

Key Points
-
•
MF hematopoietic stem cells aberrantly secrete CXCL8 and exhibit enhanced cell growth/output in response to CXCL8 in vitro.
-
•
Genetic deletion or inhibition of Cxcr2 in the hMPLW515L-adoptive transfer model ameliorates fibrosis and improves hematologic parameters.
Visual Abstract
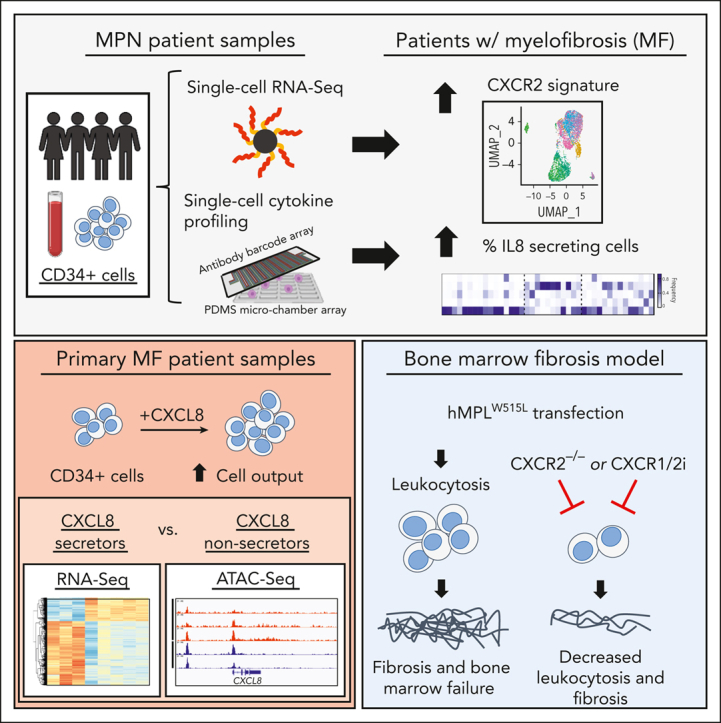
Abstract
Proinflammatory signaling is a hallmark feature of human cancer, including in myeloproliferative neoplasms (MPNs), most notably myelofibrosis (MF). Dysregulated inflammatory signaling contributes to fibrotic progression in MF; however, the individual cytokine mediators elicited by malignant MPN cells to promote collagen-producing fibrosis and disease evolution are yet to be fully elucidated. Previously, we identified a critical role for combined constitutive JAK/STAT and aberrant NF-κB proinflammatory signaling in MF development. Using single-cell transcriptional and cytokine-secretion studies of primary cells from patients with MF and the human MPLW515L (hMPLW515L) murine model of MF, we extend our previous work and delineate the role of CXCL8/CXCR2 signaling in MF pathogenesis and bone marrow fibrosis progression. Hematopoietic stem/progenitor cells from patients with MF are enriched for a CXCL8/CXCR2 gene signature and display enhanced proliferation and fitness in response to an exogenous CXCL8 ligand in vitro. Genetic deletion of Cxcr2 in the hMPLW515L-adoptive transfer model abrogates fibrosis and extends overall survival, and pharmacologic inhibition of the CXCR1/2 pathway improves hematologic parameters, attenuates bone marrow fibrosis, and synergizes with JAK inhibitor therapy. Our mechanistic insights provide a rationale for therapeutic targeting of the CXCL8/CXCR2 pathway among patients with MF.
Cytokines are known to be proinflammatory messengers in myeloproliferative neoplasms (MPNs) and are suspected to be important in the late stages of these diseases. Using primary human samples, single-cell cytokine profiling, and murine models, Dunbar and colleagues reveal that the CXCL8/CXCR2 cytokine-signaling axis is a key mediator of bone marrow fibrosis in MPN, providing a therapeutically tractable avenue to prevent, or treat, this lethal complication of MPN
Introduction
Primary myelofibrosis (MF) is a clonal myeloproliferative neoplasm (MPN) characterized by constitutional symptoms, progressive cytopenias, splenomegaly, and an increased risk of evolution to acute leukemia.1 Overt MF can also evolve from prefibrotic MPN, which includes polycythemia vera (PV) and essential thrombocythemia (ET). Gain-of-function mutations of the JAK/STAT pathway occur frequently in MPN, highlighting the role of constitutive JAK/STAT activation in disease initiation and maintenance.2, 3, 4, 5, 6, 7 Although the recurrent mutations in MPNs have been extensively studied,8 the phenotypic and prognostic pleiotropy observed suggests that biologic factors, in addition to activated JAK/STAT, contribute to MF progression and leukemic transformation.
Aberrant proinflammatory cytokine signaling is an important mediator of fibrosis across multiple tissue types, including the bone marrow (BM).9 Recent single-cell studies have provided insight into how chronic inflammation within the BM compartment promotes mesenchymal stromal cell (MSC) remodeling to drive fibrosis in MF.10, 11, 12 BM-derived fibrocytes are also thought to represent an alternative source for myofibroblasts during wound healing and in lung and kidney fibrosis as well as in the stromal reaction to MF.13,14 Importantly, the proinflammatory pathways found to contribute to MSC differentiation in MF are also frequently implicated in MPN hematopoietic stem/progenitor cell (HSPC) expansion/differentiation, including transforming growth factor β (TGFβ),15, 16, 17 JAK/STAT,18 and tumor necrosis factor α (TNFα),19,20 highlighting a likely important cross talk between mutant clonal hematopoietic stem cells (HSCs) and the BM stroma.21,22
Previously, we and others have shown that the JAK/STAT and TNFα/NF-κB inflammatory pathways cooperate to promote marrow fibrosis in MPNs.23,24 Canonical NF-κB signaling elicits a myriad of chemokines/cytokines that contribute to acute- and chronic-phase inflammation, including interleukin-6 (IL-6), CXCL8 (IL-8), and macrophage inflammatory protein-1α (MIP-1α), among others.25 These cytokines, in addition to other profibrogenic cytokines, are elevated in patients across the spectrum of MPNs and in MF and have prognostic relevance.26,27 Recent studies have highlighted the contribution of specific cytokines in MF progression28,29; however, additional cytokine pathways are likely involved, and the full spectrum of inflammatory mediators playing causative roles in MF have yet to be identified. Notably, increased serum CXCL8 levels were previously found to correlate with adverse clinical outcomes in patients with MF26; however, to our knowledge, a functional assessment of CXCL8-CXCR2 signaling in disease progression has not been done. We hypothesized that specific cytokines/chemokines elicited by MPN HSCs, including CXCL8, promote MF development and predict the likelihood of disease progression. Using both single-cell transcriptional and cytokine platforms, we identify the enrichment in CXCL8/CXCR2 signaling in MF and assess the role of CXCL8/CXCR2 in MF pathogenesis and therapeutic response.
