

Significance
Carbohydrates (or glycans) play crucial roles in biological systems ranging from energy storage to pathogen evasion. However, the contributions of specific glycans to brain functions such as emotion and cognition remain largely unknown. Here, we show that 4-O-sulfated chondroitin sulfate (CS) regulates perineuronal nets (PNNs) and excitatory–inhibitory synapses in the mouse CA2 (cornu ammonis 2) hippocampus, a brain region critical for social memory. Ablation of CS 4-O-sulfation in adult mice caused malformation of PNNs, elevated anxiety, and impaired social memory. Modulations that reversed the PNN abnormalities or replenished 4-O-sulfation restored normal mood and social cognition. These findings identify roles for chondroitin 4-O-sulfation in higher-order brain functions and suggest a potential strategy to address neurological disorders with social cognitive dysfunction.
Keywords: glycosaminoglycans (GAGs), glycans, chondroitin sulfate (CS), perineuronal nets (PNNs), social memory
Abstract
Glycan alterations are associated with aging, neuropsychiatric, and neurodegenerative diseases, although the contributions of specific glycan structures to emotion and cognitive functions remain largely unknown. Here, we used a combination of chemistry and neurobiology to show that 4-O-sulfated chondroitin sulfate (CS) polysaccharides are critical regulators of perineuronal nets (PNNs) and synapse development in the mouse hippocampus, thereby affecting anxiety and cognitive abilities such as social memory. Brain-specific deletion of CS 4-O-sulfation in mice increased PNN densities in the area CA2 (cornu ammonis 2), leading to imbalanced excitatory-to-inhibitory synaptic ratios, reduced CREB activation, elevated anxiety, and social memory dysfunction. The impairments in PNN densities, CREB activity, and social memory were recapitulated by selective ablation of CS 4-O-sulfation in the CA2 region during adulthood. Notably, enzymatic pruning of the excess PNNs reduced anxiety levels and restored social memory, while chemical manipulation of CS 4-O-sulfation levels reversibly modulated PNN densities surrounding hippocampal neurons and the balance of excitatory and inhibitory synapses. These findings reveal key roles for CS 4-O-sulfation in adult brain plasticity, social memory, and anxiety regulation, and they suggest that targeting CS 4-O-sulfation may represent a strategy to address neuropsychiatric and neurodegenerative diseases associated with social cognitive dysfunction.
Glycans, like nucleic acids and proteins, are ubiquitous in nature and play crucial roles in biological processes such as development, host–pathogen interactions, and immune regulation (1–4). The mammalian central and peripheral nervous systems provide a rich source of diverse glycans. Large-scale analyses have revealed spatial and temporal variations in glycan expression across the brain (5) and identified over 4,000 N-glycosylation sites on more than 1,500 glycoproteins (6, 7). Despite being the most structurally diverse and rapidly evolving class of macromolecules (8), glycans remain understudied, and their biological functions in the nervous system are insufficiently understood.
In the brain, a complex meshwork of interwoven glycans and proteins in the extracellular matrix (ECM) provides structural support and mediates important neural functions (9–11). Lattice-like ECM structures known as perineuronal nets (PNNs) have been proposed to act as extracellular scaffolds for ligands and to stabilize synapses, thereby modulating brain plasticity and physiological processes (10, 11). Emerging evidence suggests that PNNs contribute not only to critical period plasticity during development but also to learning and memory processing, psychiatric diseases, drug addiction, and neurodegeneration in adulthood (12–14). PNNs predominantly surround parvalbumin-expressing (PV+), fast-spiking inhibitory interneurons in cortical areas (15, 16). They also enwrap excitatory pyramidal neurons in brain regions important for emotional learning and memory, such as the amygdala, entorhinal cortex, and area cornu ammonis 2 (CA2) (17, 18).
The CA2 subregion of the hippocampus has unique molecular, synaptic, and morphological characteristics (19) and is essential for social recognition memory (20, 21). Consistent with this function, alterations in the cellular structure and circuitry in the area CA2 have been observed in neuropsychiatric disorders associated with cognitive and social dysfunction (22, 23). Although recent studies suggest that PNNs in the CA2 can contribute to both PV+ interneuron and excitatory pyramidal neuron plasticity (24, 25), the molecules and mechanisms that regulate PNNs and CA2-dependent functions such as social memory remain unclear.
PNNs are highly enriched in chondroitin sulfate proteoglycans (CSPGs), a series of core proteins decorated with chondroitin sulfate (CS) polysaccharides (26). The repeating d-glucuronic acid (GlcA) and N-acetyl-d-galactosamine (GalNAc) disaccharide units of CS polysaccharide chains display a variety of sulfation motifs (Fig. 1A) that dynamically change throughout development into adulthood (27). Notably, increased expression of the 4-O-sulfated motif CS-A coincides with PNN maturation and the end of critical period plasticity (16, 28). By adulthood, the CS-A motif represents nearly 90% of the total CS in the brain. The 4-O-sulfated CS-A and CS-E motifs have also been reported to play important roles in axon regeneration after central nervous system (CNS) injury (29–31). These motifs are up-regulated in the glial scar upon injury and limit axon regeneration by interacting with protein receptors such as the tyrosine phosphatase receptor σ (PTPσ) and Nogo receptor (29, 32–34). Loss of 4-O-sulfated CS motifs via knockdown of the chondroitin-4-O-sulfotransferase Chst11 in zebrafish enhanced regeneration after spinal cord injury (31). Based on these and other observations, chondroitin 4-O-sulfation has traditionally been viewed as a “molecular brake” that inhibits neuroplasticity (35, 36). However, its functions in PNNs and in the uninjured adult brain have not been directly investigated.
Fig. 1.
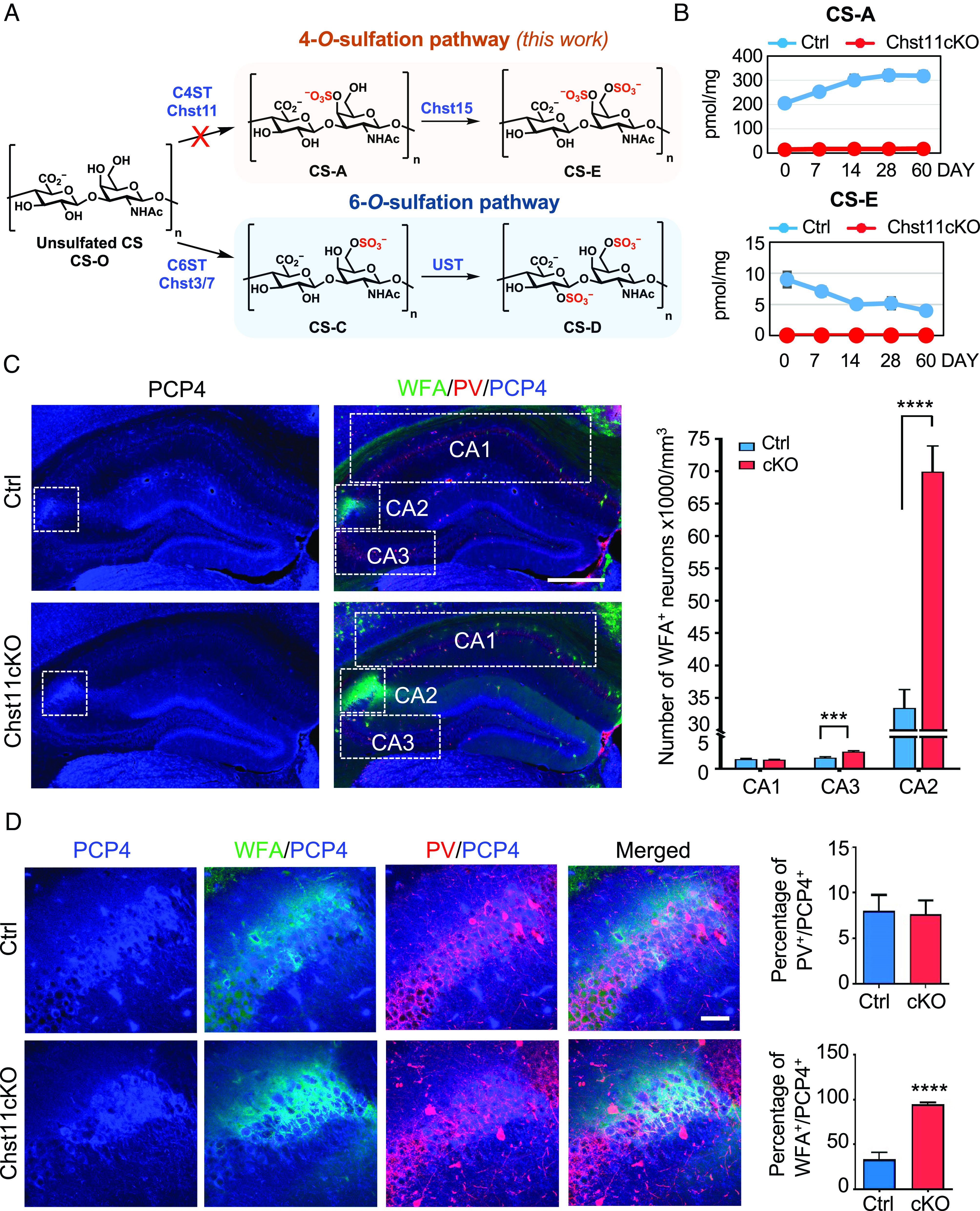
Chst11cKO mice lacking chondroitin 4-O-sulfation have a striking increase in PNNs surrounding excitatory neurons in the CA2 hippocampus. (A) Biosynthetic pathways leading to 4-O- and 6-O-sulfation of chondroitin sulfate (CS). CS consists of the repeating disaccharide unit N-acetyl-d-galactosamine-β(1,3)-d-glucuronic acid. n = 20 to 200. In the 4-O-sulfation pathway (orange, studied in this work), the disaccharide units are monosulfated at the 4-O-position of GalNAc to generate CS-A. CS-A is subsequently sulfated at the 6-O-position of GalNAc to produce CS-E. In the 6-O-sulfation pathway (blue), the disaccharide units are monosulfated at the 6-O-position of GalNAc to generate CS-C, which is subsequently sulfated at the 2-O-position of GlcA to form CS-D. (B) Disaccharide analysis shows loss of the CS-A and CS-E motifs in the brains of Chst11cKO mice. Amounts of CS-A and CS-E in the cortex of Ctrl (blue) and Chst11cKO (red) mice were quantified at postnatal day (P) 0, P7, P14, P28, and P60 by dividing the number of picomoles (pmol) of CS by the total weight (mg) of the dried cortex homogenate. (C) Representative images showing PNN-enwrapped (WFA+, green), PV+ interneurons (red), and PCP4+ excitatory pyramidal neurons (blue) in the hippocampus of Ctrl and Chst11cKO mice. Hippocampal CA1, CA2, and CA3 regions are marked with dotted white rectangles. (Scale bar, 500 μm.) Quantification shows the number of WFA+ neurons in each region. ***P < 0.001, ****P < 0.0001 vs. Ctrl, Student’s t test; n = 17 slices from 7 Ctrl mice and 14 slices from 5 Chst11cKO mice. (D) Magnified images showing the CA2 regions of Ctrl and Chst11cKO mice, stained for PCP4 (blue), PNNs (WFA, green), and PV (red). (Scale bar, 50 μm.) Quantification of the ratios of PV+ neurons (Upper) and WFA+ neurons (Lower) among PCP4+ CA2 neurons. ****P < 0.0001 vs. Ctrl, Student’s t test; n = 10 and 8 slices from 4 pairs of Ctrl and Chst11cKO mice, respectively. All data are shown as the mean ± SEM.
