

Abstract
Oxalate homeostasis is maintained through a delicate balance between endogenous sources, exogenous supply and excretion from the body. Novel studies have shed light on the essential roles of metabolic pathways, the microbiome, epithelial oxalate transporters, and adequate oxalate excretion to maintain oxalate homeostasis. In patients with primary or secondary hyperoxaluria, nephrolithiasis, acute or chronic oxalate nephropathy, or chronic kidney disease irrespective of aetiology, one or more of these elements are disrupted. The consequent impairment in oxalate homeostasis can trigger localized and systemic inflammation, progressive kidney disease and cardiovascular complications, including sudden cardiac death. Although kidney replacement therapy is the standard method for controlling elevated plasma oxalate concentrations in patients with kidney failure requiring dialysis, more research is needed to define effective elimination strategies at earlier stages of kidney disease. Beyond well-known interventions (such as dietary modifications), novel therapeutics (such as small interfering RNA gene silencers, recombinant oxalate-degrading enzymes and oxalate-degrading bacterial strains) hold promise to improve the outlook of patients with oxalate-related diseases. In addition, experimental evidence suggests that anti-inflammatory medications might represent another approach to mitigating or resolving oxalate-induced conditions.
Introduction
Oxalate, a seemingly inconspicuous dicarboxylic acid, is one of the 20 uraemic toxins with the highest relative increase in uraemia1. Oxalate homeostasis is maintained by a complex interplay of supply, metabolic pathways and excretion (Fig. 1). Consequently, oxalate homeostasis can be disturbed by numerous factors. Genetic mutations can disrupt endogenous oxalate biosynthesis and transport, whereas dietary and microbial factors as well as gastrointestinal pathologies can affect oxalate absorption. Kidney disease can also impair the elimination of oxalate, thereby turning a harmless, low-concentration metabolite into a serious multisystemic threat that can affect nearly every organ and tissue in the body, including the cardiovascular system2–4.
Fig. 1 |. Oxalate homeostasis.
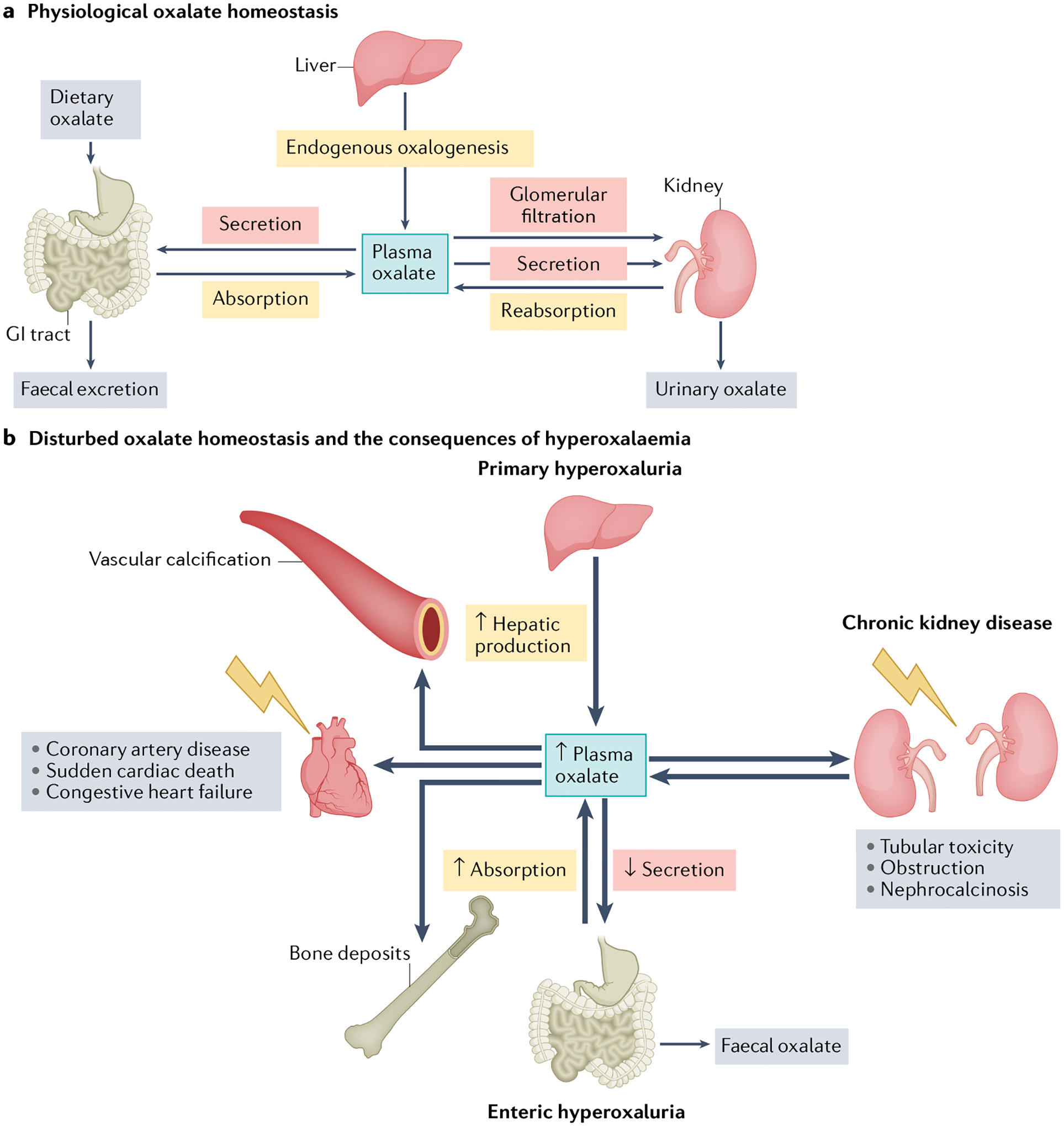
a, Physiological oxalate homeostasis. Oxalate homeostasis is maintained by a delicate interplay of supply (that is, hepatic production, gastrointestinal (GI) absorption of dietary oxalate and tubular reabsorption of circulating oxalate) and excretion (GI secretion and faecal oxalate, glomerular filtration, tubular secretion and urinary oxalate). Physiological plasma oxalate concentrations of 1–5 μM have no known negative effects on the cardiovascular system. b, Disturbed oxalate homeostasis and the consequences of hyperoxalaemia. Oxalate homeostasis might be disturbed by alterations in numerous pathways. Plasma oxalate concentrations can increase owing to decreased urinary excretion in chronic kidney disease, increased hepatic production in primary hyperoxaluria or increased GI absorption in enteric hyperoxaluria. When kidney function is still sufficiently high to enable compensatory oxalate excretion in the kidney, hyperoxaluria can result in nephrocalcinosis, tubular toxicity and obstruction. Loss of kidney excretory function can lead to supersaturation of plasma with oxalate, which can have severe adverse effects on the cardiovascular system. High plasma oxalate is associated with sudden cardiac death, coronary artery disease, congestive heart failure and vascular calcification. Oxalate can also deposit in other tissues such as bone, thyroid, spleen and lungs.
Increased serum or urine oxalate concentrations have been associated with progressive kidney disease5,6, cardiovascular conditions2,3, and cellular and systemic inflammation7–9. A 2021 study also identified elevated serum oxalate concentrations as a novel risk factor for sudden cardiac death in patients receiving dialysis3. Novel translational research findings, for example, regarding the pathogenesis of atherosclerosis10, are currently helping to move the field from mere association to causation. Further discoveries about the pathophysiology of uraemic toxins such as oxalate might help reduce the still unacceptably high excess mortality of patients with kidney and cardiovascular disease. As new evidence on the clinical implications of excess oxalate accumulates, it is crucial to understand the basic principles governing oxalate homeostasis. New studies have, for example, further defined the crucial involvement of epithelial transport proteins and the gut microbiome in oxalate regulation11,12.
In this Review, we will examine the pathways of oxalate in the body, highlighting core mechanistic steps and their clinical implications. This information will provide the groundwork for a discussion of the pathophysiological consequences of oxalate accumulation. We will also consider novel interventional, pharmacological and microbial approaches to prevent, mitigate or resolve oxalate-related conditions that affect the kidney, heart and other organs.
Oxalate sources and metabolism
Oxalate is the ionized conjugate base of oxalic acid, which is the simplest dicarboxylic acid13,14. Oxalate is either produced endogenously as a metabolic end-product or can be ingested as a component of numerous foods, drinks or chemicals (Fig. 1a). Healthy individuals typically maintain a plasma oxalate concentration of 1–5 μM. Of note, the ‘normal’ plasma oxalate range can vary depending on the measurement method; for example, one study reported that concentrations up to 11 μM were normal15–17.
Hepatic oxalate biosynthesis has been estimated to contribute 50–80% of total body oxalate levels, based on the measurement of the relative contribution of varying dietary oxalate intake on urinary oxalate levels18. Numerous molecules derived from amino acid and carbohydrate metabolism have been identified or suggested to be oxalate precursors (Fig. 2 and Table 1). For example, hydroxyproline, which derives from collagen catabolism, contributes an estimated 15% to urinary oxalate in healthy individuals19. Glycolate is another well-established oxalate precursor and has been estimated to contribute to <3% of oxalogenesis via peroxisomal metabolism20. An ongoing non-randomized clinical trial will use 13C2-glycolate infusion to estimate the contribution of glycolate to oxalate formation in healthy individuals21. Of note, although glycolate is thought to be present in most cells, its biological role is incompletely characterized22. Glyoxal is a product of carbohydrate autoxidation, lipid peroxidation and protein glycation that is primarily detoxified to glycolate by the glyoxalase system23,24. Oxalate synthesis from glyoxal in human erythrocytes and hepatocytes has been reported in vitro24,25, although a subsequent study suggested that this pathway might be of minor relevance in vivo26. Glyoxal was also proposed as the missing link between nephrolithiasis, hyperoxaluria and diabetes given that both glyoxal and oxalate excretion are elevated in patients with diabetes23,27. Finally, glycine is a small amino acid estimated to contribute <5% to urinary oxalate under physiological conditions28 (Fig. 2). Other amino acids, such as tryptophan, tyrosine and phenylalanine, are also potential oxalate precursors but are estimated to make only minor contributions to endogenous oxalate28.
Fig. 2 |. Model of endogenous oxalate synthesis pathways.
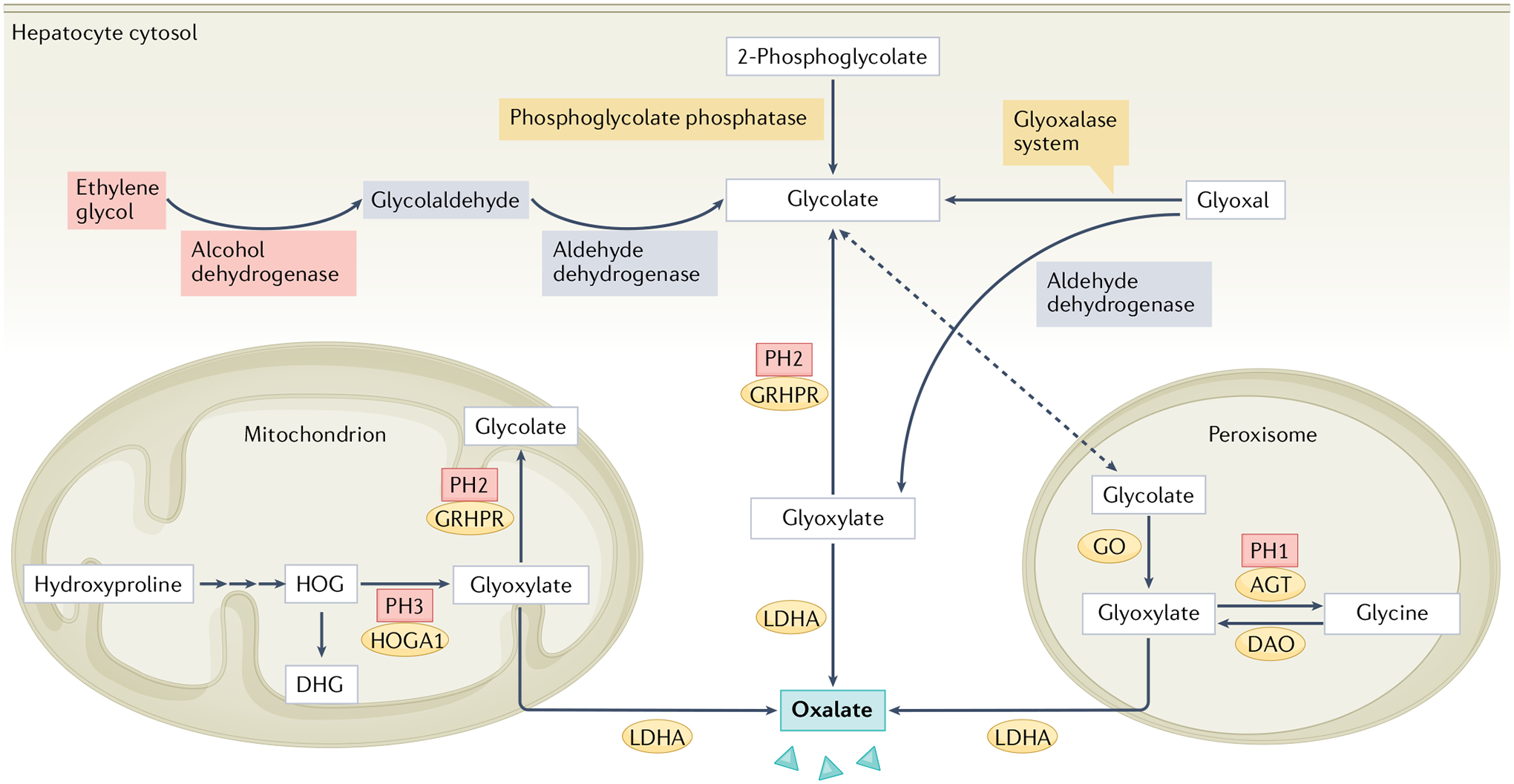
Glyoxylate links several metabolic pathways and is thought to be the principal precursor molecule of endogenous oxalate in healthy humans. Glyoxylate sources include hydroxyproline19, which is derived from collagen metabolism and is metabolized to 4-hydroxy-2-oxoglutarate (HOG) and its reduced form 2,4-dihydroxyglutarate (DHG) via three steps in the mitochondrion; HOG can be converted to glyoxylate by 4-hydroxy-2-oxoglutarate aldolase type 1 (HOGA1). Deficiency of HOGA1 results in primary hyperoxaluria type 3 (PH3) but the exact mechanism by which oxalate accumulates in this condition is not clear4. The accumulation of HOG might inhibit glyoxylate reductase/hydroxypyruvate reductase (GRHPR), which is ubiquitous in cytosol and mitochondria, and converts glyoxylate to glycolate; GRHPR deficiency causes PH2. The amino acid glycine is also converted to glyoxylate by d-amino acid oxidase (DAO) in the peroxisome. Deficiency of liver-specific, peroxisomal alanine–glyoxylate aminotransferase (AGT), which converts glyoxylate to glycine, results in PH1. In addition to glyoxylate, glycolate22 can be derived from sources such as glyoxal, which is a peroxidation product converted to glycolate by the glyoxalase system. Several other processes contribute to glycolate formation, including DNA repair (2-phosphoglycolate is converted to glycolate by phosphoglycolate phosphatase) and fructose or ethylene glycol metabolism (glycolaldehyde is converted to glycolate by aldehyde dehydrogenase). Glycolate is then converted to glyoxylate by liver-specific, peroxisomal glycolate oxidase (GO; also known as HAOX1). Glyoxylate is converted to oxalate by liver-specific lactate dehydrogenase A (LDHA).
