

Abstract
DNA-complexed heterodimers of the aryl hydrocarbon receptor (AhR) with the Ah receptor nuclear translocator (Arnt) are the molecular switches for nuclear signaling of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). AhR–Arnt heterodimers regulate genes involved in the metabolism of xenobiotics or fatty acids and various genes important for growth and differentiation. In this report several potent methods, such as the limited protease digestion, gel shift and gel shift clipping assays, allowed the investigation of ligand-stabilized conformations of AhR monomers in comparison to that of AhR–Arnt heterodimers. Interestingly, the ligand sensitivity of monomeric AhR was found to be very low at 25 nM, whereas DNA-dependent methods consistently provided EC50 values between 0.12 and 0.6 nM for AhR in a heterodimeric complex, i.e. an approximate 100-fold higher ligand sensitivity. This indicates that complex formation of AhR with Arnt on DNA is an important and critical step in transforming AhR into a high affinity receptor for TCDD. A comparison of wild-type AhR with different C-terminal receptor truncations suggests that the PAS-B subregion of its PAS domain is of central importance for stabilization of a functional, i.e. ligand-sensitive, AhR–Arnt conformation, whereas the PAS-A subregion appears to be critical for dimerization of AhR and Arnt. In conclusion, the results of this study provide important information on the ligand sensitivity of AhR and AhR–Arnt heterodimer conformations.
INTRODUCTION
The aryl hydrocarbon receptor (AhR) mediates a broad range of toxic and biochemical responses to the ubiquitous environmental pollutant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). These responses include, but are not limited to, immunosuppression, tumor promotion, teratogenicity and cachexia. Animals lacking AhR die prematurely and display a number of fibrotic responses and changes in the immune system (1). The AhR is expressed in a tissue- and cell-specific manner (2). Prior to ligand binding, AhR is located in the cytoplasm and is associated with two 90 kDa heat shock protein (hsp90) molecules. After ligand binding, AhR dissociates from this complex, translocates into the nucleus and subsequently dimerizes with the Ah receptor nuclear translocator (Arnt) (3). Although Arnt was designated as a translocator, it now appears that Arnt is not involved in transport of AhR into the nucleus but rather forms a complex with ligand-bound AhR that has already gained access to the nucleus (4). The ligand-bound heterodimeric AhR–Arnt complex acts as a transcription factor complex and binds to specific xenobiotic response elements (XREs), with the core sequence TNGCGTG, which are located in the promoters of dioxin responsive genes, i.e. DNA-complexed AhR–Arnt heterodimers can be considered as the molecular switches for dioxin signaling. Although many genes that are inducible by TCDD contain XREs in their promoter regions (5), the mechanism of transcriptional activation by the AhR–Arnt–TCDD complex has been studied primarily on the paradigm of the responsive gene CYP1A1 (6). Other TCDD-inducible genes have been found to be involved in fatty acid metabolism (7), cell cycle regulation (8) and the immune response (5).
AhR and Arnt are members of the PAS (Per–Arnt–Sim) domain family (9) that contains proteins of similar modular structure, such as mammalian hypoxia-inducible factor-1α (HIF-1α) and Drosophila regulatory proteins period (Per) and single-minded (Sim). PAS domain family members have a centrally located PAS domain containing two 51 amino acid imperfect repeats (PAS-A and PAS-B) separated by a variable spacer region, which at least for AhR is important for ligand and hsp90 binding (10). Moreover, they also contain a basic helix–loop–helix (bHLH) DNA-binding domain close to their N-terminus (11), which is also critical for dimerization. Finally, PAS family members contain one or more transactivation domains in their C-terminus (3). Arnt appears to be the common heterodimerization partner for all PAS domain family members, whereas AhR is presently the only family member that has a known ligand. However, the structural similarity of the PAS domain family members suggests that ligands for the presently known orphan members could exist.
Although ligand binding takes place in the cytoplasm, AhR acts as a nuclear receptor for TCDD, which is comparable to the members of the nuclear receptor superfamily, such as the steroid, retinoic acid, 1α,25-dihydroxyvitamin D3 and thyroid hormone receptors. (12). This analogy suggests that AhR also undergoes a conformational change through ligand binding, as X-ray crystallography has shown for retinoid receptors (13). Biochemical methods, such as the limited protease digestion assay (14), in which a ligand-bound nuclear receptor is incubated with an endoprotease such as trypsin, are able to document conformational changes in nuclear receptors. It is assumed that due to a conformational change in the receptor, trypsin cutting sites lose their accessibility and therefore the resulting receptor fragments can be interpreted as representatives of different ligand-induced receptor conformations. Recently, a second method was reported, referred to as the gel shift clipping assay (15,16), in which the ligand-dependent gel shift assay, as a well-established detection method for protein–DNA interactions, was combined with the limited protease digestion assay. This novel method allows the visualization of nuclear receptor heterodimer conformations.
In this study, DNA-independent and DNA-dependent limited protease digestion, gel shift and gel shift clipping assays were used for the analysis of conformational changes in the AhR in solution or as a DNA-complexed heterodimer with Arnt. Moreover, C-terminally truncated AhR proteins have defined the subdomains of the AhR that are essential to undergo a ligand-dependent conformational change. Characterization of the ligand sensitivity of the AhR in the context of AhR–Arnt–XRE conformations confirms and extends previous studies on functional AhR domains and is essential to understand the molecular switch of nuclear dioxin signaling.
