

Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) is a multidrug-resistant (MDR) bacterial pathogen of acute clinical significance. Resistance to current standard-of-care antibiotics, such as vancomycin and linezolid, among nosocomial and community-acquired MRSA clinical isolates is on the rise. This threat to global public health highlights the need to develop new antibiotics for the treatment of MRSA infections. Here, we describe a new benzamide FtsZ inhibitor (TXH9179) with superior antistaphylococcal activity relative to earlier-generation benzamides like PC190723 and TXA707. TXH9179 was found to be 4-fold more potent than TXA707 against a library of 55 methicillin-sensitive S. aureus (MSSA) and MRSA clinical isolates, including MRSA isolates resistant to vancomycin and linezolid. TXH9179 was also associated with a lower frequency of resistance relative to TXA707 in all but one of the MSSA and MRSA isolates examined, with the observed resistance being due to mutations in the ftsZ gene. TXH9179 induced changes in MRSA cell morphology, cell division, and FtsZ localization are fully consistent with its actions as a FtsZ inhibitor. Crystallographic studies demonstrate the direct interaction of TXH9179 with S. aureus FtsZ (SaFtsZ), while delineating the key molecular contacts that drive complex formation. TXH9179 was not associated with any mammalian cytotoxicity, even at a concentration 10-fold greater than that producing antistaphylococcal activity. In serum, the carboxamide prodrug of TXH9179 (TXH1033) is rapidly hydrolyzed to TXH9179 by serum acetylcholinesterases. Significantly, both intravenously and orally administered TXH1033 exhibited enhanced in vivo efficacy relative to the carboxamide prodrug of TXA707 (TXA709) in treating a mouse model of systemic (peritonitis) MRSA infection. Viewed as a whole, our results highlight TXH9179 as a promising new benzamide FtsZ inhibitor worthy of further development.
Graphical Abstract

INTRODUCTION
The persistent use of antibiotics over the past several decades has led to the development and spread of multidrug resistance among bacterial pathogens of acute clinical importance.1,2 According to the World Health Organization (WHO), resistant infections are associated with longer hospital stays and increased patient mortality.3 These infections also have a significant and direct financial impact on healthcare systems, with estimated costs of $20 billion a year in the US alone.4−6 Highlighting the magnitude of the antibiotic resistance crisis, the Centers for Disease Control and Prevention (CDC) have recently reported that over 2.8 million antibiotic-resistant infections occur each year in the US, with these infections resulting in more than 35,000 deaths annually.7 The CDC has also identified numerous bacterial pathogens as particularly significant threats to global public health, with methicillinresistant Staphylococcus aureus (MRSA) being one of those pathogens identified.7
Every year in the US, 320,000 patients are hospitalized with MRSA infections, resulting in over 10,000 deaths and incurring healthcare costs of $1.7 billion.7 Globally, over 750,000 deaths are associated with resistant S. aureus infections each year.8 In addition to being resistant to most β-lactam antibiotics, MRSA often exhibits cross-resistance to other classes of antibiotics.9−14 Even more alarming, MRSA isolates in both nosocomial and community settings are becoming increasingly resistant to current standard-of-care antibiotics, including vancomycin and linezolid.12 Combating increasingly multi- drug-resistant (MDR) MRSA infections requires the development of new antibiotics with novel antibacterial targets.
FtsZ is a highly conserved protein that is essential for bacterial cell division.15 In the early stages of cell division, FtsZ forms a structure at midcell termed the Z-ring, with this process being dependent on GTP.16,17 The Z-ring serves as a scaffold for the recruitment of other proteins important for cell division, including the penicillin binding proteins (PBPs).18,19 FtsZ is a highly druggable target, with several classes of inhibitors having been identified.15 Benzamide-based agents are a particularly promising class of inhibitors20−32 that target a specific interdomain cleft in S. aureus FtsZ (SaFtsZ).22,28,33 PC190723 (Figure 1) was one of the first lead benzamide compounds identified with potent antistaphylococcal activity.20−22,30−32,34 However, poor formulation properties and metabolic instability of the Cl functionality at the 6-position of the thiazolopyridine moiety have limited the potential clinical utility of PC190723.20,26,31 With the aim of improving the drug-like properties of benzamide-based FtsZ inhibitors, we have developed a prodrug platform for enhancing formulation and in vivo administration.26,31,32 We have also identified a second-generation FtsZ-targeting benzamide (TXA707) containing a 6-CF3 functionality (Figure 1) resistant to metabolic attack while also retaining potent antistaphylococcal activity.26 The carboxamide prodrug of TXA707 (TXA709) exhibited improved pharmacokinetics, oral bioavailability, and in vivo efficacy against MRSA compared to the corresponding prodrug of PC190723 (TXY541).26,31 TXA709 has recently completed Phase I clinical trials and has been highlighted by the WHO as one of only two novel antibiotics in the pipeline that has met all the WHO criteria for innovation.35
Figure 1.

Chemical structures of the benzamide FtsZ inhibitors PC190723, TXA707, and TXH9179 as well as their corresponding carboxamide prodrugs (TXY541, TXA709, and TXH1033, respectively).
Toward the goal of further enhancing antistaphylococcal potency and in vivo efficacy against MRSA, we have identified a next-generation benzamide-based FtsZ inhibitor (TXH9179) containing a 6-acetylene functionality (Figure 1). In this study, we show that TXH9179 is associated with superior bactericidal potency compared to TXA707 against a broad range of clinical isolates of methicillin-sensitive S. aureus (MSSA) and MRSA, including isolates resistant to vancomycin and linezolid. We present genetic, crystallographic, and microscopy results validating FtsZ as the antistaphylococcal target of TXH9179. Significantly, among most of the MSSA and MRSA isolates examined, the frequency of resistance (FOR) to TXH9179 was reduced relative to TXA707. We demonstrate that TXH9179 is not toxic to mammalian cells even at concentrations exceeding that associated with antistaphylococcal activity. We also show that the carboxamide prodrug of TXH9179 (TXH1033) is rapidly converted to TXH9179 by acetylcholinesterases present in serum and exhibits superior in vivo efficacy compared to TXA709 in a mouse model of MRSA infection. Viewed as a whole, our results highlight TXH9179 as a new benzamide FtsZ inhibitor worthy of further preclinical development.
RESULTS
Antibacterial Activity of TXH9179 against Clinical Isolates of MSSA, MRSA, VISA, VRSA, and LRSA.
We examined the antibacterial potency of TXH9179 against a total of 55 clinical S. aureus isolates, including 10 MSSA isolates, 13 MRSA-USA100 isolates, 11 MRSA-USA300 isolates, 6 vancomycin-intermediate S. aureus (VISA) isolates, 11 vancomycin-resistant S. aureus (VRSA) isolates, and 4 linezolid-resistant S. aureus (LRSA) isolates. In these MIC characterizations, we used the FtsZ inhibitor TXA707 as well as the antibiotics vancomycin and linezolid as comparator agents. For each set of isolates, the range of MIC values and the mode MIC (that which occurs most often) associated with each antibacterial agent tested are listed in Table 1. For all the MSSA, MRSA, VISA, VRSA, and LRSA isolates examined, TXH9179 exhibited a mode MIC of 0.25 μg/mL, a value 4- fold lower than the corresponding mode MIC of 1 μg/mL exhibited by TXA707. The mode MIC of TXH9179 was also lower than the corresponding values exhibited by vancomycin and linezolid, even against isolates sensitive to the comparator antibiotics. In this connection, the mode MIC of vancomycin ranged from 1 to 2 μg/mL versus the MSSA, MRSA, and LRSA isolates (corresponding to a 4–8-fold higher mode MIC than TXH9179) and from 4 to >512 μg/mL versus the VISA and VRSA isolates (corresponding to a 16- to >2048-fold higher mode MIC than TXH9179). The mode MIC of linezolid ranged from 1 to 2 μg/mL versus the MSSA, MRSA, VISA, and VRSA isolates (corresponding to a 4–8-fold higher mode MIC than TXH9179) and 32 μg/mL versus the LRSA isolates (corresponding to a 128-fold higher mode MIC than TXH9179). Additional studies with representative isolates of MRSA-USA100 and MRSA-USA300 revealed that the MIC of TXH9179 (0.25 μg/mL) was also 2–4-fold lower than the corresponding MIC of PC190723, which ranged from 0.5 to 1 μg/mL (Table S1).
Table 1.
Activities of TXH9179 and Comparator Agents against Clinical Isolates of MSSA, MRSA, VISA, VRSA, and LRSA1
| MIC (μg/mL) | ||
|---|---|---|
|
|
||
| isolate and agent | range | mode2 |
| MSSA (n = 10) | ||
| TXH9179 | 0.25–0.5 | 0.25 |
| TXA707 | 1–2 | 1 |
| VAN | 1–2 | 1 |
| LZD | 2 | 2 |
| MRSA-USA100 (n = 13) | ||
| TXH9179 | 0.25 | 0.25 |
| TXA707 | 1–2 | 1 |
| VAN | 1–2 | 2 |
| LZD | 1–2 | 2 |
| MRSA-USA300 (n = 11) | ||
| TXH9179 | 0.25–0.5 | 0.25 |
| TXA707 | 1–2 | 1 |
| VAN | 1–2 | 1 |
| LZD | 1–2 | 2 |
| VISA (n = 6) | ||
| TXH9179 | 0.25 | 0.25 |
| TXA707 | 1 | 1 |
| VAN | 4–8 | 4 |
| LZD | 1–2 | 1 |
| VRSA (n = 11) | ||
| TXH9179 | 0.125–0.5 | 0.25 |
| TXA707 | 1–2 | 1 |
| VAN | 16–>512 | >512 |
| LZD | 0.5–2 | 1 |
| LRSA (n = 4) | ||
| TXH9179 | 0.25–0.5 | 0.25 |
| TXA707 | 1–2 | 1 |
| VAN | 2 | 2 |
| LZD | 8–64 | 32 |
VAN, vancomycin; LZD, linezolid.
Mode reflects the MIC value that occurs most often in each set.
We also characterized the antibacterial activity of TXH9179 versus a representative isolate of MSSA (NRS232), MRSA-USA100 (NRS705), MRSA-USA300 (NRS643), VISA (NRS27), VRSA (VRS5), and LRSA (NRS127) using a time-kill approach. In these studies, we evaluated the activity of TXH9179 at 0.25 μg/mL (which corresponds to the MIC of the compound). For comparative purposes, we also included corresponding characterizations of TXA707 at the same concentration as well as DMSO vehicle. As expected, each isolate treated with vehicle exhibited 3–4 logs of growth within 9 to 24 h (Figure 2). By contrast, TXH9179 treatment resulted in approximately 1–6 logs of kill within 9 to 24 h, with the 24-h magnitude of kill being 2 logs for the MSSA isolate (Figure 2A), 3 logs for the MRSA-USA300, VISA, and LRSA isolates (Figure 2C, D, and F, respectively), 4 logs for the VRSA isolate (Figure 2E), and a complete kill of 6 logs for the MRSA-USA100 isolate (Figure 2B). Treatment of each isolate with the equivalent concentration of TXA707 did not result in any kill but instead yielded growth at a rate comparable with vehicle treatment. This result is consistent with the 4-fold higher MIC of TXA707 (1 μg/mL) compared with the MIC of TXH9179.
Figure 2.