Table 1 |.
Major sources of endogenous and exogenous oxalate
| Source | Pathway | (Patho)physiological roles |
|---|---|---|
| Endogenous sources | ||
| Glycolate | Derived from glyoxal and glyoxylate metabolism, DNA repair, fructose, ethylene glycol22,25 | Contributes to 1.3% of oxalogenesis in healthy individuals and 47.3% in PH1; diagnostic marker in PH1 (ref.110); metabolite involved in ethylene glycol poisoning |
| Hydroxyproline | Derived from collagen catabolism19 | Contributes to 15% of urinary oxalate in healthy individuals, and to 18%, 47% and 33% in PH1, PH2 and PH3, respectively19 |
| Glycine | Amino acid28 | Contributes to <5% of urinary oxalate in healthy individuals28 |
| Glyoxal | Advanced glycation end-product; also derived from glucose metabolism and lipid peroxidation23,24 | Potential link to diabetes23,27 |
| Glyoxylate | Metabolic product generated from 4-hydroxy-2-oxoglutarate, glycolate, glyoxal or glycine19,25 | Liver intermediary metabolism linking several metabolic pathways193 |
| Exogenous sources (via excessive ingestion) | ||
| Vegetables | Spinach, rhubarb, beets, sweet potato, Chaga mushroom | Oxalate nephropathy |
| Beverages | Cocoa powder, coffee, some types of tea | Can cause ice (black) tea-induced oxalate nephropathy |
| Proteins and carbohydrates | Soy, legumes, rice bran, wheatgerm, cornmeal, wholegrain flour | Diet-induced oxalate nephropathy in patients with diabetes194 |
| Fruits | Starfruit, guava, watermelon, raspberries | Starfruit-induced oxalate nephropathy |
| Seeds and nuts | Chia seeds, peanuts, sesame seeds, almonds, amaranth, hazelnuts, pistachios | Oxalate nephropathy induced by peanut, chia and almond consumption |
| Supplements | Vitamin C (ascorbic acid) | Vitamin C-induced hyperoxaluria, nephrolithiasis and oxalate nephropathy |
PH1, primary hyperoxaluria 1.
Although endogenous oxalogenesis is incompletely understood, many pathways converge in glyoxylate as the immediate oxalate precursor19,25 (Fig. 2). In a physiologically balanced state, glyoxylate is enzymatically converted to either glycine or glycolate. However, excess glyoxylate is converted into oxalate by liver-specific lactate dehydrogenase A (LDHA)19,20. Exogenous substances that can be metabolized to oxalate include large quantities of vitamin C supplements or of the colourless toxic alcohol ethylene glycol29. Notably, the measurement of oxalate in biological fluids in vitro is complicated by the non-enzymatic conversion of ascorbic acid to oxalate under non-acidic conditions30. This process might also contribute to the elevated plasma oxalate concentrations measured in patients who receive vitamin C supplements30. Ethylene glycol, which is often used as antifreeze owing to its low freezing point, is most commonly ingested with suicidal intent, as a cheap substitute for alcohol or accidentally by children31. After rapid gastrointestinal absorption, the majority of ethylene glycol is metabolized to glycolaldehyde by alcohol dehydrogenase in the liver and then converted to oxalate through several oxidative steps31 (Fig. 2). Extremely elevated plasma and urine concentrations of ethylene glycol and of its metabolites glycolic acid and lactic acid result in severe anion-gap acidosis as well as neurological, cardiopulmonary and kidney impairment31.
Despite a low bioavailability of only 5–15%32,33, dietary oxalate is estimated to contribute to 20–50% of total body oxalate18,34 (Table 1). Oxalate ingestion varies widely across culinary styles and diets in different regions of the world29,35. The typical intake of 100–200 mg/day (with a wide range) in Western diets is presumed to be harmless in patients with normal kidney function29,36. However, numerous studies have reported cases of acute nephropathy induced by the intake of extreme quantities of oxalate-rich foods (for example, starfruit)29. Transit studies suggest that physiological oxalate absorption mainly takes place in the small intestine (and, to a smaller extent, in the stomach and colon) and is modified by the presence of other faecal components37. Oxalate is an ionized conjugate base and is therefore highly susceptible to complexation with divalent cations such as Mg2+ and Ca2+, which bind oxalate in the faecal mass and reduce intestinal absorption14,37.
Based on radioisotope-labelled oxalate infusion studies, >90% of oxalate is estimated to be eliminated via the kidney, unchanged32,38,39. A physiological oxalate excretion of 10–40 mg/day (0.1–0.45 mmol/day) is maintained by a combination of glomerular filtration and net tubular secretion (mainly in the proximal tubule)33,34,40. Only a small fraction of endogenous oxalate is excreted faecally34, but enteric oxalate secretion is upregulated in murine chronic kidney disease (CKD) models, which might help prevent hyperoxalaemia11,41,42.
Epithelial oxalate handling
As discussed earlier, plasma oxalate derives from metabolic production (mainly in the liver) and net gastrointestinal absorption of the dietary oxalate that is ingested under normal conditions14,37,43. When kidney function is normal, oxalate is mainly excreted via the kidney through glomerular filtration and net tubular secretion14,37,43. Thus, steady-state urinary oxalate excretion is in balance with the sum of metabolic oxalate production and net gastrointestinal absorption of dietary oxalate.
Epithelial transport of oxalate mediates its absorption and secretion, and several approaches have been used to characterize the transport pathways involved. For example, studies using membrane vesicles from kidney or intestine identified apical and basolateral membrane anion exchange activities through which oxalate can be reversibly exchanged with anions such as Cl−, OH−, HCO3−, sulfate and formate44–50. Subsequently, cloned transporters from the solute carrier family 26 (SLC26) and SLC4 were found to mediate oxalate transport by some of the same modes of anion exchange. Specifically, functional expression of SLC26 member 1 (SLC26A1), SLC26A2 and SLC26A6 in Xenopus oocytes revealed that each solute carrier is capable of mediating the uptake or efflux of oxalate at high rates above baseline51–53. Functional expression studies also reported detectable oxalate transport through SLC26A3, SLC26A5, SLC26A7, SLC26A8, SLC26A9 and SLC26A11 (refs.54–57); SLC4A1 (also known as AE1) and SLC4A2 (also known as AE2) could also mediate oxalate transport58,59. Of note, determining the physiological roles of these transporters in transepithelial oxalate transport and oxalate homeostasis in vivo has been challenging but studies with knockout mice have provided some insights.
Gastrointestinal tract
Ingested oxalate is absorbed in multiple portions of the gastrointestinal tract, including the stomach, small intestine and large intestine14,37,43. Importantly, this absorption depends on its availability in soluble form because the formation of insoluble oxalate–calcium complexes blocks oxalate absorption14,37,43. Oxalate absorption in the gastrointestinal tract is largely passive (driven by the lumen-to-basolateral concentration gradient) and correlates positively with the amount of soluble oxalate ingested37. Transcellular absorption of oxalate in the stomach due to non-ionic diffusion across the apical membrane (driven by the extreme luminal acidity) is possible but, in the intestine, absorption seems to be predominantly paracellular given that the transepithelial oxalate permeability was similar to that of mannitol, which is a marker of paracellular permeability, across different segments of mouse intestine60. However, the finding that net oxalate absorption occurs in the absence of a passive driving force in studies of mouse ileum and large intestine suggests that transcellular absorption can also occur61 (Fig. 3). Moreover, oxalate absorptive fluxes were reduced in tissues from Slc26a3-null mice, which correlated with a reduced urinary oxalate excretion compared with that of wild-type mice61. Similarly, in wild-type mice with hyperoxaluria induced by an oral oxalate load, a small-molecule inhibitor of SLC26A3 significantly decreased oxalate absorption in the colon and greatly reduced urinary oxalate and oxalate nephropathy in response to oxalate feeding62. These studies not only demonstrated transcellular oxalate absorption but also demonstrated that it is, at least partly, dependent on the expression and activity of the apical membrane transporter SLC26A3. However, although functional expression studies of SLC26A3 demonstrated robust activity as a Cl−–HCO3− exchanger63, the ability of SLC26A3 cloned from multiple species (including humans) to transport oxalate was modest at best54,57,63,64. These findings suggest that SLC26A3 might only contribute to oxalate absorption as a consequence of interactions with other transporters that are present in native tissue but that are not co-expressed with SLC26A3 in the reported functional expression studies.
Fig. 3 |. Oxalate transport in the small intestine.
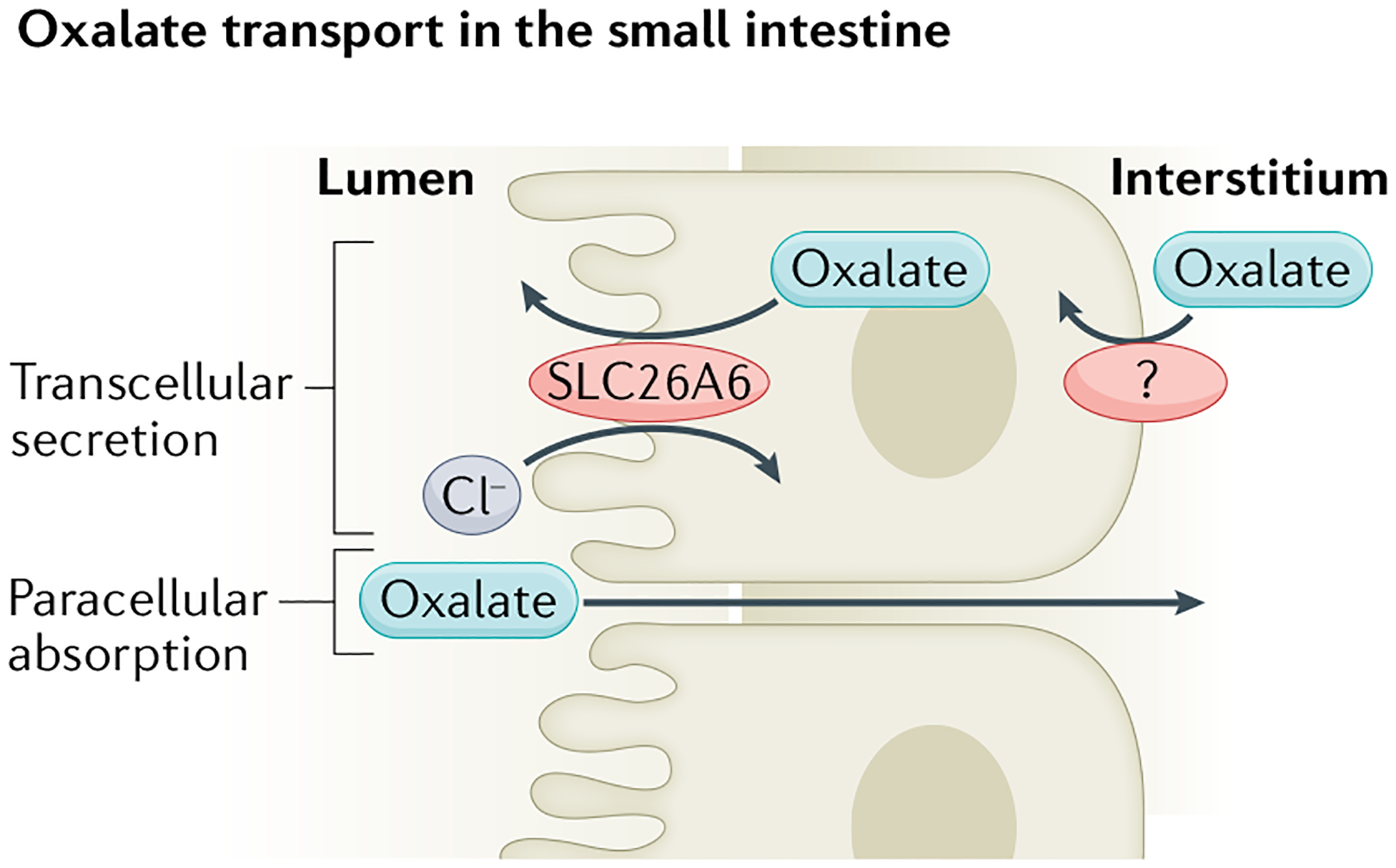
Most oxalate absorption in the small intestine occurs passively through the paracellular pathway. Transcellular oxalate secretion is mediated by apical membrane Cl−–oxalate exchange through the transporter solute carrier family 26 member 6 (SLC26A6). The transporter on the basolateral membrane that operates in combination with apical SLC26A6 to mediate transcellular oxalate secretion in the small intestine has not yet been identified.
