


Abstract
A highly integrated monolithic device was developed that automatically carries out a complex series of molecular processes on multiple samples. The device is capable of extracting and concentrating nucleic acids from milliliter aqueous samples and performing microliter chemical amplification, serial enzymatic reactions, metering, mixing and nucleic acid hybridization. The device, which is smaller than a credit card, can manipulate over 10 reagents in more than 60 sequential operations and was tested for the detection of mutations in a 1.6 kb region of the HIV genome from serum samples containing as few as 500 copies of the RNA. The elements in this device are readily linked into complex, flexible and highly parallel analysis networks for high throughput sample preparation or, conversely, for low cost portable DNA analysis instruments in point-of-care medical diagnostics, environmental testing and defensive biological agent detection.
INTRODUCTION
Both DNA microarray-based analysis and gel-based sequencing require the execution of multiple process steps including nucleic acid purification and amplification, enzymatic reactions, molecular separation and signal detection. Manual operations are labor intensive and subject to errors in handling. Recent applications of robotics can improve consistency and reduce labor but call for dedicated personnel and are susceptible to contamination. Previous efforts towards integration and miniaturization have addressed only one or two operations at a time (1–4).
In a more manual format, high density GeneChip® polynucleotide arrays have been used to analyze genetic sequences, among them the yeast genome (5) and the human mitochondrial genome (6), to characterize HIV virus polymorphisms (7), to assay RNA expression profiles (8), to detect cystic fibrosis (9) and to identify Mycobacterium tuberculosis (10) and cancer-related mutations (11). As with most genetic analysis methods, a series of preparative reactions and processes, including enzymatic amplification and modification of a purified sample, must be carried out before hybridization analysis on the polynucleotide array.
We have developed a miniature integrated device capable of performing serial and parallel multistep molecular operations, including nucleic acid hybridization to GeneChip® oligonucleotide microarrays (Fig. 1 and Table 1). The elements in this device can be easily reconfigured so that a wide range of process steps and parallel reactions with complex protocols can be integrated. Our initial work has used nucleic acid arrays for detection, but we recognize that this device could be adapted to electrophoresis, fluorescence and other analysis methods.
Figure 1.
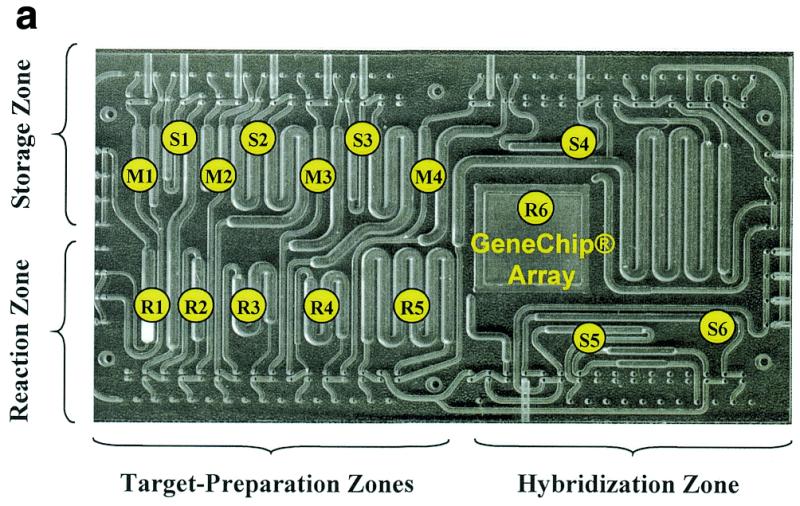
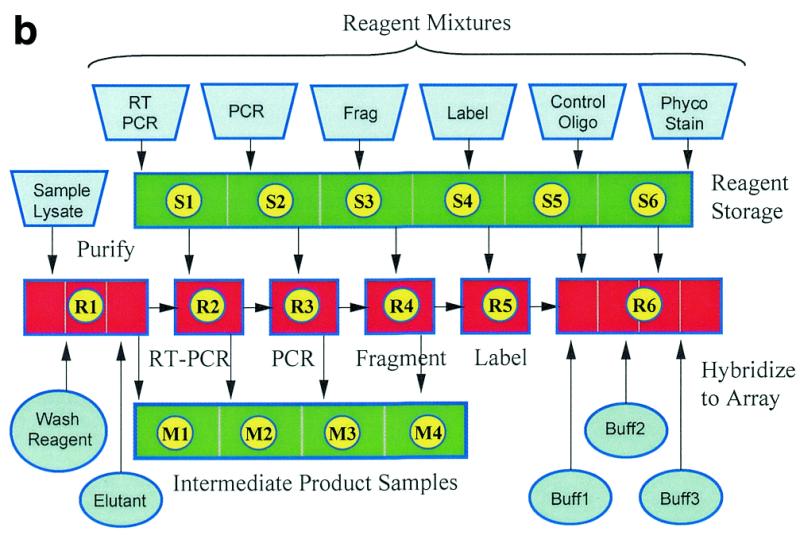
