

Stabilization of the transcription factor NRF2 promotes oncogene-driven tumor initiation but blocks tumor progression, indicating distinct, threshold-dependent effects of the KEAP1/NRF2 pathway in different stages of lung tumorigenesis.
Abstract
Mutations in the KEAP1–NRF2 (Kelch-like ECH-associated protein 1–nuclear factor-erythroid 2 p45-related factor 2) pathway occur in up to a third of non–small cell lung cancer (NSCLC) cases and often confer resistance to therapy and poor outcomes. Here, we developed murine alleles of the KEAP1 and NRF2 mutations found in human NSCLC and comprehensively interrogated their impact on tumor initiation and progression. Chronic NRF2 stabilization by Keap1 or Nrf2 mutation was not sufficient to induce tumorigenesis, even in the absence of tumor suppressors, p53 or LKB1. When combined with KrasG12D/+, constitutive NRF2 activation promoted lung tumor initiation and early progression of hyperplasia to low-grade tumors but impaired their progression to advanced-grade tumors, which was reversed by NRF2 deletion. Finally, NRF2 overexpression in KEAP1 mutant human NSCLC cell lines was detrimental to cell proliferation, viability, and anchorage-independent colony formation. Collectively, these results establish the context-dependence and activity threshold for NRF2 during the lung tumorigenic process.
Significance:
Stabilization of the transcription factor NRF2 promotes oncogene-driven tumor initiation but blocks tumor progression, indicating distinct, threshold-dependent effects of the KEAP1/NRF2 pathway in different stages of lung tumorigenesis.
Graphical Abstract
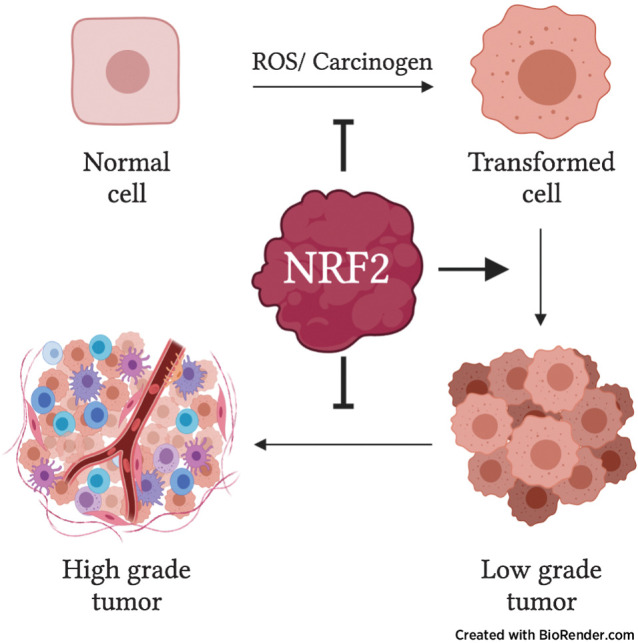
Introduction
NRF2 (nuclear factor-erythroid 2 p45-related factor 2) is a stress-responsive transcription factor that regulates the detoxification of reactive oxygen species, maintains cellular homeostasis, and regulates many facets of metabolism (1, 2). NRF2 is negatively regulated by KEAP1 (Kelch-like ECH-associated protein 1), a substrate adaptor protein for the cullin 3 (CUL3)-based E3 ubiquitin ligase that facilitates NRF2 ubiquitination and proteasomal degradation in the absence of oxidative or xenobiotic stress (3). NRF2 promotes the detoxification of carcinogens to limit deleterious mutations that initiate cancer (4–7) and NRF2 activators are being explored as chemopreventative agents (8–10). However, NRF2 is frequently stabilized in many cancers, particularly non–small cell lung cancer (NSCLC), where mutations in the KEAP1–NRF2 pathway are found in up to 30% of cases (11, 12). NRF2 stabilization is associated with poor prognosis (13–20), resistance to chemo- and radiotherapy (21, 22), cancer cell survival (22), proliferation (23), metabolic reprogramming (20, 24–26), and metastasis (27). It remains unclear, however, whether chronic NRF2 stabilization transforms normal cells. Thus, it is important to understand the contexts and mechanisms by which NRF2 can prevent and promote cancer phenotypes.
Preclinical genetically engineered mouse models (GEMM) have advanced our understanding of the role of NRF2 in lung tumorigenesis (23, 27–37). NRF2 activation in NSCLC has been modeled in GEMMs by inactivating Keap1 via conditional knockout (28–31, 33, 35) or CRISPR-mediated deletion (27, 34, 36, 37), in contrast with the KEAP1 mutations found in human lung cancer. These studies have shown that Nrf2 promotes lung tumor initiation (29, 30), tumor size (34, 38), progression (33, 34), and metastasis (27). However, other studies have failed to see an effect of Nrf2 on lung tumor initiation (38, 39) or size (36, 37, 39), and we reported that Nrf2 activation significantly decreases tumor size (32). Study conditions, time points and phenotypes assayed varied across these studies. Therefore, the role NRF2 activation plays at distinct stages of tumor initiation and progression remains to be determined.
In the present study, we generated lung cancer GEMMs expressing Keap1R554Q and Nrf2D29H mutations to comprehensively investigate how Nrf2 activation affects each stage of the tumorigenic process. These models also exhibit a series of graded NRF2 activation, allowing us to ask how different levels of Nrf2 influence lung tumor progression. We found that constitutive Nrf2 stabilization induced by these mutations was insufficient for lung tumor development, even in the context of tumor-suppressor loss. In contrast, these mutations promoted lung tumor initiation in the KrasG12D/+ model of early lung adenocarcinoma, consistent with previous studies (23, 29, 30).
Using the KrasG12D/+; p53fl/fl adenocarcinoma model, we found that homozygous Keap1 mutation unexpectedly blocked tumor progression. Supportingly, we found that Nrf2 expression and activity were downregulated in advanced tumors, and Nrf2 deletion could rescue the Keap1 mutation–mediated progression impairment. Overall, our data suggest that NRF2 has distinct, threshold-dependent effects during lung tumor initiation and progression.
Materials and Methods
Mice
Mice were housed and bred in accordance with the ethical regulations and approval of the Institutional Animal Care and Use Committee (protocols #: IS00003893R and IS00007922R). Generation of the Keap1 targeting vector was previously described (32). Briefly, the CA-Keap1R554Q allele (Keap1tm1Gmdn, MGI: 7327097) was made by inserting a wild-type (WT) Keap1 cDNA containing exons 3–5 flanked by loxP sites upstream of the R554Q mutation in exon 4 of the Keap1 gene. Keap1 was targeted in C10 murine ES cells and cells were selected with blasticidin. To make the LSL-Nfe2l2D29H allele (Nfe2l2tm1Gmdn, MGI: 7327101), a STOP cassette flanked by loxP sites was inserted into intron 1 and codon 29 in endogenous exon 2 was mutated from an aspartic acid to a histidine. The endogenous Nfe2l2 locus was targeted in C10 murine ES cells and puromycin was used to select positive cells. For both alleles, positive clones were screened by copy-number real-time PCR and injected into blastocysts. Genotyping primers were as follows: For the Keap1R554Q allele: Mutant forward: 5′-ATGGCCACACTTTTCTGGAC-3′; WT forward: 5′-GGGGGTAGAGGGAGGAGAAT-3′; Common reverse: 5′-GCCACCCTATTCACAGACCA-3′. The WT PCR product was 326 bp and the mutant PCR product 584 bp. For the Nfe2l2D29H allele: WT forward: 5′-GAGGCAGGTAGTTCTCTGAGTTTG-3′; Common reverse: 5′-GCAAATGCACTGAGACACTCAT-3′; Mutant forward: 5′-CTAGCCACCATGGCTTGAGT-3′. The WT PCR product was 189 bp and the mutant PCR product 282 bp. All mice were maintained on a mixed C57BL/6 genetic background. In addition to Keap1R554Q and Nrf2D29H mice, p53flox (RRID:IMSR_JAX:008462); Lkb1flox (RRID:IMSR_JAX:014143); Nrf2flox (RRID:IMSR_JAX:025433); and LSL-KrasG12D/+ (RRID:IMSR_JAX:008179) mice were used. For mouse lung tumor studies, intranasal installation of 2.5 × 107 PFU adenoviral-Cre (University of Iowa) was used to induce lung tumors as previously described (40). Adenoviral infections were performed under isofluorane anesthesia.
