

Abstract
The maintenance of phospholipid homeostasis is increasingly being implicated in metabolic health. Phosphatidylethanolamine (PE) is the most abundant phospholipid on the inner leaflet of cellular membranes, and we have previously shown that mice with a heterozygous ablation of the PE synthesizing enzyme, Pcyt2 (Pcyt2+/−), develop obesity, insulin resistance, and NASH. Skeletal muscle is a major determinant of systemic energy metabolism, making it a key player in metabolic disease development. Both the total PE levels and the ratio of PE to other membrane lipids in skeletal muscle are implicated in insulin resistance; however, the underlying mechanisms and the role of Pcyt2 regulation in this association remain unclear. Here, we show how reduced phospholipid synthesis due to Pcyt2 deficiency causes Pcyt2+/- skeletal muscle dysfunction and metabolic abnormalities. Pcyt2+/- skeletal muscle exhibits damage and degeneration, with skeletal muscle cell vacuolization, disordered sarcomeres, mitochondria ultrastructure irregularities and paucity, inflammation, and fibrosis. There is intramuscular adipose tissue accumulation, and major disturbances in lipid metabolism with impaired FA mobilization and oxidation, elevated lipogenesis, and long-chain fatty acyl-CoA, diacylglycerol, and triacylglycerol accumulation. Pcyt2+/− skeletal muscle exhibits perturbed glucose metabolism with elevated glycogen content, impaired insulin signaling, and reduced glucose uptake. Together, this study lends insight into the critical role of PE homeostasis in skeletal muscle metabolism and health with broad implications on metabolic disease development.
Keywords: skeletal muscle, non-alcoholic steatohepatitis, Pcyt2, phosphatidylethanolamine, phospholipids, metabolism
Graphical Abstract
Graphical Abstract.
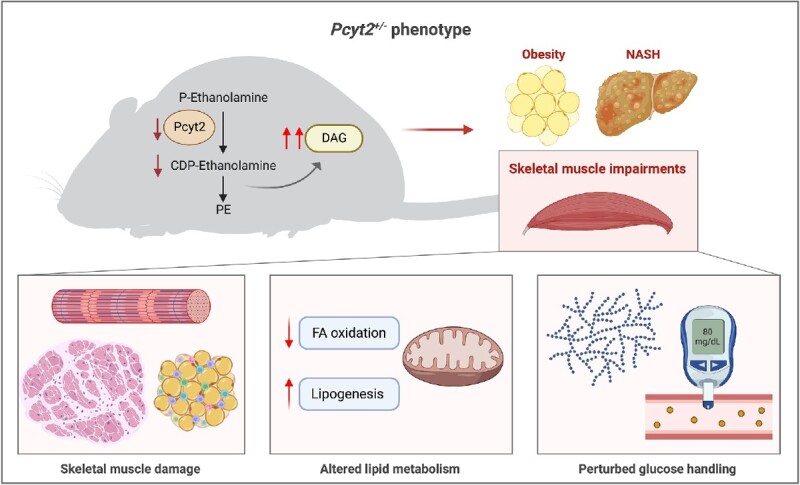
Introduction
Membrane phospholipid homeostasis is an increasingly recognized factor in metabolic health and disease.1 Specifically, we and others have shown that the dysregulation of phosphatidylethanolamine (PE) metabolism is repeatedly linked to obesity, diabetes, and fatty liver diseases.1–4 Phosphatidylethanolamine is the most abundant phospholipid on the inner leaflet of cellular membranes, where it is involved with essential processes such as cell signaling, membrane fission and fusion, autophagy, and the regulation of glucose, lipid, and systemic energy metabolism.5 Phosphatidylethanolamine is mainly synthesized de novo through the CDP-ethanolamine pathway, in which CTP: phosphoethanolamine cytidylyltransferase (Pcyt2) is the rate-limiting enzyme. We have shown that homozygous deletion of Pcyt2 is embryonically lethal,6 and generated a global heterozygous Pcyt2 knockout (Pcyt2+/−). Pcyt2+/− mice exhibit diminished Pcyt2 enzymatic activity and PE production, and in the homeostatic response, PE turnover is reduced to preserve total PE levels.2,6 As a result, Pcyt2+/− mice show compensatory metabolic mechanisms and develop age-dependent obesity, insulin resistance, dysregulated hepatic glucose and lipid homeostasis, and non-alcoholic steatohepatitis (NASH) with steatosis, hepatocellular damage, inflammation, and fibrosis.2,7
Skeletal muscle occupies a central position in the maintenance of metabolic health through its role as the principal site of insulin-stimulated glucose disposal and as a major regulator of systemic energy metabolism and inflammation; key processes that influence the pathobiology of metabolic-associated diseases.8,9 Specifically, skeletal muscle insulin resistance is a primary deficit in T2DM and is thought to be a key driver in the development and advancement of non-alcoholic fatty liver disease (NAFLD).10 Skeletal muscle insulin resistance, muscle fat infiltration (myosteatosis), and sarcopenia are linked to poor metabolic, functional, and clinical outcomes, and an increased risk for the advancement of NASH to cirrhosis and death.11–13 Myosteatosis has been shown as both a precursor and a propagator of skeletal muscle insulin resistance and impaired muscle function, and its association with perturbations in the muscle secretome is believed to promote inflammation and hepatocellular injury, directly linking it to extra-musculoskeletal metabolic disorders.13,5