(a) An integrated device that automatically performs a multistep HIV genotyping assay. At the beginning of the process reagent mixtures are loaded into storage chambers S1–S6. The device automatically carries out RNA purification from a serum lysate (extraction), RT–PCR, nested PCR, DNase fragmentation and dephosphorylation, terminal transferase labeling, dilution and hybridization, washing, phycoerythrin staining and washing (in chambers R1, R2, R3, R4, R5 and R6, respectively). Intermediate product samples are stored in chambers M1–M4 for later analysis. Three independently controlled temperature zones are indicated as Storage Zone, Reaction Zone and Hybridization Zone on this polycarbonate device with dimensions of 8 × 40 × 70 mm. (b) Process flow schematic of the automated HIV assay. The introduction of bulk reagents (washes and buffers) from externally connected vials is controlled using cartridge-based valves. Frag refers to the DNase fragmentation and dephosphorylation reaction, Phyco Stain to the streptavidin–phycoerythrin conjugate and Buff1, Buff2, and Buff3 to buffers for hybridization, washing and staining, respectively.
Table 1. The integrated device technology has been configured to automatically perform reactions and process steps for several GeneChip array applications.

Multiple samples are prepared simultaneously on one device for the P53 and CYP450 genotyping and gene expression analysis applications.
MATERIALS AND METHODS
Cartridge fabrication, assembly and operation
The assembled polycarbonate cartridge measures 8 × 40 × 70 mm and has channels and chambers that range from 0.25 to 1.5 mm in depth and 0.25 to 10 mm in length, with 0.4 mm holes for the pneumatic controls. The two polycarbonate parts were fabricated using conventional computer-controlled machining. The valve material consists of a laminate with a 0.01 mm thick mylar layer sandwiched between two 0.25 mm thick silicone layers. The parts can be held together using ultrasonic welding or adhesives; in this study small screws and a clamping fixture were used. Hydrophobic vent materials include, for example, Versapore 200 (Gelman Sciences, Ann Arbor, MI) and Durapel (Millipore, Bedford, MA). The extraction chamber was created by press fitting ~2 mg of no. 903 cellulose paper (Schleicher & Schuell, Keene, NH) into a 5 µl chamber. We found that a 0.01 mm thickness of parylene-C (Specialty Coatings, Ontario, CA) will create reproducible enzymatic performance, although satisfactory results have also been obtained using uncoated, injection molded polycarbonate. All results presented in this paper were generated using parylene-coated devices. Fluids are moved between chambers using an air pressure of 24 kPa. Diaphragm valves are closed and opened using 345 kPa and a vacuum, respectively.
Reagents and reaction conditions
Enzymatic reactions include reverse transcription, PCR, DNase digestion and terminal transferase labeling. The reaction conditions were modified from the protocols detailed in Affymetrix GeneChip HIV PRT Plus Target Preparation Instructions for Use, Analyte Specific Reagent.