Murine embryonic fibroblast generation and culture
Mouse embryonic fibroblasts (MEF) were isolated from E13.5 to 14.5-day old embryos and maintained in pyruvate-free DMEM (Corning) containing 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco) in a humidified incubator with 5% CO2 and 95% air at 37°C. MEFs were used within four passages and infected with control empty adenovirus or adenoviral-Cre (University of Iowa) at an approximate multiplicity of infection of 500.
RNA sequencing preparation and analysis
Samples were prepared using the RNeasy plus mini kit (Qiagen, 74134). RNA quality was checked with the QIAxcel RNA QC kit (Qiagen, 929104). Additional RNA QC, sequencing, mapping to the mouse genome, and analysis were performed by Novogene. Differentially expressed genes (DESeq2) with a P value of <0.05 were included in the volcano plot.
IHC
Mouse lung tissue was fixed with 10% formalin overnight, transferred to 70% ethanol, and paraffin embedded to be sectioned. Unstained tissue sections were deparaffinized in xylene, followed by rehydration in a graded alcohol series. Antigen retrieval was performed by boiling in 10 mmol/L citrate buffer (pH 6). Antibodies used for IHC include affinity-purified NRF2 (1:150 or 1:300; ref. 41), NQO1 (Sigma-Aldrich, RRID:AB_1079501, 1:500), Ki-67 (Cell Signaling Technology, RRID:AB_2620142, 1:200), and cleaved caspase-3 (Cell Signaling Technology, RRID:AB_2070042, 1:1,000). Following overnight incubation at 4°C in primary antibody, the ImmPRESS HRP (horseradish peroxidase) goat anti-rabbit kit (Vector Laboratories, RRID:AB_2631198) was used as directed by the manufacturer's instructions. DAB peroxidase (HRP) substrate (Vector Laboratories, SK-4105) was used to develop IHC staining, followed by counterstaining with hematoxylin (Vector Laboratories, H-3404). Slides were scanned with the Aperio imager at 20× and the H-score of at least five representative regions/mouse was analyzed with QuPath software (42). Representative images were captured using the Axio Lab.A1 microscope at ×40 (Carl Zeiss Microimaging Inc.).
Tumor-grading analysis and histology
Lung tumor grading was performed manually as previously described (43). Tumor-grading distribution percentages were calculated by dividing the number of tumors in a specific grade by the total number of tumors per mouse. Tumor burden by grade was calculated by dividing the area of the lung covered by a specific tumor grade by the total lung area.
NSCLC cell lines and culture
Human lung cancer cell lines used include H1944 (RRID:CVCL_1508), H322 (RRID:CVCL_1556), A549 (RRID:CVCL_0023), HCC15 (RRID:CVCL_2057), and H460 (RRID:CVCL_0459) and were previously described (24). All cell lines were acquired from an authentic source (Hamon Cancer Center Collection). Cells were cultured in RPMI-1640 (Gibco) containing 5% FBS without antibiotics in a humidified incubator with 5% CO2 and 95% air at 37°C. Cells were confirmed to be free of Mycoplasma with the MycoAlert kit (Lonza) immediately upon receipt and aliquots were frozen. Cell lines were tested monthly for Mycoplasma and used within 10–20 passages.
Lentivirus generation and infection of NSCLC cells
Lentiviruses were made by transfecting Lenti-X 293T cells (Takara 632180) overnight with polyethylenimine (PEI), lentiviral plasmid [pLX317-NRF2 (44) or the control pLX317 empty vector (32)], and packaging plasmids pCMV-dR8.2 dvpr (RRID:Addgene_8455) and pCMV-VSV-G (RRID:Addgene_8454) in DMEM containing 10% FBS. To generate NRF2-overexpressing cells, NSCLC cells were transduced for 24 hours with lentiviruses in medium containing polybrene (8 μg/mL). After transduction, infected cells were selected with 0.5 μg/mL (H1944, H322, H460) or 1 μg/mL (A549, HCC15) puromycin for 72 hours. Immediately following selection, cells were seeded in respective puromycin concentrations for the indicated assays.
Cell proliferation and cell death assays
NSCLC cells were monitored with the CELLCYTE X live cell imaging instrument (Cytena) over the course of 96 hours. Before imaging, SYTOX Green nucleic acid stain (Thermo Fisher Scientific, S7020) was added to medium at a final concentration of 20 nmol/L. Images were acquired from each well at 8-hour intervals and analyzed using CellCyte Studio (CELLINK). Cell confluency was represented as the percentage of the image covered by cells. The number of dead cells was normalized to cell confluency [number of Sytox Green positive cells/mm2/cell confluency]. The AUC values were calculated by summing the proliferation or normalized dead cell number at each time point.
Western blotting
Cells were lysed in ice-cold RIPA buffer with protease inhibitors (Thermo Fisher Scientific, PIA32955) followed by sonication in a water bath sonicator (Diagenode). Protein was quantified using the DC protein assay (Bio-Rad). Lysates were prepared with 6X SDS sample buffer containing 12% (v/v) β-ME (VWR) and separated on Bolt or NuPAGE 4%–12% Bis-Tris gels (Invitrogen). SDS-PAGE separation was followed by transfer to 0.45-μm nitrocellulose membranes (GE Healthcare). The membranes were blocked in 5% non-fat milk in TBS with 0.1% Tween 20 (TBST). For immunoblotting, the following antibodies diluted in 5% milk in TBST were used: KEAP1 (Millipore Sigma, RRID:AB_2921362, 1:2,000), NRF2 (Cell Signaling Technology, D1Z9C, RRID:AB_2715528, 1:1,000), NQO1 (Sigma-Aldrich, RRID:AB_1079501, 1:1,000), GCLC (Sana Cruz Biotechnology, H-5, RRID:AB_2736837, 1:1,000), xCT (Abcam, RRID:AB_778944, 1:1,000), GSR (Santa Cruz Biotechnology, RRID:AB_2295121, 1:1,000), β-actin (Invitrogen AM4302, RRID:AB_2536382, 1:100,000). HRP secondary antibodies used include goat anti-rabbit IgG (Jackson ImmunoResearch Labs, RRID: AB_2313567), goat anti-mouse IgG (Jackson ImmunoResearch Labs, RRID:AB_10015289), and goat anti-rat IgG (Jackson ImmunoResearch Labs, RRID:AB_2338128). Membranes were developed with Clarity ECL substrate (Bio-Rad) or a luminol-based homemade ECL substrate.
Soft agar colony formation assays
6-well plates were coated with a 0.8% agar prepared in RPMI. NSCLC cells were then seeded in 0.4% agar in RPMI. After the cell/ agar mixture solidified, RPMI medium containing 10% FBS, Pen/Strep and puromycin was added to each well and replenished every few days. Colonies were allowed to form for 10 to 16 days, and wells were stained with 0.01% crystal violet in a 4% paraformaldehyde in PBS solution. Plates were scanned on a flatbed scanner and ImageJ was used to quantify colonies.
DepMap analysis
NFE2L2 dependency scores were downloaded from the DepMap database v. 22Q2 (45). Values were plotted from CRISPR (DepMap 22Q2 Public+Score, Chronos) for NSCLC cell lines that we previously evaluated for high or low NRF2 activity (24).
Quantification and statistical analysis
GraphPad Prism9 software was used for statistical analyses and P values of <0.05 were considered significant, with symbols as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****P < 0.0001. All data are represented as mean ± SD unless otherwise stated. For all experiments, similar variances between groups were observed.
Material availability
All unique reagents generated in this study are available from the corresponding author without restriction.
Data availability
The data generated in this study are included in the article and its Supplementary Figures. Raw data are available upon request without restriction from the corresponding author. The gene expression data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE212942. The data for the NFE2L2 dependency scores analyzed in this study were obtained from the DepMap database v. 22Q2 at https://depmap.org/portal/download/all/. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Gina M. DeNicola (gina.denicola@moffitt.org).
