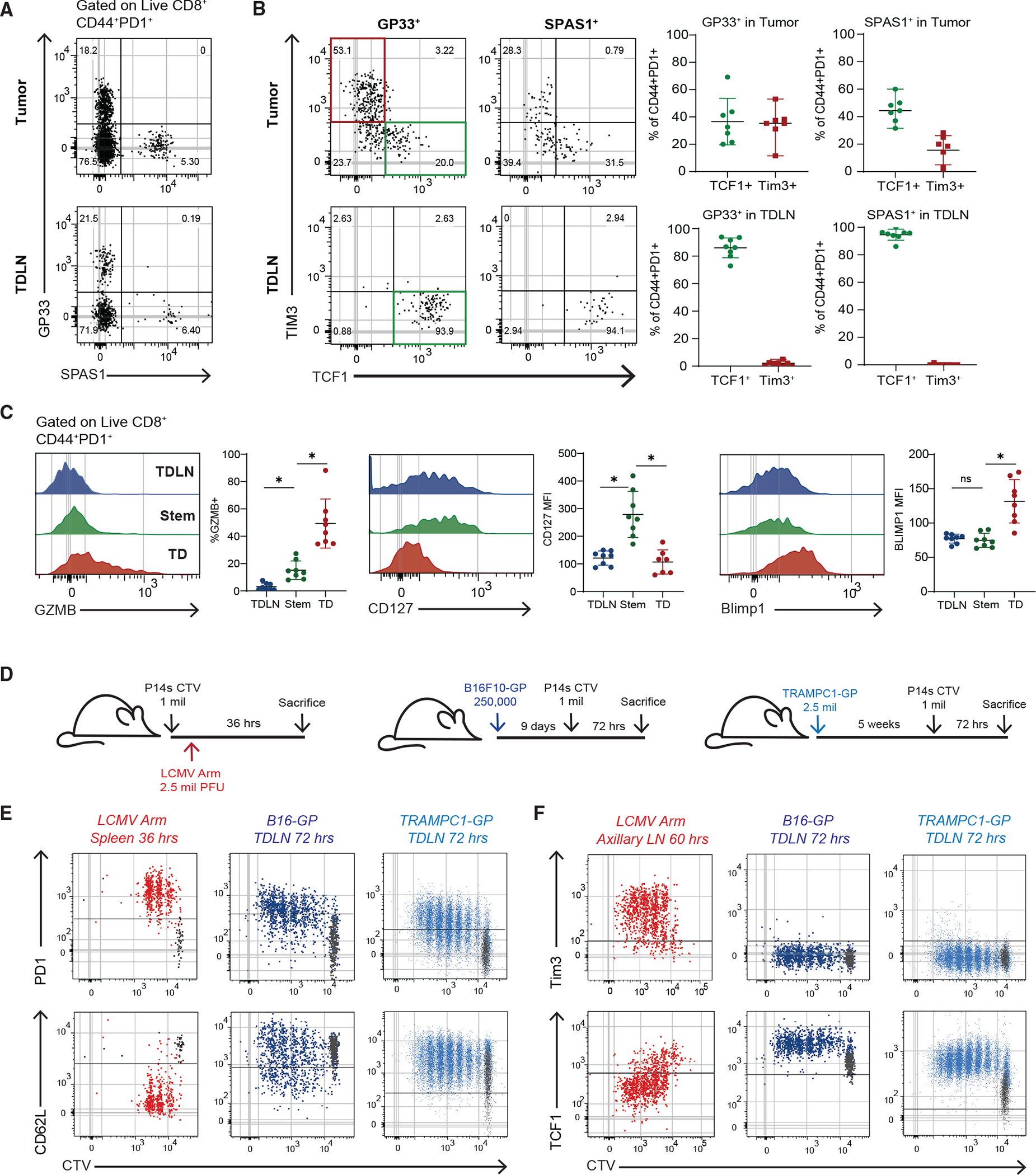
(A) Representative plot of tumor-specific CD8s in 5-week TRAMPC1-GP tumors and TDLNs.
(B) Phenotype of CD44+PD1+ tumor-specific CD8+ T cells in tumor and TDLN.
(C) Phenotype of CD44+PD1+ CD8+ T cell populations.
(D) Experimental setup of early activation of P14 CD8+ T cells.
(E) Flow cytometry of PD1 and CD62L expression by CTV of P14s activated in each respective model.
(F) Flow cytometry of TIM3 and Tcf1 expression by CTV of P14s activated in each respective model. Median and 95% confidence intervals (CIs) are shown. *p < 0.05 determined by Mann-Whitney test.
T cell activation in TDLNs is distinct from the response to a viral infection
Since these mouse models replicated what we found in human cancers, we designed an experiment to test which of the proposed models (Figures S2A–S2C) best explains how T cell activation occurs in cancer. In this experiment, we transferred CTV-labeled tumor antigen-specific P14 T cells into mice bearing B16-GP or TRAMPC1-GP tumors to track the kinetics and location of CD8+ T cell activation by division (Figure 2D). As a positive control to represent canonical T cell activation, we also transferred CTV-labeled P14 T cells into mice and then infected them with acute LCMV Arm. P14 cells responding to LCMV Arm in the spleen and axillary LNs (AxLNs) adopted a typical effector CD8+ T cell activation program, increasing expression of PD1, CD25, Gzmb, and Prf and decreasing CD62L and Tcf1 (Figures 2E and S2G). This phenotypic change was rapid and observed prior to the P14 cells even undergoing division. In comparison, in both B16-GP and TRAMPC1-GP TDLNs, P14 cells underwent many rounds of division and turned on genes associated with activation, such as PD1 and CD44, but failed to turn off CD62L (Figures 2E and S2H). When we compared P14s activated in LCMV Arm AxLN with TDLNs, we found that in LCMV Arm, P14s rapidly expressed Tim3 and lost the expression of Tcf1. In TDLNs, P14s never turned off Tcf1 and never expressed Tim3 in any of the early divisions.
We next wanted to test whether CD8+ T cells in the TDLNs might be dividing and expressing various activation markers because of local cytokines from the tumor rather than exposure to tumor antigen. P14 cells transferred into TRAMPC1-bearing mice with no GP antigen remained naive in TDLNs (CD44−PD1−CD62L+), showing that the expression of CD44 and PD1 was antigen dependent (Figure S2I). We next considered if low amounts of antigen in TDLNs might be the reason for failure of CD8+ T cells to acquire effector function upon initial activation, as several studies have suggested that amount or affinity of antigen is related to differentiation state.32–35 To investigate, we transferred P14s into B16-GP-bearing mice and injected (i.v.) 100 μg of cognate GP33 peptide at the time of transfer and every other day for 5 days. P14s in mice treated with peptide expressed far higher PD1 and Irf4, both of which are driven by TCR signaling36 (Figure S2J). Activated P14s in TDLNs remained Tcf1+ and failed to express Tim3 even after 5 days of GP33 peptide treatment (Figure S2K). This implies that lack of antigen is not the reason CD8+ T cells fail to acquire effector function within the TDLN.
Together, these data rule out model A, where T cells prime directly in the tumor, and model B, where both stem and effector cells are activated in TDLN. Only model C, where cells become stem-like cells on initial activation and then migrate to the tumor, fits with the data presented (Figures S2A–S2C).
CD8+ T cells initially activated in TDLN fail to undertake a canonical effector transcriptional or epigenetic program
We next sought to understand the early transcriptional and epigenetic events in these tumor-specific CD8+ T cells and how they compare with a canonical acute viral effector program. To do this, we sorted activated, undivided P14s as they underwent division either in response to LCMV Arm in the spleen or in TDLNs of TRAMPC1-GP tumor-bearing mice (Figure 3A). PCA analysis found that P14s activated in the spleen of LCMV and in TDLNs of tumor-bearing mice cluster separately (Figure 3B and Table S3), and cells were mostly similar between the undivided and later divisions. P14s responding to LCMV Arm immediately acquired an effector transcriptional program before even undergoing division, with high expression of the effector molecules Ifng, Tnf, and Gzmb. In comparison, P14 cells responding to the tumor underwent a completely different program. These cells failed to increase expression of any of the genes usually associated with a typical LCMV Arm CD8+ effector response (Figure 3C). Gzmb transcription, for example, went from 41 normalized counts in naive cells to 375,000 in LCMV Arm, but only to 1,200 counts in TDLNs. Other effector molecules like Tnf, Il2, and Ifng followed a similar trend. Many transcription factors associated with the stem-program like Tcf7 (encoding TCF1) and Bach2 remained higher.37–40 Using GSEA, we found that P14s from TDLNs significantly enrich for the tumor-specific stem-like program, compared with P14s activated in LCMV Arm (Figure 3D). When these data were clustered using K-means, 4 main clusters were found that showed differences in gene expression patterns between P14s from LCMV Arm and TDLNs (Figure S3A). Comparing these clusters with canonical effector signatures from LCMV Arm, Listeria monocytogenes expressing OVA (LM-OVA), and vesicular stomatitis virus expressing OVA (VSV-OVA) infections using GSEA, we found that genes rapidly turned on only during activation in LCMV Arm enriched with genes found in effector CD8+ T cells from these acute infections (Figure S3A). Finally, we performed pathway analysis using reactome gene sets that showed that pathways related to interleukin signaling were increased in P14s from LCMV Arm, while pathways related to TCR signaling were increased in P14s from TDLNs (Figures S3B and S3C).
Figure 3. Tumor-specific CD8+ T cells activated in TDLNs do not acquire an effector transcriptional or epigenetic program.
