

Abstract
While it is experimentally supported that impaired myocardial vascularization contributes to a mismatch between myocardial oxygen demand and supply, a mechanistic basis for disruption of coordinated tissue growth and angiogenesis in heart failure remains poorly understood. Silencing strategies that impair microRNA biogenesis have firmly implicated microRNAs in the regulation of angiogenesis, and individual microRNAs prove to be crucial in developmental or tumor angiogenesis. A high-throughput functional screening for the analysis of a whole-genome microRNA silencing library with regard to their phenotypic effect on endothelial cell proliferation as a key parameter, revealed several anti- and pro-proliferative microRNAs. Among those was miR-216a, a pro-angiogenic microRNA which is enriched in cardiac microvascular endothelial cells and reduced in expression under cardiac stress conditions. miR-216a null mice display dramatic cardiac phenotypes related to impaired myocardial vascularization and unbalanced autophagy and inflammation, supporting a model where microRNA regulation of microvascularization impacts the cardiac response to stress.
Keywords: microRNAs, angiogenesis, endothelial cells, cardiac remodeling, heart failure, autophagy
Graphical abstract
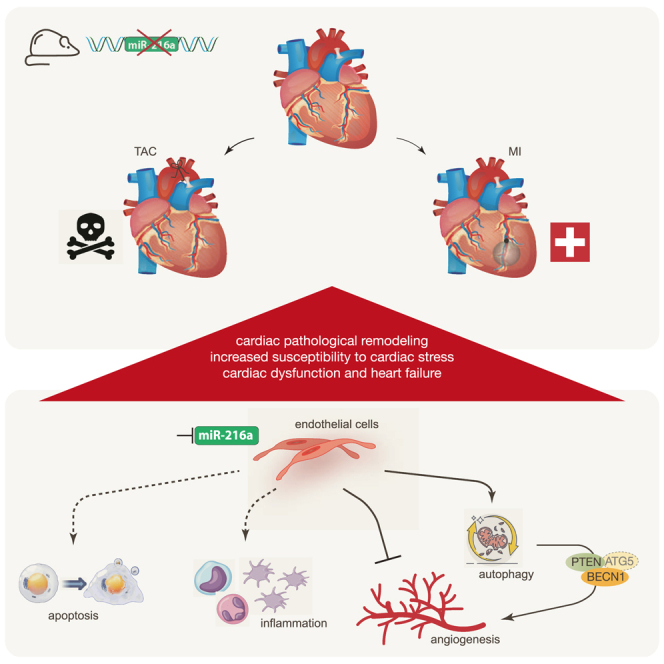
Da Costa Martins and colleagues identified miR-216a as a pro-angiogenic microRNA, enriched in cardiac microvascular endothelial cells and downregulated under cardiac stress conditions. miR-216a null mice display dramatic cardiac phenotypes related to impaired myocardial vascularization due to unbalanced autophagy and inflammation, supporting a model where microRNA regulation of microvascularization impacts the cardiac response to stress.
Introduction
Angiogenesis, the formation of new blood vessels from pre-existing vessels, is a vital adaptive mechanism of the myocardium to either ischemic injury or to conditions of sustained increased hemodynamic demand. While pathological cardiac remodeling is characterized by dysfunctional blood vessel formation, deficient oxygen supply with subsequent loss and degeneration of cardiomyocytes (CMs), atrophy, and interstitial fibrosis, the molecular mechanisms underlying the contribution of impaired myocardial vascularization toward development of heart failure (HF) remain unclear.1,2 Chronic exposure of the myocardium to pressure overload induces pathological hypertrophy with increased myocardial oxygen demand, which is not accompanied with sufficient vascularization, generating a mismatch in supply and demand. After a myocardial infarction (MI), rapid formation of collateral vessels that bypass the obstructed coronary artery is necessary for the survival of the myocardial region that surrounds the necrotic infarct.3 While CMs have been the main subject of extensive research in the past, the involvement of other cardiac cell types, including fibroblasts, endothelial cells (ECs), vascular smooth muscle cells, and various immune-competent cells, in the pathogenesis of HF only recently received more attention. The non-myocyte compartment also reacts to the same cardiac stress situations, affects neighboring cells, and alters their surrounding environment.4,5,6,7,8
Besides transcriptional activation, gene expression is also regulated at the post-transcriptional level by various classes of non-coding RNAs (ncRNAs). Notably, microRNAs (miRNAs), a class of small ncRNAs that function as guide molecules in RNA silencing, target most protein-coding transcripts, and are involved in nearly all developmental and pathological processes by coordinately targeting pathways of functionally related genes. One single miRNA can simultaneously target multiple genes with related functions, acting as a fine tuner of entire signaling pathways. While perturbation of cardiac miRNA biogenesis results in HF9,10 and dysregulated angiogenesis,11 several individual miRNAs were implicated in the development of tumors and the supporting angiogenesis.12 To date, only a select number of miRNAs have been documented to participate in post-MI angiogenesis,11,13,14,15 while to date no miRNA has been identified that regulates cardiac angiogenesis in non-ischemic HF.
Here, we report on a high content screening to identify miRNAs that regulate the proliferative capacity of ECs, from a whole-genome miRNA library of 750 different miRNA inhibitory molecules, with the ultimate goal of better understanding the regulatory mechanisms driving angiogenesis under physiological and pathological conditions. We identified dozens of miRNAs with as of yet undescribed anti- or pro-proliferative capacity in ECs, and discovered miR-216a, a known regulator of carcinoma growth and development,16,17,18 as an endogenous small ncRNA with strong proliferative effects. Gene deletion strategies of miR-216a in mice revealed spontaneous cardiac remodeling under baseline conditions, characterized by capillary rarefaction, reduced myocardial tissue oxygenation, rapid onset of HF, and strongly reduced survival following MI or under chronic pressure overload conditions. Mechanistically, lower expression levels of miR-216a directly result in increased EC autophagy and dysfunction by targeting phosphatase and tensin homolog (PTEN) and beclin-1 (BECN1), but also affect apoptosis, cell proliferation, cell-cell adhesion, cytoskeleton dynamics, and inflammatory response, supporting a mechanistic model of pathological cardiac remodeling where ECs play an essential role in the myocardial stress response.
Results
Identification of miR-216a as a regulator of EC proliferation
To identify miRNAs that alter the proliferative capacity of the endothelium we performed a fluorescence microscopy-based high-throughput screening in human umbilical vein endothelial cells (HUVECs). By using a locked nucleic acid (LNA) miRNA inhibitor library targeting 753 different miRNAs (listed in miRBase v.19, http://mirbase.org; Figure 1A), we identified miR-216a, the inhibition of which impaired EC proliferation 52 h after inhibitor transfection (Figure 1B; Table S1). From the many candidates, we have previously observed miR-216a to be downregulated in gene microarrays comparing myocardial tissue under healthy and under stress conditions.19 Although not specifically expressed in the heart (Figure S1A), miR-216a baseline levels were easily detectable in cardiac tissue by northern blotting, but significantly downregulated under cardiac stress conditions induced by sustained pressure overload (Figure 1C) or MI (Figure 1D), which suggests its contribution in maintaining cardiac homeostasis. Validation experiments confirmed the initial screening showing that inhibition of miR-216a leads to inhibition of EC proliferation while its overexpression significantly increases cell proliferative capacity (Figures 1E–1G). This was assessed by staining with two different proliferation markers Ki67, which is expressed in cells throughout the cell cycle and absent in G0, and 5-ethanyl-2′-deoxyuridine (EdU), which is incorporated in cells during the S-phase.20
Figure 1.

High-content screening identifies microRNAs regulating endothelial cell proliferation
(A) Screening strategy and workflow used to detect microRNAs that influence endothelial cell proliferation. (B) Distribution of standardized enrichment scores (Z scores) for the entire library of 753 miRNA-LNA inhibitors, calculated based on the EdU-positive human umbilical vein endothelial cells (HUVECs) for each LNA inhibitor and relative to the scrambled LNA. Inhibition of miR-216a-5p was found to significantly reduce the proliferation rate and is marked with a red dot. (C) Northern blotting analysis of miR-216a-5p expression in hearts of mice subjected to transverse aortic constriction (TAC) (upper panel) and quantification of the corrected northern blot signal (lower panel). Rnu6-2 was used as a loading control. n refers to number of hearts: n = 4 (sham) and n = 6 (TAC). (D) Northern blotting analysis of miR-216a-5p expression in hearts of mice subjected to myocardial infarction (MI) (upper panel) and quantification of the corrected northern blot signal (lower panel). Rnu6-2 was used as a loading control. n refers to number of hearts: n = 4 (sham) and n = 6 (MI). (E) Representative images of cultured HUVECs stained with Hoechst (blue) and 5-ethanyl-2′-deoxyuridine (Edu) (red) 24 h after transfection with miR-216a-5p-LNA-inhibitor or precursor-miR-216a-5p. Scale bar, 100 μm. (F and G) Quantification of HUVEC proliferation 24 h after transfection with an LNA or a precursor molecule either scrambled or specific for miR-216a-5p, represented as percentage of Edu-positive cells (F) and proliferation antigen Ki67-positive cells (G) to total cell count; n refers to independent experiments, n = 3 (10 microscopic fields/condition/experiment). In all panels numerical data are presented as mean (error bars show SEM); statistical significance was calculated using two-tailed unpaired t test. ∗p < 0.05 vs. control group.
Genomic structure of miR-216a and targeted deletion in mice
miR-216a is highly conserved across vertebrates, located in an intergenic region on chromosome 2 in the human genome, clustered together with miR-216b and miR-217, and transcribed together with a long ncRNA (lncRNA) (MIR217HG-001, Figure 2A) within the opposite strand of a longer lncRNA (RP11-481J13.1-001, Figure 2A). Although downregulation of each of the three miRNAs revealed to have an effect on EC proliferation (Table S1), only miR-216a expression was altered after exposing wild-type (WT) mice to cardiac stress conditions such as pressure overload and MI (Figures 1C, 1D, and S1B–S1E). To explore the biological role of miR-216a in vivo we generated a targeted deletion of miR-216a in mice by employing a Cre-Lox recombination strategy21 (Figures 2B and 2C). The generated homozygous miR-216a null mice (knockout [KO]) were born at Mendelian ratios and were fertile. Northern blotting analysis confirmed the absence of miR-216a expression in the heart (Figure 2D) of KO mice, while the expression of miR-216b, miR-217, and the two host lncRNAs in the mouse genome remained unaltered (Figure S1F). Intriguingly, gross examination of the hearts from miR-216a KO mice at 8–12 weeks at baseline revealed hypertrophic phenotypes reflected by increased heart weight to body weight ratios and increased CM surface areas when compared with WT hearts (Figures 2E–2G). Moreover, this cardiac hypertrophic growth was accompanied by decreased cardiac ejection fraction (EF) (Figure 2H) and increased left ventricular internal diameters (Figure 2I). These data indicate that miR-216a is necessary to maintain cardiac homeostasis and suggest that any stress condition leading to its reduced expression will result in pathological cardiac remodeling.
Figure 2.

miRNA-216a genomic location and targeting strategy
(A) Schematic representation of hsa-miR-216a-5p genomic localization (top panel) and precursor sequence (bottom panel). In the human genome, miR-216a is in a cluster together with miR-216b and miR-217, which is transcribed together within a long non-coding RNA MIR217HG-001 within the opposite strand of a longer long non-coding RNA RP11-481J13.1-001. The mature miR-216a-5p strand is conserved among species (bottom panel). (B) Strategy for targeting of miR-216a. The targeting vector, targeted allele, and null allele for miR-216a are shown. LoxP sites were introduced flanking the genomic region encompassing both the 71-bp pre-miR-216a as well as a neomycin resistance cassette flanked by two FRT sites, which allowed for bFLP recombinase-mediated excision and Cre-mediated removal of the targeted region of the miR-216a locus. Sizes of probes for Southern blotting analysis are shown. (C) Verification of homologous recombination strategy by Southern blot analysis based on BgII digestion. The wild-type (WT) allele yielded a 5.1 kb DNA fragment, whereas successful targeting (mut) results in both 5.1 and 11.9 kb fragments in heterozygous miR-216aneo/+. Removal of the neomycin cassette and Cre-mediated excision of the floxed miR-216a target region, an expected fragment of 7.9 kb was observed (null allele). (D) Northern blotting analysis of miR-216a-5p expression in hearts from WT and miR-216a knockout (KO) mice. Rnu6-2 was used as a loading control. (E) Representative images of whole hearts (top panel) and wheat germ agglutinin (WGA)-stained (bottom panel) histological sections of WT and KO hearts. Scale bars, 5 mm (top panel) and 50 μm (bottom panel). (F) Gravimetric analysis of corrected heart weights in WT and KO mice; n refers to number of hearts: n = 4 (WT) and n = 5 (KO). (G) Quantification of cardiomyocyte surface area from WT and KO mice, based on 150 cells measured per heart; n refers to number of hearts: n = 4 (WT) and n = 5 (KO). (H) Quantification of ejection fraction (EF) and (I) left ventricular internal diameter (LVID) during systole in WT and KO mice; n refers to number of hearts: n = 4 (WT) and n = 5 (KO). In all panels, numerical data are presented as mean (error bars show SEM); statistical significance was calculated using two-tailed unpaired t test. ∗p < 0.05 vs. control group.
miR-216a-null mice display severe cardiac dysfunction and reduced survival following cardiac pressure overload
To correlate the observed baseline phenotypes to the function of miR-216a in cardiac disease, we subjected miR-216a null mice to transverse aortic constriction (TAC) to induce pressure overload of the left ventricle. While we initially intended to follow up the animals for 4 weeks after TAC surgery, we reduced the length of the study to 2 weeks (Figure 3A) due to a severe and unexpected mortality of 53% observed in the KO mice in the first 2 days of pressure overload, which increased to 69% in the following 3 days (Figure 3B). Remarkably, miR-216a ablation sufficed to provoke signs of cardiac disease as demonstrated by the development of cardiac hypertrophy in sham-operated mice (Figures 3C and 3D). While the hearts of the miR-216a-KO mice at baseline seem to have a comparable size with the WT mice subjected to TAC, the cardiac four-chamber view reveals the development of much thicker ventricular walls under pressure overload, supporting the heart weight to body weight ratios observed (Figures 3C and 3D). To further characterize the cardiac phenotype of miR-216a KO mice at baseline, we examined CM size and the formation of fibrotic lesions. Wheat germ agglutinin (WGA) staining of myocardial sections showed a clear increase in CM size in KO mice compared with WT controls after sham surgery (Figures 3C and 3E). Sirius Red staining also revealed areas of fibrosis in the miR-216a KO mice in contrast to WT mice, where no fibrosis was detected (Figures 3F and 3G). Ablation of miR-216a also resulted in decreased number of capillaries per CM reflecting some degree of capillary rarefaction in these animals at baseline (Figures 3F and 3H). These results were corroborated by a decrease in Pecam1 mRNA in the KO mice at baseline and with further decrease under stress conditions (Figure 3I).