Time-kill curves for MSSA NRS232 (A), MRSA-USA100 NRS705 (B), MRSA-USA300 NRS643 (C), VISA NRS27 (D), VRSA VRS5 (E), and LRSA NRS127 (F) isolates showing the enhanced bactericidal activity of TXH9179 relative to TXA707. Bacteria were treated with DMSO vehicle, TXA707 at 0.25 μg/mL, or TXH9179 at 0.25 μg/mL.
Impact of TXH9179 Treatment on MRSA Cell Morphology, Cell Division, and FtsZ Localization.
Toward the goal of establishing FtsZ as the antibacterial target of TXH9179, we determined the impact of TXH9179 treatment on cell morphology, cell division, and the localization of FtsZ in the MRSA-USA100 isolate NRS705 using DMSO vehicle and the FtsZ inhibitor TXA707 as comparator controls. In these studies, cells were treated for 3 h with vehicle, TXA707 (at 4× MIC = 4 μg/mL), or TXH9179 (at 4× MIC = 1 μg/mL) and then labeled with a FtsZ-specific fluorescent probe (BOFP) we have previously developed.36 Both differential interference contrast (DIC) and fluorescence micrographs of the resulting cells are shown in Figure 3. Treatment with vehicle control results in cells with normal morphology and size (average diameter = 0.76 ± 0.08 μm, n = 324), with a significant percentage (41.0%) of the observed cells undergoing division, as reflected by the midcell presence of septa in varying stages of formation (Figure 3A and B). In the dividing cells, FtsZ is localized to the septa (represented by the white arrows in Figure 3B). Some of the dividing cells in Figure 3B (represented by the red arrows) are present in a transverse orientation that reveal the circular structure of the Z-ring. As expected, this behavior in response to vehicle treatment is consistent with that previously observed.22,36,37 In striking contrast to cells treated with vehicle, cells treated with the FtsZ inhibitor TXA707 appear approximately twice as large (average diameter = 1.53 ± 0.20 μm, n = 293), with a much smaller percentage (1.4%) of observed cells undergoing division (Figure 3C and D). In these enlarged cells, FtsZ tends to localize to one or more large foci at the cell perimeter (represented by the yellow arrows in Figure 3D). This pattern of behavior in S. aureus is a typical manifestation of treatment with a FtsZ inhibitor.20,22,26,27,32,36−39 Significantly, the impact on morphology, division, and FtsZ localization of treatment with TXH9179 is similar to that resulting from TXA707 treatment, with TXH9179-treated cells exhibiting an average diameter of 1.54 ± 0.20 μm (n = 353) and only 0.6% of observed cells undergoing division (Figure 3E and F).
Figure 3.

DIC and fluorescence micrographs of MRSA-USA100 NRS705 cells treated for 3 h with DMSO vehicle (A and B), 4 μg/mL TXA707 (C and D), or 1 μg/mL TXH9179 (E and F). The cells were also labeled with the fluorescent FtsZ probe BOFP to yield the fluorescence micrographs shown in B, D, and F. The white arrows in B highlight normally dividing cells with FtsZ localized to the midcell septa. The red arrows in B highlight cells presenting in a transverse orientation that reveal the circular nature of the FtsZ Z-ring. The yellow arrows in D and F highlight enlarged cells resulting from TXA707 or TXH9179 treatment in which FtsZ is mislocalized to one or more large foci at the cell periphery. Scale bars reflect 1 μm.
To afford increased granularity in our characterizations, we complemented our DIC and fluorescence microscopy studies with corresponding transmission electron microscopy (TEM) studies. Consistent with the DIC and fluorescence microscopy results, vehicle-treated cells appear normal in morphology and size (average diameter = 0.67 ± 0.07 μm, n = 89), with 42.7% of the cells undergoing division (Figure 4A). Particularly noteworthy is the definition of septal structures evident in the dividing cells (highlighted by the white arrows in Figure 4A), which reveal not only fully formed septa but also septa in the earlier stages of formation beginning with invaginations in the cell membrane and proceeding toward the center of the cell with new cell wall synthesis. Our fluorescence microscopy results are consistent with FtsZ localizing to these septal structures throughout the stages of septum formation (compare Figures 3B and 4A). Also consistent with the DIC and fluorescence microscopy results, treatment with either TXA707 or TXH9179 results in enlarged cells (average diameter of TXA707-treated cells = 1.23 ± 0.13 μm with n = 36; average diameter of TXH9179-treated cells = 1.24 ± 0.16 μm with n = 75), with none of the observed cells undergoing normal division (Figure 4B and C). Instead, most of the TXA707- and TXH9179-treated cells appear to exhibit aberrant septal structures mislocalized to large foci along the outer cell wall (highlighted by the yellow arrows in Figure 4B and C). FtsZ appears to localize to these large foci in TXA707and TXH9179-treated cells, as reflected by the corresponding fluorescence microscopy results shown in Figure 3D and F.
Figure 4.

TEM micrographs of MRSA-USA100 NRS705 cells treated for 3 h with DMSO vehicle (A), 4 μg/mL TXA707 (B), or 1 μg/mL TXH9179 (C). The white arrows in A highlight normally dividing cells exhibiting different stages of septum formation. The yellow arrows in B and C highlight enlarged cells resulting from TXA707 or TXH9179 treatment in which aberrant septal structures are mislocalized to large foci along the outer cell wall. Scale bars reflect 0.5 μm.
Frequency of Resistance (FOR) to TXH9179 in MSSA, MRSA, VISA, VRSA, and LRSA Isolates.
We used a large inoculum approach to determine the frequency of resistance (FOR) to TXH9179 compared with TXA707 in the same six representative isolates of MSSA (NRS232), MRSA-USA100 (NRS705), MRSA-USA300 (NRS643), VISA (NRS27), VRSA (VRS5), and LRSA (NRS127) used in our time-kill studies. The resulting FOR values observed for TXA707 in each isolate ranged from (1.00 ± 0.16) × 10−8 to (1.99 ± 0.56) × 10−8, with the corresponding FOR values for TXH9179 falling in a lower range of only (0.39 ± 0.16) × 10−8 to (1.09 ± 0.16) × 10−8 (Table S2). A statistical analysis of the differences in FOR between TXH9179 and TXA707 revealed that TXH9179 was associated with a statistically significant (P ≤ 0.05) reduced FOR relative to TXA707 in the MSSA, MRSA-USA100, VISA, VRSA, and LRSA isolates, while being similar in only the MRSA-USA300 isolate (Figure 5).
Figure 5.