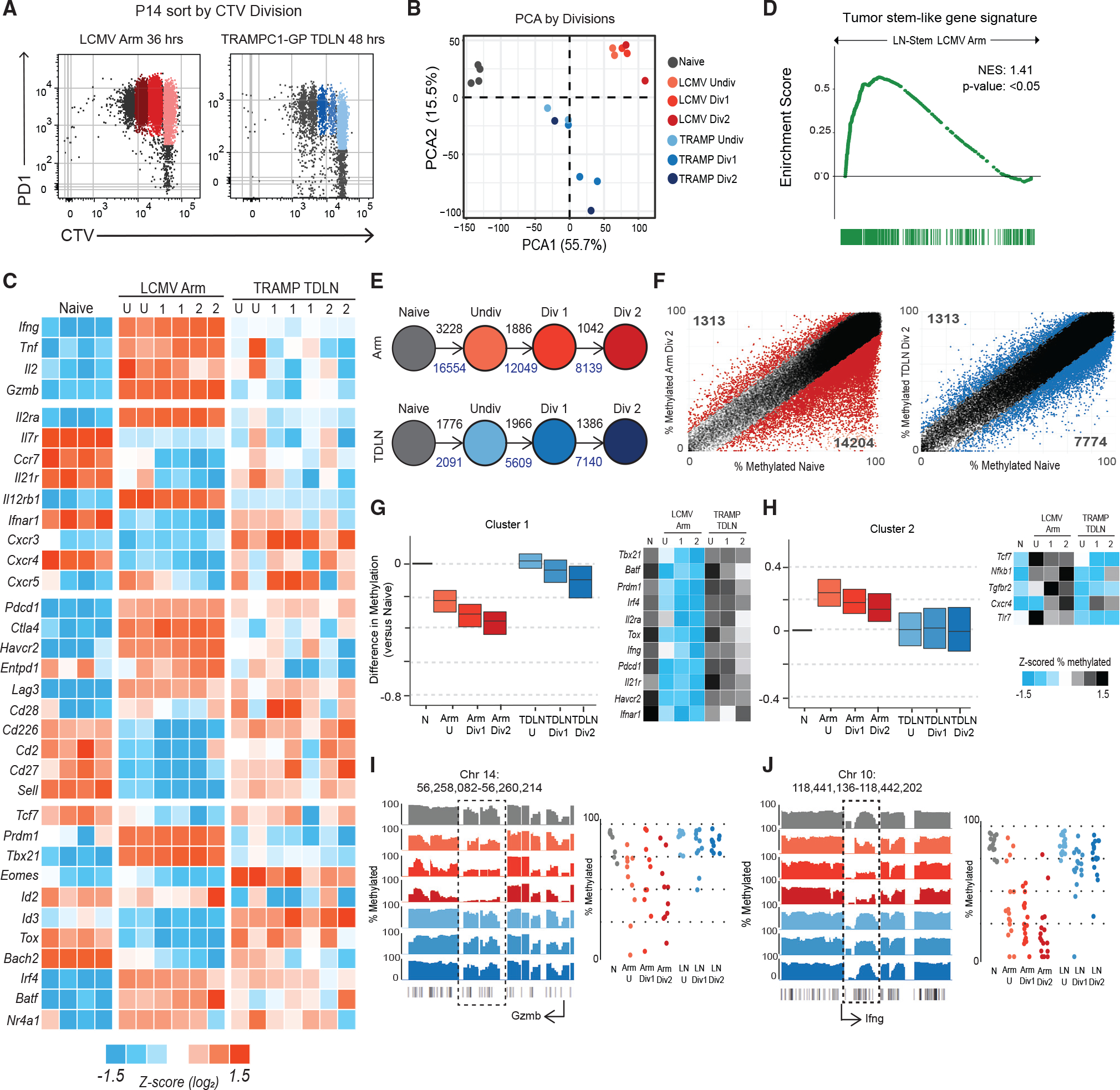
(A) Sorting scheme to isolate P14s by division in LCMV Armstrong spleen and TDLNs from TRAMPC1-GP-bearing mice.
(B) PCA of naive P14s, P14s activated in LCMV Arm, and P14s activated in TRAMPC1-GP TDLNs by division.
(C) Heatmap of Z score log2 expression of selected genes.
(D) GSEA using the gene signature from mouse tumor-specific stem-like CD8+ T cells, compared with P14s from LCMV Arm and TDLN. Enrichment score is plotted.
(E) Schematic of number of regions with at least 15% difference in methylation from naive P14s, as P14s divide; black numbers represent methylated regions, and blue numbers represent de-methylated regions.
(F) Regions of methylation in P14s Div2 from either LCMV Arm or TRAMPC1-GP TDLN plotted versus naive P14s. Colored regions represent at least 15% difference in methylation, compared with naive P14s.
(G and H) Clustering using unbiased K-means of regions demethylated in P14s from LCMV Arm, compared with naive; the same regions were plotted in P14s from TRAMPC1-GP TDLNs. Cluster 1 shown in (G), Cluster 2 shown in (H). Heatmap of Z scored %methylation of select genes is shown.
(I and J) Traces show total methylation from 0% to 100% in regions near Gzmb (G) and Ifng (H). Boxed regions show significantly differentially methylated regions. Dot plots showed methylation of each CpG in boxed region.
When we analyzed the DNA methylation changes of these cells, we found a similar trend. Even before undergoing division, activated P14 cells responding to LCMV Arm acquired many DNA methylation changes, with 3,228 methylated and 16,554 demethylated regions, compared with naive cells. In comparison, P14 T cells activated in TDLNs underwent far fewer epigenetic changes (Figures 3E, 3F,S3D, and Table S4). We analyzed methylation changes further by unbiased K-means clustering and found two dominant clusters and two additional minor clusters (Figures 3G, 3H, and S3E). The first dominant cluster represented regions that were progressively demethylated in P14 cells responding to LCMV Arm but were unchanged in activated P14 cells from TDLNs. This included genes related to effector CD8+ T cells like Prdm1, Il2ra, Havcr2, Gzmb, and Ifng loci (Figures 3G, 3I, and 3J). The second cluster identified the relatively small number of genes that became methylated in P14 cells responding to LCMV Arm but were unchanged in cells isolated from TDLNs. Tcf7 became methylated in LCMV Arm but remained demethylated in TDLNs (Figure 3H). Overall, these data suggest that the early priming conditions in TDLNs imprint an alternative transcriptional and epigenetic program whereby tumor-specific CD8+ T cells do not go through an effector stage in TDLNs but instead retain something similar to the stem-like program identified in other chronic antigen models.
Tumor-specific CD8+ T cells migrate to the tumor in the stem-like state and only acquire effector function within tumors
Our data suggest CD8+ T cells responding to tumors in both humans and mice initiate an alternate program than the classic effector program that occurs in response to acute infections such as LCMV Arm. Based on these observations, we were interested in the kinetics of cells moving from TDLN to eventually establish the stem-like and TD cells in tumors. To investigate, we transferred P14s into either B16-GP or TRAMPC1-GP tumor-bearing mice and analyzed TDLNs and tumors at varying time points post transfer (Figures 4A, S4B, and S4C). In both tumor models, P14 cells were found in TDLNs and maintained the LN-stem phenotype throughout the time course (Figures 4A and 4B) and were even detected 5 weeks after initial transfer (Figure S4A). Furthermore, 4 days post transfer P14 T cells could be detected in the tumors. At this early time point, all the P14s in the tumor were Tcf1+Tim3−, suggesting that after activation in TDLNs they can establish the stem-like CD8+ response within the tumor (Figure 4A). Between days 5 and 10 after transfer, the P14s in the tumor began to differentiate toward a TD phenotype (Tcf1−Tim3+) in both TRAMPC1-GP and B16-GP tumors, with a gradual increase in the relative frequency of TD P14s in the tumor (Figures 4A, 4B, and S4B). P14s within the tumor maintain Ki67 and lose the expression of CD62L (Figure S4D). Together, these data show that CD8+ T cells move from TDLNs to tumor in the stem-like state, and only once they are established in the tumor can they fully differentiate to acquire a TD effector phenotype.
Figure 4. Tumor-specific CD8+ T cells only acquire effector phenotype after migration into the tumor.

(A) Experimental setup for P14 transfers to study kinetics of tumor infiltration. Analysis of P14 phenotype in both TRAMPC1-GP and B16-GP tumors over the time course. P14s gated on CD44+PD1+.
(B) Summaries of total P14s in TRAMPC1-GP TDLNs and tumors over time course.
(C) Representative flow cytometry of intracellular cytokine staining (ICCS) of IFNγ and TNF-α, gated on CD44+PD1+ P14s.
(D) Experimental setup for P14 TDLN re-transfer into tumor-matched mice.
(E) Flow cytometry gating of re-transferred P14s in TDLNs and tumors of congenically mismatched recipients.
(F) Analysis of phenotype of re-transferred P14s.
(G and H) Summary plot of CD62L (G) and GzmB (H) expression of re-transferred P14s. Different shapes (circle/square/triangle) indicate to which of the 3 separate experiments each sample belongs.
(I) Heatmap of Z scored log2 expression of genes in naive P14s, sorted activated P14s D7 post transfer from TRAMPC1-GP TDLNs, endogenous tumor-stem-like CD8s, and endogenous TD CD8s.
(J) PCA of T cell subsets.
(K) Analysis of differentially methylated regions, using previously defined cluster of genes demethylated in P14s from LCMV Arm (cluster 1, Figure 3G). Plot shows cluster 1 differentially methylated regions’ T cells from various conditions.
(L) Specific methylation changes in Gzmb and Ifng. Traces show total methylation from 0% to 100% in regions near both genes. Boxed regions show significantly differentially methylated regions. Dot plots show methylation of each CpG in boxed region. Median and 95% confidence intervals (CIs) are shown. *p < 0.05 determined by Mann-Whitney test.
One possible explanation for the alternate activation program in these tumor-specific CD8+ cells is that they were anergic or dysfunctional in some way. To test this, we first performed intracellular cytokine staining (ICCS) on these cells. Then, 2 weeks after transfer, P14s in both TDLNs and tumor produced high IFNγ, TNF-α, and IL-2 after peptide stimulation (Figure 4C). We next determined if the tumor antigen-specific cells in TDLNs had functional traits of stem-like cells−the ability to regenerate their own population and both the stem and effector populations in the tumor. We generated activated LN-stem CD8+ T cells in TDLNs by inoculating mice with TRAMPC1-GP tumors and 1 × 106 P14 cells 3 weeks later. Subsequently, 2 weeks after the P14 transfer, CD44+PD1+ P14s were sorted from TDLNs and transferred into TRAMPC1-GP tumor-bearing mice, who were treated with intra-tumoral CpG to promote migration of the transferred cells to the tumor (Figure 4E). The LN-stem cells repopulated the TDLNs and were almost entirely Tcf1+ (Figures 4E and 4F). These cells did not express Gzmb or Tim3 but retained expression of CD62L (Figures 4F–4H). In comparison, the transferred cells that migrated to the tumor were found in both the Tcf1+ stem-like and Tcf1−Tim3+ TD states, further demonstrating that effector differentiation only occurs within tumors (Figures 4F–4H). Together, these experiments demonstrate that although CD8+ T cells primed in TDLNs do not undergo normal effector differentiation, they retain functionality and most importantly, retain the ability to regenerate their own population as well as the stem and effector populations within tumors.
The canonical effector transcriptional and epigenetic programs are initiated in tumors
Given that tumor-specific CD8+ T cell differentiation only occurs within the tumor, we were interested in how the transcriptional and epigenetic features of the LN-stem CD8+ T cells evolved within the tumor. We sorted LN-stem P14s from TDLNs 7 days after transfer into established TRAMPC1-GP tumor-bearing mice and sorted endogenous stem-like (CD44+PD1+Tim3−CD127+) and TD (CD44+PD1+Tim3+CD127−) CD8s from tumors. PCA analysis showed that P14s from TDLNs cluster away from both naive and tumor CD8+ T cells (Figure 4J), with 912 genes specifically increased in tumor-stem-like CD8+ T cells and 560 genes increased in LN-stem P14s (Figure S4E). These data support the notion that LN-stem P14s have a distinct transcriptional program, compared with tumor CD8+ T cell subsets. We next performed unbiased K-means clustering and found 5 distinct gene expression clusters. These clusters were used to perform GSEA on previously described effector CD8+ T cell gene sets. Gene clusters that were increased in tumor subsets were enriched for effector CD8+ T cell signatures, demonstrating the acquisition of an effector program of CD8+ T cells within the tumor (Figure S4F). Pathway analysis indicated enrichment of co-stimulation, IFN signaling, and chemokine signaling in the tumor-stem population when compared with LN-stems, suggesting potential signals that might drive the differentiation in this location (Figure S4G).