Stock reagent mixtures. All reagents were from the HIV PRT Plus reagent kit of Affymetrix unless otherwise specified. Stock reagent mixtures were kept at –20°C until needed. Reverse transcription reagent mixture (RTM) consisted of 50 µl of 5× first strand cDNA buffer (Life Technology), 20 µl of 0.1 mM DTT (Life Technology), 4 µl of 25 mM dNTP (Pharmcia), 8 µl of RT primer, 8 µl of nested primer and 20 µl of water. Reverse transcription enzyme mixture (RTE) consisted of 4 µl of RNAguard (1 U/µl; Pharmacia), 3 µl of SuperScript II reverse transcriptase (200 U/µl; Life Technology) and 3 µl of rTth DNA polymerase (2 U/µl; Perkin-Elmer). PCR reagent mixture (PCRM) was composed of 127 µl of 3.3× rTth buffer (Perkin-Elmer), 3.36 µl of 25 mM dNTP (Pharmacia), 42 µl of PCR primer, 30 µl of 25 mM MgCl2 and 122 µl of water. Fragmentation reagent mixture (FrgM) comprised 49 µl of 10× fragmentation buffer, 24.5 µl of 20× MnCl2 solution, 12 µl of 40 µg/µl BSA (Molecular Biology Grade; Fisher Scientific) and 54.5 µl of water. Terminal deoxynucleotide transferase reaction reagent mixture (TdTM) was prepared from 82 µl of 5× TdT buffer (Promega) and 24.5 µl of 25 mM CoCl2 (Boehringer Mannheim), 4 µl of biotin-ddATP (NEN), 5 µl of 40 µg/ml BSA and 7.5 µl of water. The oligo buffer mixture (Om) consists of 20 µl of oligo B1 (Affymetix) and 200 µl of 6× SSPET (1× SSPE = 150 mM NaCl, 10 mM NaH2PO4, 1 mM EDTA; T = 0.005% Triton X-100) and the staining reagent mixture (Sm) consists of 80 µl of 6× SSPET and 150 µl of 40 µg/ml BSA.
Working reagent mixtures. All working reagents were prepared on ice and loaded onto the integrated device prior to automatic operation. They are: (i) 12 µl of R reagent for reverse transcription (11 µl of RTM and 1 µl of RTE); (ii) 33.2 µl of P reagent for PCR (32.4 µl of PCRM and 0.8 µl of rTth DNA polymerase); (iii) 19 µl of F reagent for DNase fragmenation [14 µl of FrgM, 4 µl of CIAP (Life Technology) and 1 µl of diluted DNase, made by mixing 0.5 µl of the DNase (1.7 µg/µl; Affymetrix) with 10 µl of CIAP dilution buffer (Life Technology)]; (iv) 13.5 µl of T reagent for the TdT reaction [12.3 µl of TdTM, 1.2 µl of TdT (20 µg/µl; Promega)]; (v) 22 µl of Om for hybridization control; (vi) 25 µl of S reagent for staining [23 µl of Sm and 1.25 µl of 1 µg/µl streptavidin–phycoerythrin (Molecular Probes)].
Reactions. After reagent loading and sample purification, reverse transcription starts by mixing 10 µl of RNA sample with 9 µl of R reagent and incubating the mixture at 38, 42 and 45°C, each for 20 min. The cDNA is then typically amplified by 25 cycles of thermal cycling: 95°C for 20 s, 58°C for 40 s and 72°C for 90 s. About 8 µl of the product is mixed with 30 µl of P reagent and nested PCR is performed for 45 cycles using 93.5°C for 20 s, 63°C for 40 s and 72°C for 90 s. About 25 µl of the PCR product is mixed with 16 µl of D reagent to fragment the DNA and degrade dNTP at 37°C for 15 min. The enzymes are then inactivated by heating to 95°C for 10 min and then returning the temperature to 25°C. About 22 µl of fragmented DNA is end-labeled after being mixed with 10 µl T reagent and incubation at 37°C for 60 min, followed by heating at 95°C for 5 min. For hybridization, about 15 µl of the labeled DNA is then combined with 10 µl of O reagent and 70 µl of 6× SSPET.
Hybridization and staining
Hybridization is performed for 20 min at 37°C. After the completion of hybridization, the sample is removed and the chip is washed twice with buffer A (2× SSPE, 0.005% Triton X-100) at 23°C. The chip is then stained after mixing 20 µl of S reagent with 70 µl of 5× SSPET (10 min, 25°C). The program ends after washing the chip twice with buffer A and six times with buffer B (6× SSPE, 0.005% Triton X-100), both at 25°C. Other conditions are detailed in the Instructions for use of Affymetrix GeneChip HIV PRT Plus product information. Air pressure is applied to one side of the hybridization chamber, forcing the target into a secondary chamber containing trapped gas that in turn becomes pressurized. The target liquid returns to the hybridization chamber when the air pressure is released. An air pressure of 41 kPa is used to move liquid out of the hybridization chamber and to pressurize vials for buffer introduction. In the current design the hybridization chamber requires 100 µl of reagent.