Methods
Human/patient experiments
Patient samples were provided through the MPN Research Consortium and Memorial Sloan Kettering Cancer Center. Written informed consent was obtained per the institutional review boards of Memorial Sloan Kettering Cancer Center, Icahn School of Medicine, and individual MPN Research Consortium member institutions. Fibrosis was quantified using the current clinical World Health Organization criteria, previously described by Thiele et al.30 The presence of fibrosis in each patient sample was independently verified by a hematopathologist (W.X.) or pulled directly from clinical pathology reports at the time of sample collection. Deidentified, healthy CD34+ cells were purchased from AllCells. CD34+ selection was carried out using Ficoll-Paque separation and MicroBead column filtration (Miltenyi) per protocol. For in vitro experiments, CD34+ cells were cultured in StemSpan serum-free medium with stem cell factor, thrombopoietin, FMS-like tyrosine kinase 3 ligand, and IL-3 at 20 ng/mL, to which varying doses of CXCL8 was added.
Mouse models and in vivo experiments
Mouse experiments were performed in accordance with Memorial Sloan Kettering Cancer Center institutional animal care and use committee–approved protocols. Cxcr2f/f mice have been described previously.31 Human MPLW515L (hMPLW515L) experiments were performed as described previously.32 Briefly, prestimulated, lineage-negative BMs from Cxcr2f/f;VavCre+ or Cxcr2f/f;VavCre– mice were transduced via cosedimentation, with viral supernatant containing either murine stem cell virus (MSCV)-hMPLW515L–internal ribosome entry site (IRES) green fluorescent protein (GFP) or MSCV-MigR1-IRES-GFP empty vector (EV) control plasmid and transplanted into lethally irradiated C57BL/6J mice. For in vivo hMPLW515L drug trial experiments, BALB/c mice were used. Approximately 2 or 3 weeks after the transplant of MSCV-hMPLW515L-IRES-GFP retrovirally transduced lineage-negative BMs, mice were subjected to bleeding and randomly assigned to ruxolitinib (60 mg/kg orally, twice daily), reparixin (60 mg/kg subcutaneously, twice daily), their combination, or vehicle arms based on mutant cell fraction (GFP percentage) and leukocyte parameters to ensure consistency across arms. Histopathology was photographed using a BX53 Olympus microscope and DP74 camera.
Single-cell transcriptional/cytokine profiling
Single-cell messenger RNA sequencing (scRNA-seq) was conducted as previously described.33 Reverse transcription, library construction, and sequencing were carried out using reported protocols.34,35 Hg19 using STARv (2.5.2b) was used for alignment based on the Dropseq method.34 Seurat (version 3.1.2)36 was used to identify differentially expressed genes (DEGs). SingleR (version 1.0.1)37 was used to annotate cell types.38,39 Functional enrichment analysis was conducted using Databases for Annotation, Visualization and Integrated Discovery.40 Gene correlations were assessed using Scran (version 1.12.1). Single-cell cytokine profiling was carried out on CD34+ cells isolated from patients using previously described methods.41
Methylcellulose assays
CD34+ cells were plated at 500 cells per replicate in 30 mm dishes containing 1mL of StemSpan serum-free medium with 1.1% methylcellulose containing stem cell factor, thrombopoietin, FMS-like tyrosine kinase 3 ligand, granulocyte-macrophage colony-stimulating factor, IL-3, and erythropoietin, with or without CXCL8. For reparixin/ladarixin studies, dimethyl sulfoxide was added in control wells. The colonies were enumerated on day 14.
Plasma cytokine analysis
Patient CXCL8 plasma levels were determined using the CXCL8 Quantikine enzyme-linked immunosorbent assay kit per the manufacturer’s protocol. Assays for murine serum were carried out using the Millipore Mouse Cytokine 32-plex kit and FlexMAP 3D platform (Luminex).
RNA/ATAC-Seq analysis
STAR(v2.6.0a)42 and featureCounts(version 1.6.3)43 were used to align Fastq files to hg19 and determine the number of reads per gene. Differential expression analysis was performed using DESeq2,44 with a fold change cutoff of ±2 and a false discovery rate (FDR) of 1%. Motif signatures were obtained using de novo Homer approach (version 4.11).45 Gene set enrichment analysis (GSEA) was performed using GSEA version 3.046 with mSigDB (version 6.0) pathway database. For protein-protein interaction network analysis, STRING was used to visualize differentially upregulated genes with a fold change ≥4 and an FDR ≤1%.47 The degree and betweenness centrality were calculated for each node using NetworkAnalyzer (version 4.4.6).48 The STRING network was filtered for nodes having a degree ≥5 and betweenness centrality score ≥0.005. For Assay for Transposase-Accessible Chromatin with high-throughput Sequencing (ATAC-Seq), trimmed reads were mapped to hg19 using Bowtie2 (version 2.3.4.1).49 Peak calling was performed using MACS2 (version 2.1.2) against standard input (fold change >2; P < .001).50 Peaks from all samples were merged within a 500 bp window to create a peak atlas. Raw read counts were then tabulated using featureCounts (version 1.6.3).43 Peaks were annotated using genomic distance, with genes assigned to a peak if they were within 50kb upstream/downstream of the start/end site. Raw read counts were normalized using the median of ratios normalization method (DESeq2).44 Promoters were defined as being within 2kb of the transcription start site. Known motif enrichment was used for the ATAC-Seq analysis. Differential accessibility of peaks was calculated using a fold change cutoff of ±2 and an FDR of 1%.
Splenic stromal/fibroblast cells and endothelial cells preparation
Stromal cells were isolated from healthy spleen donors from the National Disease Research Interchange. To generate fibroblasts, digested splenic cells were cultured in minimal essential medium α with 5% human platelet lysates (2-5 × 104 cells per mL) in culture flasks for 24 hours. To generate endothelial cells, digested splenic cells were cultured in endothelial cell growth medium–2 (2 × 104 to 5 × 104 cells per mL) in fibronectin-coated culture flasks for 24 hours. For coculture experiments, 5 × 104 megakaryocyte (MK)–enriched cells were directly seeded onto 12-well plates over adherent mesenchymal stromal cells at a 50% confluency.
Results
CXCL8/CXCR2 signaling is enriched in CD34+ HSPCs of fibrotic MPN
To investigate the extent of proinflammatory cytokine expression among individual MPN HSPC cell populations, we carried out single-cell gene expression profiling (scRNA-seq) on CD34+ cells isolated from a small set of patients with MPN with varying degrees of fibrosis (supplemental Table 1; supplemental Figure 1A-C, available on the Blood website). Using quality control filtering, a total of 25 288 genes in 5199 single cells were included for final analysis. Visualization with uniform manifold approximation and projection analysis of HSCs revealed that patients demonstrating increased fibrosis, such as MF1 and ET with MF, were clustered and exhibited higher levels of expression of fibrosis-related genes, including TNFα/NF-κB and inflammatory response pathway genes (Figure 1A-B; supplemental Figure 1D; supplemental Table 4). Similar patterns were evident across multiple different cell types, including common myeloid progenitors and granulocyte-monocyte progenitors, suggesting shared transcriptional programs throughout myeloid lineage commitment (supplemental Figure 1E). In addition to TNFα/NF-κB–regulated gene expression programs, we also observed an increase in the expression of CXCR2-mediated chemokines, including CXCL2 (MIP-2α), CXCL3 (MIP-2β), and CXCL8 (IL-8; Figure 1B; supplemental Figure 1F).51 These data expand on those described in previous studies52, 53, 54 and validate that many CD34-expressing cells within patients with fibrotic MPN express CXCL8 and have the capacity to signal through CXCR2. Notably, CXCR2 signaling is well-known to promote mature myeloid cell activation,55 and elevated serum CXCL8 levels were previously shown to be associated with increased risk of leukemic transformation and reduced overall survival in MF26; however, the role of CXCL8 signaling in MF progression and whether CXCL8-secreting cells were enriched in MF compared with other MPN subtypes had not been previously investigated.