We next performed whole-genome DNA methylation analysis on LN-stem P14s from TDLNs at day 7 post transfer and on endogenous tumor stem and TD subsets. We used the previously defined cluster of genes that were demethylated in P14s activated in LCMV Arm (Figure 3H) to compare both TDLN D7 P14s and tumor-infiltrating subsets. P14 cells in TDLNs still had not epigenetically changed most of these loci. In comparison, once these cells reached the tumor, the stem and TD CD8+ T cells showed more demethylation of genes associated with effector function and differentiation (Figures 4K, S4H, and S4I), such as Gzmb and Ifng loci (Figure 4L). These transcriptional and epigenetic data demonstrate that LN-stem CD8s maintain an activated but undifferentiated state. Upon migration into the tumor, these cells establish the stem-like CD8+ tumor subset and can further differentiate into TD CD8+ TILs to acquire the effector program.
Co-stimulatory molecules and cytokines in the tumor microenvironment contribute to stem to effector CD8+ differentiation
CD8+ T cells acquiring an effector-like epigenetic and transcriptional program in the tumor implies something within the tumor microenvironment must drive this phenotypic change. In our previous work, we had found that stem-like CD8+ T cells in tumors always reside near areas of high antigen-presenting cell (APC) density.1 In addition, transcriptional analysis above suggested that pathways related to co-stimulation and cytokine signaling were most disparate between tumor-stem-like cells and those in the TDLNs. Based on these data, we hypothesized that APCs in the tumor might provide these important signals to cause stem-like CD8+ T cells to differentiate into TD CD8+ TILs. To investigate this idea, we first characterized the APCs from naive lymph nodes, TDLNs, and TRAMPC1-GP tumors, using scRNA-seq. Seven clusters were identified based on expression of lineage-specific markers (Figures 5A, 5B, and S5A). This clustering designation was confirmed by comparison with known myeloid cell signatures41 (Figure S5B).
Figure 5. Co-stimulation from antigen-presenting cells promotes tumor-specific CD8+ T cell differentiation.
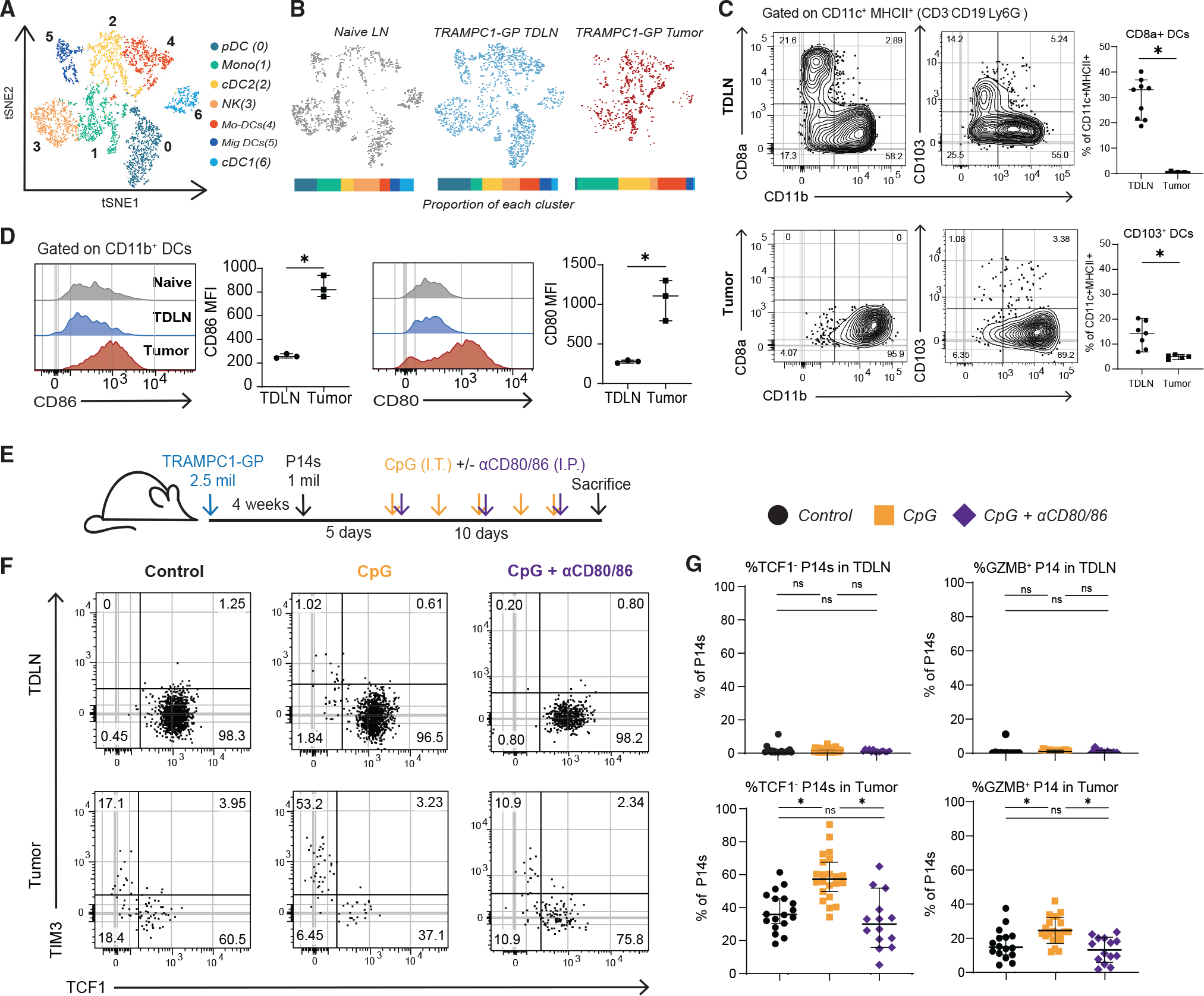
(A) tSNE clustering of antigen-presenting cells (APCs) sorted from naive LN, TDLN, and tumors of 5-week TRAMPC1-GP-bearing mice.
(B) tSNE clustering split by tissue origin, with proportions of each cluster from each tissue represented in a bar graph; each APC population is significantly different between TDLN and tumor.
(C) Flow cytometry analysis of DC subsets from TDLN and tumor from TRAMPC1-GP-bearing mice.
(D) Flow cytometry analysis of co-stimulatory molecules in CD11b+ DCs in naive LNs, TDLNs, and tumors from TRAMPC1-GP-bearing mice.
(E) Experimental setup. P14s were transferred into 4-week TRAMPC1-GP tumor-bearing mice, and 5 days post transfer, they were treated with CpG and/or CD80/CD86 blocking antibodies for 10 days.
(F and G) Phenotype of transferred PD1+ CD44+ P14s 15 days post transfer in the TDLNs and tumors of all treatment groups (F) and summaries of P14 phenotype (G). Median and 95% confidence intervals (CIs) are shown. *p < 0.05 determined by Mann-Whitney test.
The primary DC populations present in the tumor were cDC2s and mo-DCs, while all APC subsets were found in the TDLNs (Figure 5B). We further characterized these DC populations by flow cytometry, using CD11b as a combined cDC2 and mo-DC marker.42 As in the scRNA-seq analysis, in TDLNs both cDC1s (CD8a+) and CD11b+ DCs were found, while in the tumor only CD11b+ DCs were present (Figures 5C and S5C). We also analyzed migratory DCs by using CD103 and CD40 and found a small percentage in the tumor, mostly within the CD11b+ subset (Figures 5C and S5D). We further characterized CD11b+ DCs and found higher expressions of CD80 and CD86 in the tumor when compared with the TDLN (Figure 5D). These data indicate the primary DCs within the tumor are CD11b+ cDC2/mo-DCs that have a high expression of co-stimulatory molecules.
Based on the observation that DCs in tumors expressed more co-stimulatory molecules and predicted co-stimulatory pathways enriched in tumor-stem-like CD8+ T cells, we hypothesized that these signals might be the cause of differentiation from a stem-like CD8+ T cell to a TD CD8+. To test this, we transferred P14s into TRAMPC1-GP tumor-bearing mice, and 5 days post transfertreated mice with 10 μg CpG intratumorally (i.t.) (Figure 5E). CpG was used to activate APCs and determine whether increased APC activation can alter CD8+ T cell differentiation within the tumor. After CpG treatment, P14s in TDLNs maintained an LN-stem phenotype (Tcf1+); meanwhile, in the tumor, there were more differentiated (Tcf1−) P14s and Gzmb+ P14s (Figures 5F and 5G). To test if co-stimulatory molecules were involved in this process of differentiation, we blocked CD80 and CD86 during CpG treatment (Figure 5E). Blockade of these critical molecules returned differentiation and numbers of P14s to a similar amount as for control mice, and the frequency of Gzmb+ P14s decreased compared with CpG alone, suggesting co-stimulation is one of the important signals for stem to effector differentiation in the tumor (Figures 5G, S5I, and S5J).
We were next interested to see how cytokines contributed to this process. In comparison to co-stimulation blockade, blocking IL-12 or type I IFN had no significant impact on the proportion of Tcf1− P14s within the tumor or TDLNs, indicating that these cytokines are not the main drivers of CD8+ T cell differentiation with CpG treatment (Figures S5E–S5H). However, IFNAR blockade significantly decreased Gzmb expression by Tcf1− P14s, suggesting this cytokine is important for effector function (Figure S5H). Together, these data indicate co-stimulation in the tumor microenvironment is one of the major contributors to the differentiation of Tcf1+ stem-like cells to the effector state in the tumor.
Co-stimulation in human tumors correlates with the presence of TD CD8+ cells
Based on what we had observed in mouse models, we wanted to know if co-stimulatory molecule expression on DCs in human tumors might serve a similar function of regulating the stem to TD transition. We first performed scRNA-seq to broadly determine what APC populations infiltrate human kidney tumors. Four clusters of APCs were identified based on differences in gene expression using tSNE clustering: monocytes, DCs, NK cells, and B cells (Figure 6A). Using published human APC cell signatures and previously defined markers, we confirmed our clustering of APCs from human kidney tumors (Figures 6B, 6C, and S6A).41,43–50 To expand this characterization, we analyzed 152 kidney tumors and 34 prostate tumors by flow cytometry for the DC populations we identified by scRNA-seq. We defined cDC2s as CD11c+CD1c+ cells, while cDC1s and mo-DCs were defined as CD11c+CD1c− and expressed CD141 and CD11b, respectively (Figure 6D). In both kidney and prostate tumors, cDC2s and mo-DCs were the dominant DC populations (Figures 6D and 6E). All DC subsets had high expression of FLT3 as well as PDL1, further validating that they are activated DCs, as others have reported.43 Mo-DCS were the only DC subset expressing CD14, corresponding to derivation from the monocyte lineage (Figure S6B). The presence of all DC subsets correlated with higher CD8+ T cell infiltration (Figures S6C and S6D).
Figure 6. Co-stimulation from dominant CDC2s and mo-DCs in human tumors predicts CD8+ T cell infiltration.