Results
Keap1R554Q and Nfe2l2D29H alleles activate the Nrf2 transcriptional program
To study the role of NRF2 activation in lung cancer, we developed alleles harboring either the Keap1R554Q or the Nrf2D29H mutation found in human NSCLC (Fig. 1A and B). Both Keap1R554Q and Nrf2D29H mutations prevent Keap1-mediated ubiquitination of Nrf2, allowing for constitutive expression of Nrf2 and transcription of Nrf2 target genes (46, 47). To generate the conditionally active (CA)-Keap1R554Q allele, we inserted a WT Keap1 cDNA containing exons 3–5 flanked by loxP sites upstream of the R554Q mutation in endogenous exon 4 of the Keap1 gene (Fig. 1A; ref. 32). For the Lox-STOP-Lox (LSL)-Nfe2l2D29H allele, we inserted a loxP-flanked transcriptional and translational STOP (LSL) cassette upstream of the D29H mutation in exon 2 of the endogenous Nfe2l2 gene (Fig. 1B). For both alleles, Cre-mediated excision of loxP-flanked cassettes allows for physiological expression of Keap1R554Q or Nrf2D29H, recapitulating NRF2 activation in human NSCLC. To validate the functionality of these alleles, we first generated MEFs, which allowed the switching from an Nrf2-deficient state (Nrf2LSL/LSL) to an Nrf2-stabilized state (Nrf2D29H/D29H), or from a basal Nrf2 state (Keap1+/+) to a stabilized Nrf2 state (Keap1R554Q/R554Q) in an isogenic system. Using these MEFs, we performed transcriptomic profiling (Fig. 1C and D). RNA-seq indicated that both Keap1R554Q/R554Q and Nrf2D29H/D29H MEFs demonstrated increased transcription of canonical Nrf2 target genes, including Nqo1, Srxn1, Txnrd1, and Gclc (Fig. 1C and D). Prior targeting of the murine Keap1 locus to generate a Keap1flox allele resulted in the generation of a hypomorphic allele before Cre-mediated recombination, leading to decreased Keap1 levels and increased Nrf2 transcriptional activity throughout the whole animal (48). Importantly, we found no differences in expression of Keap1, Nrf2, or Nrf2 target proteins Nqo1 and Gclc between CA-Keap1R554Q and WT Keap1 MEFs, indicating that the CA-Keap1R554Q allele is not hypomorphic (Supplementary Fig. S1A–S1D). Collectively, these results indicate that the mutant Keap1R554Q and Nfe2l2D29H alleles activate the Nrf2 transcriptional program.
Figure 1.

Development of mutant Keap1 and Nfe2l2 alleles found in human lung cancer. A, The conditionally active (CA)-Keap1R554Q allele was generated by inserting a loxP-flanked, WT Keap1 cDNA containing exons 3–5 into intron 2 and introducing the R554Q mutation into endogenous exon 4 of the Keap1 gene. Before intranasal installation of adenoviral-Cre recombinase Keap1 is WT. Once the floxed cargo is excised, mutant Keap1R554Q is expressed. PA, poly A signal. B, The Lox-STOP-Lox (LSL)-Nfe2l2D29H allele was created by inserting a STOP cassette flanked by loxP sites into intron 1 and introducing the D29H mutation in endogenous exon 2 of the Nfe2l2 gene. Following Cre-mediated excision of the STOP cassette, mutant Nrf2D29H is expressed. C, Volcano plot of RNA-seq data from MEFs expressing Keap1R554Q/R554Q compared with Keap1+/+. N = 3, representative of two individual MEF lines. D, Volcano plot of RNA-seq data from MEFs expressing Nrf2D29H/D29H compared with Nrf2LSL/LSL, which lack Nrf2 expression. N = 3, representative of two individual MEF lines.
Keap1 or Nrf2 mutation is not sufficient to initiate lung tumorigenesis
Given the importance of NRF2 in cytoprotection and redox homeostasis, there has been a longstanding interest in activating NRF2 pharmacologically for chemoprevention (8–10). The long-term safety of this approach, and whether the chronic activation of NRF2 can transform healthy cells in vivo, remains unknown. Moreover, the whole-body deletion of Keap1 in mice results in postnatal lethality because of constitutive Nrf2 activation (49). In human lung tumors, KEAP1 inactivation is frequently biallelic (13), whereas NFE2L2 mutations are frequently heterozygous (50). Therefore, we induced the recombination of Keap1 and Nfe2l2 alleles in the lungs of Keap1R554Q/+, Keap1R554Q/R554Q, or Nrf2D29H/+ mice using adenoviral-Cre to test whether constitutive Nrf2 activation is sufficient to initiate lung tumor formation (Fig. 2). First, we analyzed the overall survival (OS) between the different groups. The median survival across genotypes ranged from approximately 650 to 750 days, with no significant survival differences observed between WT and Keap1/Nrf2 mutant groups (Fig. 2A). Although mice did develop tumors, they comprised age-associated tumors like lymphoma. Upon examination of mouse lung histology for the presence of lung tumors, lung tumor-free survival was also not different between the groups (Fig. 2B). Finally, histological analysis of lung tissues revealed that both alveolar and bronchiolar cells appeared phenotypically normal across the genotypes (Fig. 2C). These results indicate that constitutive Nrf2 activation is not sufficient to induce lung tumor formation.
Figure 2.
![Figure 2. Mutation of Keap1 or Nrf2 is not sufficient to initiate lung tumorigenesis. A, Overall survival of Keap1/Nrf2 mutant mice. Keap1/Nrf2+/+ (n = 17), Keap1R554Q/+ (n = 8); Keap1R554Q/R554Q (n = 11); Nrf2D29H/+ (n = 15). ns, not significant [log-rank (Mantel–Cox) test]. B, Lung tumor-free survival of Keap1/Nrf2 mutant mice. Keap1/Nrf2+/+ (n = 11); Keap1R554Q/+ (n = 4); Keap1R554Q/R554Q (n = 8); Nrf2D29H/+ (n = 11). C, Representative hematoxylin and eosin staining of mouse lung depicting normal bronchiolar and alveolar cells. Scale bars, 100 μm (top) and 20 μm (bottom).](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10267679_1953fig2.jpg)
Mutation of Keap1 or Nrf2 is not sufficient to initiate lung tumorigenesis. A, Overall survival of Keap1/Nrf2 mutant mice. Keap1/Nrf2+/+ (n = 17), Keap1R554Q/+ (n = 8); Keap1R554Q/R554Q (n = 11); Nrf2D29H/+ (n = 15). ns, not significant [log-rank (Mantel–Cox) test]. B, Lung tumor-free survival of Keap1/Nrf2 mutant mice. Keap1/Nrf2+/+ (n = 11); Keap1R554Q/+ (n = 4); Keap1R554Q/R554Q (n = 8); Nrf2D29H/+ (n = 11). C, Representative hematoxylin and eosin staining of mouse lung depicting normal bronchiolar and alveolar cells. Scale bars, 100 μm (top) and 20 μm (bottom).
Keap1 or Nrf2 mutation is not sufficient to initiate lung tumorigenesis in combination with tumor-suppressor loss
To determine whether tumor-suppressor loss was required for mutant Keap1 or Nrf2 to initiate lung tumor formation, we crossed Keap1 and Nrf2 mutant mice with p53flox and Lkb1flox alleles to concomitantly activate Nrf2 and delete these tumor suppressors in the lung (Fig. 3). We first examined the consequence of Nrf2D29H/+, Keap1R554Q/+, or Keap1R554Q/R554Q in combination with p53 deletion. Mice were aged to 500 days, at which time all mice were euthanized and examined for evidence of lung tumor formation. Although a small number of these mice did succumb to disease before 500 days, they developed age-associated tumors, including lymphoma, and we did not observe any differences in overall or lung tumor-free survival between genotypes (Fig. 3A and B). However, when examining the lung tissue histology, we observed dysplasia in Keap1R554Q/R554Q bronchioles following loss of p53 (Fig. 3C). This observation is consistent with previous work showing that tracheospheres derived from Keap1−/−;Trp53−/− cells had an aberrant morphology (31). We next examined the consequence of Nrf2D29H/+, Keap1R554Q/+ or Keap1R554Q/R554Q in combination with Lkb1 deletion. Similar to what was observed with p53, we also did not find any differences in overall or lung tumor-free survival between cohorts (Fig. 3D and E). Moreover, the bronchiolar and alveolar morphology was normal across genotypes, in contrast with what was observed upon p53 loss (Fig. 3F). Our findings indicate that Keap1/Nrf2 mutation is not sufficient to initiate lung tumor formation in combination with tumor-suppressor loss.
Figure 3.