Pcyt2 regulation has the capacity to influence skeletal muscle function through the critical role PE plays in the maintenance of sarcolemmal integrity, cell signaling events including glucose and lipid metabolic pathways, mitochondrial dynamics and respiration, and SERCA function.7,14,15 Both the total PE levels and the ratio of PE to phosphatidylcholine (PC) within the skeletal muscle are associated with insulin sensitivity in humans16,17 and primary myocytes18; however, the underlying mechanisms and the influence of Pcyt2, the rate-limiting PE biosynthetic enzyme, on this association remain unclear. Interestingly, mice with a liver-specific knockout of Pcyt2 exhibit hepatic lipid accumulation but do not develop a hepatic injury or systemic consequences such as insulin resistance or obesity,19 indicating that the development of the metabolic syndrome and NASH in global heterozygous knock-out mice (Pcyt2+/−) likely involves multi-organ dysfunction. Given that the skeletal muscle is a major determinant of systemic energy metabolism, we hypothesize a specific role for Pcyt2 regulation in the skeletal muscle that contributes to the development of Pcyt2+/− obesity, metabolic perturbations, and ultimately NASH.
In this study, we probe the consequences of Pcyt2 deficiency in skeletal muscle, with respect to muscle structure, inflammation, fatty acid (FA), and glucose homeostasis in the Pcyt2+/- model. First, because PE is a critical component of cellular membranes and has specifically been shown to influence the proper functioning of sarcolemmal and mitochondrial membranes through its role in defining membrane curvature, we conducted histopathological analysis to determine if structural abnormalities exist that may influence skeletal muscle function. Because disrupted muscle structure often indicates degenerative processes, we further determined markers of muscle damage and inflammation. We have previously demonstrated that Pcyt2 deficiency causes adult-onset obesity and dysregulated hepatic lipid and glucose homeostasis.2,3 To examine whether excess adiposity stems from decreased FA catabolism, we measured systemic free FA release using epinephrine to stimulate lipolysis and skeletal muscle FA oxidation. Finally, we characterized perturbations to FA synthesis and glucose control. Together, this study lends specific insight into the critical role of PE homeostasis in skeletal muscle metabolism and a broad understanding of the influence of phospholipid metabolism on whole-body energy regulation.
Methods
Animals
Heterozygous Pcyt2 mice (Pcyt2+/−) of mixed genetic background (C57BL/6 × 129/Sv) were generated and genotyped as previously described.6 All procedures were approved by the University of Guelph’s Animal Care Committee and were in accordance with the guidelines of the Canadian Council on Animal Care (CCAC). The ARRIVE guidelines were followed for reporting results. Mice were housed in a temperature-controlled facticity and exposed to a 12 h light/12 h dark cycle, beginning with light at 7:00 am mice were fed a standardized chow diet (Harlan Teklad S-2335) and had free access to water. Wild-type littermates (Pcyt2+/+) and Pcyt2+/− mice of both sexes (n = 3–12/per group) were euthanized by CO2 under fasted conditions at 6–8 mo. For experiments, immediately after euthanization, whole muscles were excised from the quadricep, deep proximal and medial portion of the gastrocnemius, and the soleus. Samples were trimmed of connective tissue and immediately submerged in fixative or assay buffers, or were stored at -80°C until analyzed. Quadricep was used for imaging, and both gastrocnemius and soleus were used for assays. No differences were observed between age-matched males and females, and both sexes were used for the final analysis.
