

Key Points
-
•
CAR T-cell engineering at GSH6 achieves long-term tumor control.
-
•
Validated criteria identify targetable extragenic GSHs.
Visual Abstract
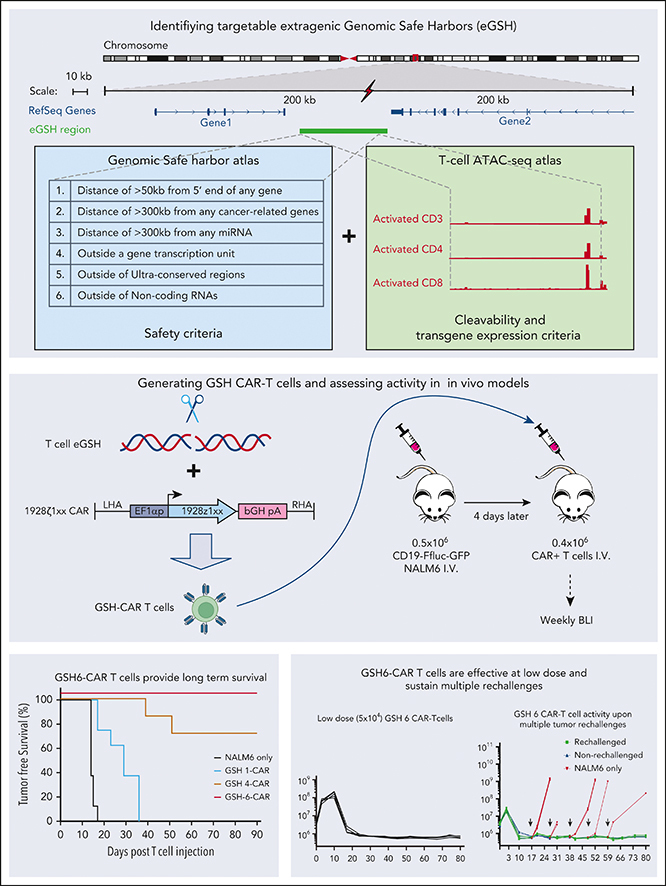
Abstract
Cell therapies that rely on engineered immune cells can be enhanced by achieving uniform and controlled transgene expression in order to maximize T-cell function and achieve predictable patient responses. Although they are effective, current genetic engineering strategies that use γ-retroviral, lentiviral, and transposon-based vectors to integrate transgenes, unavoidably produce variegated transgene expression in addition to posing a risk of insertional mutagenesis. In the setting of chimeric antigen receptor (CAR) therapy, inconsistent and random CAR expression may result in tonic signaling, T-cell exhaustion, and variable T-cell persistence. Here, we report and validate an algorithm for the identification of extragenic genomic safe harbors (GSH) that can be efficiently targeted for DNA integration and can support sustained and predictable CAR expression in human peripheral blood T cells. The algorithm is based on 7 criteria established to minimize genotoxicity by directing transgene integration away from functionally important genomic elements, maximize efficient CRISPR/Cas9–mediated targeting, and avert transgene silencing over time. T cells engineered to express a CD19 CAR at GSH6, which meets all 7 criteria, are curative at low cell dose in a mouse model of acute lymphoblastic leukemia, matching the potency of CAR T cells engineered at the TRAC locus and effectively resisting tumor rechallenge 100 days after their infusion. The identification of functional extragenic GSHs thus expands the human genome available for therapeutic precision engineering.
Targeted integration of chimeric antigen receptor (CAR) transgenes in defined genetic locations is anticipated to improve sustained expression and reduce risks associated with conventional gene transfer methods, such as insertional mutagenesis. Using a CRISPR-Cas9–assisted knock-in technique, Odak et al investigated locations in the noncoding genome that provide stable expression of CAR transgenes in human T cells, identifying GSH6 as such a site that can achieve long-term tumor control. The authors’ data expand our ability to engineer potent CAR T-cell products and provide resources for the identification of other extragenic genomic safe harbor locations.
Introduction
The therapeutic use of genetically engineered human cells is rapidly expanding beyond gene therapy for inherited monogenic disorders to a range of acquired disorders. Alterations of the human genome are not only undertaken to treat inherited mutations1 but also to introduce natural or synthetic genes to reprogram cell function, as exemplified in chimeric antigen receptor (CAR) therapy.2,3 Effective genetic engineering requires predictable and dependable transgene expression, sustaining an optimal level over time without incurring genotoxic adverse events. Albeit effective in achieving stable genetic modifications,1 γ-retroviral, lentiviral, and transposon-based vectors all afford semirandom integration, resulting in obligate insertional mutagenesis4, 5, 6 and variegated transgene expression.7,8 The integration of γ-retroviral and lentiviral vectors is biased toward gene loci, increasing the probability of transgene expression as well as the risk of disrupting the expression of endogenous genes,9,10 the most dreaded consequence of which is oncogene activation,11 as was originally observed in some patients treated with retrovirus-mediated gene therapy for X-linked severe combined immunodeficiency (X-SCID).12, 13, 14 Clonal expansions stopping short of leukemic transformation have also occurred in hematopoietic stem cell therapies for globin gene disorders15 and CAR T-cell therapy.16,17 The other major detrimental consequences of semirandom integration are variegated and unpredictable transgene expression, owing to chromosomal position effects that include transcriptional silencing because of heterochromatinization.8 In regard to T-cell engineering, suboptimal CAR expression may impair T-cell function via tonic signaling induction and terminal T-cell differentiation and exhaustion.18
In principle, these challenges could be overcome if the transgene were integrated at a defined genomic site that reliably provides safe and stable gene expression, referred to as a genomic safe harbor (GSH). Intra or juxtagenic sites have been proposed as potential GSHs in human cells, including the adeno-associated virus site 1, the chemokine receptor 5 locus, and the human orthologue of the mouse ROSA26 locus.19, 20, 21, 22, 23, 24 These sites lie either within a gene that is thought to be dispensable or in close proximity to genes that are deemed not to pose an oncogenic threat. Their vicinity, however, is gene-rich, which raises the risk of endogenous gene transactivation by ectopic enhancer or promoter elements, which may vary in different cell types.
To avert such shortcomings, one may alternatively search for gene-remote, extragenic GSH (eGSH).19 Informed by an extensive literature on retroviral integration sites and insertional mutagenesis, we previously proposed criteria for identifying safe viral vector integrations.25 The advent of site-specific nucleases now makes it possible to prospectively direct transgene integration to such locations, provided that the latter are accessible for efficient cleavage and homologous recombination. Focusing on T-cell engineering, here, we explore the potential of CRISPR/Cas9 and recombinant adeno-associated virus type 6 (rAAV6) donor vectors to target a CAR transcription unit to candidate eGSHs in order to achieve sustained and predictable transgene expression. Using a CAR specific for CD19, we demonstrate that one such site, termed as GSH6, directs optimal CAR expression and potent antitumor activity in vivo. The identification of accessible eGSHs in primary T cells will facilitate the generation of T cells that predictably and homogeneously express their therapeutic gene cargo, including CARs or other immunomodulatory molecules, thereby enhancing the efficacy and safety of cellular immunotherapy.