Studies aimed at understanding PNNs have employed either chondroitinase ABC (ChABC) to enzymatically digest CS polysaccharides (37–39) or transgenic mice lacking PNN proteins such as tenascin-R and link protein (HAPLN1) (40, 41). While these approaches have revealed important insights, they also drastically disrupt PNNs and render them indistinguishable from the diffuse ECM. Modulation of the sulfation patterns on CSPGs provides a less perturbative, complementary approach to manipulate and study PNNs. For example, overexpression of chondroitin-6-O-sulfotransferase-1 (C6ST-1) in transgenic mice modulated 6-O-sulfation and PNN formation in the developing visual cortex (VC), leading to persistent ocular dominance plasticity (16). However, the role of CS sulfation in PNNs has been examined primarily in this context of 6-O-sulfotransferase overexpression.
In this study, we investigated the impact of CS 4-O-sulfation, the dominant form of sulfation and the most abundant glycosaminoglycan structure in the adult mammalian brain, on PNNs, plasticity, and higher-order brain functions such as mood and cognition. We found that brain-specific deletion of the chondroitin 4-O-sulfotransferase gene Chst11 in mice significantly perturbed PNN levels surrounding excitatory CA2 pyramidal neurons. The resulting increase in PNNs led to reduced CREB activation, an imbalance of excitatory and inhibitory synapses, as well as anxiety and social memory dysfunction—phenotypes that were rescued by treatment with ChABC or 4-O-sulfated CS polysaccharides. In agreement with these findings, a chemical inhibitor developed by our lab to reduce CS 4-O-sulfation levels recapitulated the malformation of PNNs and synaptic defects in hippocampal neurons. Moreover, viral-mediated CA2 region-specific deletion of Chst11 in adult mice also increased PNN densities, inhibited CREB activity, and impaired social memory. Together, these data reveal important roles for CS 4-O-sulfation in adult brain plasticity, social memory, and anxiety regulation, and they suggest CS polysaccharides as targets for the study and potential treatment of neurological diseases characterized by mood disorders and social dysfunction.
Results
Chst11cKO Mice Lacking 4-O-Sulfation Show Increased WFA+ PNN Densities in the CA2 Hippocampus.
The sulfotransferase Chst11 selectively transfers a sulfate group from 3′-phosphoadenosine 5′-phosphosulfate to the 4 O-position of GalNAc residues in CS polysaccharides (42). Genetic disruption of Chst11 in mice led to severe chondrodysplasia and neonatal lethality, demonstrating essential roles for 4-O-sulfation in embryonic development (43, 44). To study the roles of CS 4-O-sulfation in the adult brain, we generated a brain-specific Chst11 conditional knockout (cKO) mouse line by crossing Chst11-floxed mice with nestin-Cre transgenic mice (45). The nestin-Cre transgene restricts Chst11 deletion to neural precursor cells starting at embryonic day 10.5 (E10.5), circumventing complications associated with constitutive Chst11 ablation during prenatal development (43). To validate the gene deletion efficiency, we analyzed the sulfation patterns of CS in the cortex of Chst11lx/−; nestin-Cre+ (Chst11cKO) mice and Chst11lx/−; nestin-Cre− (Ctrl) mice as a control. CS chains were isolated from Chst11cKO and Ctrl mouse cortices at different ages (P0, P7, P14, P28, and P60), digested with ChABC, and the resulting disaccharides were quantified by high-performance liquid chromatography. No 4-O-sulfated CS-A and CS-E disaccharide units were detected in Chst11cKO brains, indicating disruption of the 4-O-sulfation pathway (Fig. 1 A and B). Loss of chondroitin 4-O-sulfation was accompanied by an increase in unsulfated CS levels and a decrease in overall CS levels in the brain (SI Appendix, Fig. S1A), consistent with reports that 4-O-sulfation facilitates CS chain elongation (46). In contrast, no loss of 6-O-sulfation was observed: CS-D levels remained similar, and CS-C levels increased slightly in Chst11cKO mice compared to Ctrl mice after P14, presumably due to compensatory effects (SI Appendix, Fig. S1A). Disruption of the 4-O-sulfation pathway in Chst11cKO mice was further confirmed by immunohistochemical analysis. A marked reduction in CS-E immunostaining was observed in the cortex and hippocampus of Chst11cKO mice relative to Ctrl littermates (SI Appendix, Fig. S1 B and C). Collectively, these results indicate efficient deletion of the 4-O-sulfation pathway in Chst11cKO mice.
We next examined the PNN levels in the VC and hippocampus of adult Chst11cKO and Ctrl mice using the well-established marker, Wisteria floribunda agglutinin (WFA) (16, 18, 37, 39). Consistent with previous observations, WFA+ PNNs exhibited region-specific expression patterns (47). In the VC, an increase in PNN-enwrapped (WFA+) neurons was observed in adult Chst11cKO mice compared to Ctrl mice (36% ± 11%), and this increase occurred primarily in PNNs surrounding the PV+ population (SI Appendix, Fig. S2 A–C). No change was observed in the total number of PV+ neurons or its percentage within the WFA+ population (SI Appendix, Fig. S2 D and E). Notably, the greatest change in WFA+ PNN-enwrapped neurons was observed in the area CA2 of the hippocampus. Both Chst11cKO and Ctrl adult mice showed high PNN levels in the CA2 region, whereas the CA1 and CA3 areas had fewer WFA+ PNNs (Fig. 1C). A striking increase in the number of WFA+ PNNs was observed in the CA2 region (109% ± 15%) and, to a lesser extent, in the CA3 region (55% ± 13%) of Chst11cKO compared to Ctrl mice (Fig. 1C). Given the substantial increase in WFA+ PNNs detected in the CA2 and the limited understanding of CS function in this region, we focused our studies hereafter on the CA2 hippocampus.
As PNNs condense around PV+ inhibitory neurons in many brain regions (16, 48), we investigated whether the WFA+ PNNs in the CA2 surround PV+ neurons. Adult hippocampal sections were costained for PV and the CA2 marker Purkinje cell protein 4 (PCP4) (49). The PCP4+ neurons in the CA2 region of both Chst11cKO and Ctrl mice contained only a small percentage of PV+ neurons (8% ± 2%; Fig. 1 D, Upper Right). The majority of the PCP4+ neurons in Chst11cKO mice were enwrapped by WFA+ PNNs (95% ± 2%; Fig. 1 D, Lower Right), whereas fewer PCP4+ neurons in Ctrl mice were surrounded by WFA+ PNNs (33% ± 8%). These findings are consistent with previous reports that PNN-enwrapped excitatory neurons are prevalent in the CA2 hippocampus (47, 49) and indicate that CS 4-O-sulfation regulates the density of PNNs surrounding excitatory CA2 neurons.
Next, we examined potential mechanisms by which loss of 4-O-sulfation could alter PNNs in the adult hippocampus. Previous studies have shown that increasing CS 6-O-sulfation by overexpression of C6ST-1 in transgenic mice decreases accumulation of the CSPG aggrecan in PNNs, possibly by accelerating its proteolysis by a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) protease (50). However, immunohistochemical analysis of Chst11cKO and Ctrl mice revealed high, comparable expression levels of aggrecan in the CA2 region (SI Appendix, Fig. S3). The homeobox protein Otx2 localizes to PNNs by binding to the CS-E motif on CSPGs and regulates PNN assembly and maintenance, as well as PV maturation in the VC (51, 52). Comparable but low levels of Otx2 expression were observed in the hippocampus of both Chst11cKO and Ctrl mice (SI Appendix, Fig. S4). Together, these findings suggest that CS 4-O-sulfation likely regulates PNN densities in the CA2 hippocampus via mechanisms independent of both aggrecan stability and Otx2.
CS 4-O-Sulfation Regulates Dendritic Spine Maturation and Excitatory-to-Inhibitory Synaptic Ratios.
PNNs influence a broad range of synaptic functions, thereby affecting dendritic spine morphology, synapse stability, and both excitatory and inhibitory neurotransmission (53). To examine the effects of CS 4-O-sulfation on dendritic spine morphology, GFP-expressing lentivirus was injected into the hippocampal CA1, CA2, or CA3 regions of Chst11cKO or Ctrl mice to sparsely label dendrites and dendritic spines (Fig. 2A and SI Appendix, Fig. S5A). Notably, the density of mature spines on both CA1 and CA2 neurons was significantly decreased in Chst11cKO mice compared to Ctrl mice (Fig. 2 B and D), whereas the density of dendritic protrusions was unchanged (SI Appendix, Fig. S5 B and C). Furthermore, the head width of dendritic spines was significantly reduced on CA1 but not CA2 neurons in Chst11cKO compared to Ctrl mice (Fig. 2 C and E). Interestingly, CA3 neurons in Chst11cKO mice had an enlarged average spine head width, but no difference in mature spine or protrusion density, compared to CA3 neurons in Ctrl mice (SI Appendix, Fig. S5 D–G). As CA2 pyramidal cells are known to gate hippocampal CA3 excitability via feedforward inhibition (54), it is conceivable that the excess PNNs surrounding CA2 neurons in Chst11cKO mice may inhibit CA2 output and thus enhance synaptic transmission in the CA3 area, leading to enlarged dendritic spine heads on CA3 neurons. Taken together, these findings indicate that CS 4-O-sulfation is required for the proper maturation of dendritic spines in the hippocampus.
Fig. 2.