Samples and extraction
The DNA samples were prepared by suspending in 100 µl of fetal bovine serum (Life Technologies) partial HIV clones from RT–PCR products of clinical samples BCP98-24 (clone 24) and BCP98-39 (clone 39) obtained from BioClinical Partners Inc. These DNA clones were sequenced on the sense and antisense strands with an ABI sequencer. The serum sample consisted of an RNA transcript suspended in 100 µl of fetal bovine serum (Life Technologies), where the RNA was the in vitro transcribed product of a DNA clone generated from HIV-1 subtype B isolate HXB2.
Details for RNA and DNA extraction. Reagent solutions include: (i) denaturant solution containing 4 M guanidine isothiocyanate, 50 mM Tris–HCl (pH 7.5), 25 mM EDTA (Life Technologies), supplemented with 2-mercaptoethanol (Sigma) to 1% (v/v) before use; (ii) cleaning solution consisting of denaturant diluted 1:1 with water; (iii) washing solution containing 50% ethanol, 50 mM NaCl, 10 mM Tris–HCl, pH 7.0, and 0.001% Triton X-100; (iv) elution solution containing RNase-free water. The procedure consists of the following. (i) To 100 µl of serum sample add 500 µl of denaturant. For RNA recovery testing, template RNA and 1 µg yeast tRNA was added to the denaturant-containing mixture. (ii) Add 400 µl of ethanol to the mixture and vortex the mixture for 30 s. (iii) Clean the extraction module with 500 µl of cleaning solution. (iv) Rinse the extraction module with 500 µl of washing solution. (v) Load the 1 ml sample into the extraction module. (vi) Wash the extraction module with 500 µl of washing solution. (vii) Dry the matrix in the extraction module by passing clean air for 100 s. (viii) Hydrate the matrix with 25 µl of water and increase the chamber temperature to 70°C for 1 min. (ix) Recover the sample.
Gel-based analysis
The PCR products and fragmentation products were analyzed with 1% (SeaKem Le agarose; FMC) and 4% (NuSieve 3:1 agarose; FMC) agarose gels, respectively. Agarose gels were prepared in 0.5× TBE (1× TBE = 100 mM Tris, 90 mM boric acid and 1 mM EDTA) plus ethidium bromide at 0.5 µg/ml. Samples were separated in the gels by electrophoresis for 30 min at 130 V. DNA fragment sizes were determined by comparison to 500, 50 and 10 bp markers, all obtained from Life Technologies. The volumes of PCR and fragmentation products loaded into each lane were 1 and 4 µl, respectively.
RESULTS AND DISCUSSION
Integration of multiple enzymatic reactions with reagent handling
The formidable task of combining these reactions together in a single device requires precise manipulation of reagents and control of multiple temperature regions in close proximity. These complexities have limited reports of integrated reactions to a single reaction combined with analysis (3,4). To achieve full integration we have employed batch reactions, sensorless reagent positioning and careful choice of materials. Our newly developed device effectively combines an entire genetic assay into a single package by providing: (i) precise reagent handling, including metering, moving, positioning and mixing; (ii) thermal isolation between cooled enzymatic storage and heated reactions on the same device; (iii) extraction and purification of nucleic acids from a serum sample; (iv) hybridization, washing and staining on a DNA array (12,13). Fluid barriers are used in conjunction with pneumatically controlled diaphragm valves and gas pressure to position and manipulate reagents. Each barrier is a porous hydrophobic membrane that allows the passage of gas but not of aqueous liquid. A reagent bolus guided into the chamber will stop moving when it reaches the fluid barrier, or hydrophobic membrane. This approach allows us to position and meter reagents without using sensors or feedback. Fluid positioning using hydrophobic barriers was reported previously (14) and has been adapted by other workers using microfabricated structures (4,15). These functions are achieved with a disposable cartridge, or biochemical processing unit (BPU, Fig. 1), that is held between a thermal element and an array of pneumatic ports during operation. A computer is used to program up to 144 different gas pressure lines and up to four different thermal zones. The integrated device (BPU) is assembled from five polymeric parts that are formed using either computer-controlled machining or injection molding.