Materials and methods
In vivo assessment of GSH-CARs in CD19-CAR stress-test model of B-cell acute lymphoblastic leukemia
Six to 12-week-old NOD/SCID/IL-2Rγ null (NSG) male mice (The Jackson Laboratory) were used under an Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee approved protocol. All relevant animal use guidelines and ethical regulations were followed. Mice were infused with 0.5 × 106 CD19-FfLuc–green flourescent protein (GFP) NALM6 cells via tail vein injection, followed by injection of 5 × 104, 1 × 105, 2 × 105, or 4 × 105 CAR+ T cells 4 days later (4 days after transfection, unpurified, CAR+ cell number was calculated using flow cytometry). Tumor rechallenge experiments were performed through IV administration of 1 × 106 CD19-FfLuc-GFP Nalm6 cells at intervals of 10 days, at indicated time points. No mice were excluded before treatment. No randomization or blinding methods were used. Bioluminescence imaging was performed using the IVIS Imaging System (PerkinElmer) and analyzed with the Living Image software (PerkinElmer). Tumor burden was assessed as average total flux signal of dorsal and ventral images per mouse.
Statistical analyses
All statistical analyses were performed using the Prism 7 (GraphPad) software. No statistical methods were used to predetermine sample size.
Results
Identification of efficiently targeted eGSH in human primary T cells
We previously proposed 5 safety criteria to safely position transgenes away from endogenous genes, including cancer-related genes25 (criteria 1-5; Figure 1A), to which we have added a sixth safety criterion to exclude disruption of noncoding RNAs26,27 (Figure 1A). A seventh criterion addresses the need for efficient site-specific transgene integration, which requires effective access and cleavage by a nuclease and subsequent homologous recombination. An eighth criterion addresses local chromatin structure and the need for dependable and sustained transgene function (Figure 1A). Because cleavage efficiency is difficult to predict based on the guide RNA (gRNA) sequence alone,28 cleavage needs to be empirically tested. We hypothesized that accessible chromatin would be more susceptible to efficient cleavage by Cas9, as corroborated via an analysis of cleavage efficiency using assay for transposase-accessible chromatin sequencing (ATAC-seq) peak signal intensity (supplemental Figure 1, available on the Blood website). We thus performed ATAC-seq analyses in activated peripheral blood T cells obtained from healthy human donors, generating an atlas comprising all 21 566 reproducible ATAC-seq peaks (details are presented in supplemental Methods). Another genome atlas was independently established to identify genomic regions satisfying the first 6 eGSH criteria (Figure 1A). Overlapping the 2 atlases yielded 379 candidate eGSHs comprising at least 1 ATAC-seq peak inside or within 5 kb of the eGSH boundaries. These 379 eGSHs were then ranked based on the average signal intensity at the summit of the associated ATAC-seq peak (Figure 1A-B).
Figure 1.

eGSHs that satisfy criteria 1 to 7 are efficiently targeted by CRISPR/Cas9 to express a functional CAR. (A) GSH atlas safety criteria (blue box) and ATAC-seq atlas criteria (green box). The plot represents ranked average maximum signal intensities of ATAC-seq peaks associated with 379 eGSHs (without pseudogenes atlas) across 7 cell replicates. The top 6 highest-intensity GSH peaks are highlighted. Bars show mean ± standard deviation (SD) of n = 7 cell replicates. (B) Volcano plot depicting the 379 eGSHs centered on the GSH peak with a 5-kb region on each side of the peak in 7 cell samples. Peaks are arranged in decreasing order of their maximum (peak summit) signal intensities. Color indicates the value of signal intensities. The eGSH coverage column depicts the region that falls under GSH criteria 1 to 6 in yellow and the region that falls outside the criteria in blue. (C) Cleavage efficiency at top 6 candidate eGSHs. Above, a zoomed-in view of an example candidate eGSH peak spanning 1865 bps and the 4 gRNAs (indicated in red flash symbols) tested for the eGSH at the summit of the peak. Below, CRISPR/Cas9 cleavage efficiencies of 4 independent gRNAs (each independent symbol) at the 6 top eGSHs. (D) (top) rAAV6 gene cassette for targeted integration with the 1928ζ-1xx CAR construct (EF1α promoter + intron, flanked by 300-bp homology arms). (bottom) Experimental schema for CAR integration and preparation of CAR T cells for in vitro cytotoxicity assays. (E) Flow plots of CAR expression from T cells transduced with GSH-CARs at day 3 after transduction before CAR purification. (F) Cytotoxicity assay for the CD19-CAR targeted at GSH 1, 2, and 3 and the TRAC locus (Firefly luciferase-expressing NALM6 as target cells). Data are shown as mean ± SD of 3 technical replicates from the same donor. RPM, reads per million.
We first evaluated the cleavage efficiency at the 6 top eGSHs, designing 4 gRNAs per site at the ATAC-seq peak summit, all of which had a Doench score ≥50 and a specificity score >0.229,30 (see supplemental Table 1 for gRNA details). Electroporation of Cas9 messenger RNA and chemically modified single guide RNAs31 resulted in >80% cleavage efficiencies at all 6 sites (Figure 1C; supplemental Figure 2A). To assess whether high cleavage efficiency is limited to the summit or extends to its immediate vicinity, we randomly chose 2 eGSHs (GSH1 and GSH5) from the top 6 and designed gRNAs spanning 2.5 kb on either side of the peak, followed by quantification of their cleavage efficiencies in peripheral blood T cells (supplemental Figure 2B-C). Although cleavage efficiency diminished 1000 bp and 500 bp away from the edge of the GSH1 and GSH5 peaks, respectively, high efficiency was generally maintained within the peak.
To correlate chromatin accessibility and cleavage efficiency, we tested 2 gRNAs per eGSH at the peak summit for 6 eGSHs that had low ATAC-seq peak signal intensities and 3 previously identified eGSHs25 that had no associated ATAC-seq peaks (supplemental Figure 2D). We additionally included a multiple target site-specific gRNA32 that targets 8 different loci which have different associated ATAC-seq peak signal intensities (supplemental Figure 2E). Although a direct correlation between ATAC-seq peak signal intensity and cleavage efficiency could not be established, these controls further corroborated that an extragenic site with an associated high signal intensity ATAC-seq peak had a higher probability of consistent, efficient cleavage than a site associated with low intensity or no ATAC-seq peak.
eGSHs differentially regulate CAR expression and CAR T-cell function in vitro
We initially targeted the 1928ζ-1XX CAR33 driven by the elongation factor 1α (EF1α) promoter34 to GSH 1-3 (Figure 1D). TRAC-integrated 1928ζ-1XX has been shown to support robust in vivo CAR T-cell function18,33,35, 36, 37, 38 and was therefore used as a gold standard reference for effective intragenic integration and expression. GSH 1-3 were effectively targeted, albeit less effectively than the optimized TRAC targeting, yielding readily detectable CAR expression (Figure 1E). Commensurate with their respective CAR expression levels (Figure 1E), sorted GSH1 and GSH2-CAR T cells displayed greater cytolytic activity against CD19+ NALM6 leukemia cells than GSH3-CAR T cells (Figure 1F). To further analyze GSH-CAR T-cell function and CAR expression, we measured T-cell proliferation induced by repeated antigen encounter over 2 weeks, examining cell-surface CAR expression at regular time intervals (Figure 2A-C). After the first exposure to antigen, CAR expression was maintained at GSH 1 but diminished at GSH2 and GSH3 (Figure 2B). After the second exposure to antigen, CAR expression also diminished at GSH1, in contrast to the stable, sustained level measured in TRAC-CAR T cells (Figure 2B). Therefore, at all 3 eGSHs, CAR expression eventually declined, most rapidly at GSH3, followed by GSH2 and finally GSH1 (Figure 2B). The proliferation capacity of the GSH-CAR T cells over these 2 weeks was also less than that of TRAC-CAR T cells (Figure 2C).