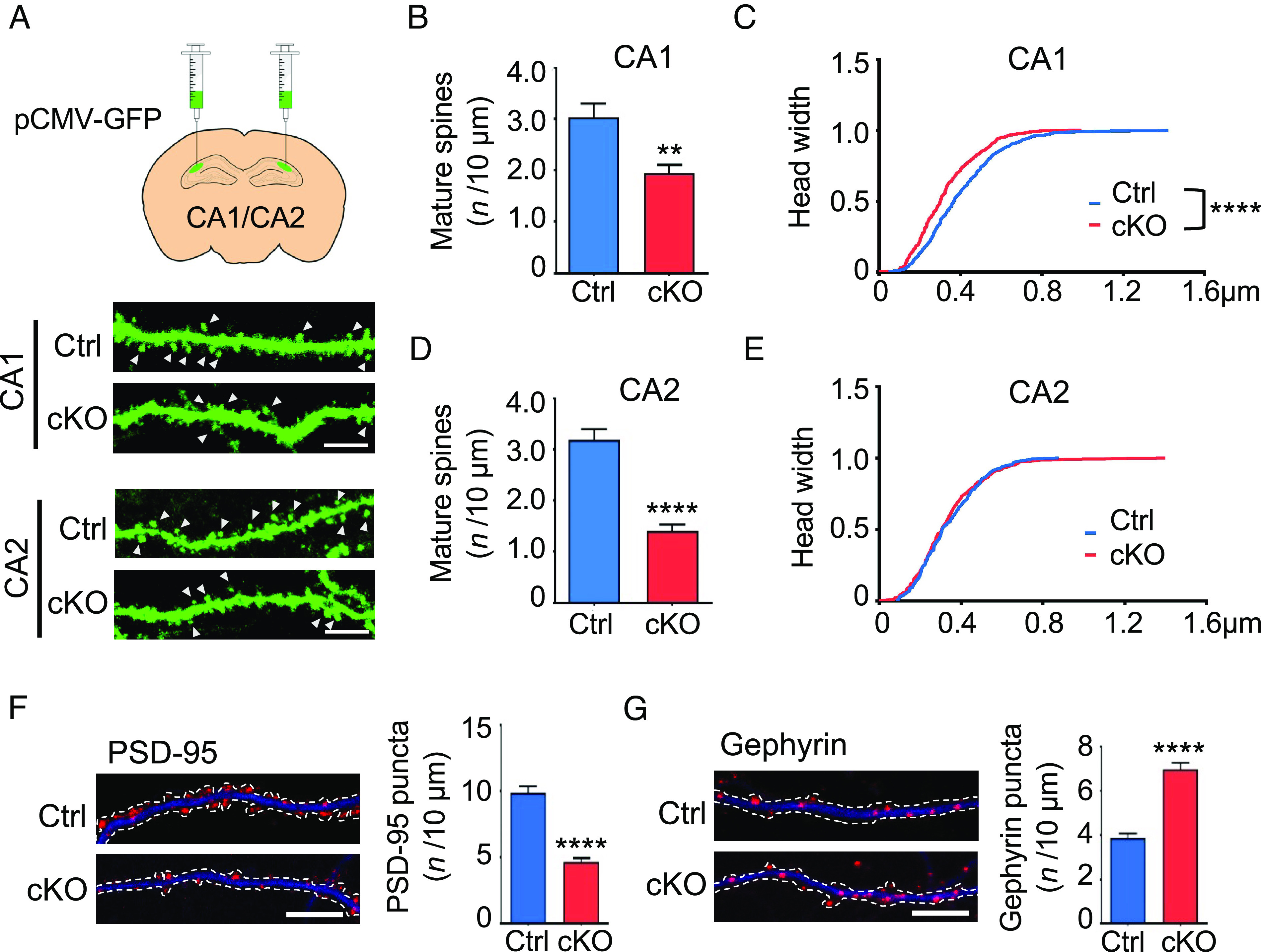
Impaired synapse development in Chst11cKO hippocampal neurons. (A) Ctrl and Chst11cKO mice were injected with GFP-expressing lentivirus into the hippocampal regions. Representative images showing dendritic spines of viral-infected CA1 and CA2 neurons. Mature spines are indicated by white arrowheads. (Scale bar, 5 μm.) (B and C) CA1 neurons from Chst11cKO mice display fewer mature spines with smaller head widths compared to Ctrl mice. Quantification of the mature spine density is shown in (B). **P < 0.01 vs. Ctrl, Student’s t test. The cumulative distribution curve of spine head widths is shown in (C). ****P < 0.0001 vs. Ctrl, Kolmogorov–Smirnov test. n = 23 and 22 dendrites from 4 pairs of Ctrl and Chst11cKO mice, respectively. (D and E) CA2 neurons from Chst11cKO mice display fewer mature spines with similar head widths compared to Ctrl mice. Quantification of the mature spine density is shown in (D). ****P < 0.0001 vs. Ctrl, Student’s t test. Cumulative distribution curve of spine head widths is shown in (E). n = 13 and 16 dendrites from 4 pairs of Ctrl and Chst11cKO mice, respectively. (F and G) Hippocampal neurons from Chst11cKO mice have fewer excitatory synapses and more inhibitory synapses compared to Ctrl mice. Representative images and quantification of PSD-95 puncta (red) distributed along the dendrites (MAP2, blue) of Ctrl and Chst11cKO neurons are shown in (F). Representative images and quantification of gephyrin puncta (red) of Ctrl and Chst11cKO neurons are shown in (G). Dendrites were traced with dotted lines. (Scale bar, 10 μm.) ****P < 0.0001 vs. Ctrl, Student’s t test; n = 20 neurons each for Ctrl and Chst11cKO mice. All data are shown as the mean ± SEM.
Dendritic spines harbor most excitatory synapses (55), and the excitatory-to-inhibitory (E/I) synaptic ratio can serve as a good indicator of synaptic balance (56). Thus, we investigated whether loss of CS 4-O-sulfation altered the development of excitatory and inhibitory synapses. Chst11cKO mice were crossed with Chst11loxP/loxP mice, and hippocampal neurons from each mouse embryo were cultured separately and genotyped (SI Appendix, Fig. S6A). We observed a large increase in WFA+ PNNs surrounding hippocampal neurons cultured from Chst11cKO mice compared to Ctrl mice (SI Appendix, Fig. S6 B and C), similar to observations in hippocampal brain sections (Fig. 1C). Hippocampal neurons from Chst11cKO mice also had fewer mature excitatory synapses relative to hippocampal neurons from Ctrl mice, as measured by a reduction in the density and size of PSD-95-expressing puncta (Fig. 2F and SI Appendix, Fig. S6D). A concomitant increase in inhibitory synapses was observed, as quantified from the density and size of gephyrin-expressing puncta (Fig. 2G and SI Appendix, Fig. S6E). Thus, loss of CS 4-O-sulfation in Chst11cKO hippocampal neurons attenuates excitatory synapse development and promotes the formation of inhibitory synapses.
We next asked whether these synaptic defects were the result of 1) the higher PNN densities observed in the hippocampus of Chst11cKO mice and/or 2) loss of the CS-A and CS-E sulfation motifs in PNNs. To reduce PNN density, we treated hippocampal neurons from Chst11cKO or Ctrl mice with ChABC. Efficient removal of PNNs on cultured hippocampal neurons was achieved after 2 h of ChABC treatment (SI Appendix, Fig. S7 A–C). Notably, ChABC treatment fully restored the density and size of both PSD-95 and gephyrin puncta to Ctrl levels (Fig. 3 A and B and SI Appendix, Fig. S7 D and E). To investigate the effects of CS sulfation on synapses, we treated Chst11cKO neurons with exogenous, natural polysaccharides enriched in specific sulfation motifs. Treatment of Chst11cKO neurons with either CS-A- or CS-E-enriched polysaccharides restored the number of WFA+ PNN-enwrapped neurons (SI Appendix, Fig. S8 A and B), as well as the density and size of excitatory synapses (Fig. 3C and SI Appendix, Fig. S8F), back to Ctrl levels. In contrast, CS-C polysaccharides enriched in 6-O-sulfation had no effect on PNN levels or excitatory synapses (Fig. 3C and SI Appendix, Fig. S8 B and F), demonstrating the importance of the sulfation pattern. Interestingly, none of the CS polysaccharides rescued the inhibitory synaptic defects of Chst11cKO hippocampal neurons (SI Appendix, Fig. S8 C–E), suggesting that CSPGs and PNNs regulate excitatory and inhibitory synapses via distinct mechanisms. Importantly, the effects of Chst11 genetic deletion on PNN density and E/I synaptic ratio were recapitulated by chemical inhibition of CS 4-O-sulfation using a small-molecule sulfotransferase inhibitor (57) (Fig. 3D and SI Appendix, Fig. S8 G and H). Thus, chemical, enzymatic, or genetic manipulation of the 4-O-sulfation motifs on CSPGs can modulate PNN densities surrounding hippocampal neurons and the balance of excitatory and inhibitory synapses.
Fig. 3.