Figure 3.

miR-216a ablation induces a spontaneous pathological cardiac phenotype that is aggravated under pressure overload
(A) Design of the study. WT and miR-216a KO mice were subjected to TAC or sham surgery and cardiac geometry and function were determined by serial Doppler echocardiography at 2 weeks after surgery. (B) Kaplan-Meier survival curves for WT and KO mice subjected to sham or transverse aortic constriction (TAC) for 2 weeks, clearly showing high mortality rates in KO mice under cardiac pressure overload induced by TAC. (C) Representative images of whole hearts (top panel) and four-chamber view (second panel), haematoxylin and eosin (H&E)-stained (third panel) histological sections from WT and KO hearts from mice subjected to either sham or TAC surgery. Scale bars, 5 mm (top two panels) and 50 μm (bottom two panels). (D) Gravimetric analysis of corrected heart weights in WT and KO mice subjected to either sham or TAC surgery; n refers to number of hearts: n = 5 WT sham, n = 7 KO sham, n = 6 WT TAC, and n = 3 KO TAC. (E) Quantification of cardiomyocyte surface area from WT and KO mice subjected to either sham or TAC surgery based on 150 cells measured per heart; n refers to number of hearts: n = 3 (WT sham), n = 3 (KO sham), n = 5 (WT TAC), and n = 3 (KO TAC). (F) Representative image of Sirius Red-stained (upper panels), WGA-stained, and IB4-stained (second panel) histological sections from WT and KO hearts from mice subjected to either sham or TAC surgery. Scale bars, 50 μm (Sirius Red) and 100 μm (IB4). (G) Quantification of collagen deposition from images of histological sections stained for Sirius Red, based on 30 microscopic fields/heart, n = 3 hearts/group. (H) Capillaries in myocardial sections of the different animal groups were identified by isolectin B4 immunohistochemistry combined with WGA and, from the images obtained, we determined the ratio of capillaries per cardiomyocyte ratios based on 30 microscopic fields/heart, n = 3 hearts/group. (I) Real-time PCR analysis of transcript abundance for pecam1 in hearts from WT and KO mice subjected to either sham or TAC, n refers to number of hearts: n = 8 WT sham, n = 10 KO sham, n = 8 WT TAC, and n = 3 KO TAC. (J and K) Quantification of ejection fraction (EF) (J) and left ventricular internal diameter during systole (LVIDs) (K) in WT and KO mice subjected to either sham or TAC surgery, n refers to number of hearts: n = 8 WT sham, n = 10 KO sham, n = 8 WT TAC, and n = 3 KO TAC. (L and M) Quantitative real-time PCR analysis of atrial natriuretic peptide (nppa) (L) and β-myosin heavy chain 7 (myh7) (M) in hearts of WT and KO mice subjected to either sham or TAC surgery; n refers to number of hearts: n = 5 WT sham, n = 7 KO sham, n = 6 WT TAC, and n = 3 KO TAC. In all panels numerical data are presented as mean (error bars show SEM); statistical significance was calculated using two-tailed unpaired t test when comparing two experimental groups or two-way ANOVA followed by Tukey’s multiple comparison test when comparing more than two experimental groups. ∗p < 0.05 vs. corresponding control group, #p < 0.05 vs. experimental group.
While pressure overload induced pathological cardiac remodeling in WT hearts, as reflected by increased myocyte disarray, CM hypertrophy, and collagen deposition, all these underlying defects were exacerbated in pressure-overloaded miR-216a KO mice that were able to survive similar cardiac stress conditions (Figures 3C–3G). Furthermore, non-invasive echo/Doppler echocardiography demonstrated that targeted deletion of miR-216a provoked systolic contractile defects already at baseline (Figures 3J and 3K; Table 1). Exposing KO mice to pressure overload remarkably aggravated cardiac dysfunction as confirmed by the severe systolic contractile cardiac defects observed (Figure 3J; Table 1). The underlying defects in the hearts of miR-216a KO mice may be related to the re-activation of the “fetal” cardiac genes natriuretic peptide type A (Nppa) and myosin heavy polypeptide 7 (Myh7), as both genes were upregulated in the KO hearts under sham conditions, even though WT and KO mice show similar increased levels under stress conditions (Figures 3L and 3M). Taken together, these findings indicate that miR-216a ablation triggered CM hypertrophy, cardiac dysfunction, and cardiac capillary rarefaction, a response that is aggravated under pressure overload-induced cardiac stress.
Table 1.
Morphometric and echocardiographic characteristics of WT and miRNA-216a KO mice subjected to 4 weeks of sham or TAC
| Sham |
TAC |
|||
|---|---|---|---|---|
| WT | miR-216a KO | WT | miR-216a KO | |
| N | 6 | 10 | 8 | 3 |
| TL (mm) | 19.71 ± 0.46 | 19.58 ± 0.15 | 19.29 ± 0.42 | 19.32 ± 0.46 |
| BW (g) | 21.85 ± 1.50 | 21.97 ± 1.22 | 21.48 ± 1.56 | 21.93 ± 1.10 |
| HW (g) | 0.12 ± 0.009 | 0.15 ± 0.01 | 0.21 ± 0.02a | 0.19 ± 0.02a |
| HW/BW | 0.006 ± 0.0001 | 0.007 ± 0.0002 | 0.01 ± 0.0004a | 0.01 ± 0.0006a |
| HW/TL (g/mm) | 0.006 ± 0.0004 | 0.008 ± 0.0004 | 0.01 ± 0.001a | 0.01 ± 0.0001a |
| LV mass (mg) | 90.01 ± 4.28 | 93.29 ± 0.61 | 166.43 ± 14.84a | 214.36 ± 42.16a |
| LV mass/BW (mg/g) | 4.07 ± 0.39 | 4.06 ± 0.20 | 7.18 ± 0.34a | 10.0 ± 2.45a,b |
| LV mass/TL (mg/mm) | 4.48 ± 0.39 | 4.47 ± 0.27 | 7.98 ± 0.63a | 11.05 ± 2.007a,b |
| IVSd (mm) | 0.96 ± 0.04 | 0.80 ± 0.04b | 1.18 ± 0.05a | 1.82 ± 0.18a,b |
| IVSs (mm) | 1.40 ± 0.05 | 1.21 ± 0.05b | 1.49 ± 0.03 | 1.98 ± 0.15a,b |
| LVIDd (mm) | 3.57 ± 0.06 | 3.94 ± 0.12 | 3.73 ± 0.15 | 3.06 ± 0.23a,b |
| LVIDs (mm) | 2.11 ± 0.08 | 2.47 ± 0.14a | 2.38 ± 0.09a | 2.79 ± 0.22b |
| LVPWd (mm) | 0.81 ± 0.03 | 0.83 ± 0.05 | 1.07 ± 0.03a | 1.67 ± 0.14a,b |
| LVPWs (mm) | 1.28 ± 0.04 | 1.08 ± 0.05 | 1.32 ± 0.05a | 1.78 ± 0.16a,b |
| FS (%) | 40.96 ± 1.82 | 30.24 ± 1.66b | 32.55 ± 1.23a | 14.70 ± 2.69a |
| EF (%) | 72.42 ± 2.13 | 57.87 ± 2.60b | 61.08 ± 3.38a | 32.45 ± 5.55a |
Data are expressed as means ± SEM. BW, body weight; LV, left ventricular; IVSd, interventricular septal thickness at end-diastole; IVSs, interventricular septal thickness at end-systole; LVIDd, left ventricular internal dimension at end-diastole; LVIDs, left ventricular internal dimension at end-systole; LVPWd, left ventricular posterior wall thickness at end-diastole; LVPWs, left ventricular posterior wall thickness at end-systole; FS, fractional shortening; EF, ejection fraction.
Indicates p < 0.05 vs. WT sham group.
Indicates p < 0.05 vs. experimental group.
miR-216a-null mice are sensitized to MI
To assess whether miR-216a ablation incites a different remodeling response to ischemic cardiac injury, we induced MI in both WT and KO mice by occluding the left anterior descending coronary artery (Figure 4A). Mortality at 4 weeks after sham or MI surgery was similar for WT and KO mice, suggesting a distinct remodeling response induced by MI compared with TAC (Figure 4B). Myocardial sections WT and KO mice subjected to MI were taken sequentially from the ligation site in the left ventricle toward the apex, and stained with Sirius Red to evaluate differences in the extension of the infarct area. miR-216a deficiency triggered extensive fibrosis leading to larger infarcts (Figures 4C and 4D), and resulted in larger and more dilated hearts compared with WT (Figures 4E–4G). Furthermore, the extension of the infarct area directly correlated with the degree of cardiac dysfunction as KO mice showed aggravated contractile defects compared with WT animals after MI (Figures 4H and 4I; Table 2). The severity of the cardiac phenotypes after MI was confirmed by stronger re-activation of the “fetal” cardiac genes Nppa and Myh7 compared with WT animals, and the exacerbated phenotypes observed in the KO mice was reflected by greater upregulation of Myh7 (Figures 4J and 4K). These data reinforced our initial observations that miR-216a ablation provokes cardiac defects and sensitized the heart to ischemia-induced cardiac stress.
Figure 4.

miR-216a silencing increases susceptibility to myocardial infarction
(A) Design of study. WT and miR-216a KO mice were subjected to myocardial infarction (MI) or sham surgery and cardiac geometry and function were determined by serial Doppler echocardiography at 4 weeks after surgery. (B) Kaplan-Meier survival curve for WT and KO mice subjected to sham or MI. (C and D) Representative images of sequential transversal sections of hearts from WT and KO mice subjected to MI (C), stained with Sirius-red for visualization and quantification of the infarct areas (relative to left ventricle area) in the different groups (D) showing significantly larger infarct areas in the KO mice hearts; n refers to number of hearts: n = 4 WT MI, n = 3 KO MI. Scale bar, 5 mm. (E) Gravimetric analysis of corrected heart weights in WT and KO mice subjected to either sham or MI surgery; n refers to number of hearts: n = 5 WT sham, n = 9 KO sham, n = 6 WT MI, and n = 8 KO MI. (F) Representative images of WGA-stained histological cardiac sections from WT and KO mice subjected to sham or MI. Scale bar, 50 μm. (G) Quantification of cardiomyocyte surface area from WT and KO mice subjected to either sham or MI surgery based on 150 cells measured per heart; n = 3 hearts per group. (H and I) Quantification of EF (H) and LVIDs (I) in WT and KO mice subjected to either sham or MI; n refers to number of hearts: n = 6 WT sham, n = 11 KO sham, n = 8 WT MI, and n = 12 KO MI). (J and K) Quantitative real-time PCR analysis of atrial natriuretic peptide (Nppa) (J) and β-myosin heavy chain 7 (Myh7) (K) in hearts of WT and KO mice subjected to either sham or MI surgery; n refers to number of hearts (n = 6 WT sham, n = 8 KO sham, n = 8 WT MI, and n = 8 KO MI). In all panels numerical data are presented as mean (error bars show SEM); statistical significance was calculated using two-tailed unpaired t test when comparing two experimental groups or two-way ANOVA followed by Tukey’s multiple comparison test when comparing more than two experimental groups. ∗p < 0.05 vs. corresponding control group, #p < 0.05 vs. experimental group.
Table 2.