Figure 2.

eGSHs differentially regulate CAR expression and CAR T-cell function in vitro. (A) Experimental schema for weekly antigenic stimulation of purified CAR+ T cells starting 7 days after transduction. Flow cytometry for CAR expression on day 0, 7, and 14 was performed before plating onto CD19+ aAPCs. (B) CAR expression profile of CAR+ T cells over 2 weeks of antigenic stimulation. (C) Proliferation in response to weekly antigenic stimulation over 2 weeks for cells shown in Figure 2B. Data are shown as cumulative fold change in T-cell numbers, mean, and range of 2 technical replicates. (D) rAAV6 cassette design incorporating chromatin insulator element C1. (E) CAR expression profile of all CAR+ T cells over 2 weeks of antigenic stimulation. Data shown are a representative example of 2 technical replicates. See supplemental Figure 3A-B for quantification of data. (F) Cytotoxicity assay for all CARs shown in panel E, at day 0. Data are shown as mean ± SD of 3 technical replicates. (G) Proliferation of GSH-CAR+ cells shown in panel E over 2 weeks in culture. Data are shown as mean and range from 2 technical replicates. Data from E-G are a representative example from 1 T-cell donor. All experiments have been performed for each construct with at least 2 T-cell donors. aAPCs, artificial antigen presenting cells; UT, untransduced cells used as controls.
We hypothesized that a chromatin insulator element with barrier function may offset silencing and sustain CAR expression. We thus flanked the CAR transcription unit at GSH1 with the human C1 insulator39 (Figure 2D) and tested 3 additional eGSHs (GSHs 4-6; Figure 1C). Initial CAR expression levels varied among the sites, with GSH6 yielding expression most similar to TRAC, either with or without the insulator (Figure 2E; supplemental Figure 3A-B). All GSHs showed comparable cytolytic potential against NALM6 on day 0, except for GSH3, which showed lower cytolysis (Figure 2F). After 2 weekly exposures to CD19, GSH2 and GSH3-CAR T cells showed the least expansion (Figure 2G). The C1 insulator had a variable, site-specific impact, increasing CAR expression at GSH1 but not at GSH 4, 5, and 6 (Figure 2E). These observations are consistent with previous observations on the site-specific dependence of insulator activity.40,41 Interestingly, uninsulated GSH6-CAR T cells still maintained their CAR expression levels and displayed proliferation capacity closest to TRAC-CAR T cells. Long-term expression and activity were observed in GSH6-CAR T cells after 3 weeks, with and without antigenic stimulation, further emphasizing the ability of GSH6 to support long-term transgene expression (supplemental Figure 3C-F). A nonsignaling transgene (Humanized Renilla reniformis GFP) integrated at GSH6 also similarly showed sustained transgene expression for 2 weeks in culture (supplemental Figure 4).
In vivo antitumor efficacy depends on eGSH-mediated maintenance of CAR expression
We proceeded to functionally test 3 representative GSH-CAR T cells (GSH 1, 4, and 6, with and without the C1 insulator element) in a pre–B-acute lymphoblastic leukemia NALM6 mouse model. GSH-CAR T cells were assessed under stress-test conditions,42 in which CAR T-cell dosing is lowered to test the functional limits of the CAR T cells (Figure 3A). GSH1 and GSH4±C1-CAR T cells only transiently controlled tumor burden before relapse (Figure 3B-C; supplemental Figure 5). Consistent with the improved CAR expression of the C1 element at GSH1 (Figure 2E), GSH1+C1-CAR T cells displayed improved functional capacity relative to that of GSH1-CAR T cells (Figure 3 B-C). GSH6-CAR T cells, with or without the C1 element, displayed equivalent potency to TRAC-CAR T cells, mediating long-term tumor control (Figure 3 B-C), consistent with their sustained CAR expression in vitro (Figure 2E).
Figure 3.

In vivo anti-tumor efficacy is dependent upon the ability of the targeted GSH to maintain CAR expression over time. (A) Experimental schema for in vivo assessment of tumor burden using CD19-CAR stress-test model for B-acute lymphoblastic leukemia in NSG mice. CAR+ cells were not purified but CAR+ cell numbers were calculated based on flow cytometry. (B) Kaplan Meier tumor-free survival curves for mice administered with GSH 1, 4, and 6 CARs ± insulator C1 and TRAC-CARs. Combined results from 2 experiments with 2 independent T-cell donors (n = 7-12). (C) Tumor burden curves over 90 days in the group of mice from panel B. Some mice with no tumor burden had to be euthanized owing to severe graft-versus-host disease. (D) Total CAR+ T-cell number in the BM of mice 10 days after infusion (results from one representative donor, n = 4-8 mice per group). Data are shown as mean ± SD. (E) Mouse cells were depleted from the BM cells of all mice illustrated in panel D, the remaining cells were pooled by group and an 18 hour cytotoxicity assay was performed with CD19-Ffluc-GFP NALM6 cells at a ratio of 3:1, T cells:NALM6 cells. Cell number calculation was done based on flow cytometry data after mouse cell depletion (supplemental Figures 6B and 8A). Data are not shown for GSH1 CARs because measurement of luciferase was skewed because of the presence of NALM6+ cells (not eliminated by mouse cell depletion) in BM at day 10. Data are shown as mean ± SD of 3 technical replicates from the pooled cells. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; Mann-Whitney U test. BM, bone marrow; BLI, bioluminescent imaging; Ffluc, firefly luciferase; ns, not significant.