Figure 1.

CD34+ cells secreting only CXCL8 are enriched in a subset of patients with MF, and this correlates with clinical features, including grade reticulin fibrosis. (A) Uniform manifold approximation and projection visualization of individual HSCs colored based on the patient (see supplemental Table 1). (B, top) GSEA of differentially expressed genes (DEGs) based on clustering of patient HSCs; (B, bottom) most DEGs and their pathway associations (percent expressed: percentage of cells expressing listed gene; average expression scale: Z score of normalized read counts, with blue, positive values and gray, negative values). UPR, unfolded protein response. (C) Heatmap demonstrating frequency of individual cytokine-secreting CD34+ cells detected across MPN subtypes MF, PV, and ET among individual patients as a percentage of total cytokine-secreting cells (from 0% in gray to 100% in dark blue). Four cytokines presented: IL-6, MIP-1β, TNFα, and CXCL8. (D) Violin plot depicting the correlation between MPN subtype and percent fraction of cells secreting only CXCL8, as detected via single-cell cytokine analysis. (E) Ratio of total cell output relative to untreated cultured healthy donor (HD) (light blue) vs MF (dark blue) CD34+ cells in response to exogenous CXCL8 (50 or 100 ng). Representative of triplicate experiments from N = 3 HD and N = 6 MF samples. Data shown represent mean ± standard deviation (SD. (F) Percent of total CD34+ cells expressing CXCR1 (left) or CXCR2 (right) via flow cytometry of HD (control [CTRL]; N = 13) vs patients with MF (N = 15). Data shown represent mean ± SD. ∗P < .05; ∗∗∗P < .001. NES, normalized enrichment score.
To understand whether messenger RNA expression of CXCL8 manifested into functional CXCL8/IL8 secretion, we performed single-cell cytokine-secretion profiling41 on circulating CD34+ cells isolated from a larger cohort of patients with MPN spanning all 3 clinical MPN subtypes (N = 11, MF; N = 13, PV; and N = 14, ET; supplemental Table 2). Among 5 cytokines assessed, we observed the expansion of a monomorphic, CXCL8-only secreting cell population enriched in MF compared with PV and ET (54% MF vs 31% PV vs 0% ET; Figure 1C). This also correlated with detectable CXCL8 levels in MPN plasma samples (supplemental Figure 2A). Intriguingly, RANTES was significantly enriched in ET (supplemental Figure 2B), but given the previous data highlighting the adverse clinical implications of CXCL8 in MF, we focused our subsequent studies on CXCL8 specifically. Consistent with our cohort that underwent scRNA-seq, the frequency of CXCL8-secreting CD34+ cells correlated not only with the MF subtype (Figure 1D) but also with the degree of reticulin fibrosis and leukocytosis (supplemental Figure 2C-D), suggesting that the presence of circulating CXCL8-secreting CD34+ cells in blood may serve as a biomarker for the presence of significant BM fibrosis (supplemental Figure 2E).
To functionally assess the impact of CXCL8/CXCR2 signaling on human MPN HSCs, we cultured MF CD34+ cells with exogenous CXCL8. This revealed enhanced proliferation and total cell output of treated cells, including increases in CD33+ monocytic and CD41+ MK cell numbers (Figure 1E; supplemental Figure 2F). Via flow cytometry, we also observed an increase in both the fraction of CXCR1/2-expressing MF CD34+ cells and CXCR1/2 surface expression intensity compared with healthy CD34+ control cells, consistent with the enhanced response of MF hematopoietic cells to CXCL8 (Figure 1F; supplemental Figure 2G). Furthermore, colony-forming assays also revealed enhanced colony-forming unit–GM colony output relative to the degree of CXCR2 surface receptor expression (supplemental Figure 2H). Notably, both CXCR1/2 surface expression and CXCL8 single-cell cytokine enumeration correlated with JAK2V617F variant allele frequency (supplemental Figure 2I-J), consistent with recent single-cell studies,56 suggesting that the magnitude of JAK/STAT signaling corresponds with CXCL8/CXCR2 output. Together, these data show that CXCL8 is enriched and aberrantly secreted by multiple cell populations in MF and promotes cell growth and proliferation of MF HSPCs.
Integrated ATAC/RNA-Seq reveals enriched pathways in CXCL8 secretor vs nonsecretor MPNs
Our cytokine data demonstrated a correlation between CXCL8-secreting CD34+ cells and BM reticulin fibrosis, suggesting that the CXCL8-CXCR2 pathway signaling promotes fibrotic progression in a subset of patients with MF. Given our prior data demonstrating alterations in enhancer landscapes of TNFα/NF-κB–enriched murine MF models, we investigated whether the transcriptional and chromatin proinflammatory states of MF CD34+ cells varied in the context of CXCL8 cytokine secretion. We performed bulk RNA-Seq and assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-Seq) on CD34+ cells isolated from patients with MPN with varying degrees of fibrosis and stratified the samples as CXCL8 secretors vs nonsecretors based on single-cell cytokine-secretion profiling data (supplemental Table 3). Gene expression analysis revealed general clustering of CXCL8-secretors vs nonsecretors, irrespective of the MPN subtype (supplemental Figure 3A). Review of the most DEGs (supplemental Figure 3B) between CXCL8 secretors vs nonsecretors revealed an enrichment in genes encoding neutrophil markers and those involved in the activated innate immune response (eg, CTSG, AZU1, MPO, PRTN3, ELANE, and RNASE2/3), suggesting a CD34+ population that skewed toward enhanced mature myeloid differentiation (Figure 2A; supplemental Table 5). Consistent with our scRNA-Seq, GSEA showed enrichment in TNFα/NF-κB and hallmark interferon alfa or gamma (IFN-α/γ) response gene sets (Figure 2B), and network analysis revealed other gene oncology processes indicative of mature myeloid/neutrophil differentiation/activation and toll-like receptor (TLR) signaling (Figure 2C; supplemental Figure 3C; supplemental Table 5), increasingly implicated in MF.10,20,52 Integrated analysis of gene expression and chromatin accessibility data in a subset of patients (N = 5; supplemental Table 3) confirmed increased expression/accessibility of genes involved in innate immune response, neutrophil activation/differentiation, and TLR signaling (eg, S100A8/A9, CCL3, KLF4, CEBPB, and TLR4); FOS/JUN activation (eg, FOS/FOSB); extracellular matrix remodeling (eg, PLAUR); and type I IFN-α response (eg, OAS1/L and IFI6; Figure 2D). These findings were further supported by the results from the analysis of an extended cohort of patients with MF from a publicly available transcription microarray data set stratified based on CXCL8 gene expression (supplemental Figure 3D-F), including similar enrichment in TNFα/NF-κB and IFN-α/-γ proinflammatory gene sets and those associated with mature myeloid cell activation and TLR signaling (supplemental Figure 3G-H). These observations, coupled with an increased expression of known reciprocal negative regulators of these proinflammatory pathways, including DUSP1/2 and ZFP36, support an important role for neutrophil activation, alarmin overexpression, and the acute phase inflammatory response in CXCL8-secreting MF CD34+ cells.