Transcellular oxalate secretion in multiple intestinal segments opposes oxalate absorption37. The importance of oxalate secretion in limiting net oxalate absorption has been most clearly demonstrated in mouse small intestine. The apical membrane oxalate transporter SLC26A6, which is expressed in the kidneys, pancreas and small intestine, has robust activity as a Cl−–oxalate exchanger and would therefore be predicted to mediate oxalate efflux across cell membranes51,52,65. Accordingly, net oxalate secretion in the duodenum and ileum of Slc26a6-knockout mice was greatly reduced compared with that of wild-type mice66,67. Moreover, Slc26a6-null mice had hyperoxalaemia, which led to hyperoxaluria and urolithiasis; the development of urolithiasis was mouse-strain dependent66,67. Removal of oxalate from the diet greatly reduced both hyperoxalaemia and hyperoxaluria in Slc26a6-null mice, indicating that the source of excess urinary oxalate was predominantly dietary67. These findings support a role for SLC26A6 in back-secreting oxalate that is passively absorbed through tight junctions (Fig. 3). Moreover, a 2021 study suggests that short-chain fatty acids can reduce urinary oxalate levels by upregulating SLC26A6 (ref.68). In a mouse model of CKD11, faecal oxalate excretion increased following induction of CKD, suggesting that intestinal oxalate secretion might be enhanced in response to reduced kidney excretion, yet this increase was absent in Slc26a6-null mice, in which CKD-induced hyperoxalaemia was exacerbated compared with wild-type mice11. Of note, epithelial oxalate secretion can also occur in the mouse large intestine in the absence of SLC26A6, indicating the presence of alternative pathways for apical membrane oxalate secretion, at least in this tissue69. The relevance of intestinal SLC26A6 expression to oxalate homeostasis in humans is thus far only anecdotal. In a patient with subclinical coeliac disease and without fat malabsorption, hyperoxaluria correlated with a markedly reduced expression of SLC26A6 in the small intestine70 whereas, in another patient, enteric hyperoxaluria and nephrolithiasis were associated with a dominant-negative SLC26A6 mutation71. Knockout mice studies also demonstrated a role for SLC26A6 in epithelial oxalate secretion in the salivary gland72.
SLC26A1 is a basolateral sulfate–HCO3−–oxalate anion exchanger that is expressed in several tissues including liver, kidney and intestine52,73–77. The initial report that Slc26a1-null mice had hyperoxalaemia, hyperoxaluria and crystal deposition in the kidneys (similar to the phenotype of Slc26a6-null mice) suggested comparable participation of SLC26A1 and SLC26A6 in intestinal oxalate secretion78. However, subsequent studies found that knockout of Slc26a1 did not impair intestinal oxalate secretion79,80 but rather reduced urinary oxalate excretion80. Biallelic SLC26A1 mutations that impaired the transport function (assayed as sulfate flux) were identified in two patients with calcium oxalate nephrolithiasis, although only one of the patients had hyperoxaluria (mild)81. Consequently, whether loss of function of SLC26A1 can cause substantial hyperoxaluria owing to defective intestinal oxalate secretion remains highly uncertain.
Liver
In addition to variable expression along the gastrointestinal tract, SLC26A6 and SLC26A1 are both expressed in the liver and proximal tubule73,74,82–85. In the liver, the kinetic properties of basolateral SLC26A1 indicate that it is a strong candidate mediator of the efflux of metabolically produced oxalate into the plasma pool77, whereas apical SLC26A6 might contribute to the reported biliary excretion of oxalate86. However, direct evidence for a role of these transporters in mediating oxalate transport in hepatocytes is lacking.
Kidneys
In the kidneys, oxalate is filtered and then undergoes passive absorption and active secretion in the proximal tubule, resulting in net secretion40,87. Net kidney oxalate secretion is dependent on SLC26A6 as the fractional excretion of oxalate decreased from >1 in wild-type mice to <1 in Slc26a6-null mice88. A defect in kidney oxalate secretion in Slc26a6-null mice would therefore be expected to lower urinary oxalate. However, Slc26a6-null mice have hyperoxaluria, suggesting that the aforementioned defect in intestinal oxalate secretion leading to enhanced net oxalate absorption causes sufficient hyperoxalaemia to result in hyperoxaluria even when urinary oxalate secretion is reduced. SLC26A1 is a plausible candidate to mediate basolateral membrane influx of oxalate in combination with apical SLC26A6 to accomplish oxalate secretion in the proximal tubule. However, the kinetic properties of SLC26A1 might preclude it from mediating a substantial influx of oxalate under physiological conditions77. Specifically, based on the relative affinities for HCO3− and oxalate compared with their plasma concentrations, SLC26A1 was predicted to predominantly mediate HCO3− rather than oxalate influx in exchange for intracellular sulfate in the proximal tubule77. No direct evidence of a role for SLC26A1 in mediating oxalate secretion by the proximal tubule is yet available, although the reduced urinary oxalate excretion observed in mice with global knockout of Slc26a1 (ref.80) could hint at a role in kidney oxalate secretion.
Interestingly, human studies demonstrated that, under fasting conditions, fractional excretion of oxalate is ~1, indicating little or no net secretion, although appreciable and rapid net oxalate secretion occurs after ingestion of an oxalate load89. Similarly, substantial net oxalate secretion is observed in patients with elevated plasma oxalate owing to primary hyperoxaluria (PH) but not in patients who form stones without PH90. The mechanisms underlying the upregulation of kidney oxalate secretion in response to acute or chronic oxalate loads remain to be defined. Another aspect of kidney oxalate handling that is incompletely understood is transepithelial transport beyond the proximal tubule. For example, active net absorption of oxalate has been described across the kidney papillary epithelium91. Although this process might not substantially modify urinary oxalate excretion, it might modulate the local interstitial concentration of oxalate and thus affect crystal formation91.
Of note, independently of its role as an oxalate transporter, SLC26A6 might also be an important modifier of the risk of stone formation owing to its ability to interact with and inhibit the activity of the citrate transporter NADC1 (also known as SLC13A2)92. Knockout of Slc26a6 leads to increased activity of NADC1, greater reabsorption of filtered citrate, reduced excretion of urinary citrate and greater risk of calcium nephrolithiasis92.
Role of the microbiome in oxalate homeostasis
Trillions of bacteria colonize the human gut and are involved in diverse functions, including the metabolism of dietary components93,94. These bacteria are increasingly recognized to make important contributions to health and disease. Humans and other mammals lack the enzymes to metabolize oxalate and therefore rely on bacterial degradation to reduce intestinal oxalate, potentially reducing its absorption95. Oxalobacter formigenes is a specialist oxalate degrader and induces colonic oxalate secretion96–98. Epidemiological data have linked antibiotic use with an increase in the incidence of kidney stones99,100, and disturbance of oxalate-degrading microbiota might be a contributing factor101.
Numerous oxalate-degrading microbes have been identified in vitro, but the relative importance of each species in vivo is debated102. A 2021 systematic analysis used high-throughput metagenomic and metatranscriptomic data to investigate bacterial oxalate degradation in vivo. The study showed that the human gut microbiota in healthy adults comprises a diverse community that actively transcribes oxalate-degrading genes12. In healthy individuals, oxalate degradation is primarily performed by O. formigenes, which represents the largest reservoir of oxalate-metabolizing genes at the transcriptional level, greater than all other oxalate-degrading organisms combined, including Escherichia coli, Bifidobacterium spp. and Lactobacillus spp.12 (Fig. 4).
Fig. 4 |. Microbial modulation of oxalate homeostasis.
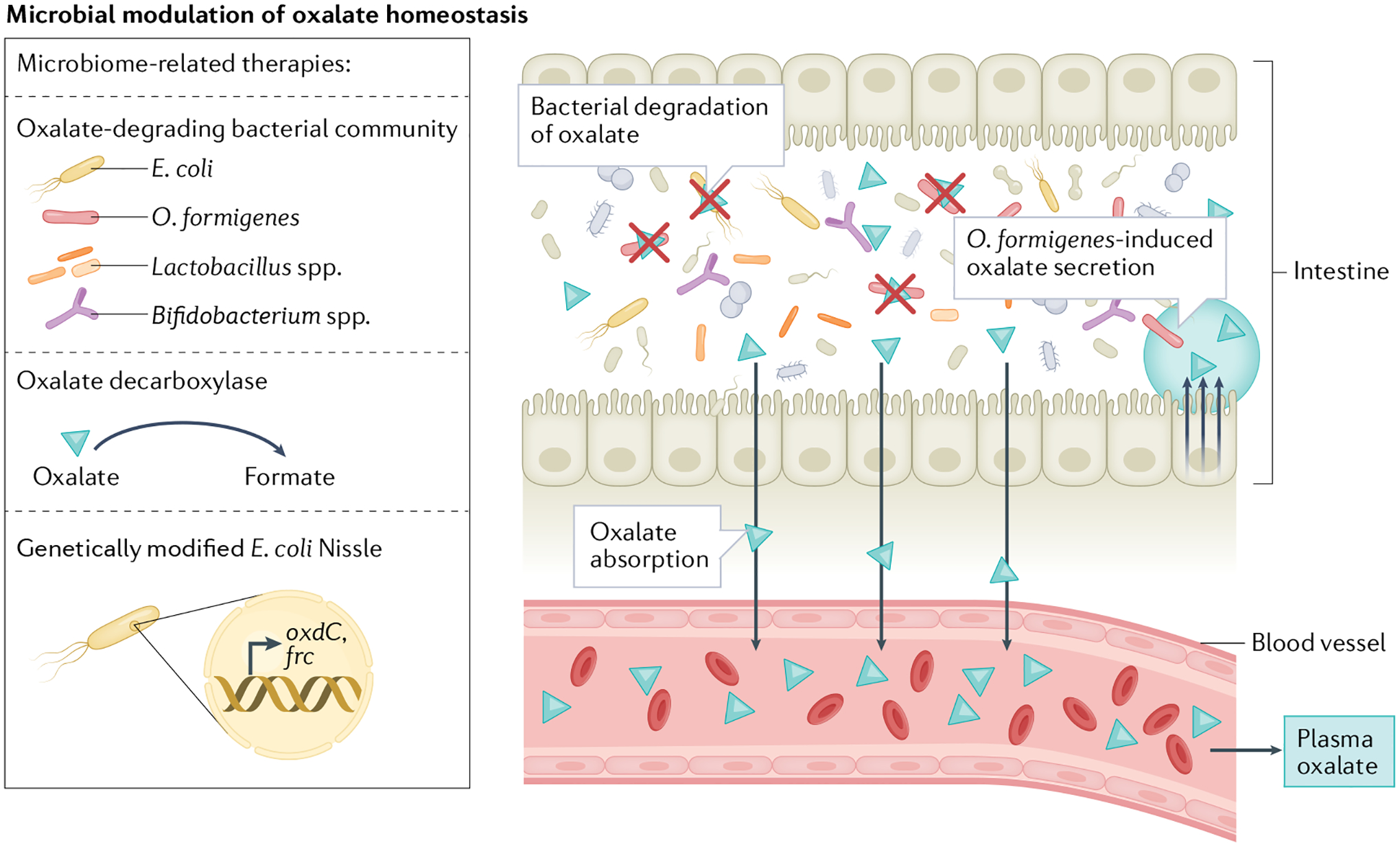
Dietary oxalate is degraded by several species of the intestinal microbiota, which can reduce the amount of oxalate available for intestinal absorption. Several bacterial species can degrade oxalate, including Oxalobacter formigenes, Escherichia coli, Bifidobacterium spp. and Lactobacillus spp. O. formigenes also releases a secretagogue that induces oxalate secretion into the gut. Several microbiome-based therapies are being developed to enhance oxalate degradation and reduce oxalate absorption in the gastrointestinal tract. These therapies include supplementation with oxalate-degrading bacterial communities, use of the enzyme oxalate decarboxylase, which metabolizes oxalate, and use of the E. coli Nissle strain, which has been genetically modified to encode the oxalate degradation pathway genes that encode oxalyl-CoA decarboxylase (oxdC) and formyl-CoA:oxalate CoA-transferase (frc).
Most studies linking microbial oxalate metabolism and disease were performed by comparing the gut microbiome of individuals who form kidney stones to that of healthy individuals103–105. For example, analysis of stool cultures showed that the prevalence of O. formigenes was lower in individuals who form stones than in healthy individuals106, but this finding was not consistent with subsequent studies that used microbiome sequencing data107,108. Comparing the gut microbiome of individuals who form stones with that of cohabitating healthy adults revealed that healthy individuals had a more extensive microbial network with a higher abundance of connected species centred around O. formigenes, which suggests that they might have a greater capacity for microorganism-driven oxalate metabolism108. Levels of bacterial genes involved in oxalate degradation were also significantly lower in the microbiome of individuals who form stones, which correlated inversely with 24-h oxalate excretion. Similarly, the cumulative abundance of oxalate-degrading bacteria correlated inversely with urinary oxalate103. One study also reported that oxalate-degrading bacteria of three taxa — Enterococcus faecalis, Enterococcus faecium and Bifidobacterium animalis — were less abundant in children with kidney stones than in healthy children105. By contrast, an earlier study showed that patients with hyperoxaluria and kidney stones had a selective enrichment of oxalate-metabolizing bacterial species compared with healthy controls104. More translational and clinical studies are needed to establish the causal relationship between the abundance and function of oxalate-degrading microbiota and urinary oxalate.
Dysregulated oxalate homeostasis
Dysfunction in key steps of physiological oxalate homeostasis can lead to the development of different types of primary and secondary hyperoxaluria (Figs. 1b and 2). Hyperoxaluria is defined as a urinary oxalate excretion of >40–45 mg/day (0.45–0.5 mmol/day), which is associated with various systemic manifestations34,109.