![Figure 3. Mutation of Keap1 or Nrf2 is not sufficient to initiate lung tumorigenesis with p53 or Lkb1 loss. A, Overall survival of p53fl/fl mice expressing WT or mutant Keap1/Nrf2. Keap1/Nrf2+/+ (n = 15); Keap1R554Q/+ (n = 7); Keap1R554Q/R554Q (n = 18); Nrf2D29H/+ (n = 18). ns, not significant [log-rank (Mantel–Cox) test]. B, Lung tumor-free survival of p53fl/fl mice expressing WT or mutant Keap1/Nrf2. Keap1/Nrf2+/+ (n = 11); Keap1R554Q/+ (n = 6); Keap1R554Q/R554Q (n = 16); Nrf2D29H/+ (n = 10). C, Representative hematoxylin and eosin staining of mouse lung depicting bronchiolar and alveolar cells of the p53fl/fl models. Scale bars, 100 μm (top), 20 μm (middle), and 10 μm (bottom). D, Overall survival of Lkb1fl/fl mice expressing WT or mutant Keap1/Nrf2. Keap1/Nrf2+/+ (n = 11); Keap1R554Q/+ (n = 7); Keap1R554Q/R554Q (n = 11); Nrf2D29H/+ (n = 5). ns, not significant [log-rank (Mantel–Cox) test]. E, Lung tumor-free survival of Lkb1fl/fl mice expressing WT or mutant Keap1/Nrf2. Keap1/Nrf2+/+ (n = 11); Keap1R554Q/+ (n = 6); Keap1R554Q/R554Q (n = 9); Nrf2D29H/+ (n = 4). F, Representative hematoxylin and eosin staining of mouse lung depicting bronchiolar and alveolar cells of the Lkb1fl/fl models. Scale bars, 100 μm (top), 20 μm (middle), and 10 μm (bottom). A, B, D, and E, Mice were infected intranasally with adenoviral-Cre, followed by collection at 500 days to analyze lung tissue histology.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10267679_1953fig3.jpg)
Mutation of Keap1 or Nrf2 is not sufficient to initiate lung tumorigenesis with p53 or Lkb1 loss. A, Overall survival of p53fl/fl mice expressing WT or mutant Keap1/Nrf2. Keap1/Nrf2+/+ (n = 15); Keap1R554Q/+ (n = 7); Keap1R554Q/R554Q (n = 18); Nrf2D29H/+ (n = 18). ns, not significant [log-rank (Mantel–Cox) test]. B, Lung tumor-free survival of p53fl/fl mice expressing WT or mutant Keap1/Nrf2. Keap1/Nrf2+/+ (n = 11); Keap1R554Q/+ (n = 6); Keap1R554Q/R554Q (n = 16); Nrf2D29H/+ (n = 10). C, Representative hematoxylin and eosin staining of mouse lung depicting bronchiolar and alveolar cells of the p53fl/fl models. Scale bars, 100 μm (top), 20 μm (middle), and 10 μm (bottom). D, Overall survival of Lkb1fl/fl mice expressing WT or mutant Keap1/Nrf2. Keap1/Nrf2+/+ (n = 11); Keap1R554Q/+ (n = 7); Keap1R554Q/R554Q (n = 11); Nrf2D29H/+ (n = 5). ns, not significant [log-rank (Mantel–Cox) test]. E, Lung tumor-free survival of Lkb1fl/fl mice expressing WT or mutant Keap1/Nrf2. Keap1/Nrf2+/+ (n = 11); Keap1R554Q/+ (n = 6); Keap1R554Q/R554Q (n = 9); Nrf2D29H/+ (n = 4). F, Representative hematoxylin and eosin staining of mouse lung depicting bronchiolar and alveolar cells of the Lkb1fl/fl models. Scale bars, 100 μm (top), 20 μm (middle), and 10 μm (bottom). A, B, D, and E, Mice were infected intranasally with adenoviral-Cre, followed by collection at 500 days to analyze lung tissue histology.
Nrf2 activation cooperates with mutant Kras to promote lung tumor initiation and early progression
We and others have reported that Nrf2 activation is important for Kras mutant lung tumorigenesis (23, 29, 30). To understand how Keap1 or Nrf2 mutation affect lung tumor initiation and early progression, we crossed Keap1/Nrf2 mutant mice with the KrasG12D/+ model of early lung adenocarcinoma (40). Although all mice succumbed to lung tumors with a median survival of around 200 days, we observed no difference in survival between cohorts (Fig. 4A and B). We next validated that these mutations were activating toward Nrf2 in tumors by performing IHC staining for Nrf2 and the Nrf2 target Nqo1. We observed that Keap1R554Q/R554Q expression resulted in the greatest degree of Nrf2 activation, followed by Nrf2D29H/+, and then Keap1R554Q/+ compared with Keap1/Nrf2+/+ expression (Fig. 4C–F). To examine the influence of Nrf2 activation on tumor initiation, we quantified tumor number across the genotypes and found that Keap1R554Q/R554Q and Nrf2D29H/+ significantly increased tumor number in the KrasG12D/+ model (Fig. 4G and H), consistent with prior reports using Keap1 deletion models (29, 30). We then examined the influence of Nrf2 on tumor progression by analyzing tumor grade. The distribution of atypical adenomatous and bronchiolar hyperplasia (AAH and BH, respectively) and tumors from grades 1 (adenoma) to 5 (adenocarcinoma) was determined. We observed an increase in the proportion and number of grade 1 tumors in Keap1R554Q/R554Q and Nrf2D29H/+ mice compared with Keap1R554Q/+ and Keap1/Nrf2+/+ mice (Fig. 4I; Supplementary Fig. S2A). We also examined tumor size by grade and found that the median tumor size did not differ across genotypes (Supplementary Fig. S2B), although there were some modest differences across the individual grades, including decreased AAH size in all Keap1/Nrf2 mutant models (Supplementary Fig. S2C). Moreover, we found that there was a decrease in grade 3 tumor burden across all Keap1 and Nrf2 mutant models (Fig. 4J), which resulted in a decreased overall tumor burden (Supplementary Fig. S2D) because grade 3 tumors were much larger than other grades (Supplementary Fig. S2C). Grade 3 tumors were extremely rare in the KrasG12D/+ model, however, limiting our ability to draw conclusions on the influence of Nrf2 activation on a limited number of tumors. Our findings indicate that KrasG12D/+ mutation cooperates with Keap1/Nrf2 mutation to promote formation of lung tumors and early progression to low-grade tumors.
Figure 4.
![Figure 4. Keap1/Nrf2 mutation cooperates with KrasG12D/+ to promote lung tumor initiation and early progression. A, Overall survival of Keap1 mutant mice with KrasG12D/+ mutation. Keap1+/+ (n = 16); Keap1R554Q/+ (n = 18); Keap1R554Q/R554Q (n = 18). B, Overall survival of Nrf2 mutant mice with KrasG12D/+ mutation. Nrf2+/+ (n = 14); Nrf2D29H/+ (n = 12). ns, not significant [log-rank (Mantel–Cox) test]. C, Representative IHC staining of Nrf2 in Keap1/Nrf2 mutant tumors with KrasG12D/+ mutation. Scale bars, 20 μm. D, H-scores for Nrf2 (nuclear) IHC staining. E, Representative IHC staining of Nrf2 target Nqo1. Scale bars, 20 μm. F, H-scores for Nqo1 (whole cell) IHC staining. C–F, N = 3 mice per genotype and >20,000 tumor cells per mouse. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA). G, Representative whole-lung hematoxylin and eosin–stained section. Scale bars, 2,000 μm. H, Tumor number per mouse in Keap1/Nrf2 mutant models normalized to lung area. *, P < 0.05; **, P < 0.01 (one-way ANOVA). I, Distribution of tumor grades across Keap1/Nrf2 mutant models. *, P < 0.05; **, P < 0.01 (unpaired t test with Holm–Sidak multiple comparisons test). AAH, atypical adenomatous hyperplasia; BH, bronchiolar hyperplasia. J, Fraction of lung tumor burden by grade (lung tumor area/total lung area per grade). *, P < 0.05 (unpaired t test with Holm–Sidak multiple comparisons test). I and J, n = 10 mice and ≥2,000 tumors per genotype.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10267679_1953fig4.jpg)
Keap1/Nrf2 mutation cooperates with KrasG12D/+ to promote lung tumor initiation and early progression. A, Overall survival of Keap1 mutant mice with KrasG12D/+ mutation. Keap1+/+ (n = 16); Keap1R554Q/+ (n = 18); Keap1R554Q/R554Q (n = 18). B, Overall survival of Nrf2 mutant mice with KrasG12D/+ mutation. Nrf2+/+ (n = 14); Nrf2D29H/+ (n = 12). ns, not significant [log-rank (Mantel–Cox) test]. C, Representative IHC staining of Nrf2 in Keap1/Nrf2 mutant tumors with KrasG12D/+ mutation. Scale bars, 20 μm. D, H-scores for Nrf2 (nuclear) IHC staining. E, Representative IHC staining of Nrf2 target Nqo1. Scale bars, 20 μm. F, H-scores for Nqo1 (whole cell) IHC staining. C–F,N = 3 mice per genotype and >20,000 tumor cells per mouse. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA). G, Representative whole-lung hematoxylin and eosin–stained section. Scale bars, 2,000 μm. H, Tumor number per mouse in Keap1/Nrf2 mutant models normalized to lung area. *, P < 0.05; **, P < 0.01 (one-way ANOVA). I, Distribution of tumor grades across Keap1/Nrf2 mutant models. *, P < 0.05; **, P < 0.01 (unpaired t test with Holm–Sidak multiple comparisons test). AAH, atypical adenomatous hyperplasia; BH, bronchiolar hyperplasia. J, Fraction of lung tumor burden by grade (lung tumor area/total lung area per grade). *, P < 0.05 (unpaired t test with Holm–Sidak multiple comparisons test). I and J,n = 10 mice and ≥2,000 tumors per genotype.