MATERIALS AND METHODS
Generation of in vitro translated wild-type and truncated AhR protein
The cDNAs for mouse AhR (AhR1–805) (17) and Arnt (18) were subcloned into the vector pRc/CMV (19). To generate the C-terminal truncations AhR1–491, AhR1–418, AhR1–410, AhR1–320 and AhR1–167 the cDNA of AhR was internally digested by the restriction enzymes NotI, BamHI, BglII, PstI and EcoRI, respectively. Linearized cDNAs were then transcribed with T7 RNA polymerase and translated as recommended by the supplier (Promega, Mannheim, Germany).
Limited protease digestion
In DNA-independent limited protease digestion assays 5 µl of in vitro translated, [35S]methionine-labeled wild-type AhR and 5 µl of unprogrammed lysate were incubated with 0.5 µl of graded concentrations of TCDD (Ökometric, Bayreuth, Germany) or DMSO as a solvent control in a total volume of 20 µl of binding buffer [10 mM HEPES, pH 7.9, 1 mM DTT, 0.2 µg/µl poly(dI·dC) and 5% glycerol] for 60 min at room temperature. The buffer was adjusted to 220 mM monovalent cations by addition of KCl. For DNA-dependent limited protease digestion assays 5 µl of in vitro translated wild-type Arnt was used instead of unprogrammed lysate and both receptors were allowed to form heterodimers in the presence of TCDD by incubation for 60 min at room temperature. After complex formation, ~1 ng of non-labeled double-stranded oligonucleotide containing the mouse CYP1A1 XRE (core sequence TAGCGTGACA) (20) was added to the receptor/ligand mixture and incubation was continued for another 15 min. In both cases, the endoprotease trypsin (final concentration 10–12 µg/ml; Promega) was then added and incubation was continued for 10 min at room temperature. The digestion reactions were stopped by adding 1 vol of protein gel loading buffer (0.25 M Tris, pH 6.8, 20% glycerol, 5% mercaptoethanol, 2% SDS, 0.025% w/v bromophenol blue) and denaturation at 95°C for 5 min. Protease-resistant AhR fragments were electrophoresed through 15% SDS–polyacrylamide gels, exposed to a Fuji MP2040S imager screen and quantified on a Fuji FLA2000 reader (Tokyo) using Image Gauge software (Raytest, Sprockhövel, Germany).
Gel shift and gel shift clipping assays
For both type of assays comparable amounts (2.5 µl each) of in vitro translated wild-type AhR protein (or indicated AhR truncations) and in vitro translated wild-type Arnt protein were mixed and incubated in the presence of the indicated concentrations of TCDD (or DMSO as solvent control) for 60 min at room temperature in a total volume of 20 µl binding buffer. The buffer was adjusted to 220 mM monovalent cations by addition of KCl. The mouse CYP1A1 XRE was labeled by a fill-in reaction using [32P]dCTP and the Klenow fragment of DNA polymerase I (Promega). Approximately 1 ng of labeled probe (50 000 c.p.m.) was then added to the receptor/ligand mixture and incubation was continued for a further 15 min. For gel shift clipping assays, trypsin was added (final concentration 10 µg/ml) and incubation was further continued for 10 min at room temperature. In both cases protein–DNA complexes were resolved through 8% non-denaturing polyacrylamide gels (at room temperature) in 0.5× TBE (45 mM Tris, 45 mM boric acid, 1 mM EDTA, pH 8.3) and exposed to a Fuji MP2040S imager screen. The ratio of protein-complexed probe to free probe was quantified on a Fuji FLA2000 reader using Image Gauge software.
RESULTS
Limited protease digestion assays were performed with in vitro translated, 35S-labeled AhR and the endoprotease trypsin (Fig. 1). In the presence of graded concentrations of TCDD an AhR fragment of a molecular mass of ~35 kDa was found to be most prominent and was interpreted as being representative of the ligand-induced AhR conformation (cLPD). TCDD was found to stabilize the cLPD of monomeric AhR with a half-maximal effective concentration (EC50) value of 25 nM. In this reference experimental series 14% of the AhR input was stabilized in cLPD, which, when compared with the solvent control, represents a ligand inducibility factor of approximately 8.
Figure 1.
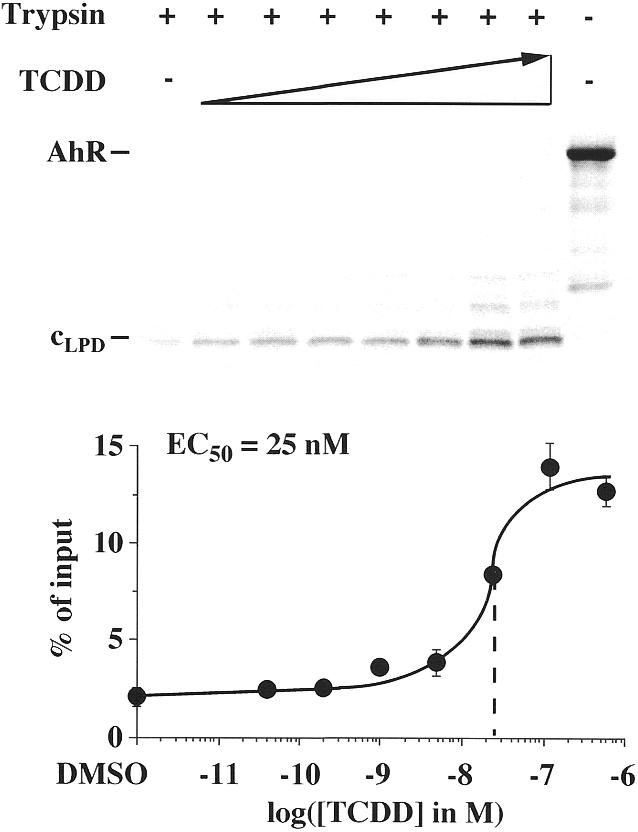
Ligand-dependent stabilization of AhR conformations in solution. DNA-independent limited protease digestion assays were performed by preincubating in vitro translated, [35S]methionine-labeled AhR with graded concentrations of TCDD for 60 min at room temperature. Then trypsin was added (final concentration 12 µg/ml) and incubation was continued for 10 min. Samples were electrophoresed through 15% SDS–polyacrylamide gels. A representative experiment is shown, where full-length AhR and the protease-resistant AhR fragment (interpreted as a representative of AhR conformation cLPD) are indicated. The amount of cLPD was quantified in relation to AhR input by phosphorimaging. Each data point represents the average of triplicate determinations and bars indicate standard deviations. The EC50 value for stabilization of cLPD was determined from the dose–response curve.