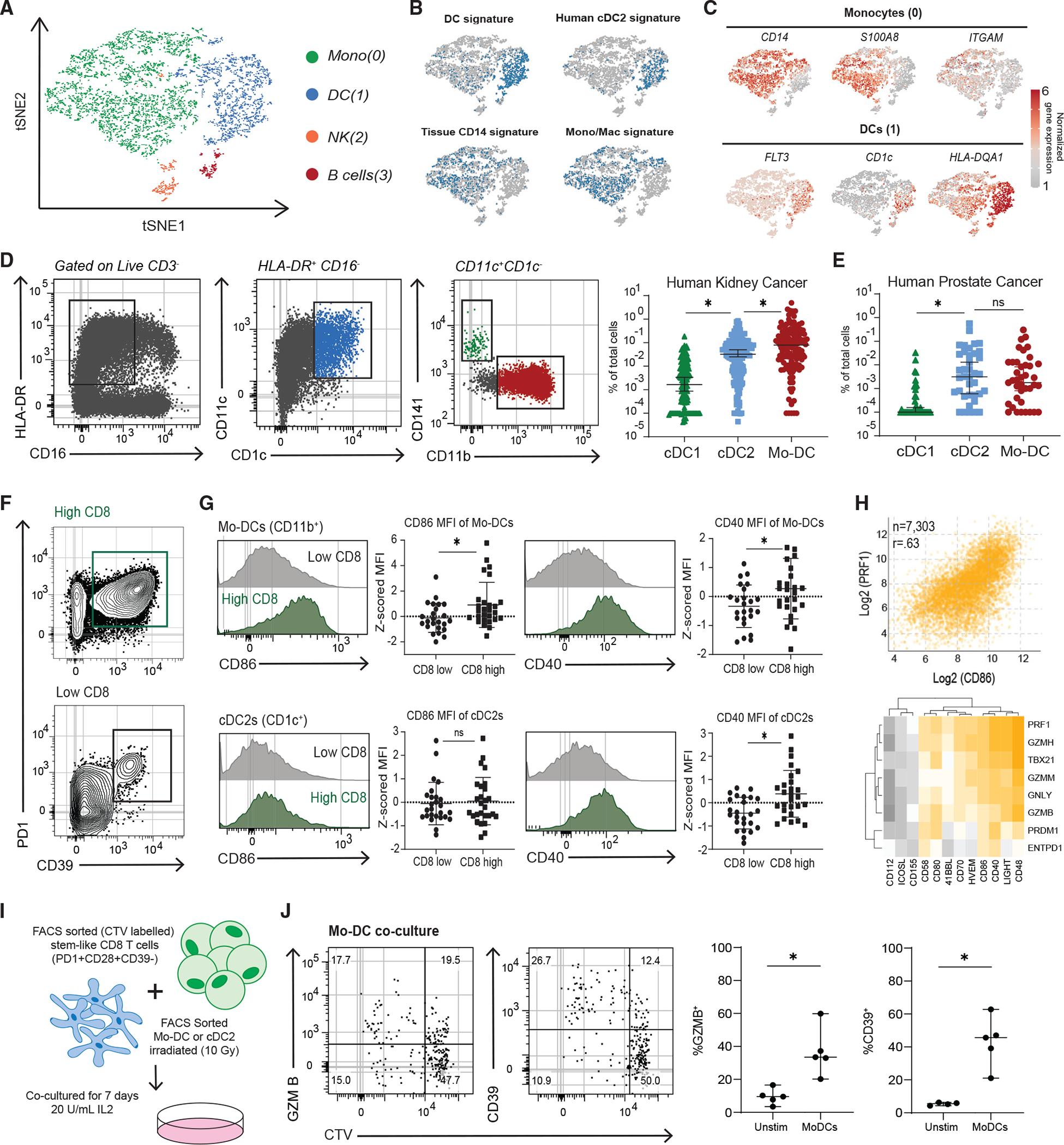
(A) tSNE clustering of single-cell RNA-seq of APCs from two patients’ kidney tumor samples.
(B) VISION analysis of gene signatures associated with APC subsets from human tissues. tSNE plots show the top quintile of cells enriched for the signature highlighted in blue.
(C) Normalized gene expressions of selected genes that define the monocyte and DC cluster are shown.
(D) Flow cytometry analysis of 152 kidney tumors, gating to distinguish three DC subsets.
(E) Summary of three DC subsets in 34 prostate tumors.
(F) Representative flow cytometry plots of high and low CD8+-infiltrated kidney tumors. PD1 and CD39 expression shown to denote TD CD8+ TILS.
(G) Expression of co-stimulatory molecules on mo-DCs and cDC2s in kidney tumors from high and low CD8+ T cell tumors.
(H) TCGA data from top 8 represented tumor types. Correlation of CD86 expression versus perforin (PRF1) expression shown. Heatmap of correlations between other co-stimulatory molecules versus effector and cytotoxic genes.
(I) Experimental layout to test the capacity of different DC subsets from tumors to induce differentiation of autologous tumor-stem-like CD8+ T cells.
(J) Representative plots showing CTV dilution and phenotype after 7 days of stem-like CD8+ T cell co-culture with irradiated mo-DCs. Medians and 95% CIs are represented. *p < 0.05 determined by Mann-Whitney test.
We were next interested in expression of co-stimulatory molecules by DCs in human kidney tumors and how it might correlate with generating TD CD8+ TILs. In our prior work, we found that patients with higher total CD8+ T cell infiltration in tumors also have more TD CD8+ TILs within the tumor.1 We therefore split patients into high and low CD8+ infiltration, based on the median CD8+ infiltration of the patients analyzed, to investigate how co-stimulation might correlate with CD8+ differentiation in the tumor (Figure 6F). Mo-DCs from tumors with higher total CD8+ T cell infiltration expressed higher CD86 and CD40 (Figure 6G), and cDC2s expressed more CD40 in tumors with higher CD8+ T cell infiltration (Figure 6G). By contrast, expression of co-stimulatory molecules on cDC1s was not significantly different between low and high CD8+ T cell-infiltrated tumors (Figure S6E). We next used the TCGA database to determine how CD8+ effector molecules might correlate with co-stimulation in the tumor across the top 8 most represented tumor types. We compared expression of various co-stimulatory molecules with cytotoxic genes that are typically only expressed by TD CD8s in tumors; we found that CD80, CD86, and CD40 as well as CD48, CD58, and LIGHT expression correlated with expression of the cytotoxic genes (Figures 6H and S6F). Overall, in mouse and human tumors, cDC2s and mo-DCs make up most of the DCs, and expression of co-stimulatory molecules correlates with CD8+ T cell differentiation.
To determine if these major populations found in the tumor had the functional capacity to cause differentiation of stem-like CD8s, mo-DCs and cDC2s were sorted from tumors, as shown in Figure 6D, and co-cultured with patient-matched, CTV-labeled tumor-stem-like CD8+ cells (Figure 6I). In all patients analyzed, both mo-DCs and cDC2s were capable of inducing proliferation of the stem-like CD8s (Figures 6J, S6G, and S6H). Both DC subsets were also capable of inducing differentiation in stem-like CD8+ T cells, as shown by an increase in GZMB and CD39 (Figures 6J and S6H). These data demonstrate the capacity of mo-DCs and, to an extent, cDC2s to induce stem-like CD8+ T cell differentiation.
Co-stimulation is required for human stem-like CD8+ T cells to differentiate to the effector state
These data suggest that co-stimulation is a major signal that drives T cells to acquire cytotoxic function in tumors. To functionally test this, we sorted CTV-labeled stem-like TILs (PD1+ CD39−CD28+) from human kidney tumors and stimulated them in vitro with either anti-CD3 alone, anti-CD3/CD28, or anti-CD3/CD28/CD2 beads (Figure 7A). After 5 days in vitro, stem-like CD8+ T cells that received only TCR stimulation (anti-CD3) did not proliferate and did not acquire a TD CD8 phenotype (Figures 7B and 7C). In comparison, stem-like CD8s stimulated with anti-CD3/CD28 proliferated and increased proteins related to effector differentiation (Figures 7B and 7C). When we provided additional co-stimulation with both anti-CD28 and anti-CD2 beads, more stem-like cells divided and differentiated into a TD, effector-like state (Figures 7B and 7C). These data illustrate that co-stimulatory molecules are required to induce stem-like CD8+ T cell differentiation and that TCR signaling alone is not sufficient. Furthermore, and in line with the correlative data we collected in human tumors (Figures 6F–6H), more co-stimulation accelerated this process.
Figure 7. Co-stimulation is necessary for the differentiation of human tumor-stem-like CD8+ T cells.
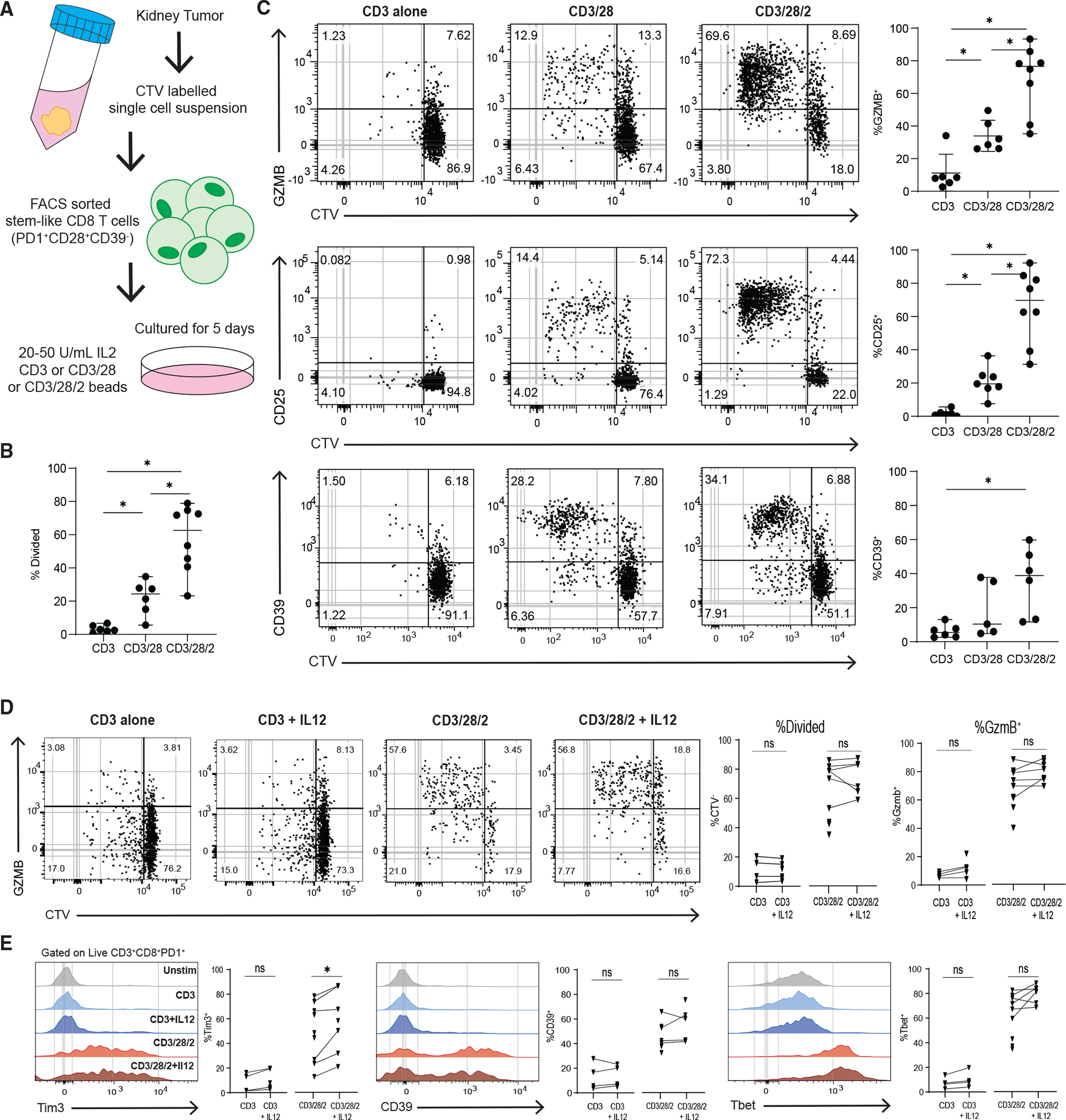
(A) Experimental layout to test the requirement of co-stimulation to differentiate sorted tumor-stem-like CD8+ T cells.