Nrf2 activation impairs lung adenocarcinoma progression
The decrease in grade 3 tumor burden in the Keap1/Nrf2 mutant models suggested that Nrf2 activation may impair progression to higher grade tumors. We next used the KrasG12D/+; p53fl/fl (KP) model, which develops advanced-grade lung adenocarcinomas (43). We previously reported that the expression of Keap1R554Q/R554Q dramatically decreased overall tumor size in this model (32), but other groups have found differing effects of Nrf2 activation. Although some studies reported that Keap1 inactivation promoted adenocarcinoma progression (33, 34), others reported that Keap1 deletion did not affect tumor size (36, 37), but the conditions used and phenotypes assayed varied across these studies. Thus, we decided to perform comprehensive phenotyping on KP tumors following Nrf2 activation. Similar to what we observed in the KrasG12D/+ model, we found that OS of the KP model was not affected by Keap1 or Nrf2 mutation (Fig. 5A and B). Moreover, we found that Keap1/Nrf2 mutation affected Nrf2 activation in a similar manner to the KrasG12D/+ model, with Keap1R554Q/R554Q being the most activating, followed by Nrf2D29H/+, and then Keap1R554Q/+ compared with Keap1/Nrf2+/+ mice (Fig. 5C–F). From the histology images, it was very apparent that Keap1R554Q/R554Q mice had decreased overall tumor burden (Fig. 5G), which was confirmed upon quantitation (Supplementary Fig. S3A). Next, we examined tumor progression by tumor grading and found a significant decrease in the proportion, number, and burden of grade 3 and 4 tumors in the Keap1R554Q/R554Q cohort (Fig. 5H and I; Supplementary Fig. S3B). We also observed a modest, nonsignificant decrease in the proportion, number, and burden of grade 3 and 4 tumors in the Nrf2D29H/+ cohort (Fig. 5H and I; Supplementary Fig. S3B), further supporting a threshold for Nrf2 to impair tumor progression. In addition, we saw an increase in the proportion, number, and burden of grade 1 tumors in the Keap1R554Q/R554Q group, suggesting that there may be a threshold for Nrf2 activation to promote early progression, but impair late progression (Fig. 5H and I; Supplementary Fig. S3B). Although there were no differences in overall median tumor size (Supplementary Fig. S3C), Keap1R554Q/R554Q alveolar hyperplasia and grade 1 tumors were significantly smaller compared with their WT counterparts (Supplementary Fig. S3D). Consistently, we found a decrease in tumor cell proliferation in the Keap1R554Q/R554Q cohort, specifically in AAH and grade 1/2 tumors, and no tumor cell death was observed (Supplementary Fig. S3E–S3H). Importantly, despite the reduction in grade 1 size, the higher grade 1 burden in Keap1R554Q/R554Q mice was accounted for by the increase in the total number of grade 1 tumors (Supplementary Fig. S3B and S3I), highlighting that burden is a complex measurement influenced by multiple variables. We next investigated whether tumors that progressed to the adenocarcinoma stage altered Nrf2 expression and/or activity. To this end, we analyzed Nrf2 and Nqo1 levels across all tumor grades and hyperplasia (AAH and BH). We found that Nrf2 and Nqo1 levels were highly elevated in Keap1/Nrf2 mutant grade 1 tumors, with Nqo1 demonstrating increased nuclear localization in homozygous Keap1 mutant tumors compared with Nrf2 mutant tumors (Fig. 6A). However, as tumors progressed to higher grades, Nrf2 and Nqo1 expression was reduced in the Keap1/Nrf2 mutant models (Fig. 6A–C). These results suggest that Nrf2 activation beyond a certain threshold impairs advanced-grade tumor progression, requiring selection for a more tolerable level of Nrf2 expression and activity in high-grade tumors.
Figure 5.
![Figure 5. Homozygous Keap1R554Q impairs adenocarcinoma progression in the KrasG12D/+; p53fl/fl model. A, Overall survival of KrasG12D/+; p53fl/fl mice with Keap1 mutation. Keap1+/+ (n = 20); Keap1R554Q/+ (n = 11); Keap1R554Q/R554Q (n = 25). B, Overall survival of KrasG12D/+; p53fl/fl mice with Nrf2 mutation. Nrf2+/+ (n = 25); Nrf2D29H/+ (n = 29). ns, not significant [log-rank (Mantel–Cox) test]. C, Representative IHC staining of Nrf2 in KrasG12D/+; p53fl/fl mice with Keap1/Nrf2 mutation. Scale bars, 20 μm. D, H-scores for Nrf2 (nuclear) IHC staining. E, Representative IHC staining of Nrf2 target Nqo1. Scale bars, 20 μm. F, H-scores for Nqo1 (whole cell) IHC staining. C–F, N = 3 mice per genotype and >20,000 tumor cells per mouse. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA). G, Representative whole-lung hematoxylin and eosin–stained section. Scale bars, 2,000 μm. H, Distribution of tumor grades across Keap1/Nrf2 mutant models. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (unpaired t test with Holm–Sidak multiple comparisons test). $, fewer than three tumors detected across all mice. I, Fraction of lung tumor burden by grade (lung tumor area per grade/total lung area). *, P < 0.05; ***, P < 0.001 (unpaired t test with Holm–Sidak multiple comparisons test). $, fewer than three tumors detected across all mice. H and I, n ≥ 9 mice and ≥1,900 tumors per genotype. Only one grade 5 tumor was found in the Keap1R554Q/R554Q cohort, and therefore was excluded from these analyses.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10267679_1953fig5.jpg)
Homozygous Keap1R554Q impairs adenocarcinoma progression in the KrasG12D/+; p53fl/fl model. A, Overall survival of KrasG12D/+; p53fl/fl mice with Keap1 mutation. Keap1+/+ (n = 20); Keap1R554Q/+ (n = 11); Keap1R554Q/R554Q (n = 25). B, Overall survival of KrasG12D/+; p53fl/fl mice with Nrf2 mutation. Nrf2+/+ (n = 25); Nrf2D29H/+ (n = 29). ns, not significant [log-rank (Mantel–Cox) test]. C, Representative IHC staining of Nrf2 in KrasG12D/+; p53fl/fl mice with Keap1/Nrf2 mutation. Scale bars, 20 μm. D, H-scores for Nrf2 (nuclear) IHC staining. E, Representative IHC staining of Nrf2 target Nqo1. Scale bars, 20 μm. F, H-scores for Nqo1 (whole cell) IHC staining. C–F,N = 3 mice per genotype and >20,000 tumor cells per mouse. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA). G, Representative whole-lung hematoxylin and eosin–stained section. Scale bars, 2,000 μm. H, Distribution of tumor grades across Keap1/Nrf2 mutant models. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (unpaired t test with Holm–Sidak multiple comparisons test). $, fewer than three tumors detected across all mice. I, Fraction of lung tumor burden by grade (lung tumor area per grade/total lung area). *, P < 0.05; ***, P < 0.001 (unpaired t test with Holm–Sidak multiple comparisons test). $, fewer than three tumors detected across all mice. H and I,n ≥ 9 mice and ≥1,900 tumors per genotype. Only one grade 5 tumor was found in the Keap1R554Q/R554Q cohort, and therefore was excluded from these analyses.
Figure 6.

Nrf2 expression and activity are reduced in higher-grade tumors. A, Representative Nrf2 and Nqo1 IHC staining in grade 1 and 4 tumors from KrasG12D/+; p53fl/fl mice with Keap1 or Nrf2 mutation. Scale bars, 20 μm. B and C, Heatmaps depicting the H-scores per grade from IHC staining for Nrf2 (nuclear; B) and the Nrf2 target Nqo1 (whole cell; C). N = 3 mice per genotype, >20,000 tumor cells per mouse. Only one grade 5 tumor was found in the Keap1R554Q/R554Q cohort, and therefore was excluded from these analyses.