Figure 2.

Integrated transcriptional (RNA-Seq)/chromatin accessibility (ATAC-Seq) profiling identifies pathways enriched in CXCL8-secretor MF. (A) Volcano plot demonstrating most DEGs in CXCL8 secretor (N = 3) vs nonsecretor (N = 5) patients with MPN via RNA-Seq. The significant events with an inclusion level >0.5 log fold change and an FDR-corrected P < .0001 are shown in blue. (B) GSEA demonstrating enriched pathways of CXCL8 secretors vs nonsecretors plotted as NES vs FDR q value. (C) Table depicting results of enriched pathways from optimized subnetwork gene expression analysis in CXCL8 secretor vs nonsecretor patients with MPN. (D) Waterfall plot with integrated gene expression and chromatin accessibility showing most differentially regulated genes (represented as log2 fold change) in CXCL8 secretors (N = 3) vs nonsecretors (N = 2) and their corresponding degree of changes in accessibility peaks (represented as log2 fold change and -log10[padj]; red, positive values; blue, negative values). (E) Tornado plot and heatmaps depicting accessibility at promoter regions of the top 500 leading-edge genes in the hallmark TNFα/NF-κB gene set of CXCL8 nonsecretor (N = 2) vs CXCL8-secretor (N = 3) patients with MPN. (F) Known Homer motif analysis from ATAC-Seq data demonstrating increased accessibility of CEBP, AP-1 (and AP-1 related), interferon-regulatory factor (and interferon-regulatory factor–related), NF-κB, and STAT5 motif signatures among enhancer regions of CXCL8-high/fibrotic MPN.
Given the degree of enrichment in TNFα/NF-κB in CXCL8 secretors and previous associations of this gene set/pathway in MF, we sought to determine whether the chromatin accessibility states of CXCL8-secreting MF represented a distinct proinflammatory MPN entity or all MPN CD34+ cells were poised for inflammatory signaling via NF-κB. Assessment of the accessibility landscape surrounding the leading-edge genes most responsible for driving the hallmark TNFα and IFN-α/-γ pathways revealed a marked increase in the ATAC signal in patients who were classified as CXCL8 secretor vs nonsecretor, concordant with their transcriptional output, suggesting that, rather than being inherently poised for NF-κB inflammatory signaling, additional epigenetic changes are required to engage this proinflammatory program to promote MF (Figure 2E). In addition to the alterations at critical TNFα/NF-κB gene loci, we also observed a strong, global positive enrichment in FOS/JUN (AP-1), C/EPBE, and interferon-regulatory factor motif signatures. When segregating between promoter and enhancer regions, accessibility alterations in the enhancer regions drove most of the enrichment observed, including a STAT5 motif signature (Figure 2F; supplemental Figure 3I), consistent with our prior studies in murine MF models.23 Surprisingly, the overall accessibility at the CXCL8 locus was unchanged, despite known NF-κB, AP-1, STAT, and C/EBPE motifs at the CXCL8 promoter (supplemental Figure 3J),57 a finding that is also true for other canonical NF-κB– and CXCR2-mediated cytokines, including TNFα itself, IL-6, IL-10, and CXCL1 (supplemental Table 6). These data suggest that cytokine promoter regions within MPN CD34+ cells, including at CXCL8, might be inherently poised for inflammatory-mediated regulation and that differential enhancer activity and/or the lineage-specific chromatin states themselves are the primary drivers of the increased proinflammatory signaling observed in CXCL-8 high/fibrotic MPN.
Cxcr2 deletion improves hematologic parameters and reduces fibrosis in the hMPLW515L adoptive transfer model
Our genomic/epigenomic studies and in vitro data suggested that MF HSCs are competent to signal via CXCR2 and that CXCL8-CXCR2 signaling might be relevant in a subset of MF. To explore the CXCR2 pathway signaling in fibrotic progression, we investigated the effects of genetic deletion of Cxcr2 within the murine hematopoietic compartment using the hMPLW515L fibrosis mouse model.32 Although mice lack the analog equivalent of human CXCL8 (hCXCL8),58 murine CXCR1/2 receptors share close homology to those of humans, bind to hCXCL8, and activate similar downstream mediators.59 Consistent with this, culture of cKit+ murine BM cells in the presence of hCXCL8 demonstrated enhanced signaling through pERK and plasmid STAT3 that was abrogated in the setting of Cxcr2 knockout (supplemental Figure 4A). Isolated lineage-negative VavCre-Cxcr2–/– or Cre–wild-type (WT) Cxcr2f/f BM cells were then transduced with either MSCV-hMPLW515L-IRES-GFP or MSCV-MigR1-IRES-GFP EV control plasmid and transplanted into lethally irradiated C57BL/6J recipient mice and monitored for the development of MF (supplemental Figure 4B). Cxcr2 knockout was validated by loss of surface expression on myeloid cells in primary VavCre-Cxcr2–/– mice (supplemental Figure 4C) and in transplant recipients. Mice that received transplantation with Cxcr2–/– hMPLW515L-expressing cells displayed reductions in white blood cell (WBC; mean 28.0 × 103/μL vs 104.1 × 103/μL; P < .01) and platelet parameters (mean 773.6 × 103/μL vs 1933.75 × 103/μL; P < .01) compared with WT Cxcr2f/f-expressing hMPLW515L mice (Figure 3A). Further, absolute CD11b+Gr1+ neutrophil numbers were also reduced in the peripheral blood of Cxcr2–/– hMPLW515L mice (supplemental Figure 4D). In addition to improvements in blood cell count parameters, we observed a significant reduction in the mutant GFP+ cell fraction (mean 63.5% vs 93.3%; P = .031; Figure 3B) and liver weights (mean 1234 vs 2783 mg; P < .01) with Cxcr2–/– hMPLW515L mice; however, spleen weights were not convincingly reduced (mean 269.2 vs 436.3 mg; P = .19; Figure 3C; supplemental Figure 4E). These phenotypic changes did not appear to be related to an inherent homing/engraftment defect of Cxcr2–/– vs Cxcr2f/f cells or to hematopoietic dysfunction induced by Cxcr2 loss (supplemental Figure 4F-G). Consistent with our MF patient culture data, flow cytometric analysis also revealed reductions in CD41+ BM MK cell fractions (mean 7.5% vs 1%; P < .01; supplemental Figure 4H), and histopathologic sections of Cxcr2–/– hMPLW515L BM confirmed reductions in observable MKs (supplemental Figure 4I). Notably, reticulin staining revealed significant improvements in fibrosis in both the BM and spleen (Figure 3D; supplemental Figure 4J-K), which, together with the reductions in observable MKs, translated into significant improvements in the overall survival (median 84 vs 42 days; P < .01; Figure 3E).
Figure 3.