Morphometric and echocardiographic characteristics of WT and miRNA-216a KO mice subjected to 4 weeks of sham or MI
| Sham |
TAC |
|||
|---|---|---|---|---|
| WT | miR-216a KO | WT | miR-216a KO | |
| N | 6 | 11 | 8 | 12 |
| TL (mm) | 19.85 ± 0.10 | 19.38 ± 0.27 | 19.40 ± 0.35 | 19.33 ± 0.31 |
| BW (g) | 22.18 ± 1.49 | 28.45 ± 0.59b | 20.25 ± 1.26 | 23.73 ± 0.87a |
| HW (g) | 0.14 ± 0.01 | 0.20 ± 0.01 | 0.18 ± 0.02 | 0.25 ± 0.03 |
| HW/BW | 0.006 ± 0.0005 | 0.007 ± 0.0003 | 0.009 ± 0.001 | 0.010 ± 0.002 |
| HW/TL (g/mm) | 0.007 ± 0.0004 | 0.011 ± 0.0006b | 0.009 ± 0.0008 | 0.013 ± 0.0014b |
| LV mass (mg) | 97.48 ± 6.84 | 112.31 ± 4.01 | 112.04 ± 6.98 | 106.78 ± 6.15 |
| LV mass/BW (mg/g) | 4.36 ± 0.53 | 3.92 ± 0.19 | 5.81 ± 0.53 | 4.56 ± 0.33 |
| LV mass/TL (mg/mm) | 0.22 ± 0.03 | 0.2 ± 0.01 | 0.3 ± 0.03 | 0.24 ± 0.02 |
| IVSd (mm) | 0.89 ± 0.06 | 0.86 ± 0.03 | 0.72 ± 0.12 | 0.46 ± 0.10a |
| IVSs (mm) | 1.40 ± 0.07 | 1.27 ± 0.03 | 0.99 ± 0.15 | 0.57 ± 0.12a,b |
| LVIDd (mm) | 3.79 ± 0.14 | 4.22 ± 0.07 | 4.48 ± 0.30 | 5.32 ± 0.21b |
| LVIDs (mm) | 2.19 ± 0.13 | 2.90 ± 0.07b | 3.39 ± 0.33a | 4.53 ± 0.31a,b |
| LVPWd (mm) | 0.87 ± 0.04 | 0.86 ± 0.04 | 0.89 ± 0.10 | 0.78 ± 0.05 |
| LVPWs (mm) | 1.45 ± 1.89 | 1.29 ± 0.04 | 1.22 ± 0.10 | 1.06 ± 0.08 |
| FS (%) | 42.37 ± 1.89 | 31.33 ± 0.87b | 25.20 ± 1.26a | 15.94 ± 2.50a,b |
| EF (%) | 73.90 ± 2.10 | 59.44 ± 1.30b | 49.49 ± 4.17a | 32.44 ± 4.80a,b |
Data are expressed as means ± SEM. BW, body weight; LV, left ventricular; IVSd, interventricular septal thickness at end-diastole; IVSs, interventricular septal thickness at end-systole; LVIDd, left ventricular internal dimension at end-diastole; LVIDs, left ventricular internal dimension at end-systole; LVPWd, left ventricular posterior wall thickness at end-diastole; LVPWs, left ventricular posterior wall thickness at end-systole; FS, fractional shortening; EF, ejection fraction.
Indicates p < 0.05 vs. WT-sham group.
Indicates p < 0.05 vs. experimental group.
miR-216a-null mice display impaired cardiac endothelial function
miR-216a is barely detected in human CMs but expressed in human endothelium,22 and we were able to demonstrate a similar expression pattern for miR-216a in the murine heart when comparing primary cardiac ECs with other primary non-EC types such as CMs, fibroblasts, and inflammatory cells (CD45+) (Figure 5A). To address whether the effects observed upon modulation of miR-216a levels are mostly due to its effect on ECs, we assessed whether it can also affect the CM phenotype, as the second most expressing cells. When treating neonatal rat CMs with either specific inhibitor or precursor molecules, and their respective controls, we did not observe any affected phenotype, as revealed by no signs of hypertrophic growth determined by cell surface area and expression of Nppa (Figures 5B–5D). Furthermore, myocardial tissue sections derived from WT hearts were used for miR-216a detection by in situ hybridization and revealed a distinct capillary staining confirming confinement or enrichment of miR-216a expression to ECs in the murine heart (Figure 5E). Evaluation of cardiac capillary density by staining microvessels with Griffonia simplicifolia I (GSI), revealed a significant reduction in the KO hearts already at baseline, resembling the effect observed in WT animals after MI (Figures 5F and 5G). A further decline in cardiac capillary density was observed after subjecting KO mice to ischemic injury (Figures 5F and 5G). These effects were accompanied by capillary/CM mismatch since the number of capillaries per CM was lower in the KO mice at baseline and worsened after MI (Figure 5H). A decrease in Pecam1 expression observed in the KO mice at baseline and after MI further support cardiac microvascular impairment upon absence of miR-216a expression in the heart (Figure 5I).
Figure 5.

miR-216a silencing promotes capillary rarefaction and endothelial dysfunction
(A) Quantitative real-time PCR analysis of miR-216a expression in mouse primary cardiac cells. All different cell fractions, endothelial cells (EC); fibroblasts (FB), and inflammatory cells (CD45+) were compared with the cardiomyocyte fraction (CM); n refers to independent experiments (n = 4). (B) Confocal microscopy images of neonatal rat cardiomyocytes transfected with precursor molecules or LNA inhibitors specific for miR-216a, or their respective controls. Cells are stained for actinin alpha 1 (ACTN1) and with DAPI for nuclei visualization. Fibroblasts are marked in red. (C) Quantification of cell surface area from conditions in b based on 100 cells per condition, n = 3 independent experiments. (D) Real-time PCR analysis of transcript abundance for the fetal marker nppa in conditions in b (n = 3 independent experiments). (E) Whole-mount in situ hybridization (ISH) for miR-216 detection on KO mouse-derived cardiac tissue. Scale bar, 50 μm. (F) Representative images of histological sections of hearts from WT and KO mice subjected to sham or MI, where capillaries were stained with Griffonia simplicifolia I (GSI). Scale bar, 50 μm. (G) Quantification of cardiac capillary density based on 30 microscopic field/heart, n = 3 hearts/group and (H) cardiac capillary to cardiomyocyte ratios in histological sections of hearts from WT and KO mice subjected to sham or MI, based on 30 microscopic field/heart, n = 3 hearts/group. (I) Real-time PCR analysis of transcript abundance for pecam1 in hearts from WT and KO mice subjected to either sham or MI, n refers to number of hearts: n = 5 per group. (J–L) Myocardial tissue oxygenation analysis as assessed by combining photoacoustics (PA) with high frequency ultrasound. (J) Representative real-time images of focal tissue oxygen saturation in the myocardium of WT and KO mice. (K) PA intensity and (L) myocardial tissue oxygen saturation (stO2) in WT and KO mice. n refers to number of mice (n = 3 per group). (M) Quantitative real-time PCR analysis of miR-216a expression in different primary endothelial cell lines: human umbilical endothelial cells (HUVEC), human lung microvascular endothelial cells (HLMEC), human dermal microvascular endothelial cells (HDMEC), human cardiac microvascular endothelial cells (HCMEC), and human retinal microvascular endothelial cells (HRMEC), n = 4 independent experiments. (N) Representative images of the wound healing/migration assay on HUVECs after transfection with scrambled precursor or LNA, a miR-216a precursor molecule or an LNA-miR-216, for modulation of miR-216a expression, and (O) quantification of respective wound closure rate (n = 3 independent experiments for each condition). Scale bar, 200 μm. (P) Representative images of tubulogenesis assay on HUVECs after transfection with scrambled precursor or LNA, a miR-216a precursor molecule or an LNA-miR-216, and (Q) respective quantification (n = 3 independent experiments for each condition). Scale bar, 500 μm. In all panels numerical data are presented as mean (error bars show SEM); statistical significance was calculated using two-tailed unpaired t test when comparing two experimental groups or two-way ANOVA followed by Tukey’s multiple comparison test when comparing more than two experimental groups. ∗p < 0.05 vs. corresponding control group, #p < 0.05 vs. experimental group.
The heart has a high oxygen demand and any alterations in the cardiac microvascular network will directly affect cardiac function and performance.23 To address whether capillary rarefaction in miR-216a mutant mice limits cardiac oxygen supply, we used a combination of photoacoustics (PA) and high-frequency ultrasound (US) for real-time assessment of cardiac tissue oxygenation status in vivo. This technique is based on the different absorption spectra of hemoglobin upon oxygenation and calculates focal tissue oxygen saturation after using dual-wavelength PA to separately measure tissue concentration of hemoglobin and deoxyhemoglobin.24,25,26 Scanning of the hearts with PA and B-mode US detected a different PA pattern in WT and KO hearts, with the latter displaying much lower PA intensities as depicted in the B-mode and oxygen-saturation images. These measurements further revealed significantly lower myocardial oxygen saturation in the KO compared with the WT mice (Figures 5J–5L).
As the specific function of miR-216a in cardiac angiogenesis is still unknown, we next analyzed the contribution of miR-216a in maintaining basic angiogenic endothelial properties by modulating its expression on ECs and assessing how it affected their functionality. Interestingly, when comparing miR-216a expression levels among different types of human microvascular ECs in relation to the commonly used HUVECs, we observed that, overall, microvascular ECs display higher levels of miR-216a at baseline (Figure 5M). Moreover, human cardiac microvascular ECs (HCMECs) showed the highest miR-216a expression levels compared with microvascular ECs derived from other vascular beds (Figure 5M). We found that LNA inhibitor-mediated repression of endogenous miR-216a expression blocked EC migration as well as vascular network formation in Matrigel assays (Figures 5N–5Q). In agreement, forced overexpression of miR-216a boosted the migratory and network formation capacity of ECs (Figures 5N–5Q).
Altogether, our results imply that miR-216a, by promoting endothelial angiogenic capacity, sustains a functional cardiac microvascular network that is able to supply the myocardium with sufficient oxygen and nutrients in both healthy and disease conditions, and that alterations in miR-216a expression levels have dramatic consequences in cardiac tissue oxygenation leading to adverse cardiac remodeling.
The mir-216a KO mouse generated in this study is a full-body KO and therefore all cells display depletion of this specific miRNA. Because miR-216a is mostly expressed on cardiac ECs and we relate the cardiac phenotypes observed to impaired endothelial function, it would still be reasonable to consider whether other organs are affected by the depletion of miR-216a. To determine this, we have analyzed the mRNA expression of several endothelial markers in different tissues: heart, liver, lung, kidney, and skin. While we observed reduced expression of Pecam1, Vwf, and Flt1 in the myocardium, no changes were observed in the other tissues (Figures S2A–S2C). For Kdr no differences were observed, except in the liver where there was reduced expression (Figure S2D). These data suggest that the most effects of silencing miR-216a on endothelial function are cardiac and that the disease phenotypes that are observed in these mice are mainly cardiac related.
miR-216a links cardiac endothelial angiogenic capacity with autophagy via direct regulation of PTEN and BECN1
miR-216a has been associated with tumor growth and progression by regulating cell proliferation and angiogenesis,17 via direct binding to the PTEN gene, a tumor suppressor. To investigate whether similar processes take place in the heart, we first confirmed the presence of an evolutionary conserved miR-216a seed region in the 3′ UTR sequence of PTEN (Figure S3A). The functionality of this seed region was established by a reduction in PTEN 3′ UTR luciferase reporter activity after transient transfection of HUVECs with precursors for miR-216a, an effect that was absent when co-transfecting a PTEN 3′ UTR sequence harboring a mutated miR-216a seed region (Figure S3B). To further establish the functional interaction between miR-216a and PTEN, we performed quantitative real-time PCR and western blot analysis and observed derepression of PTEN, both at mRNA and protein level, upon inhibition of miR-216a in HUVECs (Figures 6A–6C).
Figure 6.

miR-216a links cardiac endothelial function and autophagy via regulation of PTEN
(A) Quantitative PCR analysis of PTEN in HUVECs transfected either with a scrambled LNA or an LNA-miR-216a (n = 4 independent experiments including each condition). (B) Western blot analysis of PTEN and two autophagy surrogate markers, LC3 and p62, in HUVECs transfected either with a scrambled LNA or an LNA-miR-216a. GAPDH was used as loading control. (C–E) Quantification of PTEN expression (C), p62 (D), and LC3-II (E) from conditions in (B); n = 3 independent experiments including each condition. (F) Western blot analysis of PTEN, LC3, and p62 in myocardial tissue of WT and KO mice. GAPDH was used as loading control. (G–I) Quantification of PTEN expression (G), p62 (H), and LC3-II (I) in experimental groups from (G); n = 3 independent experiments including each condition. (J) Quantitative PCR analysis of Pten in myocardial tissue of WT and KO mice subjected to sham or MI; n = 5 hearts per group. (K) Western blot analysis and (L) quantification of PTEN expression in myocardial tissue of WT and KO mice subjected to sham or MI; n = 3 hearts per group. (M) Experimental setup of modulation of PTEN expression levels in HUVECs, using a specific siRNA to achieve downregulation and an adenovirus for upregulation. (N) Quantification of HUVEC proliferation 48 h after transduction with an Ad-GFP (control) or an Ad-PTEN, represented as percentage of Edu-positive cells; n = 3 independent experiments (10 microscopic fields for each condition/experiment). (O) Quantification of HUVEC proliferation 48 h after transfection with a scrambled- or PTEN-siRNA, represented as percentage of Edu-positive cells; n = 3 independent experiments (10 microscopic fields for each condition/experiment). (P) Quantitative PCR analysis of BECN1 in HUVECs transfected either with a scrambled LNA or an LNA-miR-216a (n = 4 independent experiments including each condition). (Q) Quantitative PCR analysis of Becn1 in myocardial tissue of WT and KO mice subjected to sham or MI; n = 5 hearts per group. (R) Quantitative PCR analysis of ATG5 in HUVECs transfected either with a scrambled LNA or an LNA-miR-216a (n = 4 independent experiments including each condition). (S) Quantitative PCR analysis of Atg5 in myocardial tissue of WT and KO mice subjected to sham or MI; n = 5 hearts per group. (T) Experimental setup to inhibit either BECN1 or ATG5 expression levels in HUVECs upon downregulation of miR-216a, using a specific siRNA and an LNA probe, respectively. (U) Quantification of HUVEC proliferation 48 h after transfection with scrambled LNA, or an LNA-miR-216, and ctrl-sirna, BECN1-sirna or ATG5-siRNA, represented as percentage of Edu-positive cells; n = 3 independent experiments (10 microscopic fields for each condition/experiment). (V) Quantification of wound healing/migration assay on HUVECs after transfection with scrambled LNA, or an LNA-miR-216, and ctrl-sirna, BECN1-sirna or ATG5-siRNA, represented as wound closure rate (n = 3 independent experiments for each condition). In all panels numerical data are presented as mean (error bars show SEM); statistical significance was calculated using two-tailed unpaired t test when comparing two experimental groups or two-way ANOVA followed by Tukey’s multiple comparison test when comparing more than two experimental groups. ∗p < 0.05 vs. corresponding control group.