NRF2 overexpression impairs NSCLC cell proliferation, viability, and anchorage-independent colony formation
KEAP1 has other substrates (51–56), raising the question of whether NRF2 plays a causal role in tumor suppression. It was previously reported that KEAP1 mutant lung cancer cell lines are “NRF2 addicted” and dependent on NRF2 for proliferation (57). Supportingly, analysis of DepMap data (45) revealed that NSCLC cell lines with high NRF2 activity (24), which were enriched for KEAP1 mutations, exhibited NRF2 dependence (Fig. 7A). To directly test the hypothesis that excessive NRF2 activation above a specific threshold is detrimental to lung tumor cell growth, we used lentiviral transduction to overexpress NRF2 in five KEAP1 mutant lung cancer cell lines (H1944, H322, A549, HCC15, and H460). We confirmed overexpression of NRF2 by Western blot analysis of NRF2 and target genes GCLC, xCT, and GSR (Fig. 7B). Lentiviral transduction increased the expression of NRF2 in all cell lines, and increased the expression of NRF2 targets, demonstrating that NRF2-binding sites were not saturated by the level of NRF2 in these cell lines (Fig. 7B). Next, we determined the influence of NRF2 overexpression on cellular proliferation and death over the course of four days using live cell imaging (Fig. 7C–G). Like what we observed in the KrasG12D/+; p53fl/fl model upon Keap1R554Q/R554Q expression, we found that NRF2 overexpression decreased cell proliferation in all cell lines (Fig. 7F). Moreover, Nrf2 overexpression increased cell death in 4 out of 5 cell lines (Fig. 7G). Finally, we observed impaired anchorage-independent growth in soft agar in all cell lines (Fig. 7H and I). These results indicate that there is an optimal threshold of NRF2 activity, and that excess NRF2 activation can impair lung cancer phenotypes.
Figure 7.

NRF2 overexpression impairs lung cancer cell proliferation, viability, and soft agar colony formation. A, Dependency scores obtained from DepMap (45) and represented as NFEL2 22Q2 Public+Score, Chronos for NSCLC cell lines previously determined to have high or low NRF2 activity (24). Yellow symbols, NRF2 mutant line; dark red or dark blue symbols, KEAP1 mutant lines; green symbol, KEAP1-deleted lines. ****, P < 0.0001 (unpaired t test). B, Western blot analysis of NRF2, β-actin, and NRF2 target GCLC, xCT, and GSR expression in KEAP1 mutant lung cancer cell lines transduced with PLX317-empty vector (EV) or PLX317-NRF2 (NRF2). C, Representative images of HCC15 cells transduced with empty vector or NRF2 demonstrating cell confluency (enhanced contour) and cell death (Sytox Green). Scale bars, 100 μm. D and E, Analysis of empty vector and NRF2 HCC15 cell proliferation and death over 96 hours. Proliferation is represented as the percentage of confluency at each time point, and cell death as the number of Sytox Green–positive cells per area normalized to the percentage of confluency. N = 3 technical replicates per cell line and two independent experiments. F and G, AUC analysis of cell proliferation (F) and Sytox Green–positive cell death (G) in KEAP1 mutant lung cancer cells lines ± NRF2, normalized to empty vector control. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA). C–G, NSCLC cells were seeded in triplicate in 96-well plates at a density of 2,500 cells/well. H, Representative images of H460 and HCC15 soft agar colony formation ± NRF2. I, Quantification of soft agar colony number of KEAP1 mutant lung cancer cell lines. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA). N = 3 technical replicates per cell line; two independent experiments. H and I, 5,000 cells per well were seeded in 6-well plates in triplicate.
Single-copy Nrf2 deletion rescues homozygous Keap1R554Q-mediated tumor progression impairment
To directly examine whether reducing Nrf2 levels could alleviate the block in adenocarcinoma progression in the Keap1R554Q/R554Q model, we crossed an Nrf2flox allele into both our KrasG12D/+; p53fl/fl and KrasG12D/+; p53fl/fl; Keap1R554Q/R554Q models. Because we previously found that complete Nrf2 deficiency impairs tumor initiation (23), we examined the consequence of single copy Nrf2 deletion on tumor phenotypes. Again, we found no difference in OS between groups (Fig. 8A). We also observed that Nrf2 deletion in the Keap1R554Q/R554Q model significantly decreased expression of Nrf2 and Nqo1 (Fig. 8B–E; Supplementary Fig. S4A and S4B). Moreover, histological examination of the lungs revealed a striking difference in tumor burden, with Nrf2 heterozygous deletion having a minimal effect on the KrasG12D/+; p53fl/fl model while dramatically increasing burden in the KrasG12D/+; p53fl/fl; Keap1R554Q/R554Q model (Fig. 8F; Supplementary Fig. S5A). Tumor size was not affected (Supplementary Fig. S5B and S5C). Next, we examined tumor progression in these models. In agreement with our previous experiment (Fig. 5H), we found that the Keap1R554Q/R554Q cohort had a significant reduction in adenocarcinoma progression upon Nrf2WT expression (Fig. 8G). However, upon single-copy Nrf2 deletion (Nrf2flox/+), Keap1R554Q/R554Q failed to suppress tumor progression (Fig. 8G). Moreover, the decrease in grade 3 tumor number and burden induced by Keap1R554Q/R554Q was alleviated by single-copy deletion of Nrf2 (Fig. 8H; Supplementary Fig. S5D and S5E). These results demonstrate that there is a threshold by which Nrf2 activation can promote or impair tumor initiation or progression.
Figure 8.
![Figure 8. Single-copy Nrf2 deletion rescues homozygous Keap1R554Q-mediated adenocarcinoma progression impairment in the KrasG12D/+; p53fl/fl model. A, Overall survival of KrasG12D/+; p53fl/fl mice with Keap1 mutation and/or single-copy Nrf2 deletion. Keap1+/+; Nrf2+/+ (n = 16), Keap1+/+; Nrf2fl/+ (n = 7), Keap1R554Q/R554Q; Nrf2+/+ (n = 10), Keap1R554Q/R554Q; Nrf2fl/+ (n = 11). ns, not significant [log-rank (Mantel–Cox) test]. B, Representative IHC staining of Nrf2 in KrasG12D/+; p53fl/fl mice with Keap1 mutation and/or heterozygous Nrf2 deletion. Scale bars, 20 μm. C, H-scores for Nrf2 (nuclear) IHC staining. D, Representative IHC staining of Nrf2 target Nqo1. Scale bars, 20 μm. E, H-scores for Nqo1 (whole-cell) IHC staining. B–E, N = 3 mice per genotype and >20,000 tumor cells per mouse. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 (one-way ANOVA). F, Representative whole-lung hematoxylin and eosin–stained sections. Scale bars, 2,000 μm. G, Distribution of tumor grades across Keap1 mutant/Nrf2-deleted models. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (unpaired t test with Holm–Sidak multiple comparisons test). $, fewer than three tumors detected across all mice. H, Fraction of lung tumor burden by grade (lung tumor area per grade/total lung area). *, P < 0.05; **, P < 0.01; ****, P < 0.0001 (unpaired t test with Holm–Sidak multiple comparisons test). $, fewer than three tumors detected across all mice. G and H, n = 7 mice and ≥1,000 tumors per genotype were analyzed. Only one grade 4 and one grade 5 tumor were found in the Keap1R554Q/R554Q cohort, and therefore were excluded from these analyses.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10267679_1953fig8.jpg)
Single-copy Nrf2 deletion rescues homozygous Keap1R554Q-mediated adenocarcinoma progression impairment in the KrasG12D/+; p53fl/fl model. A, Overall survival of KrasG12D/+; p53fl/fl mice with Keap1 mutation and/or single-copy Nrf2 deletion. Keap1+/+; Nrf2+/+ (n = 16), Keap1+/+; Nrf2fl/+ (n = 7), Keap1R554Q/R554Q; Nrf2+/+ (n = 10), Keap1R554Q/R554Q; Nrf2fl/+ (n = 11). ns, not significant [log-rank (Mantel–Cox) test]. B, Representative IHC staining of Nrf2 in KrasG12D/+; p53fl/fl mice with Keap1 mutation and/or heterozygous Nrf2 deletion. Scale bars, 20 μm. C, H-scores for Nrf2 (nuclear) IHC staining. D, Representative IHC staining of Nrf2 target Nqo1. Scale bars, 20 μm. E, H-scores for Nqo1 (whole-cell) IHC staining. B–E,N = 3 mice per genotype and >20,000 tumor cells per mouse. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 (one-way ANOVA). F, Representative whole-lung hematoxylin and eosin–stained sections. Scale bars, 2,000 μm. G, Distribution of tumor grades across Keap1 mutant/Nrf2-deleted models. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (unpaired t test with Holm–Sidak multiple comparisons test). $, fewer than three tumors detected across all mice. H, Fraction of lung tumor burden by grade (lung tumor area per grade/total lung area). *, P < 0.05; **, P < 0.01; ****, P < 0.0001 (unpaired t test with Holm–Sidak multiple comparisons test). $, fewer than three tumors detected across all mice. G and H,n = 7 mice and ≥1,000 tumors per genotype were analyzed. Only one grade 4 and one grade 5 tumor were found in the Keap1R554Q/R554Q cohort, and therefore were excluded from these analyses.