CAR expression and T-cell differentiation and exhaustion markers, were examined in T cells retrieved from bone marrow (BM) 10 days after infusion (supplemental Figures 6, 7, and 8A). GSH6±C1-CAR T cells were the most abundant (Figure 3D; supplemental Figure 7A-B). The C1 insulator significantly improved CAR T-cell numbers at GSH1 but not at GSH4. GSH1-CAR T cells displayed an overall naïve phenotype, consistent with their low CAR expression and limited target engagement, whereas GSH6±C1-CAR T cells exhibited differentiation and exhaustion profiles overall similar to that of TRAC-CAR T cells (supplemental Figure 7C-F). When CAR T cells were freshly retrieved from BM and were cocultured with CD19+ NALM6, GSH6-CAR T cells, with or without C1, showed upregulation in CAR expression upon exposure to CD19 and lysed NALM6 cells most effectively (Figure 3E; supplemental Figure 8A). GSH1+C1, GSH4, and GSH4+C1-CAR T cells also showed upregulation in CAR expression but to a lesser extent than that of GSH6-CAR T cells, in line with their lesser lytic activity. Taken together, our observations suggest that the inability to sustain CAR expression at GSH1 and GSH4 accounted for the failure to control NALM6 leukemia. GSH6-CAR T cells, however, sustained CAR expression and prevailed over NALM6.
GSH6 supports long-term tumor control at low T-cell dose and upon multiple rechallenges
CAR expression at GSH6 was maintained upon re-exposure to antigen and over a span of time, whether in the presence or absence of C1, in vitro and in vivo (Figure 2E; supplemental Figure 8A). These remarkable attributes prompted us to closely compare GSH6-CAR T cells to TRAC-CAR T cells.18,33,35, 36, 37, 38 Monitoring CAR expression after exposure to CD19 showed an initial dip, as expected from CAR internalization,18 followed by the recovery of cell-surface expression after 36 hours (Figure 4A-B). In contrast to the consistent high levels of CAR expression observed with the TRAC-EF1α CAR, CAR expression at GSH6 returned to a baseline level, similar to that with TRAC-CAR.18
Figure 4.


GSH6 supports long-term tumor control at low T-cell doses and upon multiple rechallenges. (A) Representative flow plots depicting CAR expression on CAR+ T cells over a week after stimulation with CD19+ aAPCs in vitro. (B) Quantified median fluorescence intensity (MFI) of CAR expression on CAR+ T cells shown in panel A (2 technical replicates each of 2 independent T-cell donors). Data are shown as mean ± SD. (C) Tumor burden in tumor bearing mice administered with GSH6-CARs without insulator C1 and TRAC-CARs at doses 2 × 105, 1 × 105, and 5 × 104 monitored over a period of 80 days after T-cell injection, n = 5. (D) Tumor burden of mice comparing the in vivo efficacy of GSH6-CAR and TRAC-CAR upon 5 tumor rechallenges 10 days apart starting at day 17 after T-cell injection vs no further rechallenge, followed for 80 days after CAR T-cell administration, n = 5 per group. The NALM6 only group represents treatment naïve, age-matched mice injected with NALM6 cells at the rechallenge timepoints (n = 2 mice each for the first 3 time points; n = 1 for the last 2 time points). (E) Tumor burden over 40 days after rechallenge with 1 × 106 NALM6 cells in mice surviving at day 100 after CAR T-cell administration (from Figure 3C). Some mice had to be euthanized or died of graft-vs-host disease, but none of them had tumor at the time of rechallenge.
GSH6-CAR T cells were further compared with TRAC-CAR T cells in vivo, lowering the CAR T-cell dose to 2 × 105, 1 × 105, and 5 × 104. GSH6±C1-CAR T cells effectively controlled tumor burden at all T-cell doses, equally to TRAC-CAR T cells but for a slight kinetic difference (Figure 4C; supplemental Figure 8B). To further pressure-test GSH6-CAR T cells, we rechallenged mice treated with 4 × 105 GSH6-CAR T cells with CD19+NALM6 cells administered at 10-day intervals. GSH6-CAR T cells completely controlled every challenge, matching TRAC-CAR T cells (Figure 4D).
Finally, to assess whether GSH6-CAR T cells could control tumor burden long after initial tumor eradication, we rechallenged long-term surviving mice (from Figure 3C) with CD19+ NALM6, 100 days after CAR T-cell infusion. One tumor-free GSH1+C1 mouse surviving at day 100 was also similarly rechallenged. The GSH1+C1–CAR T-cell recipient was unable to control the challenge, but 6 of 8 rechallenged GSH6±C1–CAR T-cell treated mice remained tumor free (Figure 4E). BM analyses performed 40 days after rechallenging showed persistence of the adoptively transferred CAR T cells (supplemental Figure 8C). We measured CAR expression in T cells at various differentiation states between the time of infusion, at day 10 after infusion and day 40 after rechallenge or day 140 after infusion in nonrechallenged mice (supplemental Figure 9). These data establish that CAR expression at GSH6 remained stable between days 10 and 140 in vivo.
eGSH architecture and association with function
Low CAR expression or rapid silencing was observed at most eGSHs apart from GSH6 and was associated with treatment failure. We expanded our functional studies to include 4 additional sites, including 2 (GSH20 and GSH30) that were located within or very close to a pseudogene, because GSH6 lies within a pseudogene (Figure 5A). All 4 additional sites comprised at least 1 high intensity ATAC-seq peak and showed high cleavage efficiency (criterion 7; supplemental Figure 10A). Although GSH20 seemed most similar to GSH6 in terms of presence of endogenous gene proximity and fell within a pseudogene, CAR expression at GSH20 was consistently lower than at GSH6, and GSH20-CAR T cells failed to express the CAR to the same extent as GSH6-CAR T cells upon multiple antigen stimulations (supplemental Figure 10B-C). Cytotoxicity assays and CAR expression showed that GSH 7, 12, and 20 belonged to the intermediate performing eGSH group, whereas GSH30 was among the lowest, similar to GSH 2, 3, and 4 (supplemental Figure 10C-E). These data argue against encoding a CAR within a pseudogene being sufficient to afford sustained CAR expression and CAR T-cell function.
Figure 5.

Examination of surrounding genomic features of eGSHs and association with functional activity. (A) 400-kbp region centered on the GSH peak for GSHs 1 to 6 and GSHs 7, 12, 20, and 30 are shown. The red flash mark indicates the position of the gRNA targeted at each GSH. Indicated are Refseq coding genes in dark blue, noncoding genes in light blue, pseudogenes in orange, and eGSH region in light green, ATAC-seq peaks in activated cells obtained from our data (donor 2 is used as a representative) in red, and in resting cells, obtained from data in the study by Corces et al,58 in dark green. The signal intensity for both sets of data are scaled to the same range for all panels. (B) The 263 kb genomic region around GSH6 is shown encompassing the 2 closest genes on either side of GSH6. Genes are illustrated in blue and the corresponding polymerase chain reaction (PCR) amplicons used for quantitative reverse transcriptase polymerase chain reaction are shown in red along with their names below. The red flash symbol indicates the gRNA cut site. (C) RNA expression of genes around GSH6 represented as ΔCt in comparison to 18s rRNA. Two primer pairs were used for ZNF746 and 1 each for KRBA1 and ZNF767P. Data are shown as mean ± SD of n = 5 to 9 technical replicates from RNA of CAR T cells at day 0 and day 7 after stimulation on CD19+ aAPCs (CAR protein expression of the same cells is shown in Figure 2E). Dotted line indicates minimum nontemplate control ΔCt value, that is, no expression (taken from CAR expression values in untransduced cells); ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001; 2-way ANOVA with Dunnett multiple comparison test.