As autophagy and angiogenesis are normally associated, besides being anti-angiogenic, PTEN is also a pro-autophagic factor in different cell types,27,28,29 with a controversial role in the maintenance of cardiac homeostasis. To determine whether miR-216a inhibition in ECs modulates autophagic processes in vitro, we investigated the expression of autophagic marker genes in HUVECs after transfection with a specific miR-216a LNA inhibitor and found a dramatic increase in LC3 lipidation, whereas the autophagy substrate p62 levels showed a tendency to decrease (Figures 6B–6E). These results were further confirmed in vivo in miR-216a KO hearts, which, at baseline, already display elevated levels of PTEN accompanied by increased autophagy, as reflected by elevated LC3-II and reduced p62 expression levels (Figures 6F–6I). PTEN expression levels were further raised in miR-216a KO mice after subjection to MI but remained similar to the ones observed in WT animals under the same conditions (Figures 6J–6L). To determine whether we could mimic the effect of miR-216a inhibition by upregulation of PTEN we infected HUVECs with an Ad-PTEN but no effect on proliferation (Figures 6M and 6N) nor on migration or tube formation was observed (data not shown). In contrast, when inhibiting PTEN in HUVECs with a specific siRNA, EC proliferation was increased (Figures 6M and 6O), without additional effects on migration or tube formation (data not shown). Although these results demonstrate that the reduction of miR-216a stimulates cardiac autophagic processes in cardiac ECs by upregulation of PTEN, they also suggest that targeting of PTEN is not the main mechanism by which miR-216a regulates EC function and that other targets may be involved in the process.
The fact that miR-216a alters autophagy on ECs has been reported previously. In the aging endothelium, miR-216a was shown to directly target the essential autophagy gene Beclin1 (BECN1), known to control the early stages of autophagic vesicle formation.30 Furthermore, increased levels of BECN1 have been associated with impaired cell proliferation in cancer cells31 and excessive autophagy has been linked to impaired angiogenesis.32 To determine whether this pathway is also contributing to the cardiac phenotypes observed by us, we firstly confirmed the presence of an evolutionary conserved miR-216a seed region in the 3′ UTR sequence of BECN1 (Figure S3C). The functionality of this seed region was established by a reduction in BECN1 3′ UTR luciferase reporter activity after transient transfection of HUVECs with precursors for miR-216a, an effect that was absent when co-transfecting a BECN1 3′ UTR sequence harboring a mutated miR-216a seed region (Figure S3D). To further establish the functional interaction between miR-216a and BECN1, we performed quantitative real-time PCR analysis and observed derepression of BECN1 upon inhibition of miR-216a in HUVECs (Figure 6P) and in the hearts of miR-216a KO mice at baseline. This effect was even stronger when the animals were subjected to MI (Figure 6Q).
Although not a direct target of miR-216a, regulation of ATG5, a gene involved in autophagosomal precursor formation,33 was previously shown to be indirectly affected by miR-216a.34 Indeed, we observed an increase in ATG5 expression after inhibiting miR-216a in ECs (Figure 6R). Although we could observe a clear increase in Atg5 in the KO mice subjected to MI, we did not observe differences in these mice at baseline compared with the WT animals (Figure 6S). Interestingly, regulation of all three genes of interest, Pten, Becn1, and Atg5, was only affected on ECs and not on CMs that were derived from the miR-216a KO hearts (Figures S3E and S3F), supporting endothelial specificity of this regulatory pathway.
Moreover, as excessive autophagy is linked to impaired angiogenesis,31 we tested the effect of modulating BECN1 and ATG5 on HUVECs. To counteract the effect of decreasing miR-216a expression on the expression levels of BECN1 and ATG5, we inhibited each gene in HUVECs that were treated with a specific miR-216a LNA inhibitor (Figure 6T). At baseline levels of miR-216a, downregulation of each of these genes resulted in increased EC proliferation and migration capacity. Upon inhibition of miR-216a, when expression of both genes is elevated, downregulation of either BECN1 or ATG5 by specific siRNAs resulted in restored endothelial angiogenic capacity, reflected by increased proliferation and migration rates (Figures 6U and 6V).
An exacerbated upregulation of Pten, Becn1, and Atg5 was also observed in miR-216a KO mice that were subjected to TAC, suggesting that the severe phenotype observed in those mice may also be related to excessive autophagy on ECs. There, however, we did not observe an increase of Atg5 in the KO mice at baseline (Figures S3G–S3I).
Other mechanisms underlying the role of miR-216a in the cardiac response to ischemic injury
Next, and to better understand how LV remodeling is affected by disturbed levels of miR-216a, we performed RNA sequencing to assess the transcriptomic changes in total RNA from ECs transfected with either scrambled or miR-216a-specific precursor molecules. Transcriptome analysis identified 194 transcripts potentially regulated by miR-216a (Figure 7A; Table S2). Generation of a heatmap representing the most differentially expressed transcripts between the 2 experimental groups revealed 25 downregulated and 12 upregulated genes upon overexpression of miR-216a in ECs (Figure 7A). Genes that were downregulated, and therefore considered potential miR-216a direct target genes, were mostly associated with cellular components related to cell-cell adhesion and cytoskeletal dynamics (SCL1a1, SRGAP1, ANKRD), TGFB signaling (TGFBR2), apoptosis and/or autophagy (PIR, SDC4, BCL10), cell-cycle and DNA damage regulation (CCNJ, HDAC3, UBN1), leukocyte adhesion (ANKRD, ICAM1), EC dysfunction (SDC4, SDCBP), exosome biogenesis (SDC4, ANKRD, SLC1A1), and cardiac remodeling (ANKRD, HDAC3) (Figures 7A and 7B). The upregulated genes are associated with inflammation and proliferation of inflammatory cells (ANGPTL2, NARF), (cancer) cell proliferation (CYBRD1, ANGPTL, NARF), autophagy (BBC3, ANGPTL2), resistance to apoptosis (CYBRD1), and CM death (SNF791) (Figure 7A). Furthermore, gene ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis associated the observed gene expression patterns with regulation of exosome assembly, apoptosis-related signaling, cell-cell and leukocyte adhesion, inflammatory response, response to shear stress, and regulation of different signaling pathways involved in cell proliferation and angiogenesis such as the Hippo pathway, NF-κB, and AGE-RAGE signaling pathways (Figures 7B and 7C). While sequencing data are derived from HUVECs, we were able to validate similar expression profiles in HCMECs upon increased expression of miR-216a (Figures 7D and 7E). Although we identified several genes as potential targets of miR-216a from the sequencing results, we were not able to demonstrate direct target binding (data not shown). Nevertheless, the expression pattens observed in the hearts of the KO mice are, to a certain extent, in agreement with the results, with expression levels in the opposite direction of the sequencing data (Figures 7F and 7G).
Figure 7.

miR-216a-dependent transcriptional changes in endothelial cells
RNA sequencing was performed to assess the transcriptomic changes in endothelial cells treated with scrambled or miR-216a precursor molecules. (A) Heatmap of the top differentially expressed genes in endothelial cells treated with precursor-miR-216a compared with endothelial cells treated with scrambled precursor showing log2 FPKM (color scale) values of dysregulated genes, with yellow and blue colors representing increased and decreased expression, respectively. (B) Number of differentially expressed genes enriched in GO terms and (C) KEGG pathway analysis of differentially expressed genes. N refers to the number of genes involved in the pathway and DE refers to the genes in our dataset of differently expressed genes, therefore DE/N stands for the gene ratio and P.DE the p value for over-representation of the GO/KEGG term in the set. (D and E) Real-time PCR analysis of the expression of five representative (D) downregulated and (E) upregulated genes, n = 3 independent experiments. Numerical data are presented as mean (error bars show SEM); statistical significance was calculated using two-tailed unpaired t test. ∗p < 0.05 vs. corresponding control group. (F and G) Real-time PCR analysis of the expression of five representative (F) downregulated and (G) upregulated genes in myocardial tissue of WT and KO mice, n refers to number of hearts (n = 4 for each condition). Model depicting the regulatory mechanisms by which miR-216a controls physiological cardiac angiogenesis levels and regulates cardiac function. Decreased mir-216a expression levels in (cardiac) endothelial cells not only increases autophagic activity through direct upregulation of PTEN/BECN1 and indirectly ATG5 expression with subsequent impairment of angiogenesis, but also majorly impacts on apoptosis signaling and cardiac inflammatory response, leading to cardiac pathologic remodeling, susceptibility to cardiac stress, and ultimately to cardiac dysfunction and heart failure.
By acting upon specific molecular and cellular processes such as autophagy, angiogenesis, and inflammatory response, alterations in cardiac miR-216a expression levels result in cardiac pathologic remodeling, increased susceptibility to cardiac stress and ultimately cardiac dysfunction and HF (Figure 7H).
Discussion
In pathological cardiac remodeling, the hypertrophied myocardium develops disproportionally to neovascularization, generating a mismatch between oxygen demand and supply that results in tissue hypoxia and contributes to the transition to HF.35,36 Here, we report the discovery of a miR-216a as a posttranscriptional regulator of EC function, myocardial capillary network formation and adaptation. Our data show for the first time that the cardiac microvasculature is tightly regulated by miRNAs and interference with these processes induces susceptibility to cardiac stress conditions. Remarkably, in contrast to the paucity of adverse phenotypes observed at baseline in most mouse miRNA gene knockouts,37,38,39,40,41,42 miR-216a ablation in mice suffices to provoke pathological cardiac remodeling and HF. The myocardium is highly dependent on oxidative energy production and displays a very high capillary density, with each myocyte being supplied with oxygen and nutrients by roughly 1.0–1.6 capillaries on average.43,44,45 miR-216a deficiency was associated with reduced capillary density and capillary to CM ratios, and resulted in severe impairment of myocardial tissue oxygenation, as determined by state-of-the-art PA with high-frequency US. miR-216a impaired the microvascular network formation by reducing the capacity of ECs to proliferate, migrate, and form tube networks. The contribution of miR-216a to maintain proper cardiac geometry and function becomes even more prominent under conditions of cardiac stress, as pressure overload provoked high mortality rates associated with severe cardiac remodeling and failure, and ischemic injury resulted in larger infarcts and more extensive fibrotic areas compared with WT animals under similar stress conditions. The fact that maintenance of proper and sufficient cardiac microvascularization is tightly regulated by miRNA-dependent mechanisms identifies a potential new therapeutic target for interventional strategies in HF, with cardiac ECs rather than CMs as the main cellular target. The pro-angiogenic vascular endothelial growth factor (VEGF) is a known key player in postnatal myocardial angiogenesis. Counter-intuitively, clinical trials with VEGF gene therapy in patients with coronary or peripheral artery disease have only yielded ambiguous long-term results with disputable clinical benefit.46,47,48,49,50 One potential reason for this discrepancy is that the delivery of a single growth factor may not suffice to establish a functional vascular network, suggesting that fully matured cardiac neovascularization likely relies on multiple factors acting simultaneously and/or sequentially, requiring a combinatorial therapy to achieve angiogenesis in vivo. In this context, modulation of angiogenesis by miRNAs may represent a novel biological entity to coordinately regulate proteins with related functions (e.g., cell growth, intracellular signaling, and contractility), in contrast to classical drugs, which act on specific cellular targets.51 The next step should be the development of specific delivery methods to cardiac ECs and assess the therapeutic effect of increasing miR-216a in animal models of pathological cardiac remodeling or HF. However, no viral vector was yet developed to specifically target ECs in myocardial tissue in vivo, even though certain viruses show distinct tissue tropism.