Discussion
Using GEMMs of Keap1/Nrf2 mutation, we find that Keap1 or Nrf2 mutations alone are insufficient to cause lung tumor formation. Even in combination with tumor-suppressor loss, we did not observe lung tumor formation after 500 days, suggesting that chronic NRF2 activation would be a safe strategy for chemoprevention. These findings corroborate previous studies showing that Keap1 deletion does not induce lung tumor development (28), even in the absence of tumor-suppressors p53 or Lkb1 (29), out to 12–15 months. In combination with KrasG12D/+ mutation, we found that Nrf2 activation promoted tumor initiation. Our results are consistent with previous work, indicating that Nrf2 activation via Keap1 deletion promotes KrasG12D/+ tumor initiation (29, 30). Surprisingly, we find that Nrf2 activation impairs tumor progression, which is correlated with Nrf2 dosage. In our previous study with Keap1R554Q/R554Q mice, we reported smaller tumors in the KP model (32), which we now find is due to impaired tumor progression mediated by Nrf2 hyperactivation. Because most prior studies from other groups did not specifically analyze tumor grade (27, 29, 36, 37), they may not have captured this effect of Nrf2 on tumor progression. Alternatively, there may be biological differences between Keap1 deletion and mutation. Moreover, the dosage-dependent effects of Nrf2 that we found are also reminiscent of what has been observed for oncogenes like Ras and Myc, where low levels promote transformation and proliferation, and high levels promote senescence or death (58, 59).
Our findings that tumors downregulate Nrf2 to select for a level permissive for tumor progression and that ectopic NRF2 expression antagonizes the proliferation and viability of human NSCLC cells are supported by our finding that single-copy deletion of Nrf2 can rescue the homozygous Keap1R554Q-mediated block in tumor progression. This result demonstrates a direct role for Nrf2 hyperactivation but we cannot exclude the possibility that alternative KEAP1 substrates, such as PGAM5 (51), PALB2 (52), MCM3 (53) or EMSY (54), contribute to the block in tumor progression. In agreement with our findings, a Keap1-binding defective Nrf2E79Q mouse model of small-cell lung cancer (SCLC) with p53/ p16 inactivation (60) also displayed Nrf2 downregulation in aggressive SCLC tumors. The exact mechanism(s) by which high NRF2 activity impairs tumor cell proliferation and tumor progression remains to be determined. NRF2 activation is associated with multiple metabolic liabilities that could explain this phenotype, including CDO1-mediated toxic metabolite formation and NADPH depletion (32), xCT-mediated glutamate depletion (26, 34, 61), and ALDH3A1-mediated reductive stress (62). Future studies are needed to determine whether these or other mechanisms play a role in NRF2-mediated proliferation and progression impairment.
Given that KEAP1 and NRF2 mutations are found with a high frequency in human NSCLC and are associated with poor outcomes, these findings raise the question of under which contexts NRF2 activation provides an advantage. Our findings that NRF2/KEAP1 mutation promotes tumor initiation are consistent with recent results from the TRACERx study, where KEAP1 mutations were found to be an initiating driver together with KRAS and TP53 mutations in lung adenocarcinoma (63), and suggest that additional genetic events may be needed to overcome NRF2-mediated inhibition of tumor progression. We did not find that these mutations conferred poor outcomes in our mouse models, but there are multiple features of patient tumors not captured by our models that remain to be examined. They did not develop metastases with sufficient frequency, precluding an examination of the influence of Nrf2 activation on metastasis as reported previously (27). Moreover, mice are not exposed to smoking and other environmental toxins under which NRF2 activation may promote survival (4–7). We have also not tested the response of these models to therapy. Additional mechanistic studies are warranted to clarify the clinical translation of these approaches, including understanding how NRF2 is downregulated upon progression and the consequences of NRF2 pharmacological activation and inhibition on the various tumor grades in our GEMM models. These studies will be important to compare the level of NRF2 achieved with pharmacological manipulation to the level induced by KEAP1/NRF2 mutation and where that falls on the threshold spectrum. Moreover, our models are designed to only activate NRF2 in cancer cells whereas these pharmacological agents will modulate NRF2 in the microenvironment, which will also influence tumor growth (30, 64, 65). These future studies will further clarify the context-dependent role of NRF2 during the complex stages of tumorigenesis.
Supplementary Material
Supplementary Fig. S1 shows that the CA-Keap1R554Q allele does not exhibit hypomorphism in mouse embryonic fibroblasts.
Supplementary Fig. S2 shows lung tumor analysis data for Keap1/Nrf2 mutant mice in the KrasG12D/+ model.
Supplementary Fig. S3 shows analysis of lung tumor burden and tumor cell proliferation/death in the Kras; p53 model with Keap1/Nrf2 mutation.
Supplementary Fig. S4 shows the H-scores by tumor grade of Nrf2 and Nqo1 immunohistochemical staining in KrasG12D/+; p53fl/fl and KrasG12D/+; p53fl/fl; Keap1R554Q/R554Q mouse models with single copy Nrf2 deletion.
Supplementary Fig. S5 shows lung tumor analysis data in the KrasG12D/+; p53fl/fl model with single copy Nrf2 deletion.
Acknowledgments
This work was supported by grants from the NIH/NCI (R37-CA230042), the American Lung Association (LCDA-498544), and the American Cancer Society's Institutional Research grant (to G.M. DeNicola). This work was also supported by the Analytical Microscopy and the Tissue Core Facilities at the Moffitt Cancer Center, an NCI-designated Comprehensive Cancer Center (P30-CA076292). The authors would like to thank the DeNicola laboratory members and Dr. A. Gomes for the helpful discussions. They thank Cheyenne Schneider for proofreading.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
E.E. Schmidt reports grants from Montana State University during the conduct of the study. G.M. DeNicola reports grants from NIH/NCI, American Lung Association, and American Cancer Society during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
J.M. DeBlasi: Data curation, formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. A. Falzone: Data curation, investigation, methodology, writing–review and editing. S. Caldwell: Data curation, investigation, methodology, writing–review and editing. N. Prieto-Farigua: Data curation, investigation, methodology, writing–review and editing. J.R. Prigge: Resources, writing–review and editing. E.E. Schmidt: Resources, writing–review and editing. I.I.C. Chio: Resources, writing–review and editing. F.A. Karreth: Resources, methodology, writing–review and editing. G.M. DeNicola: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. DeBlasi JM, DeNicola GM. Dissecting the crosstalk between NRF2 signaling and metabolic processes in cancer. Cancers 2020;12:3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci 2020;21:4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999;13:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramos-Gomez M, Dolan PM, Itoh K, Yamamoto M, Kensler TW. Interactive effects of nrf2 genotype and oltipraz on benzo[ a]pyrene–DNA adducts and tumor yield in mice. Carcinogenesis 2003;24:461–7. [DOI] [PubMed] [Google Scholar]
- 5. Ramos-Gomez M, Kwak M-K, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2transcription factor-deficient mice. Proc Natl Acad Sci USA 2001;98:3410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol 2001;173:154–60. [DOI] [PubMed] [Google Scholar]
- 7. Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res 2004;64:6424–31. [DOI] [PubMed] [Google Scholar]
- 8. Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis 2010;31:90–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmidlin CJ, Shakya A, Dodson M, Chapman E, Zhang DD. The intricacies of NRF2 regulation in cancer. Semin Cancer Biol 2021;76:110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Telkoparan-Akillilar P, Suzen S, Saso L. Pharmacological applications of Nrf2 inhibitors as potential antineoplastic drugs. Int J Mol Sci 2019;20:2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Network CGAR. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Network CGAR. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1–NRF2 interaction in non–small cell lung cancer. PLoS Med 2006;3:e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci USA 2008;105:13568–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang R, An J, Ji F, Jiao H, Sun H, Zhou D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun 2008;373:151–4. [DOI] [PubMed] [Google Scholar]
- 16. Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol 2010;220:446–51. [DOI] [PubMed] [Google Scholar]