Comparing ATAC-seq peak distribution within 50 kb on either side at all eGSHs, GSH6 presented with the greatest bilateral peak abundance in both activated and resting T cells (Figure 5A; Figure 6). In addition, GSH6 also exhibited a higher number of active genes within 200 kb of the integration site. Integration of the EF1α enhancer/promoter driven CAR at GSH6 without the C1 insulator did not alter their expression apart from slightly reducing the number of ZNF767P pseudogene transcripts (Figure 5B-C). On comparing the structural features of all 10 eGSHs, GSH6 was found to distinguish itself in possessing the following attributes: (1) proximity to multiple peaks on both sides of the targeted peak, (2) greater intensity of proximal peaks, (3) presence of proximal and targeted peaks in both resting and activated states, and (4) presence of and expression of surrounding genes in resting as well as activated state (albeit at a distance, respecting criteria 1-6; Figure 5A, Figure 6, supplemental Table 2). None of the other 9 eGSH met all 4 attributes.
Figure 6.

CAR expression, function, and surrounding genomic features at 10 candidate eGSHs. Summary of CAR expression over multiple weekly stimulations, surrounding ATAC-seq peaks, gene presence (including pseudogenes), and expression at all 10 eGSHs. Column 2: Expression in the immediate (Imm.) or day 0, early or day 7, and late or day 14 time points of antigenic stimulation as per data in Figure 2E and supplemental Figure 10C; Column 3: Number of ATAC-seq peaks within 50 kb in activated (A), resting (R), or activated and resting (A+R) state excluding targeted peak upstream (5′) and downstream (3′) of eGSH; Column 4: Presence of targeted ATAC-seq peaks in A, R, or A+R state; Column 5: Presence of surrounding ATAC-seq peaks on one side (upstream/downstream) or both sides of the eGSH within 50 kb; Column 6: Number of genes (coding/noncoding/pseudogene) within 200 kb of eGSH; Column 7: Presence of surrounding genes on one side (upstream/downstream) or both sides of the eGSH; Column 8: Gene expression in T cells (either A or R); Column 9: Average gene expression level of surrounding genes, <100 = Lo and >100 = Hi, according to supplemental Table 2. The GSHs are highlighted with colors based on their CAR T-cell functional activity over time, considering expression and proliferation over 14 days as per the key. A, activated; A+R, activated and resting; exp, expression; Hi, high; Lo, low; Imm., immediate; N, no; ND, not detected; R, resting; Y, yes.
To further support this proposition, we sought to prospectively test this criterion in a collection of adoptively transferred, clonally traceable T cells, annotated for their vector integration site. To identify integration sites permitting long-term vector expression, we turned to a genetic correction model, in which sustained vector expression is required for long-term T-cell persistence and vector silencing would predictably result in T-cell loss. To this end, we investigated a data set generated in patients with X-linked severe combined immunodeficiency (SCID-X1) treated with a γ-retroviral vector encoding the interleukin receptor common γ-chain, which is required to restore T-cell responsiveness to γ-chain cytokines and for T-cell survival.43 We thus assessed the distributions of eGSH integrations in patients who underwent SCID-X1 gene therapy44 under the hypothesis that vector integrations in eGSH regions that persisted continued to express the transgene product and would be enriched over time for those bearing proximity to ATAC-seq peaks relative to their initial preinfusion distribution.
We thus assessed the enrichment of integration sites that fall within eGSHs and that have ATAC-seq peaks within specified distances from the integration site, comparing their relative abundance over time (median follow-up = 1 year). Enrichment (fold change) was observed to be highest when the eGSH integration site was located within 50 kb of an activated or resting ATAC-seq peak (supplemental Table 3; Table 1). Location beyond these distances from the ATAC-seq peak was associated with reduced degree of enrichment (Table 1). Presence of ATAC-seq peaks on both sides of the integration site was associated with the greatest enrichment. Enrichment was also greater (fold change, 2.64; P = 1.69 × 10-13; Table 1, section “GSHs with ATAC-seq peaks both upstream and downstream within specified distances from GSH boundary”) for eGSH integration sites that were proximal to resting ATAC-seq peaks compared with the activated-only ATAC-seq peaks that were within 50 kb. These findings are thus consistent with observations in our 10 targeted eGSH (Figure 6) and further support that proximity to ATAC-seq peaks in the resting cell state is important for sustaining vector activity over time. Based on these findings, we formulated a final, seventh structural criterion for the identification of functional extragenic GSHs (Table 2) ie, ”Location within an ATAC-seq peak in the activated state and proximity to ATAC-seq peaks (<50 kb) and active genes in resting T-cell state (<200 kb),“ fulfilling the 2 functional goals we set out to address for the identification of functional extragenic GSHs, efficient cleavability and reliable transgene expression and regulation.
Table 1.
Long-term eGSH distribution in SCID-X1 trial samples
| eGSHs located within ATAC-seq peaks or within 5 kb of ATAC-seq peak boundary∗ | ||||||
|---|---|---|---|---|---|---|
| Condition | T0 (165) |
Tl (1430) |
Fold change | P value | ||
| No. | % | No. | % | |||
| Activated | 14 | 8.50 | 423 | 29.60 | 3.48 | 5.14E-10 |
| Resting | 36 | 21.80 | 733 | 51.30 | 2.35 | 2.54E-13 |
| eGSHs with ATAC-seq peaks both upstream and downstream within specified distances from integration site† | ||||||
|---|---|---|---|---|---|---|
| Condition_distance | T0 (165) |
Tl (1430) |
Fold change | P value | ||
| No. | % | No. | % | |||
| ACT_50 kb | 16 | 9.7 | 244 | 17.1 | 1.76 | 1.41E-02 |
| ACT_100 kb | 26 | 15.8 | 452 | 31.6 | 2.01 | 1.39E-05 |
| ACT_200 kb | 32 | 19.4 | 522 | 36.5 | 1.88 | 6.30E-06 |
| REST_50 kb | 29 | 17.6 | 664 | 46.4 | 2.64 | 1.69E-13 |
| REST_100 kb | 41 | 24.8 | 752 | 52.6 | 2.12 | 6.99E-12 |
| REST_200kb | 60 | 36.4 | 843 | 59.0 | 1.62 | 4.81E-08 |
| eGSHs with ATAC-seq peaks only upstream within specified distances from integration site‡ | ||||||
|---|---|---|---|---|---|---|
| Condition_distance | T0 (165) |
Tl (1430) |
Fold change | P value | ||
| No. | % | No. | % | |||
| ACT_50 kb | 12 | 7.3 | 94 | 6.57 | 0.90 | 7.41E-01 |
| ACT_100 kb | 16 | 9.7 | 116 | 8.11 | 0.84 | 4.57E-01 |
| ACT_200 kb | 26 | 15.8 | 176 | 12.31 | 0.78 | 2.16E-01 |
| REST_50 kb | 13 | 7.9 | 91 | 6.36 | 0.81 | 4.09E-01 |
| REST_100 kb | 18 | 10.9 | 143 | 10.00 | 0.92 | 6.83E-01 |
| REST_200 kb | 25 | 15.2 | 188 | 13.15 | 0.87 | 4.69E-01 |
| eGSHs with ATAC-seq peaks only downstream within specified distances from integration site§ | ||||||
|---|---|---|---|---|---|---|
| Condition_distance | T0 (165) |
Tl (1430) |
Fold change | P value | ||
| No. | % | No. | % | |||
| ACT_50 kb | 8 | 4.85 | 241 | 16.85 | 3.48 | 1.23E-05 |
| ACT_100 kb | 12 | 7.27 | 74 | 5.17 | 0.71 | 2.72E-01 |
| ACT_200 kb | 20 | 12.12 | 148 | 10.35 | 0.85 | 5.02E-01 |
| REST_50 kb | 23 | 13.94 | 130 | 9.09 | 0.65 | 5.07E-02 |
| REST_100 kb | 23 | 13.94 | 73 | 5.10 | 0.37 | 7.33E-05 |
| REST_200 kb | 21 | 12.73 | 124 | 8.67 | 0.68 | 8.74E-02 |
Table shows the number of eGSH insertion sites that have ATAC-seq peaks within the distances and direction specified in the table headers. Upstream and downstream are determined in relation to the direction of the integrated transgene. A 2-tailed Fisher exact test is used to compute the P value for the enrichment between time zero (before infusion, T0) and later (Tl) time points.