Here, we show that Pten is involved in the regulation of endothelial angiogenic function and autophagy in the development of HF in miR-216a null mice. Autophagy is a double-edged sword and its dysregulation in the endothelium contributes to the pathogenesis of disease affecting various organ systems.28 Within the cardiovascular system, however, most work has highlighted the role of autophagy in CMs while neglecting other cardiac cell types including ECs. Although it negatively regulates cell-cycle progression and is therefore an established tumor suppressor, the specific functions of PTEN in cardiac endothelial behavior and vascular patterning remain unknown, as well as the underlying mechanisms implicating PTEN in the maintenance of cardiac homeostasis. While CM-specific loss-of-function studies have associated Pten with cardiac hypertrophic growth and cardiac dysfunction in mice,27,52 counter studies induced similar cardiac phenotypes in mice overexpressing Pten also in a cardiac-specific manner. The finding that Pten is de-repressed in miR-216a null mice and accompanied by decreased capillary density and increased autophagy indicates that cardiac angiogenesis and autophagy are two closely linked biological processes, essential for normal cardiac homeostasis and its adaptive response under stress conditions (Figure 7F).53,54 Nevertheless, Pten does not seem to be the only player responsible for the observed effects on the cardiac vasculature. This is not surprising, as most miRNAs do not act only on one single mRNA, and miR-216a is not an exception. Our results show that also Becn1 expression, an autophagy-promoting gene and previously identified direct target of miR-216a, was dysregulated upon modulation of miR-216a in both in vitro and in vivo models. Inhibition of miR-216a seems to result in exacerbated autophagy on ECs, which is reflected by increased levels of not only Becn1 but also Atg5, an autophagy gene that is important for the phagophore membrane.55,56 In contrast to its protective role in maintaining basal cellular homeostasis, excessive autophagy is associated with dysregulation of catabolic activity and maladaptation to cellular stress.57 Dysregulation of such processes on ECs could explain the severe phenotypes observed in the miR-216a KO mice, where excessive autophagy directly impairs endothelial function and angiogenic capacity. Whereas a considerable body of literature documenting that deficiency of autophagy elicits endothelial dysfunction is available,58,59,60 there is very scarce information on how overstimulation of this process will affect cellular and organ function, specifically on ECs and in the heart. It has been shown that excessive autophagy can disrupt vascular homeostasis to similar extent as loss of autophagy and lead to inflammation and atherosclerosis.61,62 In the heart, increased autophagy was shown to induce cell death and exacerbate cardiac remodeling and fibrosis in diabetes-associated cardiomyopathy and inhibition of Becn1 resulted in improved cardiac structure and function in diabetic mice.63 Although our results support dysregulation of EC function through excessive autophagy induced by inhibition of miR-216a in cardiac ECs, it remains unclear how the process leads to such pronounced differences in cardiac phenotypes following pressure overload or infarction.
Autophagy is not the only cellular process affected by altered levels of miR-216a. In fact, sequencing analysis revealed several genes that were previously associated with cellular processes such as apoptosis, cell proliferation, cell-cell adhesion, cytoskeleton dynamics, and inflammatory response to be under control of this miRNA. Although alterations of endogenous miR-216a levels have not yet been directly associated with microvascular remodeling in HF, circulating miR-216a has been proposed as a diagnostic biomarker for this condition.64 In a study comparing expression levels of different miRNAs in end-stage failing hearts with and without left ventricular (LV) assist device unload, miR-216a was shown to be downregulated in the LVAD group and negatively correlated with cardiac index.65 In line, CM proliferation was shown to be regulated by an lncRNA, NC_045363, that acts as an endogenous sponge by binding to miR-216a and preventing its downregulation of target genes within the JAK2/STAT3 pathway.66 While these studies seem to contradict our own results regarding the observed cardiac phenotypes, other studies have shown that microvascular ECs are sensitive to lower levels of miR-216a,67 which decreases their viability and angiogenic capacity, similar to what we observe in hearts where miR-216a was depleted. Moreover, increased levels of miR-216a have been associated with macrophage polarization and progression of atherosclerosis68 as well as with endothelial senescence and inflammation.69 Altogether, these studies suggest distinct roles of miR-216a in different cardiac cell types and, to better understand the complexity of the pathological cardiac response and phenotypes observed, more research to modulate miR-216a in a cell-specific manner in the heart, would be required. Nevertheless, the current study highlights the way miR-216a tightly controls cardiac microvascular integrity and function, and how alterations in its expression levels results in adverse cellular and molecular responses ultimately leading to extreme cardiac phenotypes and HF.
Our results underline miR-216a as a pro-angiogenic miRNA, increased expression levels in vivo of which could be beneficial in different settings of cardiac disease. Altogether, this study sets the stage for future pre-clinical advances in the modulation of miR-216a in vivo for the treatment of impaired microvascular remodeling upon prolonged cardiac stress and eventually HF.
Materials and methods
Study design
The purpose of this study was to identify a miRNA that plays an important role during cardiac microvascular remodeling under cardiac stress conditions. Furthermore, we aimed at understanding the molecular mechanism by which such miRNA interferes with the angiogenic capacity of the cardiac endothelium and, thus, regulates cardiac hemostasis. Ultimately, and as capillary rarefaction and endothelial dysfunction are hallmarks of patients with HF, we aimed at facilitating the translation of our findings into a therapeutic approach. By performing a high-throughput functional screen of a whole-genome miRNA silencing library with regard to the phenotypic effect on EC proliferation as a key parameter, we identified miR-216a as a potential regulator of endothelial function. To assess the relevance of this miRNA for EC proliferation in vitro we performed gain- and loss-of-function studies and quantified the expression levels of specific markers of active DNA synthesis and cell cycle.
For in vivo experiments, we generated miR-216a-deficient mice (miR-216a KO) to evaluate the influence of this miRNA in cardiac pathologic remodeling induced by stress conditions such as pressure or volume overload. The TAC model to induce cardiac pressure overload was chosen as a well-established model of pathological cardiac hypertrophy. The model of MI, induced by LAD ligation, is a well-established model of ischemic heart disease that closely resembles the disorder in humans with respect to structural and functional characteristics. We included miR-216a KO mice as an experimental group and their WT littermates as a control group in our studies. Except for their phenotype, animals were further randomly assigned to the different surgical groups and, wherever applicable, experimental interventions were kept blinded until statistical analysis. Group sizes were chosen based on our previous experience with both the TAC and MI models, which indicates that a minimal group size of five animals is sufficient to distinguish meaningful changes in cardiac morphology after 4 weeks of cardiac stress. For the TAC study, however, we had to adapt our initial design and use all the KO animals that survived 2 weeks of cardiac pressure overload. In both models, cardiac remodeling was assessed by (1) measuring cardiac hypertrophy, quantified by heart weight to body weight ratios; cardiac fibrosis by quantification of Sirius Red staining; and quantification of CM cell surface area by WGA staining, (2) determining cardiac function by echocardiography, and (3) real-time qPCR measurements of cardiac stress marker expression. Molecular analysis of the MI-subjected hearts was always performed on RNA derived from the remote zone of the infarcted heart. In the animals subjected to MI, we have also determined the extension of the infarcted areas to evaluate the severity of MI on the different animal groups. Capillary density and capillary/CM ratios were determined by capillary staining with Isolectin B4. To reveal the consequences of reduced capillary densities in the miR-216a KO mice we evaluated myocardial tissue oxygenation levels in vivo by combining high-frequency US with photoacoustic imaging. To determine the expression pattern of miR-216a we have performed (1) northern blotting analysis on different murine tissues, (2) isolation of different cardiac cell types to determine respective miR-216a expression levels by qRT-PCR, and (3) in situ hybridization on murine myocardial sections. Proliferation, wound-healing, and tube-formation assays were performed in vitro to assess the effect of modulating miR-216a on the angiogenic properties of ECs. Additional bioinformatics studies revealed several potential miR-216a target genes, including Pten, which, by displaying a conserved seed region for miR-216a on its 3′ UTR, could account for the effects of miR-216a on microvascular remodeling. Direct binding between miR-216a and Pten was validated by luciferase activity assays, and shown to be functional by western blotting analysis of PTEN expression upon modulation of miR-216a expression both in vitro and in vivo.
MicroRNA screening
The miRNA LNA inhibitor library (miRCURY LNA miRNA Inhibitor Library) targeting 753 miRNAs (listed in miRBase v.19, http://mirbase.org) was obtained from Exiqon. Data for all miRNAs and their respective miRBase accession numbers are included in Table S1. HUVECs were seeded in 96-well plates (5,000 cells/well) and transfected with LNA at a final concentration of 50 nM by using a standard forward transfection protocol with oligofectamine (Life Technologies). Each 96-well plate included control wells where HUVECs were transfected with scrambled LNA inhibitors (Exiqon). Twenty-four hours after transfection, cells were transferred to starvation medium (EBM2 0.1% FBS, Lonza) for 12 h and afterward to condition medium containing 5% FBS, 50 ng/μL VEGF (Peprotech), 10 ng/μL bFGF and 5 mM EdU (Life Technologies). Sixteen hours later the cells were fixed, further processed by combining the Click-IT imaging kit with Alexa 594 fluorochrome (Life Technologies) to reveal EdU incorporation, and stained with Hoechst (Life Technologies). Image acquisition was performed using a Nikon Eclipse Ti_E epi-fluorescent inverted microscope with motorized stage at 10× magnification. Five to six images were acquired per wavelength and well, which corresponded to approximately 3,000 cells analyzed per condition. Image analysis was performed using FIJI/ImageJ software (NIH) and CellProfiler (Broad Institute).70
Generation of miR-216a gene targeted mice
Our targeting strategy introduced a neomycin resistance cassette and loxP sites flanking the genomic region encompassing the 71-bp pre-miRNA-216a (Figures 2A and 2B). For the targeting vector, a 4.4-kb fragment upstream of and a 2.5-kb fragment immediately downstream of the pre-miR-216a were generated as the 5′ arm and the 3′ arm by PCR amplification of B6CBAF1/J genomic DNA. After vector linearization and electroporation of ESCs, ES clones were isolated and analyzed for homologous recombination by Southern blotting and PCR. miR-216a targeted ES clones were used for blastocyst injection. The resulting chimeric mice were bred to homozygosity and miR-216 aF/F mice were bred with CAG-Cre mice71 to obtain a miR-216a null allele. Mouse genotypes were determined by PCR on tail DNA using primers specific to the miR-216a locus: 5′-GGGTGGGAGAAAACAAATGGTCTGG-3′ and 5′-AACAGGGATGGTAAGTTCATGGCAACC-3′). All experimental procedures involving animals in this study were performed according to institutional guidelines and reviewed and approved by local Animal Care and Use Committees.
Aortic banding, MI, and transthoracic echocardiography
TAC or sham surgery was performed in 2- to 3-month-old miR-216a null mice in a BL6CBAF1 background and WT mice in a BL6CBAF1 background by subjecting the aorta to a defined 27 gauge constriction between the first and second truncus of the aortic arch as described previously.72,73 For MI, a permanent ligation of the left coronary artery was performed as described previously.74 For both surgeries, sham-operated animals underwent similar procedures without ligation of the aorta or occlusion of the left coronary artery. For Doppler echocardiography, mice were shaved and lightly anesthetized with isoflurane (mean 3% in oxygen) and allowed to breathe spontaneously via a nasal cone. Non-invasive, echocardiographic parameters were measured using a RMV707B (15–45 MHz) scan-head interfaced with a VisualSonics Vevo 2100 high-frequency US system (VisualSonics, Toronto, Canada). Long-axis ECG-triggered cine loops of the LV contraction cycle were obtained in B-mode to assess end-diastolic/systolic volume. Short-axis recordings of the LV contraction cycle were taken in M-mode to assess the wall thickness of the anterior/posterior wall at the mid-papillary level. Doppler was used to calculate the pressure gradient between the proximal and distal sites of the TAC and only mice with a pressure gradient >50 mm Hg were included. From B-mode recordings, LV length from basis to apex, LV internal diameter in systole (LVIDs) and diastole (LVIDd) were determined. From M-mode recordings, LV posterior wall thickness in systole (LV PWs) and diastole (LV PWd) were determined. LV mass was calculated with the following formula: 0.8 × (1.04 × ((LVIDd + LV PWd + IVSd)3) – (LVIDd)3) + 0.6); fractional shortening was calculated with the following formula: (LVIDd – LVIDs)/LVIDd × 100. EF was calculated as (SV/Vd) × 100 with Vs, systolic volume (3.1416 × (LVIDs3)/6), Vd, diastolic volume (3.1416 × (LVIDd3)/6), and SV, stroke volume (Vd – Vs).
