

Significance
A hallmark of tumor growth is the formation of immunosuppressive tumor milieu termed tumor immune microenvironment (TIME). Tumors create a favorable environment for their own propagation by expressing or releasing immunosuppressive molecules. In this study, we report that tumor cells release endogenous polyamine spermidine to suppress T cell activation in the TIME. Mechanistically, spermidine down-regulates plasma membrane cholesterol levels, thereby suppressing T cell antigen receptor clustering. This study raises the prospect of targeting the spermidine–TIME axis as a promising strategy for invigorating otherwise hyporesponsive T cells for an improved tumor immunotherapy.
Keywords: cell death, T cell, cancer immunotherapy, oncometabolite, spermidine
Abstract
The activation and expansion of T cells that recognize cancer cells is an essential aspect to antitumor immunity. Tumors may escape destruction by the immune system through ectopic expression of inhibitory immune ligands typically exemplified by the PD-L1/PD-1 pathway. Here, we reveal another facet of tumor evasion from T cell surveillance. By secretome profiling of necrotic tumor cells, we identified an oncometabolite spermidine as a unique inhibitor of T cell receptor (TCR) signaling. Mechanistically, spermidine causes the downregulation of the plasma membrane cholesterol levels, resulting in the suppression of TCR clustering. Using syngeneic mouse models, we show that spermidine is abundantly detected in the tumor immune microenvironment (TIME) and that administration of the polyamine synthesis inhibitor effectively enhanced CD8+ T cell–dependent antitumor responses. Further, the combination of the polyamine synthesis inhibitor with anti-PD-1 immune checkpoint antibody resulted in a much stronger antitumor immune response. This study reveals an aspect of immunosuppressive TIME, wherein spermidine functions as a metabolic T cell checkpoint that may offer a unique approach for promoting tumor immunotherapy.
It is now widely appreciated that the immune system is critical to prevent tumor pathogenesis (1). CD8+ T cells are best known as critical players in the eradication of tumor cells; however, they often fall into a hyporesponsive state, as characterized by weak proliferation and reduced production of tumoricidal effector molecules (e.g., IFN (Interferon)-γ, TNF (Tumor necrosis factor)-α, and granzyme B) (2). There exists a variety of mechanisms for this immunosuppressive state, of which perhaps the best known is the upregulation of checkpoint molecules such as programmed death-1 (PD-1) and T cell immunoglobulin and mucin domain–containing protein 3 within tumor-specific effector T cells (3). This multifacet nature of the immunosuppressive mechanisms may be underscored by insufficient efficacy of tumor immunotherapies including immune checkpoint blockade (ICB) (4).
In recent years, substantial efforts have been devoted to deciphering the underlying molecular mechanisms of T cell hyporesponsiveness within the tumor immune microenvironment (TIME). Many of these have mainly focused on characterizing the molecular features of intratumoral CD8+ T cells, such as transcriptomic and metabolic states, and successfully identified multiple T cell–intrinsic factors responsible for their curtailed tumoricidal activities (5, 6). On the contrary, accumulating evidence suggests that the functional fate of T cells is determined by the integration of numerous environmental cues (7, 8). In tumor-bearing hosts, tumors themselves constitute immunosuppressive niches that favor their escape from immune destruction (9, 10). In this context, attention has been focused on metabolites that regulate T cells acting as metabolic checkpoints (11, 12).
It has been known that tumor necrotic cell death promotes immune suppression. Tumor cell necrosis is caused by several stress stimuli including nutrient deprivation and hypoxia (13, 14), and expansion of areas of necrotic tumor is a pathological hallmark of advanced-stage solid tumor and indicator of poor prognosis (15). Immunosuppression mediated by necrotic tumor in the context of tumor immunity has been ascribed to the release of intracellular components termed damage-associated molecular patterns (DAMPs) or alarmins, originally defined as mediators of sterile inflammation (16, 17). Among the factors described includes translationally controlled tumor protein which promotes tumor growth through the induction of chemokines to recruit myeloid-derived suppressor cells (MDSCs) to the TIME (18). In addition to those DAMPs, potassium ions (K+) extracellularly released upon tumor necrosis have been shown to accelerate T cell dysfunction within tumors (19). These reports underscore the complexity of immunosuppression in the TIME and clearly point to the importance of better understanding for how the cell death affects TIME.
In the present study, we identified spermidine, a natural mammalian polyamine described as being released upon cell death of stressed tumor cells, as an oncometabolite of T cell suppression in the TIME. The underlying mechanism for the suppression is unique in that spermidine suppresses plasma membrane cholesterol levels of T cells, thereby inhibiting the T cell receptor (TCR) clustering. We discuss our findings in the context of approaches for tumor immunotherapy.
Results
Biochemical Analysis of Immunosuppressive Molecules from Necrotic Tumor Cells.
We first examined the relationship between tumor necrosis and antitumor T cell responses. As reported previously, we found dense areas of cellular necrosis in both mouse and human solid tumors by histopathological analysis (SI Appendix, Fig. S1 A and B) (9, 15, 18). In addition, analysis using TCGA (The Cancer Genome Atlas) pathological and gene expression data revealed a negative correlation between the size of necrotic cell area and the transcriptomic signature of effector CD8+ T cell responses, including CD8A and TBX21 expression (SI Appendix, Fig. S1C). These observations suggest that necrotic tumor cells may release a molecule(s) that suppress antitumor CD8+ T cell responses in the tumor milieu.