Limb Clasping Test
Clasping test and scoring system were performed following the protocol of Guyenet et al.20 Briefly, mice were grasped near the tail base and lifted for 10 s to observe clasping behavior. Mice were scored based on the degree and length of time the limbs are retracted toward the abdomen.
Immunoblotting
Soleus and gastrocnemius muscles (n = 3–4 per group) were immediately frozen in liquid nitrogen and stored at -80°C. Frozen tissues were homogenized in 50 m m HEPES [pH 7.4], 1% TritonX-100, 50 m m sodium pyrophosphate, 0.1 m sodium fluoride, 10 m m EDTA, 10 m m sodium orthovanadate, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 2 m m benzamidine, and 2 m m PMSF using a polytron homogenizer. The protein content was determined using the BCA protein assay kit (Pierce). Proteins were resolved on a 5% and 10% denaturing SDS-PAGE gels and semi-dry transferred to PVDF membranes. Following transfer, proteins were visualized using Ponceau S staining to ensure proper transfer and equal loading. Membranes were blocked at room temperature for 2 h in 5% bovine serum albumin (BSA) in TBS-T, followed by incubation with primary antibodies Irs1, pSer307-Irs1 (Millipore); Pi3k p85, Akt, pThr308-Akt, pSer473-Akt, Ampkα, p-Ampkα, p-Acc, Acc, Sirt1, p-p38 Mapk, Pkcα, p38 Mapk, Cpt1, Glut4, pSer563-Hsl, Hsl, and β-Tubulin (Cell signaling); Srebp1c (Invitrogen) at a 1:1000 dilution in %5 BSA at 4°C overnight. Membranes were washed 3x in TBS-T and incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (1:10 000) in 5% BSA in TBS-T for 1 h at room temperature and visualized using chemiluminescent substrate (Sigma). β-Tubulin was used as a loading control. Soleus and gastrocnemius muscle samples were mixed and analyzed together. The intensity of specific bands was quantified using NIH ImageJ software.
Tissue Sectioning and Staining
Quadricep muscles (n = 5 per group) were excised and immediately fixed in 10% neutral buffered formalin for 12–16 h and embedded in paraffin until histopathologic examination. Quadriceps were sectioned in the transverse planes in the middle of the quadricep muscle to better view skeletal muscle cell morphology, nuclei, and collagen deposition between adjacent muscle fibers. Sections were de-waxed in xylene and rehydrated in a series of ethanol washes. Sections of 10 μm were stained with hematoxylin and eosin (H&E) to examine the general structure and lipid droplets or with Masson’s trichrome stain for fibrosis. Samples were also subject to immunohistochemistry using F4/80 antibody for macrophages (Abcam) and CD3 antibody for T-cells (Abcam). All samples were visualized with light microscopy using standard techniques, and staining was performed at the Ontario Veterinary College, Department of Pathobiology, University of Guelph.
Transmission Electron Microscopy
Muscle tissues were prepared as recommended by the Electron Microscopy Facility of the University of Guelph. Briefly, quadricep muscle (n = 4 per group) were quickly immersed in fixing buffer (2.5% glutaraldehyde and 1.0% paraformaldehyde in phosphate-buffered saline (PBS)) and incubated at 4°C overnight. Samples were washed in 0.1 m HEPES and suspended in 1.0% osmium tetroxide for 4 h. Tissues were washed 3 times in 100 m MHEPES, suspended in 2% uranyl acetate for 3 h, washed 3 times in 0.1% HEPES, and dehydrated by incubation in a graded ethanol series (ie, 25%–100% ethanol). Tissue was infiltrated with resin by suspending the tissue in 50% ethanol–50% resin (LR White; London Resin Company) for 4 h and then in pure resin for 4 h, using a rotating mixer. Tissues were embedded in pure resin overnight at 60°C to polymerize the resin. Quadricep was sectioned longitudinally in the center of the muscle to view the sarcomere structure. Sections (100 nm) were laid onto 200-mesh Formvar-carbon copper grids and stained with 2% uranyl acetate and Reynolds lead citrate. A minimum of three sections were placed onto each grid. Images were obtained from each sample in a randomized systematic order. Samples were viewed on a Philips CM 10 transmission electron microscope (TEM) at 80 kV, and images were obtained with an Olympus/SIS Morada charge-coupled device (CCD) camera using Olympus/SIS iTEM software.