High-frequency US and photoacoustic imaging
All US and PA scanning was performed on a VisualSonics Vevo 2100 LAZR Imaging Station (VisualSonics).75 Mice were anesthetized using 1.5% isoflurane with medical air at a flow of 2 L/min. Hair was removed over areas of interest using a depilatory cream. US gel during image acquisition, ECG, respiration, blood pressure, and body temperature were monitored, and anesthesia depth was adjusted if necessary. To collect anatomical information at high resolution, B-mode imaging was acquired using a high-frequency US probe (MS550D, VisualSonics, broadband frequency: 22–55 MHz) at 40 MHz. Blood flow in small blood vessels was imaged using Power-Doppler mode at 16 MHz, 100% power, using an LZ250 transducer (VisualSonics, broadband frequency: 13–24 MHz). During PA imaging, a tunable laser (680–970 nm) was used as described previously.75 Pulse-to-pulse energy fluctuation was continuously monitored, and variations exceeding 25% led to recalibration of the system before further imaging. Oxygen saturation and hemoglobin concentration were measured at a frequency of 21 MHz (LZ250, VisualSonics). In Oxy-Hemo mode, PA dual-wavelength imaging at 750 and 850 nm creates images of oxygenated hemoglobin and deoxygenated hemoglobin, which are co-registered with grayscale B-mode imaging. Within a region of interest, oxygen saturation (StO2) was calculated as the percentage of oxygenated hemoglobin in total hemoglobin. Quantification of total hemoglobin and oxygen saturation was achieved using the HemoMeaZure tool (VisualSonics) and OxyZated tool (VisualSonics), respectively, and as described previously.24
Histological analysis and (immunofluorescence) microscopy
Hearts were arrested in diastole, perfused, and fixed with 4% paraformaldehyde (PFA)/PBS solution, embedded in paraffin and sectioned at 4 μm thickness. Paraffin sections were stained with hematoxylin and eosin (H&E) for routine histological analysis, Sirius Red for the detection of fibrillar collagen, and FITC-labeled WGA (Sigma-Aldrich, 1:100) to visualize and quantify myocyte cross-sectional area. Cell surface areas were determined using ImageJ imaging software (http://rsb.info.nih.gov/ij/). Isolectin B4 staining (GSI-biotin, Vector, 1:100) was performed to visualize the capillaries in cardiac tissue. Slides were visualized using a Zeiss Axioskop 2Plus with an AxioCamHRc. Modification of Isolectin B4 staining with additional fluorescence-labeled streptavidin (Dylight 595-conjugated streptavidin, Jackson Thermo, 1:100) and counterstaining with FITC-labeled WGA was performed to assess capillary to CM ratios. The fluorescent signals were visualized using a DMI3000B Leica microscope (Leica).
Infarct size measurements
Consecutive microsections of the heart were made from the ligation site to the apex and stained with Sirius Red to assess the infarct area. The LV infarct and the total area were manually traced on digital images and measured automatically with ImageJ imaging software (http://rsb.info.nih.gov/ij/). Infarct size, expressed as percentage of total LV area, was calculated by dividing the sum of infarct areas by the sum of LV areas (from all sections, including those without infarct scar). Slides were visualized using the bright-field Leica microscope DM 2000 (Leica).
In situ hybridization
mmu-miR-216a was detected using the miRCURY LNA miRNA detection probe miR-216 (Exiqon). Procedures for in situ hybridization were performed as described previously73 with minor modifications. Heart tissue was rehydrated and treated for 1 h at 37°C with 10 μg mL−1 proteinase K, washed for 5 min in PBS and then dehydrated with gradually increasing concentrations of ethanol (70%–100%). The tissues were air-dried for 15 min and hybridization overnight was performed with in situ hybridization buffer (Exiqon) containing 40 nM digoxigenin-labeled antisense miR-216a probe (Exiqon) at 50°C. Post-hybridization washings were performed at 50°C by successive 5-min incubations in decreasing stringency of SSC solutions (5×, 1×, 0.2×, and 0.1×). Tissues were further washed with PBS for 5 min at room temperature and blocked with blocking solution (2% sheep serum and 5% BSA, both Sigma-Aldrich) prior to incubation with anti-digoxigenin-AP Fab fragments (Roche, 1:800) in blocking solution for 2 h at room temperature. The tissues were washed with PBS containing 0.1% Tween 20 (PBST) for 5 min at room temperature and the signal was detected with AP substrate NBT/BCIP (Roche) at 30°C. The tissues were counterstained with nuclear fast red and dehydrated with increasing concentrations of ethanol and xylene and signals were visualized at 40× magnification by using a DM2000 Leica microscope (Leica).
Western blot analysis
SDS-PAGE electrophoresis and blotting were performed as described previously.76 In short, whole tissue or cell lysates were produced in RIPA buffer supplemented wtih PhosSTOP (Roche) and protease inhibitor cocktail (Roche). Subsequently, samples were boiled in 4× Leammli buffer, including 2% β-mercaptoethanol, for 5 min at 95°C. SDS-PAGE and western blotting were performed using the Mini-PROTEAN 3 system (Bio-Rad). Blotted membranes were blocked in 5% BSA/TBS-Tween. Primary antibody labeling was performed overnight at 4°C while secondary IgG-horseradish peroxidase (HRP)-conjugated antibodies were applied for 2 h at room temperature. After each antibody incubation, blots were washed for 3 × 10 min in TBS-Tween. Images were generated using Supersignal West Dura Extended Duration ECL Substrate (Pierce) and the LAS-3000 documentation system (FujiFilm Life Science). Stripping was performed with Restore western blot stripping buffer (Pierce). Outputs were normalized for loading and results are expressed as an n-fold increase over the values of the control group in densitometric arbitrary units. Primary antibodies that were used included rabbit monoclonal anti-PTEN (Cell Signaling Technology, 1:1,000), rabbit polyclonal anti-LC3A/B (Cell Signaling Technology, 1:1,000), purified mouse monoclonal anti-p62 lck ligand (BD Biosciences, 1:1,000). Secondary antibodies included mouse monoclonal anti-GAPDH (Millipore, 1:10,000), polyclonal rabbit anti-mouse IgG-HRP (DAKO, 1:5,000), and polyclonal swine anti-rabbit IgG–HRP (DAKO, 1:5,000).
Northern blot analysis
Northern blotting was performed as described previously19 with minor modifications. In short, 10 μg of total RNA was fractionated on denaturing 12% polyacrylamide (40% acrylamide/bis solution 19:1, Roche) gels with 8 M urea TBE (Tris/borate/EDTA). After electrophoresis, RNA was transferred to a positively charged nylon membrane (Roche) using a semidry blotter (Bio-Rad), and fixed by UV crosslinking according to the manufacturer’s instructions. After pre-hybridization with CHURCH hybridization buffer (0.36 M Na2HPO4 ·2H2O, 0.14 M NaH2PO4·2H2O, 1 mM EDTA, and 7% SDS) for 1 h at 55°C, the membranes were hybridized overnight with specific miRCURY LNA miRNA detection probes for miR-216a or U6 small nuclear RNA (Rnu6-2) (QIAGEN). Detection was achieved with Fab fragments from a polyclonal antibody to 3′ digoxigenin conjugated to alkaline phosphatase (Roche, 1:5,000) and CDP star chemiluminescent substrate (Roche).
Quantitative real-time PCR
Total RNA (1 μg) was applied to either miR-based or mRNA-based reverse transcription. Real-time PCR was performed on a Bio-Rad iCycler (Bio-Rad) using SYBR Green. Transcript quantities were compared using the relative Ct method, where the amount of target normalized to the amount of endogenous control (L7) and relative to the control sample is given by 2−ΔΔCt. For miRNA real-time PCR, miRNAs were isolated with TRIzol reagent (Invitrogen) and cDNA was generated with the miScript Reverse Transcription Kit (QIAGEN). For real-time PCR detection of miRNAs, specific miRNA primers and the ExiLENT SYBR green master mix (QIAGEN) were used. Primer sequences are provided in Table S3.
Differential cell isolation
Isolation of different cell populations from murine hearts was performed using a Lagendorff-free method, as described previously.77 In brief, mouse hearts were perfused and digested with collagenase II (Worthington LS004176), IV (Worthington LS004188), and protease (Sigma-Aldrich P5147), cut into small pieces, and completely dissociated by pipetting. Cell suspensions were passed through a strainer and myocytes settled for 20 min through gravity. Cells from the supernatant, which contained the non-myocyte cell population, were incubated on ice for 20 min with different antibody-labeled magnetic beads: to isolate cardiac ECs, the non-CMs cell suspension was incubated for 15 min at 4°C with magnetic CD146-labeled MicroBeads (Miltenyi 130-092-007).
Cell culture
Pooled HUVECs were purchased from Lonza (CC-2519). Cells were cultured in EGM-2 medium consisting of EBM-2 Basal Medium (Lonza CC-3156) supplemented with Endothelial Cell Growth Medium-2 Bullet Kit (CC-4176) and cultured at 37°C with 5% CO2. Cells in passage numbers 6–7 were used. A total of 15 × 104 HUVECs was cultured in 6-well plates and, upon reaching 70% confluency, were transfected and/or used for functional assays.
Neonatal rat CMs (NRCMs) were isolated from hearts collected from 1- to 2-day-old neonatal rats, atrial tissue was removed, and the ventricles were cut in 1 mm3 before enzymatic digestion.78 Tissue was subjected to multiple rounds of enzymatic digestion using 0.05% pancreatin (Sigma P3292) and 290 units/mg of collagenase (Gibco 17100-017). Cells were collected by centrifugation at 1,500 rpm for 6 min and resuspended in DMEM medium (Gibco 21885-025) supplemented with 15% FBS (Gibco 10270-106) and gentamicin (10 mg/mL Gibco 15710). Subsequently, cells were differentially plated for 1 h on uncoated cell culture dishes to remove non-myocytes. The CMs were then plated on 0.2% gelatin-coated cell culture dishes and cultured the following day in serum-free DMEM containing 20% M199 (Gibco 31153-026), 15% FBS, 100 units/mL penicillin/streptomycin (Gibco 15140-122), and gentamicin (10 mg/mL). After 24 h, CMs were maintained in serum-free DMEM and treated with 10 μM phenylephrine (Sigma P6126) and 10 μM isoproterenol (Sigma I5627) for 48 h. For visualization of CM size a monoclonal antibody against α2-actinin (ACTN2, 1:500; Sigma) was used, followed by Fluor-488 goat anti-mouse IgG (1:250; Jackson ImmunoResearch). For detecting any fibroblasts remaining in culture a prolyl 4-hydroxylase beta antibody (1:200, Acris) was used, together with an Alexa 568-coupled anti-mouse antibody (1:400, Invitrogen). Nuclear staining was performed with 4′,6-diamidino-2-phenylindole (Molecular Probes).
Transient cell transfection, adenovirus
Precursor molecules (10 nM, Life Technologies) and miRCURY LNA inhibitor (50 nM; QIAGEN) specific for hsa-miR-216a or scrambled were transfected into HUVECs or NRCMS with 0.5 μL oligofectamine (Invitrogen) according to the manufacturer’s instructions. For siRNA transfection, selected siRNAs (IDT) at a final concentration of 50 nM were transfected with lipofectamine (RNAiMAX, Life Technologies). AdGFP was generated as described previously.79 AdPTEN was purchased by Vector Biolabs (ADV-269728). All initial recombinants were plaque purified, expanded, and titered by duplicate plaque assays in monolayers of HEK293. Recombinants were tested for appropriate expression in HCMECs by semi-quantitative RT-PCR or western blotting when appropriate. Adenoviral infection of ECs was performed at a multiplicity of infection of 100 plaque-forming units (pfu) in 2 mL (6-cm culture dishes) of EGM-2 medium (Lonza) supplemented with 2% FBS for 2 h at 37°C in a humidified, 5% CO2 incubator.
Tube formation assay
HUVECs, transfected with either scrambled precursor or precursor-216a (both 10 nM, Life Technologies), scrambled LNA, or LNA-216a (both 50 nM, QIAGEN) for 24 h were cultured in a 96-well plate coated with 50 μL Matrigel (BD Biosciences) at a density of 1.5 × 104 cells per well in EGM-2 medium (Lonza). The cells were incubated for 16 h at 37°C and imaged with a digital microscope camera EVOSxl (AMG). Quantitative analysis of network structure formation was performed with ImageJ software (http://rsbweb.nih.gov/ij/) by counting the number of junctions and measuring the total length of tube structures.
Scratch wound migration assay
HUVECs were plated in 48-well culture plates after 24-h transfection with scrambled precursor, precursor-216a, scrambled LNA, or LNA-216a, at a density of 80,000 cells per well in EGM-2 medium (Lonza). Upon total confluence, a scratch was made in the middle of the cell monolayer with a pipette tip. The cells were washed with EBM-2 (Lonza) and incubated with EBM-2 medium supplemented with 0.5% FBS, with or without 50 ng/mL recombinant human VEGF (Peprotech) for 8 h at 37C°. The distance between the edges of the scratch was captured immediately after making the scratch and 8 h later with a Canon EOS600D camera (Canon) linked to an inverted microscope (Eclipse TS100, Nikon) and measured with ImageJ software (http://rsbweb.nih.gov/ij/). The difference in distance between the edges of the scratch at the two time points is presented as percentage of migration.
Cell proliferation assay
Twenty-four hours after transfection with scrambled precursor, precursor-216a, scrambled LNA, or LNA-216a, HUVECs were plated in 96-well plates at a density of 2 × 103 cells per well in EGM-2 medium (Lonza). Six hours after seeding, cells were incubated with 5 μM Edu (Life Technologies) for 16 h at 37°C. After cell fixation in PFA 4%, EdU detection was performed with a Click-iT EdU Alexa Fluor 594 Imaging Kit (Life Technologies) according to the manufacturer’s instructions. Detection of Ki67 was performed with rabbit anti-human Ki67 polyclonal antibody (Monosan, 1:200) and nuclear counterstaining was done with Hoechst (Life Technologies, 1:1,000).
pMIR reporter assays
Human PTEN 3′UTR (873 bp) containing a potential miR-216a binding site was generated by PCR as a SacI-NheI fragment and sub-cloned into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega). Similarly, human BECN1 3′ UTR (661 bp) containing a potential miR-216a binding site was generated by PCR as an SgfI-MluI fragment and sub-cloned into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega). HUVECs were transfected with pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega) harboring the entire WT 3′ UTR of mouse Pten, or with a mutation on the miR-216a binding site, followed by transfection with precursor miR-216a or scrambled precursor. Firefly luciferase activity was measured 72 h after transfection by using a dual luciferase assay kit (Promega) and Renilla luciferase as internal control, on a 96-well Victor4 multilabel counter (PerkinElmer).