In an attempt to identify a molecule(s) that suppresses the CD8+ T cell responses, we utilized conditioned media prepared by inducing necrosis in B16F10 melanoma cells, which are known to induce hyporesponsiveness of intratumoral CD8+ T cells when grown in vivo (20). Interestingly, necrotic cell supernatant prepared from freeze-and-thaw-induced B16F10 cells (F–T sup) significantly suppressed TCR-induced production of effector cytokines and proliferation of mouse CD8+ T cells in vitro in a dose-dependent manner (Fig. 1 A and B and SI Appendix, Fig. S2A). Similar results were also obtained when necrotic B16F10 cell supernatants were prepared following glucose starvation or hypoxic stress (Fig. 1 C and D). Since the supernatant derived from normally cultured intact B16F10 cells did not affect T cell activity (Fig. 1 C and D), it is most likely that B16F10-derived T cell inhibitory molecule(s) is released upon necrotic cell death.
Fig. 1.

Necrotic B16F10 cell supernatant suppresses CD8+ T cell activation. (A–G) Effect of necrotic B16F10 cell supernatant on in vitro CD8+ T cell activation and proliferation induced by anti-CD3/CD28 antibodies (Abs). (A) Representative flow cytometry (FCM) plots (Left) and summary (Right) of TNF-α, IFN-γ, and IL-2 (Interleukin-2) expression in CD8+ T cells left untreated (Ctrl) or treated with supernatant prepared from freeze–thawed B16F10 cells (F–T sup). The numbers in the FCM plots indicate the percentage of cells within the CD8+ T cell population. (B) CD8+ T cells were treated as in (A), and Ki-67 expression was analyzed by FCM. (C and D) CD8+ T cells were treated with supernatants prepared from glucose-starved (Glucose starv.) or oxygen-deprived (Hypoxia) necrotic B16F10 cells. Culture supernatant of B16F10 cells grown under normal culture condition (Normal) was also used. Expression of IFN-γ, IL-2, and TNF-α (C), and Ki-67 (D) was examined by FCM. (E and F) CD8+ T cells were treated with F–T sup fractionated into MW >3 kDa and <3 kDa by ultrafiltration filters. Expression of IFN-γ, IL-2 and TNF-α (E), and Ki-67 (F) was examined by FCM. (G) CD8+ T cells were treated with the aqueous metabolite fraction extracted from <3 kDa F–T sup in (E and F). IFN-γ expression was analyzed by FCM. *P < 0.05; **P < 0.01; ***P < 0.001 compared with the control (Ctrl) group unless otherwise indicated; ns, not significant; data are presented as the mean ± SD.
In order to further characterize the molecular nature of the inhibitory molecule(s), we treated B16F10 F–T sup with heat or proteinase K; however, neither of these treatments affected the inhibitory activity on T cells (SI Appendix, Fig. S2 B and C). Further, molecular weight (MW) fractionation of the F–T sup revealed that T cell inhibitory activities were detectable in the fraction with MW smaller than 3 kDa (<3 kDa) (Fig. 1 E and F and SI Appendix, Fig. S2D). Of note, this fraction also suppressed the cytokine-producing activities of CD4+ T cells (SI Appendix, Fig. S2 E and F). These data suggest the presence of nonprotein molecule(s) such as metabolites that affect both CD8+ and CD4+ T cells. Consistent with this, the metabolite-enriched fraction of B16F10 F–T sup also suppressed IFN-γ production in in vitro cultured CD8+ T cells (Fig. 1G).
Identification of the T Cell Suppressive Molecule.
We then attempted to identify the T cell inhibitory molecule(s) by conducting capillary electrophoresis-time-of-flight mass spectrometry (CE-TOFMS)–based comprehensive metabolome analysis. Nine metabolites were identified as candidates for immunosuppression when the metabolite-enriched fractions of a variety of B16F10 conditioned media (Fig. 2A andB and Materials and Methods) were each subjected to the analysis: adenosine monophosphate (AMP), guanosine monophosphate (GMP), cytidine monophosphate (CMP), uridine monophosphate (UMP), nicotinamide, creatine, β-alanine, taurine, and spermidine (Fig. 2 C and D).
Fig. 2.

Comprehensive metabolome analysis identifies necrotic cell–released spermidine as a modulator of CD8+ T cell activation. (A) Schematic for assessing the metabolite secretome of necrotic B16F10 cells. (B) Principal component analysis on the metabolomics data of the supernatant samples in (A). (C) Venn diagrams illustrating metabolomics data in (B). Nine overlapped metabolites were identified according to the criteria described in Materials and Methods. (D) Concentrations of the metabolites in (C) in the supernatants of live (Normal) or necrotic (Glucose starv., Hypoxia, and F–T#) B16F10 cells. (E) CD8+ T cells were left untreated or treated with each of the metabolites shown in (C and D) at different concentrations, and anti-CD3/CD28-induced IFN-γ production was analyzed by FCM. The ratio of IFN-γ expression to untreated (Ctrl) condition is depicted in the line graph. (F) CD8+ T cells were treated with F–T sup prepared from B16F10 cells left untreated or treated with eflornithine, and then anti-CD3/CD28-induced IFN-γ expression was analyzed by FCM. (G) Spermidine concentrations in serum from normal and B16F10 tumor–bearing mice (N = 8 to 13/group, Left panel), or serum and TIF of B16F10 tumor–bearing mice (N = 5/group, Right panel), were examined. Identical symbols indicate that samples originated from the same mouse. *P < 0.05; **P < 0.01; ns, not significant; data are presented as the mean ± SD.
To narrow down the identity of the specific T cell inhibitory metabolite, each of these was administered in the in vitro T cell culture system at various concentrations to examine the effect on the TCR activation–induced IFN-γ production. As shown in Fig. 2E, spermidine exhibited potent inhibitory effects on IFN-γ production at low concentration ranges (≤ 12.5 µM), while other metabolites showed little or modest effect even at a high concentration of up to 100 µM. Notably, F–T sup prepared from B16F10 cells treated with eflornithine, an inhibitor of spermidine synthesis targeting ornithine decarboxylase 1 (ODC1) (21), showed little, if any, inhibitory effect on IFN-γ production (Fig. 2F and SI Appendix, Fig. S3A), supporting the notion that spermidine is responsible for the T cell suppression. Perhaps expectedly, spermidine was also detected in the necrotic cell supernatants of other tumor cell lines, such as mouse SL4 and human Caco-2 and A549 (SI Appendix, Fig. S3B).