Another critical operation within the device is removal of the air separating each reagent bolus. We have accomplished reagent joining or linking using either porous hydrophobic vents alone or in combination with valves (12,14). Once combined into a single bolus, the reagents must be mixed to achieve homogeneity. Mixing fluids in small channels with laminar flow is generally considered difficult but propelling a bolus or packet of fluid through a channel creates an advantageous recirculation pattern along the axis of the channel. This is a consequence of non-slip boundary conditions superimposed with the requirement that the leading and trailing menisci move as one. (This flow pattern has been described previously in the context of larger fluid handling systems; see for example 16.) This recirculation effect facilitates effective blending of elongated aqueous reagent packets.
In addition to guiding each reagent bolus to the appropriate chamber, the diaphragm valves physically prevent reagent migration or evaporation during high temperature reactions such as PCR, enzyme inactivation and DNA denaturation, where temperatures of 95°C are common. These reaction networks generally require independent control of multiple temperature zones so that reactions can be effected without damaging temperature-sensitive reagents and materials stored on the same substrate. Thermally isolated systems are more readily fashioned from polymeric substrates, because they are five times more insulating than glass and 800 times more insulating than silicon [the thermal conductivities of polycarbonate, glass and silicon are 0.16, 0.95, 124 W/(m K), respectively]. Silicon and glass substrates are often used as substrates for miniature laboratories and thermal requirements are managed by either operating isothermally and avoiding multiple steps or limiting protocols to low temperature reactions. We have selected polycarbonate as the substrate because it provides excellent thermal isolation and can be manufactured inexpensively as a low cost disposable. Reaction temperatures are controlled by pressing thermal elements against the thin wall of a polycarbonate cartridge. Thermoelectric coolers (Peltier junctions) are used to both heat and cool. For the application shown in Figure 1 there are three thermoelectric coolers that control the reaction, storage and hybridization zones indicated.
Enzymatic reactions such as PCR can be extremely sensitive to material surfaces. As reaction chambers are miniaturized, the surface-to-volume ratio increases. Previous workers have focused on surface coatings to improve reaction performance (17). We have successfully performed many enzymatic reactions in polymeric cartridge reactors with 5–30 µl volumes (Table 2) in which some reformulation of reagents combined with a parylene surface coating proved successful. Enzyme concentrations were generally increased 1.5- to 3-fold to produce yields equivalent to the tube-based reactions. The DNA digestion reaction is sensitive to enzymatic activity, so BSA was used to further stabilize this enzyme. The stability of reaction cocktails was also tested for storage on the automated platform. Successful reactions were achieved with storage times of up to 7 h at 4°C.
Table 2. Reagent compositions are slightly altered for consistent performance in the integrated device.
| Reaction | Enzymes | Modifications for cartridge |
|---|---|---|
| PCR (multiplex from genomic DNA) | Taq Gold | 1.5× Taq concentration |
| PCR using rTth | rTth polymerase | 1.5× rTth concentration |
| Reverse transcription | rTth polymerase | None |
| Second strand cDNA | DNA polymerase I | None |
| T4 ligase | ||
| RNase H | ||
| Taq polymerase | ||
| In vitro transcription | T7 RNA polymerase or T3 RNA polymerase | None |
| Labeling using terminal transferase | TdT | 2–3× TdT concentration |
| Fragmentation and dephosphorylation | DNase | 2× DNase concentration |
| CIAP | 4 µg/µl BSA added |
These enzymatic reactions were performed successfully in microliter volumes. In these experiments chambers were coated with parylene.
Nucleic acid purification and concentration
We have also developed a nucleic acid extraction module using a solid phase purification method (18) and integrated it into our BPU device. Extraction generally includes releasing the nucleic acid from the sample (lysis), removing contaminants (purification) and recovering the nucleic acid. To accommodate the wide variety of sample types that might be generally encountered (e.g. blood, tissue, etc.), we require a homogeneous lysate formed by mixing the sample with a chaotropic salt (4 M guanidine isothiocyanate). We have focused initially on purifying DNA and RNA from serum samples. On the integrated device, the extraction chamber is packed with cellulose. The 1 ml lysate mixture is drawn by vacuum through the extraction chamber, which is then automatically washed with an ethanol:water wash solution. After a brief air dry, the captured nucleic acid is eluted from the cellulose into a bolus of water. Purification performance was evaluated using serum samples spiked with different copy numbers (0, 50, 100, 300 and 500 copies) of an RNA transcript that contained a partial HIV sequence. We obtained good amplification using serum samples spiked with 300 RNA copies and automatically processed from extraction through nested PCR in our device (as a control, direct spikes into a bench top amplification protocol could detect as few as 30 copies).