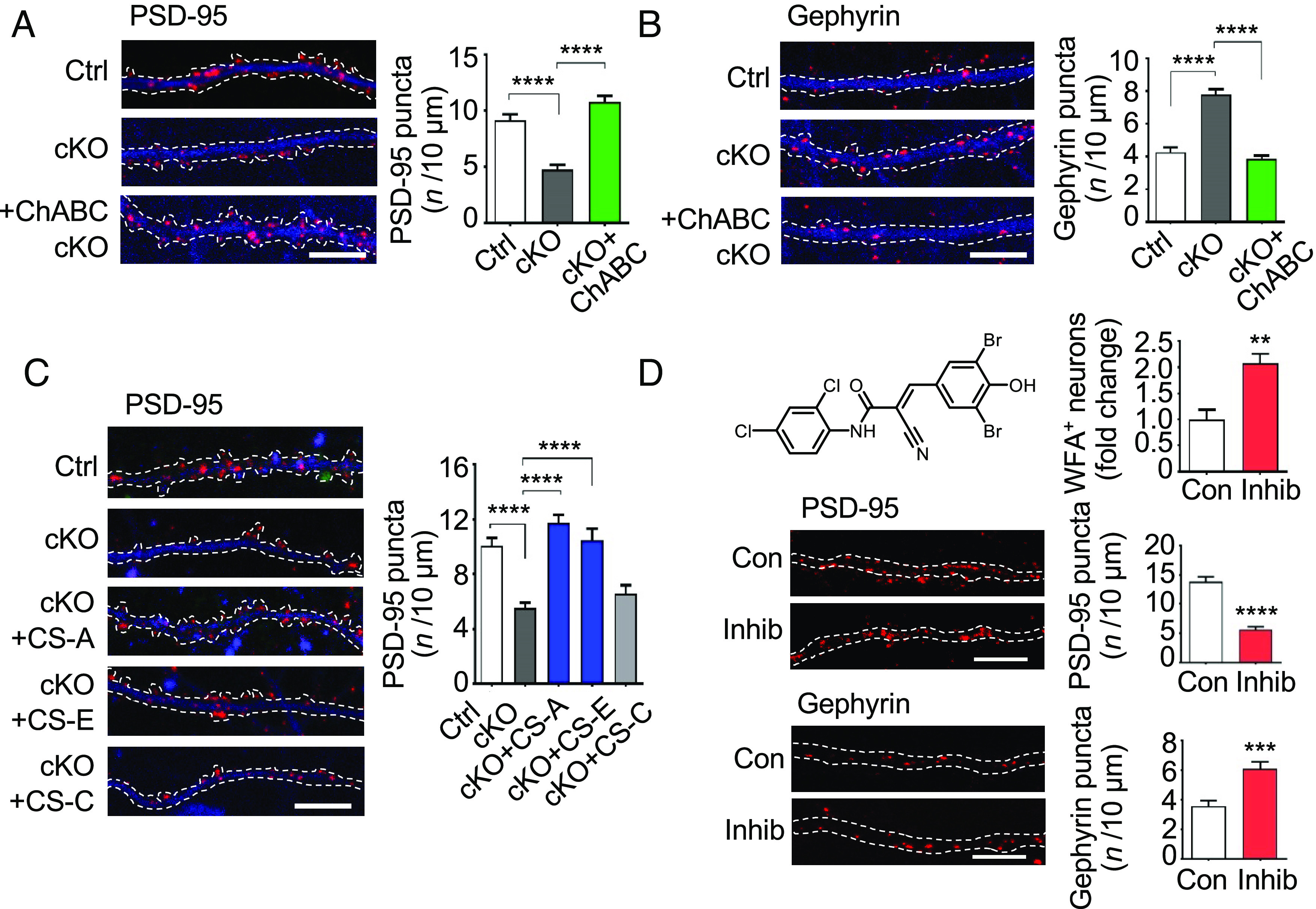
Modulation of PNN density or CS 4-O-sulfation levels regulates hippocampal synapses. (A) ChABC treatment rescued the decreased density of excitatory synapses in Chst11cKO neurons. Representative images and quantification of PSD-95 puncta (red) distributed along the dendrites (MAP2, blue) of the indicated cultured neurons. (Scale bar, 10 μm.) ****P < 0.0001 vs. Chst11cKO, one-way ANOVA followed by Tukey’s multiple comparisons test; n = 10, 11, and 10 neurons for Ctrl, Chst11cKO, and ChABC-treated Chst11cKO conditions, respectively. (B) ChABC treatment rescued the increased density of inhibitory synapses in Chst11cKO neurons. Representative images and quantification of gephyrin puncta (red) distributed along the dendrites (MAP2, blue) of the indicated cultured neurons. (Scale bar, 10 μm.) ****P < 0.0001 vs. Chst11cKO, one-way ANOVA followed by Tukey’s multiple comparisons test; n = 10 neurons per condition. (C) CS-A or CS-E, but not CS-C, polysaccharides rescued the decreased density of excitatory synapses in Chst11cKO neurons. Representative images and quantification of PSD-95 puncta (red) distributed along the dendrites (MAP2, blue) of the indicated cultured neurons. (Scale bar, 10 μm.) Quantification of PSD-95 puncta number per 10 μm. ****P < 0.0001 vs. Chst11cKO, one-way ANOVA followed by Tukey's multiple comparisons test; n = 15, 13, 14, 10, and 12 neurons for Ctrl, Chst11cKO, and Chst11cKO neurons treated with CS-A, CS-E or CS-C, respectively. (D) A chemical inhibitor of the CS-E sulfotransferase Chst15 (Inhib, 10 µM in DMSO, 24 h) (57) elevated PNN levels (Top), decreased the density of excitatory synapses (Middle) and increased the density of inhibitory synapses (Bottom). Hippocampal neurons were cultured from wild-type C57 mice. DMSO was used as a control (Con). Quantification of the number of WFA+ neurons (normalized to Con) is shown in (D). **P < 0.01 vs. Con, Student’s t test; n = 7 regions each for cultured neurons treated with Con or Inhib. Representative images and quantification of both PSD-95 and gephyrin puncta distributed along the dendrites (MAP2, blue) of the indicated cultured neurons are also shown in (D). (Scale bar, 10 μm.) ***P < 0.001, ****P < 0.0001 vs. Con, Student’s t test. For PSD-95, n = 9 and 10 neurons treated with Con or Inhib, respectively. For gephyrin, n = 8 and 9 neurons treated with Con or Inhib, respectively. All data are shown as the mean ± SEM.
To assess whether the observed synaptic defects alter E/I synaptic transmission, we performed whole-cell patch clamp recordings on acute hippocampal slices from Chst11cKO and Ctrl mice. As reported previously (58), patch clamping of CA2 neurons is technically challenging due to the high PNN densities surrounding these neurons. Nonetheless, CA2 Chst11cKO neurons showed a trend toward increased frequency of miniature inhibitory postsynaptic currents (mIPSCs) compared to Ctrl neurons (P = 0.07; SI Appendix, Fig. S9 A and C), consistent with the increased number of inhibitory synapses observed in Chst11cKO neurons. The amplitude of mIPSC and miniature excitatory postsynaptic currents (mEPSCs) was similar for both CA2 Chst11cKO and Ctrl neurons (SI Appendix, Fig. S9 D and F), and no significant change in mEPSC frequency was detected (SI Appendix, Fig. S9 B and E). Our ability to measure decreases in mEPSC frequency may have been hindered by the low frequencies of CA2 pyramidal neurons (59). Overall, the data suggest that deletion of CS 4-O-sulfation alters dendritic spine maturation and E/I synapse ratios and leads to functional deficits in synaptic transmission.
Loss of CS 4-O-Sulfation and Increased PNNs Reduce CREB Activity in Hippocampal Neurons.
Previously, we had shown that neurotrophins such as brain-derived neurotrophic factor (BDNF) bind preferentially to 4-O-sulfated CS motifs and form ternary CS-BDNF-TrkB receptor complexes (60, 61). Cell surface engineering of cultured hippocampal neurons to display CS polysaccharides enriched in the CS-A or CS-E motifs led to enhanced neurotrophin signaling and neurite outgrowth (62), suggesting that CS-A/E polysaccharides can function as coreceptors to activate neurotrophin signaling. On the other hand, CSPGs have also been shown to modulate BDNF-induced dendritic spine growth in cultured cortical neurons (63) and TrkB phosphorylation in PV+ neurons in vitro (64) via the PTPσ receptor. We therefore hypothesized that the synaptic defects in Chst11cKO neurons might stem from dysregulation of BDNF signaling. To test this, we examined cyclic AMP-response element binding protein (CREB), an important regulator of synapse development and plasticity whose phosphorylation is induced by BDNF (65, 66). Indeed, the average levels of CREB phosphorylated at Ser133 (p-CREB) were significantly reduced in the CA2 neurons of Chst11cKO mice compared to Ctrl mice, whereas total CREB levels remained unchanged (Fig. 4 A and B and SI Appendix, Fig. S10 A–C). This reduction was specific to the area CA2, consistent with the marked change in PNN densities observed in this hippocampal subregion (Fig. 1C). To assess whether the observed inhibition of CREB activity was due to the increase in PNN densities surrounding CA2 neurons, we stereotaxically delivered ChABC or penicillinase (Pen) as a control into the CA2 hippocampus of adult Chst11cKO and Ctrl mice. WFA staining after 2 wk revealed that the PNN levels of Chst11cKO mice injected with ChABC were comparable to those of Ctrl mice injected with Pen (Fig. 4C). Importantly, ChABC-mediated pruning of PNNs in the CA2 significantly enhanced the p-CREB levels in Chst11cKO mice (Fig. 4D). Interestingly, Ctrl mice injected with ChABC showed an increase in p-CREB levels in the CA2 (Fig. 4D), suggesting that altering basal PNN levels in the brains of normal adult mice can modulate CREB activity.
Fig. 4.

Reduced p-CREB levels in Chst11cKO hippocampal neurons can be rescued by treatment with ChABC or 4-O-sulfated CS polysaccharides. (A) Representative images showing hippocampal slices of Ctrl and Chst11cKO mice stained with a phospho-Ser133 CREB antibody (red) and WFA (green). The Left panels show images of the CA2 and CA3 regions. (Scale bar, 200 μm.) The Right panels show the cropped and magnified images of the CA1, CA2, and CA3 regions used for quantifications. (Scale bar, 50 μm.) (B) Quantification of the average p-CREB fluorescence intensity per neuron in the indicated hippocampal regions (normalized to Ctrl CA1 region). *P < 0.05 vs. Ctrl, Student’s t test; n = 6 pairs of Ctrl and Chst11cKO mice. (C) ChABC or penicillinase (Pen) was injected bilaterally into hippocampal CA2 regions. Representative images of Ctrl and Chst11cKO hippocampal slices 2 wk after injection. Slices were stained with WFA (green) and an anti-PCP4 antibody (blue; lower panels in gray scale). (Scale bar, 500 μm.) (D) Quantification of the average p-CREB fluorescence intensity per neuron (normalized to Pen-treated Ctrl mice) in the CA2 region for each condition.**P < 0.01, ****P < 0.0001, two-way ANOVA, Genotype × Treatment: F(1,33) = 1.181, P = 0.2850 for CA2; n = 10, 9, 9, and 9 slices from 3 mice each for Ctrl Pen, Ctrl ChABC, Chst11cKO Pen, and Chst11cKO ChABC conditions, respectively. (E) Quantification of p-CREB levels for Ctrl, Chst11cKO, and ChABC-treated Chst11cKO neuronal cultures (normalized to Ctrl). ****P < 0.0001 vs. Chst11cKO, one-way ANOVA followed by Tukey’s multiple comparisons test; n = 300 to 400 neurons for each condition. (F) Treatment with 4-O-sulfated CS-A or CS-E polysaccharides, but not 6-O-sulfated CS-C polysaccharides, rescued the p-CREB levels in Chst11cKO neurons. Quantification of p-CREB levels for Ctrl, Chst11cKO, and CS polysaccharide–treated (20 μg/mL, 24 h) Chst11cKO neurons. *P < 0.05, ****P < 0.0001 vs. Chst11cKO, one-way ANOVA followed by Tukey's multiple comparisons test; n = ~100 to 150 neurons for each condition. All data are shown as the mean ± SEM.
We also studied the regulation of p-CREB levels using hippocampal neuronal cultures. Neurons from wild-type C57 mice enwrapped by PNNs had lower p-CREB levels (SI Appendix, Fig. S11 A and B), and PNN removal using ChABC significantly increased p-CREB levels, but not total CREB levels, for at least 24 h (SI Appendix, Fig. S11C). Consistent with the in vivo data, Chst11cKO neurons had more WFA+ PNNs (SI Appendix, Fig. S6 B and C) and reduced p-CREB levels (Fig. 4E and SI Appendix, Fig. S11D) compared to Ctrl neurons. Moreover, ChABC digestion of the PNNs surrounding Chst11cKO neurons led to a significant increase in p-CREB levels, returning them to Ctrl levels (Fig. 4E). Notably, treatment of Chst11cKO neurons with exogenous natural polysaccharides enriched in the 4-O-sulfated CS-A or CS-E motifs, but not the 6-O-sulfated CS-C motif, restored both the PNN (SI Appendix, Fig. S8 A and B) and p-CREB (Fig. 4F) levels to Ctrl levels. Taken together, these data suggest that 4-O-sulfation of CS polysaccharides regulates the density of PNNs surrounding hippocampal neurons, thereby modulating CREB activity and synapse development.