Spermidine is a polyamine, a ubiquitous and naturally occurring polycationic alkylamine characterized as an oncometabolite whose abnormal intracellular accumulation is triggered by oncogenic signaling (21–23). Accumulating evidence indicates that polyamines are released extracellularly, with high concentrations of polyamines detected in urine and saliva of cancer patients; they can thus be used as potential tumor biomarkers (23–25). Indeed, we found that spermidine concentration was remarkably up-regulated in the tumor interstitial fluid (TIF) of the B16F10 tumor–bearing mice, while the serum spermidine levels were low and similar between normal and B16F10 tumor–bearing mice (Fig. 2G). The results lend support to the notion that spermidine is locally released by tumor cell death. Since TIF represents an informative surrogate for the extracellular milieu of the local tumor site (26, 27), it is likely that the high spermidine levels in TIF provided by the tumor are critical to the regulation of intratumoral immune responses.
Suppression of T Cell Activation by Spermidine.
We next examined whether spermidine affects T cell activation in vitro. As shown in Fig. 3 A and B, spermidine suppressed the production of cytokines and expression of Ki-67, a hallmark of T cell proliferation, in CD8+ T cells stimulated by anti-CD3/CD28 antibodies. Similar observations were also made with CD4+ T cells, indicating that spermidine affects the activation of both types of T cells in this experimental setting (SI Appendix, Fig. S4A).
Fig. 3.
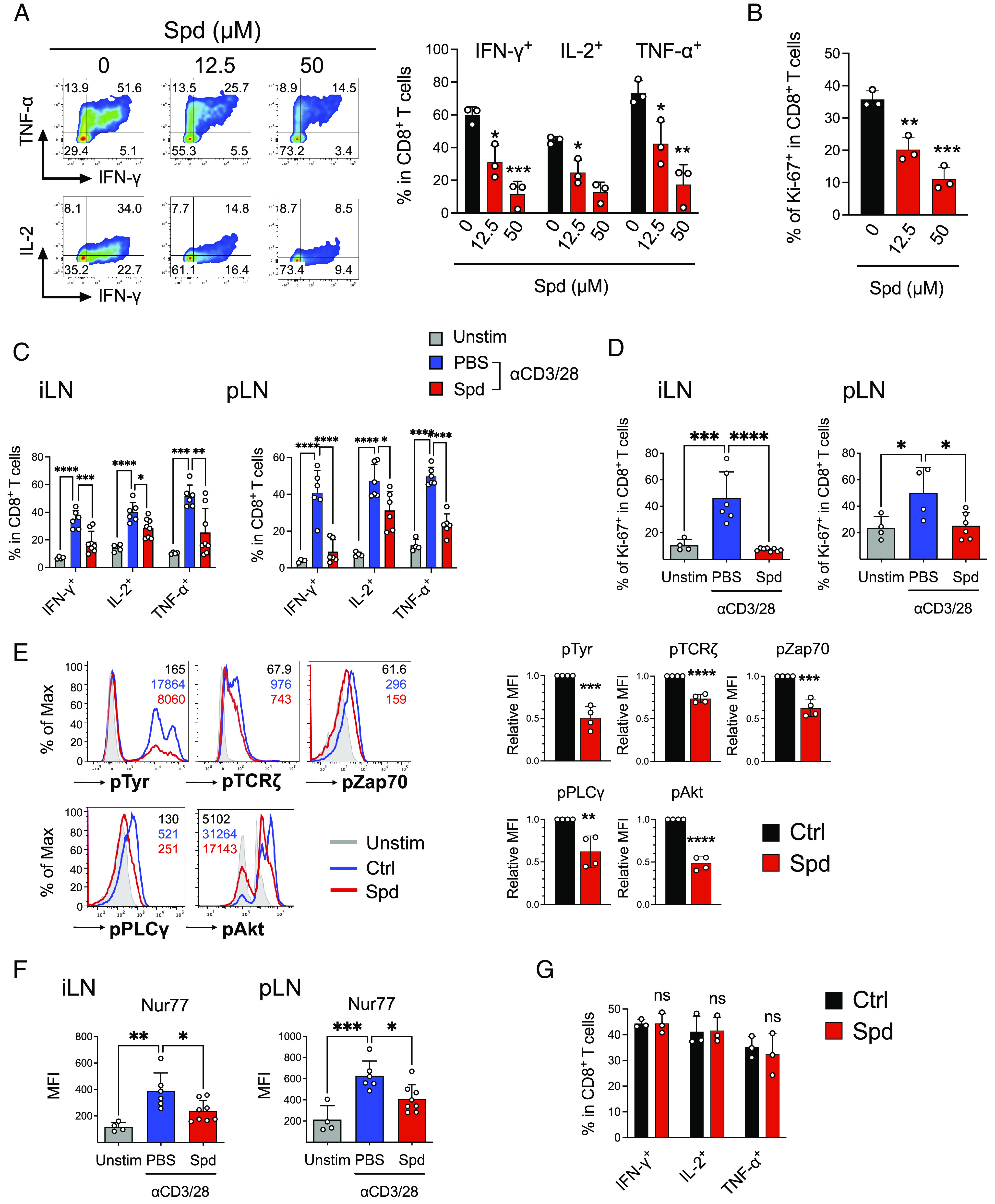
Spermidine selectively suppresses TCR-induced activation of T cells. (A–F) Effect of spermidine on anti-CD3/CD28 Abs–induced CD8+ T cell activation in vitro (A, B, and E) and in vivo (C, D, and F). (A) Representative FCM plots (Left) and summary (Right) of IFN-γ, IL-2, and TNF-α expression in CD8+ T cells left untreated (control) or treated with spermidine (Spd; 12.5 µM, 50 µM) for 3 d. The numbers in the FCM plots indicate the percentage of cells within the CD8+ T cell population. (B) CD8+ T cells were treated as in (A). Ki-67 expression was analyzed by FCM. (C and D) PBS (Phosphate-buffered saline) (Unstim) or anti-CD3/CD28 Abs (αCD3/28) were injected into the footpads of C57BL/6 mice, followed by intraperitoneal (i.p.) administration of PBS or spermidine (Spd; 75 mg/kg). Eighteen hours after anti-CD3/CD28 Abs injection, iLNs and pLNs were harvested and subjected to FCM analysis to examine the expression of IFN-γ, IL-2, TNF-α (C), and Ki-67 (D) within the CD8+ T cells (Left; iLNs, Right; pLNs, N = 4 to 8/group). (E) CD8+ T cells were left untreated (Ctrl) or treated with spermidine (Spd; 25 µM) for 3 d, and phosphorylation levels of tyrosine, TCRζ, Zap70, PLCγ, and Akt were examined by FCM. Unstimulated CD8+ T cells (Unstim) were also analyzed. The numbers in the histograms (Left) indicate mean fluorescence intensities (MFIs) within the CD8+ T cell population. Summaries of MFI values (relative to Ctrl) are shown in the right panels. (F) Expression of Nur77 in (C and D) was analyzed by FCM. Summaries of MFIs are shown (N = 4 to 8/group). (G) CD8+ T cells were stimulated with PMA (10 ng/mL) and IM (500 ng/mL) in the absence (Ctrl) or presence of spermidine (Spd; 50 µM) for 3 d. Expression of IFN-γ, IL-2, and TNF-α was examined by FCM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 compared with the control (Ctrl) group unless otherwise indicated; ns, not significant; data are presented as the mean ± SD.