The ligand sensitivity of the AhR was then tested within a heterodimeric complex with Arnt. Gel shift assays were performed for this purpose with in vitro translated AhR–Arnt heterodimers on the mouse CYP1A1 XRE (20). A specific AhR–Arnt heterodimer complex was obtained on this XRE (Fig. 2), whereas AhR or Arnt homodimer formation was not observed (data not shown). Graded concentrations of TCDD increased the amount of the protein–DNA complex by a factor of approximately 5 and provided an EC50 value of 0.4 nM. This suggested that the ligand sensitivity of the AhR is increased more than 60-fold through complex formation with Arnt and DNA.
Figure 2.
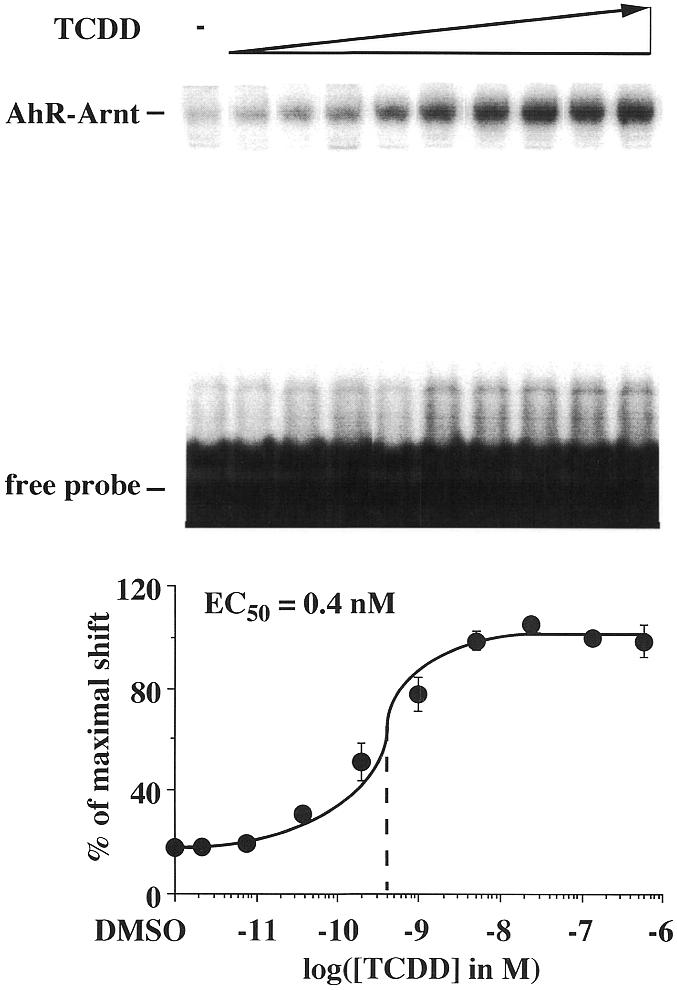
Ligand-triggered stabilization of AhR–Arnt heterodimers on DNA. Gel shift assays were performed by preincubating heterodimers of in vitro translated AhR and Arnt proteins for 60 min at room temperature and then adding graded concentrations of TCDD for 15 min. Heterodimers were further incubated with the 32P-labeled mouse CYP1A1 XRE for 15 min at room temperature. Protein–DNA complexes were separated from free probe through 8% non-denaturing polyacrylamide gels. A representative experiment is shown. The amount of AhR–Arnt heterodimer–DNA complex was quantified by phosphorimaging. Each data point represents the average of triplicate determinations and bars indicate standard deviations. The EC50 value for stabilization of AhR–Arnt heterodimers on DNA was determined from the dose–response curve.
In order to challenge the observation of an increased ligand sensitivity through complex formation on DNA, limited protease digestion assays were performed with DNA-complexed AhR–Arnt heterodimers and graded concentrations of TCDD (Fig. 3). In these DNA-dependent assays the same 35 kDa AhR fragment, cLPD, was observed as in the DNA-independent assays (Fig. 1), but in the DNA-dependent assay lower amounts of intermediary AhR fragments were obtained. Interestingly, the sensitivity of the AhR conformation, cLPD, for TCDD (EC50 value 0.12 nM) was found to be 200-fold higher than that of monomeric AhR (EC50 value 25 nM; Fig. 1). In parallel, at saturating concentrations of TCDD the percentage of AhR molecules that were stabilized in cLPD was increased to 27% of AhR input.
Figure 3.