(B) Summary plot shows the proportion of divided CD8+ T cells in each in vitro condition, based on CTV dilution.
(C) Representative plots showing CTV dilution and phenotype after 5 days of culture in each condition.
(D) Representative plots showing CTV dilution and expression of GZMB of control in vitro stimulations or with added IL-12.
(E) Histograms showing expression of TIM3, CD39, and Tbet for all conditions. Medians and 95% CIs are represented. *p < 0.05 determined by Mann-Whitney test. *p < 0.05 determined by Wilcoxon test when sufficient paired samples were analyzed, shown as connected by a line.
Since signal 3 cytokines are an important mechanism regulating CD8+ T cell function and the immune response to cancer, we assessed the contribution of these cytokines in stem to effector differentiation.51,52 We found that IL-12 with TCR alone (anti-CD3) was not sufficient to cause any division or increase in GZMB, TIM3, and TBET expression of the stem-like CD8+ T cells (Figures 7D and 7E). Similarly, when IL-12 was added to anti-CD3/28/2, there was no increase in proliferation or differentiation, with a minor increase in Tim3 expression (Figures 7D and 7E). When IL-12 was added to a lower co-stimulatory condition, anti-CD3/28 stimulation, there were still no differences in proliferation or GZMB expression, but both Tim3 and TBET underwent a small but significant increase (Figures S7A and S7B). The addition of IL-12 to tumor TD (TIM3+CD39+) stimulations did not induce proliferation or alter the expression of TIM3 (Figure S7C). We also analyzed the effects of IL-12 on naive CD8+ T cells stimulated with anti-CD3 or anti-CD3/28/2 (Figure S7D). Similarly to stem-like CD8+ T cells, naive CD8+ T cells do not proliferate with CD3 stimulation alone or with the addition of IL-12, but there was an increase in GZMB expression when naive cells were stimulated with anti-CD3/28/2 with IL-12 (Figures S7D and S7E). We next analyzed the amount of IFNγ in supernatants of in vitro cultures of cells stimulated in these conditions and found that there was no significant increase in IFNγ with the addition of IL-12 (Figure S7F). We next tested the effect of type I IFN on stem-like CD8+ T cell differentiation by adding IFNb to the anti-CD3/28/2 stimulations. Unexpectedly, we found that addition of IFNb significantly reduced the proliferation of stem-like CD8+ T cells and TBET expression, but not GZMB, CD39, or TIM3 expression (Figures S7G–S7I).
From these data, we conclude that for stem-like CD8+ T cells to undergo differentiation to the effector state, both TCR stimulation and co-stimulation are required. In addition, signal 3 cytokines like type I IFNs and IL-12 may contribute to regulating some genes associated with effector function. Together, these data highlight co-stimulation as a critical signal in the tumor microenvironment that regulate anti-tumor CD8+ T cell differentiation and acquisition of an effector phenotype.
DISCUSSION
The goal of this work was to investigate how CD8+ T cells differentiate in response to tumor antigens. We found that unlike canonical CD8+ T cell activation seen against viral infections where T cells rapidly acquire effector function, tumor-specific CD8+ T cells proliferate in TDLNs but fail to express effector molecules. In human TDLNs, these cells could have been in this state for years. These undifferentiated cells instead turn on co-stimulatory receptors and several chemokine receptors and slowly migrate to the tumor. Only once in the tumor do these cells acquire a transcriptional and epigenetic program that resembles effector cells. As others have recently described, the majority of activated CD8+ T cells in TDLNs maintain a stem-like phenotype.53,54 The differentiation in the tumor from the stem to effector state required both TCR signaling and co-stimulation, which are unexpected requirements for T cells to need again several days after their initial activation. Based on these observations, we propose the 2-step activation model as the best explanation for how CD8+ T cells respond to cancer. This model has several implications for how we understand the T cell response to cancer and how we develop future therapies.
The most important point that this work demonstrates is that the signals that drive differentiation of T cells to a cytotoxic state in cancer do not occur during the initial activation phase, as we and many others have found in viral infections.12–14,55,56 Notably, the canonical 3 signals that are described in viral infections are required, but only once these cells have previously been activated in TDLNs and migrated to the tumor.56–58 This finding emphasizes a critical role of the tumor microenvironment to provide the correct signals to cause this stem to effector differentiation, and it points to an obvious defect that can occur during the immune response to cancer—the breakdown of these signals in the tumor microenvironment, resulting in a loss of cytotoxic CD8+ response to the cancer.
The two-step model also suggests a hypothesis about why anti-PD1 therapy has sporadic success in patients. Tcf1+ CD8+ T cells are the primary cells that proliferate in response to anti-PD1 therapy, and CD28 is necessary for CD8+ T cells to respond to PD1 blockade.19,21,28,59,60 LN-stem CD8+ T cells can respond to PD1 blockade, and they promote anti-tumor immunity.61 Given our data that stem-like CD8+ T cells need co-stimulation to proliferate and differentiate, PD1 blockade alone may not be sufficient to drive continuous effector differentiation. Our data are in line with this hypothesis, where patients with low T cell infiltration had far lower co-stimulatory molecules in their tumors, and low T cell infiltration prior to PD1 blockade predicts a poor response to therapy.27,62,63 Our model implies that in situations like these, treatment with drugs like TLR ligands directed to the tumor could help provide the second signal needed to the stem-like CD8s.
Finally, the mechanisms that instruct T cells to undertake this alternate program are still to be determined. We found that addition of an excessive amount of antigen could not force the cells to acquire effector function in TDLNs, suggesting that they were not lacking TCR signal. Given the critical role of co-stimulation in the tumor, we think an obvious answer is that in the tumor, the danger signals needed to activate dendritic cells are greatly reduced. The expected result from strong TCR signaling but lack of co-stimulation would be T cells entering an anergic state.64,65 However, we find that these cells persist for at least 5 weeks in TDLNs in this state. In humans, the cells have presumably been in the TDLNs for as long as the patient has had cancer, yet they retain proliferative capacity, so it is clearly a different process from anergy. Our ongoing work aims to further understand the signals that cause T cells to undergo this alternate program and, most importantly, to determine if this is an optimal or erroneous response to the tumor.
Limitations of the study
In this study, we only analyzed TDLNs from human kidney and prostate cancer and 3 sub-cutaneous mouse models. Other cancer types and mouse models need to be investigated to better understand the generalizability of our observations. Additionally, although we found minor effects of in vivo blockade of IL-12 and type I IFN, our results do not rule out that the addition of these cytokines in vivo could regulate CD8+ T cell response, and more investigation of how different cytokines regulate this differentiation process is required.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests should be directed to Haydn Kissick.
Materials availability
Materials generated by this study are available upon request.
Data and code availability
Raw fastq files and associated RNA, whole genome bisulphite sequencing, and single cell RNA sequencing have been uploaded to the NCBI Gene Expression Omnibus (GEO) database under identifier GSE216731. Custom python and R scripts for TCR, RNA, and DNA analysis are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
Patients were recruited in accordance with an approved IRB protocol, and all patients provided informed consent.
Mice
All mice were used in accordance with National Institutes of Health and the Emory University Institutional Animal Care and Use Committee guidelines. C57BL/6J and BALB/CJ mice were purchased from Jackson laboratories at 7–8 weeks. LCMV DbGP33-specific TCR transgenic P14 were bred in house and backcrossed to CD45.1+ (B6.SJL-Ptprca Pepcb/BoyJ) purchased from Jackson laboratories. Male mice were used for TRAMPC1/TRAMPC1-GP, since it is a prostate cancer model. Female C57BL/6J mice were used for B16-GP and female BALB/CJ for RENCA-HA experiments.
Tumor cell lines
TRAMPC1 cells expressing full length LCMV GP were made using a lentiviral transduction. The codon optimized LCMV GP was cloned into the pLVX-IRES-ZsGreen plasmid. Lentivirus was made by transfecting 293 T cells and harvested. TRAMPC1 cells were infected with concentrated lentivirus. After several days of infection TRAMPC1-GP ZSgreen+ cells were sorted on the Becton Dickinson FACS Aria II Cell Sorter. Cells were re-sorted to achieve 99% purity, and to check for stable TRAMPC1-GP transduction. RENCA cells were transduced using the same protocol except with influenza HA, instead of LCMV GP, to produce the RENCA-HA cell line. B16F10-GP were previously made using the same pLVZ-IRES-GP-Zsgreen plasmid as previously described.68 All cells were cultured at 37 C in 5% Co2, in their respective media.
METHOD DETAILS
Human sample collection, processing, and flow staining
Tumor samples were collected after patients underwent partial or radical nephrectomy, prostatectomy or undergoing transurethral resection of a bladder tumor (TURBT). Tumor draining lymph nodes were collected directly after resection. Samples were collected immediately after tumor resection in Hank’s Balanced Salt Solution. The samples were then processed by getting cut into small pieces, digested with a Liberase enzyme cocktail, and then homogenized using a MACS Dissociator, digested tumor was washed through a 70 um filter to get a single cell suspension. Red blood cells were lysed using RBC ACK Lysis buffer, fat was removed using a 44% Percoll/RPMI gradient, and samples were frozen in freezing media (FBS+10% DMSO) at −80C.
Single cell suspensions from processed human tumor samples were stained with antibodies listed in STAR Methods. Live/dead staining was done using fixable near-IR or aqua dead cell staining kit (Invitrogen). Cells were permed using the FOXP3 Fixation/Permeabilization kit (eBioscience) for 45 minutes with fixation/permeabilization buffer at 4C and stained with intracellular antibodies in permeabilization buffer for 30 mins at 4C. Samples were acquired on Becton Dickinson LSRII and Symphony instruments and analyzed using Flowjo (v10).
In vitro stimulation and co-cultures
Single cell suspension from fresh and frozen human tumor samples were stained with CellTrace violet (CTV) (Thermo), using 1 ul of CTV per 10 million cells. CTV labelled cell suspension were sorted on Live CD8+ PD1+CD39−Tim3−CD28+ for stem-like CD8 T cells, and Live CD8+PD1+CD39+ or Tim3+ for TD CD8s, on the Becton Dickinson FACS Aria II Cell Sorter. Sorted CD8 T cells were cultured in U-bottom plates in T cell media (RPMI. 10% FBS, 1% Pen-Strep, 1% L-glutamine, 1% sodium pyruvate, 1% non-essential amino acids, 0.0005% 2-Mercaptoethanol) and 20–50 U/mL of IL2 (Peprotech). Stimulation assays were performed using anti-CD3, anti-CD3/28, anti-CD3/28/2 beads at a ratio of 1 bead for 2 CD8 T cells. For in-vitro stimulations with cytokines, either 10ng/ml of human Il12 or human IFNb (Peprotech) were added to cultures D0 of culture. Samples were analyzed by flow cytometry for proliferation and expression of various proteins are 5–7 days after stimulation. IFN-gamma production was measured by taking supernatants at day 5 post stimulation. Using the high sensitivity Human IFN-gamma ELISA kit, we measured IFN-gamma production above the limit of detection of 0.16pg/mL (Thermo Fisher).