- 17. Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, et al. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther 2010;9:336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, et al. Nrf2 and Keap1 abnormalities in non–small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res 2010;16:3743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Q, Xu L, Wang G, Chen L, Li C, Jiang X, et al. Prognostic and clinicopathological significance of NRF2 expression in non–small cell lung cancer: a meta-analysis. PLoS ONE 2020;15:e0241241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao J, Lin X, Meng D, Zeng L, Zhuang R, Huang S, et al. Nrf2 mediates metabolic reprogramming in non–small cell lung cancer. Front Oncol 2020;10:578315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer 2012;12:564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singh A, Bodas M, Wakabayashi N, Bunz F, Biswal S. Gain of Nrf2 function in non–small cell lung cancer cells confers radioresistance. Antioxidants and Redox Signaling 2010;13:1627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Denicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011;475:106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. NRF2 regulates serine biosynthesis in non–small cell lung cancer. Nat Genet 2015;47:1475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012;22:66–79. [DOI] [PubMed] [Google Scholar]
- 26. Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 2017;6:e28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell 2019;178:316–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Best SA, De Souza DP, Kersbergen A, Policheni AN, Dayalan S, Tull D, et al. Synergy between the KEAP1/NRF2 and PI3K pathways drives non–small cell lung cancer with an altered immune microenvironment. Cell Metab 2018;27:935–43. [DOI] [PubMed] [Google Scholar]
- 29. Best SA, Ding S, Kersbergen A, Dong X, Song JY, Xie Y, et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat Commun 2019;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hayashi M, Kuga A, Suzuki M, Panda H, Kitamura H, Motohashi H, et al. Microenvironmental activation of Nrf2 restricts the progression of Nrf2-activated malignant tumors. Cancer Res 2020;80:3331–44. [DOI] [PubMed] [Google Scholar]
- 31. Jeong Y, Hoang NT, Lovejoy A, Stehr H, Newman AM, Gentles AJ, et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov 2017;7:86–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang YP, Torrente L, Falzone A, Elkins CM, Liu M, Asara JM, et al. Cysteine dioxygenase 1 is a metabolic liability for non–small cell lung cancer. eLife 2019;8:e45572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Romero R, Sánchez-Rivera FJ, Westcott PMK, Mercer KL, Bhutkar A, Muir A, et al. Keap1 mutation renders lung adenocarcinomas dependent on Slc33a1. Nature Cancer 2020;1:589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, Leboeuf SE, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 2017;23:1362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singh A, Daemen A, Nickles D, Jeon S-M, Foreman O, Sudini K, et al. NRF2 activation promotes aggressive lung cancer and associates with poor clinical outcomes. Clin Cancer Res 2021;27:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Foggetti G, Li C, Cai H, Hellyer JA, Lin W-Y, Ayeni D, et al. Genetic determinants of EGFR-driven lung cancer growth and therapeutic response in vivo. Cancer Discov 2021;11:1736–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rogers ZN, McFarland CD, Winters IP, Seoane JA, Brady JJ, Yoon S, et al. Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat Genet 2018;50:483–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cai H, Chew SK, Li C, Tsai MK, Andrejka L, Murray CW, et al. A functional taxonomy of tumor suppression in oncogenic KRAS-driven lung cancer. Cancer Discov 2021;11:1754–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rogers ZN, McFarland CD, Winters IP, Naranjo S, Chuang C-H, Petrov D, et al. A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat Methods 2017;14:737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001;15:3243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McLoughlin MR, Orlicky DJ, Prigge JR, Krishna P, Talago EA, Cavigli IR, et al. TrxR1, Gsr, and oxidative stress determine hepatocellular carcinoma malignancy. Proc Natl Acad Sci USA 2019;116:11408–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 2005;65:10280–8. [DOI] [PubMed] [Google Scholar]
- 44. Berger AH, Brooks AN, Wu X, Shrestha Y, Chouinard C, Piccioni F, et al. High-throughput phenotyping of lung cancer somatic mutations. Cancer Cell 2016;30:214–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DepMap, Broad (2022): DepMap 22Q2 Public. figshare. Dataset. doi: https://doi.org/10.6084/m9.figshare.19700056.v2.
- 46. Hast BE, Cloer EW, Goldfarb D, Li H, Siesser PF, Yan F, et al. Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res 2014;74:808–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, et al. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol 2007;27:7511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Taguchi K, Maher JM, Suzuki T, Kawatani Y, Motohashi H, Yamamoto M. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol Cell Biol 2010;30:3016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 2003;35:238–45. [DOI] [PubMed] [Google Scholar]
- 50. Huppke P, Weissbach S, Church JA, Schnur R, Krusen M, Dreha-Kulaczewski S, et al. Activating de novo mutations in NFE2L2 encoding NRF2 cause a multisystem disorder. Nat Commun 2017;8:818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lo SC, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res 2008;314:1789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol Cell Biol 2012;32:1506–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mulvaney KM, Matson JP, Siesser PF, Tamir TY, Goldfarb D, Jacobs TM, et al. Identification and characterization of MCM3 as a kelch-like ECH-associated protein 1 (KEAP1) substrate. J Biol Chem 2016;291:23719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marzio A, Kurz E, Sahni JM, Di Feo G, Puccini J, Jiang S, et al. EMSY inhibits homologous recombination repair and the interferon response, promoting lung cancer immune evasion. Cell 2022;185:169–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang Y, Shi Z, Zhou Y, Xiao Q, Wang H, Peng Y. Emerging substrate proteins of kelch-like ECH associated protein 1 (Keap1) and potential challenges for the development of small-molecule inhibitors of the keap1–nuclear factor erythroid 2-related factor 2 (Nrf2) protein–protein interaction. J Med Chem 2020;63:7986–8002. [DOI] [PubMed] [Google Scholar]
- 56. McCutcheon DC, Lee G, Carlos A, Montgomery JE. Moellering RE. photoproximity profiling of protein–protein interactions in cells. J Am Chem Soc 2020;142:146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kitamura H, Motohashi H. NRF2 addiction in cancer cells. Cancer Sci 2018;109:900–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004;5:375–87. [DOI] [PubMed] [Google Scholar]
- 59. Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, et al. Distinct thresholds govern Myc's biological output in vivo. Cancer Cell 2008;14:447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hamad SH, Montgomery SA, Simon JM, Bowman BM, Spainhower KB, Murphy RM, et al. TP53, CDKN2A/P16, and NFE2L2/NRF2 regulate the incidence of pure- and combined-small cell lung cancer in mice. Oncogene 2022;41:3423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, Vander Heiden MG. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 2017;6:e27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weiss-Sadan T, Ge M, Hayashi M, Gohar M, Yao C-H, de Groot A, et al. NRF2 activation induces NADH-reductive stress, providing a metabolic vulnerability in lung cancer. Cell Metab 2023;35:487–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non–small cell lung cancer. N Engl J Med 2017;376:2109–21. [DOI] [PubMed] [Google Scholar]
- 64. Satoh H, Moriguchi T, Saigusa D, Baird L, Yu L, Rokutan H, et al. NRF2 intensifies host defense systems to prevent lung carcinogenesis, but after tumor initiation accelerates malignant cell growth. Cancer Res 2016;76:3088–96. [DOI] [PubMed] [Google Scholar]
- 65. Satoh H, Moriguchi T, Taguchi K, Takai J, Maher JM, Suzuki T, et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 2010;31:1833–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1 shows that the CA-Keap1R554Q allele does not exhibit hypomorphism in mouse embryonic fibroblasts.
Supplementary Fig. S2 shows lung tumor analysis data for Keap1/Nrf2 mutant mice in the KrasG12D/+ model.
Supplementary Fig. S3 shows analysis of lung tumor burden and tumor cell proliferation/death in the Kras; p53 model with Keap1/Nrf2 mutation.
Supplementary Fig. S4 shows the H-scores by tumor grade of Nrf2 and Nqo1 immunohistochemical staining in KrasG12D/+; p53fl/fl and KrasG12D/+; p53fl/fl; Keap1R554Q/R554Q mouse models with single copy Nrf2 deletion.
Supplementary Fig. S5 shows lung tumor analysis data in the KrasG12D/+; p53fl/fl model with single copy Nrf2 deletion.
Data Availability Statement
The data generated in this study are included in the article and its Supplementary Figures. Raw data are available upon request without restriction from the corresponding author. The gene expression data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE212942. The data for the NFE2L2 dependency scores analyzed in this study were obtained from the DepMap database v. 22Q2 at https://depmap.org/portal/download/all/. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Gina M. DeNicola (gina.denicola@moffitt.org).