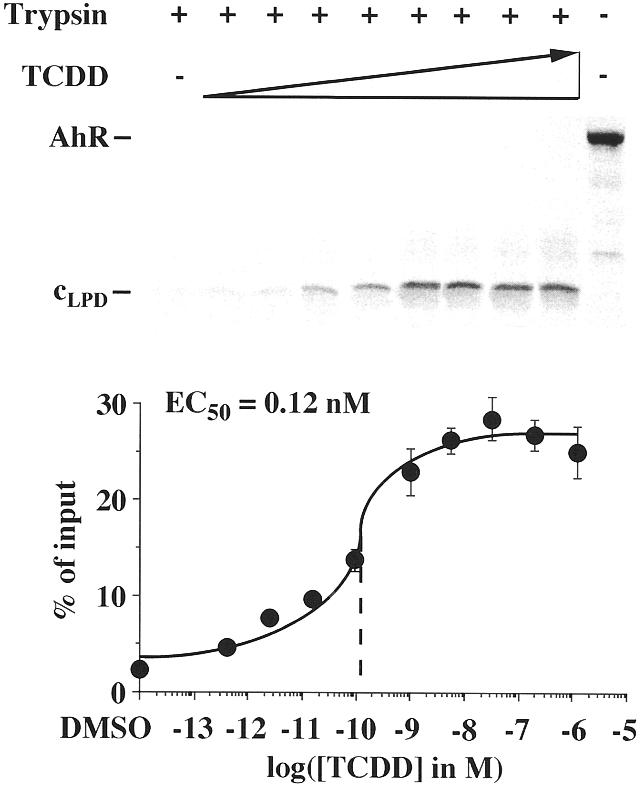
AhR–Arnt heterodimer complex formation on DNA enhances ligand sensitivity of the AhR cLPD conformation. DNA-dependent limited protease digestion assays were performed by preincubating in vitro translated, [35S]methionine-labeled AhR with in vitro translated Arnt for 60 min at room temperature and then adding graded concentrations of TCDD for 15 min. AhR–Arnt heterodimers were further incubated with the non-labeled mouse CYP1A1 XRE for 15 min. Trypsin (final concentration 10 µg/ml) was then added and incubation was continued for an additional 10 min. Samples were electrophoresed through 15% SDS–polyacrylamide gels. A representative experiment is shown. The amount of the cLPD AhR fragment was quantified in relation to full-length AhR input by phosphorimaging. Each data point represents the average of triplicate determinations and bars indicate standard deviations. The EC50 value for stabilization of cLPD was determined from the dose–response curve.
In order to merge the results from the DNA-independent (Fig. 1) and the DNA-dependent limited protease digestion assays (Fig. 3) with those from the gel shift assay (Fig. 2), a novel method, referred to as the gel shift clipping assay (15,16), was applied to AhR–Arnt heterodimers (Fig. 4A). In this assay AhR–Arnt heterodimers were formed in the presence of a saturating concentration of TCDD (10 nM) on the mouse CYP1A1 XRE, as in the gel shift assays, and then trypsin was added, as in the limited protease digestion assay. Two protein–DNA complexes were obtained that were interpreted as the representatives of two different AhR–Arnt heterodimer conformations (c1GSC and c2GSC). The amounts of both conformations decreased with increasing incubation time, but only c1GSC appeared to be stabilized in a ligand-dependent fashion, whereas c2GSC showed no significant ligand sensitivity. The quantification of c1GSC indicated that even after a 60 min incubation with trypsin a reasonable amount of AhR–Arnt heterodimers (~30% of input) remained resistant. However, a 10 min incubation time appeared to show the best signal to noise ratio and was therefore taken as standard for the following assays. Gel shift clipping assays were then performed with graded concentrations of TCDD (Fig. 4B). The amount of stabilized AhR–Arnt conformation c1GSC was found to be enhanced with increasing TCDD concentration and provided an EC50 value of 0.6 nM, i.e. a value that appeared to be similar to those observed in other DNA-dependent assays (Figs 2 and 3).
Figure 4.
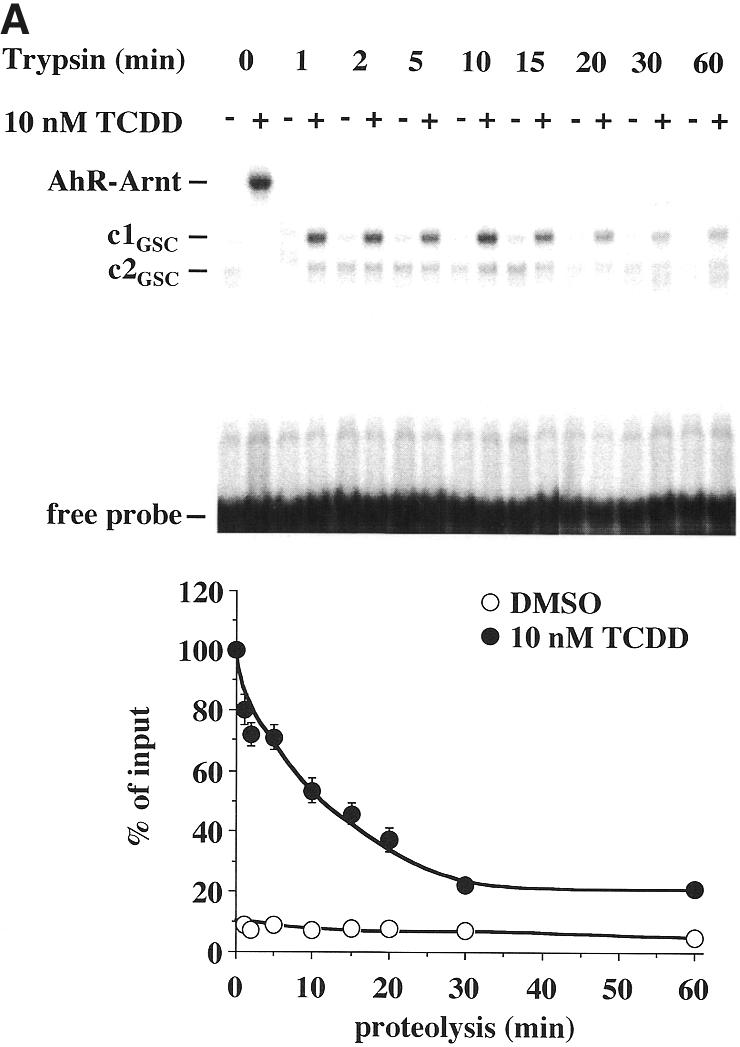
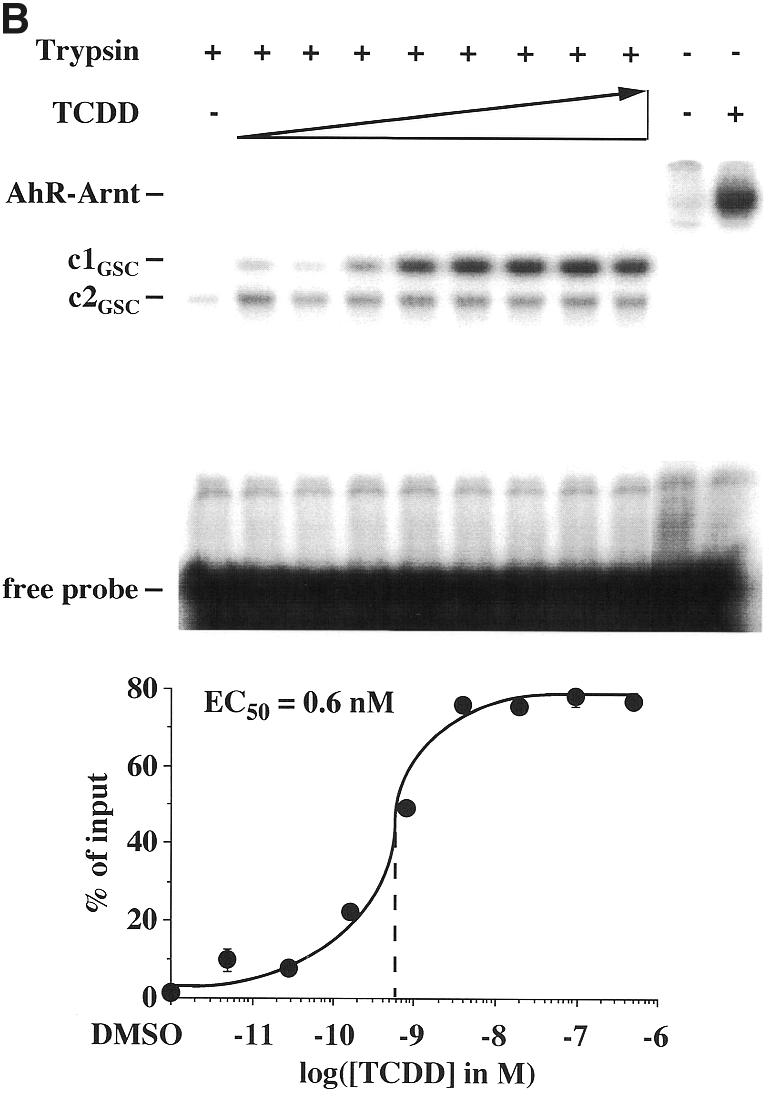