We then focused on the effects of spermidine on in vivo activation of CD8+ T cells. Polyclonal T cell activation was induced by injecting anti-CD3/CD28 antibodies into the hind footpad of C57BL/6 mice (28), followed by intraperitoneal administration of spermidine. As shown in Fig. 3 C and D, spermidine treatment significantly inhibited anti-CD3/CD28-induced production of effector cytokines and proliferation of CD8+ T cells within the draining lymph nodes (iLNs; inguinal lymph nodes, pLNs; popliteal lymph nodes) (SI Appendix, Fig. S4B). Thus, spermidine inhibits TCR-induced activation of CD8+ T cells both in vitro and in vivo.
To understand the molecular basis of the spermidine-induced CD8+ T cell suppression, we further examined its effect on downstream signaling events following TCR stimulation. Notably, spermidine potently inhibited TCR-induced global tyrosine phosphorylation and CD3/CD28 proximal phosphorylation including TCRζ, Zap70, PLC (Phospholipase C)-γ1, and Akt (Fig. 3E and SI Appendix, Fig. S4C). Additionally, the spermidine-mediated inhibition of TCR signaling was also confirmed in vivo by examining the expression of Nur77, a specific indicator of TCR signaling (29) (Fig. 3F and SI Appendix, Fig. S4D). On the contrary, spermidine did not affect phorbol 12-myristate 13-acetate (PMA)/ionomycin (IM)-induced production of cytokines by CD8+ T cells (Fig. 3G). Taken together, these data indicate that spermidine exerts its T cell inhibitory effects by suppressing proximal TCR signal transducing machinery.
Suppression of Plasma Membrane Cholesterol Levels by Spermidine in CD8+ T Cells.
In order to gain further insights into the spermidine-mediated inhibition of CD8+ T cell signaling, we conducted a comprehensive gene expression analysis of the in vitro cultured mouse CD8+ T cells treated with or without spermidine (Fig. 4A). Gene set enrichment analysis (GSEA) showed that spermidine treatment resulted in suppression of the genes involved in DNA replication and cell cycle (Fig. 4B). Further, the expression of genes involved in cholesterol homeostasis was also inhibited by spermidine (Fig. 4 B andC). Specifically, expression of genes involved in the regulation of cholesterol biosynthesis, namely, 3-hydroxy-3-methylglutaryl-CoA reductase (Hmgcr) and squalene epoxidase (Sqle), and cholesterol influx such as low-density lipoprotein receptor (Ldlr) was affected, while genes associated with cellular cholesterol efflux were unaffected (Fig. 4D and SI Appendix, Fig. S5A). Additionally, time-course expression analyses revealed that spermidine interfered with TCR stimulation–induced augmentation of cholesterol biosynthesis and import, while it did not affect its efflux process that is generally blocked by the TCR activation (SI Appendix, Fig. S5B).
Fig. 4.