Integration of DNA array processing
In addition to extraction, we have integrated array-based hybridization in our BPU device. The hybridization process is generally comprised of the hybridization reaction, washing, label binding (staining) and washing. Each of these processes must be carried out under conditions of controlled temperature and convection. The first wash step removes material that is not specifically bound to the array and the stain step is used to react a fluorescent label with a hapten tag placed on the sample target during amplification or by end-labeling. Fresh buffers and wash solutions are automatically introduced into the cartridge from external pressurized vials connected to valves on the disposable cartridge. Uniform convection during hybridization and washing steps is produced by filling and draining the sample target from the hybridization chamber, repeatedly sweeping an air–liquid meniscus across the surface of the GeneChip® array. The entire hybridization, washing, staining and washing process is carried out by a series of chamber filling, meniscus sweeping and waste discharge steps.
System performance
These hybridization, enzymatic and extraction processes are effectively integrated together on a single device using our fluid manipulation methods. For example, the cartridge in Figure 1a carries out purification through hybridization automatically for the GeneChip® HIV PRT Plus assay. The process steps depicted in Figure 1b are completely integrated together and automated. The user injects the reagent mixtures for RT–PCR, nested PCR, labeling and fragmentation, labeled oligonucleotide and streptavidin-conjugated phycoerythrin into the disposable cartridge using a hand pipette. The 1 ml lysate (serum sample mixed with chaotropic salt and ethanol) is then drawn through the extraction chamber. After reagent loading, the system is operated automatically by computer. During each reaction, the storage and hybridization regions are held at 4°C. After each reaction, a portion of the product is diverted to a sampling chamber for later analysis and the remainder is combined with the next reagent cocktail.
Serum samples with HIV virus were simulated by mixing the in vitro transcribed 1.6 kb RNA product from a well-characterized HIV cDNA clone with fetal bovine serum. Typical reaction results are shown in Figure 3, where an agarose gel is used to analyze the BPU-generated PCR and fragmentation products. Analysis of the GeneChip image shown in Figure 2 yielded the correct sequence as determined by independent sequencing with an accuracy of 99.7%. (In seven test reactions, miscalls plus no-calls were 1, 1, 1, 5, 5, 6 and 27. A median of five errors in 1.6 kb yields 99.7%. The reaction with 27 errors had a low GeneChip fluorescence intensity resulting from a low PCR yield. We anticipate that optimization of this system would result in improved reproducibility and performance.) To demonstrate that our system is not only sensitive but also accurate and versatile we injected RNA-free lysate mixtures containing DNA with known HIV sequence. (Bench tests of the purification stage using carrier DNA indicated a yield of ~50%.) The identical protocol was carried out skipping the RT–PCR step (through a software modification). The final sequence data match the original DNA clone 1.6 kb sequence with an accuracy of better than 98.5% (Fig. 2).
Figure 3.
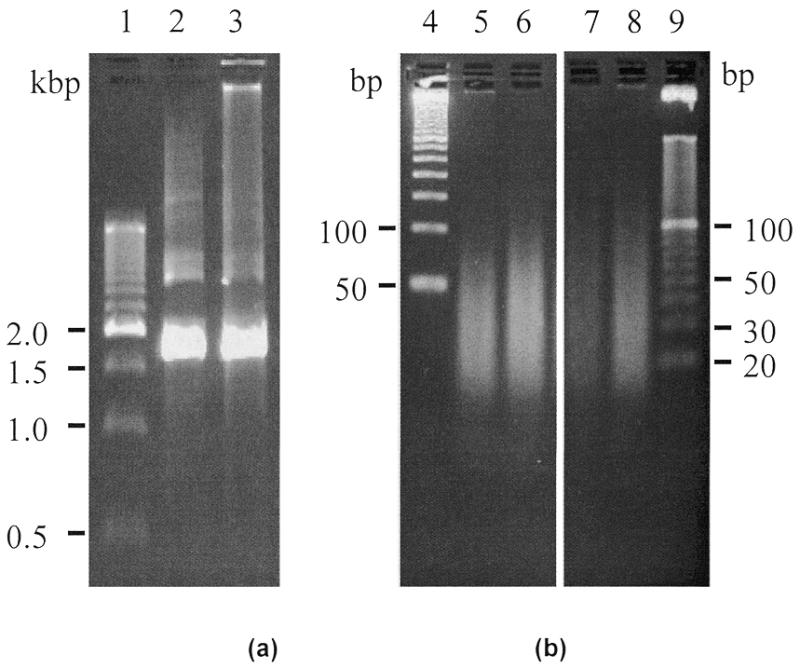
Automated target preparation for HIV assay. (a) Fluorescence image of an agarose gel showing products of nested PCR after RT–PCR, from reactions carried out in the tube control (lane 2) and on the integrated device (lane 3). (b) Fluorescence image of an agarose gel showing fragmented PCR products from endonuclease reactions carried out in the tube control (lanes 5 and 6) and on the integrated device (lanes 7 and 8). Lanes 1, 4 and 9 are 500, 50 and 10 bp markers, respectively. Note that the images in (b) are from the same gel with the intervening lanes cropped to save space.