Ligand-triggered stabilization of AhR–Arnt heterodimers on DNA. Gel shift clipping assays were performed by preincubating heterodimers of in vitro translated AhR and Arnt proteins for 60 min at room temperature and then adding saturating (10 nM) (A) or graded (B) concentrations of TCDD for 15 min. AhR–Arnt heterodimers were further incubated with the 32P-labeled mouse CYP1A1 XRE for 15 min at room temperature. Trypsin was then added (final concentration 10 µg/ml) and incubation was continued for the indicated times (A) or 10 min (B) at room temperature. Protein–DNA complexes were separated from free probe through 8% non-denaturing polyacrylamide gels. Representative experiments are shown. The digested AhR–Arnt heterodimer–DNA complexes are interpreted as representatives of AhR–Arnt heterodimer conformations c1GSC and c2GSC. The amount of the ligand-dependent heterodimer conformation c1GSC was quantified in relation to respective ligand-induced, non-digested AhR–Arnt heterodimers by phosphorimaging. Each data point represents the average of triplicate determinations and bars indicate standard deviations. The EC50 value for stabilization of c1GSC (B) was determined from the dose–response curve.
A more detailed investigation of AhR–Arnt conformations utilized the C-terminally truncated AhR proteins AhR1–491, AhR1–418, AhR1–410, AhR1–320 and AhR1–167. These truncated proteins were produced by in vitro transcription/translation of AhR cDNA digested with the restriction enzymes NotI, BamHI, BglII, PstI and EcoRI, respectively. Gel shift and gel shift clipping assays were performed with in vitro translated AhR–Arnt heterodimers, which were formed by wild-type AhR (AhR1–805) protein or the indicated truncated AhR proteins and wild-type Arnt proteins on the mouse CYP1A1 XRE in the presence of a saturating concentration of TCDD (10 nM) or solvent (DMSO) (Fig. 5). The proteins AhR1–491, AhR1–418 and AhR1–410 (C-terminal truncations of the PAS domain) were found to react similarly to wild-type AhR, as they showed comparable ligand-dependent effects in the gel shift and gel shift clipping assays. The respective non-digested heterodimeric complexes migrated faster than wild-type AhR–Arnt complexes due to a lower molecular mass of the truncated proteins, but the AhR–Arnt heterodimer conformations c1GSC and c2GSC of AhR1–805, AhR1–491, AhR1–418 and AhR1–410 appeared to be identical. Interestingly, the AhR protein AhR1–320 demonstrated heterodimer formation with Arnt (even stronger than wild-type AhR) and stabilized the conformation c2GSC (but not c1GSC); however, this C-terminal truncation of the PAS-B domain had completely lost ligand sensitivity. Finally, the AhR protein AhR1–167 (C-terminal truncation of the bHLH domain) appeared to have additionally lost the ability to form heterodimers with Arnt.