Analysis of Triglyceride Content
Soleus and gastrocnemius muscle samples (n = 6 per group) were homogenized in 500 μL of PBS and 5% Tween 20. Samples were heated for 5 mins at 95°C and cooled to room temperature. The heating/cooling process was repeated, and insoluble material was removed through centrifugation. A triglyceride (TAG) assay kit (Wako Diagnostics 994–02 891 and 998–02 992) was used to quantify TAG content.
Analysis of Diacylglyceride Content
Soleus and gastrocnemius muscle samples (n = 3 per group) were homogenized for the measurement of diacylglyceride (DAG) content as previously described.21 Lipids were extracted from muscle using chloroform: methanol: PBS + 0.2% SDS (1:2:0.8). Diacylglycerol kinase and [γ-32P] ATP (15 μCi/μmol cold ATP) were added to the extracts, and the reaction was stopped using chloroform: methanol (2:1). Samples were run on thin layer chromatography plates in chloroform: acetone: methanol: acetic acid: water (100:40:20:20:10). The DAG bands were scraped and counted by liquid scintillation using the Beckman instrument.
Determination of protein kinase C (PKC) Activity in Membrane and Cytosol Fractions
Soleus and gastrocnemius muscle samples (n = 3 per group) were homogenized, and the cytosolic and particulate fractions were separated. PKC activity was determined by using the PKC enzyme assay system (Amersham life science, RPN77) as previously described.22 Briefly, 25 μL of cytosolic or membrane fraction were added to assay tubes containing 25 μL of the component mixture (3 m m Ca (C3H302)2, 75 μg/mL L-α-phosphatidyl-L-serine, 6 μg/mL phorbol 12-myristate 13-acetate, 225 μm substrate peptide, and 7.5 m m dithiothreitol in 50 m m Tris–HCl containing 0.05% sodium azide). Reaction was initiated by adding 25 μL of Mg-ATP buffer (10 μCi/mL [γ32P]ATP, 1.2 m m ATP, 72 m m MgC12, and 30 m m HEPES). Samples were incubated at 37°C for 15 min, and the reaction was terminated by adding 100 μL of stop reagent (300 m m orthophosphoric acid). The phosphorylated peptide was separated by blotting 35 μL onto peptide-binding papers. Papers were washed with 75 m m phosphoric acid twice for 5 min, and disks were counted by liquid scintillation using the Beckman instrument.
Malonyl-CoA and Long-Chain Fatty acyl-CoA Assay
To measure the levels of malonyl-CoA and long-chain fatty acyl-CoA (LCFA), soleus and gastrocnemius muscle samples (n = 10 per group) were homogenized in 1 mL 6% perchloric acid and centrifuged at 5000 g for 15 min at 4°C. The supernatant containing acid-soluble metabolites was collected and neutralized with 2 mol/L KHCO3, pH 7.0, and centrifuged at 2000 g for 10 min at 4°C. The clear supernatant was used for malonyl-CoA assays following the protocol of Antinozzi et al. 1998 by measuring the malonyl-CoA-dependent incorporation of labeled acetyl-CoA into palmitic acid. To measure LCFA, the acid-insoluble precipitates were washed with twice with 1 mL diethyl ether and 200 μL H20. Dithiothreitol was added to the pellet, followed by 1 mol/L KOH to adjusted to pH 11.5. Sample was heated for 10 min at 55°C to hydrolyze the thioester bonds of LCFA and assayed for released CoA following the protocol of Antinozzi et al. 1998. The change in fluorescence was used to calculate the LCFA concentration in the samples.23
Free FA and Glycerol Measurements
For the measurement of serum free fatty acid (FFA) and glycerol with and without epinephrine stimulation, mice (n = 5 per group) were fasted overnight and injected intraperitoneally with a weight-adjusted bolus of epinephrine (20 μg/100 g body weight) or an equivalent volume of sterile saline. After 15 mins, mice were euthanized, and blood was collected via cardiac exsanguination. Blood clotted at room temperature and was centrifuged at 2000 g to collect serum. Free fatty acid (Wako Bioproducts) and glycerol (Millipore Sigma) were determined using commercially available kits according to the manufacturer’s instructions.