For DC co-culture experiments, DCs were sorted as shown in Figure 6F. DC populations were irradiated with 10 Gy before being put in culture. Co-cultures were plated at 2 DCs per 1 T cell, as much as possible based on cell number recovered, with CTV labelled stem-like CD8 T cells sorted from matching tumors. Samples were analyzed 7 days post co-culture on the Beckton Dickinson Symphony.
Human CD8 T cell RNA-seq and TCR analysis
Single cell suspensions from human tumor and TDLN samples were stained with antibodies, using near-IR live/dead cell staining kit to discern live cells. Populations were sorted on the Becton Dickinson FACS Aria II Cell Sorter, for tumor stem-like CD8s; Live CD8+ PD1+CD39−Tim3−CD28+, for TD tumor CD8s; Live CD8+PD1+CD39+ or Tim3+, and LN-stems; Live CD8+PD1+CD45RA−CD28+CD39−. DNA and RNA were isolated using the Qiagen RNA/DNA micro kit. RNA was used for TCR analysis and bulk RNA-seq. TCR libraries were made using the SMARTer Human TCR a/b Profiling Kit (Takara Biosciences) following the manufacturer’s instructions. TCR libraries were sequenced using a Miseq. Complete T cell receptor V(D)J clonotype sequences were obtained using MIXCR. Aligned CDR3 regions and frequency of clonotypes were exported for further evaluation. In post processing steps, TCR analysis was performed using the immunarch R package (v 0.6.5) and custom R scripts. TCR beta chains were used for population overlap and diversity analysis, given its uniqueness to each cell and higher combinatorial potential. CDR3 amino acid sequences from sorted human TILS (PD1+CD28+CD39- and PD1+CD39+) and LN PD1+ CD8 T cells were cross referenced with known published iNKT and MAIT cell TCRs.69–72 The CDR3 regions used for the overlap of LN activated CD8s with CD28+ stem-like TILS did not contain CDR3 regions that match iNKT or MAIT cell TCRs.
RNA libraries were prepared using the clontech SMART-Seq V4 and sequenced HiSeq1000. Data was normalized and differentially expressed genes were determined using DeSeq.67 RNA-seq analysis was done using custom R scripts. Briefly, gene expression patterns were determined using K means clustering based on the fold change from naïve generated by Deseq. Normalized gene counts, heatmaps and volcano plots were generated using ggplot2 R package and excel.
Whole genome bisulfite sequencing (WGBS)
DNA was isolated using the AllPrep DNA/RNA Mini kit (Qiagen) and sonicated to generate random 300 to 500-bp fragments. DNA was end-repaired and A-tailed using the Hyper Prep Kit (KAPA Biosystems) following the manufacturer’s protocol. Sequencing adapters that contained fully methylated cytosine residues (Integrated DNA Technologies) were ligated using the Hyper Prep Kit (KAPA Biosystems). Adapter-ligated DNA was bisulfite converted using the EpiTect Bisulfite Kit (Qiagen), with a denaturation time of 10 min. Final libraries were PCR amplified 8–11 times using Tru-seq (Illunima)-compatible custom index primers, as previously described in Barwick et al.73, and HiFi Uracil (KAPA Biosystems). The resulting WGBS libraries were quality checked by Bioanalyzer, pooled at equimolar ratios and sequenced on a NovaSeq S4.
WGBS analysis
Sequenced data was aligned to the human or mouse genome using Hisat2 and analyzed using custom R and Python scripts. Fisher’s exact test was used to define regions of statistically (p value less that 1×10−4) differentially methylated CpG motifs. Continuous regions of differentially methylated CpGs were identified by finding regions where at least 6 out of 10 CpGs in a continuous stretch were differentially methylated. These regions were then collapsed and analyzed as single ‘differentially methylated regions’ (DMRs).
10x sorting for Human APC and Mouse APC and analysis
Single cell suspensions were stained and sorted on the Beckton Dickinson FACS Aria II Cell Sorter. For mouse single cell RNAseq, APCs were sorted on Live,CD3−CD19−Ly6G−I-A/I-E+ from naïve LN, TDLN, and Tumor. For human single cell RNAseq, APC were sorted Live,CD45+CD3−HLA-DR+ from kidney tumors. Single cell RNAseq libraries were made using the Chromium single cell 5’ Library and Gel Bead Kit (10x Genomics). Sorted cells were sorted and captured into the Gel Beads-in-emulsion (GEMs). After the reverse transcription GEMs were disrupted and cDNA was isolated and pooled. The barcoded cDNA was fragmented, end repair and A-tailing was done, followed by sample index PCR. The purified libraries were sequenced to 50,000 reads/cell on a HisSeq300 (Illumina) with 26 cycles for read 1, 8 cycles for index (i7) and 91 cycles for read2.
Cellranger v3.1 was used to align, filter, count the barcodes and unique molecular identifiers (UMI). Data was then analyzed using Seurat v3.0.74 Briefly, cells with less than 5% mitochondrial genes were used. Cells that expressed less than 200 genes or more than 2000 were excluded from analysis. Raw counts were then normalized for each UMI based on total expression, scaled by multiplying by 10,000 and then log transformed. Variable genes were determined based on average expression and dispersion, then used to perform principal component (PCA) analysis. Selected PCAs were used to generate clusters and t-SNE plots. Heatmaps were generated using scaled expression data of marker genes, using the FindAllMarkers function in Seurat. Normalized gene expression data was also shown as feature plots. Gene set scoring was performed using VISION R package V2.1. Gene sets were used from Heidkamp et al., Banchereau et al., and Carpentier et al., for human APC analysis, and from Miller et al., and Helft et al. for mouse APC analysis. 28,48–50,75
Tumor cell lines and injection
All cells were cultured at 37 C in 5% Co2, in their respective media. For tumor inoculation all cell lines were taken out of culture using 0.05% Trypsin, washed once with respective media and 2x with PBS. Cells were resuspended in PBS at different concentrations based on cell line for a 100 ul injection volume. 2.5 million TRAMPC1-GP/TRAMPC1, 250k-500k B16-GP or 250k RENCA-HA cells were injected subcutaneously. Mice were monitored and sacrificed in accordance with Emory University Institutional Animal Care and Use Committee guidelines.
Mouse tissue processing and flow staining
Tumor, axillary and inguinal TDLNs, and spleens were harvested and digested in Collagenase D (2 mg/mL) for 30 minutes in a shaker at 37C. Digested tissue was washed through a 70um filter using wash buffer (RPMI + 2% FBS) to produce a single cell suspension. Tumor and spleen were RBC ACK lysed and resuspended in FACS wash (PBS + 2%FBS + EDTA). Tumor samples underwent an additional step of a 44% Percoll/RPMI gradient to get rid of excess fat before staining. Mouse tissues were stained with antibodies listed in the antibody table. Extracellular staining was done in FACS wash for 30 min at 4C using antibodies described in Table 1. For intracellular staining the FOXP3 fixation/permeabilization kit (eBiosciences) was used. Cells were fixed in fixation/permeabilization buffer for 1–4 hours at 4C, and then stained with intracellular antibodies in permeabilization buffer for 30 min at 4C. Data was acquired on the Beckton Dickinson LSRII and analyzed in Flowjo (v10).
Intracellular cytokine staining of P14s
For intracellular cytokine analysis, after processing TDLNs and tumors, 106 cells were cultured with GP33 peptide (0.2 ug/mL) with Golgi-plug and Golgi-stop (BD) for 5 hours at 37C in RPMI with 10% FBS. Extracellular staining was done as described above, followed by intracellular staining using the Cytofix/Cytoperm kit (BD). Briefly, after a 20 min permeabilization at 4C, staining of IFNg, TNFa, and IL2 was done in permeabilization buffer for 30 min at 4C. Samples were acquired on Becton Dickinson LSRII and Symphony instruments and analyzed using Flowjo (v10).
Adoptive transfers and LCMV Armstrong infection
Spleens from CD45.1+ LCMV DbGP33-specific TCR transgenic P14 mice were isolated. Cells were washed through a 70 um filter with wash buffer, RBC ACK lysed and resuspended in FACS wash. CD8 T cells were isolated using EasySep CD8 negative selection kit (StemCell). 1 mil P14 CD8 T cells were transferred intravenously directly after isolation, into either naïve CD45.1+ recipients or tumor-bearing CD45.2+ mice. For LCMV infection, 2.5 mil PFU of LCMV Armstrong was injected intravenously after P14 CD8 T cell transfer. For proliferation studies P14 CD8 T cells were CTV labelled at 1 ul of CTV per 10 million cells, and 1 million CTV labelled P14s were transferred intravenously.
For LN-stem re-transfer experiments, P14s were transferred into 2 to 3-week TRAMPC1-GP tumor bearing mice. Two weeks later CD44+PD1+ P14s were sorted from TDLN and re-transferred into congenically mismatched 2-week TRAMPC1-GP tumor bearing mice. Recipient mice were treated with CpG, intratumorally every other day for 2.5 weeks then sacrificed and TDLNs and Tumors were analyzed. Samples with less than 5 P14s acquired or when P14s were less than 0.00015% of total cells were counted as below limit of detection (LOD) and were not used for phenotype analysis.