Figure 2.
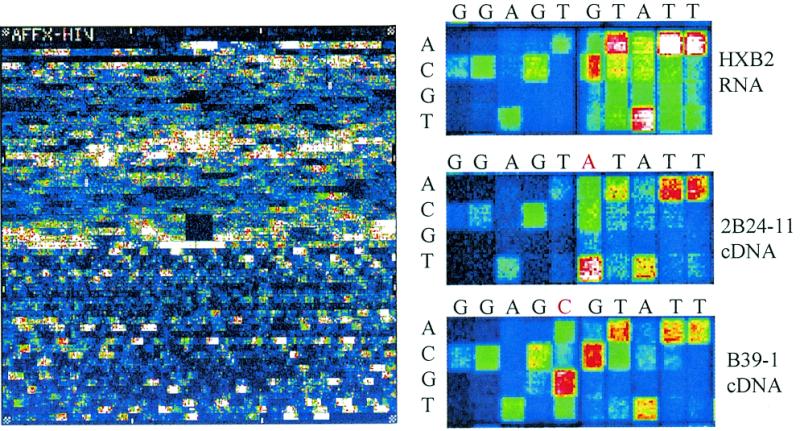
Hybridization images of HIV PRT Plus® GeneChip® arrays fully processed on an integrated device. (a) Full image (HXB2 1.6 kb transcript). (b) Focus region illustrating correctly called mutations with samples derived from three different strains of HIV (HXB2, 2B24-11 and B39-1). The sense sequence is indicated above the images.
This device has also been configured for other applications, including multistep, multisample target preparation for polymorphism screening of the human P53 tumor suppressor gene, human cytochrome p450 genes and quantitative eukaryotic gene expression (Table 1). In each case the results were comparable to the bench top control experiments (unpublished material; 12,13). These experiments demonstrate the flexibility and broad applicability of this automated platform.
The device described here represents an extremely versatile technology that promises to provide low cost automation for multistep sample preparation through entire assays. [The cartridge consists of injection molded parts that are inexpensive to manufacture and the instrument is not complex. Initial estimates of the cartridge and instrument costs are less than $10 and $20 000, respectively (excluding the GeneChip array and scanner).] While these demonstrations have focused on preparation and analysis using GeneChip nucleic acid arrays, the methods and structures described in this work can be generally applied to other types of genetic analysis, including those employing electrophoretic separations such as gel-based sequencing. With this technology, scientists and technicians will be able to accurately perform protocols with increasing levels of complexity and throughput while consuming less sample and reagents.
Acknowledgments
ACKNOWLEDGEMENTS
The advice and assistance of scientists at Affymetrix Inc. were invaluable in performing these experiments. The authors would like to particularly thank Kai Wu and Qing Yang for developing and validating the benchtop HIV assay and for providing the sequenced cDNA and RNA samples, Tom Ryder, Clotilde Perbost and Weiwei Liu for evaluating the CYP450 results, Sami Hussain and Hajime Matsuzake for evaluating the P53 results and John Oliner, Janet Warrington, Robert Lipshutz, Tom Gingeras, Dick Watts and Lubert Stryer for their assistance in the preparation of this manuscript. This work was supported by NIST cooperative agreement 70NANB5H031.