Frequency of resistance (FOR) to TXH9179 relative to TXA707 in MSSA NRS232, MRSA-USA100 NRS705, MRSA-USA300 NRS643, VISA NRS27, VRSA VRS5, and LRSA NRS127 isolates. Each FOR value represents the mean of three replicates, with the indicated uncertainty reflecting the standard deviation from the mean. The double asterisk (**) denotes a statistically significant difference in FOR between the two FtsZ inhibitors (with a corresponding P ≤ 0.05), while n.s. denotes not significant.
We sequenced the ftsZ gene in 5 different TXH9179-resistant clones for each of the 6 isolate types. All 30 resistant clones bore mutations in their ftsZ genes, with the specific mutations observed and their relative overall frequencies being listed in Table 2. G196S was the mutation observed most frequently (43.3%), followed by G196A (23.3%), M262I/N263Y (13.3%), G196C (10.0%), and N263K (10.0%). We also noted significant differences in mutation frequency between the different isolate types (Figure S1). For example, the only mutation detected in the resistant LRSA clones was G196S, while the G196A mutation was detected in 80% of the resistant VRSA clones. The M262I/N263Y double mutation was detected in 80% of the resistant VISA clones, with these clones being the only ones in which the double mutation was detected. The only mutations observed in the MSSA clones were G196S (60%) and G196A (40%), with G196S and G196C each being observed in 40% of the MRSA-USA100 clones. The mutation observed most (40%) in the MRSA-USA300 clones was N263K.
Table 2.
Relative Frequencies of ftsZ Mutations That Confer Resistance to TXH9179 in MSSA, MRSA-USA100, MRSA-USA300, VISA, VRSA, and LRSA Isolates1
| ftsZ mutation | relative frequency (%) |
|---|---|
| G196S | 43.3 |
| G196A | 23.3 |
| M262I/N263Y | 13.3 |
| G196C | 10.0 |
| N263K | 10.0 |
Frequencies were derived from sequence analyses of 30 different TXH9179-resistant clones. Of the 30 clones, 5 were derived from each of the following 6 isolates: MSSA NRS232, MRSA-USA100 NRS705, MRSA-USA300 NRS643, VISA NRS27, VRSA VRS5, and LRSA NRS127.
Crystal Structure of TXH9179 in Complex with SaFtsZ.
To explore potential molecular factors contributing to the enhanced antibacterial potency of TXH9179 relative to TXA707, we determined the crystal structure of the SaFtsZ enzymatic domain (residues 12–316) in complex with TXH9179 at 1.3 Å resolution. The overall SaFtsZ monomeric structure showed the canonical T-state conformation, and the F0−Fc omit map clearly represents the presence of TXH9179 in the binding cleft (Figure 6). TXH9179 interacts with surrounding residues Gln192, Gly196, Leu200, Val203, Met226, Gly227, Val297, Thr309, and Ile311 via hydrophobic interactions. The acetylene group on the thiazolopyridine moiety forms contacts with Gln192, Met226, and Gly227. The benzamide moiety of TXH9179 forms two hydrogen bonds with the main chain of Leu209 and the side chain of Asn263, and the thiazolopyridine moiety interacts with Asp199 via a water-mediated hydrogen bond. TXH9179 adopts a straight conformation similar to that previously reported for PC190723 in complex with SaFtsZ (PDB entries: 3VOB and 4DXD),22,33 while maintaining the interactions with the surrounding residues as well as the orientations of those residues (Figure 7A). The straight conformation adopted by TXH9179 contrasts the bent binding mode we previously determined for TXA707 in complex with SaFtsZ (PDB entry: 5XDT),28 in which the side chains of Met226, Thr309, and Ile311 changed orientations to maximize hydrophobic contacts with TXA707 (Figure 7B).
Figure 6.

Close-up view of the inhibitor binding site in the SaFtsZ-TXH9179 complex. The F0−Fc omit map is contoured at 3.0 σ as a gray mesh. Hydrogen bonds are depicted as yellow dotted lines. Water molecules are depicted as red spheres.
Figure 7.

Comparison of the TXH9179, PC190723, and TXA707 complexes with SaFtsZ illustrating the straight or bent binding mode adopted by the different FtsZ inhibitors. (A) Superposition of the TXH9179 (orange) and PC190723 (blue, PDB entry: 4DXD) complexes. (B) Superposition of the TXH9179 (orange) and TXA707 (pink, PDB entry: 5XDT) complexes. Only one alternative conformation is shown for Met226 and Ile311 in the TXA707 complex.
Cytotoxicity of TXH9179 versus Mammalian Cells.
In advance of in vivo characterizations, we sought to determine the potential of TXH9179 for cytotoxicity versus mammalian cells. To this end, we used a 4-day MTT colorimetric assay to evaluate the cytotoxicity of TXH9179 at concentrations of 1 and 2.5 μg/mL (4× and 10× the antistaphylococcal MIC, respectively) versus opossum kidney (OK) cells (ATCC CRL-1840). In this assay, we included DMSO vehicle as a negative control and 1 μg/mL cycloheximide (CHX) as a positive control. As expected, CHX reduced the absorption signal at 570 nm (A570) by approximately 50% relative to DMSO vehicle control (Figure S2). In marked contrast to CHX, neither concentration of TXH9179 resulted in a significant reduction in A570 compared to vehicle.
Kinetics Associated with the Conversion of the Carboxamide Prodrug TXH1033 to the Active Product TXH9179 in the Presence of Mouse Serum.
Toward the goal of facilitating the formulation and in vivo administration of TXH9179, we synthesized a carboxamide prodrug of TXH9179 (which we designate as TXH1033) similar to the corresponding carboxamide prodrugs of TXA707 (designated TXA709) and PC190723 (designated TXY541) we previously developed (Figure 1).26,31 In our initial characterizations, we used HPLC to monitor the conversion of TXH1033 to TXH9179 in mouse serum at 37 °C as well as the kinetics associated with that conversion. TXH1033 is rapidly converted to TXH9179, with the half-life (t1/2) of conversion being 1.7 ± 0.1 min (Figures S3 and S4). This conversion t1/2 is approximately twice as rapid as that (3.0 ± 0.2 min) we previously determined for the conversion of TXA709 to TXA707 in mouse serum at 37 °C.26
We sought to determine if the rapid conversion of TXH1033 to TXH9179 was catalyzed by acetylcholinesterases present in serum. To this end, we explored the impact of the acetylcholinesterase inhibitor neostigmine on the conversion kinetics of TXH1033 in mouse serum at 37 °C. Significantly, the presence of 75 mM neostigmine slowed the conversion of TXH1033 to TXH9179 by more than 5-fold, yielding a conversion t1/2 of 8.9 ± 0.3 min (Figures S3 and S4). Similar studies with TXA709 in mouse serum revealed that neostigmine also slows the conversion of TXA709 to TXA707 (Figure S5).
Impact of Mouse Serum on the Activity of TXH9179 and TXH1033 against MRSA.
In advance of in vivo efficacy determinations in a mouse model of MRSA-USA100 infection, we sought to examine the impact of 50% mouse serum on the activity of both TXH9179 and TXH1033 against the MRSA-USA100 isolate NRS705. The presence of 50% mouse serum increased the MIC of TXH1033 by only 2-fold (from 0.5 to 1 μg/mL), while increasing the MIC of TXH9179 by 4-fold (from 0.25 to 1 μg/mL) (Table S3). It is likely that the observed activity of TXH1033 in the absence of mouse serum (MIC = 0.5 μg/mL) reflects some pH-induced conversion to TXH9179 in CAMH media, a behavior we have previously shown to occur with TXA709, albeit at a significantly slower rate than the corresponding conversion rate in the presence of serum.26
In Vivo Efficacy of Intravenously and Orally Administered TXH1033 Against MRSA in a Mouse Model of Systemic Infection.
We sought to determine whether the enhanced antistaphylococcal activity of TXH9179 relative to TXA707 observed in vitro is also observable in vivo. To this end, we used a mouse model of systemic (peritonitis) infection with the MRSA-USA100 isolate NRS705 to compare the in vivo efficacies of the TXH1033 and TXA709 prodrugs administered intravenously or orally. In these experiments, groups of four mice were infected intraperitoneally with a lethal inoculum of MRSA, followed by intravenous or oral administration of either vehicle (10 mM citrate, pH 2.6), TXA709, or TXH1033. Both prodrugs were given at doses ranging from 15 to 60 mg/kg when administered intravenously and from 24 to 96 mg/kg when administered orally.
Intravenous treatment of MRSA-infected mice with vehicle was associated with 0% survival (0 out of 4 mice) (Figure 8A and B). By contrast, intravenous administration of either TXA709 or TXH1033 resulted in a dose-dependent impact on survival, with the impact of TXH1033 being manifested at a lower dose than TXA709. In this connection, intravenous treatment with TXA709 at doses of 15, 30, 45, and 60 mg/kg resulted in 0%, 25%, 75%, and 100% survival, respectively (Figure 8A). Intravenous treatment with TXH1033 at the corresponding doses resulted in 0%, 50%, 100%, and 100% survival, respectively (Figure 8B). A similar pattern of behavior was observed when all agents were administered orally. As expected, orally administered vehicle was associated with 0% survival (Figure 8C and D). Oral treatment with TXA709 at doses of 24, 48, 72, and 96 mg/kg resulted in 0%, 0%, 25%, and 100% survival, respectively (Figure 8C). Oral treatment with TXH1033 at the corresponding doses resulted in 0%, 0%, 100%, and 100% survival, respectively (Figure 8D).
Figure 8.