Figure 5.
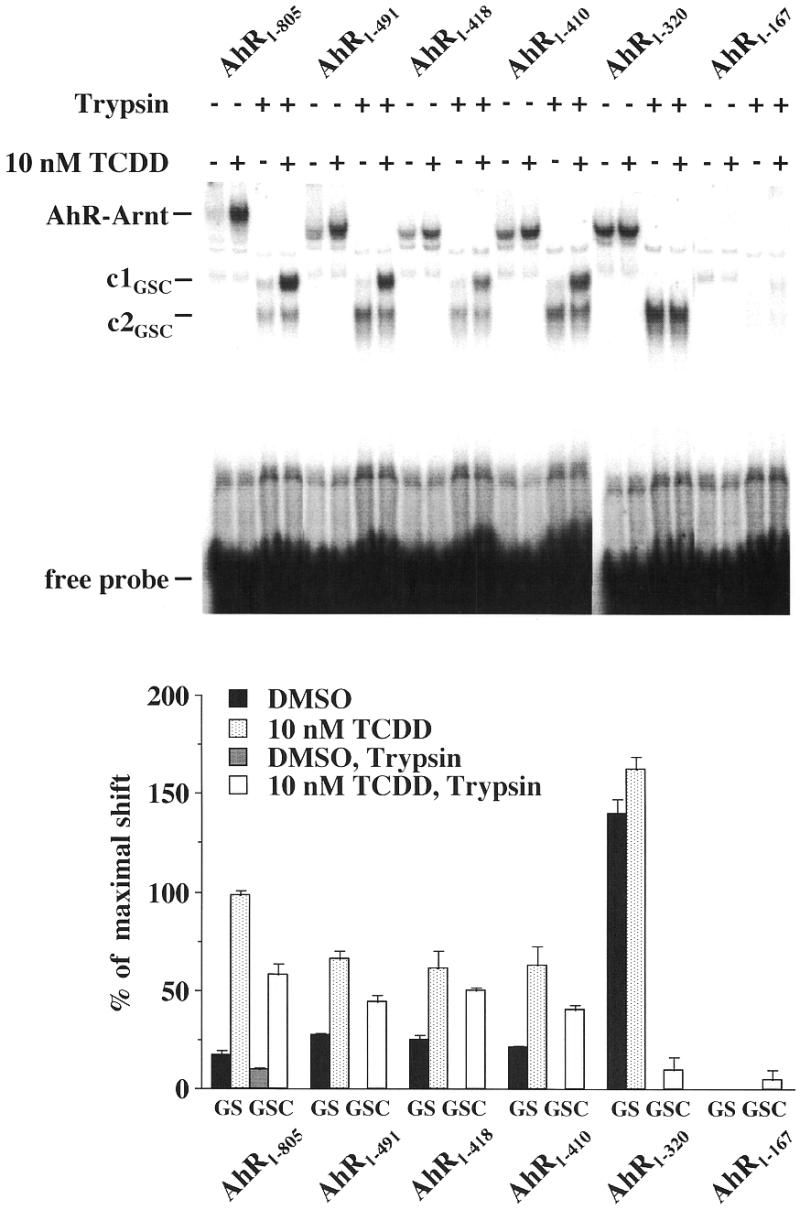
The essential AhR subdomain for stabilization of AhR–Arnt conformations. C-terminally truncated AhR proteins AhR1–491, AhR1–418, AhR1–410, AhR1–320 and AhR1–167 were generated by in vitro transcription/translation of AhR cDNA digested by the restriction enzymes NotI, BamHI, BglII, PstI and EcoRI, respectively. Gel shift and gel shift clipping assays were performed by preincubating AhR–Arnt heterodimers, which were formed by wild-type AhR protein (AhR1–805) or the indicated truncated AhR proteins and wild-type Arnt protein, for 60 min at room temperature and then adding 10 nM TCDD or solvent (DMSO) for 15 min. Heterodimers were further incubated with the 32P-labeled mouse CYP1A1 XRE for 15 min at room temperature. For gel shift clipping assays, trypsin was then added (final concentration 10 µg/ml) to one aliquot of the samples and incubation continued for 10 min. Protein–DNA complexes were separated from free probe through 8% non-denaturing polyacrylamide gels. A representative experiment is shown. The amount of ligand-dependent, digested AhR–Arnt heterodimer conformation c1GSC was quantified by phosphorimaging. Each column represents the average of triplicate determinations and bars indicate standard deviations.