In vivo treatments (CpG, anti-CD80/CD86, anti-IL12, anti-IFNAR)
For CpG treatments, 10ug was injected intra-tumorally (I.T.) at week 4 of tumor growth. CpG was injected every other day for 10 days. For combination CpG and CD80/86 blockade, anti-CD80 (B7.1) (200 ug) and anti-CD86 (B7.2) (200ug) antibodies were injected every 3 days IP, as previously described.59 For cytokine blocking experiments anti-IL12p40 (250ug) or anti-IFNAR (250ug) antibodies were injected every other day IP, as previously described.76,77
Mouse T cell sorting for RNA and DNA methylation
Single cell suspension from processed mouse tissues were stained with antibodies as described in Key Resources. Tumor and TDLN samples were sorted on the Becton Dickinson FACS Aria II Cell Sorter. For RNA-seq and DNA methylation analysis, RNA and DNA were extracted using the Qiagen RNA/DNA micro Kit. RNA libraries were prepared and sequenced as previously described. Data was normalized and differentially expressed genes were determined using DeSeq. Data was then analyzed using custom R scripts and visualized using ggplot2 R package, as previously described. For Gene Set Enrichment Analysis (GSEA) each subset was analyzed using the pre-ranked list mode with 1,000 permutations. The gene sets that were used for GSEA and gene set enrichment were from available Reactome gene sets as well as the following published works.19,78–82 Enrichment scores were visualized using the corrplot and ggplot2 R packages. Pathway analysis was visualized using Cytoscape.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data is shown from a representative experiment, of at least two independent experiments. Summaries show medians with 95% confidence intervals, unless noted otherwise. Statistical analysis was done using Graphpad Prism software. Significance was determined by using Mann-Whitney test, unless noted otherwise. All statistical tests were described in figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENTor RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-mouse CD11b (Fitc) | Biolegend | clone: M1/70, RRID: AB_312789 |
| anti-mouse MHCII (I-A/I-E) (A700) | Biolegend | clone: M5/114.15.2, RRID: AB_493727 |
| anti-mouse CD86 (BV421) | Biolegend | clone: GL-1, RRID: AB_10898329 |
| anti-mouse CD80 (BV605) | Biolegend | clone: 16-10A1, RRID: AB_11126141 |
| anti-mouse CD19 (BV650) | Biolegend | clone: 6D5, RRID: AB_11204087 |
| anti-mouse CD3 (BV650) | Biolegend | clone: 17A2, RRID: AB_11204249 |
| anti-mouse CD11c (BV711) | Biolegend | clone: N418, RRID: AB_2563905 |
| anti-mouse CD8a (Percp-Cy5.5, Pe-Cy7) | Biolegend | clone: 53–6.7, RRID: AB_2075238 |
| anti-mouse IRF4 (FITC, A647) | Biolegend | clone: IRF4.3E4, RRID: AB_2563266 |
| anti-mouse TCF1 (PE) | BD | clone: S33–966, RRID: AB_2687845 |
| anti-mouse CD127 (PE-Dazzle) | Biolegend | clone: A7R34, RRID: AB_2564216 |
| anti-mouse Tim3 (Pe-cy7, BV421) | Biolegend | clone: RMT3–23, RRID: AB_2571933 |
| anti-mouse Blimp1 (APC) | Biolegend | clone: 5E7, RRID: AB_2565618 |
| anti-mouse CD44 (A700, BV510) | Biolegend | clone: IM7, RRID: AB_2561391 |
| anti-mouse CD62L (BV605) | Biolegend | clone: MEL-14, RRID: AB_11125577 |
| anti-mouse CD45.1 (BV711) | Biolegend | clone: A20, RRID: AB_2562605 |
| anti-mouse PD1 (BV786) | Biolegend | clone: 29E.1A12, RRID: AB_2563680 |
| anti-mouse Ki67 (FITC, BV650) | BD | clone: B56, RRID: AB_2688008 |
| anti-mouse/human Gzmb (A700, BV421) | BD | clone: GB11, RRID: AB_1645453 |
| anti-mouse IFNy (APC) | Biolegend | clone: XMG1.2, RRID: AB_315403 |
| anti-mouse TNFa (FITC) | Biolegend | clone: MP6-XT22, RRID: AB_315425 |
| anti-mouse IL2 (PE) | Biolegend | clone: JES6-5H4, RRID: AB_315302 |
| Fixable Live Dead | Thermo-Fisher | Near IR, Aqua |
| anti-mouse Fc block | Biolegend | clone: 93, RRID: AB_1574975 |
| anti-human CD141 (FITC) | Miltenyi Biotec | clone: REA674, RRID: AB_2751167 |
| anti-human HLA-DR (PE, APC-Cy7) | Biolegend | clone: L243, RRID: AB_493586 |
| anti-human CD1c (BUV395, APC) | Biolegend/BD | clone: F10/21A3, RRID: AB_2741017 |
| anti-human CD11b (BUV737, BV605) | Biolegend/BD | clone: ICRF44, RRID: AB_2562020 |
| anti-human CD14 (BUV661) | BD | clone: M5E2, RRID: AB_2871011 |
| anti-human CD86 (BV786) | Biolegend | clone: IT2.2, RRID: AB_2616793 |
| anti-human CD11c (BV711) | Biolegend | clone: 3.9, RRID: AB_11219609 |
| anti-human CD40 (A700) | Biolegend | clone: 5C3, RRID: AB_2563921 |
| anti-human CD16 (Pe-Dazzle, BUV661) | Biolegend/BD | clone: 3G8, RRID: AB_2563638 |
| anti-human CD135 (PE) | BD | clone: 4G8, RRID: AB_397175 |
| anti-human PDL1 (BV605) | Biolegend | clone: 29E2A3, RRID: AB_2565926 |
| anti-human Tim3 (PE) | R&D | clone: FAB2365P, RRID: AB_2232901 |
| anti-human CD45 (APC) | Biolegend | clone: 2D1, RRID: AB_2566372 |
| anti-human CD3 (BUV395, BV510) | Biolegend/BD | clone: UCHT1, RRID: AB_2563468 |
| anti-human CD28 (Pe-Cy7) | Thermofisher | clone: CD28.2, RRID: AB_1944363 |
| anti-human PD1 (BUV737, BV786) | Biolegend/BD | clone: EH12.1, RRID: AB_2870118 |
| anti-human CD39 (BV421, BV786) | Biolegend | clone: A1, RRID: AB_2564575 |
| anti-human TCF1 (A488) | Cell Signaling | clone: C63D3, RRID: AB_2797627 |
| anti-human Ki67 (A647, FITC) | BD | clone: B56, RRID: AB_647087 |
| anti-human CD69 (Pe-Dazzle) | Biolegend | clone: FN50, RRID: AB_2564276 |
| anti-human CD127 (BV711) | Biolegend | clone: A019D5, RRID: AB_2562908 |
| anti-human CD45RA (APC, BV510) | Biolegend | clone: HI100, RRID: AB_314415 |
| anti-human CCR7 (APC, BV421) | Biolegend | clone: G043H7, RRID: AB_10915474 |
| anti-human CD25 (A647) | Biolegend | clone: M-A251, RRID: AB_2904482 |
| anti-human CD8 (Percp-Cy5.5, BUV395) | Biolegend/BD | clone: RPA-T8, RRID: AB_2874820 |
| human FcR Blocking Reagent | Miltenyi Biotec | clone: 130-059-901, RRID: AB_2892112 |
|
| ||
| Biological samples | ||
|
| ||
| Human Tumor Samples | Emory University | N/A |
| Mouse Tissue | Emory University | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Collagenase D | Sigma | 11088882001 |
| GP33 peptide | Genscript | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| eBiosciences Foxp3/Transcription Factor Staining Kit | Thermo Fisher | 00–5523-00 |
| EasySep CD8 Mouse Negative Selection Kit | Stem Cell | 19853 |
| IFNy Human ELISA kit | Thermo Fisher | KHC4021 |
|
| ||
| Deposited data | ||
|
| ||
| Human and mouse genomics data | This Paper | GEO: GSE216731 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| TRAMPC1-LCMV-GP | Emory University | N/A |
| B16F10-LCMV-GP | Emory University | N/A |
| TRAMPC1 | Emory University | N/A |
| RENCA-HA | Emory University | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse:C57BL/6J | The Jackson Laboratory | 000664 |
| Mouse:P14(B6;D2-Tg(TcrLCMV)327Sdz/JDvsJ) | The Jackson Laboratory | 004694 |
|
| ||
| Software and algorithms | ||
|
| ||
| R: The Project for Statistical Computing | N/A | https://www.r-project.org/ |
| Seurat | Satija et al.66 | https://satijalab.org/seurat/ |
| DeSeq2 | Love et al.67 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Graphpad Prism | GraphPad Prism | N/A |
Highlights.
CD8 T cells in tumor-draining lymph nodes maintain a TCF1+ stem-like program
Tumor-specific CD8+ T cells migrate to the tumor in the stem-like state
CD8+ T cells only acquire the canonical effector program within the tumor
Effector program acquisition requires co-stimulation in the tumor microenvironment
ACKNOWLEDGMENTS
This work was supported by funding from the Prostate Cancer Foundation, the James M. Cox Foundation, and James C. Kennedy; pilot funding from the Winship Cancer Institute supported by the Dunwoody Country Club Senior Men’s Association; NCI grants R00-CA197804 (to H.K.) and U01-CA113913 (to M.S.); and DOD grant W81XWH-20-1-0525 (to H.K. and V.M.). We would like to acknowledge the Yerkes NHP Genomics Core, which is supported in part by NIH P51 OD011132 and NIH S10 OD026799, and the Emory Flow Cytometry Core under award number UL1TR002378.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2022.12.002.