Cxcr2 deletion in murine BM improves counts and reticulin fibrosis in the hMPLW515L adoptive transfer model of MF. (A) WBC counts (×103/μL), hematocrit levels (%), and platelet counts (×103/μL) of Cxcr2f/f;Cre+ knockout (KO) hMPLW515L mice compared with Cxcr2f/f;Cre– WT hMPLW515L or MSCV-MigR1-IRES-GFP EV control mice at timed euthanization 9 weeks after transplant. N = 4 or 5 per arm; ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001. Data shown represent mean ± standard error of mean (SEM). Two-way analysis of variance was used to compare groups. (B) Peripheral blood mutant cell fraction vs GFP percentage in Cxcr2f/f;Cre+ hMPLW515L mice vs Cxcr2f/f;Cre– WT hMPLW515L or EV control mice. N = 4 or 5 per arm; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001. Data shown represent mean ± SEM. (C) Liver weights (mg) of Cxcr2f/f;Cre+ KO vs Cxcr2f/f;Cre– WT hMPLW515L mice compared with EV controls. ∗∗P < .01. Data shown represent mean ± SEM. (D) Representative H&E and reticulin images of BM from Cxcr2f/f;Cre+ KO vs Cxcr2f/f;Cre– WT hMPLW515L mice at timed euthanization 9 weeks after transplant. Representative images of N = 6 mice per arm. (E) Kaplan-Meier survival analysis of Cxcr2f/f;Cre+ KO hMPLW515L mice (N = 16) vs Cxcr2f/f;Cre– WT hMPLW515L mice (N = 13). ∗∗P < .01 (log-rank test). (F) Fold change in serum cytokine levels of IL-6, IL-10, and TNFα of Cxcr2f/f;Cre+ KO compared with Cxcr2f/f;Cre– WT hMPLW515L mice. N = 8 per arm. ∗P < .05. Data shown represent mean ± SEM. (G) Western blot analysis of the alarmins S100a8/a9 from the harvested splenocytes of Cxcr2f/f;Cre+ KO vs Cxcr2f/f;Cre– WT hMPLW515L mice. Original magnification ×20 (D). H&E, hematoxylin and eosin; ns, not significant.
We also examined the effect of Cxcr2 loss on proinflammatory cytokine enumeration and TLR signaling with Luminex bead-based serum cytokine profiling. Notably, Cxcr2–/– hMPLW515L mice displayed reductions in critical TLR-mediated cytokines, specifically IL-6, IL-10, and TNFα in comparison with Cxcr2f/f WT hMPLW515L mice (Figure 3F). In further support of this, and consistent with our in vitro and patient expression data, a western blot analysis of harvested splenocytes from Cxcr2–/– hMPLW515L mice revealed reductions in detectable levels of TLR agonists S100a8/a9 (Figure 3G), further suggesting a role for Cxcr2 in modulating TLR-mediated proinflammatory signaling.
CXCR1/2 inhibition improves hematologic parameters and fibrosis in the hMPLW515L model
We then sought to validate our genetic deletion results by evaluating pharmacologic inhibition of the CXCR1/2 pathway in the hMPLW515L model. Balb/C mice that received transplantation with hMPLW515L-transfected BM cells displaying evidence of disease were assigned to 4 separate treatment arms based on the WBC count: vehicle, ruxolitinib (60 mg/kg orally twice daily), the CXCR1/2 inhibitor reparixin (60 mg/kg subcutaneously twice daily), or combination therapy (supplemental Figure 5A). Consistent with our genetic deletion studies, mice treated with reparixin, either alone or in combination with ruxolitinib, demonstrated improved leukocytosis (mean 251 × 103/μL vehicle vs 80.4 × 103/μL reparixin vs 30.7 × 103/μL combo; P < .05) and platelet counts (mean 3437.8 × 103/μl vehicle vs 1244.2 × 103/μL reparixin vs 742 × 103/μL combo; P < .05) in comparison with vehicle-treated mice (Figure 4A). Minimal toxicity was observed with reparixin, including a separate cohort of WT mice that had received transplants with EV-transfected BM cells and treated with an identical duration and dosing schedule of the drug (supplemental Figure 5B-F). Consistent with our genetic deletion studies, reparixin monotherapy modestly reduced spleen weights (mean, 585.8 vs 490.5 mg; P = .35; supplemental Figure 5G), and, like ruxolitinib, had only minimal effects on GFP+ and Mac1+Gr1+ peripheral blood neutrophil fractions (mean, 75.2% vs 67.9%; P = .3; Figure 4B; supplemental Figure 5H). Notably, however, reparixin monotherapy or combined reparixin/ruxolitinib resulted in a significant reduction in BM MK number, consistent with that reported in our genetic knockout studies (Figure 4C). Most importantly, reparixin monotherapy resulted in a significant reduction in reticulin fibrosis, both in the BM and spleen (Figure 4D-E; supplemental Figure 5I), which was further enhanced when combined with ruxolitinib. Importantly, the reduction in BM and spleen fibrosis with reparixin therapy was also observed in the Gata1low model of MF,60 validating CXCR1/2 as a potential target for the treatment of fibrosis irrespective of MPN driver mutation status.
Figure 4.

Pharmacologic inhibition of CXCR1/2 improves hematologic parameters and reticulin fibrosis in the hMPLW515L-adoptive transfer model of MF. (A) WBC counts (×103/μL), hematocrit levels (%), and PLT (×103/μL) of hMPLW515L-affected mice treated with vehicle, ruxolitinib (60 mg/kg twice daily), the CXCR1/2 inhibitor reparixin (60 mg/kg twice daily), or combination therapy at timed euthanization after 21 days of treatment. N = 6 mice per arm. ∗P < .05; ∗∗P < .01. The t test (unpaired, two-tailed) was used to compare the mean of 2 groups. Data shown represent mean ± SEM. (B) Peripheral blood mutant cell fraction by GFP percentage of treated mice. Data shown represent mean ± SEM. (C) MKs number per high powered field (HPF) observed in BM of hMPLW515L mice in response to treatment. ∗P < .05. Data shown represent mean ± SEM. (D) BM reticulin scores of hMPLW515L-diseased mice treated with either vehicle, ruxolitinib, reparixin, or combination therapy. N = 6 mice per arm. ∗P < .05; ∗∗P < .01. (E) Representative H&E and reticulin images of hMPLW515L-diseased BM treated with ruxolitinib, reparixin, or combination therapy compared with vehicle-treated mice. N = 6 mice per condition. (F) Colony-forming unit (CFU) assay demonstrating total granulocyte-macrophage progenitor (CFU-GM) colony number as a ratio to control of untreated healthy human donor (light blue) vs MF (dark blue) CD34+ cells with exogenous CXCL8 and its response to the second-generation CXCR1/2 antagonist ladarixin (10 μM) in vitro. ∗P < .05; ∗∗P < .01. Representative of duplicate experiments from 5 healthy donor (HD) and 13 individual MF cases. (G) Fold change in detectable CXCL8 levels in conditioned media (CM) elicited by either HD vs MF MKs with or without the addition of reparixin (10 μM). Representative of duplicate experiments from 3 HD and 6 individual MF cases. ∗P < .05. Data shown represent mean ± SD. (H) Total levels of CXCL8 in conditioned media of cultured stromal cells, either alone or together with healthy vs MF MKs with or without the addition of reparixin (10 μM). ∗P < .05. Data shown represent mean ± SD. Representative of duplicate experiments from 3 HD and 3 individual MF cases. Original magnification ×20 (E).