Primary hyperoxaluria
PH comprises autosomal recessive inherited enzyme deficiencies that result in increased hepatic oxalogenesis (Table 2 and Fig. 2; reviewed in ref.110). The diagnosis of PH is primarily based on clinical presentation, including elevated oxalate concentrations in plasma and urine, and can be confirmed with genetic analyses110–113. The most common manifestations of PH include urolithiasis, nephrocalcinosis and urinary tract infections111,112,114. The carrier frequency in the population is estimated at 1:70, and estimates of disease prevalence range between 1:58,000 and <3:1,000,000 (ref.113). PH type 1 (PH1) is characterized by a deficiency of alanine–glyoxylate aminotransferase (AGT; encoded by AGXT). More than 200 different AGXT mutations have been identified, with a highly variable genotype–phenotype correlation (see the Human Gene Mutation Database)110. A 2022 study used non-canonical splicing site and copy number variant sequencing to improve the diagnostic accuracy from 26% to 35% in patients with suspected PH115. Of note, another study implemented bioinformatics to identify the microRNA miR-4660, which repressed AGT activity in a subgroup of patients with mutation-negative oxalosis116. Several mutations lead to protein aggregation and mitochondrial mistrafficking of AGT (in humans, AGT needs to be peroxisomal to function properly)117, which results in decreased or diminished enzyme activity118–120. A 2021 report suggested that lack of protein dimerization might be one of the mechanisms underlying AGT mistrafficking121.
Table 2 |.
Primary hyperoxaluria types, diagnostics and manifestations
| Type (% of PH cases) | Affected enzyme | Diagnostics | Manifestations |
|---|---|---|---|
| PH1 (refs.110,113) (~80%) | AGT; >200 AGXT mutations described | Clinical presentation, high glycolate (non-specific) and high oxalate in urine and plasma, genetic analysis | Reported age of onset 0.1–53 years113; median urinary oxalate excretion: 1.8 mmol/1.73 m2/day113 (vitamin B6-responsive PH1: 0.94 mmol/1.73 m2/day; vitamin B6-non-responsive PH1: 1.37 mmol/1.73 m2/day111); progression to CKD (>73% of patients) and kidney failure (>50% of patients)114 |
| PH2 (refs.111–113) (7.9–10%) | GRHPR; at least 39 GRHPR mutations described | Clinical presentation, high l-glycerate (non-specific) in urine and high oxalate in plasma and urine, genetic analysis | Reported age of onset 0.6–42 years113; median urinary oxalate excretion: 1.4–1.7 mmol/1.73 m2/day111,113; progression to CKD (50% of patients) and kidney failure (25% of patients)111,112 |
| PH3 (refs.111,113) (8.4–17%) | HOGA1; at least 37 HOGA1 mutations described | Clinical presentation, high 4-hydroxy-2-oxoglutarate or 2,4-dihydroxyglutarate in urine and high oxalate in plasma and urine, genetic analysis | Reported age of onset 0.3–48 years111,113; median urinary oxalate excretion: 1.1–1.16 mmol/1.73 m2/day111,113; progression to CKD (~20% of patients) and kidney failure (one case reported)111,113 |
AGT, alanine–glyoxylate aminotransferase; CKD, chronic kidney disease; GRHPR, glyoxylate reductase/hydroxypyruvate reductase; HOGA1, 4-hydroxy-2-oxoglutarate aldolase type 1; PH1, primary hyperoxaluria type 1.
PH type 2 (PH2) accounts for approximately 7.9–10% of PH cases and results from mutations in GRHPR, which encodes the enzyme glyoxylate reductase/hydroxypyruvate reductase (GRHPR). This enzyme detoxifies glyoxylate to glycolate in the cytosol and mitochondria of hepatocytes111,112. In GRHPR deficiency, glyoxylate and hydroxypyruvate accumulate and are subsequently converted to oxalate and l-glycerate by hepatic LDHA112. At least 39 GRHPR mutations have been described112. PH type 3 (PH3) is caused by a mutation in the gene encoding 4-hydroxy-2-oxoglutarate aldolase type 1 (HOGA1), which is primarily expressed in hepatocyte mitochondria, and accounts for 8.4–17% of PH cases111. HOGA1 catalyses the last step of hydroxyproline metabolism by converting 4-hydroxy-2-oxoglutarate (HOG) into glyoxylate and pyruvate; in HOGA1 deficiency, HOG accumulation has been reported to inhibit GRHPR, resulting in increased oxalate production19. A 2021 report described at least 37 HOGA1 mutations111. The age of disease onset was reportedly earlier in PH3 than in PH2 (ref.113), but several studies report a median age of onset <10 years of age for all three PH types, with a range reaching into late adulthood4,111–113 (Table 2).
The natural history of PH evolves in two phases and their exact timelines depend on the severity and type of the underlying mutation. In the first phase, increased oxalate synthesis is compensated by hyperoxaluria that can reach extremes of up to 1–2 mmol/1.73 m2/day and eventually results in oxalate deposition in the kidney and progressive kidney damage. The second phase unravels as kidney function declines, oxalate excretion becomes insufficient and systemic oxalate deposition ensues, which leads to secondary organ damage4. Although urinary oxalate concentrations have been suggested to correlate positively with PH progression, the most common PH1-causing AGXT mutation, G170R, typically correlates with less severe progression of kidney disease, compared with other PH1-driving AGXT mutations. This difference might be partially explained by the ability to reduce enzyme mistargeting in the G170R genotype through treatment with the cofactor pyridoxine, also known as vitamin B6 (only a few other PH1-causing mutations, such as AGXTF152I, respond to pyridoxine)113. Of note, in the largest study of PH2 to date, the progression of CKD was not associated with genotype or urinary oxalate excretion in an age-corrected analysis112. In this study, patients with PH2 were also less likely than patients with PH1 to present with CKD stage V at diagnosis or before 15 years of age112. In the largest study to date of patients with PH3, PH3 genotype and phenotype did not correlate, and urinary oxalate excretion was not significantly different between vitamin B6-non-responsive PH1, PH2 and PH3 (ref.111). Although kidney failure due to PH3 has been reported113, most studies describe milder kidney disease in patients with PH3 than in those with PH1 or PH2. However, these data might be biased owing to the scarcity of long-term outcome monitoring data and the low penetrance of HOGA1 mutations111.
Secondary hyperoxaluria
Secondary hyperoxalurias are broadly classified as enteric or dietary29. The most common aetiologies of enteric hyperoxaluria are Roux-en-Y gastric bypass and pancreatic insufficiency122; others include malabsorptive conditions, such as short bowel syndrome, coeliac disease, Crohn’s disease and cystic fibrosis, or the use of medications that interfere with intestinal oxalate absorption such as octreotide, which is a somatostatin analogue, or orlistat, which is a lipase inhibitor36. Dietary hyperoxaluria is most often caused by excessive intake of vitamin C or oxalate (discussed above)122.
All types of hyperoxaluria are characterized by three histopathological hallmarks: calcium oxalate crystal deposition, damage to the tubular epithelium and interstitial alterations in the kidney29. However, these patterns might differ between different aetiologies of oxalate nephropathy. For example, in one study, patients with enteric hyperoxaluria were more likely to have tubulointerstitial atrophy and fibrosis, whereas those with non-enteric hyperoxaluria had more pronounced tubular crystal accumulation and interstitial inflammation122. The authors hypothesized that the severity of the kidney outcomes might depend on the latency period following oxalate exposure. However, the mechanisms underlying these differences in the clinical manifestations of enteric and non-enteric hyperoxaluria are still unknown. In vivo findings suggest that crystal deposition is crucial to hyperoxaluria-induced inflammation123.
Chronic kidney disease
In PH, increased endogenous hepatic synthesis of oxalate can lead to extreme plasma oxalate concentrations ranging from 80 μM to 125 μM in advanced stages of disease124. In people with common forms of CKD who do not have PH, plasma or serum oxalate is elevated owing to kidney impairment, especially in patients with kidney failure undergoing dialysis. Nonetheless, plasma or serum oxalate levels in these patients are not as high as those observed in patients with PH, except in patients with Crohn’s disease and ileocecal resections that are treated with maintenance haemodialysis124–126. In these patients, serum oxalate levels correlate with calcium oxalate supersaturation in serum samples and, in certain cases, might reach the serum calcium oxalate supersaturation threshold of 30 μM124,125 (Fig. 1b). Although high plasma oxalate was associated with kidney function decline in primary hyperoxaluria127, data on plasma oxalate and outcomes in more common forms of CKD are currently limited. Nonetheless, although classically described as a risk factor for nephrolithiasis and acute kidney injury owing to intratubular crystal obstruction, the role of oxalate in the pathogenesis of CKD has been further defined in the past few years. Mice fed a diet high in oxalate developed a reproducible CKD phenotype characterized by hypertension, hyperkalaemia, metabolic acidosis, anaemia and hyperphosphataemia; kidney histopathology revealed fibrosis, tubular injury, atubular glomeruli and inflammation128. Epidemiological studies also showed that higher urinary and plasma oxalate levels might be associated with adverse outcomes in kidney disease. A prospective study of adults with common forms of CKD (Chronic Renal Insufficiency Cohort) in the USA showed an independent association between 24-h urinary oxalate excretion and both CKD progression and kidney failure6. Participants in the highest quintile of urinary oxalate excretion had a 33% increased risk of CKD progression and 45% increased risk of kidney failure; 24-h urinary oxalate excretion was also positively associated with greater levels of proteinuria and lower estimated glomerular filtration rate (eGFR)6. Importantly, plasma oxalate levels can be significantly elevated in patients with kidney failure on dialysis because dialysis cannot completely compensate for lost kidney excretion. In a post-hoc analysis of data from a randomized controlled study of patients with diabetes receiving dialysis in Germany, the highest quartile of blood oxalate level was associated with a 40% increased risk of combined cardiovascular events (composite of death from cardiac causes, fatal or non-fatal stroke, or non-fatal myocardial infarction)3. Blood oxalate levels were also significantly associated with sudden cardiac death in secondary analyses.
The role of oxalate as a biomarker in kidney transplant recipients is also unclear. Urinary oxalate excretion was not associated with graft survival129, but the presence of calcium oxalate deposits in allograft biopsy samples might be associated with delayed graft function and decline in allograft function130,131. In 167 kidney transplant recipients followed for 15 years who had plasma oxalate measured 10 weeks after transplantation, plasma levels in the upper quartile were associated with lower long-term patient survival and graft loss; however, these associations were not significant when examining death-censored graft loss132.
Metabolic diseases
Several metabolic diseases are associated with mild hyperoxaluria. For example, individuals with obesity have a high incidence of nephrolithiasis and hyperoxaluria, which correlates with decreased eGFR and increased risk of CKD133. One potential culprit is the pro-inflammatory effect of obesity. Obese mice had a 3.3-fold increase in urinary oxalate excretion (adjusted for creatinine) compared with lean control mice; this increase was accompanied by a significant reduction in intestinal oxalate secretion. This change in secretion might be due to reduced expression of SLC26A6 in obese mice compared with lean mice134. In vitro, pro-inflammatory cytokines suppress Slc26a6 mRNA and protein expression134. Another group identified glyoxylate pathway modifications in the hepatocytes of obese mice that led to the hypermethylation and downregulation of Agxt. These changes resulted in a significant increase in hepatic oxalogenesis after hydroxyproline challenge135. Moreover, the pathophysiological overlap between oxalate excretion and diabetes is notable. Individuals with diabetes have an 11% increase in urinary oxalate excretion compared with individuals without diabetes6, which might be partly attributable to elevated concentrations of the oxalate precursors glyoxal and glyoxylate27. Of note, the presence of diabetes was associated with a 44% increase in the risk of a 50% reduction in eGFR or kidney failure among patients with urinary oxalate excretion of ≥16.2 mg/day compared with those with urinary oxalate excretion of <16.2 mg/day6.
Cellular effects of excess oxalate
As discussed earlier, oxalate can form complexes with positively charged minerals. These complexes can grow in size and form oxalate kidney stones that obstruct urinary flow and cause kidney damage133. In addition, oxalate can affect cellular function directly. In vitro, oxalate inhibits the proliferation of kidney epithelial cells, stimulates fibrotic transformation and calcification, and induces cell death128,136. Oxalate might also promote epithelial-to-mesenchymal transformation137. Incubating mouse inner medullary collecting duct cells with oxalate increased expression of mesenchymal markers such as α-smooth muscle actin (also known as aortic smooth muscle actin) and reduced E-cadherin expression138. Moreover, oxalate activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in kidney epithelial cells and triggers the release of reactive oxygen species, which promotes oxidative stress139, and has been associated with reduced glutathione and impaired mitochondrial function140.
In addition to the effect of oxalate on epithelial cells, several studies have suggested that oxalate crystals activate inflammatory cells141. For example, oxalate crystals stimulated dendritic cells and macrophages to synthesize and release IL-1β via activation of the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome5,9. Moreover, exposure of human monocytes to a low dose of oxalate crystals disrupted mitochondrial function142. The investigators hypothesized that these biological events might predispose to recurrent stone formation. A subsequent study revealed that macrophages treated with oxalate also had decreased cellular bioenergetics, mitochondrial complex I and IV activity, and ATP levels compared with control cells143. These cells had impaired metabolism, redox homeostasis and cytokine signalling, which compromised their antibacterial response in vitro and increased the risk of bacterial infection143.
A 2021 study demonstrated that the glycine-to-oxalate ratio in blood is lower in both patients and mice with atherosclerosis than in controls10. This effect was due to suppressed activity of AGT, similar to what is observed in patients with PH1. Mice deficient for AGT and apolipoprotein E (ApoE) had increased atherosclerosis compared with mice that were only deficient for ApoE, which was accompanied by the induction of hepatic pro-atherogenic pathways associated with increased inflammation (based on changes in cytokine and chemokine signalling). Macrophages from AGT-deficient mice exposed to oxalate also showed mitochondrial dysfunction and superoxide accumulation, which led to increased release of the pro-atherogenic CC chemokine ligand 5 (CCL5). The observed phenotype could be reversed with AGXT overexpression. One of the limitations of the study was the high concentration of oxalate used to stimulate macrophages; although non-cytotoxic, a concentration of 750 μM is still several times higher than that observed in the blood of patients, even in those with impaired kidney excretion. Nevertheless, this study suggests a mechanistic link between increased plasma oxalate and atherosclerosis.