EC lines
Different primary EC lines were purchased from Lonza: HUVECs (HUVEC, CC_2519), human lung microvascular endothelial cells (HMVEC-L, CC-2527), human dermal microvascular endothelial cells (HMVEC-dAd, CC-2543), and human cardiac microvascular endothelial cells (HMVEC-C, CC-7030). Primary human retinal microvascular ECs were purchased from Cell Biologicals (H-6065). All cells were cultured and grown to confluency according to the manufacturer’s instructions.
RNA sequencing
Deep sequencing of total RNA, isolated from HUVECs, was performed 24 h after ransfection with 10 nM of either hsa-miR-216a-5p or scrambled (control) precursor molecules (Life Technologies). RNA purity and integrity were determined with an Agilent RNA Nano 6000 kit for the 2100 Bioanalyzer (Agilent Technologies). Ribosomal RNA (rRNA) deletion and subsequent NGS library preparation was performed according to the manufacturer’s instructions using the NEBNext rRNA depletion and RNA Ultra II library prep kit (NEB). Quality control of the resulting DNA library was carried out using the Agilent DNA 1000 kit (2100 Bioanalyzer, Agilent Technologies). Library molarity was determined by qPCR (NEBNext library quant kit, NEB) for subsequent equimolar pooling and sequenced on the NextSeq500 instrument using v2 chemistry in a single-read mode 75 cycles. The Bcl2fastq tool (Illumina, v.2.20.0.422) was applied to compile raw fastq data per sample. Raw sequence files were quality checked using FASTQC software (www.bioinformatics.babraham.ac.uk/projects/fastqc) and trimmed to remove Illumina adaptor using Cutadapt software. The raw sequencing reads were then mapped to Ensembl Homo sapiens reference genome (Ensembl GRCh37 PAR-masked) using STAR software. Rounded gene counts were normalized to RPKM (reads per kilobase of exon model per million mapped reads) using the rpkm function in the Bioconductor package edgeR. Genes with RPKM values greater than 2.00 in both miRNA and scramble transfected HUVECs were considered as expressed genes. Fold changes were taken with respect to the expression upon scramble transfection. Genes whose expression fold change was greater than 1.3 were considered as differentially expressed.
Bioinformatics analyses
Generalized hypergeometric tests for enrichment of GO terms and KEGG pathways were used for genes represented by transcripts differentially expressed in each treatment group following the kallisto > sleuth pipeline using the goana and kegga functions in the limma software package in R,80 filtered by significance (p < 0.05). N stands for the number of genes involved in the pathway, DE stands for genes in our dataset of differently expressed genes, therefore DE/N stands for the gene ratio, and P.DE the p value for over-representation of the GO/KEGG term in the set.
Statistical analysis
The results are presented as mean ± standard error of the mean. Statistical analyses were performed using Prism software (GraphPad Software), and consisted of one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test when comparing more than two experimental groups, or unpaired Student’s t test when comparing two experimental groups. For the Kaplan-Meier survival assay, the log rank (Mantel-Cox) test was used. Differences were considered significant when p < 0.05.
Data availability statement
The data that support the findings of this study are available from the corresponding author on reasonable request: P.d.C.M., p.dacostamartins@maastrichtuniversity.nl.
Acknowledgments
We acknowledge Genoway (Lyon, France) for helping with the strategy and generation of the miR-216a KO mice. Funding: A.V. acknowledges the financial support of the European Union’s Seventh Framework (FP7/2007-2013) grant agreement 289720. J.D.B. acknowledges the financial support of the Dutch province of Limburg. L.D.W. acknowledges support from the Netherlands Cardiovascular Research Initiative and the Dutch Heart Foundation, the Netherlands. L.D.W. was further supported by grant 311549 from the European Research Council (ERC) and a VICI award 918-156-47 from The Netherlands Organization for Scientific Research (NWO). P.C.M. is supported by a MEERVOUD grant from The Netherlands Organization for Scientific Research (NWO), a Dutch CardioVascular Alliance (DCVA) awarded to the Phaedra consortium, as well as the Impulse Grant 2018 awarded to the Phaedra IMPACT consortium (CVON-2018-29), and is an Established Investigator of the Dutch Heart Foundation (NHS2015T066). P.C.M. and R.C.A. were further supported by the H2020 Twinning project RESETageing (GA 952266). J.M.M.K. was supported by a Dutch CardioVascular Alliance (DCVA) awarded to the Phaedra consortium as well as the Impulse Grant 2018 awarded to the Phaedra IMPACT consortium (CVON-2018-29).
Author contributions
R.P.J., B.D., S.O., J.M.M.K., R.C.A., E.M.P., T.D., and P.A.d.C.M. performed northern blots, qRT-PCR, western blots experiments, and angiogenic functional assays. R.P.J., R.C.A., and A.V. performed the functional screening and respective validation. R.J.P., J.M.M.K., B.D., and M.A. performed primary cell isolations. R.J.P., D.M.S., and N.B. performed surgical procedures and echocardiography in mouse models. R.J.P. and J.M.M.K. performed histology in the different mouse models, the in situ hybridizations, and generated the luciferase reporter constructs used in the luciferase assays. R.J.P., T.D., and R.C.A. performed the photoacoustics combined with ultrasound analysis of the mouse models. L.O., J.C.H., and M.S. performed the sequencing and the bioinformatics analysis. P.A.d.C.M. and L.J.d.W. generated miR-216a gene-targeted mouse mice. J.D.B. and L.J.d.W. provided reagents, models or data, and critically reviewed the manuscript before submission. P.A.d.C.M. acquired funding for the study.
Declaration of interests
L.d.W. and P.d.C.M. are cofounders of Mirabilis Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.04.007.
Supplemental information
References
- 1.Diaz-Sandoval L.J., Losordo D.W. Gene therapy for cardiovascular angiogenesis. Expert Opin. Biol. Ther. 2003;3:599–616. doi: 10.1517/14712598.3.4.599. [DOI] [PubMed] [Google Scholar]
- 2.Itescu S., Kocher A.A., Schuster M.D. Myocardial neovascularization by adult bone marrow-derived angioblasts: strategies for improvement of cardiomyocyte function. Heart Fail. Rev. 2003;8:253–258. doi: 10.1023/a:1024721717926. [DOI] [PubMed] [Google Scholar]
- 3.Wang X., Huang W., Liu G., Cai W., Millard R.W., Wang Y., Chang J., Peng T., Fan G.-C. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J. Mol. Cell. Cardiol. 2014;74:139–150. doi: 10.1016/j.yjmcc.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van der Laan A.M., Piek J.J., Van Royen N. Targeting angiogenesis to restore the microcirculation after reperfused MI. Nat. Rev. Cardiol. 2009;6:515–523. doi: 10.1038/nrcardio.2009.103. [DOI] [PubMed] [Google Scholar]
- 5.Bang C., Batkai S., Dangwal S., Gupta S.K., Foinquinos A., Holzmann A., Just A., Remke J., Zimmer K., Zeug A., et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Invest. 2014;124:2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halkein J., Tabruyn S.P., Ricke-Hoch M., Haghikia A., Nguyen N.Q.N., Scherr M., Castermans K., Malvaux L., Lambert V., Thiry M., et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J. Clin. Invest. 2013;123:2143–2154. doi: 10.1172/JCI64365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hergenreider E., Heydt S., Tréguer K., Boettger T., Horrevoets A.J.G., Zeiher A.M., Scheffer M.P., Frangakis A.S., Yin X., Mayr M., et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012;14:249–256. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 8.Khan M., Nickoloff E., Abramova T., Johnson J., Verma S.K., Krishnamurthy P., Mackie A.R., Vaughan E., Garikipati V.N.S., Benedict C., et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res. 2015;117:52–64. doi: 10.1161/CIRCRESAHA.117.305990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J.F., Murchison E.P., Tang R., Callis T.E., Tatsuguchi M., Deng Z., Rojas M., Hammond S.M., Schneider M.D., Selzman C.H., et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA. 2008;105:2111–2116. doi: 10.1073/pnas.0710228105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Da Costa Martins P.A., Bourajjaj M., Gladka M., Kortland M., van Oort R.J., Pinto Y.M., Molkentin J.D., De Windt L.J. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation. 2008;118:1567–1576. doi: 10.1161/CIRCULATIONAHA.108.769984. [DOI] [PubMed] [Google Scholar]
- 11.Kuehbacher A., Urbich C., Zeiher A.M., Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ. Res. 2007;101:59–68. doi: 10.1161/CIRCRESAHA.107.153916. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki H.I., Katsura A., Matsuyama H., Miyazono K. MicroRNA regulons in tumor microenvironment. Oncogene. 2015;34:3085–3094. doi: 10.1038/onc.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suárez Y., Fernández-Hernando C., Yu J., Gerber S.A., Harrison K.D., Pober J.S., Iruela-Arispe M.L., Merkenschlager M., Sessa W.C. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc. Natl. Acad. Sci. USA. 2008;105:14082–14087. doi: 10.1073/pnas.0804597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greco S., De Simone M., Colussi C., Zaccagnini G., Fasanaro P., Pescatori M., Cardani R., Perbellini R., Isaia E., Sale P., et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 2009;23:3335–3346. doi: 10.1096/fj.08-128579. [DOI] [PubMed] [Google Scholar]
- 15.Hu R., Wallace J., Dahlem T.J., Grunwald D.J., O'Connell R.M. Targeting human microRNA genes using engineered Tal-effector nucleases (TALENs) PLoS One. 2013;8:e63074. doi: 10.1371/journal.pone.0063074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R.T., Xu M., Xu C.X., Song Z.G., Jin H. Decreased expression of miR216a contributes to non-small-cell lung cancer progression. Clin. Cancer Res. 2014;20:4705–4716. doi: 10.1158/1078-0432.CCR-14-0517. [DOI] [PubMed] [Google Scholar]
- 17.Xia H., Ooi L.L., Hui K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629–641. doi: 10.1002/hep.26369. [DOI] [PubMed] [Google Scholar]
- 18.Bofill-De Ros X., Gironella M., Fillat C. miR-148a- and miR-216a-regulated oncolytic adenoviruses targeting pancreatic tumors attenuate tissue damage without perturbation of miRNA activity. Mol. Ther. 2014;22:1665–1677. doi: 10.1038/mt.2014.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Da Costa Martins P.A., Salic K., Gladka M.M., Armand A.S., Leptidis S., El Azzouzi H., Hansen A., Coenen-de-Roo C.J., Bierhuizen M.F., van der Nagel R., et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat. Cell Biol. 2010;12:1220–1227. doi: 10.1038/ncb2126. [DOI] [PubMed] [Google Scholar]
- 20.Salic A., Mitchison T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodríguez C.I., Buchholz F., Galloway J., Sequerra R., Kasper J., Ayala R., Stewart A.F., Dymecki S.M. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 22.Greco S., Fasanaro P., Castelvecchio S., D’Alessandra Y., Arcelli D., Di Donato M., Malavazos A., Capogrossi M.C., Menicanti L., Martelli F. MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes. 2012;61:1633–1641. doi: 10.2337/db11-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffman J.I.E., Buckberg G.D. The myocardial oxygen supply:demand index revisited. J. Am. Heart Assoc. 2014;3:e000285. doi: 10.1161/JAHA.113.000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerling M., Zhao Y., Nania S., Norberg K.J., Verbeke C.S., Englert B., Kuiper R.V., Bergström A., Hassan M., Neesse A., et al. Real-time assessment of tissue hypoxia in vivo with combined photoacoustics and high-frequency ultrasound. Theranostics. 2014;4:604–613. doi: 10.7150/thno.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao J., Maslov K.I., Zhang Y., Xia Y., Wang L.V. Label-free oxygen-metabolic photoacoustic microscopy in vivo. J. Biomed. Opt. 2011;16:076003. doi: 10.1117/1.3594786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laufer J., Johnson P., Zhang E., Treeby B., Cox B., Pedley B., Beard P. In vivo preclinical photoacoustic imaging of tumor vasculature development and therapy. J. Biomed. Opt. 2012;17:056016. doi: 10.1117/1.JBO.17.5.056016. [DOI] [PubMed] [Google Scholar]