ACT, ATAC-seq peaks in the activated T-cell data set; REST, ATAC-seq peaks in the resting T-cell data set within the specified distances.
Number of eGSH integration sites that are located in an ATAC-seq peak or within 5 kb from an ATAC-seq peak boundary.
Number of eGSH integration sites with at least 1 ATAC-seq peak upstream and at least 1 ATAC-seq peak downstream considering a distance of 50 kb, 100 kb, or 200 kb from integration site.
Number of eGSH integration sites with at least 1 ATAC-seq peak upstream and no peaks downstream direction considering a distance of 50 kb, 100 kb, or 200 kb from integration site.
Number of eGSH integration sites with at least 1 ATAC-seq peak downstream direction and no peaks upstream direction considering a distance of 50 kb, 100 kb, or 200 kb from integration site.
Table 2.
Seven criteria for identifying cell type–specific functional extragenic genomic safe harbors (eGSHs)
| 1. | Distance of >50 kb from 5′ end of any gene |
| 2. | Distance of >300 kb from any cancer-related genes |
| 3. | Distance of >300 kb from any miRNA |
| 4. | Outside a gene transcription unit |
| 5. | Outside of ultraconserved regions |
| 6. | Outside of noncoding RNAs |
| 7. | Within ATAC-seq peak in activated state and proximity to ATAC-seq peaks (<50 kb) and active genes in resting state (<200 kb) |
Discussion
Therapeutic cell engineering based on transgene integration requires the identification of safe genomic sites that afford dependable transgene expression. To achieve this goal, one may elect to target gene loci that provide desirable transcriptional regulation, eg, the TRAC locus to express CARs18 or loci encoding genes deemed to be dispensable, eg, the Adeno-associated virus site 1.23,45 As an alternative, we sought to identify extragenic regions that did not disrupt conserved genetic elements but still afforded sustained transgene expression. Reviewing extensive insertional mutagenesis data accumulated in a number of clinical trials that used γ-retroviral and lentiviral vectors,6,11, 12, 13, 14, 15 we previously proposed safety criteria for identifying suitable integration sites in clonal stem cells25 (Figure 1A; criteria 1-5), to which we have now added 2 criteria for averting the disruption of noncoding RNAs and favoring nuclease accessibility and transgene expression (Table 2; criteria 6-7). Here, we demonstrate the feasibility of prospectively targeting such extragenic genomic regions and support CAR expression to afford long-term and potent antitumor T-cell efficacy.
Recent studies have established the impact of chromatin accessibility in determining cleavage activity in the context of a living cell.46, 47, 48, 49, 50, 51, 52 We, therefore, hypothesized that Cas9 would efficiently bind and cleave candidate eGSH presenting with high ATAC-seq peak signal intensity. Adding this requirement to our 6 safety criteria reduced the number of candidate eGSHs in human primary T cells to 379. All 10 sites that we tested at the ATAC-seq peak summit were efficiently cleaved by CRISPR/Cas9 (Figure 1C; supplemental Figure 10A). In addition, gRNAs located anywhere within ATAC-seq peak boundaries afforded equally high cleavage (supplemental Figure 2B-C), allowing for flexibility in selecting gRNAs with optimal off-target profiles for eventual clinical use. However, upon integration of a CAR transgene, CAR expression varied among sites (Figure 2E; supplemental Figure 10C). Although all targeted sites initially expressed the CAR, only 1 (GSH6) maintained expression after 2 weekly exposures to antigen (Figure 2E). The other sites silenced within days or weeks. The incorporation of a chromatin insulator element with barrier function39 partially rescued CAR expression at some sites but not others (Figure 2E), which is consistent with earlier studies describing the context dependence of chicken β-globin 5' DNase I-hypersensitive site (cHS4) insulator activity.40,41 At GSH6, flanking the CAR transcription unit with the barrier element slightly diminished CAR expression in vitro and CAR T-cell expansion in vivo (Figures 2E and 3D).
Our study highlights the profound impact of integration site and variegated gene expression on CAR T-cell function. Approaches based on the use of retroviral vectors (γ-retroviral, lentiviral, or spumaviral) and DNA transposons are all subject to position effects.7 These approaches also expose the risk of gene disruption, as shown with CAR lentiviral vectors integrated at the TET2 or CBL-B loci.16,17 As anticipated from the eGSH criteria, integration of the CAR transcription unit at GSH6 including a strong enhancer/promoter did not perturb expression of endogenous genes within 150 kb on either side, in resting or in activated T cells (Figure 5B-C). GSH6 is located within a pseudogene, ZNF767P, which is transcribed but is noncoding and lacks any known function. ZNF767P RNA is expressed at very low levels in T cells and its expression is further slightly reduced upon integration of the CAR transgene without the insulator. Incorporation of the insulator, resulted in greater alteration of expression of surrounding genes upon activation, possibly as a consequence of insulator-mediated forced chromatin looping.53
When expressed with GSH6, the CD19 CAR proved to be as effective as the TRAC-encoded CAR under stringent stress-test conditions.33 Therefore, after low-dose CAR T-cell infusion, GSH6-CAR treated mice were protected from 5 consecutive rechallenges with tumor cells and even a late rechallenge 100 days after the initial, single CAR T-cell infusion. CAR expression at GSH6 with and without antigen exposure remained in a relatively narrow range and at a moderate expression level, unlike the less optimal patterns of retroviral long terminal repeats or the EF1α promoter, which predispose to tonic signaling18 (Figure 4 A-B). Kinetic analysis of CAR expression, reflecting the function of the EF1α enhancer/promoter and the bovine growth hormone polyadenylation signal within the GSH6 chromosomal context, revealed that although the time to recovery of expression after antigen stimulation is similar to that after TRAC-EF1α-CAR, CAR expression at GSH6 did not remain elevated, as observed at the TRAC locus, but rather promptly returned to baseline levels. Upon re-exposure to antigen, CAR expression was upregulated and allowed CAR induced killing (supplemental Figure 8A), potentially avoiding the tonic signaling and premature exhaustion brought on by consistently high levels of expression.18 Although both TRAC-CAR and GSH6-CAR T cells performed in a similar manner in the in vivo therapeutic model, they differ fundamentally in that TRAC-CAR T cells lack a T-cell receptor, whereas GSH-CAR T cells retain it, which may be beneficial to increase in vivo expansion of CAR T cells in some settings.54, 55, 56 Other eGSHs that show short-term activity may be useful if extinction of gene expression within a certain time frame were desirable, harnessing chromatin regulation for the temporal control of CARs, T-cell receptors, and other therapeutic molecules.