In vivo efficacy of TXA709 (A and C) and TXH1033 (B and D) against MRSA-USA100 NRS705 in a mouse peritonitis model of systemic infection. Compounds and vehicle (10 mM citrate, pH 2.6) were administered either intravenously (A and B) or orally (C and D) at the indicated doses.
DISCUSSION
We describe studies of TXH9179, a next-generation thiazolopyridine-benzamide FtsZ inhibitor containing a 6-acetylene group on the thiazolopyridine moiety (Figure 1). Our initial characterizations focused on comparing the antistaphylococcal potency of TXH9179 to the corresponding potencies of two previously identified thiazolopyridine-benzamide FtsZ inhibitors containing either a 6-CF3 group (TXA707) or a 6-Cl group (PC190723). In these characterizations, we used a library of 55 total clinical isolates of MSSA (n = 10), MRSA-USA100 (n = 13), MRSA-USA300 (n = 11), VISA (n = 6), VRSA (n = 11), and LRSA (n = 4). As reflected by its consistently lower MIC values versus all the isolates examined, TXH9179 exhibited enhanced antistaphylococcal potency relative to both TXA707 and PC190723 (Tables 2 and S1). Significantly, this superior potency was maintained even versus isolates resistant to the current standard-of-care antibiotics vancomycin and linezolid (Table 2). Time-kill studies with representative isolates of MSSA, MRSA-USA100, MRSA-USA300, VISA, VRSA, and LRSA revealed that TXH9179 was rapidly bactericidal, with the extent of kill being greater for the MRSA-USA100, MRSA-USA300, VISA, VRSA, and LRSA isolates than for the MSSA isolate (Figure 2).
To validate FtsZ as the antistaphylococcal target of TXH9179, we conducted three sets of studies designed to (i) determine the impact of TXH9179 on MRSA cell morphology, cell division, and FtsZ localization; (ii) determine the FOR to TXH9179 in MSSA, MRSA-USA100, MRSA-USA300, VISA, VRSA, and LRSA isolates and map the resistance mutations to the ftsZ gene; and (iii) establish the direct interaction of TXH9179 with SaFtsZ by determining the crystal structure of the TXH9179-SaFtsZ complex.
In our initial target validation studies, DIC, fluorescence, and electron microscopy characterizations demonstrated that treatment of a representative MRSA-USA100 isolate with TXH9179 resulted in an enlarged cell morphology and few if any cells exhibiting normal septal structures, consistent with a marked inhibition of cell division (Figures 3 and 4). We used fluorescence microscopy with a FtsZ-specific fluorescent probe (BOFP) we have previously developed36 to monitor the impact of TXH9179 treatment on FtsZ localization in the MRSA isolate. TXH9179 treatment caused the mislocalization of FtsZ to one or more large foci at the cell periphery (Figure 3B, D, and F), with TEM studies suggesting that these large foci reflect aberrant septal structures mislocalized along the outer cell wall (Figure 4). These types of induced changes in S. aureus cell morphology, cell division, and FtsZ localization are characteristic of previously established benzamide inhibitors of FtsZ20,22,26,27,32,36−39 and, as such, are fully consistent with FtsZ being the antistaphylococcal target of TXH9179.
In our subsequent target validation studies, we conducted a sequencing analysis of the ftsZ gene from 30 resistant clones grown in the presence of TXH9179 at 4× MIC (1 μg/mL), with 5 of the 30 clones being derived from each of 6 different representative isolate types (MSSA, MRSA-USA100, MRSA-USA300, VISA, VRSA, and LRSA). All 30 resistant clones bore mutations in their ftsZ genes (Table 2). These genetic observations provide compelling evidence for FtsZ serving as the antistaphylococcal target of TXH9179. Among the ftsZ mutations observed, mutations of G196 were the most prevalent of all, with G196S being observed most frequently (43.3%), followed by G196A (23.3%) and G196C (10.0%). Previous studies of the prototypical benzamide FtsZ inhibitors PC19072322 and TXA70726 also revealed a predominance of G196 mutations in resistant MRSA clones. Significantly, however, the overall FOR to TXH9179 relative to TXA707 was lower in 5 of the 6 isolate types (MSSA, MRSA-USA100, VIDSA, VRSA, and LRSA), while being similar in the MRSA-USA300 isolate (Figure 5 and Table S2). Thus, the general propensity for staphylococcal isolates to develop mutational resistance to TXH9179 is reduced compared to TXA707. It is of interest to note that the frequency of specific resistant ftsZ mutations differed significantly among the six isolate types examined (Figure S1), suggesting that the propensities for genetic variability in the ftsZ genes can vary widely among different S. aureus strains.
In our third set of target validation studies, we determined the crystal structure of the TXH9179-SaFtsZ complex (Figure 6). These studies provide not only evidence for the direct interaction of TXH9179 with SaFtsZ but also an indication of potential molecular factors contributing to the enhanced antistaphylococcal potency of TXH9179 relative to TXA707 and PC190723. The only difference in chemical structure between PC190723, TXA707, and TXH9179 is the substituent at the 6-position of the thiazolopyridine moiety (Figure 1). As we described previously, TXA707 adopts a bent binding orientation so as to accommodate the large CF3 group in an inner hydrophobic pocket (Figure 7B).28 However, the comparatively smaller Cl group of PC190723 and acetylene group of TXH9179 are not able to form sufficient hydrophobic interactions in the inner pocket, with both compounds therefore adopting a straight binding orientation (Figure 7A). The bent orientation of TXA707 requires conformational changes on the part of residues Met226, Thr309, and Ile311 for the 6-CF3 group of the compound to access the inner hydrophobic pocket (Figure 7B). The straight binding orientations of TXH9179 and PC190723 do not require reorientation of these residues for them to be engaged in hydrophobic contacts, with these contacts being enhanced by the larger van der Waals surface of the 6-acetylene group of TXH9179 relative to the Cl group of PC190723. In addition, the 6-acetylene group of TXH9179 is highly polarizable, with the potential for engaging in enhanced dipole−dipole interactions with the side chain of Met226, which is also highly polarizable. These collective structural properties would bolster TXH9179 binding and likely contribute to the superior antistaphylococcal potency of the compound.
To explore the potential of TXH9179 for mammalian cytotoxicity, we used a 4-day MTT colorimetric assay to determine if exposure to TXH9179 at concentrations equivalent to 4× and 10× the antistaphylococcal MIC of the compound (1 and 2.5 μg/mL, respectively) impacted the growth of OK cells. Significantly, neither concentration of TXH9179 resulted in a significant reduction in A570 (Figure S2). Thus, TXH9179 is not associated with any mammalian cytotoxicity, even at a concentration 10-fold greater than that generating antistaphylococcal activity.
Armed with the gratifying results of our in vitro characterizations, we sought to evaluate antistaphylococcal efficacy of TXH9179 in vivo. Toward this goal, we synthesized the carboxamide prodrug of TXH9179 (designated TXH1033) to facilitate formulation in acidic vehicles suitable for both oral and intravenous administration. TXH1033 is similar in chemical structure to the corresponding carboxamide prodrugs of TXA707 (designated TXA709) and PC190723 (designated TXY541) we have previously developed (Figure 1).26,31 Prior to murine in vivo efficacy characterizations, we first evaluated the kinetics of TXH1033 conversion to TXH9179 in mouse serum. TXH1033 is rapidly converted to TXH9179 in serum, with a conversion t1/2 of 1.7 ± 0.1 min (Figures S3 and S4). This conversion kinetics is approximately twice as fast as the corresponding kinetics we have previously determined for conversion of the TXA709 and TXY541 prodrugs to their active products.26,31 Note that the conversion of TXH1033 to TXH9179 in mouse serum was slowed significantly by the presence of the acetylcholinesterase inhibitor neostigmine at a concentration of 75 mM, with the conversion t1/2 increasing approximately 5-fold to 8.9 ± 0.3 min (Figures S3 and S4). Thus, the rapid serum-induced conversion of TXH1033 to TXH9179 is catalyzed to a significant degree by acetylcholinesterases present in serum. We observed a similar behavior for the serum-induced conversion of TXA709 to TXA707 (Figure S5).
Extensive binding to serum proteins can limit the efficacy of an antibacterial agent. We therefore also examined the impact of mouse serum on the antistaphylococcal activity of both TXH1033 and TXH9179. Importantly, both TXH9179 and TXH1033 retained significant potency against a representative MRSA-USA100 isolate in the presence of 50% mouse serum, with both compounds exhibiting an MIC of 1 μg/mL under these conditions (Table S3).
With the above characterizations of TXH1033 boding well for in vivo studies, we examined the efficacy of both intravenously and orally administered TXH1033 in a mouse model of systemic (peritonitis) infection with a MRSA-USA100 isolate (Figure 8). We used TXA709 as our comparator FtsZ inhibitor in these dose escalation bioassays, in which a survival of ≥50% was viewed as an efficacious response. Significantly, the efficacious dose of TXH1033 was consistently lower than that of TXA709 via both routes of administration. In this connection, TXH1033 was efficacious at an intravenous dose of 30 mg/kg and oral dose of 72 mg/kg, while TXA709 was efficacious at an intravenous dose of 45 mg/kg and oral dose of 96 mg/kg. Thus, the enhanced antistaphylococcal activity of TXH9179 relative to TXA707 observed in vitro was also observable in vivo with administration of the corresponding TXH1033 and TXA709 prodrugs.
Stemming the ever-present tide of antibiotic resistance among clinically significant bacterial pathogens requires an ongoing effort to develop new antibacterial agents and identify novel antibacterial targets. The studies described herein represent an important step toward the development of new FtsZ inhibitors with the potential for translation into clinical use for the treatment of MRSA infections. In the aggregate, our results highlight TXH9179 (and its prodrug TXH1033) as an exciting new lead FtsZ inhibitor in the antibiotic development pipeline.
METHODS
Bacterial Isolates and Other Reagents.
Clinical isolates of MSSA, MRSA-USA100, MRSA-USA300, VISA, VRSA, and LRSA were provided by the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) for distribution by the Biodefense and Emerging Infections Research Resources Repository (BEI Resources), NIAID, NIH. The BEI Resources Web site (https://www.beiresources.org ) describes the location of origin of each isolate. Luria−Bertani (LB) media was obtained from Millipore. Cation-adjusted Mueller Hinton (CAMH) media and tryptic soy agar (TSA) were obtained from Becton Dickson. Phosphate-buffered saline (PBS) was obtained from BioWhittaker and dimethyl sulfoxide (DMSO) was obtained from Fluka Analytical. High-resolution agarose, acetonitrile, trifluoroacetic acid (TFA), mouse serum, porcine mucin, neostigmine bromide, and vancomycin hydrochloride were obtained from Sigma. Linezolid was obtained from LKT Laboratories. TXA707, TXA709, and BOFP were synthesized as described previously.26,36
Synthesis of TXH9179 and TXH1033.
TXH9179 and TXH1033 were synthesized via Scheme 1 as shown.
Scheme 1.