These observations support previous studies suggesting that oxalate might promote atherosclerosis. In vitro, supraphysiological extracellular oxalate concentrations inhibit the proliferation and induce oxidative stress in endothelial cells8,144. Moreover, the oxalate crystal-induced release of pro-inflammatory cytokine IL-1α from monocytes has been associated with increased risk of atherosclerotic cardiovascular events in patients with acute myocardial infarction or CKD, possibly by promoting leukocyte–endothelial adhesion and inflammation through the expression of vascular cell adhesion molecule 1 on endothelial cells7. Importantly, future clinical studies will need to address whether oxalate concentrations observed in humans with hyperoxalaemia can have the same effects as exposure to supraphysiological amounts of oxalate in vitro.
Therapies for oxalate dysregulation
Many conservative treatment options are available for mild types of hyperoxaluria, including the hyperoxaluria observed in patients with nephrolithiasis. These therapeutic approaches are based on the prevention of urinary calcium oxalate crystallization and might be as simple as adequate hydration, which has proven useful for the prevention of kidney stones; hyperhydration is recommended in patients with PH1 (>3 l/1.73 m2 body surface area/day)110,145,146. In addition, a phase I trial of tolvaptan has shown promise in reducing urinary calcium oxalate, calcium phosphate and uric acid supersaturation by increasing urine volume147. A similar effect was reported for thiazide diuretics, whose hypocalciuric and hypermagnesiuric properties might reduce calcium oxalate kidney stone formation148. Citrate administration has also been suggested to prevent the formation of kidney stones149,150. Moreover, a 2016 study showed that citrate and hydroxycitrate can dissolve calcium oxalate monohydrate crystals in vitro150. Even at concentrations three times lower than that of the solute, citrate and hydroxycitrate adsorption to oxalate crystal surfaces led to the localized dissolution of calcium and oxalate ions with comparable efficacy150. Of note, a 2022 report from a phase II clinical trial suggested that lemon juice might be a viable alternative to pharmaceutical citrate formulations, which are frequently discontinued by patients owing to adverse gastrointestinal effects151.
Targeting oxalate absorption
In enteric hyperoxaluria, several therapeutic approaches are based on the binding of oxalate in the intestine to prevent hyperabsorption. As described earlier, the bioavailability of oxalate is influenced by gut health and the presence of different faecal components (for example, cations, fat or medication)14,36,37. Consequently, direct supplementation of calcium and fat limitation (which has been reported to increase intestinal calcium–oxalate binding) might decrease oxalate hyperabsorption146,152. Based on the same principle of limiting oxalate absorption, the bile acid-binding resin cholestyramine reduced colonic oxalate absorption in rats by blocking the binding of oxalate to bile acids and subsequent intestinal reabsorption but its efficacy was variable in humans153. However, in a case study of a patient with short bowel syndrome, cholestyramine reduced both faecal fat and urinary oxalate excretion154. Calcium-containing phosphate binders, which might promote the formation of calcium–oxalate complexes, have also been suggested as potential oxalate-lowering agents155. However, current KDIGO guidelines recommend limiting the use of calcium-based phosphate binders in patients with CKD stages G3a–G5D owing to the increased mortality risk associated with these compounds compared with non-calcium-based phosphate binders156. Following encouraging experimental results, the non-calcium-based phosphate binder lanthanum carbonate is currently being tested in a phase III trial in patients with secondary hyperoxaluria and nephrolithiasis157. Treatment with the non-calcium phosphate binder sevelamer hydrochloride for 1 week also led to a non-significant reduction in urinary oxalate in an open-label study of patients with enteric hyperoxaluria158. Of note, a 2021 quantum chemical study suggested that trivalent cations, such as Fe3+, Al3+ or La3+, rather than divalent cations as well as the chemical element neodymium, might be interesting oxalate-binding candidates for preclinical testing159.
Microbiome-related therapies
The effect of microbiome manipulation on oxalate homeostasis using single organisms or bacterial communities has been evaluated extensively in animal models and a few human studies (Fig. 4). Consequently, several therapeutic formulations are based predominantly on the oxalate-degrading capacity of certain bacteria. In rodent models of hyperoxaluria, O. formigenes colonization consistently led to a reduction in urinary oxalate97,160. In addition to oxalate degradation, O. formigenes produced a secretagogue, yet to be characterized, that induces oxalate secretion into the gut97. However, although the O. formigenes derivatives OC3 and OC5 (lyophilized O. formigenes in enteric-coated capsules, the latter with a higher viable cell count) are well tolerated by humans, they had mixed efficacy in phase I–III trials161,162.
Colonization with Bifidobacterium and Lactobacillus species also reduced urinary oxalate in animals97,163. Oxadrop, which is a probiotic composed of Lactobacillus acidophilus, Lactobacillus brevis, Streptococcus thermophilus and Bifidobacterium infantis, resulted in a short-lived reduction in urinary oxalate in patients with enteric hyperoxaluria164. Importantly, none of these trials evaluated bacterial viability in the gut following their ingestion. Other bacterial strains currently being investigated in early-stage trials include Nov-001, UBLG-36 and SYNB8802. Nov-001 is a therapeutically engineered microbial combination product that includes NB1000S, which is an oxalate-degrading bacterium, and NB2000P, a prebiotic control molecule used to control the abundance of NB1000S; a phase II trial is currently recruiting patients with enteric hyperoxaluria after an encouraging phase I study165. UBLG-36, which is a Lactobacillus paragasseri strain, is highly effective in degrading oxalate in vitro166. Finally, orally administered SYNB8802, which is a synthetic oxalate-degrading E. coli Nissle strain, significantly reduced urinary oxalate in mice and non-human primates. In silico modelling based on human gastrointestinal physiology predicted that this strain could lower urinary oxalate by up to 71% in patients with enteric hyperoxaluria167.
Whole microbial transfers have also been investigated as potential approaches to modulate oxalate metabolism. In rats, a faecal transplant from a mammalian herbivore whose whole microbiome community is adapted to degrading high amounts of dietary oxalate led to lower urinary and faecal oxalate levels compared with the ingestion of oxalate-degrading isolates168; the decrease in urinary oxalate persisted for 9 months after the transfer169. Similarly, a faecal transplant from conventional rats, which have a gut microbiome similar to that of humans, into germ-free mice reduced urinary oxalate170. This transfer caused a reduction in SLC26A6 expression in the kidney and colon and increased expression in the caecum. To date, no studies have evaluated the effect of microbiome community transfer in humans.
Instead of administering bacterial strains, several investigators have attempted direct supplementation with oxalate decarboxylase (OxDC), which is the enzyme used by O. formigenes to degrade oxalate. A double-blind randomized controlled crossover trial of a proprietary OxDC in healthy individuals showed a 24% urinary oxalate reduction compared with placebo171. In a phase I trial, recombinant OxDC cloned from Bacillus subtilis significantly reduced urinary oxalate in patients with Roux-en-Y gastric bypass (mean 66.3 mg/day, standard deviation (SD) 28.0 mg/day at baseline; 44.5 mg/day, SD 23.7 mg/day at 4 weeks; P = 0.018), but this reduction was not significant in patients with idiopathic calcium oxalate stone formation (43.2 mg/day, SD 5.9 mg/day at baseline; 32.3 mg/day, SD 3.2 mg/day at 4 weeks; P = 0.06)172. Another OxDC formulation (ALLN-177) significantly reduced urinary oxalate excretion in patients with preserved kidney function, including a phase III trial in patients with enteric and idiopathic hyperoxaluria (urinary oxalate excretion for all participants 77.7 mg/day, SD 55.9 mg/day at baseline; 63.7 mg/day, SD 40.1 mg/day after 4 days of treatment; P < 0.05)173. In a pilot study of patients with CKD and severe enteric hyperoxaluria, ALLN-177 reduced plasma oxalate by 29% from baseline174. However, a phase III study of ALLN-177 in patients with enteric hyperoxaluria over a 4-week period demonstrated that the reduction in urinary oxalate was only 14.3% greater than that achieved with placebo175. Determining whether a modest lowering of urinary oxalate has a clinical benefit with regard to stone disease progression will require long-term follow-up studies.
Treatments for primary hyperoxaluria
Therapeutic options for primary hyperoxaluria have traditionally been scarce and the only curative option was combined liver and kidney transplantation110. Vitamin B6 supplementation reduces hepatic oxalate production by restoring AGT activity and reducing mitochondrial mistargeting in patients with PH1 owing to specific AGXT mutations, such as G170R or F152I, in vivo and clinically118–120. More specifically, in vitro studies suggest that the monomer form of AGT is unstable and prone to misfolding and aggregation, which impedes peroxisomal transport; vitamin B6 promotes dimerization, which appears to stabilize the enzyme and may facilitate proper peroxisomal transport119,121.
In vitamin B6-non-responsive PH1, therapeutic approaches aim to inhibit the key hepatic enzymes that produce oxalate (glycolate oxidase, also known as hydroxyacid oxidase, and LDHA), for example, by using RNA interference (reviewed in refs.110,176). RNA interference is an innate mechanism of most eukaryotic cells that allows for sequence-specific inhibition of gene expression via non-coding double-stranded RNA fragments (20–25 bp long)177,178. These small interfering RNAs (siRNAs) are recognized by an RNA-induced silencing complex, which induces degradation of the target mRNA177,178. Exogenously synthesized siRNAs have been celebrated as a major therapeutic breakthrough that allows the potent repression of disease-causing genes. However, researchers are still improving chemical siRNA modifications to optimize organ-specific drug delivery and reduce off-target effects, including unintended immune stimulation177,178.
siRNA-based Lumasiran is the first EMA-approved and FDA-approved PH1-specific treatment and must be administered subcutaneously110. This treatment silences the glycolate oxidase gene (HAO1) and was shown to reduce urinary oxalate by 64.1–70% compared with placebo in phase III trials of adults and children; the first results from another phase III trial (NCT04152200) are currently undergoing quality control179,180. The resulting increase in plasma glycolate concentrations did not appear to be associated with adverse events179, in contrast to the severe acidosis observed in patients with ethylene glycol poisoning leading to elevated plasma glycolate concentrations181. Case reports demonstrate tolerability and efficacy as early as the second week of life, and suggest that pre-treatment with Lumasiran might enable the use of isolated kidney transplantation (rather than combined with liver transplantation) in PH1 (refs.182,183). Nedosiran, another subcutaneously administered siRNA-based drug, silences LDHA and was shown to reduce urinary oxalate excretion by 68% after multiple doses184. The medication was also successfully used to reduce dialysis frequency and defer combined liver and kidney transplantation in a patient with PH1 (ref.184); in a phase I study, nedosiran also normalized urinary oxalate excretion in one-third of patients with PH2 (ref.185).
Gene editing has also been used to knock out Hao1 and Ldha in animal studies. The CRISPR–Cas9 system was delivered by an adenovirus vector to knock out Hao1 in Agxt1−/− mice, which are used as a PH1 model; this treatment normalized urinary oxalate excretion and prevented nephrocalcinosis without toxicity186. Administration of an Ldha-targeting adenovirus-coupled CRISPR–Cas9 system to rats with a PH1-causing AgxtD205N mutation reduced LDHA expression by 50% and significantly lowered urinary oxalate excretion and kidney calcium oxalate deposits after an ethylene glycol challenge compared with controls; the treatment also caused mild changes in hepatic glycolytic and triglyceride pathways187. Another study of Agxt1−/− mice treated with a single dose of an adenovirus-coupled CRISPR–Cas9 system reduced LDHA expression by >95% and reduced urine oxalate to levels similar to those of wild-type animals after ethylene glycol exposure without significant toxic off-target effects or hepatotoxicity188. In a PH3 model (Hoga1−/− mice), the same CRISPR–Cas9 cleavage vector system was slightly less effective than in the PH1 model but also induced a significant reduction in LDHA expression and urine oxalate levels were comparable to those of wild-type animals after hydroxyproline challenge188. Of note, none of the mice in the PH3 model developed kidney damage upon hydroxyproline exposure (in contrast to PH1 mice challenged with ethylene glycol)188. More research is needed to test the applicability of these findings and potential off-target effects in humans.
Immune modulation
Immune modulation is another potential approach for treating oxalate-related diseases. For example, inhibition of NLRP3 inflammasome activation might reduce cleavage of the pro-inflammatory cytokines IL-1β and IL-18 via caspase 1 and inhibit inflammatory cell death (pyroptosis)189. The microRNA miR-22-3p binds to NLRP3 as well as to long intergenic non-protein-coding RNA 339, which is highly upregulated in human kidney proximal epithelial cells treated with calcium oxalate monohydrate. This microRNA reduced calcium oxalate-induced inflammasome activation and pyroptosis in human kidney proximal epithelial cells in vitro190. In a study of male hyperoxaluric rats, inhibition of the transient receptor potential vanilloid 1 channel, which functions upstream of NLRP3, mitigated reactive oxygen species-induced NLRP3 activation and calcium oxalate-induced nephropathy but did not reduce hyperoxaluria191. Other NLRP3 inhibitors with promising murine in vivo results include the diarylsulfonylurea-based CP-456773, which mitigates crystal-induced kidney fibrosis192.
Conclusions
An abundance of evidence supports a pathological role of excess oxalate when the balance between oxalate intake, production and excretion is disrupted. Overall, 50–80% of oxalate is endogenously produced and derived from amino acid and carbohydrate metabolism, whereas 20–50% derives from dietary intake and is absorbed predominantly in the small intestine. Epithelial transport of oxalate via anion exchange mediates oxalate absorption and secretion along the gastrointestinal tract, in the liver and in the kidneys, with SLC26A6 being the best-established oxalate transporter. The human gut harbours important microbial networks that help regulate oxalate levels, most notably the specialist oxalate degrader bacterium O. formigenes. Inherited enzyme defects that lead to increased endogenous oxalate production or increased intestinal absorption of oxalate observed in secondary hyperoxaluria can lead to oxalate nephropathy and kidney failure. Moreover, elevated urinary oxalate concentrations are associated with CKD progression, and increased plasma oxalate levels correlate with cardiovascular mortality in patients with kidney failure requiring dialysis. In addition, the presence of metabolic diseases, such as obesity and diabetes mellitus, might increase the risk of developing nephrolithiasis and hyperoxaluria or losing kidney function in the setting of high oxalate excretion. At a cellular level, oxalate inhibits proliferation, stimulates fibrotic transformation and calcification, leads to the upregulation of pro-atherogenic and inflammatory pathways by promoting inflammasome activation and oxidative stress, and induces cell death.