Spermidine down-regulates T cell cholesterol levels and thereby impairs TCR clustering. (A–D) CD8+ T cells were activated by anti-CD3/CD28 Abs in the absence (Ctrl) or presence of spermidine (Spd; 25 µM) in vitro for 3 d. A whole transcriptome analysis was performed (n = 2). (A) Differentially expressed genes (DEGs) represented in red or green were obtained. (B) DEGs were functionally annotated by GSEA using MsigDB “hallmark” gene sets. The top five hallmark gene sets up-regulated (red) or down-regulated (green) by spermidine treatment were ordered by normalized enrichment score (NES). (C) The GSEA plot “HALLMARK_CHOLESTEROL_HOMEOSTASIS” is shown. (D) Heat map showing the expression of genes associated with cellular cholesterol homeostasis is presented. (E and F) Effect of spermidine treatment on T cell cholesterol levels in vitro (E) and in vivo (F). (E) CD8+ T cells were activated and treated as in (A–D), and then stained using cholesterol probe Filipin III. The fluorescence intensity of Filipin III was quantified by FCM, and the numbers in the histogram (Left) indicate Filipin III MFIs within the CD8+ T cell population. Unstimulated CD8+ T cells (Unstim) were also examined. Summaries of MFI values (relative to Ctrl) are shown in the right panel. (F) C57BL/6 mice were treated as in Fig. 3 C andD. iLNs (Left) and pLNs (Right) were subjected to Filipin III staining. The fluorescence intensity of Filipin III within the CD8+ T cells was quantified, and summaries of MFI values are shown (N = 4 to 8/group). (G) CD8+ T cells were cultured in vitro as in (E) with or without MβCD-coated cholesterol (2 µg/mL). Expression of IFN-γ, IL-2, and TNF-α and phosphorylation levels of tyrosine and TCRζ were examined by FCM. The numbers next to the histograms (Left) indicate the percentage of cytokine-producing cells and MFIs of the phosphoprotein expression within the CD8+ T cells, and summaries are shown in the right panels. Summaries of MFIs are represented as relative values to Ctrl. (H) CD8+ T cells cultured in vitro as in (G) were rested for 6 h, then left untreated (Unstim) or restimulated with plate-coated anti-CD3 Ab (10 µg/mL) for 1 h. Representative confocal images (Upper) and quantification of cells with CD3ε capping (Lower) are shown. **P < 0.01; ***P < 0.001; ****P < 0.0001 compared with the control (Ctrl) group unless otherwise indicated; ns, not significant; data are presented as the mean ± SD.
To further assess the spermidine’s effect on the CD8+ T cell cholesterol levels, its quantification and visualization were performed by Filipin III staining. Cholesterol was mainly distributed in the plasma membranes in CD8+ T cells and its level was markedly increased after TCR activation (30); however, the increase was markedly inhibited by spermidine treatment (Fig. 4E and SI Appendix, Fig. S5C). Notably but consistently, similar observations were made with CD8+ T cells in vivo (Fig. 4F).
Does suppression of cholesterol by spermidine solely account for the impaired CD8+ T cell activation? To address this issue, we examined whether exogenously added cholesterol affects the spermidine-mediated suppression of anti-CD3/CD28-induced CD8+ T cell activation. We then found that both methyl-beta-cyclodextrin (MβCD)-coated water-soluble cholesterol and LDL (Low-density lipoprotein) cholesterol almost completely restored the T cell inhibitory effects of spermidine (Fig. 4G and SI Appendix, Fig. S5D). Further, depletion of plasma membrane cholesterol by MβCD severely disrupted TCR signal transduction, but had no effect on PMA/IM-induced CD8+ T cell activation, as confirmed by the status of the common downstream molecules S6 ribosomal protein and Nur77 (SI Appendix, Fig. S5 E andF). Altogether, these data further lend support to the notion that spermidine suppresses CD8+ T cell effector function by decreasing the plasma membrane cholesterol level.
Cholesterol is a major structural component of the plasma membrane and has been known to play an indispensable role in TCR clustering and the formation of the T cell immunological synapse (30–32). Therefore, we sought to examine the effect of spermidine on TCR clustering on the plasma membrane of activated CD8+ T cells by using CD3ε macrocluster formation (called “capping”) as an indicator (33). As shown in Fig. 4H, the anti-CD3 stimulation triggered the formation of CD3ε macroclusters, but spermidine treatment significantly abrogated this process. Perhaps expectedly, this inhibition was relieved by addition of cholesterol to the assay (Fig. 4H). Thus, the downregulation of plasma membrane cholesterol accounts for the impaired clustering of TCRs on CD8+ T cells induced by spermidine treatment.
Effect of Polyamine Blockade on In Vivo Antitumor CD8+ T Cell Response.
We next examined the effect of eflornithine to antitumor CD8+ T cell responses in vivo. As shown in Fig. 5A, eflornithine injection to B16F10 tumor–bearing mice resulted in a strong suppression of the tumor growth, accompanied by a ca. threefold increase of CD8+ T cell infiltrated in the tumor tissue (Fig. 5B). Filipin III staining revealed that cholesterol content of these CD8+ T cells was significantly increased by eflornithine treatment, but this increase was abolished by spermidine supplementation (SI Appendix, Fig. S6A). Perhaps expectedly, the suppression was significantly attenuated when eflornithine and anti-CD8α antibody were coinjected (Fig. 5A). Further, about twofold increase of Ki-67 expression levels was observed in CD8+ T cells in the eflornithine-treated mice, whereas such effect was abolished by cotreatment with spermidine (Fig. 5B). Of note, CD4+ T cells, including regulatory T cells, were not affected by the eflornithine treatment, and neither natural killer cells nor myeloid cells was affected in this analysis (SI Appendix, Fig. S6B). This may be due to the different metabolic programs of CD4+ and CD8+ T cells, at least in part (34).
Fig. 5.

Polyamine synthesis blockade unleashes antitumor CD8+ T cell responses in vivo. (A–D) B16F10 cells were subcutaneously transplanted into C57BL/6 mice on day 0. PBS (Ctrl) or eflornithine (500 mg/kg) was intraperitoneally (i.p.) injected daily from day 7 to day 13. (A) For in vivo depletion of CD8+ T cells, anti-CD8α Ab (100 µg/mouse) was administered on days 6, 9, and 12 in parallel with eflornithine or alone (N = 5 to 8/group). Tumor growth rate was monitored throughout the experimental period (Upper), and tumor volume (mm3) at day 15 was summarized (Lower). (B) Spermidine (Spd; 50 mg/kg) was i.p. administered daily from day 7 to day 13 in parallel with eflornithine or alone (N = 7 to 8/group). Single-cell suspensions prepared from tumor tissue (at day 15) were subjected to FCM analyses. Frequencies of CD8+ T cells within CD45+ cells (Left) and Ki-67+ cells within CD8+ T cells (Right) were examined. (C and D) Anti-PD-1 Ab (200 µg/mouse) was i.p. administered on days 7, 10, and 13 in parallel with eflornithine or alone (N = 5 to 8/group). (C) Tumor growth rate was monitored throughout the experimental period (Upper), and tumor volume (mm3) at day 15 was summarized (Lower). (D) Single-cell suspensions prepared from tumor tissue (at day 15) were subjected to FCM analyses. Frequencies of CD8+ T cells within CD45+ cells, and IFN-γ+, IL-2+, and CD107a+ cells within CD8+ T cells were examined. *P < 0.05; **P < 0.01; ns, not significant; data are presented as the mean ± SD.