FA Oxidation
Soleus and gastrocnemius muscles were isolated from fasted Pcyt2+/- and Pcyt2+/+ control littermates (n = 9 per group). Fatty acid oxidation was assessed as previously described.24 Briefly, muscles were immediately submerged in ice-cold medium containing 250 m m sucrose, 1 m m EDTA, 10 m m Tris–HCl, and 10 m m Tris–HCl (pH 7.4) and homogenized. Next, 40 μL of a 20-fold diluted muscle homogenate were preincubated with a 95% O2-5% CO2 mixture at 30°C for 15 min. The 160 μL reaction mixture containing Krebs-Ringer-Henseleit buffer containing 2.4% BSA and 0.4 m m [3H] palmitate (1 μCi/mL) was added to preincubated and incubated at 30°C for 15 min, and 100 μL of 4 N sulfuric acid were injected to stop the reaction. Palmitate oxidation was assessed by measuring the quantity of tritiated water released into the medium by liquid scintillation counting.
ATP Determination Assay
ATP content was measured in the soleus and gastrocnemius (n = 12 per group) with an ATP determination kit (Invitrogen, CAT#A22066) that uses recombinant firefly luciferase. The manufacturer’s instructions were followed.
Glucose Tolerance Test
For glucose tolerance test (GTT), mice (n = 3 per group) were fasted 6 h before intraperitoneal injection (I.P.) of 2 mg/kg of body weight of glucose in 0.9% saline. Blood glucose level was measured through the tail vein by glucometer (Freestyle lite, Abbott Laboratories, Saint-Laurent, QC, Canada) immediately before injection and 30, 60, and 120 mins after glucose injection.
Glucose Uptake in Isolated Muscle
Soleus and gastrocnemius muscles were isolated from mice that were fasted for 12 h. Muscles (n = 8 per group) were submerged in a pre-equilibration in Krebs-Henseleit bicarbonate buffer for 2 h and then incubated for 20 min in the absence or presence of insulin (10 mU/mL). The 0.5 μCi 2-deoxy[14C]glucose and 0.3 μCi 14C-mannitol tracers were added, and muscles were incubated for an additional 20 min with the tracers and quickly blotted on filter paper and frozen in liquid nitrogen. Muscles were digested for 1 h at 50°C in 1 N NaOH and then counted by liquid scintillation for radioactive tracer incorporation.
Glycogen Content
Glycogen content was determined as previously described.25 In brief, soleus and gastrocnemius muscles (n = 12 per group) were digested 500 μL 30% KOH saturated with NaSO4 and boiled for 30 mins until a homogenous solution was obtained. Cold 95% ethanol was added to precipitate glycogen, and glycogen was separated by centrifugation. Glycogen was dissolved in distilled H2O. Glycogen content was determined at 490 nm after the addition of 5% phenol and 95% sulfuric acid. The standard curve was generated using pure glycogen (Roche).
Glycogen Synthesis
Glycogen synthesis was assessed by measuring the incorporation of [U-14C]D-glucose into glycogen, as previously described.26 Briefly, soleus and gastrocnemius muscles (n = 10 per group) were pre-incubated in Krebs-Ringer bicarbonate buffer with 1% defatted BSA and 5.6 m m glucose for 30 min. After pre-incubation, samples were incubated in the same Krebs-Ringer bicarbonate buffer with 0.3 μCi [U-14C]D-glucose added. Samples were washed in ice-cold PBS, blotted on filter paper, frozen in liquid nitrogen, and digested in 0.5 mL of 1 m KOH at 70°C for 1 h. Aliquots were taken for protein quantification (Bradford method). Carrier (10 mg) was added to the hydrolysates, and glycogen was precipitated at -20°C overnight with 100% ethanol and was resuspended in 0.5 mL of H2O. The incorporation of [U-14C]D-glucose into glycogen was determined using liquid scintillation counting.
Statistical Analysis
Sample analysis was blinded, and data was analyzed using a two-tailed unpaired t-test to test for differences between the Pcyt2+/+ and Pcyt2+/− groups. Mixed sexes were used for the analysis. Significance was rejected at P ≥ 0.05. Results are represented as the mean ± standard deviation (SD). All statistical tests were performed with Graphpad Prism 9 software.