Therapies for oxalate dysregulation aim to re-establish the physiological components necessary to maintain oxalate homeostasis (Table 3). Modulation of free oxalate availability in urine and the gut has been used as a successful strategy to mitigate nephrolithiasis and secondary hyperoxaluria. One promising approach on the horizon are allogeneic transfers of oxalate-degrading microbial communities to reduce urinary oxalate. By contrast, treatments for PH focus on restoring the function of enzymes involved in hepatic oxalogenesis such as AGT, for example, through vitamin B6 supplementation in select PH subtypes, or inhibition of oxalate-producing LDHA or glycolate oxidase through siRNAs or CRISPR–Cas9 technology. Finally, immune modulation is another potential approach to prevent and treat the severe consequences of oxalate-related diseases.
Table 3 |.
Promising novel therapeutic options targeting oxalate dysregulation
| Strategy | Primary indication | Clinical evidence | Experimental evidence only |
|---|---|---|---|
| Prevention of urinary calcium oxalate crystallization | Nephrolithiasis | Hyperhydration110,145,146, tolvaptan147, thiazides148, (hydroxy)citrate149,150, lemon juice151,a, active Agropyron repens extract195 | NA |
| Prevention of intestinal hyperabsorption | All types of hyperoxaluria (by limiting oxalate intake); secondary hyperoxaluria | Limited oxalate intake109,196, calcium146,152, limited fat intake146,152, cholestyramine153,154,a, lanthanum carbonate157, sevelamer158 | NA |
| Administration of oxalate-degrading microbes | Different types of hyperoxaluria | OC5 (refs.161,162,197), Nov-001 (ref.165) | Lactobacillus paragasseri UBLG-36 (ref.166), SYNB8802 (ref.167) |
| Oxalate decarboxylase supplementation | Secondary hyperoxaluria | Proprietary oxalate decarboxylase171, recombinant oxalate decarboxylase cloned from Bacillus subtilis172, ALLN-177 (refs.173,174) | NA |
| Ensure proper folding and/or relocation of AGT | Primary hyperoxaluria type 1 | Vitamin B6 (refs.118–120,198,199) | NA |
| Protein inhibition or knockout of genes encoding oxalate-producing hepatic enzymes | Primary hyperoxaluria type 1 | Lumasiran179,180,182,183, nedosiran184 | CRISPR–Cas9 (refs.186–188) |
| Inhibition of NLRP3 and pro-inflammatory cytokines | Experimental only | NA | miR-22-3p190, CP-456773/CRID3/MCC950 (ref.192) |
AGT, alanine–glyoxylate aminotransferase; NA, not applicable; NLRP3, NACHT, LRR and PYD domains-containing protein 3.
Ongoing clinical trial.
Further research is needed to better characterize the role and therapeutic potential of different transporters from the SLC26 anion exchanger family and microbial communities in maintaining oxalate homeostasis. Important open questions include the extent to which oxalate is causally involved in the progression of CKD and CKD-related cardiovascular disease as well as effective therapeutic strategies to achieve sustained lowering of oxalate concentrations. Additionally, more detailed studies of the inflammatory pathways through which oxalate exerts its detrimental effects will likely reveal new treatment options. Robust clinical studies are an essential next step to translate in vitro, in vivo and observational clinical findings into the human setting and leverage this knowledge for the prevention and treatment of oxalate-related conditions in clinical practice.
Key points.
| • Oxalate homeostasis is maintained through a combination of endogenous biosynthesis, exogenous supply, and renal and faecal excretion. Disruptions to these mechanisms caused by genetic mutations (primary hyperoxaluria) or increased oxalate absorption (secondary hyperoxaluria) result in oxalate nephropathy. |
| • Solute carrier family 26 member 6 anion transporter has an important role in the maintenance of oxalate homeostasis by facilitating transcellular oxalate secretion in the intestine (in contrast to paracellular absorption). |
| • Among several oxalate-degrading bacteria in the human gut microbiota, Oxalobacter formigenes represents a major reservoir of oxalate-metabolizing genes. |
| • Oxalate inhibits kidney epithelial cell proliferation, promotes fibrotic transformation, calcification and atherosclerosis, and induces cell death. These pathological pathways are probably mediated, at least in part, through NLRP3 inflammasome stimulation and mitochondrial disruption. |
| • Urinary oxalate excretion is independently associated with the progression of chronic kidney disease and kidney failure. Elevated blood oxalate is also associated with increased risk of cardiovascular events, in particular sudden cardiac death. |
| • Oxalate-balancing treatment options include O. formigenes analogues and oxalate decarboxylase supplements, small interfering RNA therapeutics that target oxalate-producing hepatic enzymes, and experimental CRISPR–Cas9 approaches and anti-inflammatory strategies. |
Acknowledgements
The work of F.K. was supported by the German Research Foundation (DFG) — KN1148/4-1 and Project-ID 394046635, SFB 1365.
Competing interests
F.K. reports personal fees from Allena, Oxthera, Sanofi, Fresenius Medical Care, Alnylam Pharmaceuticals, Advicenne, Medice and Zai, and grant support from Alnylam and Dicerna Pharmaceuticals. S.W. reports personal fees from Public Health Advocacy Institute, CVS, Roth Capital Partners, Kantum Pharma, Mallinckrodt, Wolters Kluewer, GE Health Care, GSK, Allena Pharmaceuticals, Mass Medical International, Barron and Budd (versus Fresenius), JNJ, Venbio, Strataca, Takeda, Cerus, Pfizer, Bunch and James, Harvard Clinical Research Institute (also known as Baim Institute for Clinical Research), Oxidien, Sironax, Metro Biotechnology, Biomarin, Bain and Regeneron. L.N. reports personal fees from Oxthera, Dicerna, Federation Bio, Allena, Novome and Synlogic. All other authors declare no competing interests.
References
- 1.Duranton F et al. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol 23, 1258–1270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gulhan B et al. The relationship between serum oxalic acid, central hemodynamic parameters and colonization by Oxalobacter formigenes in hemodialysis patients. Cardiorenal Med 5, 164–174 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfau A et al. High oxalate concentrations correlate with increased risk for sudden cardiac death in dialysis patients. J. Am. Soc. Nephrol 10.1681/ASN.2020121793 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cochat P & Rumsby G Primary hyperoxaluria. N. Engl. J. Med 369, 649–658 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Knauf F et al. NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int 84, 895–901 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waikar SS et al. Association of urinary oxalate excretion with the risk of chronic kidney disease progression. JAMA Intern. Med 179, 542–551 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schunk SJ et al. Interleukin-1α is a central regulator of leukocyte-endothelial adhesion in myocardial infarction and in chronic kidney disease. Circulation 144, 893–908 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Sun K et al. Hyperoxalemia leads to oxidative stress in endothelial cells and mice with chronic kidney disease. Kidney Blood Press. Res 46, 377–386 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Mulay SR et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J. Clin. Invest 123, 236–246 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y et al. Dysregulated oxalate metabolism is a driver and therapeutic target in atherosclerosis. Cell Rep 36, 109420 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neumeier LI et al. Enteric oxalate secretion mediated by Slc26a6 defends against hyperoxalemia in murine models of chronic kidney disease. J. Am. Soc. Nephrol 31, 1987–1995 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M et al. Microbial genetic and transcriptional contributions to oxalate degradation by the gut microbiota in health and disease. eLife 10.7554/eLife.63642 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams HE Oxalic acid and the hyperoxaluric syndromes. Kidney Int 13, 410–417 (1978). [DOI] [PubMed] [Google Scholar]
- 14.Ermer T, Eckardt KU, Aronson PS & Knauf F Oxalate, inflammasome, and progression of kidney disease. Curr. Opin. Nephrol. Hypertens 25, 363–371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ladwig PM, Liedtke RR, Larson TS & Lieske JC Sensitive spectrophotometric assay for plasma oxalate. Clin. Chem 51, 2377–2380 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Porowski T et al. Reference values of plasma oxalate in children and adolescents. Pediatr. Nephrol 23, 1787–1794 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Elgstoen KBP Liquid chromatography–tandem mass spectrometry method for routine measurement of oxalic acid in human plasma. J. Chromatogr. B 873, 31–36 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Holmes RP, Goodman HO & Assimos DG Contribution of dietary oxalate to urinary oxalate excretion. Kidney Int 59, 270–276 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Fargue S et al. Hydroxyproline metabolism and oxalate synthesis in primary hyperoxaluria. J. Am. Soc. Nephrol 29, 1615–1623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garrelfs S et al. Endogenous oxalate production in primary hyperoxaluria type 1 patients. J. Am. Soc. Nephrol 22, 3175–3186 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/study/NCT04437225 (2020). [DOI] [PubMed]
- 22.Knight J, Hinsdale M & Holmes R Glycolate and 2-phosphoglycolate content of tissues measured by ion chromatography coupled to mass spectrometry. Anal. Biochem 421, 121–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lange JN, Wood KD, Knight J, Assimos DG & Holmes RP Glyoxal formation and its role in endogenous oxalate synthesis. Adv. Urol 2012, 819202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knight J, Wood KD, Lange JN, Assimos DG & Holmes RP Oxalate formation from glyoxal in erythrocytes. Urology 10.1016/j.urology.2015.10.014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knight J, Assimos DG, Easter L & Holmes RP Metabolism of fructose to oxalate and glycolate. Horm. Metab. Res 42, 868–873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X et al. Generation of a GLO-2 deficient mouse reveals its effects on liver carbonyl and glutathione levels. Biochem. Biophys. Rep 28, 101138 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Efe O, Verma A & Waikar SS Urinary oxalate as a potential mediator of kidney disease in diabetes mellitus and obesity. Curr. Opin. Nephrol. Hypertens 28, 316–320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knight J, Assimos DG, Callahan MF & Holmes RP Metabolism of primed, constant infusions of [1,2-13C2] glycine and [1-13C1] phenylalanine to urinary oxalate. Metabolism 60, 950–956 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glew RH et al. Nephropathy in dietary hyperoxaluria: a potentially preventable acute or chronic kidney disease. World J. Nephrol 3, 122–142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y et al. Plasma oxalate levels in prevalent hemodialysis patients and potential implications for ascorbic acid supplementation. Clin. Biochem 49, 1133–1139 (2016). [DOI] [PubMed] [Google Scholar]
- 31.McQuade DJ, Dargan PI & Wood DM Challenges in the diagnosis of ethylene glycol poisoning. Ann. Clin. Biochem 51, 167–178 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Chai W, Liebman M, Kynast-Gales S & Massey L Oxalate absorption and endogenous oxalate synthesis from ascorbate in calcium oxalate stone formers and non-stone formers. Am. J. Kidney Dis 44, 1060–1069 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Sikora P et al. [13C2]oxalate absorption in children with idiopathic calcium oxalate urolithiasis or primary hyperoxaluria. Kidney Int 73, 1181–1186 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Marengo SR & Romani AMP Oxalate in renal stone disease: the terminal metabolite that just won’t go away. Nat. Clin. Pract. Nephrol 4, 368–377 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Avila-Nava A et al. Oxalate content and antioxidant activity of different ethnic foods. J. Ren. Nutr 31, 73–79 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Asplin J The management of patients with enteric hyperoxaluria. Urolithiasis 10.1007/s00240-015-0846-5 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Whittamore JM & Hatch M Oxalate flux across the intestine: contributions from membrane transporters. Compr. Physiol 12, 2835–2875 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Elder TD & Wyngaarden JB The biosynthesis and turnover of oxalate in normal and hyperoxaluric subjects. J. Clin. Invest 39, 1337–1344 (1960). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hautmann R & Osswald H Pharmacokinetic studies of oxalate in man. Invest. Urol 16, 395–398 (1979). [PubMed] [Google Scholar]
- 40.Senekjian HO & Weinman EJ Oxalate transport by proximal tubule of the rabbit kidney. Am. J. Physiol 243, F271–F275 (1982). [DOI] [PubMed] [Google Scholar]
- 41.Costello JF, Smith M, Stolarski C & Sadovnic MJ Extrarenal clearance of oxalate increases with progression of renal failure in the rat. J. Am. Soc. Nephrol 3, 1098–1104 (1992). [DOI] [PubMed] [Google Scholar]
- 42.Hatch M, Freel RW & Vaziri ND Intestinal excretion of oxalate in chronic renal failure. J. Am. Soc. Nephrol 5, 1339–1343 (1994). [DOI] [PubMed] [Google Scholar]
- 43.Robijn S, Hoppe B, Vervaet BA, D’Haese PC & Verhulst A Hyperoxaluria: a gut-kidney axis? Kidney Int 80, 1146–1158 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Low I, Friedrich T & Burckhardt G Properties of an anion exchanger in rat renal basolateral membrane vesicles. Am. J. Physiol 246, F334–F342 (1984). [DOI] [PubMed] [Google Scholar]
- 45.Knickelbein RG, Aronson PS & Dobbins JW Oxalate transport by anion exchange across rabbit ileal brush border. J. Clin. Invest 77, 170–175 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karniski LP & Aronson PS Anion exchange pathways for Cl− transport in rabbit renal microvillus membranes. Am. J. Physiol 253, F513–F521 (1987). [DOI] [PubMed] [Google Scholar]
- 47.Kuo SM & Aronson PS Oxalate transport via the sulfate/HCO3 exchanger in rabbit renal basolateral membrane vesicles. J. Biol. Chem 263, 9710–9717 (1988). [PubMed] [Google Scholar]
- 48.Knickelbein RG & Dobbins JW Sulfate and oxalate exchange for bicarbonate across the basolateral membrane of rabbit ileum. Am. J. Physiol 259, G807–G813 (1990). [DOI] [PubMed] [Google Scholar]
- 49.Yamakawa K & Kawamura J Oxalate:OH exchange across rat renal cortical brush border membrane. Kidney Int 37, 1105–1112 (1990). [DOI] [PubMed] [Google Scholar]
- 50.Kuo SM & Aronson PS Pathways for oxalate transport in rabbit renal microvillus membrane vesicles. J. Biol. Chem 271, 15491–15497 (1996). [DOI] [PubMed] [Google Scholar]