Preparation of N-(3-((6-Ethynylthiazolo[5,4-b]pyridin-2-yl)methoxy)-2,6-difluorobenzoyl)-1-methylpiperidine-4-carboxamide (TXH1033): (a) 2-(Benzyloxy)acetyl chloride, Et3N, Dichloromethane; (b) Lawesson Reagent, Toluene; (c) BBr3, Dichloromethane; (d) 2,6-Difluoro-3-hydroxybenzamide, K2CO3, NaI, N,N-Dimethylformamide; (e) Ethynyltrimethylsilane, Pd(PPh3)4, CuI, N,N-Diisopropylethylamine, N,N-Dimethylformamide; (f) 1-Methylpiperidine-4-carbonyl chloride hydrochloride, NaH, Tetrahydrofuran, Water1
1The percentage indicated at each step reflects the yield at that step.
Synthetic Procedures. N-(3-((6-Ethynylthiazolo[5,4-b]pyridin-2yl)methoxy)-2,6-difluorobenzoyl)-1-methylpiperidine-4-carboxamide (TXH1033).
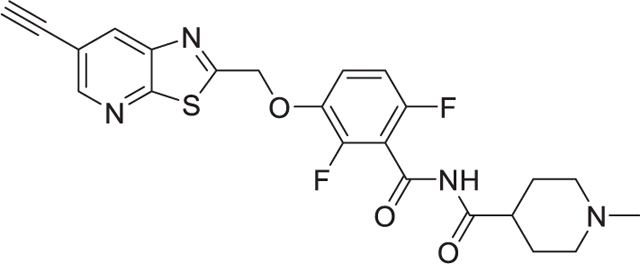
To a solution of 3-((6-ethynylthiazolo[5,4-b]pyridin-2-yl)methoxy)-2,6-difluorobenzamide (150 mg, 0.43 mmol) in tetrahydrofuran (10 mL), 1-methylpiperidine-4-carbonyl chloride hydrochloride (150 mg, 0.76 mmol) was added followed by sodium hydride (150 mg, 3.75 mmol). To the mixture, water (68 μL, 3.75 mmol) was added, and the mixture was stirred for 10 min at RT. The reaction mixture was diluted with dichloromethane and was washed with water and brine. The organic layer was dried under reduced pressure, and the residue was purified on an ISCO chromatograph (0−10% methanol/dichloromethane) to give the product as a white solid (59 mg, 29%); mp 203−204 °C; 1H NMR (300 MHz, CDCl3) 8.65 (d, J = 2 Hz, 1H), 8.30 (d, J = 2 Hz, 1H), 7.15–7.13 (m, 1H), 6.89–6.86 (m, 1H), 5.46 (s, 2H), 3.29 (s, 1H), 2.90–2.87 (m, 2H), 2.75 (m, 1H), 2.26 (s, 3H), 2.11–2.04 (m, 2H), 1.95–1.90 (m, 2H), 1.85–1.77 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 175.4, 170.2, 166.3, 160.8, 157.7, 150.3, 145.2, 141.8, 140.2, 133.5, 117.4, 112.5, 106.7, 105.5, 85.0, 69.5, 54.9, 46.5, 42.4, 28.1; HRMS (ESI) for C23H20F2N4O3S: [M + H]+ calculated 471.1297, found 471.1302.
Requisite Intermediates Were Prepared As Follows. 2-(Benzyloxy)-N-(5-bromo-2-chloropyridin-3-yl)acetamide.

To a solution of 5-bromo-2-chloropyridin-3-amine (2.0 g, 9.64 mmol) in dichloromethane (50 mL), triethylamine (2.02 mL, 14.46 mmol) was added at 0 °C followed by 2-(benzyloxy)acetyl chloride (2.28 mL, 14.46 mmol). The reaction mixture was warmed to RT and was stirred for 2 h at that temperature. The mixture was then diluted with dichloromethane and was washed with water followed by brine. The organic layer was dried over sodium sulfate and was concentrated under reduced pressure. The residue was purified on an ISCO chromatograph (0–10% methanol/dichloromethane) to give the product as a white solid (3.30 g, 96%); 1H NMR (300 MHz, DMSOd6) 9.52 (s, 1H), 8.59 (s, 1H), 8.38 (s, 1H), 7.41−7.31 (m, 5H), 4.65 (s, 2H), 4.21 (s, 2H).
2-((Benzyloxy)methyl)-6-bromothiazolo[5,4-b]pyridine.

To a solution of 2-(benzyloxy)-N-(5-bromo-2-chloropyridin-3-yl)-acetamide (1.0 g, 2.82 mmol) in toluene (10 mL), Lawesson reagent (1.71 g, 4.23 mmol) was added. The reaction mixture was refluxed overnight. After the mixture was cooled to RT, it was then diluted with dichloromethane. The organic layer was washed with water followed by brine and was then dried over sodium sulfate. The organic layer was then concentrated under reduced pressure, and the residue was purified on an ISCO chromatograph (0–30% ethyl acetate/hexane) to give the product as a white solid (945 mg, 100%); 1H NMR (300 MHz, CDCl3) 8.64 (s, 1H), 8.35 (s, 1H), 7.40–7.36 (m, 5H), 4.90 (s, 2H), 4.73 (s, 2H).
6-Bromo-2-(bromomethyl)thiazolo[5,4-b]pyridine.

To a solution of 2-((benzyloxy)methyl)-6-bromothiazolo[5,4-b]-pyridine (600 mg, 1.79 mmol) in dichloromethane (20 mL), a solution of boron tribromide (1.0M, 3.58 mL) was added at 0 °C. The reaction mixture was warmed to RT and then stirred for overnight at that temperature. The mixture was then diluted with dichloromethane and was washed with saturated sodium carbonate followed by brine. The organic layer was dried over sodium sulfate and was concentrated under reduced pressure. The residue was purified on an ISCO chromatograph (0–20% ethyl acetate/hexane) to give the product as a white solid (524 mg, 95%); 1H NMR (300 MHz, DMSO-d6) 8.77 (m, 2H), 5.14 (s, 2H).
3-((6-Bromothiazolo[5,4-b]pyridin-2-yl)methoxy)-2,6-difluoro-benzamide.

To a solution of 6-bromo-2-(bromomethyl)thiazolo[5,4-b]pyridine (496 mg, 1.61 mmol) in N,N-dimethylformamide (10 mL), potassium carbonate (334 mg, 2.42 mmol) and sodium iodide (24 mg, 0.16 mmol) were added. The mixture was stirred for 15 min at RT and 2,6-difluoro-3-hydroxybenzamide (279 mg, 1.61 mmol) was then added. The mixture was stirred at RT for 2 h. The mixture was then diluted with ethyl acetate and was then washed with water followed by brine. The organic layer was dried over sodium sulfate and then concentrated under reduced pressure. The residue was suspended in dichloromethane, and the suspension was filtered to give the product as a white solid (486 mg, 75%); 1H NMR (300 MHz, DMSO-d6) 8.77−8.75 (m, 2H), 8.17 (bs, 1H), 7.89 (bs, 1H), 7.42–7.35 (m, 1H), 7.12−7.07 (m, 1H).
2,6-Difluoro-3-((6-((trimethylsilyl)ethynyl)thiazolo[5,4-b]pyridin2-yl)methoxy)benzamide.

To a solution of 3-((6-bromothiazolo[5,4-b]pyridin-2-yl)methoxy)-2,6-difluorobenzamide (100 mg, 0.25 mmol), ethynyltrimethylsilane (0.18 mL, 1.25 mmol), tetrakis(triphenylphosphine)palladium(0) (21 mg, 0.03 mmol), cupper iodide (6 mg, 0.03 mmol), and N,N-diisopropylethylamine (0.13 mL, 0.75 mmol) were dissolved in N,N-dimethylformamide (5 mL) and then stirred at 60 °C overnight. The mixture was cooled to RT and then diluted with ethyl acetate. The organic layer was washed with water followed by brine and then dried over sodium sulfate. The organic layer was then concentrated under reduced pressure, and the residue was purified on an ISCO chromatograph (0–100% ethyl acetate/hexane) to give the product as a white solid (63 mg, 61%); 1H NMR (300 MHz, DMSO-d6) 8.69 (s, 1H), 8.53 (s, 1H), 8.18 (bs, 1H), 7.90 (bs, 1H), 7.43−7.39 (m, 1H), 7.14−7.11 (m, 1H), 5.70 (s, 2H), 0.26 (s, 9H).
3-((6-Ethynylthiazolo[5,4-b]pyridin-2-yl)methoxy)-2,6-difluoro-benzamide (TXH9179).