Our initial hypothesis was that accessible eGSHs would display efficient transgene integration and expression, forming the basis for criteria 7 and 8 (Figure 1A). However, although cleavage efficiency was high as expected from high accessibility at all eGSHs, integration efficiency varied among sites, and further research is required to identify other factors that affect homologous recombination efficiency. In terms of transgene expression, the surrounding ATAC-seq peaks and gene expression profiles in resting and activated T cells provide some insights into what may constitute a more favorable site for sustained expression in T cells. Active peaks in both resting and activated T-cell states were found at GSH6 (Figure 6). A combination of factors affecting three-dimensional chromatin architecture likely plays an important role in regulating transgene expression from eGSHs, including distance and DNA scaffolding in different cell states. Although further analyses are needed, our data for 10 eGSHs and the SCID-X1 clinical trial data indicate that the presence of ATAC-seq peaks in close proximity (≤50 kb from integration site), especially in resting T-cell state and on both sides of the integration site as well as the presence of active genes in the resting T-cell state within a broader vicinity (∼200 kb), also on both sides of the integration site, are more likely to ensure dependable transgene expression from an eGSH and can serve as criteria for selecting dependable eGSHs for therapeutic cell engineering.
T-cell engineering at GSH6 is promising, given the sustained in vivo expression we observed over 100 days and the lack of perturbation of surrounding gene expression. In addition, GSH6 is also located away from the centromere and telomeres, which, in principle, reduces the risk of aneuploidy induced via CRISPR/Cas9 editing.57 Predictable transgene expression is essential for the regulation of a variety of immunotherapeutic molecules other than CARs, including cytokines, chemokines, single-chain variable fragments, bispecific T-cell engagers, and antibodies. Our approach for identifying dependable extragenic genomic regions for foreign DNA integration is applicable in principle to other clinically relevant cell types. We anticipate that the identification of eGSH based on the criteria we propose here will expand the functional human genome for the development of safe cell therapies based on precision engineering.
Conflict-of-interest disclosure: A.O. and M.S. have submitted a patent application partially based on data reported in this manuscript. J.F., J. Eyquem, J.M.-S., and M.S. are named inventors on unrelated patent applications in the field of T-cell engineering. Patent applications are submitted by MSKCC. The remaining authors declare no competing financial interests.
The current affiliation for A.O. is Strand Therapeutics Inc, Boston, MA.
The current affiliation for H.Y. is Calico Life Sciences, San Francisco, CA.
The current affiliation for J.F. is Cluster of Excellence iFIT, University Children's Hospital Tübingen, Tübingen, Germany.
The current affiliation for J. Eyquem is University of Califormia San Francisco, Department of Medicine, Division of Hemato-Oncology, San Francisco, CA.
Acknowledgments
The authors thank George Stamatoyannopoulos for providing us the sequence of the C1 insulator. The authors thank Maria Lemdal-Sjӧstrand and Archana Iyer for the RNA-seq data set and Anton Dobrin, Elisa De Stanchina, Kvin Lertpiriyapong, Alessandra Piersigilli, and Pallavi Vedantam for their support in performing in vivo experiments. The authors thank Lee Zamparo for his support in analyzing the resting T-cell ATAC-seq data set. The authors thank the following Memorial Sloan Kettering Cancer Center (MSKCC) core facilities for their excellent support: The Cell Therapy and Cell Engineering Facility (CTCEF), the SKI Flow Cytometry core facility; animal facility; animal imaging core; laboratory for comparative pathology; bioinformatics core; and integrated genomics operation core. A part of the visual abstract was created using BioRender.com.
This work was partially supported by MSKCC core grant P30 CA008748.
Authorship
Contribution: A.O. designed the study, performed experiments, analyzed and interpreted the data, and wrote the manuscript; H.Y. performed the computational analysis for all ATAC-seq data; J.F. helped design and perform experiments; J.M.-S., F.K. and J. Eyquem helped to design and perform in vitro and in vivo experiments; V.A.C. and J. Everett performed analysis of SCID-X1 clinical trial data; F.D.B. assisted in the generation of the list of oncogenes used for identification of eGSH and analysis of SCID-X1 clinical trial data; C.S.L. designed, analyzed, and interpreted the computational analysis; M.S. designed the study, analyzed and interpreted the data, and wrote the manuscript.