- 51.Jiang Z, Grichtchenko II, Boron WF & Aronson PS Specificity of anion exchange mediated by mouse Slc26a6. J. Biol. Chem 277, 33963–33967 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Xie Q, Welch R, Mercado A, Romero MF & Mount DB Molecular characterization of the murine Slc26a6 anion exchanger: functional comparison with Slc26a1. Am. J. Physiol. Ren. Physiol 283, F826–F838 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Heneghan JF et al. Regulated transport of sulfate and oxalate by SLC26A2/DTDST. Am. J. Physiol. Cell Physiol 298, C1363–C1375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silberg DG, Wang W, Moseley RH & Traber PG The Down Regulated in Adenoma (dra) gene encodes an intestine-specific membrane sulfate transport protein. J. Biol. Chem 270, 11897–11902 (1995). [DOI] [PubMed] [Google Scholar]
- 55.Bai JP et al. Prestin’s anion transport and voltage-sensing capabilities are independent. Biophys. J 96, 3179–3186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lohi H et al. Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J. Biol. Chem 277, 14246–14254 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Stewart AK et al. SLC26 anion exchangers of guinea pig pancreatic duct: molecular cloning and functional characterization. Am. J. Physiol. Cell Physiol 301, C289–C303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jennings ML & Adame MF Characterization of oxalate transport by the human erythrocyte band 3 protein. J. Gen. Physiol 107, 145–159 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reimold FR et al. Substitution of transmembrane domain Cys residues alters pH(o)-sensitive anion transport by AE2/SLC4A2 anion exchanger. Pflug. Arch 465, 839–851 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knauf F et al. Net intestinal transport of oxalate reflects passive absorption and SLC26A6-mediated secretion. J. Am. Soc. Nephrol 22, 2247–2255 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Freel RW, Whittamore JM & Hatch M Transcellular oxalate and Cl− absorption in mouse intestine is mediated by the DRA anion exchanger Slc26a3, and DRA deletion decreases urinary oxalate. Am. J. Physiol. Gastrointest. Liver Physiol 305, G520–G527 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cil O, Chu T, Lee S, Haggie PM & Verkman AS Small-molecule inhibitor of intestinal anion exchanger SLC26A3 for treatment of hyperoxaluria and nephrolithiasis. JCI Insight 10.1172/jci.insight.153359 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chernova MN et al. Acute regulation of the SLC26A3 congenital chloride diarrhoea anion exchanger (DRA) expressed in Xenopus oocytes. J. Physiol 549, 3–19 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wasiluk T et al. Simultaneous expression of ClopHensor and SLC26A3 reveals the nature of endogenous oxalate transport in CHO cells. Biol. Open 10.1242/bio.041665 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chernova MN et al. Functional comparison of mouse slc26a6 anion exchanger with human SLC26A6 polypeptide variants: differences in anion selectivity, regulation, and electrogenicity. J. Biol. Chem 280, 8564–8580 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Freel RW, Hatch M, Green M & Soleimani M Ileal oxalate absorption and urinary oxalate excretion are enhanced in Slc26a6 null mice. Am. J. Physiol. Gastrointest. Liver Physiol 290, G719–G728 (2006). [DOI] [PubMed] [Google Scholar]
- 67.Jiang Z et al. Calcium oxalate urolithiasis in mice lacking anion transporter Slc26a6. Nat. Genet 38, 474–478 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Liu Y et al. Short-Chain fatty acids reduced renal calcium oxalate stones by regulating the expression of intestinal oxalate transporter SLC26A6. mSystems 6, e0104521 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Whittamore JM & Hatch M The anion exchanger PAT-1 (Slc26a6) does not participate in oxalate or chloride transport by mouse large intestine. Pflug. Arch 473, 95–106 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Capolongo G, Abul-Ezz S, Moe OW & Sakhaee K Subclinical celiac disease and crystal-induced kidney disease following kidney transplant. Am. J. Kidney Dis 60, 662–667 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cornière N et al. Dominant negative mutation in oxalate transporter SLC26A6 associated with enteric hyperoxaluria and nephrolithiasis. J. Med. Genet 10.1136/jmedgenet-2021-108256 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mukaibo T et al. The apical anion exchanger Slc26a6 promotes oxalate secretion by murine submandibular gland acinar cells. J. Biol. Chem 293, 6259–6268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bissig M, Hagenbuch B, Stieger B, Koller T & Meier PJ Functional expression cloning of the canalicular sulfate transport system of rat hepatocytes. J. Biol. Chem 269, 3017–3021 (1994). [PubMed] [Google Scholar]
- 74.Karniski LP et al. Immunolocalization of sat-1 sulfate/oxalate/bicarbonate anion exchanger in the rat kidney. Am. J. Physiol 275, F79–F87 (1998). [DOI] [PubMed] [Google Scholar]
- 75.Lee A, Beck L & Markovich D The mouse sulfate anion transporter gene Sat1 (Slc26a1): cloning, tissue distribution, gene structure, functional characterization, and transcriptional regulation thyroid hormone. DNA Cell Biol 22, 19–31 (2003). [DOI] [PubMed] [Google Scholar]
- 76.Quondamatteo F et al. Localization of the sulfate/anion exchanger in the rat liver. Am. J. Physiol. Gastrointest. Liver Physiol 290, G1075–G1081 (2006). [DOI] [PubMed] [Google Scholar]
- 77.Krick W, Schnedler N, Burckhardt G & Burckhardt BC Ability of sat-1 to transport sulfate, bicarbonate, or oxalate under physiological conditions. Am. J. Physiol. Ren. Physiol 297, F145–F154 (2009). [DOI] [PubMed] [Google Scholar]
- 78.Dawson PA et al. Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J. Clin. Invest 120, 706–712 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ko N, Knauf F, Jiang Z, Markovich D & Aronson PS Sat1 is dispensable for active oxalate secretion in mouse duodenum. Am. J. Physiol. Cell Physiol 303, C52–C57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Whittamore JM, Stephens CE & Hatch M Absence of the sulfate transporter SAT-1 has no impact on oxalate handling by mouse intestine and does not cause hyperoxaluria or hyperoxalemia. Am. J. Physiol. Gastrointest. Liver Physiol 316, G82–G94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gee HY et al. Mutations in SLC26A1 cause nephrolithiasis. Am. J. Hum. Genet 98, 1228–1234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lohi H et al. Mapping of five new putative anion transporter genes in human and characterization of SLC26A6, a candidate gene for pancreatic anion exchanger. Genomics 70, 102–112 (2000). [DOI] [PubMed] [Google Scholar]
- 83.Waldegger S et al. Cloning and characterization of SLC26A6, a novel member of the solute carrier 26 gene family. Genomics 72, 43–50 (2001). [DOI] [PubMed] [Google Scholar]
- 84.Knauf F et al. Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc. Natl Acad. Sci. USA 98, 9425–9430 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang Z, Petrovic S, Mann E & Soleimani M Identification of an apical Cl−/HCO3− exchanger in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol 282, G573–G579 (2002). [DOI] [PubMed] [Google Scholar]
- 86.Sugimoto T et al. Fate of circulating oxalate in rats. Eur. Urol 23, 485–489 (1993). [DOI] [PubMed] [Google Scholar]
- 87.Weinman EJ, Frankfurt SJ, Ince A & Sansom S Renal tubular transport of organic acids. Studies with oxalate and para-aminohippurate in the rat. J. Clin. Invest 61, 801–806 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Knauf F, Velazquez H, Pfann V, Jiang Z & Aronson PS Characterization of renal NaCl and oxalate transport in Slc26a6−/− mice. Am. J. Physiol. Ren. Physiol 316, F128–F133 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Holmes RP, Ambrosius WT & Assimos DG Dietary oxalate loads and renal oxalate handling. J. Urol 174, 943–947 (2005). [DOI] [PubMed] [Google Scholar]
- 90.Worcester EM et al. A test of the hypothesis that oxalate secretion produces proximal tubule crystallization in primary hyperoxaluria type I. Am. J. Physiol. Ren. Physiol 305, F1574–F1584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chandhoke PS & Fan J Transport of oxalate across the rabbit papillary surface epithelium. J. Urol 164, 1724–1728 (2000). [PubMed] [Google Scholar]
- 92.Ohana E, Shcheynikov N, Moe OW & Muallem S SLC26A6 and NaDC-1 transporters interact to regulate oxalate and citrate homeostasis. J. Am. Soc. Nephrol 24, 1617–1626 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lloyd-Price J et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.The Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project. Nature 569, 641–648 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Abratt VR & Reid SJ Oxalate-degrading bacteria of the human gut as probiotics in the management of kidney stone disease. Adv. Appl. Microbiol 72, 63–87 (2010). [DOI] [PubMed] [Google Scholar]
- 96.Daniel SL et al. Forty years of Oxalobacter formigenes, a gutsy oxalate-degrading specialist. Appl. Environ. Microbiol 87, e0054421 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arvans D et al. Oxalobacter formigenes-derived bioactive factors stimulate oxalate transport by intestinal epithelial cells. J. Am. Soc. Nephrol 28, 876–887 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hatch M & Freel RW A human strain of Oxalobacter (HC-1) promotes enteric oxalate secretion in the small intestine of mice and reduces urinary oxalate excretion. Urolithiasis 41, 379–384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tasian GE et al. Oral antibiotic exposure and kidney stone disease. J. Am. Soc. Nephrol 29, 1731–1740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ferraro PM, Curhan GC, Gambaro G & Taylor EN Antibiotic use and risk of incident kidney stones in female nurses. Am. J. Kidney Dis 74, 736–741 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nazzal L & Blaser MJ Does the receipt of antibiotics for common infectious diseases predispose to kidney stones? A cautionary note for all health care practitioners. J. Am. Soc. Nephrol 29, 1590–1592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miller AW & Dearing D The metabolic and ecological interactions of oxalate-degrading bacteria in the Mammalian gut. Pathogens 2, 636–652 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ticinesi A et al. Understanding the gut-kidney axis in nephrolithiasis: an analysis of the gut microbiota composition and functionality of stone formers. Gut 67, 2097–2106 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Suryavanshi MV et al. Hyperoxaluria leads to dysbiosis and drives selective enrichment of oxalate metabolizing bacterial species in recurrent kidney stone endures. Sci. Rep 6, 34712 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Denburg MR et al. Perturbations of the gut microbiome and metabolome in children with calcium oxalate kidney stone disease. J. Am. Soc. Nephrol 31, 1358–1369 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaufman DW et al. Oxalobacter formigenes may reduce the risk of calcium oxalate kidney stones. J. Am. Soc. Nephrol 19, 1197–1203 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ticinesi A, Nouvenne A & Meschi T Gut microbiome and kidney stone disease: not just an Oxalobacter story. Kidney Int 96, 25–27 (2019). [DOI] [PubMed] [Google Scholar]
- 108.Miller AW, Choy D, Penniston KL & Lange D Inhibition of urinary stone disease by a multi-species bacterial network ensures healthy oxalate homeostasis. Kidney Int 96, 180–188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dill H, Martin-Higueras C & Hoppe B Diet-related urine collections: assistance in categorization of hyperoxaluria. Urolithiasis 10.1007/s00240-021-01290-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shee K & Stoller ML Perspectives in primary hyperoxaluria — historical, current and future clinical interventions. Nat. Rev. Urol 10.1038/s41585-021-00543-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Martin-Higueras C et al. A report from the European Hyperoxaluria Consortium (OxalEurope) Registry on a large cohort of patients with primary hyperoxaluria type 3. Kidney Int 100, 621–635 (2021). [DOI] [PubMed] [Google Scholar]
- 112.Garrelfs SF et al. Patients with primary hyperoxaluria type 2 have significant morbidity and require careful follow-up. Kidney Int 96, 1389–1399 (2019). [DOI] [PubMed] [Google Scholar]
- 113.Hopp K et al. Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J. Am. Soc. Nephrol 26, 2559–2570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mandrile G et al. Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int 86, 1197–1204 (2014). [DOI] [PubMed] [Google Scholar]
- 115.Zhao Y et al. Extended genetic analysis of exome sequencing for primary hyperoxaluria in pediatric urolithiasis patients with hyperoxaluria. Gene 10.1016/j.gene.2021.146155 (2022). [DOI] [PubMed] [Google Scholar]
- 116.Tu X et al. Human MiR-4660 regulates the expression of alanine-glyoxylate aminotransferase and may be a biomarker for idiopathic oxalosis. Clin. Exp. Nephrol 23, 890–897 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Belostotsky R et al. Translation inhibition corrects aberrant localization of mutant alanine-glyoxylate aminotransferase: possible therapeutic approach for hyperoxaluria. J. Mol. Med 96, 621–630 (2018). [DOI] [PubMed] [Google Scholar]
- 118.Lorenz EC et al. Recovery from dialysis in patients with primary hyperoxaluria type 1 treated with pyridoxine: a report of 3 cases. Am. J. Kidney Dis 77, 816–819 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dindo M, Oppici E, Dell’Orco D, Montone R & Cellini B Correlation between the molecular effects of mutations at the dimer interface of alanine-glyoxylate aminotransferase leading to primary hyperoxaluria type I and the cellular response to vitamin B(6). J. Inherit. Metab. Dis 41, 263–275 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Monico CG, Rossetti S, Olson JB & Milliner DS Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int 67, 1704–1709 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Dindo M et al. Dimerization drives proper folding of human alanine:glyoxylate aminotransferase but is dispensable for peroxisomal targeting. J. Pers. Med 10.3390/jpm11040273 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reddy S et al. Clinical outcomes and histological patterns in oxalate nephropathy due to enteric and nonenteric risk factors. Am. J. Nephrol 10.1159/000520286 (2021). [DOI] [PubMed] [Google Scholar]
- 123.Joshi S, Wang W & Khan SR Transcriptional study of hyperoxaluria and calcium oxalate nephrolithiasis in male rats: Inflammatory changes are mainly associated with crystal deposition. PLoS ONE 12, e0185009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ogawa Y et al. Calcium oxalate saturation in dialysis patients with and without primary hyperoxaluria. Urol. Res 34, 12–16 (2006). [DOI] [PubMed] [Google Scholar]