Loss of CS 4-O-Sulfation and High PNN Densities Impair Social Memory and Elevate Anxiety Levels.
Neurological disorders such as autism, schizophrenia, and fragile X syndrome are characterized by structural alterations in dendritic spines and synapses (67, 68). Moreover, mice with targeted CREB mutations or gene deletion exhibit deficiencies in long-term memory (69, 70). As silencing of CA2 neurons disrupts social recognition memory (20), we examined the effects of PNNs and CS 4-O-sulfation on CA2-dependent social memory. Chst11cKO and Ctrl mice received bilateral stereotaxic injections of ChABC or Pen in the CA2 region, followed by a two-trial social memory test (71). In the first trial, the subject mouse was allowed to interact with a stimulus mouse for 2 min (Fig. 5A). After 30 min of separation, the subject mouse was then reexposed in the second trial to the previously encountered stimulus mouse for 2 min. For Ctrl mice injected with Pen, the interaction time with the stimulus mouse was significantly reduced from the first trial to the second, indicative of normal social recognition memory (Fig. 5 B and D). In contrast, Chst11cKO mice injected with Pen exhibited no significant reduction in average interaction time between the two trials (Fig. 5 C and D). Importantly, Chst11cKO mice injected with ChABC showed a decrease in average interaction time during the second trial, suggesting that removal of the excess PNNs rescued the social memory deficits (Fig. 5 D and F). Interestingly, Ctrl mice injected with ChABC showed no reduction in interaction time from the first trial to the second (Fig. 5 D and E). Thus, while higher PNN densities in the area CA2 impair social memory, lower PNN densities caused by ChABC overdigestion also appear to be detrimental. These findings suggest that PNN levels are finely balanced in vivo under physiological conditions to achieve normal social memory function.
Fig. 5.

Loss of CS 4-O-sulfation and high PNN densities in the area CA2 impair social memory and increase anxiety. (A) Schematic depicting the two-trial social memory test employed in this study. (B and C) Chst11cKO mice exhibit impairments in social memory that are not observed in Ctrl mice. Interaction times are shown for Ctrl mice (B, **P < 0.01 vs. trial 1, paired Student’s t test; n = 9) or Chst11cKO mice (C, not significant; n = 11) injected with Pen. Each circle represents an individual subject. (D–F) Injection of ChABC into the CA2 region rescued the social memory deficits in Chst11cKO mice and impaired social memory in Ctrl mice. The average ratio of the 2nd to the 1st interaction time for each condition is shown in (D). **P < 0.01, two-way ANOVA, Genotype × Treatment: F(1,36) = 29.90, P < 0.0001; n = 9, 11, 10, and 10 mice for Ctrl Pen, Chst11cKO Pen, Ctrl ChABC, and Chst11cKO ChABC groups, respectively. Interaction times are shown for Ctrl mice (E, not significant; n = 10) or Chst11cKO mice (F, ***P < 0.001 vs. trial 1, paired Student’s t test; n = 10) injected with ChABC. (G–I) Chst11cKO mice exhibit elevated anxiety levels compared to Ctrl mice. The average percentage of time spent in the center of the arena during the OFT is shown in (G). **P < 0.01 vs. Ctrl, Student’s t test; n = 12 Ctrl and 10 Chst11cKO mice. The average percentage of time spent in the open arms of the EPM is shown in (H). *P < 0.05 vs. Ctrl, Student’s t test; n = 10 Ctrl and 11 Chst11cKO mice. The average percentage of time spent in the lighted box of the LDB is shown in (I). *P < 0.05 vs. Ctrl, Student’s t test; n = 10 Ctrl and 11 Chst11cKO mice. (J–L) PNN digestion in the CA2 region restored the anxiety levels of Chst11cKO mice back to Ctrl levels, as shown in the OFT and LDB tests. For the OFT (J), **P < 0.01 vs. Chst11cKO Pen, two-way ANOVA, Genotype × Treatment: F(1,54) = 3.253, P = 0.0769; n = 13, 13, 16, and 16 mice for Ctrl Pen, Chst11cKO Pen, Ctrl ChABC, and Chst11cKO ChABC groups, respectively. For the EPM (K), *P < 0.05, ***P < 0.001 vs. Ctrl Pen, two-way ANOVA, Genotype x Treatment: F(1,48) = 11.32, P = 0.0015; n = 13, 12, 13, and 14 mice for Ctrl Pen, Chst11cKO Pen, Ctrl ChABC, and Chst11cKO ChABC groups, respectively. For the LDB (L), *P < 0.05, **P < 0.01 vs. Chst11cKO Pen, two-way ANOVA, Genotype × Treatment: F(1,52) = 11.75, P = 0.0012; n = 14, 13, 15, and 14 mice for Ctrl Pen, Chst11cKO Pen, Ctrl ChABC, and Chst11cKO ChABC groups, respectively. All data are shown as the mean ± SEM.
Next, we assessed whether loss of CS 4-O-sulfation in Chst11cKO mice was associated with other brain dysfunctions. Specifically, we monitored the anxiety level and general locomotor activity of Chst11cKO mice using the open-field test (OFT). While the total distance traveled by Chst11cKO mice was comparable to Ctrl mice (SI Appendix, Fig. S12A), Chst11cKO mice spent less time in the center of the open field arena relative to Ctrl mice (Fig. 5G), suggesting a higher level of anxiety. To verify further the increased anxiety levels of these animals, we performed the elevated plus maze (EPM) and light–dark box (LDB) tests (72, 73). Consistent with the enhanced anxiety levels shown in the OFT, Chst11cKO mice spent less time in the open arm of the EPM despite traveling comparable distances (Fig. 5H and SI Appendix, Fig. S12B) and less time in the brightly lit chamber of the LDB compared to Ctrl mice (Fig. 5I).
To probe whether PNNs contribute to the observed anxiety defects, we injected ChABC or Pen bilaterally into the CA2 region as before. Indeed, PNN digestion in the CA2 region of Chst11cKO mice decreased the higher anxiety levels, restoring them to similar levels as Pen- or ChABC-injected Ctrl mice (Fig. 5 J–L) without affecting locomotor activity (SI Appendix, Fig. S12 C and D). Interestingly, contrary to what we observed for social memory (Fig. 5 D and E), no significant differences in anxiety-related OFT and LDB behavior were detected between Pen- and ChABC-injected Ctrl mice (Fig. 5 J and L), although a reduction in the open arm duration was observed in the EPM (Fig. 5K). Thus, PNN digestion solely in the CA2 region of Ctrl mice appears to be sufficient to alter social recognition memory but not anxiety levels. Nonetheless, restoring PNN densities in the CA2 hippocampus of Chst11cKO mice to normal Ctrl levels is sufficient to rescue the observed anxiety-like behavior.
Hippocampal-Specific Deletion of Chst11 Increases PNN Densities in the CA2 Region and Leads to Impaired Social Memory.
As previous studies have suggested that inactivation of CA2 pyramidal neurons does not influence anxiety-like behavior (20), we further explored the region-specific effects of Chst11 deletion. A Cre-recombinase-expressing virus was employed to knock out Chst11 with high regional and temporal specificity in the CA2 region. Adult Chst11loxP/loxP mice were stereotaxically injected with viruses expressing either mCherry-tagged Cre recombinase (AAV5-CaMKIIɑ-mCherry-Cre, Cre) or only mCherry (AAV5-CaMKIIɑ-mCherry, Con) under the control of the calcium/calmodulin-dependent protein kinase II (CaMKII) promoter (Fig. 6A). This restricted Cre expression and hence Chst11 gene deletion specifically to excitatory neurons in the CA2 hippocampus (74). Accordingly, we observed a robust mCherry signal in the CA2 region of Cre-injected Chst11loxP/loxP mice three weeks after virus injection, indicative of region-specific Cre expression in this area (Fig. 6A). The CA2-specific loss of CS 4-O-sulfation was confirmed in mCherry-positive neurons by immunostaining with an anti-CS-E antibody (SI Appendix, Fig. S13A). Similar to Chst11cKO mice, Cre-injected Chst11loxP/loxP mice showed a significant increase in WFA+ PNN-enwrapped neurons (Fig. 6A) and a reduction in p-CREB levels in the CA2 region compared to Con-injected mice (Fig. 6B). These results demonstrate that modulation of CS 4-O-sulfation specifically in the CA2 region of adult mice is sufficient to alter PNN densities and CREB activity in the hippocampus.
Fig. 6.