Since eflornithine enhances antitumor CD8+ T cell response by a mechanism distinct from immune checkpoint inhibitors, we also examined the effect of combined treatment of eflornithine with an anti-PD-1 antibody on B16F10 tumor growth. As previously reported (35, 36), anti-PD-1 is ineffective in the subcutaneous B16F10 tumor; however, combinational administration of anti-PD-1 with eflornithine potently suppressed tumor growth that was significantly stronger than eflornithine alone (Fig. 5C). Immunophenotyping of tumor-infiltrating T cells revealed that, although anti-PD-1 monotherapy significantly increased the proportion of CD8+ T cells, it failed to increase its subpopulation with effector cytokine–producing capacity and cytotoxic activity (Fig. 5D). On the contrary, anti-PD-1 and eflornithine combination therapy synergistically augmented the accumulation of cytotoxic CD8+ T cells within the tumor, as measured by an increased population of the cells expressing CD107a+, a marker of CD8+ T cell degranulation (Fig. 5D). Further, similar antitumor effect of the combined therapy was also seen with SL4 colon tumor–bearing mice (SI Appendix, Fig. S6 C andD).
Correlation Analysis of Tumor Tissue Polyamine Synthesis and CD8+ T Cell Activity in Human Cancers.
The above results prompted us to analyze whether these findings made by our mouse models are correlated with human cancers. By analyzing the TCGA gene expression data, we found a negative correlation between the expression levels of ODC1, the determinant enzyme for spermidine biosynthesis, and transcriptomic signature of effector CD8+ T cell responses, such as CD8A and TBX21, in lung cancer and pan-cancer cohorts (SI Appendix, Fig. S7 A andB). On the contrary, expression of genes involved in the degradation of spermidine, such as ornithine decarboxylase antizyme 1 (OAZ1) and spermidine/spermine N1-acetyltransferase 1 (SAT1), showed a significant positive correlation with effector CD8+ T cell signature (SI Appendix, Fig. S7 A andB). With some exceptions, similar findings were also made for several other cancer types (SI Appendix, Fig. S7C). These data suggest a mouse-to-human concordance in which cancer cell–derived spermidine also functions as a suppressor of antitumor CD8+ T cell immunity in some, if not all, human cancers.
Discussion
Intracellular polyamines, such as spermidine, are well characterized as an essential metabolic fuel in growing tumor cells and have long been attracting attention as an optimal target for cancer therapy (21, 23). In this study, we report a unique tumor cell–extrinsic role of spermidine; tumor cell–derived spermidine suppresses T cell activation and contributes to tumor growth in the TIME by rendering tumor-reactive CD8+ T cells anergic through cholesterol deprivation. Indeed, we found that i) exogenously added spermidine could potently suppress TCR-induced T cell effector function both in vitro and in vivo, ii) a substantial concentration of spermidine could be detected in the extracellular fluid of in vivo established tumor tissue, and iii) polyamine synthesis inhibitor eflornithine restored the antitumor CD8+ T cell responses, which was counteracted by exogenously administered spermidine. Our results are consistent with an enhancement of antitumor CD8+ T cell response by inhibiting cholesterol esterification (30). Of note, while spermidine suppresses anti-CD3/CD28-induced activation of both CD8+ and CD4+ T cells (Fig. 3 and SI Appendix, Fig. S4), eflornithine selectively affected tumor-infiltrating CD8+ T cells in the mouse model (SI Appendix, Fig. S6B). This could be explained by the fact that each T cell subset utilizes distinct metabolic adaptation programs in the TIME (34, 37); therefore, one may speculate that spermidine selectively affects a part of the programs.
In view of the importance of “lipid raft,” for which cholesterol is an essential structural component in a variety of cellular signaling (38), our observation suggests that spermidine release by tumors may affect signaling pathways other than TCR signaling in the TIME such as those for IFN-γ receptor (39) and Toll-like receptors (40, 41). Therefore, how spermidine is involved in other pathological conditions such as inflammatory diseases will be an interesting issue. Also, a remaining issue is the underlying mechanism for how spermidine inhibits cholesterol levels in CD8+ T cells. According to previous reports, both endogenous and exogenous polyamines profoundly affect cellular cholesterol homeostasis in the mammalian system (42, 43). Spermidine is also known to stabilize Z-DNA formation (44), suggesting that spermidine may be involved in the transcriptional regulation of cholesterol synthesis via chromatin control. The detailed molecular mechanisms of polyamine-mediated cellular cholesterol regulation need to be further elucidated clearly. Of additional note, it has been reported that spermidine supplementation could break therapeutic resistance to ICB by directly activating CD8+ T cells in aged mice (45). Thus, it is likely that the action of spermidine is bifunctional and context dependent.
What determines the immunological consequences of cell death has long been a somewhat controversial issue. Massive cell death induced by chemotherapy or radiation therapy is thought to stimulate adaptive immune responses, e.g., through the release of tumor antigens and others, the event so-called immunogenic cell death (46, 47). On the contrary, we and others have shown other immunological facets of tumor cell death that suppress antitumor immunity via the release of DAMPs (17, 18, 48, 49). It is possible that rapid, massive tumor cell death during the above therapy may result in the activation of antitumor immunity, whereas chronic, slow tumor cell death during tumor progression would result in immune suppression in the TIME as exemplified here.