DISCUSSION
The AhR is the nuclear receptor for TCDD and most probably the only target of the toxic xenobiotic compound within cells (3). AhR knockout mice have been demonstrated to be resistant to TCDD effects (21,22), which suggests that TCDD toxicity largely depends on the ligand–receptor interaction of TCDD and AhR, thus making studies directed towards defining the ligand sensitivity of the AhR very important. In this study several potent methods, such as limited protease digestion and the gel shift and gel shift clipping assays, have been used to describe the response of AhR monomers and of AhR–Arnt heterodimers to TCDD. The assays differ in their perspectives on ligand-induced AhR conformations. The DNA-independent limited protease digestion assay studies monomeric AhR in solution, whereas the gel shift and the gel shift clipping assays analyse DNA-bound AhR–Arnt heterodimers. Moreover, in this study limited protease digestion was also performed with DNA-bound AhR–Arnt heterodimers, i.e. in an Arnt- and DNA-dependent fashion.
The ligand sensitivity of monomeric AhR was found to be very low at 25 nM, whereas the three DNA-dependent methods consistently provided EC50 values between 0.12 and 0.6 nM, i.e. an ~100-fold higher ligand sensitivity of AhR–Arnt heterodimers. This is in good agreement with earlier results where apparent dissociation constants for the TCDD–AhR interaction were determined by competition assays and photoaffinity labeling to be 1–2 nM (23). It has to be noted that the assays used in this study do not measure ligand binding per se, as known from traditional ligand binding assays that use radiolabeled ligand, but detect the effect of the ligand on stabilization of AhR conformations and heterodimeric AhR–Arnt complexes. Moreover, a comparable observation was made for HIF-1α–Arnt heterodimers, where HIF-1α was found to be stabilized through heterodimerization with Arnt (24).
Approximately 40–50% of the in vitro translated receptors within AhR–Arnt complexes or other heterodimeric nuclear receptor complexes were shown to be able to bind to DNA, i.e. to participate in the gel shift (25). Up to 80% of this heterodimer pool, i.e. more than 32% of the AhR input, has been shown to bind ligand by producing a protease-resistant AhR–Arnt–XRE complex in the gel shift clipping assay. In contrast, in DNA-independent limited protease digestion assays only ~15% of the AhR input was protease resistant, which increased in DNA-dependent limited protease digestion assays to 30% and fits well with the results from the gel shift clipping experiment. This indicates that interaction with TCDD not only increases the ligand sensitivity of the AhR, but also the proportion of AhR molecules that respond to ligand, and suggests that complex formation with Arnt on DNA is an important and critical step in transforming the AhR to a high affinity receptor for TCDD. Therefore, for a more accurate analysis of the binding affinity of a AhR agonist for the receptor, the respective assays should preferably be performed with DNA-complexed AhR. Due to the fact that Arnt is located in the nucleus, the commitment of AhR as a high affinity receptor takes places after translocation into the nucleus. However, a role of TCDD in conformational changes in the AhR has recently been suggested (26,27).
TCDD exposure leads to a variety of outcomes at different levels: thymus atrophy and immunosuppression are effects common in all animal species analyzed so far and require only low doses, whereas other effects become apparent only at higher doses. Risks assessment therefore needs to take into account the pharmacological characteristics of the AhR ligand and the endogenous conditions, like AhR abundance and its tissue distribution. The results of this study suggest that TCDD will have effects on gene activation only at concentrations that are sufficient to activate the AhR within a AhR–Arnt–DNA complex. In the vitamin D endocrine system the same set of assays provided a value (~0.1 nM) for the ligand sensitivity of VDR–RXR heterodimers (the molecular switches of 1α,25-dihydroxyvitamin D3 signaling) which fits with the critical concentration that was effective in activating primary 1α,25-dihydroxyvitamin D3 responsive genes (16). For an AhR signaling system this would mean that at a concentration of 0.1 nM or higher, primary TCDD responsive genes, such as CYP1A1, will be activated, which in part results in toxic effects. This conclusion correlates with previous reports on the activation of CYP1A1 gene activity by TCDD (6). However, it is obvious that the xenobiotic compound TCDD is not the physiological ligand of AhR. Such a ligand remains elusive, although a number of possible candidates, such as dietary components (28), tryptophan derivatives (29) and products of heme metabolism (30), have been suggested. These and other putative AhR agonists can be tested in the limited protease digestion and gel shift clipping assays for their potential in stabilizing AhR–Arnt heterodimers in the same way as TCDD has been analyzed in this study. The in vitro evaluation of AhR ligands will provide fast and accurate information, both on the mechanism of action and on the critical concentration of various xenobiotic compounds.
The results of the comparison of wild-type AhR with different C-terminal receptor truncations suggest that the PAS-B subregion of the PAS domain is of central importance for stabilization of a functional, i.e. ligand-sensitive, AhR–Arnt conformation, whereas the PAS-A subregion appears to be critical for dimerization of AhR and Arnt. The latter finding correlates with a previous report on the subdomains of the AhR (31) and, in general, demonstrates the potency of the gel shift clipping assay to determine the functionality of ligand-induced nuclear receptor complexes.
In conclusion, the results of this study have provided important information on the ligand sensitivity of AhR and AhR–Arnt heterodimer conformations. One application of this more detailed understanding of the molecular switches of dioxin signaling could be to establish a fast in vitro screening system for putative toxic effects of xenobiotic and natural compounds.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank P. Polly for critical reading of the manuscript and Drs J. P. Whitlock and O. Hankinson for providing AhR and Arnt cDNAs, respectively. This work was supported by the Sonderforschungsbereich 503, projects A6 (to C.C.) and C5 (to C.E.).