To a solution of 2,6-difluoro-3-((6-((trimethylsilyl)ethynyl)thiazolo[5,4-b]pyridin-2-yl)methoxy)benzamide (63 mg, 0.15 mmol) in a mixture of methanol (5 mL) and water (0.5 mL), potassium carbonate (41 mg, 0.30 mmol) was added. The reaction mixture was stirred for 2 h. The mixture was then diluted with ethyl acetate and then washed with saturated ammonium chloride followed by brine. The organic layer was dried over sodium sulfate and then concentrated under reduced pressure. The residue was suspended in dichloromethane, and the suspension was filtered to give the product as a beige solid (44 mg, 85%); mp 199−200 °C; 1H NMR (300 MHz, DMSO-d6) 8.72 (s, 1H), 8.58 (s, 1H), 8.16 (bs, 1H), 7.88 (bs, 1H), 7.43–7.35 (m, 1H), 7.13–7.07 (m, 1H), 5.71 (s, 2H), 4.55 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 170.4, 161.5, 157.7, 151.9, 150.3, 147.3, 145.2, 142.4, 133.4, 117.4, 117.0, 111.8, 111.6, 85.0, 80.4, 69.5; HRMS (ESI) for C16H9F2N3O2S: [M + H]+ calculated 346.0456, found 346.0459.
Minimum Inhibitory Concentration (MIC) Assays.
Microtiter plates containing 2-fold serial dilutions of TXH9179, TXA707, vancomycin, and linezolid in CAMH broth were prepared in accordance with Clinical Laboratory and Standards Institute (CLSI) guidelines for the broth microdilution susceptibility method.40 Exponentially growing isolates of MSSA (n = 10), MRSA-USA100 (n = 13), MRSA-USA300 (n = 11), VISA (n = 6), VRSA (n = 11), and LRSA (n = 4) were diluted in CAMH broth, and the microtiter plates were inoculated at a final concentration of 5 × 105 CFU/mL. The volume in each well was 0.1 mL, and each test compound concentration was present in duplicate. The microtiter plates were incubated with shaking at 37 °C for 18 h and growth was measured using a VersaMax microplate reader (Molecular Devices). The MIC was defined as the lowest antibiotic concentration at which bacterial growth was inhibited by ≥90%. MIC assays probing the impact of mouse serum on the MICs of TXH9179 and TXH1033 against MRSA-USA100 (NRS705) were performed as described above, except that the CAMH was supplemented with 50% mouse serum.
Time-Kill Assays.
Overnight cultures of MSSA (NRS232), MRSA-USA100 (NRS705), MRSA-USA300 (NRS643), VISA (NRS27), VRSA (VRS5), and LRSA (NRS127) were each diluted to a final count of approximately 106 CFU/mL in CAMH broth. The colony count of each culture at time zero was verified by plating serial dilutions (in PBS) on tryptic soy agar (TSA) plates. Each culture was then distributed into 3 tubes containing DMSO vehicle, 0.25 μg/mL TXA707, or 0.25 μg/mL TXH9179. All tubes were then placed in an incubator at 37 °C with shaking, and aliquots were removed at time points of 3, 6, 9, and 24 h. The CFU/mL at each time point was determined by plating serial dilutions (in PBS) of the aliquots on TSA plates and counting colonies after incubation at 37 °C for 24 h.
Differential Interference Contrast (DIC) and Fluorescence Microscopy.
In each of 3 culture tubes, exponentially growing MRSA-USA100 (NRS705) cells were diluted to an optical density at 600 nm (OD600) of 0.1 in 5 mL of CAMH broth. The first tube contained TXA707 at 4× MIC (4 μg/mL), the second tube contained TXH9179 at 4× MIC (1 μg/mL), and the third tube contained an equivalent volume of DMSO vehicle as a negative control. The cultures were incubated with shaking at 37 °C for 3 h. One mL of each culture was then centrifuged at 15,000 × g for 3 min and the resulting pellet washed twice with 1 mL of PBS. Each pellet was then resuspended in 500 μL of PBS and 1 μg/mL of BOFP was added. The cells were then incubated in the dark for 5 min at RT, centrifuged at 15,000 × g for 3 min, and the pellets washed twice with PBS. The pellets were then resuspended in 200 μL of PBS and 8 μL of each suspension was spread on a 0.25 mm layer of 1% high-resolution agarose in PBS, which was mounted on a standard 75 × 25 × 1 mm3 microscope slide (Azer Scientific) using a 1.7 × 2.8 × 0.025 cm3 Gene Frame (ThermoFisher). A 24 × 40 mm2 coverslip (Azer Scientific) was then applied to the agarose pad to prepare the slide for microscopic visualization. The bacteria were then visualized by DIC and fluorescence microcopy utilizing an Olympus BX50 microscope equipped with an X-cite Exacte 200 W mercury lamp, a 100x Olympus UPLSAPO oil immersion objective (1.40 aperture), and a Chroma ET-EGFP (FITC/Cy2) filter. Micrographs were captured using a QImaging Retiga R3 charge-coupled device (CCD) camera and the Ocular Version 2.0 software package (QImaging). Cell sizes were quantified using the Fiji software package of ImageJ.
Transmission Electron Microscopy (TEM).
In each of 3 culture tubes, exponentially growing MRSA-USA100 (NRS705) cells were diluted to an OD600 of 0.1 in 5 mL of CAMH broth. The first tube contained TXA707 at 4× MIC (4 μg/mL), the second tube contained TXH9179 at 4× MIC (1 μg/mL), and the third tube contained an equivalent volume of DMSO vehicle as a negative control. The cultures were incubated with shaking at 37 °C for 3 h and then centrifuged at 15,000 × g for 3 min. The resulting pellets were washed once with 5 mL of PBS and then resuspended in 2.5 mL of 100 mM cacodylate buffer (pH 7.2) containing 2.5% glutaraldehyde and 4% paraformaldehyde. The fixed bacterial cells were then postfixed in buffered osmium tetroxide (1%), subsequently dehydrated in a graded series of ethanol, and embedded in Epon resin. Thin sections (90 nm) were cut on a Leica EM UC6 ultramicrotome. Sectioned grids were then stained with a saturated solution of uranyl acetate and lead citrate. Images were captured with an AMT XR111 digital camera at 80 kV on a Philips CM12 transmission electron microscope. The diameter of the bacterial cells was measured as described above.
Frequency of Resistance (FOR) Assays and ftsZ Mutant Sequence Analyses.
A large inoculum approach described previously31 was used to determine the FOR of MSSA (NRS232), MRSA-USA100 (NRS705), MRSA-USA300 (NRS643), VISA (NRS27), VRSA (VRS5), and LRSA (NRS127) to TXH9179 or TXA707. For these determinations, approximately 109−1010 CFUs of bacteria were plated on TSA plates containing TXH9179 at 4× MIC (1 μg/mL) or TXA707 at 4× MIC (4 μg/mL). All plates were incubated at 37 °C and examined for the emergence of resistant colonies after 48 h of growth. The FOR was defined as the number of resistant colonies on the TXA707 or TXH9179 plates divided by the bacterial inoculum initially transferred onto the plate.
To determine whether the genetic basis for the TXH9179 resistance was related to a mutation in the ftsZ gene, five TXH9179-resistant colonies for each of the six isolate types (30 clones in total) were added to separate culture tubes containing LB broth and then incubated overnight. The genomic DNA was then isolated using a Qiagen DNeasy UltraClean Microbial Kit. The ftsZ gene from each clone was then amplified using the Q5 High-Fidelity 2x Master Mix (New England Biolabs) and a standard polymerase chain reaction (PCR) protocol. The resulting PCR product sizes were then confirmed by gel electrophoresis. The PCR products were purified using a Monarch PCR and DNA Cleanup Kit (New England BioLabs) and the DNA concentration was quantified using a SpectraMax M2 plate reader equipped with a SpectraDrop Micro-Volume Microplate (Molecular Devices). The sequence of each ftsZ gene product was then determined by Sanger sequencing, with sequence alignments being made in Benchling.
X-ray Crystallography.
SaFtsZ (residues 12−316) was cloned, expressed, and purified as described previously.28 The protein was crystallized at 9.5 mg mL−1 and 20 °C using the sitting-drop vapor-diffusion technique under conditions of 100 mM Tris-HCl (pH 8.1), 43% (w/v) pentaerythritol propoxylate 629 (17/8 PO/OH, PEP629), and 300 mM KCl. After 3 weeks, the crystals were soaked for 2 days in the same reservoir supplemented with 5 mM TXH9179 and 10% (v/v) DMSO. Crystals were flash-frozen in a nitrogen gas stream at 100 K without cryoprotectants. X-ray diffraction data were collected at SPring-8 BL44XU (Hyogo, Japan) under a cryogenic nitrogen gas stream at 100 K. The diffraction data were processed and scaled with HKL2000.41 The phases were determined by molecular replacement with Phaser in the CCP4 suite42 using the previously reported structure of the SaFtsZ-GDP complex (PDB entry: 3VOA)33 as a search model. The model structure was refined with REFMAC543 and PHENIX,44 with manual modification using COOT.45 The refined structure was validated with MolProbity.46 Data collection and refinement statistics are summarized in Table S4. The final atomic coordinates and structure factor amplitude have been deposited in the RCSB Protein Data Bank (PDB entry: 8HTB). Figures 6 and 7 were prepared with PyMOL (Schrödinger).
Mammalian Cytotoxicity Assay.
The cytotoxicity of TXH9179 at concentrations of 1 and 2.5 μg/mL versus opossum kidney (OK) cells (ATCC CRL-1840) was determined using a 4-day MTT colorimetric assay as previously described.47 DMSO vehicle was included as a negative control and 1 μg/mL cycloheximide (CHX) was included as a positive control. Six replicates of each sample were measured, with statistically significant differences between vehicle-treated and compound-treated samples being determined by one-way ANOVA.