It is interesting to note that polyamines, including spermidine, are elevated in human cancers, and that polyamine blockade therapy has been developed, with some of these inhibitors entering clinical trials (50, 51). Therefore, tumor cell–derived extracellular spermidine may act in those cancers by the mechanism similar to what we reported here. On the contrary, it cannot be strictly ruled out at present whether or to what extent spermidine is released by nontumor cells in TIME. These issues need further elaboration.
In summary, here we describe a hitherto unknown role for the oncometabolite spermidine in the formation of inhibitory milieu for CD8+ T cells within the TIME. Tumor cell death occurring under stressed TIME triggers the extracellular release of spermidine at local tumor site, which then functions as a tumor-derived metabolite checkpoint restricting effector T cell function. Manipulating spermidine content within tumor cells by existing polyamine blockade therapies or establishing the technique for directly targeting extracellular spermidine holds promise for realizing the full potential of T cell–targeting cancer immunotherapy.
Materials and Methods
Mice.
All experiments using mice were approved by the University of Tokyo’s Animal Research Committee. C57BL/6 mice were purchased from CLEA Japan Inc. All mice were maintained in specific pathogen-free conditions, and all experiments were done in accordance with animal welfare guidelines of the University of Tokyo (P20-103 & RAC190002).
Induction of Cell Death and Preparation of Necrotic Cell Supernatants.
For the freeze-and-thaw (F–T) method, cells were suspended in serum-free RPMI (Roswell Park Memorial Institute media), frozen at −80 °C, and then thawed at 37 °C, repeated twice. Glucose starvation was induced by culturing cells in glucose-free RPMI (Nacalai Tesque). Hypoxic culture conditions (0.3% O2, 5% CO2) were generated by using the BIONIX hypoxic culture kit (Sugiyama-Gen). After each treatment, supernatant samples were centrifuged to remove cell debris and filtered through a membrane filter with 0.22 µm pore size (Corning). For heat treatment, the supernatant was incubated at 95 °C for 15 min, and the denatured protein was precipitated and removed by centrifugation. For proteinase K treatment, the supernatant was treated with proteinase K (100 µg/mL; TaKaRa Bio) at 37 °C for 1 h, followed by incubation with p-amidinophenyl methanesulfonyl fluoride (5 mM; Nacalai Tesque) on ice for 20 min to inactivate proteinase K. Fractionation of the supernatants was conducted using Amicon Ultra centrifugal filters with 3 kDa MW cutoff Ultracel membrane (Millipore). The supernatant fraction of >3 kDa remained above the filter, and the fraction of <3 kDa passed through to the lower tube. The >3 kDa fraction was resuspended in serum-free RPMI and adjusted to the volume almost equal to the <3 kDa fraction.
Additional information can be found in SI Appendix, Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We greatly thank G. Lee for his valuable comments and discussions. We also thank S. Nakagawa for metabolome analysis and M. Sugahara for technical assistance. This study was partly supported by Grants-in-Aid for Scientific Research (A) 20H00504 (T.T.), (B) 22H02887 (H.Y.), and (Young Scientists Grant) 19K16736 (S. Hibino), 21K15499 (S. Hibino), and 21K15465 (S. Hangai) from the Ministry of Education, Culture, Sports, Science of Japan (MEXT), Japan Society for the Promotion of Science Research Fellow 19J00887 (S. Hibino); Grant AMED-PRIME 20gm6110008 from the Japan Agency for Medical Research and Development (AMED) (H.Y.); the Takeda Science Foundation (S. Hibino and H.Y.); the Kowa Life Science Foundation (S. Hibino); Foundation for Promotion of Cancer Research (S. Hibino); Joint Research Program of the Institute for Genetic Medicine, Hokkaido University (H.Y.); Extramural Collaborative Research Grant of Cancer Research Institute, Kanazawa University (H.Y.); the Uehara Memorial Foundation (H.Y.); and the UTEC-UTokyo FSI (Future Society Initiative) Research Grant Program (H.Y.). The Department of Inflammology is supported by the Boostimmune Inc.
Author contributions
S. Hibino, T.T., and H.Y. designed research; S. Hibino, S.E., S. Hangai, K.E., S.A., and M.S. performed research; S. Hibino, T.O., T.S., and H.Y. analyzed data; and S. Hibino, T.T., and H.Y. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: C.B., Harvard Medical School; and R.M., Yale University.
Contributor Information
Tadatsugu Taniguchi, Email: tada@m.u-tokyo.ac.jp.
Hideyuki Yanai, Email: yanai@inflammology.rcast.u-tokyo.ac.jp.