REFERENCES
- 1.Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, Cardenas M, Wilkinson S, Lake R, Sowalsky AG, et al. (2019). An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470. 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagè s C, Tosolini M, Camus M, Berger A, Wind P, et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 3.Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW, Saw RP, and Thompson JF (2012). Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J. Clin. Oncol. 30, 2678–2683. 10.1200/JCO.2011.37.8539. [DOI] [PubMed] [Google Scholar]
- 4.Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, et al. (2005). Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 353, 2654–2666. 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 5.Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eroglu Z, Zaretsky JM, Hu-Lieskovan S, Kim DW, Algazi A, Johnson DB, Liniker E, Ben K, Munhoz R, Rapisuwon S, et al. (2018). High response rate to PD-1 blockade in desmoplastic melanomas. Nature 553, 347–350. 10.1038/nature25187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blessin NC, Spriestersbach P, Li W, Mandelkow T, Dum D, Simon R, Hube-Magg C, Lutz F, Viehweger F, Lennartz M, et al. (2020). Prevalence of CD8(+) cytotoxic lymphocytes in human neoplasms. Cell. Oncol. (Dordr) 43, 421–430. 10.1007/s13402-020-00496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, Sharpe AH, Freeman GJ, Irving BA, and Ahmed R (2018). Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA 115, 4749–4754. 10.1073/pnas.1718217115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iezzi G, Karjalainen K, and Lanzavecchia A (1998). The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity 8, 89–95. 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]
- 10.Kaech SM, and Ahmed R (2001). Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naïve cells. Nat. Immunol. 2, 415–422. 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Stipdonk MJ, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, and Schoenberger SP (2003). Dynamic programming of CD8+ T lymphocyte responses. Nat. Immunol. 4, 361–365. 10.1038/ni912. [DOI] [PubMed] [Google Scholar]
- 12.Harding FA, McArthur JG, Gross JA, Raulet DH, and Allison JP (1992). CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 356, 607–609. 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- 13.Turka LA, Linsley PS, Lin H, Brady W, Leiden JM, Wei RQ, Gibson ML, Zheng XG, Myrdal S, and Gordon D (1992). T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc. Natl. Acad. Sci. USA 89, 11102–11105. 10.1073/pnas.89.22.11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz RH (1990). A cell culture model for T lymphocyte clonal anergy. Science 248, 1349–1356. 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 15.Yu P, Spiotto MT, Lee Y, Schreiber H, and Fu Y-X (2003). Complementary role of CD4+ T cells and secondary lymphoid tissues for cross-presentation of tumor antigen to CD8+ T cells. J. Exp. Med. 197, 985–995. 10.1084/jem.20021804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson MJ, Shafer-Weaver K, Greenberg NM, and Hurwitz AA (2007). Tolerization of tumor-specific T cells despite efficient initial priming in a primary murine model of prostate cancer. J. Immunol. 178, 1268–1276. 10.4049/jimmunol.178.3.1268. [DOI] [PubMed] [Google Scholar]
- 17.Stoitzner P, Green LK, Jung JY, Price KM, Atarea H, Kivell B, and Ronchese F (2008). Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol. Immunother. 57, 1665–1673. 10.1007/s00262-008-0487-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen DS, and Mellman I (2013). Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10. 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 19.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T cell factor 1-expressing memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 21.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20. 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. (2016). Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature 537, 412–428. 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- 23.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e10. 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 24.Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, Mavilio D, Alloisio M, Ferrari F, Lopci E, et al. (2018). High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J. Exp. Med. 215, 2520–2535. 10.1084/jem.20180684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eberhardt CS, Kissick HT, Patel MR, Cardenas MA, Prokhnevska N, Obeng RC, Nasti TH, Griffith CC, Im SJ, Wang X, et al. (2021). Functional HPV-specific PD-1(+) stem-like CD8 T cells in head and neck cancer. Nature 597, 279–284. 10.1038/s41586-021-03862-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, Araki K, and Ahmed R (2018). CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu. Rev. Med. 69, 301–318. 10.1146/annurev-med-012017-043208. [DOI] [PubMed] [Google Scholar]
- 27.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, et al. (2019). Checkpoint blockade immunotherapy induces dynamic changes in PD-1−CD8+ tumor-infiltrating T cells. Immunity 50, 181–194.e186. 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336. 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krishna S, Lowery FJ, Copeland AR, Bahadiroglu E, Mukherjee R, Jia L, Anibal JT, Sachs A, Adebola SO, Gurusamy D, et al. (2020). Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 370, 1328–1334. 10.1126/science.abb9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, Schietinger A, Philip M, Schreiber H, and Fu Y-X (2004). Priming of naive T cells inside tumors leads to eradication of established tumors. Nat. Immunol. 5, 141–149. 10.1038/ni1029. [DOI] [PubMed] [Google Scholar]
- 31.Fassò M, Waitz R, Hou Y, Rim T, Greenberg NM, Shastri N, Fong L, and Allison JP (2008). SPAS-1 (stimulator of prostatic adenocarcinoma-specific T cells)/SH3GLB2: A prostate tumor antigen identified by CTLA-4 blockade. Proc. Natl. Acad. Sci. USA 105, 3509–3514. 10.1073/pnas.0712269105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivas FV, O’Herrin S, and Gajewski TF (2001). CD28 is not required for c-Jun N-terminal kinase activation in T cells. J. Immunol. 167, 3123–3128. 10.4049/jimmunol.167.6.3123. [DOI] [PubMed] [Google Scholar]
- 33.Shakiba M, Zumbo P, Espinosa-Carrasco G, Menocal L, Dündar F, Carson SE, Bruno EM, Sanchez-Rivera FJ, Lowe SW, Camara S, et al. (2022). TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J. Exp. Med. 219, e20201966. 10.1084/jem.20201966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJD, Suresh M, Altman JD, and Ahmed R (1998). Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188, 2205–2213. 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evavold BD, and Allen PM (1991). Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand. Science 252, 1308–1310. 10.1126/science.1833816. [DOI] [PubMed] [Google Scholar]
- 36.Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, et al. (2017). Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory-like T cells during chronic. Infect. Immun. 47, 1129–1141.e1125. 10.1016/j.immuni.2017.11.021. [DOI] [PubMed] [Google Scholar]
- 37.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218. 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao C, Lou G, Sun H-W, Zhu Z, Sun Y, Chen Z, Chauss D, Moseman EA, Cheng J, D’Antonio MA, et al. (2021). BACH2 enforces the transcriptional and epigenetic programs of stem-like CD8+ T cells. Nat. Immunol. 22, 370–380. 10.1038/s41590-021-00868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, Shi W, and Kallies A (2020). Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 21, 1256–1266. 10.1038/s41590-020-0760-z. [DOI] [PubMed] [Google Scholar]
- 41.Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al. (2012). Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 13, 888–899. 10.1038/ni.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, Klinakis A, Charo IF, Jung S, Gommerman JL, et al. (2011). Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35, 780–791. 10.1016/j.immuni.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dutertre CA, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, Ng PY, van den Hoogen LL, Leong JY, Lee B, et al. (2019). Single-cell analysis of human mononuclear phagocytes reveals subset-defining markers and identifies circulating inflammatory dendritic cells. Immunity 51, 573–589.e8. 10.1016/j.immuni.2019.08.008. [DOI] [PubMed] [Google Scholar]
- 44.Tang-Huau T-L, Gueguen P, Goudot C, Durand M, Bohec M, Baulande S, Pasquier B, Amigorena S, and Segura E (2018). Human in vivo-generated monocyte-derived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat. Commun. 9, 2570. 10.1038/s41467-018-04985-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, Miron M, Kumar BV, Griesemer A, Ho S-H, et al. (2017). Dendritic cells display subset and tissue-specific maturation dynamics over human life. Immunity 46, 504–515. 10.1016/j.immuni.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen C-JJ, Dunbar PR, Wadley RB, Jeet V, et al. (2010). Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 207, 1247–1260. 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Segura E, Valladeau-Guilemond J, Donnadieu M-H, Sastre-Garau X, Soumelis V, and Amigorena S (2012). Characterization of resident and migratory dendritic cells in human lymph nodes. J. Exp. Med. 209, 653–660. 10.1084/jem.20111457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heidkamp GF, Sander J, Lehmann CHK, Heger L, Eissing N, Baranska A, Lühr JJ, Hoffmann A, Reimer KC, Lux A, et al. (2016). Human lymphoid organ dendritic cell identity is predominantly dictated by ontogeny, not tissue microenvironment. Sci. Immunol. 1, eaai7677. 10.1126/sciimmunol.aai7677. [DOI] [PubMed] [Google Scholar]
- 49.Carpentier S, Vu Manh TP, Chelbi R, Henri S, Malissen B, Haniffa M, Ginhoux F, and Dalod M (2016). Comparative genomics analysis of mononuclear phagocyte subsets confirms homology between lymphoid tissue-resident and dermal XCR1(+) DCs in mouse and human and distinguishes them from Langerhans cells. J. Immunol. Methods 432, 35–49. 10.1016/j.jim.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banchereau R, Baldwin N, Cepika AM, Athale S, Xue Y, Yu CI, Metang P, Cheruku A, Berthier I, Gayet I, et al. (2014). Transcriptional specialization of human dendritic cell subsets in response to microbial vaccines. Nat. Commun. 5, 5283. 10.1038/ncomms6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horton BL, Morgan DM, Momin N, Zagorulya M, Torres-Mejia E, Bhandarkar V, Wittrup KD, Love JC, and Spranger S (2021). Lack of CD8+ T cell effector differentiation during priming mediates checkpoint blockade resistance in non-small cell lung cancer. Sci. Immunol. 6, eabi8800. 10.1126/sciimmunol.abi8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity 49, 1148–1161.e7. 10.1016/j.immuni.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Connolly KA, Kuchroo M, Venkat A, Khatun A, Wang J, William I, Hornick NI, Fitzgerald BL, Damo M, Kasmani MY, et al. (2021). A reservoir of stem-like CD8+ T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 6, eabg7836. 10.1126/sciimmunol.abg7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schenkel JM, Herbst RH, Canner D, Li A, Hillman M, Shanahan S-L, Gibbons G, Smith OC, Kim JY, Westcott P, et al. (2021). Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1+ CD8+ T cells in tumor-draining lymph nodes. Immunity 54, 2338–2353.e2336. 10.1016/j.immuni.2021.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Starbeck-Miller GR, Xue H-H, and Harty JT (2014). IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J. Exp. Med. 211, 105–120. 10.1084/jem.20130901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, and Mescher MF (2005). Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174, 4465–4469. 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 57.Valenzuela J, Schmidt C, and Mescher M (2002). The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J. Immunol. 169, 6842–6849. 10.4049/jimmunol.169.12.6842. [DOI] [PubMed] [Google Scholar]
- 58.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, and Mescher MF (1999). Inflammatory cytokines Provide a Third Signal for Activation of Naive CD4+ and CD8+ T Cells. J. Immunol. 162, 3256–3262. [PubMed] [Google Scholar]
- 59.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. (2017). Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science 355, 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. (2017). Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 114, 4993–4998. 10.1073/pnas.1705327114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, van Nimwegen M, Lau SP, Latupeirissa K, Schetters S, et al. (2020). The PD-1/PD-L1-checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell 38, 685–700.e688. 10.1016/j.ccell.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 62.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen P-L, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. (2016). Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 6, 827–837. 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwartz RH, Mueller DL, Jenkins MK, and Quill H (1989). T-cell clonal anergy. Cold Spring Harbor Symp. Quant. Biol. 54, 605–610. 10.1101/sqb.1989.054.01.072. [DOI] [PubMed] [Google Scholar]
- 65.Schwartz RH (2003). T cell anergy. Annu. Rev. Immunol. 21, 305–334. 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 66.Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buchwald ZS, Nasti TH, Lee J, Eberhardt CS, Wieland A, Im SJ, Lawson D, Curran W, Ahmed R, and Khan MK (2020). Tumor-draining lymph node is important for a robust abscopal effect stimulated by radiotherapy. J. Immunother. Cancer 8, e000867. 10.1136/jitc-2020-000867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carnero Contentti E, Farez MF, and Correale J (2019). Mucosal-associated invariant T cell features and TCR repertoire characteristics during the course of multiple sclerosis. Front. Immunol. 10, 2690. 10.3389/fimmu.2019.02690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chamoto K, Guo T, Imataki O, Tanaka M, Nakatsugawa M, Ochi T, Yamashita Y, Saito AM, Saito TI, Butler MO, and Hirano N (2016). CDR3β sequence motifs regulate autoreactivity of human invariant NKT cell receptors. J. Autoimmun. 68, 39–51. 10.1016/j.jaut.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matulis G, Sanderson JP, Lissin NM, Asparuhova MB, Bommineni GR, Schümperli D, Schmidt RR, Villiger PM, Jakobsen BK, and Gadola SD (2010). Innate-like control of human iNKT cell autoreactivity via the hypervariable CDR3β loop. PLOS Biol. 8, e1000402. 10.1371/journal.pbio.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rossjohn J, Pellicci DG, Patel O, Gapin L, and Godfrey DI (2012). Recognition of CD1d-restricted antigens by natural killer T cells. Nat. Rev. Immunol. 12, 845–857. 10.1038/nri3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barwick BG, Scharer CD, Bally APR, and Boss JM (2016). Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat. Immunol. 17, 1216–1225. 10.1038/ni.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, Brown BD, Schmolke M, Miller JC, Leboeuf M, et al. (2012). Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J. Clin. Invest. 122, 4037–4047. 10.1172/JCI60659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Danilo M, Chennupati V, Silva JG, Siegert S, and Held W (2018). Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T cell differentiation. Cell Rep. 22, 2107–2117. 10.1016/j.celrep.2018.01.072. [DOI] [PubMed] [Google Scholar]
- 77.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, and Brooks DG (2013). Blockade of chronic Type I interferon signaling to control persistent LCMV infection. Science 340, 202–207. 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, McGuire DJ, Cohen KW, et al. (2017). Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367. 10.1038/nature24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, and Goldrath AW; Immunological Genome Project Consortium (2013). Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat. Immunol. 14, 404–412. 10.1038/ni.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, and Wherry EJ (2012). Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37, 1130–1144. 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, et al. (2016). Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401. 10.1016/j.immuni.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684. 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw fastq files and associated RNA, whole genome bisulphite sequencing, and single cell RNA sequencing have been uploaded to the NCBI Gene Expression Omnibus (GEO) database under identifier GSE216731. Custom python and R scripts for TCR, RNA, and DNA analysis are available upon request.