CXCR1/2 inhibition demonstrates efficacy against primary MPN cells in vitro
We also assessed the impact of CXCR1/2 inhibitor therapy on the proliferation and colony-forming capacity of primary MF CD34+ cells in vitro. Consistent with our aforementioned liquid culture experiments, MF CD34+ cells demonstrated enhanced colony-forming capacity in the presence of CXCL8. This effect, however, was abolished with the addition of ladarixin, a second-generation CXCR1/2 inhibitor (Figure 4F). Similar effects were also observed with CD33+ and CD41+ cell output when exposed to CXCR1/2 inhibition (supplemental Figure 6A), consistent with our in vivo studies. Intriguingly, treatment with reparixin also reduced levels of both CXCL8 and vascular endothelial growth factor elaborated by cultured MF MKs, including when cocultured with BM stromal cells (Figure 4G-H; supplemental Figure 6B-C) suggesting downregulation of an autocrine feedback loop. Together, these data confirm an important role for CXCR2 pathway signaling in BM fibrosis development in both human cells and murine systems and validate CXCR1/2 as a potential target for the treatment of BM fibrosis irrespective of the MPN driver mutation status.
Discussion
Aberrant proinflammatory signaling is a hallmark feature of MPNs.18 Treatment with JAK1/2 inhibitors improves symptoms and clinical outcomes in MF, underscoring the role of constitutive JAK/STAT signaling in disease maintenance.61, 62, 63 Although many proinflammatory cytokines are reduced with JAK inhibition, others, including CXCL8, are not,52,61 suggesting that alternative sustained proinflammatory pathways play a critical role in MF progression. Previously, we and others identified an NF-κB–mediated proinflammatory signaling network promoting fibrosis in MF.23,24,52 An improved understanding of how specific cytokines drive fibrosis progression through NF-κB will provide important mechanistic insights and identify potential biomarkers predictive of treatment response.
Here, we used primary samples from patients with MF and murine models of MF to uncover an important role for CXCL8-CXCR2 signaling in fibrotic progression and add to existing literature establishing CXCL8-CXCR2 as a critical node promoting disease evolution across the spectrum of myeloid disorders. CXCL8 is one of several proinflammatory cytokines upregulated by NF-κB and a potent inducer of neutrophil differentiation and mobilization to sites of acute infection.55 Aberrant CXCL8-CXCR2 signaling is implicated in numerous proinflammatory and autoimmune phenomena, including solid organ fibrosis.64,65 In epithelial malignancies, CXCL8 enhances neoangiogenesis and extracellular matrix remodeling to promote a microenvironment conducive for furthered tumor growth and metastatic progression.55 In hematologic malignancies, CXCL8 promotes leukemic stem cell fitness in chronic myelogenous leukemia, myelodysplastic syndrome, and acute myeloid leukemia.66,67 Notably, CXCR1/2 antagonists induce apoptosis of leukemic stem cells in vitro and in vivo.66, 67, 68 Here, we used integrated single-cell transcriptional and cytokine assays and identified strong enrichment in a CXCL8-CXCR2 pathway signature in MF compared with prefibrotic MPN subtypes, PV and ET. Furthermore, we demonstrate that MF CD34+ cells display enhanced cell growth in response to CXCL8, a finding that also correlated with CXCR1/2 surface expression levels. Notably, these findings both contrast and complement the work by Emadi et al, who previously demonstrated an inhibitory role for IL8-CXCR2 signaling in MF-mediated megakaryopoiesis in vitro.54 Importantly, these studies collectively show that MF HSPCs are primed for ongoing aberrant signaling through CXCR2. Whether the differential impact of CXCL8 signaling on MF-mediated megakaryocytes is dose/time-dependent, our studies show that CXCL8 has broad effects across the MF hematopoietic hierarchy and that genetic/pharmacologic inhibition of this pathway in vivo reduced the MF hematopoietic output, myeloid expansion, and BM fibrosis. This suggests that CXCL8-secreting HSPCs might represent a circulating biomarker for BM fibrosis, and prospective studies can delineate if the emergence of a CXCL8-secreting clone over time predicts fibrotic progression in MPN and/or if this can be therapeutically targeted in the clinical context.
Performing chromatin accessibility analysis on a small cohort of patients across MPN subtypes using ATAC-Seq, we also expanded on our previous data in MF mouse models23 and demonstrated the extent to which constitutive JAK/STAT signaling influences chromatin state to promote profibrotic inflammatory programs in MF progression. We identify a strong correlation between CXCR2 cytokine expression and increased TNFα/NF-κB gene accessibility and enrichment in AP-1 or C/EBPE motif signatures in patient samples with high CXCL8 levels. AP-1 was previously found to be associated with fibrosis in different histologic contexts,69 and our findings suggest a more prominent (and perhaps underappreciated) role for FOS/JUN-mediated inflammatory signaling in MF progression, data concordant with emerging studies assessing proinflammatory signaling accessibility changes within specific MPN genotypic contexts.70 That we did not observe major accessibility changes at multiple proinflammatory cytokine loci, including CXCL8, would suggest promoter regions are epigenetically primed for inflammatory signaling in MPN HSCs and that epigenetic changes among enhancer regions, and perhaps cell lineage specificity itself, drive profibrotic inflammatory signaling in MF.
The myeloid differentiation program enriched in CXCL8-high MF is reminiscent of an acute hematopoietic stress response and adds to the expanding literature on the role of TLR signaling in MF.52,71,72 We have previously shown that NF-κB is activated in both mutant and nonmutant MPLW515L-diseased cells in vivo,53 and others have shown that healthy HSCs express TLR receptors that potently upregulate proinflammatory cytokines in response to various pathogen- and damage-associated molecular patterns, including the TLR agonists S100A8/A9, to promote myeloid cell maturation/mobilization in an autocrine and paracrine manner.73,74 In MF, HSPCs are preferentially sensitive to TLR agonists in vitro,52 and S100a8/a9 play key roles in MSC proliferation and myofibroblast differentiation,10,75 suggesting simultaneous roles for TLR signaling on MF HSPCs and their surrounding microenvironment. Together, these data suggest that CXCL8, elicited by mutant HSCs and other myeloid cells, might promote a feed-forward loop of enhanced S100A8/A9 release and TLR activation that, over time, reinforces MSC transcriptional changes that favor fibroblastic proliferation. Importantly, our inhibitor studies suggest that this cycle can be disrupted by pharmacologic inhibition of CXCR1/2, with associated improvements in the MK/neutrophil number, extramedullary hematopoiesis, and fibrosis. Given this, we believe that CXCL8/CXCR2 inhibition represents an attractive therapeutic opportunity to intercept MF progression in MPNs and therefore warrants further study in the clinical context.