Data, Materials, and Software Availability
Microarray data have been deposited in Gene Expression Omnibus (GEO) database (GSE216472) (52). All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Galon J., Bruni D., Tumor immunology and tumor evolution: Intertwined histories. Immunity 52, 55–81 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Philip M., Schietinger A., CD8+ T cell differentiation and dysfunction in cancer Nat. Rev. Immunol. 22, 209–223 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waldman A. D., Fritz J. M., Lenardo M. J., A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma P., Hu-Lieskovan S., Wargo J. A., Ribas A., Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Leun A. M., Thommen D. S., Schumacher T. N., CD8+ T cell states in human cancer: Insights from single-cell analysis Nat. Rev. Cancer 20, 218–232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reina-Campos M., Scharping N. E., Goldrath A. W., CD8+ T cell metabolism in infection and cancer Nat. Rev. Immunol. 21, 718–738 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramsay G., Cantrell D., Environmental and metabolic sensors that control T cell biology. Front. Immunol. 6, 99 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scharping N. E., et al. , Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurusamy D., Clever D., Eil R., Restifo N. P., Novel “Elements” of immune suppression within the tumor microenvironment. Cancer Immunol. Res. 5, 426–433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson K. G., Stromnes I. M., Greenberg P. D., Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell 31, 311–325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang R., Green D. R., Metabolic checkpoints in activated T cells. Nat. Immunol. 13, 907–915 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Bunse L., et al. , Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 24, 1192–1203 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Brown J. M., Wilson W. R., Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 4, 437–447 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Karsch-Bluman A., et al. , Tissue necrosis and its role in cancer progression. Oncogene 38, 1920–1935 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Richards C. H., Mohammed Z., Qayyum T., Horgan P. G., McMillan D. C., The prognostic value of histological tumor necrosis in solid organ malignant disease: A systematic review. Future Oncol. 7, 1223–1235 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Scaffidi P., Misteli T., Bianchi M. E., Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Yanai H., Hangai S., Taniguchi T., Damage-associated molecular patterns and Toll-like receptors in the tumor immune microenvironment. Int. Immunol. 33, 841–846 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Hangai S., et al. , Orchestration of myeloid-derived suppressor cells in the tumor microenvironment by ubiquitous cellular protein TCTP released by tumor cells. Nat. Immunol. 22, 947–957 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Eil R., et al. , Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 537, 539–543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng Y., Zha Y., Driessens G., Locke F., Gajewski T. F., Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J. Exp. Med. 209, 2157–2163 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arruabarrena-Aristorena A., Zabala-Letona A., Carracedo A., Oil for the cancer engine: The cross-talk between oncogenic signaling and polyamine metabolism. Sci. Adv. 4, eaar2606 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madeo F., Eisenberg T., Pietrocola F., Kroemer G., Spermidine in health and disease. Science 359, eaan2788 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Casero R. A., Murray Stewart T., Pegg A. E., Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 18, 681–695 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song X., et al. , Oral squamous cell carcinoma diagnosed from saliva metabolic profiling. Proc. Natl. Acad. Sci. U.S.A. 117, 16167–16173 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Igarashi K., et al. , High-throughput screening of salivary polyamine markers for discrimination of colorectal cancer by multisegment injection capillary electrophoresis tandem mass spectrometry. J. Chromatogr. A 1652, 462355 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Wagner M., Wiig H., Tumor interstitial fluid formation, characterization, and clinical implications. Front. Oncol. 5, 115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sullivan M. R., et al. , Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, e44235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Sullivan D., et al. , Fever supports CD8+ effector T cell responses by promoting mitochondrial translation. Proc. Natl. Acad. Sci. U.S.A. 118, e2023752118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanellopoulou C., et al. , Mg2+ regulation of kinase signaling and immune function. J. Exp. Med. 216, 1828–1842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang W., et al. , Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531, 651–655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zech T., et al. , Accumulation of raft lipids in T-cell plasma membrane domains engaged in TCR signalling. EMBO J. 28, 466–476 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molnár E., et al. , Cholesterol and sphingomyelin drive ligand-independent T-cell antigen receptor nanoclustering. J. Biol. Chem. 287, 42664–42674 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li D., Molldrem J. J., Ma Q., LFA-1 regulates CD8+ T cell activation via T cell receptor-mediated and LFA-1-mediated Erk1/2 signal pathways J. Biol. Chem. 284, 21001–21010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chapman N. M., Chi H., Metabolic adaptation of lymphocytes in immunity and disease. Immunity 55, 14–30 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curran M. A., Allison J. P., Tumor vaccines expressing flt3 ligand synergize with ctla-4 blockade to reject preimplanted tumors. Cancer Res. 69, 7747–7755 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tavazoie M. F., et al. , LXR/ApoE activation restricts innate immune suppression in cancer. Cell 172, 825–840.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manzo T., et al. , Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J. Exp. Med. 217, e20191920 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simons K., Toomre D., Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Takaoka A., et al. , Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science 288, 2357–2360 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Zhu X., et al. , Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J. Lipid Res. 51, 3196–3206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruysschaert J.-M., Lonez C., Role of lipid microdomains in TLR-mediated signalling. Biochim. Biophys. Acta (BBA) - Biomembranes 1848, 1860–1867 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Kakimoto M., Yamamoto H., Tanaka A. R., Spermine synthesis inhibitor blocks 25-hydroxycholesterol-induced- apoptosis via SREBP2 upregulation in DLD-1 cell spheroids. Biochem. Biophys. Rep. 22, 100754 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michiels C. F., Kurdi A., Timmermans J.-P., De Meyer G. R. Y., Martinet W., Spermidine reduces lipid accumulation and necrotic core formation in atherosclerotic plaques via induction of autophagy. Atherosclerosis 251, 319–327 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Brooks W. H., Increased polyamines alter chromatin and stabilize autoantigens in autoimmune diseases. Front. Immunol. 4, 91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Al-Habsi M., et al. , Spermidine activates mitochondrial trifunctional protein and improves antitumor immunity in mice. Science 378, eabj3510 (2022). [DOI] [PubMed] [Google Scholar]
- 46.Galluzzi L., Buqué A., Kepp O., Zitvogel L., Kroemer G., Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Kroemer G., Galassi C., Zitvogel L., Galluzzi L., Immunogenic cell stress and death. Nat. Immunol. 23, 487–500 (2022). [DOI] [PubMed] [Google Scholar]
- 48.Hangai S., et al. , PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc. Natl. Acad. Sci. U.S.A. 113, 3844–3849 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hou J., Greten T. F., Xia Q., Immunosuppressive cell death in cancer. Nat. Rev. Immunol. 17, 401–401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raj K. P., et al. , Role of dietary polyamines in a phase III clinical trial of difluoromethylornithine (DFMO) and sulindac for prevention of sporadic colorectal adenomas. Br. J. Cancer 108, 512–518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bassiri H., et al. , Translational development of difluoromethylornithine (DFMO) for the treatment of neuroblastoma. Transl. Pediatr. 4, 226–238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Habana S., Gene expression profiling of in vitro activated mouse CD8+ T cells treated with or without spermidine. NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE216472. Deposited 24 October 2022. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Microarray data have been deposited in Gene Expression Omnibus (GEO) database (GSE216472) (52). All study data are included in the article and/or SI Appendix.