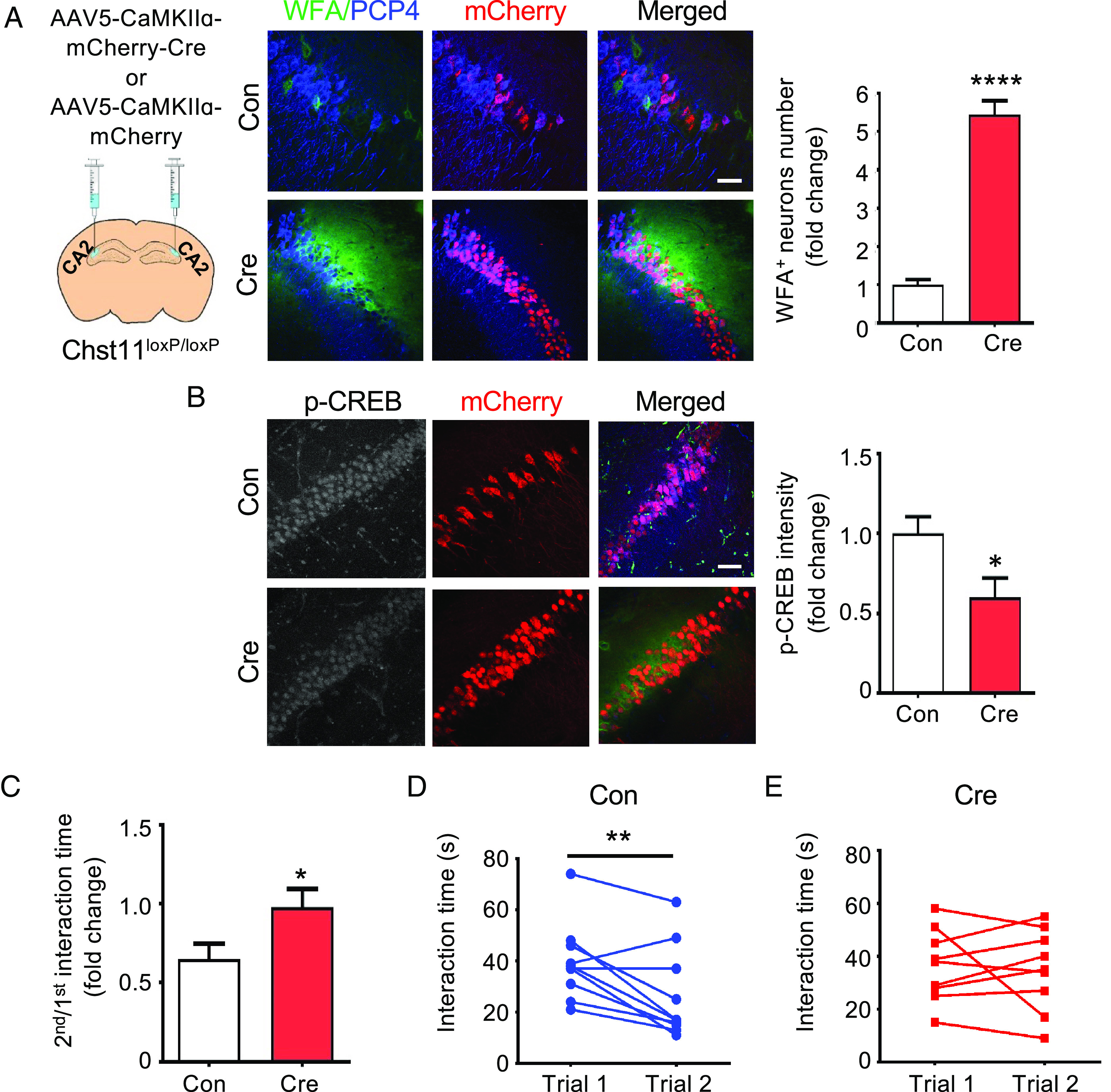
CA2-specific deletion of Chst11 increases PNN densities and impairs social memory. (A) CA2 region-specific Chst11 deletion using AAV-CaMKIIɑ-mCherry-Cre (Cre) results in increased PNN densities. Representative images of the CA2 region from Cre-injected or AAV-CamKIIɑ-mCherry (Con)-injected mice are shown. Hippocampal slices were labeled with mCherry (red), anti-PCP4 antibody (blue), and WFA (green). (Scale bar, 50 μm.) Quantification of the PNN density, measured as the number of WFA+ neurons in the CA2 region (normalized to Con). ****P < 0.0001 vs. Con, Student’s t test; n = 10 brain slices each from 4 pairs of Cre- and Con-injected mice. (B) Reduced p-CREB levels in the CA2 region of Cre-injected floxed Chst11 mice. Representative images showing the CA2 regions of Con- and Cre-injected floxed Chst11 mice, stained with p-CREB antibody (gray scale in separate image; blue in merged image), mCherry (red), and WFA (green). Quantification of p-CREB levels in the CA2 regions of Cre-injected floxed Chst11 mice and Con-injected mice. *P < 0.05 vs. Con, Student’s t test; n = 12 slices each from 4 pairs of Con- and Cre-injected animals. (C–E) Cre-injected mice displayed impaired social memory compared to Con-injected mice. The ratio of the 2nd to the 1st interaction time for Con- and Cre-injected mice is shown in (C). *P < 0.05 vs. Con, Student’s t test; n = 10 and 9 for Con-injected and Cre-injected mice, respectively. Interaction times for Con-injected mice (D, **P < 0.01 vs. trial 1, paired Student’s t test; n = 10) and Cre-injected mice (E, not significant; n = 9). Each circle represents an individual subject. All data are shown as the mean ± SEM.
Next, we examined the social memory and anxiety-like behavior of these mice. While Con-injected mice displayed a significant reduction in social interaction time in the second trial, Cre-injected mice spent an equal amount of time in both trials (Fig. 6 C–E), indicating that CA2-specific Chst11 deletion leads to social memory defects. Despite the increased anxiety-like behavior observed in Chst11cKO mice (Fig. 5 G–I), CA2-specific deletion of Chst11 at adulthood did not alter anxiety levels. Cre-injected Chst11loxP/loxP mice showed comparable anxiety levels to Con-injected Chst11loxP/loxP mice, as assessed by the OFT, EPM, and LDB tests (SI Appendix, Fig. S13 B–D). Taken together with our earlier observations that ChABC treatment did not affect anxiety levels in the Ctrl mice (Fig. 5 J –L), these findings suggest that CS 4-O-sulfation may influence anxiety-like behaviors through complex mechanisms involving CA2 neurons and other brain regions. Nevertheless, the results indicate that CA2-specific deletion of CS 4-O-sulfation at adulthood is sufficient to drive malformation of PNNs and social memory dysfunction.
Discussion
In this study, we demonstrate that 4-O-sulfated CS, the dominant sulfation motif on CSPGs in the adult brain, plays a critical role in regulating PNN levels and E/I synapse balance in the adult hippocampus. Brain-wide deletion of chondroitin 4-O-sulfation led to elevated PNNs, particularly in the CA2 hippocampus, and thereby disrupted CREB activation, synapse development, and social memory in mice. Modulation of 4-O-sulfation levels via other methods, including viral-mediated gene deletion and chemical inhibitors of CS sulfotransferases recapitulated the effects of Chst11 gene knockout, while ChABC and exogenous 4-O-sulfated, but not 6-O-sulfated, CS polysaccharides rescued the effects. Collectively, our data demonstrate that CS 4-O-sulfation is essential for the proper functioning of the hippocampus and contributes to higher brain functions.
During mouse brain development, the proportion of chondroitin 4-O-sulfation on CSPGs progressively increases, while the proportion of chondroitin 6-O-sulfation decreases (16, 28). The function of 6-O-sulfation on CSPGs has been studied in the developing VC, where it was tightly linked to PNNs surrounding PV-expressing GABAergic inhibitory interneurons (16, 75). Transgenic mice overexpressing the 6-O-sulfotransferase C6ST-1 showed fewer PNNs enwrapping PV+ interneurons and prolonged ocular dominance plasticity (16), leading to the proposal that a low 4S/6S ratio regulates the functional maturation of PV-expressing interneurons and maintains plasticity in the VC. Based on these results, chondroitin 4-O-sulfation was inferred to be a molecular brake that inhibits ocular dominance plasticity at the end of the critical period (28). Consistent with an inhibitory role, 4-O-sulfated CS motifs are also up-regulated in the glial scar after CNS injury and limit axon regeneration and neuroplasticity (29, 30). Knockdown of Chst11 in zebrafish led to loss of chondroitin 4-O-sulfation and enhanced regeneration after spinal cord injury (31). However, the precise functions of 4-O-sulfated structures such as CS-A, which represents about 90% of the total CS and is the most abundant glycosaminoglycan structure in the adult brain, are not well understood. In this study, we directly examined the roles of 4-O-sulfation in the adult uninjured brain. We observed impaired dendritic spine maturation, an imbalance of E/I synapses, together with elevated anxiety levels and social memory defects, in mice with a brain-specific deletion of the chondroitin 4-O-sulfotransferase Chst11 gene. Our findings indicate critical albeit nuanced roles for chondroitin 4-O-sulfation in the regulation of PNNs and neuroplasticity, with distinct roles depending on the cell type, developmental stage, and brain region. Thus, the sulfation patterns on CSPGs must be finely tuned in a cell-type-specific manner, both during critical periods of development and throughout adulthood.
We found that CS 4-O-sulfation in PNNs modulated CREB activity important for excitatory synapse development and maturation in the CA2 hippocampus. Brain-wide ablation of 4-O-sulfation markedly increased PNNs and caused fewer mature dendritic spines in the CA2 region. Furthermore, a decrease in E/I synapse ratio was observed in hippocampal neurons cultured from mice lacking 4-O-sulfation. Previous studies by our lab and others have shown that 4-O-sulfated CS motifs presented on cell surfaces are specifically recognized by neurotrophins such as BDNF and can stimulate neurotrophin-mediated neurite growth (60, 62, 76). Moreover, BDNF-TrkB pathway activation and enhanced CREB activity promote dendritic spine maturation (77, 78), while loss of BDNF via Cre-mediated bdnf deletion specifically in postmitotic neurons impairs dendritic spine growth (79). Thus, we postulated that the observed synaptic defects might be caused by dysregulation of BDNF-CREB signaling. In accordance with this hypothesis, CREB activity was significantly reduced in CA2 neurons of Chst11cKO mice compared to Ctrl mice, and this activity was rescued by restoring PNN levels to normal Ctrl levels. Notably, the deficits in PNNs, CREB activation, and synapses could also be recapitulated by decreasing CS 4-O-sulfation levels in wild-type hippocampal neurons using a CS sulfotransferase chemical inhibitor. Moreover, restoration of CS 4-O-sulfation levels in Chst11cKO neurons using natural CS-A or CS-E polysaccharides reduced the PNN densities and returned CREB activity to normal levels. We propose that the sulfation patterns on CSPGs in PNNs may regulate CREB activity and dendritic spine maturation via at least two distinct mechanisms. First, chondroitin 4-O-sulfation levels modulate the density of PNNs, which can act as “molecular sieves” to control diffusion and access of extracellular ligands such as BDNF to neuronal cell surfaces. Second, the presentation of 4-O-sulfated motifs on CSPGs within PNNs can act as “molecular scaffolds” for extracellular ligands (52, 80), which facilitates the specific recruitment of BDNF and possibly other ligands to the surface of hippocampal neurons and assists in ligand-receptor activation. In support of these mechanisms, we have previously shown that natural CS-E polysaccharides are capable of forming ternary CS-BDNF-TrkB complexes (60, 61) and promoting both neurotrophin pathway activation and neurite outgrowth (62). In Chst11cKO mice, the increased PNN densities, combined with the loss of 4-O-sulfation motifs important for proper BDNF function, likely account for the observed reduction in CREB activity and synaptic defects. Overall, our studies suggest that 4-O-sulfation of CSPGs plays a critical role in controlling PNN densities and maintaining optimal levels of ligand-receptor engagement at the cell surface, thereby regulating CREB activity essential for synapse development and hippocampal function. It is worth noting that additional proteins and cell types may also play a role. For example, semaphorin 3A has been shown to bind PNNs via 4-O-sulfated CS-E motifs and to promote dendritic spine development and the clustering of postsynaptic molecules (81–83). Moreover, Sema3F is a negative regulator of spine development, and the CSPG neurocan inhibited Sema3F-induced spine elimination (84, 85). Microglia, astrocytes, and oligodendrocytes have also been shown to regulate dendritic spine development, and astrocytes are a known source of BDNF (86–88). Additional studies will be needed to fully elucidate the mechanisms contributing to the synaptic defects observed in Chst11cKO mice.