Footnotes
Additional data are available on request from the corresponding author, Michel Sadelain (m-sadelain@ski.mskcc.org).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. 2018;359(6372) doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 2.Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545(7655):423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunbar CE. Stem cell gene transfer: Insights into integration and hematopoiesis from primate genetic marking studies. Ann N Y Acad Sci. 2005;1044:178–182. doi: 10.1196/annals.1349.023. [DOI] [PubMed] [Google Scholar]
- 5.Bushman F, Lewinski M, Ciuffi A, et al. Genome-wide analysis of retroviral DNA integration. Nat Rev Microbiol. 2005;3(11):848–858. doi: 10.1038/nrmicro1263. [DOI] [PubMed] [Google Scholar]
- 6.Schwarzwaelder K, Howe SJ, Schmidt M, et al. Gammaretrovirus-mediated correction of SCID-X1 is associated with skewed vector integration site distribution in vivo. J Clin Invest. 2007;117(8):2241–2249. doi: 10.1172/JCI31661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rivella S, Sadelain M. Genetic treatment of severe hemoglobinopathies: the combat against transgene variegation and transgene silencing. Semin Hematol. 1998;35(2):112–125. [PubMed] [Google Scholar]
- 8.Ellis J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum Gene Ther. 2005;16(11):1241–1246. doi: 10.1089/hum.2005.16.1241. [DOI] [PubMed] [Google Scholar]
- 9.Schroder ARW, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors sctive genes and local hotspots. Cell. 2002;110(4):521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 10.Nienhuis AW, Dunbar CE, Sorrentino BP. Genotoxicity of retroviral integration in hematopoietic cells. Mol Ther. 2006;13(6):1031–1049. doi: 10.1016/j.ymthe.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Stein S, Ott MG, Schultze-Strasser S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16(2):198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 12.Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118(9):3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohn DB, Sadelain M, Glorioso JC. Occurrence of leukaemia following gene therapy of X-linked SCID. Nat Rev Cancer. 2003;3(7):477–488. doi: 10.1038/nrc1122. [DOI] [PubMed] [Google Scholar]
- 15.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467(7313):318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraietta JA, Nobles CL, Sammons MA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558(7709):307–312. doi: 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah NN, Qin H, Yates B, et al. Clonal expansion of CAR T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Adv. 2019;3(15):2317–2322. doi: 10.1182/bloodadvances.2019000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer. 2011;12(1):51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- 20.Kotin RM, Linden RM, Berns KI. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 1992;11(13):5071–5078. doi: 10.1002/j.1460-2075.1992.tb05614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irion S, Luche H, Gadue P, Fehling HJ, Kennedy M, Keller G. Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat Biotechnol. 2007;25(12):1477–1482. doi: 10.1038/nbt1362. [DOI] [PubMed] [Google Scholar]
- 22.Lombardo A, Genovese P, Beausejour CM, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25(11):1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 23.DeKelver RC, Choi VM, Moehle EA, et al. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010;20(8):1133–1142. doi: 10.1101/gr.106773.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papapetrou EP, Schambach A. Gene insertion into genomic safe harbors for human gene therapy. Mol Ther. 2016;24(4):678–684. doi: 10.1038/mt.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papapetrou EP, Lee G, Malani N, et al. Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells. Nat Biotechnol. 2011;29(1):73–78. doi: 10.1038/nbt.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. 2016;96(4):1297–1325. doi: 10.1152/physrev.00041.2015. [DOI] [PubMed] [Google Scholar]
- 27.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 28.Verkuijl SA, Rots MG. The influence of eukaryotic chromatin state on CRISPR–Cas9 editing efficiencies. Curr Opin Biotechnol. 2019;55:68–73. doi: 10.1016/j.copbio.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Perez AR, Pritykin Y, Vidigal JA, et al. GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol. 2017;35(4):347–349. doi: 10.1038/nbt.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doench JG, Fusi N, Sullender M, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hendel A, Bak RO, Clark JT, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33(9):985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Overbeek M, Capurso D, Carter MM, et al. DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol Cell. 2016;63(4):633–646. doi: 10.1016/j.molcel.2016.06.037. [DOI] [PubMed] [Google Scholar]
- 33.Feucht J, Sun J, Eyquem J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25(1):82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eyquem J, Poirot L, Galetto R, Scharenberg AM, Smith J. Characterization of three loci for homologous gene targeting and transgene expression. Biotechnol Bioeng. 2013;110(8):2225–2235. doi: 10.1002/bit.24892. [DOI] [PubMed] [Google Scholar]
- 35.MacLeod DT, Antony J, Martin AJ, et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T Cells. Mol Ther. 2017;25(4):949–961. doi: 10.1016/j.ymthe.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roth TL, Puig-Saus C, Yu R, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559(7714):405–409. doi: 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sachdeva M, Busser BW, Temburni S, et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat Commun. 2019;10(1):5100. doi: 10.1038/s41467-019-13088-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roth TL, Li PJ, Blaeschke F, et al. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell. 2020;181(3):728–744.e21. doi: 10.1016/j.cell.2020.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu M, Maurano MT, Wang H, et al. Genomic discovery of potent chromatin insulators for human gene therapy. Nat Biotechnol. 2015;33(2):198–203. doi: 10.1038/nbt.3062. [DOI] [PubMed] [Google Scholar]
- 40.Walters MC, Fiering S, Bouhassira EE, et al. The chicken β-globin 5′HS4 boundary element blocks enhancer-mediated suppression of silencing. Mol Cell Biol. 1999;19(5):3714–3726. doi: 10.1128/mcb.19.5.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rivella S, Callegari JA, May C, Tan CW, Sadelain M. The cHS4 insulator increases the probability of retroviral expression at random chromosomal integration sites. J Virol. 2000;74(10):4679–4687. doi: 10.1128/jvi.74.10.4679-4687.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Z, Condomines M, Van Der Stegen SJC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanco E, Izotova N, Booth C, Thrasher AJ. Immune reconstitution after gene therapy approaches in patients with X-linked severe combined immunodeficiency disease. Front Immunol. 2020;11:608653. doi: 10.3389/fimmu.2020.608653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hacein-Bey-Abina S, Pai S-Y, Gaspar HB, et al. A Modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014;371(15):1407–1417. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klatt D, Cheng E, Hoffmann Di, et al. Differential transgene silencing of myeloid-specific promoters in the AAVS1 safe harbor locus of induced pluripotent stem cell-derived myeloid cells. Hum Gene Ther. 2020;31(3-4):199–210. doi: 10.1089/hum.2019.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lazzarotto CR, Malinin NL, Li Y, et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat Biotechnol. 2020;38(11):1317–1327. doi: 10.1038/s41587-020-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu X, Scott DA, Kriz AJ, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32(7):670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen X, Rinsma M, Janssen JM, Liu J, Maggio I, Goncalves MAFV. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 2016;44(13):6482–6492. doi: 10.1093/nar/gkw524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jensen KT, Fløe L, Petersen TS, et al. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017;591(13):1892–1901. doi: 10.1002/1873-3468.12707. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, Zeng S, Hu R, et al. Using local chromatin structure to improve CRISPR / Cas9 efficiency in zebrafish. PLoS One. 2017;12(8) doi: 10.1371/journal.pone.0182528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hinz JM, Laughery MF, Wyrick JJ. Nucleosomes inhibit Cas9 endonuclease activity in vitro. Biochemistry. 2015;54(48):7063–7066. doi: 10.1021/acs.biochem.5b01108. [DOI] [PubMed] [Google Scholar]
- 52.Knight SC, Xie L, Deng W, et al. Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science. 2015;350(6262):823–826. doi: 10.1126/science.aac6572. [DOI] [PubMed] [Google Scholar]
- 53.de Wit E, Vos ESM, Holwerda SJB, et al. CTCF binding polarity determines chromatin looping. Mol Cell. 2015;60(4):676–684. doi: 10.1016/j.molcel.2015.09.023. [DOI] [PubMed] [Google Scholar]
- 54.Ghosh A, Smith M, James SE, et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med. 2017;23(2):242–249. doi: 10.1038/nm.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stenger D, Stief TA, Kaeuferle T, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood. 2020;136(12):1407–1418. doi: 10.1182/blood.2020005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lapteva N, Gilbert M, Diaconu I, et al. T-cell receptor stimulation enhances the expansion and function of CD19 chimeric antigen receptor–expressing T cells. Clin Cancer Res. 2019;25(24):7340–7350. doi: 10.1158/1078-0432.CCR-18-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nahmad AD, Reuveni E, Goldschmidt E, et al. Frequent aneuploidy in primary human T cells after CRISPR–Cas9 cleavage. Nat Biotechnol. 2022;40(12):1807–1813. doi: 10.1038/s41587-022-01377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corces MR, Buenrostro JD, Wu B, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48(10):1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.