REFERENCES
- 1.Harrison D.J., Fluri,K., Seiler,K., Fan,Z., Effenhauser,C.S. and Manz,A. (1993) Science, 261, 895–897. [DOI] [PubMed] [Google Scholar]
- 2.Kopp M.U., deMello,A.J. and Manz,A. (1998) Science, 280, 1046–1048. [DOI] [PubMed] [Google Scholar]
- 3.Wooley A.T., Hadley,D., Landre,P., deMello,A.J., Mathies,R.A. and Northrup,M.A. (1996) Anal. Chem., 68, 4081–4086. [DOI] [PubMed] [Google Scholar]
- 4.Burns M.A., Johnson,B.N., Brahmasandra,S.N., Handique,K., Webster,J.R., Krishnan,M., Sammarco,T.S., Man,P.M., Jones,D., Heldsinger,D., Mastrangelo,C.H. and Burke,D.T. (1998) Science, 282, 484–487. [DOI] [PubMed] [Google Scholar]
- 5.Winzeler E.A., Richards,D.R., Conway,A.R., Goldstein,A.L., Kalman,S., McCullough,M.J., McCusker,J.H., Stevens,D.A., Wodicka,L., Lockhart,D.J. and Davis,R.W. (1998) Science, 281, 1194–1197. [DOI] [PubMed] [Google Scholar]
- 6.Chee M., Yang,R., Hubbell,E., Berno,A., Huang,X.C., Stern,D., Winkler,J., Lockhart,D.J., Morris,M.S. and Fodor,S.A. (1996) Science, 274, 610–614. [DOI] [PubMed] [Google Scholar]
- 7.Kozal M., Shah,N., Shen,N., Fucini,R., Yang,R., Merigan,T., Richman,D.D., Morris,M.S., Hubbell,E., Chee,M. and Gingeras,T.R. (1996) Nature Med., 7, 753–759. [DOI] [PubMed] [Google Scholar]
- 8.Gray N.S., Wodicka,L., Thunnissen,A.W., Norman,T.C., Kwon,S., Espinoza,F.H., Morgan,D.O., Barnes,G., LeClerc,S., Meijer,L., Kim,S., Lockhart,D. and Schultz,P. (1998) Science, 281, 533–537. [DOI] [PubMed] [Google Scholar]
- 9.Cronin M., Miyada,C., Fucini,R., Kim,S., Masino,R. and Wespi,R. (1996) Biologicals, 24, 209–209. [DOI] [PubMed] [Google Scholar]
- 10.Troesch A., Nguyen,H., Miyada,C.G., Desvarenne,S., Gingeras,T.R., Kaplan,P.M., Cros,P. and Mabilat,C. (1999) J. Clin. Microbiol., 37, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hacia J.G., Brody,L.C., Chee,M.S., Fodor,S.P.A. and Collins,F.S. (1996) Nature Genet., 14, 441–447. [DOI] [PubMed] [Google Scholar]
- 12.Anderson R.C., Bogdan,G.J., Puski,A. and Su,X. (1998) In Solid-State Sensor and Actuator Workshop, Hilton Head Island, SC, pp. 7–10.
- 13.Anderson R.C., Bogdan,G.J., Puski,A. and Su,X. (1998) In Harrison,D.J. and van den Berg,A. (eds), Micro Total Analysis Systems ’98. Kluwer, Amsterdam, The Netherlands, pp. 11–16.
- 14.Anderson R.C., Bogdan,G.J., Barniv,Z., Dawes,T., Winkler,J. and Roy,K. (1997) In Transducers 97: International Conference on Solid-State Sensors and Actuators, Chicago, IL, Vol. 1, pp. 477–480.
- 15.Hosokawa K., Fujii,T. and Endo,I. (1998) In Harrison,D.J. and van den Berg,A. (eds), Micro Total Analysis Systems ’98. Kluwer, Amsterdam, The Netherlands, pp. 307–310.
- 16.Duda J.L. and Vrentas,J.S. (1971) J. Fluid Mech., 45, 247. [Google Scholar]
- 17.Shoffner M.A., Cheng,J., Hvichia,G.E., Kricka,L.J. and Wilding,P. (1996) Nucleic Acids Res., 24, 375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su X. and Comeau,A.M. (1999) Anal. Biochem., 267, 415–418. [DOI] [PubMed] [Google Scholar]