REFERENCES
- 1.Fernandez-Salguerro P., Pineau,T., Hilbert,D.M., McPhail,T., Lee,S.S.T., Kimura,S., Nebert,D.W., Rudikoff,S., Ward,J.M. and Gonzalez,F.J. (1995) Science, 268, 722–726. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt J.V. and Bradfield,C.A. (1996) Annu. Rev. Cell Dev. Biol., 12, 55–89. [DOI] [PubMed] [Google Scholar]
- 3.Rowlands J.C. and Gustafsson,J.-A. (1997) Crit. Rev. Toxicol., 27, 109–134. [DOI] [PubMed] [Google Scholar]
- 4.Hankinson O. (1995) Annu. Rev. Pharmacol. Toxicol., 35, 307–340. [DOI] [PubMed] [Google Scholar]
- 5.Lai Z.-W., Pinea,T. and Esser,C. (1996) Chem. Biol. Interact., 100, 91–112. [DOI] [PubMed] [Google Scholar]
- 6.Whitlock J.P. (1999) Annu. Rev. Pharmacol. Toxicol., 39, 103–125. [DOI] [PubMed] [Google Scholar]
- 7.Munzel P.A., Lehmkoster,T., Bruck,M., Ritter,J.K. and Bock,K.W. (1998) Arch. Biochem. Biophys., 350, 72–78. [DOI] [PubMed] [Google Scholar]
- 8.Kolluri S.K., Weiss,C., Koff,A. and Göttlicher,M. (1999) Genes Dev., 13, 1742–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crews S.T. (1998) Genes Dev., 12, 607–620. [DOI] [PubMed] [Google Scholar]
- 10.Fukunaga B.N., Probst,M.R., Reisz-Porszasz,S. and Hankinson,O. (1995) J. Biol. Chem., 270, 29270–29278. [DOI] [PubMed] [Google Scholar]
- 11.Bacsi S.G. and Hankinson,O. (1996) J. Biol. Chem., 271, 8843–8850. [DOI] [PubMed] [Google Scholar]
- 12.Mangelsdorf D.J., Thummel,C., Beato,M., Herrlich,P., Schütz,G., Umesono,K., Blumberg,B., Kastner,P., Mark,M., Chambon,P. and Evans,R.M. (1995) Cell, 83, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wurtz J.-M., Bourguet,W., Renaud,J.-P., Vivat,V., Chambon,P., Moras,D. and Gronemeyer,H. (1996) Nature Struct. Biol., 3, 87–94. [DOI] [PubMed] [Google Scholar]
- 14.Leng X., Tsai,S., O’Malley,B.W. and Tsai,M.-J. (1993) J. Steroid Biochem. Mol. Biol., 46, 643–661. [DOI] [PubMed] [Google Scholar]
- 15.Quack M., Szafranski,K., Rouvinen,J. and Carlberg,C. (1998) Nucleic Acids Res., 26, 5372–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quack M. and Carlberg,C. (1999) Mol. Pharmacol., 55, 1077–1087. [DOI] [PubMed] [Google Scholar]
- 17.Burbach K.M., Poland,A. and Bradfield,C.A. (1992) Proc. Natl Acad. Sci. USA, 89, 8185–8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffman E.C., Reyes,H., Chu,F.-F., Sander,F., Conley,L.H., Brooks,B.A. and Hankinson,O. (1991) Science, 252, 954–958. [DOI] [PubMed] [Google Scholar]
- 19.Ma Q., Dong,L. and Whitlock,J.P. (1995) J. Biol. Chem., 270, 12697–12703. [DOI] [PubMed] [Google Scholar]
- 20.Lusska A., Shen,E. and Whitlock,P.E. (1993) J. Biol. Chem., 268, 6575–6580. [PubMed] [Google Scholar]
- 21.Staples J.E., Murante,F.G., Fiore,N.C., Gasiewicz,T.A. and Silverstone,A.E. (1998) J. Immunol., 160, 3844–3854. [PubMed] [Google Scholar]
- 22.Fernandez-Salguerro P., Hijbert,D.M., Rudikoff,S., Ward,J.M. and Gonzalez,F.J. (1996) Toxicol. Appl. Pharmacol., 140, 173–179. [DOI] [PubMed] [Google Scholar]
- 23.Okey A.B. (1992) In Kalow,A. (ed.), Pharmacogenetics of Drug Metabolism. Pergamon Press, New York, NY, pp. 549–608.
- 24.Kallio P.J., Pongratz,I., Gradin,K., McGuire,J. and Poellinger,L. (1997) Proc. Natl Acad. Sci. USA, 94, 5667–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quack M. and Carlberg,C. (2000) Mol. Pharmacol., 57, 375–384. [PubMed] [Google Scholar]
- 26.Henry E.C., Kende,A.S., Rucci,G., Toteben,M.J., Willey,J.J., Dertinger,S.D., Pollenz,R.S., Jones,J.P. and Gasiewicz,T.A. (1999) Mol. Pharmacol., 55, 716–725. [PubMed] [Google Scholar]
- 27.Lees M.J. and Whitelaw,M.L. (1999) Mol. Cell. Biol., 19, 5811–5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang Y.C., Riby,J., Chang,G.H., Peng,B.C., Firestone,G. and Bjeldanes,L.F. (1999) Biochem. Pharmacol., 58, 825–834. [DOI] [PubMed] [Google Scholar]
- 29.Wei Y.D., Helleberg,H., Rannug,U. and Rannug,A. (1998) Chem. Biol. Interact., 110, 39–55. [DOI] [PubMed] [Google Scholar]
- 30.Phelan D., Winter,G.M., Rogers,W.J., Lam,J.C. and Denison,M.S. (1998) Arch. Biochem. Biophys., 357, 155–163. [DOI] [PubMed] [Google Scholar]
- 31.Pongratz I., Antonsson,C., Whitelaw,M.L. and Poellinger,L. (1998) Mol. Cell. Biol., 18, 4079–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]