Conflict-of-interest disclosure: R.L.L. is on the supervisory board of Qiagen and is a scientific adviser to Imago, Mission Bio, Bakx, Zentalis, Ajax, Auron, Prelude, C4 Therapeutics, and IsoPlexis; has received research support from AbbVie, Constellation, Ajax, Zentalis, and Prelude; has received research support from and consulted for Celgene and Roche and has consulted for Syndax, Incyte, Janssen, Astellas, MorphoSys, and Novartis; and has received honoraria from AstraZeneca and Novartis for invited lectures and from Gilead and Novartis for grant reviews. A.J.D. has served on an advisory committee for Incyte. R.F. is cofounder and scientific adviser of IsoPlexis, Singleron Biotechnologies, and AtlasXomics with significant financial interest. A.R.M. and R.H. received funds from Dompé farmaceutici S.p.A. (Via Campo di Pile, 67100 L’Aquila, Italy). J.C., E.T., T.M., and J.Z. are employees and equity partners of IsoPlexis Corporation. M.K. is currently an employee of Imago BioSciences. R.L.B. has received honoraria from Mission Bio and is a member of the Speakers Bureau for Mission Bio. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the members of the Levine laboratory for their helpful comments and discussion. The authors also thank the members of the Hoffman laboratory and MPN-RC tissue bank as well as ISMMS tissue bank for providing samples from patients with MF. The authors thank Dompé farmaceutici S.p.A for providing the reparixin and ladarixin used in these studies.
Studies supported by the Memorial Sloan Kettering core facilities were supported in part by MSKCC Support Grant/Core Grant P30 CA008748 and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. This work was supported by National Institutes of Health (NIH), National Cancer Institute awards (P01 CA108671) (R.H., R.L.L., and A.R.M.) and (AIRC IG23525) (A.R.M.). R.L.L. was supported by a Leukemia and Lymphoma Society Scholar award. A.J.D. is a William Raveis Charitable Fund Physician-Scientist of the Damon Runyon Cancer Research Foundation (PST-24-19). He also has received funding from the American Association for Cancer Research and the American Association of Clinical Oncology. R.L.L. is also supported by a Leukemia & Lymphoma Society Specialized Center of Research grant. T.M. and J.Z. were supported by NIH Small Business Innovation Research grant R44EB023777.
Authorship
Contribution: A.J.D., D.K., M.L., A.R.M., R.F., R.L.L., and R.H. conceived the project, designed the experiments, and analyzed the data; D.K., Z.C., Y.X., and R.F performed scRNA-Seq and single-cell cytokine sample processing and analysis; M.L. was performed in vitro work with technical assistance from L.X. and N.E.; A.J.D. and M.F. performed hMPLW515L and genetic knockout mouse studies as well as patient sample collection/processing/clinical annotation with technical assistance from Y.P., A. Karzai, Z.Z., K.O., S.M., J.S., A. Krishnan, F.M., F.G., P.V., and J.Y.; J.L.Y., R.L.B., and R.K. performed bulk ATAC-Seq and RNA-Seq computational analyses; R.R., E.M., M.K., M.E.S., J.C., E.T., J.Z., C.H., A.Z., K.C., and T.M. assisted with management of clinical data and specimens; W.X. and G.S. assisted with pathological assessment of specimens; A.J.D., R.L.L., and R.H. contributed to the first and final drafts of the manuscript; and R.L.L., R.H., R.F., and A.R.M. contributed toward funding acquisition.
Footnotes
∗A.J.D., D.K., and M.L. contributed equally to this work.
†R.L.L. and R.H. jointly supervised this work.
Transcriptional and next-generation sequencing data reported in this article have been deposited in the Gene Expression Omnibus database (accession numbers GSE189980 and GSE190383).
Original data are available on request from the corresponding authors, Ross L. Levine (leviner@mskcc.org) and Ronald Hoffman (ronald.hoffman@mssm.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Ross L. Levine, Email: leviner@mskcc.org.
Ronald Hoffman, Email: ronald.hoffman@mssm.edu.
Supplementary Material
References
- 1.Tefferi A, Pardanani A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncol. 2015;1(1):97–105. doi: 10.1001/jamaoncol.2015.89. [DOI] [PubMed] [Google Scholar]
- 2.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 3.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 4.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 5.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 6.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 7.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grinfeld J, Nangalia J, Green AR. Molecular determinants of pathogenesis and clinical phenotype in myeloproliferative neoplasms. Haematologica. 2017;102(1):7–17. doi: 10.3324/haematol.2014.113845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mack M. Inflammation and fibrosis. Matrix Biol. 2018;68-69:106–121. doi: 10.1016/j.matbio.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Leimkuhler NB, Gleitz HFE, Ronghui L, et al. Heterogeneous bone-marrow stromal progenitors drive myelofibrosis via a druggable alarmin axis. Cell Stem Cell. 2021;28(4):637–652.e8. doi: 10.1016/j.stem.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider RK, Mullally A, Dugourd A, et al. Gli1(+) mesenchymal stromal cells are a key driver of bone marrow fibrosis and an important cellular therapeutic target. Cell Stem Cell. 2018;23(2):308–309. doi: 10.1016/j.stem.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Decker M, Martinez-Morentin L, Wang G, et al. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat Cell Biol. 2017;19(6):677–688. doi: 10.1038/ncb3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verstovsek S, Manshouri T, Pilling D, et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J Exp Med. 2016;213(9):1723–1740. doi: 10.1084/jem.20160283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozono Y, Shide K, Kameda T, et al. Neoplastic fibrocytes play an essential role in bone marrow fibrosis in Jak2V617F-induced primary myelofibrosis mice. Leukemia. 2021;35(2):454–467. doi: 10.1038/s41375-020-0880-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yue L, Bartenstein M, Zhao W, et al. Efficacy of ALK5 inhibition in myelofibrosis. JCI Insight. 2017;2(7) doi: 10.1172/jci.insight.90932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ponce CC, de Lourdes Lopes Ferrari Chauffaille M, Ihara SSM, Silva MRR. Increased angiogenesis in primary myelofibrosis: latent transforming growth factor-beta as a possible angiogenic factor. Rev Bras Hematol Hemoter. 2014;36(5):322–328. doi: 10.1016/j.bjhh.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chagraoui H, Komura E, Tulliez M, Giraudier S, Vainchenker W, Wendling F. Prominent role of TGF-beta 1 in thrombopoietin-induced myelofibrosis in mice. Blood. 2002;100(10):3495–3503. doi: 10.1182/blood-2002-04-1133. [DOI] [PubMed] [Google Scholar]
- 18.Fisher DAC, Fowles JS, Zhou A, Oh ST. Inflammatory pathophysiology as a contributor to myeloproliferative neoplasms. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.683401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleischman AG, Aichberger KJ, Luty SB, et al. TNFalpha facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood. 2011;118(24):6392–6398. doi: 10.1182/blood-2011-04-348144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai HY, Brooks SA, Craver BM, et al. Defective negative regulation of Toll-like receptor signaling leads to excessive TNF-alpha in myeloproliferative neoplasm. Blood Adv. 2019;3(2):122–131. doi: 10.1182/bloodadvances.2018026450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285–299. doi: 10.1016/j.stem.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arranz L, Sanchez-Aguilera A, Martin-Perez D, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512(7512):78–81. doi: 10.1038/nature13383. [DOI] [PubMed] [Google Scholar]