Emerging evidence suggests that PNNs contribute to cognitive functions in multiple brain regions, including cortical regions, the hippocampus, and the cerebellum (39, 89, 90). However, these conclusions were obtained by destroying PNNs using ChABC, and they raise another important question—do PNNs require optimal levels and precise timing to perform these specific functions? In our work, we demonstrate not only that ablation of CA2 PNNs impairs social memory but also that a CA2-specific and adult-specific increase of PNNs is sufficient to drive social memory dysfunction. We found that social memory was impaired when PNN levels were either too high, as observed in Chst11cKO mice, or too low, as observed in ChABC-treated Ctrl mice. Consistent with these latter observations, PNN digestion in the area CA2 was recently reported to disrupt long-term depression of inhibitory transmission (iLTD) and social memory formation in mice (25). Notably, we found that CA2 region-specific deletion of 4-O-sulfation was sufficient to cause malformation of PNNs and social memory deficits. Furthermore, the addition of natural CS-A or CS-E polysaccharides to cultured neurons deficient in CS 4-O-sulfation restored the PNN levels and E/I synapse ratios back to normal. Together, these observations suggest that dynamic modulation of CS 4-O-sulfation may enable the fine-tuned control of PNNs and allow for the remediation of E/I imbalances and CA2-associated dysfunctions.
In addition to affecting social memory, our studies suggest that chondroitin 4-O-sulfation and PNNs influence anxiety-related behaviors. Although Chst11cKO mice showed elevated anxiety levels, Chst11-floxed mice injected with Cre in the CA2 hippocampus did not exhibit anxiety defects, suggesting the involvement of other brain regions in anxiety regulation. Consistent with these observations, previous studies have reported that silencing of CA2 output affects social memory but not anxiety-like behaviors, spatial memory, or contextual memory (20, 25). Multiple brain areas and their interconnections have been suggested to regulate anxiety such as the amygdala, medial prefrontal cortex, hypothalamus, and the ventral CA1 region of the hippocampus (91). As the Chst11 gene deletion in Chst11cKO mice occurred in all brain regions, the elevated anxiety levels of Chst11cKO mice are likely due to the loss of 4-O-sulfation in brain regions other than CA2. Interestingly, however, restoring PNN densities only in the CA2 hippocampus to normal Ctrl levels was sufficient to rescue the anxiety-like behaviors of Chst11cKO mice. These findings suggest that while the cause of anxiety can be attributed to loss of 4-O-sulfation in multiple brain regions, release of CA2 activity by PNN removal may activate certain brain circuits and thereby produce anxiolytic effects. Transgenic animals lacking 4-O-sulfation or other CS sulfation motifs may be valuable models in this regard and contribute to a deeper understanding of anxiety, autism, and other neuropsychiatric disorders.
Pathological alterations in PNNs and CS sulfation are associated with neurodegenerative, neurodevelopmental, and mental disorders characterized by changes in cognition, emotion, and memory loss (13, 92, 93). For example, PNN numbers were significantly reduced in individuals with schizophrenia (94, 95), and altered CS sulfation patterns, particularly 4-O-sulfated motifs, were observed in the postmortem brains of human subjects with Alzheimer’s disease, bipolar disorder, and schizophrenia (92, 96). Abnormal PNN formation was also detected in the CA2 hippocampus of Rett syndrome (Mecp2-null) and autism (Black and Tan Brachyury (BTBR) T+ Itpr3tf/J strain) mouse models (97, 98). Our studies reveal that CS 4-O-sulfation regulates PNNs in the adult hippocampus and contributes to the proper balance of excitatory and inhibitory synapses, as well as hippocampal cognitive abilities such as social memory. Importantly, we demonstrate that enzymatic or chemical manipulation of chondroitin 4-O-sulfation levels specifically in adulthood may allow for the dynamic modulation of PNN levels, remediation of synaptic E/I imbalances, and CA2-dependent social behaviors. Thus, treatments that target chondroitin 4-O-sulfation may represent a strategy to address diseases with synaptic disturbances in E/I balance or PNN-associated pathologies, including autism, Rett syndrome, schizophrenia, and Alzheimer’s disease. More broadly, our studies identify an important role for the polysaccharides on CSPGs in the adult hippocampus and further highlight the importance of elucidating the roles of glycans in cognitive functions and neurological diseases.
Materials and Methods
All materials and methods are described in the SI Appendix, including Animals, Antibodies and Chemicals, Analysis of CS Disaccharide Composition, Neuronal Cultures and Chemical Treatments, Immunohistochemistry, Viruses, Stereotaxic Surgery, Image Acquisition and Quantification, Electrophysiology, Social Memory and Anxiety-Related Behavioral Tests, and Statistical Analyses.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Prof. Melitta Schachner and Dr. Nuray Akyüz for generously sharing the Chst11loxP/loxP and Chst11+/− mice, Dr. Henry Lester (Caltech) for providing the stereotactic device for mouse brain surgery, and Dr. Wei-Li Wu (Caltech, now at the National Cheng Kung University) for providing help and suggestions on the animal behavioral paradigms. This work was supported by the NIH (R01 GM093627).
Author contributions
H.H., Y.O., and L.C.H.-W. designed research; H.H., Y.Z., G.M.M., and G.C.Z. performed research; H.H., Y.Z., Y.O. and L.C.H.-W. analyzed data; and H.H., A.M.J., and L.C.H.-W. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: J.S., Case Western Reserve University; and C.-H.W., The Scripps Research Institute.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Varki A., Biological roles of glycans. Glycobiology 27, 3–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou J. Y., Cobb B. A., Glycans in immunologic health and disease. Annu. Rev. Immunol. 39, 511–536 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Ohtsubo K., Marth J. D., Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Mereiter S., Balmaña M., Campos D., Gomes J., Reis C. A., Glycosylation in the era of cancer-targeted therapy: Where are we heading? Cancer Cell 36, 6–16 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Lee J., et al. , Spatial and temporal diversity of glycome expression in mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 117, 28743–28753 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao H., Chen W., Smeekens J. M., Wu R., An enrichment method based on synergistic and reversible covalent interactions for large-scale analysis of glycoproteins. Nat. Commun. 9, 1692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleene R., Schachner M., Glycans and neural cell interactions. Nat. Rev. Neurosci. 5, 195–208 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Springer S. A., Gagneux P., Glycan evolution in response to collaboration, conflict, and constraint. J. Biol. Chem. 288, 6904–6911 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dityatev A., Schachner M., Sonderegger P., The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 11, 735–746 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Benarroch E. E., Extracellular matrix in the CNS: Dynamic structure and clinical correlations. Neurology 85, 1417–1427 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Fawcett J. W., Oohashi T., Pizzorusso T., The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat. Rev. Neurosci. 20, 451–465 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Reh R. K., et al. , Critical period regulation across multiple timescales. Proc. Natl. Acad. Sci. U.S.A. 117, 23242–23251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorg B. A., et al. , Casting a wide net: Role of perineuronal nets in neural plasticity. J. Neurosci. 36, 11459–11468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fawcett J. W., et al. , The extracellular matrix and perineuronal nets in memory. Mol Psychiatry 27, 3192–3203 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devienne G., et al. , Regulation of perineuronal nets in the adult cortex by the activity of the cortical network. J. Neurosci. 41, 5779–5790 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyata S., Komatsu Y., Yoshimura Y., Taya C., Kitagawa H., Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat. Neurosci. 15, 414–422, S1–S2 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Morikawa S., Ikegaya Y., Narita M., Tamura H., Activation of perineuronal net-expressing excitatory neurons during associative memory encoding and retrieval. Sci. Rep. 7, 46024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carstens K. E., Phillips M. L., Pozzo-Miller L., Weinberg R. J., Dudek S. M., Perineuronal nets suppress plasticity of excitatory synapses on CA2 pyramidal neurons. J. Neurosci. 36, 6312–6320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudek S. M., Alexander G. M., Farris S., Rediscovering area CA2: Unique properties and functions. Nat. Rev. Neurosci. 17, 89–102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hitti F. L., Siegelbaum S. A., The hippocampal CA2 region is essential for social memory. Nature 508, 88–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith A. S., Williams Avram S. K., Cymerblit-Sabba A., Song J., Young W. S., Targeted activation of the hippocampal CA2 area strongly enhances social memory. Mol. Psychiatry 21, 1137–1144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chevaleyre V., Piskorowski R. A., Hippocampal area CA2: An overlooked but promising therapeutic target. Trends Mol. Med. 22, 645–655 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Piskorowski R. A., et al. , Age-dependent specific changes in area CA2 of the hippocampus and social memory deficit in a mouse model of the 22q11.2 deletion syndrome. Neuron 89, 163–176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao M., Choi Y. S., Obrietan K., Dudek S. M., Synaptic plasticity (and the lack thereof) in hippocampal CA2 neurons. J. Neurosci. 27, 12025–12032 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Domínguez S., et al. , Maturation of PNN and ErbB4 signaling in area CA2 during adolescence underlies the emergence of PV interneuron plasticity and social memory. Cell Rep. 29, 1099–1112.e4 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Miller G. M., Hsieh-Wilson L. C., Sugar-dependent modulation of neuronal development, regeneration, and plasticity by chondroitin sulfate proteoglycans. Exp. Neurol. 274, 115–125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugahara K., Mikami T., Chondroitin/dermatan sulfate in the central nervous system. Curr. Opin. Struct. Biol. 17, 536–545 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Hou X., et al. , Chondroitin sulfate is required for onset and offset of critical period plasticity in visual cortex. Sci. Rep. 7, 12646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown J. M., et al. , A sulfated carbohydrate epitope inhibits axon regeneration after injury. Proc. Natl. Acad. Sci. U.S.A. 109, 4768–4773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H., et al. , Chondroitin-4-sulfation negatively regulates axonal guidance and growth. J. Cell Sci. 121, 3083–3091 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sahu S., Li R., Loers G., Schachner M., Knockdown of chondroitin-4-sulfotransferase-1, but not of dermatan-4-sulfotransferase-1, accelerates regeneration of zebrafish after spinal cord injury. FASEB J. 33, 2252–2262 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Hussein R. K., Mencio C. P., Katagiri Y., Brake A. M., Geller H. M., Role of chondroitin sulfation following spinal cord injury. Front. Cell Neurosci. 14, 208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lang B. T., et al. , Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature 518, 404–408 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dickendesher T. L., et al. , NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat. Neurosci. 15, 703–712 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bavelier D., Levi D. M., Li R. W., Dan Y., Hensch T. K., Removing brakes on adult brain plasticity: From molecular to behavioral interventions. J. Neurosci. 