HPLC Assays of TXH1033 and TXA709 Hydrolysis in Mouse Serum.
Two sets of two tubes containing 1 mL of mouse serum alone and 1 mL of mouse serum and 75 mM neostigmine were warmed to 37 °C. TXH1033 or TXA709 was added to each set of tubes at a final concentration of 75 μM. The samples were then incubated at 37 °C, with 150 μL aliquots being withdrawn at time points of 0.5, 2, 3.5, 5, and 10 min. Control samples containing 75 μM TXH9179 in mouse serum alone or mouse serum and 75 mM neostigmine were also prepared, with a single 150 μL aliquot being withdrawn after 10 min of incubation at 37 °C. 300 μL of acidified acetonitrile [acetonitrile/0.1% (v/v) TFA] was added to each aliquot to bring the final compound concentrations to 25 μM and the mixtures were then vortexed and centrifuged at 15,000 × g for 3 min. The supernatants were then characterized by reverse-phase HPLC. All HPLC measurements were performed on a Shimadzu LC-20AT liquid chromatograph equipped with a Shimadzu SPD-20AV UV/vis detector set at 296 nm and a reverse-phase SPHER-100 C18 column (Princeton Chromatography). The column size was 150 × 4.6 mm2 with a particle size of 5 μm and a pore size of 100 Å. The samples were injected at a volume of 20 μL and a flow rate of 1 mL/min was applied. A gradient of 10−90% acidified acetonitrile and water was used for the mobile phase. The total run time was 23 min, with the sampling frequency being 2 Hz and the response time being 1 s. All peak areas were measured using the Shimadzu LabSolutions Lite Version 5.82 software package.
In Vivo Efficacy Assays in a Murine Model of Peritonitis Infection with MRSA.
All murine studies were conducted in full compliance with the standards established by the US National Research Council’s Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) of Rutgers Robert Wood Johnson Medical School. Groups of 4 female Swiss-Webster mice (Taconic Biosciences, Rensselaer, NY) with an average weight of 25 g were infected intraperitoneally with a lethal inoculum (2.5 × 107 CFUs) of MRSA-USA100 isolate NRS705 in saline (0.9% NaCl) containing 5% (w/v) porcine mucin. Inocula were prepared by combining overnight cultures with sterile 10% mucin to achieve the desired bacterial CFU and mucin percentage. Colony counts for all inocula were verified by plating serial dilutions on TSA plates. Beginning 15 min after infection, all test compounds and the vehicle were administered either intravenously by tail vein injection or orally by gavage, with the vehicle being 10 mM citrate (pH 2.6) for both routes of administration. The dosing volume was 10 mL/kg for the intravenous administrations and 12 mL/kg for the oral administrations.
In the intravenous studies, nine groups of four mice were treated as follows: Group 1, citrate vehicle alone; Group 2, 15 mg/kg TXH1033; Group 3, 30 mg/kg TXH1033 (in two divided doses of 15 mg/kg given 15 min apart); Group 4, 45 mg/kg TXH1033 (in three divided doses of 15 mg/kg given 15 min apart); Group 5, 60 mg/kg TXH1033 (in four divided doses of 15 mg/kg given 15 min apart); Group 6, 15 mg/kg TXA709; Group 7, 30 mg/kg TXA709 (in two divided doses of 15 mg/kg given 15 min apart); Group 8, 45 mg/kg TXA709 (in three divided doses of 15 mg/kg given 15 min apart); and Group 9, 60 mg/kg TXA709 (in four divided doses of 15 mg/kg given 15 min apart). In the oral studies, nine groups of four mice were treated as follows: Group 1, citrate vehicle alone; Group 2, 24 mg/kg TXH1033; Group 3, 48 mg/kg TXH1033 (in two divided doses of 24 mg/kg given 15 min apart); Group 4, 72 mg/kg TXH1033 (in three divided doses of 24 mg/kg given 15 min apart); Group 5, 96 mg/kg TXH1033 (in four divided doses of 24 mg/kg given 15 min apart); Group 6, 24 mg/kg TXA709; Group 7, 48 mg/kg TXA709 (in two divided doses of 24 mg/kg given 15 min apart); Group 8, 72 mg/kg TXA709 (in three divided doses of 24 mg/kg given 15 min apart); and Group 9, 96 mg/kg TXA709 (in four divided doses of 24 mg/kg given 15 min apart).
The body temperatures of all mice were monitored for a period of 5 days after infection. Body temperatures were recorded at the Xiphoid process using a noninvasive infrared thermometer (Braintree Scientific, Inc.). Infected mice with body temperatures ≤ 28.9 °C were viewed as being unable to recover from the infection48 and were euthanized.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 AI118874 (to D.S.P.); Grant-in-Aid for the Japan Society for the Promotion of Science (JSPS) Fellows (15J00589 to J.F.); Grants-in-Aid for Scientific Research (16H00783 and 18K06094, 19H04735, 19K07582 to H.M.); Takeda Science Foundation; the Science Research Promotion Fund from the Promotion and Mutual Aid Corporation for Private Schools of Japan; the Cooperative Research Program of the Institute for Protein Research, Osaka University (CR-18-05 and CR-19-05); the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP19am0101070. The crystallographic work was performed under the approval of the SPring-8 Program Advisory Committee (Proposal Nos. 2017A6748, 2017A2570, and 2017B6748). We thank the SPring-8 BL44XU beamline staff for their assistance with the X-ray diffraction data collection. We are also grateful to Huizhou Fan and Yuxuan Wang for their assistance with the mammalian cytotoxicity studies, as well as to Longqin Hu and Yiling Wang for their assistance with the 13C NMR data acquisition.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00934.
Comparative activities of PC109723, TXA707, and TXH9179 against MRSA-USA100 and MRSA-USA300 isolates as well as the impact of 50% mouse serum on the activities of TXH9179 and TXH1033 against MRSA-USA100; frequencies of resistance of TXA707 and TXH9179 against isolates of MSSA, MRSA-USA100, MRSA-USA300, VISA, VRSA, and LRSA as well as the frequencies of specific ftsZ mutations observed in resistant clones derived from those isolates; crystallographic data collection and refinement statistics; mammalian cytotoxicity data for TXH9179; impact of neostigmine on the kinetics for conversion of the TXH1033 and TXA709 prodrugs to their active products in mouse serum (PDF)
The authors declare the following competing financial interest(s): Drs. Pilch and LaVoie are co-founders of TAXIS Pharmaceuticals and therefore have a financial interest in the company.
Contributor Information
Eric Bryan, Department of Pharmacology, Rutgers Robert Wood Johnson Medical School, Piscataway, New Jersey 08854, United States.
Edgar Ferrer-González, Department of Pharmacology, Rutgers Robert Wood Johnson Medical School, Piscataway, New Jersey 08854, United States.
Hye Yeon Sagong, Department of Medicinal Chemistry, Ernest Mario School of Pharmacy, Rutgers University, Piscataway, New Jersey 08854, United States; TAXIS, Pharmaceuticals, Inc., Monmouth Junction, New Jersey, 08852, United States.
Junso Fujita, Department of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan; Present Address: J.F., Graduate School of Frontier Biosciences, Osaka University, 1−3 Yamadaoka, Suita, Osaka 565-0871, Japan.
Lilly Mark, TAXIS Pharmaceuticals, Inc., Monmouth Junction, New Jersey 08852, United States.
Malvika Kaul, Department of Pharmacology, Rutgers Robert Wood Johnson Medical School, Piscataway, New Jersey 08854, United States.
Edmond J. LaVoie, Department of Medicinal Chemistry, Ernest Mario School of Pharmacy, Rutgers University, Piscataway, New Jersey 08854, United States
Hiroyoshi Matsumura, Department of Biotechnology, College of Life Sciences, Ritsumeikan University, Shiga 525-8577, Japan.
Daniel S. Pilch, Department of Pharmacology, Rutgers Robert Wood Johnson Medical School, Piscataway, New Jersey 08854, United States
REFERENCES
- (1).Aslam B; Wang W; Arshad MI; Khurshid M; Muzammil S; Rasool MH; Nisar MA; Alvi RF; Aslam MA; Qamar MU; Salamat MKF; Baloch Z. Antibiotic Resistance: A Rundown of a Global Crisis. Infect. Drug Resist 2018, 11, 1645–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ventola CL The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharmacol. Ther 2015, 40, 277–283. [PMC free article] [PubMed] [Google Scholar]
- (3).Global Action Plan on Antimicrobial Resistance; World Health Organization: Geneva, 2015. [DOI] [PubMed] [Google Scholar]
- (4).Antibiotic Resistance Threats in the United States, 2013; Centers for Disease Control and Prevention: Atlanta, GA, 2013. [Google Scholar]
- (5).Dadgostar P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist 2019, 12, 3903–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pulingam T; Parumasivam T; Gazzali AM; Sulaiman AM; Chee JY; Lakshmanan M; Chin CF; Sudesh K. Antimicrobial Resistance: Prevalence, Economic Burden, Mechanisms of Resistance and Strategies to Overcome. Eur. J. Pharm. Sci 2022, 170, 106103. [DOI] [PubMed] [Google Scholar]
- (7).Antibiotic Resistance Threats in the United States, 2019; Centers for Disease Control and Prevention: Atlanta, GA, 2019. [Google Scholar]