- 23.Kleppe M, Koche R, Zou L, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell. 2018;33(4):785–787. doi: 10.1016/j.ccell.2018.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fisher DAC, Malkova O, Engle EK, et al. Mass cytometry analysis reveals hyperactive NF Kappa B signaling in myelofibrosis and secondary acute myeloid leukemia. Leukemia. 2017;31(9):1962–1974. doi: 10.1038/leu.2016.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29(10):1356–1363. doi: 10.1200/JCO.2010.32.9490. [DOI] [PubMed] [Google Scholar]
- 27.Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013;24(2):133–145. doi: 10.1016/j.cytogfr.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Gleitz HFE, Dugourd AJF, Leimkuhler NB, et al. Increased CXCL4 expression in hematopoietic cells links inflammation and progression of bone marrow fibrosis in MPN. Blood. 2020;136(18):2051–2064. doi: 10.1182/blood.2019004095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rai S, Grockowiak E, Hansen N, et al. Inhibition of interleukin-1beta reduces myelofibrosis and osteosclerosis in mice with JAK2-V617F driven myeloproliferative neoplasm. Nat Commun. 2022;13(1):5346. doi: 10.1038/s41467-022-32927-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90(8):1128–1132. [PubMed] [Google Scholar]
- 31.Liu L, Li M, Spangler LC, et al. Functional defect of peripheral neutrophils in mice with induced deletion of CXCR2. Genesis. 2013;51(8):587–595. doi: 10.1002/dvg.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dura B, Choi JY, Zhang K, et al. scFTD-seq: freeze-thaw lysis based, portable approach toward highly distributed single-cell 3' mRNA profiling. Nucleic Acids Res. 2019;47(3):e16. doi: 10.1093/nar/gky1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macosko EZ, Basu A, Satija R, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gierahn TM, Wadsworth MH, 2nd, Hughes TK, et al. Seq-well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods. 2017;14(4):395–398. doi: 10.1038/nmeth.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aran D, Looney AP, Liu L, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stunnenberg HG, International Human Epigenome Consortium. Hirst M. The International Human Epigenome Consortium: a blueprint for scientific collaboration and discovery. Cell. 2016;167(7):1897. doi: 10.1016/j.cell.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 39.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 41.Lu Y, Chen JJ, Mu L, et al. High-throughput secretomic analysis of single cells to assess functional cellular heterogeneity. Anal Chem. 2013;85(4):2548–2556. doi: 10.1021/ac400082e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 44.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doncheva NT, Assenov Y, Domingues FS, Albrecht M. Topological analysis and interactive visualization of biological networks and protein structures. Nat Protoc. 2012;7(4):670–685. doi: 10.1038/nprot.2012.004. [DOI] [PubMed] [Google Scholar]
- 49.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 Axis in cancer and inflammatory diseases. Theranostics. 2017;7(6):1543–1588. doi: 10.7150/thno.15625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fisher DAC, Miner CA, Engle EK, et al. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFkappaB signaling. Leukemia. 2019;33(8):1978–1995. doi: 10.1038/s41375-019-0379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleppe M, Kwak M, Koppikar P, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015;5(3):316–331. doi: 10.1158/2159-8290.CD-14-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emadi S, Clay D, Desterke C, et al. IL-8 and its CXCR1 and CXCR2 receptors participate in the control of megakaryocytic proliferation, differentiation, and ploidy in myeloid metaplasia with myelofibrosis. Blood. 2005;105(2):464–473. doi: 10.1182/blood-2003-12-4415. [DOI] [PubMed] [Google Scholar]
- 55.Waugh DJJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14(21):6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez-Meira A, Buck G, Clark SA, et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol Cell. 2019;73(6):1292–1305.e8. doi: 10.1016/j.molcel.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72(5):847–855. [PubMed] [Google Scholar]
- 58.Modi WS, Yoshimura T. Isolation of novel GRO genes and a phylogenetic analysis of the CXC chemokine subfamily in mammals. Mol Biol Evol. 1999;16(2):180–193. doi: 10.1093/oxfordjournals.molbev.a026101. [DOI] [PubMed] [Google Scholar]
- 59.Singer M, Sansonetti PJ. IL-8 is a key chemokine regulating neutrophil recruitment in a new mouse model of Shigella-induced colitis. J Immunol. 2004;173(6):4197–4206. doi: 10.4049/jimmunol.173.6.4197. [DOI] [PubMed] [Google Scholar]
- 60.Verachi P, Gobbo F, Martelli F, et al. The CXCR1/CXCR2 inhibitor reparixin alters the development of myelofibrosis in the gata1 (low) mice. Front Oncol. 2022;12 doi: 10.3389/fonc.2022.853484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 62.Verstovsek S, Mesa RA, Gotlib J, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10(1):55. doi: 10.1186/s13045-017-0417-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R axis: a double agent in tumor immune resistance. Vaccines (Basel) 2016;4(3):22. doi: 10.3390/vaccines4030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang L, Herrera J, Gilbertsen A, et al. IL-8 mediates idiopathic pulmonary fibrosis mesenchymal progenitor cell fibrogenicity. Am J Physiol Lung Cell Mol Physiol. 2018;314(1):L127–L136. doi: 10.1152/ajplung.00200.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schinke C, Giricz O, Li W, et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood. 2015;125(20):3144–3152. doi: 10.1182/blood-2015-01-621631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Agarwal P, Li H, Choi K, et al. TNF-alpha-induced alterations in stromal progenitors enhance leukemic stem cell growth via CXCR2 signaling. Cell Rep. 2021;36(2) doi: 10.1016/j.celrep.2021.109386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abdul-Aziz AM, Shafat MS, Mehta TK, et al. MIF-induced stromal PKCbeta/IL8 Is essential in human acute myeloid leukemia. Cancer Res. 2017;77(2):303–311. doi: 10.1158/0008-5472.CAN-16-1095. [DOI] [PubMed] [Google Scholar]
- 69.Wernig G, Chen SY, Cui L, et al. Unifying mechanism for different fibrotic diseases. Proc Natl Acad Sci U S A. 2017;114(18):4757–4762. doi: 10.1073/pnas.1621375114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Myers FI RM, Kottapalli S, Prieto T, et al. Integrated single-cell genotyping and chromatin accessibility charts JAK2V617F human hematopoietic differentiation. bioRxiv. Preprint posted online 11 May 2022 doi: 10.1101/2022.05.11.491515. [DOI] [Google Scholar]
- 71.Starczynowski DT, Karsan A. Innate immune signaling in the myelodysplastic syndromes. Hematol Oncol Clin North Am. 2010;24(2):343–359. doi: 10.1016/j.hoc.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 72.Wong WJ, Baltay M, Getz A, et al. Gene expression profiling distinguishes prefibrotic from overtly fibrotic myeloproliferative neoplasms and identifies disease subsets with distinct inflammatory signatures. PLoS One. 2019;14(5) doi: 10.1371/journal.pone.0216810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zambetti NA, Ping Z, Chen S, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19(5):613–627. doi: 10.1016/j.stem.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 74.Zhao JL, Ma C, O'Connell RM, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell. 2014;14(4):445–459. doi: 10.1016/j.stem.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Y, Akada H, Nath D, Hutchison RE, Mohi G. Loss of Ezh2 cooperates with Jak2V617F in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood. 2016;127(26):3410–3423. doi: 10.1182/blood-2015-11-679431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.