30, 14964–14971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang S., et al. , Antibody recognizing 4-sulfated chondroitin sulfate proteoglycans restores memory in tauopathy-induced neurodegeneration. Neurobiol. Aging 59, 197–209 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Gogolla N., Caroni P., Luthi A., Herry C., Perineuronal nets protect fear memories from erasure. Science 325, 1258–1261 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Pizzorusso T., et al. , Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298, 1248–1251 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Carulli D., et al. , Cerebellar plasticity and associative memories are controlled by perineuronal nets. Proc. Natl. Acad. Sci. U.S.A. 117, 6855–6865 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gottschling C., Wegrzyn D., Denecke B., Faissner A., Elimination of the four extracellular matrix molecules tenascin-C, tenascin-R, brevican and neurocan alters the ratio of excitatory and inhibitory synapses. Sci. Rep. 9, 13939 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nojima K., Miyazaki H., Hori T., Vargova L., Oohashi T., Assessment of possible contributions of hyaluronan and proteoglycan binding link protein 4 to differential perineuronal net formation at the calyx of Held. Front. Cell Dev. Biol. 9, 730550 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamauchi S., et al. , Purification and characterization of Chondroitin 4-Sulfotransferase from the culture medium of a rat chondrosarcoma cell line. J. Biol. Chem. 274, 2456–2463 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Klüppel M., Wight T. N., Chan C., Hinek A., Wrana J. L., Maintenance of chondroitin sulfation balance by chondroitin-4-sulfotransferase 1 is required for chondrocyte development and growth factor signaling during cartilage morphogenesis. Development 132, 3989–4003 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Bian S., et al. , Dermatan sulfotransferase Chst14/D4st1, but not chondroitin sulfotransferase Chst11/C4st1, regulates proliferation and neurogenesis of neural progenitor cells. J. Cell Sci. 124, 4051–4063 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Zimmerman L., et al. , Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron 12, 11–24 (1994). [DOI] [PubMed] [Google Scholar]
- 46.Izumikawa T., Okuura Y., Koike T., Sakoda N., Kitagawa H., Chondroitin 4-O-sulfotransferase-1 regulates the chain length of chondroitin sulfate in co-operation with chondroitin N-acetylgalactosaminyltransferase-2. Biochem. J. 434, 321–331 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Lensjø K. K., Christensen A. C., Tennøe S., Fyhn M., Hafting T., Differential expression and cell-type specificity of perineuronal nets in hippocampus, medial entorhinal cortex, and visual cortex examined in the rat and mouse. eNeuro 4, ENEURO.0379-16.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamada J., Ohgomori T., Jinno S., Perineuronal nets affect parvalbumin expression in GABAergic neurons of the mouse hippocampus. Eur. J. Neurosci. 41, 368–378 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Kohara K., et al. , Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nat. Neurosci. 17, 269–279 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyata S., Kitagawa H., Chondroitin 6-sulfation regulates perineuronal net formation by controlling the stability of aggrecan. Neural Plast. 2016, 1305801 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beurdeley M., et al. , Otx2 binding to perineuronal nets persistently regulates plasticity in the mature visual cortex. J. Neurosci. 32, 9429–9437 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernard C., Prochiantz A., Otx2-PNN interaction to regulate cortical plasticity. Neural Plast. 2016, 7931693 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levy A. D., Omar M. H., Koleske A. J., Extracellular matrix control of dendritic spine and synapse structure and plasticity in adulthood. Front. Neuroanat. 8, 116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boehringer R., et al. , Chronic loss of CA2 transmission leads to hippocampal hyperexcitability. Neuron 94, 642–655.e9 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Berry K. P., Nedivi E., Spine dynamics: Are they all the same? Neuron 96, 43–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lauterborn J. C., et al. , Increased excitatory to inhibitory synaptic ratio in parietal cortex samples from individuals with Alzheimer’s disease. Nat. Commun. 12, 2603 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheung S. T., et al. , Discovery of a small-molecule modulator of glycosaminoglycan sulfation. ACS Chem. Biol. 12, 3126–3133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morales E., et al. , Releasing the peri-neuronal net to patch-clamp neurons in adult CNS. Pflugers Arch. 448, 248–258 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Hayani H., Song I., Dityatev A., Increased excitability and reduced excitatory synaptic input into fast-spiking CA2 interneurons after enzymatic attenuation of extracellular matrix. Front. Cell Neurosci. 12, 149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gama C. I., et al. , Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol. 2, 467–473 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Rogers C. J., et al. , Elucidating glycosaminoglycan-protein-protein interactions using carbohydrate microarray and computational approaches. Proc. Natl. Acad. Sci. U.S.A. 108, 9747–9752 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pulsipher A., Griffin M. E., Stone S. E., Brown J. M., Hsieh-Wilson L. C., Directing neuronal signaling through cell-surface glycan engineering. J. Am. Chem. Soc. 136, 6794–6797 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kurihara D., Yamashita T., Chondroitin sulfate proteoglycans down-regulate spine formation in cortical neurons by targeting tropomyosin-related kinase B (TrkB) protein. J. Biol. Chem. 287, 13822–13828 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lesnikova A., et al. , Chondroitinase and antidepressants promote plasticity by releasing TRKB from dephosphorylating control of PTPσ in parvalbumin neurons. J. Neurosci. 41, 972–980 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kandel E. R., The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 5, 14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pignataro A., Borreca A., Ammassari-Teule M., Middei S., CREB regulates experience-dependent spine formation and enlargement in mouse barrel cortex. Neural Plast. 2015, 651469 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Bartolomeis A., Latte G., Tomasetti C., Iasevoli F., Glutamatergic postsynaptic density protein dysfunctions in synaptic plasticity and dendritic spines morphology: Relevance to schizophrenia and other behavioral disorders pathophysiology, and implications for novel therapeutic approaches. Mol. Neurobiol. 49, 484–511 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Bagni C., Zukin R. S., A synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron 101, 1070–1088 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bourtchuladze R., et al. , Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79, 59–68 (1994). [DOI] [PubMed] [Google Scholar]
- 70.Kida S., et al. , CREB required for the stability of new and reactivated fear memories. Nat. Neurosci. 5, 348–355 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Leroy F., Brann D. H., Meira T., Siegelbaum S. A., Input-timing-dependent plasticity in the hippocampal CA2 region and its potential role in social memory. Neuron 102, 260–262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walf A. A., Frye C. A., The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat. Protoc. 2, 322–328 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bourin M., Hascoët M., The mouse light/dark box test. Eur. J. Pharmacol. 463, 55–65 (2003). [DOI] [PubMed] [Google Scholar]
- 74.Egashira Y., Mori Y., Yanagawa Y., Takamori S., Development of lentiviral vectors for efficient glutamatergic-selective gene expression in cultured hippocampal neurons. Sci. Rep. 8, 15156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bradshaw K. P., Figueroa Velez D. X., Habeeb M., Gandhi S. P., Precocious deposition of perineuronal nets on parvalbumin inhibitory neurons transplanted into adult visual cortex. Sci. Rep. 8, 7480 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nandini C. D., et al. , Structural and functional characterization of oversulfated chondroitin sulfate/dermatan sulfate hybrid chains from the notochord of hagfish. Neuritogenic and binding activities for growth factors and neurotrophic factors. J. Biol. Chem. 279, 50799–50809 (2004). [DOI] [PubMed] [Google Scholar]
- 77.Kellner Y., et al. , The BDNF effects on dendritic spines of mature hippocampal neurons depend on neuronal activity. Front. Synaptic Neurosci. 6, 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Serita T., Fukushima H., Kida S., Constitutive activation of CREB in mice enhances temporal association learning and increases hippocampal CA1 neuronal spine density and complexity. Sci. Rep. 7, 42528 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rauskolb S., et al. , Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J. Neurosci. 30, 1739 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boggio E. M., et al. , Inhibition of Semaphorin3A promotes ocular dominance plasticity in the adult rat visual cortex. Mol. Neurobiol. 56, 5987–5997 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Dick G., et al. , Semaphorin 3A binds to the perineuronal nets via chondroitin sulfate type E motifs in rodent brains. J. Biol. Chem. 288, 27384–27395 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morita A., et al. , Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J. Neurosci. 26, 2971–2980 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yamashita N., et al. , Regulation of spine development by semaphorin3A through cyclin-dependent kinase 5 phosphorylation of collapsin response mediator protein 1. J. Neurosci. 27, 12546–12554 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tran T. S., et al. , Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature 462, 1065–1069 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mohan V., et al. , Neurocan inhibits Semaphorin 3F induced dendritic spine remodeling through NrCAM in cortical neurons. Front. Cell Neurosci. 12, 346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paolicelli R. C., et al. , Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (2011). [DOI] [PubMed] [Google Scholar]
- 87.Zemmar A., et al. , Oligodendrocyte- and neuron-specific Nogo-A Restrict dendritic branching and spine density in the adult mouse motor cortex. Cereb. Cortex 28, 2109–2117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Pins B., et al. , Conditional BDNF delivery from astrocytes rescues memory deficits, spine density, and synaptic properties in the 5xFAD mouse model of Alzheimer disease. J. Neurosci. 39, 2441–2458 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Romberg C., et al. , Depletion of perineuronal nets enhances recognition memory and long-term depression in the perirhinal cortex. J. Neurosci. 33, 7057–7065 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hylin M. J., Orsi S. A., Moore A. N., Dash P. K., Disruption of the perineuronal net in the hippocampus or medial prefrontal cortex impairs fear conditioning. Learn. Mem. 20, 267–273 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tovote P., Fadok J. P., Lüthi A., Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci. 16, 317–331 (2015). [DOI] [PubMed] [Google Scholar]
- 92.Logsdon A. F., et al. , Decoding perineuronal net glycan sulfation patterns in the Alzheimer’s disease brain. Alzheimer’s Dement. 18, 942–954 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Takiguchi M., Morinobu S., Funakoshi K., Chondroitin sulfate expression around spinal motoneurons during postnatal development in rats. Brain Res. 1752, 147252 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Enwright J. F., et al. , Reduced labeling of parvalbumin neurons and perineuronal nets in the dorsolateral prefrontal cortex of subjects with schizophrenia. Neuropsychopharmacology 41, 2206–2214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mauney S. A., et al. , Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol. Psychiatry 74, 427–435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pantazopoulos H., et al. , Aggrecan and chondroitin-6-sulfate abnormalities in schizophrenia and bipolar disorder: A postmortem study on the amygdala. Transl. Psychiatry 5, e496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carstens K. E., et al. , Perineuronal net degradation rescues CA2 plasticity in a mouse model of Rett syndrome. J. Clin. Invest. 131, e137221 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cope E. C., et al. , Atypical perineuronal nets in the CA2 region interfere with social memory in a mouse model of social dysfunction. Mol. Psychiatry 27, 3520–3531 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.