- 125.Worcester EM, Nakagawa Y, Bushinsky DA & Coe FL Evidence that serum calcium oxalate supersaturation is a consequence of oxalate retention in patients with chronic renal failure. J. Clin. Invest 77, 1888–1896 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hueppelshaeuser R et al. Enteric hyperoxaluria, recurrent urolithiasis, and systemic oxalosis in patients with Crohn’s disease. Pediatr. Nephrol 27, 1103–1109 (2012). [DOI] [PubMed] [Google Scholar]
- 127.Shah RJ, Vaughan LE, Enders FT, Milliner DS & Lieske JC Plasma oxalate as a predictor of kidney function decline in a primary hyperoxaluria cohort. Int. J. Mol. Sci 21, 3608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mulay SR et al. Oxalate-induced chronic kidney disease with its uremic and cardiovascular complications in C57BL/6 mice. Am. J. Physiol. Ren. Physiol 10.1152/ajprenal.00488.2015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tubben A et al. Urinary oxalate excretion and long-term outcomes in kidney transplant recipients. J. Clin. Med 10.3390/jcm8122104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pinheiro HS, Camara NO, Osaki KS, De Moura LA & Pacheco-Silva A Early presence of calcium oxalate deposition in kidney graft biopsies is associated with poor long-term graft survival. Am. J. Transpl 5, 323–329 (2005). [DOI] [PubMed] [Google Scholar]
- 131.Palsson R et al. The association of calcium oxalate deposition in kidney allografts with graft and patient survival. Nephrol. Dial. Transpl 35, 888–894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Krogstad V et al. High plasma oxalate levels early after kidney transplantation are associated with impaired long-term outcomes. Transpl. Int 35, 10240 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rule AD, Krambeck AE & Lieske JC Chronic kidney disease in kidney stone formers. Clin. J. Am. Soc. Nephrol 6, 2069–2075 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Amin R et al. Reduced active transcellular intestinal oxalate secretion contributes to the pathogenesis of obesity-associated hyperoxaluria. Kidney Int 93, 1098–1107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gianmoena K et al. Epigenomic and transcriptional profiling identifies impaired glyoxylate detoxification in NAFLD as a risk factor for hyperoxaluria. Cell Rep 36, 109526 (2021). [DOI] [PubMed] [Google Scholar]
- 136.Evan AP et al. Renal histopathology and crystal deposits in patients with small bowel resection and calcium oxalate stone disease. Kidney Int 78, 310–317 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Convento MB et al. Calcium oxalate crystals and oxalate induce an epithelial-to-mesenchymal transition in the proximal tubular epithelial cells: contribution to oxalate kidney injury. Sci. Rep 7, 45740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Convento M, Pessoa E, Aragão A, Schor N & Borges F Oxalate induces type II epithelial to mesenchymal transition (EMT) in inner medullary collecting duct cells (IMCD) in vitro and stimulate the expression of osteogenic and fibrotic markers in kidney medulla in vivo. Oncotarget 10, 1102–1118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu Y et al. Role of ROS-induced NLRP3 inflammasome activation in the formation of calcium oxalate nephrolithiasis. Front. Immunol 13, 818625 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kohri K et al. Biomolecular mechanism of urinary stone formation involving osteopontin. Urol. Res 40, 623–637 (2012). [DOI] [PubMed] [Google Scholar]
- 141.Khan SR, Canales BK & Dominguez-Gutierrez PR Randall’s plaque and calcium oxalate stone formation: role for immunity and inflammation. Nat. Rev. Nephrol 17, 417–433 (2021). [DOI] [PubMed] [Google Scholar]
- 142.Patel M et al. Oxalate induces mitochondrial dysfunction and disrupts redox homeostasis in a human monocyte derived cell line. Redox Biol 15, 207–215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kumar P, Saini K, Saini V & Mitchell T Oxalate alters cellular bioenergetics, redox homeostasis, antibacterial response, and immune response in macrophages. Front. Immunol 12, 694865 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Recht PA et al. Oxalic acid alters intracellular calcium in endothelial cells. Atherosclerosis 173, 321–328 (2004). [DOI] [PubMed] [Google Scholar]
- 145.Bao Y, Tu X & Wei Q Water for preventing urinary stones. Cochrane Database Syst. Rev 2, CD004292 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kumar R et al. Fat malabsorption and increased intestinal oxalate absorption are common after roux-en-Y gastric bypass surgery. Surgery 149, 654–661 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cheungpasitporn W, Erickson SB, Rule AD, Enders F & Lieske JC Short-term tolvaptan increases water intake and effectively decreases urinary calcium oxalate, calcium phosphate and uric acid supersaturations. J. Urol 195, 1476–1481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Reilly RF, Peixoto AJ & Desir GV The evidence-based use of thiazide diuretics in hypertension and nephrolithiasis. Clin. J. Am. Soc. Nephrol 5, 1893 (2010). [DOI] [PubMed] [Google Scholar]
- 149.Phillips R et al. Citrate salts for preventing and treating calcium containing kidney stones in adults. Cochrane Database Syst. Rev 10, CD010057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chung J et al. Molecular modifiers reveal a mechanism of pathological crystal growth inhibition. Nature 536, 446–450 (2016). [DOI] [PubMed] [Google Scholar]
- 151.Ruggenenti P et al. Fresh lemon juice supplementation for the prevention of recurrent stones in calcium oxalate nephrolithiasis: a pragmatic, prospective, randomised, open, blinded endpoint (PROBE) trial. EClinicalMedicine 43, 101227 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Espino-Grosso P, Monsour C & Canales BK The effect of calcium and vitamin B6 Supplementation on oxalate excretion in a rodent gastric bypass model of enteric hyperoxaluria. Urology 124, 310.e9–310.e14 (2019). [DOI] [PubMed] [Google Scholar]
- 153.Nazzal L, Puri S & Goldfarb DS Enteric hyperoxaluria: an important cause of end-stage kidney disease. Nephrol. Dial. Transplant 31, 375–382 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Emmett M et al. Conjugated bile acid replacement therapy reduces urinary oxalate excretion in short bowel syndrome. Am. J. Kidney Dis 41, 230–237 (2003). [DOI] [PubMed] [Google Scholar]
- 155.Oka Y, Miyazaki M, Takatsu S & Matsuda H Calcium-based phosphate binders and plasma oxalate concentration in dialysis patients. J. Am. Soc. Nephrol 10.1681/ASN.2022030248 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017 clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int. Suppl 7, 1–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03346369 (2017). [DOI] [PubMed]
- 158.Lieske JC, Regnier C & Dillon JJ Use of sevelamer hydrochloride as an oxalate binder. J. Urol 179, 1407–1410 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vekeman J et al. In search of an efficient complexing agent for oxalates and phosphates: a quantum chemical study. Nanomaterials 10.3390/nano11071763 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Canales BK & Hatch M Oxalobacter formigenes colonization normalizes oxalate excretion in a gastric bypass model of hyperoxaluria. Surg. Obes. Relat. Dis 13, 1152–1157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Milliner D, Hoppe B & Groothoff J A randomised phase II/III study to evaluate the efficacy and safety of orally administered Oxalobacter formigenes to treat primary hyperoxaluria. Urolithiasis 46, 313–323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hoppe B et al. Effects of Oxalobacter formigenes in subjects with primary hyperoxaluria Type 1 and end-stage renal disease: a phase II study. Nephrol. Dial. Transplant 36, 1464–1473 (2020). [DOI] [PubMed] [Google Scholar]
- 163.Klimesova K, Whittamore JM & Hatch M Bifidobacterium animalis subsp. lactis decreases urinary oxalate excretion in a mouse model of primary hyperoxaluria. Urolithiasis 43, 107–117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lieske JC, Goldfarb DS, De Simone C & Regnier C Use of a probiotic to decrease enteric hyperoxaluria. Kidney Int 68, 1244–1249 (2005). [DOI] [PubMed] [Google Scholar]
- 165.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04909723 (2021). [DOI] [PubMed]
- 166.Mehra Y & Viswanathan P High-quality whole-genome sequence analysis of Lactobacillus paragasseri UBLG-36 reveals oxalate-degrading potential of the strain. PLoS ONE 16, e0260116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lubkowicz D et al. An engineered bacterial therapeutic lowers urinary oxalate in preclinical models and in silico simulations of enteric hyperoxaluria. Mol. Syst. Biol 18, e10539 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Miller AW, Dale C & Dearing MD The induction of oxalate metabolism in vivo is more effective with functional microbial communities than with functional microbial species. mSystems 10.1128/mSystems.00088-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Miller AW, Oakeson KF, Dale C & Dearing MD Microbial community transplant results in increased and long-term oxalate degradation. Microb. Ecol 72, 470–478 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Stern JM et al. Fecal transplant modifies urine chemistry risk factors for urinary stone disease. Physiol. Rep 7, e14012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Quintero E et al. A prospective, double-blind, randomized, placebo-controlled, crossover study using an orally administered oxalate decarboxylase (OxDC). Kidney360 1, 1284–1290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/study/NCT01127087 (2010). [DOI] [PubMed]
- 173.Lingeman JE et al. ALLN-177, oral enzyme therapy for hyperoxaluria. Int. Urol. Nephrol 51, 601–608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Pfau A et al. Pilot study of reloxaliase in patients with severe enteric hyperoxaluria and hyperoxalemia. Nephrol. Dial. Transpl 36, 945–948 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lieske John C et al. Randomized placebo-controlled trial of reloxaliase in enteric hyperoxaluria. NEJM Evid 1, EVIDoa2100053 (2022). [DOI] [PubMed] [Google Scholar]
- 176.Dejban P & Lieske JC New therapeutics for primary hyperoxaluria type 1. Curr. Opin. Nephrol. Hypertens 31, 344–350 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Aagaard L & Rossi JJ RNAi therapeutics: principles, prospects and challenges. Adv. Drug Deliv. Rev 59, 75–86 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Traber GM & Yu A-M RNAi based therapeutics and novel RNA bioengineering technologies. J. Pharmacol. Exp. Ther 10.1124/jpet.122.001234 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Garrelfs SF et al. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N. Engl. J. Med 384, 1216–1226 (2021). [DOI] [PubMed] [Google Scholar]
- 180.Sas DJ et al. Phase 3 trial of lumasiran for primary hyperoxaluria type 1: a new RNAi therapeutic in infants and young children. Genet. Med 10.1016/j.gim.2021.10.024 (2021). [DOI] [PubMed] [Google Scholar]
- 181.Leth PM & Gregersen M Ethylene glycol poisoning. Forensic Sci. Int 155, 179–184 (2005). [DOI] [PubMed] [Google Scholar]
- 182.Méaux MN et al. The effect of lumasiran therapy for primary hyperoxaluria type 1 in small infants. Pediatr. Nephrol 10.1007/s00467-021-05393-1 (2022). [DOI] [PubMed] [Google Scholar]
- 183.Joher N et al. Early post-transplant recurrence of oxalate nephropathy in a patient with primary hyperoxaluria type 1, despite pretransplant lumasiran therapy. Kidney Int 101, 185–186 (2022). [DOI] [PubMed] [Google Scholar]
- 184.Shee K et al. Nedosiran dramatically reduces serum oxalate in dialysis-dependent primary hyperoxaluria 1: a compassionate use case report. Urology 156, e147–e149 (2021). [DOI] [PubMed] [Google Scholar]
- 185.Hoppe B et al. Safety, pharmacodynamics, and exposure-response modeling results from a first-in-human phase 1 study of nedosiran (PHYOX1) in primary hyperoxaluria. Kidney Int 10.1016/j.kint.2021.08.015 (2021). [DOI] [PubMed] [Google Scholar]
- 186.Zabaleta N et al. CRISPR/Cas9-mediated glycolate oxidase disruption is an efficacious and safe treatment for primary hyperoxaluria type I. Nat. Commun 9, 5454 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zheng R et al. Knockdown of lactate dehydrogenase by adeno-associated virus-delivered CRISPR/Cas9 system alleviates primary hyperoxaluria type 1. Clin. Transl Med 10, e261 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Martinez-Turrillas R et al. In vivo CRISPR-Cas9 inhibition of hepatic LDH as treatment of primary hyperoxaluria. Mol. Ther. Methods Clin. Dev 25, 137–146 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mulay SR Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int 96, 58–66 (2019). [DOI] [PubMed] [Google Scholar]
- 190.Song Z et al. Long noncoding RNA LINC00339 promotes renal tubular epithelial pyroptosis by regulating the miR-22–3p/NLRP3 axis in calcium oxalate-induced kidney stone. J. Cell. Biochem 120, 10452–10462 (2019). [DOI] [PubMed] [Google Scholar]
- 191.Lu C-L, Teng T-Y, Liao M-T & Ma M-C TRPV1 hyperfunction contributes to renal inflammation in oxalate nephropathy. Int. J. Mol. Sci 22, 6204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ludwig-Portugall I et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int 90, 525–539 (2016). [DOI] [PubMed] [Google Scholar]
- 193.Holmes RP & Assimos DG Glyoxylate synthesis and its modulation and influence on oxalate synthesis. J. Urol 160, 1617–1624 (1998). [PubMed] [Google Scholar]
- 194.Clark B, Baqdunes MW & Kunkel GM Diet-induced oxalate nephropathy. BMJ Case Rep 12, e231284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/study/NCT04860492 (2021). [DOI] [PubMed]
- 196.Lieske JC et al. Diet, but not oral probiotics, effectively reduces urinary oxalate excretion and calcium oxalate supersaturation. Kidney Int 78, 1178–1185 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Pape L, Ahlenstiel-Grunow T, Birtel J, Krohne TU & Hoppe B Oxalobacter formigenes treatment combined with intensive dialysis lowers plasma oxalate and halts disease progression in a patient with severe infantile oxalosis. Pediatr. Nephrol 10.1007/s00467-019-04463-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lorenz EC et al. Sustained pyridoxine response in primary hyperoxaluria type 1 recipients of kidney alone transplant. Am. J. Transplant 14, 1433–1438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hoyer-Kuhn H et al. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin. J. Am. Soc. Nephrol 9, 468–477 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]