- (8).Murray CJL; Ikuta KS; Sharara F; Swetschinski L; Robles Aguilar G; Gray A; Han C; Bisignano C; Rao P; Wool E; Johnson SC; Browne AJ; Chipeta MG; Fell F; Hackett S; Haines-Woodhouse G; Kashef Hamadani BH; Kumaran EAP; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Peacock SJ; Paterson GK Mechanisms of Methicillin Resistance in Staphylococcus aureus. Annu. Rev. Biochem 2015, 84, 577–601. [DOI] [PubMed] [Google Scholar]
- (10).Guignard B; Entenza JM; Moreillon P. β-lactams Against Methicillin-Resistant Staphylococcus aureus. Curr. Opin. Pharmacol 2005, 5, 479–489. [DOI] [PubMed] [Google Scholar]
- (11).Fuda C; Suvorov M; Vakulenko SB; Mobashery S. The Basis for Resistance to β-Lactam Antibiotics by Penicillin-Binding Protein 2a of Methicillin-Resistant Staphylococcus aureus. J. Biol. Chem 2004, 279, 40802–40806. [DOI] [PubMed] [Google Scholar]
- (12).Liu W-T; Chen E-Z; Yang L; Peng C; Wang Q; Xu Z; Chen D-Q Emerging Resistance Mechanisms for 4 Types of Common Anti-MRSA Antibiotics in Staphylococcus aureus: A Comprehensive Review. Microb. Pathog 2021, 156, 104915. [DOI] [PubMed] [Google Scholar]
- (13).Vestergaard M; Frees D; Ingmer H. Antibiotic Resistance and the MRSA Problem. Microbiol. Spectr 2019, 7, 7.2.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lee AS; de Lencastre H; Garau J; Kluytmans J; MalhotraKumar S; Peschel A; Harbarth S. Methicillin-Resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 2018, 4, 18033. [DOI] [PubMed] [Google Scholar]
- (15).Silber N; Matos de Opitz CL; Mayer C.; Sass P. Cell Division Protein FtsZ: From Structure and Mechanism to Antibiotic Target. Future Microbiol. 2020, 15, 801–831. [DOI] [PubMed] [Google Scholar]
- (16).Adams DW; Errington J. Bacterial Cell Division: Assembly, Maintenance and Disassembly of the Z Ring. Nat. Rev. Microbiol 2009, 7, 642–653. [DOI] [PubMed] [Google Scholar]
- (17).Margolin W. FtsZ and the Division of Prokaryotic Cells and Organelles. Nat. Rev. Mol. Cell Biol 2005, 6, 862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lund VA; Wacnik K; Turner RD; Cotterell BE; Walther CG; Fenn SJ; Grein F; Wollman AJ; Leake MC; Olivier N; Cadby A; Mesnage S; Jones S; Foster SJ Molecular Coordination of Staphylococcus aureus Cell Division. Elife 2018, 7, No. e32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Pinho MG; Errington J. Dispersed Mode of Staphylococcus aureus Cell Wall Synthesis in the Absence of the Division Machinery. Mol. Microbiol 2003, 50, 871–881. [DOI] [PubMed] [Google Scholar]
- (20).Haydon DJ; Stokes NR; Ure R; Galbraith G; Bennett JM; Brown DR; Baker PJ; Barynin VV; Rice DW; Sedelnikova SE; Heal JR; Sheridan JM; Aiwale ST; Chauhan PK; Srivastava A; Taneja A; Collins I; Errington J; et al. An Inhibitor of FtsZ with Potent and Selective Anti-Staphylococcal Activity. Science 2008, 321, 1673–1675. [DOI] [PubMed] [Google Scholar]
- (21).Elsen NL; Lu J; Parthasarathy G; Reid JC; Sharma S; Soisson SM; Lumb KJ Mechanism of Action of the Cell-Division Inhibitor PC190723: Modulation of FtsZ Assembly Cooperativity. J. Am. Chem. Soc 2012, 134, 12342–12345. [DOI] [PubMed] [Google Scholar]
- (22).Tan CM; Therien AG; Lu J; Lee SH; Caron A; Gill CJ; Lebeau-Jacob C; Benton-Perdomo L; Monteiro JM; Pereira PM; Elsen NL; Wu J; Deschamps K; Petcu M; Wong S; Daigneault E; Kramer S; Liang L; et al. Restoring Methicillin-Resistant Staphylococcus aureus Susceptibility to β-Lactam Antibiotics. Sci. Transl. Med 2012, 4, 126ra135−126ra135. [DOI] [PubMed] [Google Scholar]
- (23).Stokes NR; Baker N; Bennett JM; Berry J; Collins I; Czaplewski LG; Logan A; Macdonald R; Macleod L; Peasley H; Mitchell JP; Nayal N; Yadav A; Srivastava A; Haydon DJ An Improved Small-Molecule Inhibitor of FtsZ with Superior In Vitro Potency, Drug-Like Properties, and In Vivo Efficacy. Antimicrob. Agents Chemother 2013, 57, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Stokes NR; Baker N; Bennett JM; Chauhan PK; Collins I; Davies DT; Gavade M; Kumar D; Lancett P; Macdonald R; MacLeod L; Mahajan A; Mitchell JP; Nayal N; Nayal YN; Pitt GRW; Singh M; Yadav A; et al. Design, Synthesis and Structure-Activity Relationships of Substituted Oxazole-Benzamide Antibacterial Inhibitors of FtsZ. Bioorg. Med. Chem. Lett 2014, 24, 353–359. [DOI] [PubMed] [Google Scholar]
- (25).Chiodini G; Pallavicini M; Zanotto C; Bissa M; Radaelli A; Straniero V; Bolchi C; Fumagalli L; Ruggeri P; De Giuli Morghen C; Valoti E. Benzodioxane-Benzamides as New Bacterial Cell Division Inhibitors. Eur. J. Med. Chem 2015, 89, 252–265. [DOI] [PubMed] [Google Scholar]
- (26).Kaul M; Mark L; Zhang Y; Parhi AK; Lyu YL; Pawlak J; Saravolatz S; Saravolatz LD; Weinstein MP; LaVoie EJ; Pilch DS TXA709, an FtsZ-Targeting Benzamide Prodrug with Improved Pharmacokinetics and Enhanced In Vivo Efficacy against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother 2015, 59, 4845–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Straniero V Pallavicini M; Chiodini G; Zanotto C; Volonte L; Radaelli A; Bolchi C; Fumagalli L; Sanguinetti M; Menchinelli G; Delogu G; Battah B; De Giuli Morghen C; Valoti E 3-(Benzodioxan-2-ylmethoxy)-2,6-Difluorobenzamides Bearing Hydrophobic Substituents at the 7-Position of the Benzodioxane Nucleus Potently Inhibit Methicillin-Resistant Sa and Mtb Cell Division. Eur. J. Med. Chem 2016, 120, 227–243. [DOI] [PubMed] [Google Scholar]
- (28).Fujita J; Maeda Y; Mizohata E; Inoue T; Kaul M; Parhi AK; LaVoie EJ; Pilch DS; Matsumura H. Structural Flexibility of an Inhibitor Overcomes Drug Resistance Mutations in Staphylococcus aureus FtsZ. ACS Chem. Biol 2017, 12, 1947–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Deng J; Zhang T; Li B; Xu M; Wang Y. Design, Synthesis and Biological Evaluation of Biphenyl-Benzamides as Potent FtsZ Inhibitors. Eur. J. Med. Chem 2022, 239, 114553. [DOI] [PubMed] [Google Scholar]
- (30).Andreu JM; Schaffner-Barbero C; Huecas S; Alonso D; Lopez-Rodriguez ML; Ruiz-Avila LB; Núñez-Ramírez R; Llorca O; Martín-Galiano AJ The Antibacterial Cell Division Inhibitor PC190723 Is an FtsZ Polymer-Stabilizing Agent That Induces Filament Assembly and Condensation. J. Biol. Chem 2010, 285, 14239–14246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kaul M; Mark L; Zhang Y; Parhi AK; LaVoie EJ; Pilch DS Pharmacokinetics and In Vivo Antistaphylococcal Efficacy of TXY541, a 1-Methylpiperidine-4-Carboxamide Prodrug of PC190723. Biochem. Pharmacol 2013, 86, 1699–1707. [DOI] [PubMed] [Google Scholar]
- (32).Kaul M; Mark L; Zhang Y; Parhi AK; LaVoie EJ; Pilch DS An FtsZ-Targeting Prodrug with Oral Antistaphylococcal Efficacy In Vivo. Antimicrob. Agents Chemother 2013, 57, 5860−5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Matsui T; Yamane J; Mogi N; Yamaguchi H; Takemoto H; Yao M; Tanaka I. Structural Reorganization of the Bacterial Cell-Division Protein FtsZ from Staphylococcus aureus. Acta Crystallogr. D Biol. Crystallogr 2012, 68, 1175–1188. [DOI] [PubMed] [Google Scholar]
- (34).Stokes NR; Sievers J; Barker S; Bennett JM; Brown DR; Collins I; Errington VM; Foulger D; Hall M; Halsey R; Johnson H; Rose V; Thomaides HB; Haydon DJ; Czaplewski LG; Errington J. Novel Inhibitors of Bacterial Cytokinesis Identified by a Cell-Based Antibiotic Screening Assay. J. Biol. Chem 2005, 280, 39709–39715. [DOI] [PubMed] [Google Scholar]
- (35).2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; World Health Organization: Geneva, 2021. [Google Scholar]
- (36).Ferrer-González E; Fujita J; Yoshizawa T; Nelson JM; Pilch AJ; Hillman E; Ozawa M; Kuroda N; Al-Tameemi HM; Boyd JM; LaVoie EJ; Matsumura H; Pilch DS StructureGuided Design of a Fluorescent Probe for the Visualization of FtsZ in Clinically Important Gram-Positive and Gram-Negative Bacterial Pathogens. Sci. Rep 2019, 9, 20092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ferrer-González E; Huh H; Al-Tameemi HM; Boyd JM; Lee SH; Pilch DS Impact of FtsZ Inhibition on the Localization of the Penicillin Binding Proteins in Methicillin-Resistant Staphylococcus aureus. J. Bacteriol 2021, 203, No. e0020421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ferrer-González E; Kaul M; Parhi AK; LaVoie EJ; Pilch DS β-Lactam Antibiotics with a High Affinity for PBP2 Act Synergistically with the FtsZ-Targeting Agent TXA707 against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother 2017, 61, No. e00863–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kaul M; Ferrer-González E; Mark L; Parhi AK; LaVoie EJ; Pilch DS Combination with a FtsZ Inhibitor Potentiates the in Vivo Efficacy of Oxacillin against Methicillin-Resistant Staphylococcus aureus. Med. Chem. Res 2022, 31, 1705–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, 2018. [Google Scholar]
- (41).Otwinowski Z; Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (42).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Emsley P; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (46).Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zhang H; Kunadia A; Lin Y; Fondell JD; Seidel D; Fan H. Identification of a Strong and Specific Antichlamydial N-acylhydrazone. PLoS One 2017, 12, No. e0185783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Stiles BG; Campbell YG; Castle RM; Grove SA Correlation of Temperature and Toxicity in Murine Studies of Staphylococcal Enterotoxins and Toxic Shock Syndrome Toxin 1. Infect. Immun 1999, 67, 1521–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.