- 27.Roe N.D., Xu X., Kandadi M.R., Hu N., Pang J., Weiser-Evans M.C.M., Ren J. Targeted deletion of PTEN in cardiomyocytes renders cardiac contractile dysfunction through interruption of Pink1-AMPK signaling and autophagy. Biochim. Biophys. Acta. 2015;1852:290–298. doi: 10.1016/j.bbadis.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi A.M.K., Ryter S.W., Levine B. Autophagy in human health and disease. N. Engl. J. Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 29.Santanam U., Banach-Petrosky W., Abate-Shen C., Shen M.M., White E., DiPaola R.S. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016;30:399–407. doi: 10.1101/gad.274134.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang R., Zeh H.J., Lotze M.T., Tang D. The beclin-1 network regulates autophagy and apoptosis. Cell Death Diff. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ki X., He S., Ma B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer. 2020;19:12–28. doi: 10.1186/s12943-020-1138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schrijvers D.M., De Meyer G.R.Y., Martinet W. Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler. Thromb. Vasc. Biol. 2011;31:2787–2791. doi: 10.1161/ATVBAHA.111.224899. [DOI] [PubMed] [Google Scholar]
- 33.Rubinsztein D.C., Shpilka T., Elazar Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012;22:R29–R34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 34.Menghini R., Casagrande V., Marino A., Marchetti V., Cardellini M., Stoehr R., Rizza S., Martelli E., Greco S., Mauriello A., et al. MiR-216a: a link between endothelial dusfunction and autophagy. Cell Death Dis. 2014;5:e1029. doi: 10.1038/cddis.2013.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Izumiya Y., Shiojima I., Sato K., Sawyer D.B., Colucci W.S., Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–893. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiojima I., Sato K., Izumiya Y., Schiekofer S., Ito M., Liao R., Colucci W.S., Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai E.C. Two decades of miRNA biology: lessons and challenges. RNA. 2015;21:675–677. doi: 10.1261/rna.051193.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ventura A., Young A.G., Winslow M.M., Lintault L., Meissner A., Erkeland S.J., Newman J., Bronson R.T., Crowley D., Stone J.R., et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuhnert F., Mancuso M.R., Hampton J., Stankunas K., Asano T., Chen C.Z., Kuo C.J. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135:3989–3993. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- 40.Miyaki S., Sato T., Inoue A., Otsuki S., Ito Y., Yokoyama S., Kato Y., Takemoto F., Nakasa T., Yamashita S., et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010;24:1173–1185. doi: 10.1101/gad.1915510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu J., Bao J., Kim M., Yuan S., Tang C., Zheng H., Mastick G.S., Xu C., Yan W. Two miRNA clusters, miR-34b/c and miR-449, are essential for normal brain development, motile ciliogenesis, and spermatogenesis. Proc. Natl. Acad. Sci. USA. 2014;111:E2851–E2857. doi: 10.1073/pnas.1407777111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu N., Bezprozvannaya S., Williams A.H., Qi X., Richardson J.A., Bassel-Duby R., Olson E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tirziu D., Chorianopoulos E., Moodie K.L., Palac R.T., Zhuang Z.W., Tjwa M., Roncal C., Eriksson U., Fu Q., Elfenbein A., et al. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J. Clin. Invest. 2007;117:3188–3197. doi: 10.1172/JCI32024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hilfiker-Kleiner D., Hilfiker A., Fuchs M., Kaminski K., Schaefer A., Schieffer B., Hillmer A., Schmiedl A., Ding Z., Podewski E., et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004;95:187–195. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 45.Heineke J. Wag the dog: how endothelial cells regulate cardiomyocyte growth. Arterioscler. Thromb. Vasc. Biol. 2012;32:545–547. doi: 10.1161/ATVBAHA.111.242784. [DOI] [PubMed] [Google Scholar]
- 46.Taimeh Z., Loughran J., Birks E.J., Bolli R. Vascular endothelial growth factor in heart failure. Nat. Rev. Cardiol. 2013;10:519–530. doi: 10.1038/nrcardio.2013.94. [DOI] [PubMed] [Google Scholar]
- 47.Tiong A., Freedman S.B. Gene therapy for cardiovascular disease: the potential of VEGF. Curr. Opin. Mol. Ther. 2004;6:151–159. [PubMed] [Google Scholar]
- 48.Zachary I., Morgan R.D. Therapeutic angiogenesis for cardiovascular disease: biological context, challenges, prospects. Heart. 2011;97:181–189. doi: 10.1136/hrt.2009.180414. [DOI] [PubMed] [Google Scholar]
- 49.Belch J., Hiatt W.R., Baumgartner I., Driver I.V., Nikol S., Norgren L., Van Belle E., TAMARIS Committees and Investigators Effect of fibroblast growth factor NV1FGF on amputation and death: a randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet. 2011;377:1929–1937. doi: 10.1016/S0140-6736(11)60394-2. [DOI] [PubMed] [Google Scholar]
- 50.Henry T.D., Annex B.H., McKendall G.R., Azrin M.A., Lopez J.J., Giordano F.J., Shah P.K., Willerson J.T., Benza R.L., Berman D.S., et al. The VIVA trial: vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation. 2003;107:1359–1365. doi: 10.1161/01.cir.0000061911.47710.8a. [DOI] [PubMed] [Google Scholar]
- 51.Krützfeldt J., Poy M.N., Stoffel M. Strategies to determine the biological function of microRNAs. Nat. Genet. 2006;38:S14–S19. doi: 10.1038/ng1799. [DOI] [PubMed] [Google Scholar]
- 52.Crackower M.A., Oudit G.Y., Kozieradzki I., Sarao R., Sun H., Sasaki T., Hirsch E., Suzuki A., Shioi T., Irie-Sasaki J., et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 53.Schwartzbauer G., Robbins J. The tumor suppressor gene PTEN can regulate cardiac hypertrophy and survival. J. Biol. Chem. 2001;276:35786–35793. doi: 10.1074/jbc.M102479200. [DOI] [PubMed] [Google Scholar]
- 54.Oudit G.Y., Penninger J.M. Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 2009;82:250–260. doi: 10.1093/cvr/cvp014. [DOI] [PubMed] [Google Scholar]
- 55.Romanov J., Walczak M., Ibiricu I., Schüchner S., Ogris E., Kraft C., Martens S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012;31:4304–4317. doi: 10.1038/emboj.2012.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsushita M., Suzuki N.N., Obara K., Fujioka Y., Ohsumi Y., Inagaki F. Structure of Atg5.Atg16, a complex essential for autophagy. J. Biol. Chem. 2007;282:6763–6772. doi: 10.1074/jbc.M609876200. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y., Shoji-Kawata S., Sumpter R.M., Jr., Wei Y., Ginet V., Zhang L., Posner B., Tran K.A., Green D.R., Xavier R.J., et al. Autosis is a Na+, K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hy[poxia-ischemia. Proc. Natl. Acad. Sci. USA. 2013;110:20364–20371. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mameli E., Martello A., Caporali A. Autophagy at the interface of endothelial cell homeostasis and vascular disease. FEBS J. 2022;289:2976–2991. doi: 10.1111/febs.15873. [DOI] [PubMed] [Google Scholar]
- 59.Lyu Z.S., Cao X.N., Wen Q., Mo X.D., Zhao H.Y., Chen Y.H., Wang Y., Chang Y.J., Xu L.P., Zhang X.H., et al. Autophagy in endothelial cells regulates their haematopoiesis-supporting ability. EBioMedicine. 2020;53:101677–101691. doi: 10.1016/j.ebiom.2020.102677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hughes W.E., Beyer A.M. Vascular autophagy in physiology and pathology. Am. J. Physiol. Heart Circ. Physiol. 2019;316:H183–H185. doi: 10.1152/ajpheart.00707.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lavandero S., Chiong M., Rothermel B.A., Hill J.A. Autophagy in cardiovascular biology. J. Clin. Invest. 2015;125:55–64. doi: 10.1172/JCI73943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park S.K., La Salle D.T., Cerbie J., Cho J.M., Bledsoe A., Nelson A., Morgan D.E., Richardson R.S., Shiu Y.T., Boudina S., et al. Elevated arterial shear rate increases indexes of endothelial cell autophagy and nitric oxide synthase activation in humans. Am. J. Physiol. Heart Circ. Physiol. 2019;316:H106–H112. doi: 10.1152/ajpheart.00561.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen W.R., Liu H.B., Chen Y.D., Sha Y., Ma Q., Zhu P.J., Mu Y. Melatonin attenuates myocardial ischemia/reperfusion injury by inhibiting autophagy via an AMPK/mTOR signaling pathway. Cell. Physiol. Biochem. 2018;47:2067–2076. doi: 10.1159/000491474. [DOI] [PubMed] [Google Scholar]
- 64.Ding H., Wang Y., Hu L., Xue S., Wang Y., Zhang L., Zhang Y., Qi H., Yu H., Aung L.H.H., et al. Combined detection of miR-21-5p, miR-30a-3p, miR-30a-5p, miR-155-5p, miR-216a and miR-217 for screening of early heart failure diseases. Biosci. Rep. 2020;40 doi: 10.1042/BSR20191653. BSR20191653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barsanti C., Trivella M.G., D’Aurizio R., El Baroudi M., Baumgart M., Groth M., Caruso R., Verde A., Botta L., Cozzi L., et al. Differential regulation of microRNAs in end-stage failing hearts is associated with left ventricular assist devoce unloading. Biomed. Res. Int. 2015;2015:595512. doi: 10.1155/2015/592512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang J., Chen X., Shen D., Ge D., Chen J., Pei J., Li Y., Yue Z., Feng J., Chu M., et al. A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J. Mol. Cell. Cardiol. 2019;127:105–114. doi: 10.1016/j.yjmcc.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 67.Wang X., Xue X., Wang H., Xu F., Xin Z., Wang K., Cui M., Qin W. Quercetin inhibits human microvascular endothelial cells viability, migration and tube-formation in vitro through restraining microRNA-216a. J. Drug Target. 2020;28:609–616. doi: 10.1080/1061186X.2019.1700263. [DOI] [PubMed] [Google Scholar]
- 68.Yang S., Li J., Chen Y., Zhang S., Feng C., Hou Z., Cai J., Wang Y., Hui R., Lv B., Zhang W. MicroRNA-216a promotes M1 macrophages polarization and atherosclerosis progression by activating telomerase via the Smad3/NF-κB pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:1772–1781. doi: 10.1016/j.bbadis.2018.06.016. [DOI] [PubMed] [Google Scholar]
- 69.Yang S., Mi X., Chen Y., Feng C., Hou Z., Hui R., Zhang W. MicroRNA-216a induces endothelial senescence and inflammation via Smad3/IκBα pathway. J. Cell. Mol. Med. 2018;22:2739–2749. doi: 10.1111/jcmm.13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J., et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sakai K., Miyazaki J.i. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem. Biophys. Res. Commun. 1997;237:318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- 72.Rockman H.A., Ross R.S., Harris A.N., Knowlton K.U., Steinhelper M.E., Field L.J., Ross J., Jr., Chien K.R. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bourajjaj M., Armand A.S., Da Costa Martins P.A., Weijts B., Vander nagel R., Heeneman S., Wehrens X.H., De Windt L.J. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J. Biol. Chem. 2008;283:22295–22303. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
- 74.Van Rooij E., Doevendans P.A., Crijns H.J., Heeneman S., Lips D.J., Van Bilsen M., Williams R.S., Olson E.N., Bassel-Duby R., Rothermel B.A., et al. MCIP1 overexpression suppresses left ventricular remodeling and sustains cardiac function after myocardial infarction. Circ. Res. 2004;94:e18–e26. doi: 10.1161/01.RES.0000118597.54416.00. [DOI] [PubMed] [Google Scholar]
- 75.Needles A., Heinmiller A., Sun J., Theodoropoulos C., Bates D., Hirson D., Yin M., Foster F.S. Development and initial application of a fully integrated photoacoustic micro-ultrasound system. IEEE Trans. Ultrason. Ferroelectr. Freq. Control. 2013;60:888–897. doi: 10.1109/TUFFC.2013.2646. [DOI] [PubMed] [Google Scholar]
- 76.Dirkx E., Gladka M.M., Philippen L.E., Armand A.S., Kinet V., Leptidis S., El Azzouzi H., Salic K., Bourajjaj M., da Silva G.J.J., et al. Nfat and miR-25 cooperate to reactivate the transcription factor Hand2 in heart failure. Nat. Cell Biol. 2013;15:1282–1293. doi: 10.1038/ncb2866. [DOI] [PubMed] [Google Scholar]
- 77.Song X., Kusakari Y., Xiao C.Y., Kinsella S.D., Rosenberg M.A., Scherrer-Crosbie M., Hara K., Rosenzweig A., Matsui T. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am. J. Physiol. Cell Physiol. 2010;299:C1256–C1266. doi: 10.1152/ajpcell.00338.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Windt L.J., Lim H.W., Haq S., Force T., Molkentin J.D. Calcineurin promotes protein kinase C and c-Jun NH2-terminal kinase activation in the heart. Cross-talk between cardiac hypertrophic signaling pathways. J. Biol. Chem. 2000;275:13571–13579. doi: 10.1074/jbc.275.18.13571. [DOI] [PubMed] [Google Scholar]
- 79.Van Oort R.J., Van Rooij E., Bourajjaj M., Schimmel J., Jansen M.A., Van der Nagel R., Doevendans P.A., Schneider M.D., van Echteld C.J.A., De Windt L.J. MEF2 activates a genetic program promoting chamber dilation nd contractlie dysfunction in calcineurin-induced heart failure. Circulation. 2006;114:298–308. doi: 10.1161/CIRCULATIONAHA.105.608968. [DOI] [PubMed] [Google Scholar]
- 80.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request: P.d.C.M., p.dacostamartins@maastrichtuniversity.nl.