Results
Pcyt2+/− Exhibit Myosteatosis and Disrupted Muscle Structure
We have previously shown that Pcyt2+/− mice develop adult-onset obesity and ectopic lipid accumulation in the liver.2,7 Here, we aimed to determine whether there is also increased infiltration of adipose tissue into the skeletal muscle. The Pcyt2+/− quadricep muscle is larger than Pcyt2+/+, and H&E staining reveals the accumulation of intramuscular adipose tissue deposition (black asterisks) (Figures 1A and B, Supplementary Figure S1). Hematoxylin and eosin staining also reveals elements of muscle degeneration, including smaller, rounded hypereosinophilic skeletal muscle cells with several cells exhibiting centralized nuclei (blue arrows), swollen hypereosinophilic cell (black arrows), and vacuolization. We further examined muscle ultrastructure using TEM (Figure 1C, Supplementary Figure S2). Transmission electron microscopy of the quadricep muscle indicates disrupted sarcomere structure with loss of proper striation, as shown by disordered myofilament arrangements and irregular or indistinct Z-lines in Pcyt2+/− (orange asterisks). Mitochondrial abundance is decreased, and mitochondria appear swollen with irregular size, shape, and cristae organization (black arrows), indicting compromised structural integrity and function. There is a large increase in the presence of vacuoles (red arrows), suggesting further evidence of skeletal muscle cell degeneration. In contrast, Pcyt2+/+ mice show no adipocyte infiltration or intracellular lipid accumulation and normal skeletal muscle cell architecture in both H&E staining and TEM. We conducted a hindlimb clasping test27,28 to determine if muscle function was impacted (Figures 1D and E). We observed that upon tail suspension, Pcyt2+/- mice exhibit mild to moderate limb clasping relative to Pcyt2+/+ controls, which can indicate muscle weakness or neuromuscular dysfunction. Together, these data show evidence for degenerative processes in the Pcyt2+/− quadricep that are linked to impaired physical function, and myosteatosis, which can cause the loss of muscle quality.5
Figure 1.

Muscle structure is altered in Pcyt2+/- mice. (A) Images and weights of quadricep muscle of Pcyt2+/- relative to Pcyt2+/+ showing enlarged quadricep muscle. (B) Hematoxylin and eosin staining of the quadriceps show intramuscular lipid accumulation and degenerating myofibers. Black asterisks show lipid accumulation, blue arrows show smaller, hypereosinophilic fibers with several cells exhibiting the internal migration of nuclei, black arrow shows a swollen hypereosinophilic cell. (C) Transmission electron microscopy of the quadriceps. Orange asterisks show disordered sarcomere structure with loss of proper striation, black arrows show swollen and irregular mitochondria, and red arrows show increased presence of vacuoles. (D) Images of hindlimb clasping in Pcyt2+/- and (E) quantification clasping using a scoring system based on the degree and length of time the limbs are retracted toward the abdomen. Pcyt2+/- mice show increased hindlimb clasping, which is indicative of muscle weakness or neuromuscular disorders. Data are derived from both male and female mice and presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Increased Macrophage Infiltration and Fibrosis
The infiltration of immune cells often accompanies degenerative muscle fibers; therefore, we performed immunohistochemistry (IHC) analysis on quadricep muscle using CD3 and F4/80 antibodies to determine T-cell and macrophage presence, respectively. In Pcyt2+/- quadricep, there were minimal detectable CD3 + T-cells. However, F4/80 + macrophage infiltration is evident in Pcyt2+/- quadricep relative to Pcyt2+/+, a hallmark of muscle injury and inflammation (Figures 2A and B, Supplementary Figures S3 and S4). Indeed, the protein content of the proinflammatory markers p-p38 Mapk (p-p38 Mapk: p38 Mapk), p65 NfκB, and Stat3 is elevated in Pcyt2+/−, by 4.9-fold, 3.2-fold, and 2.1-fold, respectively, indicating increased activation of inflammatory signaling cascades (Figure 2C). Excessive or extended infiltration of macrophages due to prolonged tissue injury and inflammation can lead to the formation of fibrotic tissue and further compromised function; thus, we measured collagen deposition using Masson’s trichrome stain. Indeed, Pcyt2+/- mice show increased levels of fibrotic tissue (blue) that surrounds and separates adjacent myofibers (Figure 2D, Supplementary Figure S5). Together, these data suggest that Pcyt2+/- mice experience ectopic lipid accumulation in skeletal muscle and tissue injury, leading to disrupted skeletal muscle cell architecture, degeneration, inflammation, fibrosis, and functional impairment.
Figure 2.

Increased macrophage infiltration and fibrosis. Images of IHC staining of the quadricep using (A) Anti-CD3 antibody to determine T-cell infiltration and (B) Anti-F4/80 antibody to determine macrophage infiltration in Pcyt2+/−relative to Pcyt2+/+ quadricep muscle. Immunohistochemistry shows normal CD3 + cell content but an increased presence of F4/80 + macrophages in Pcyt2+/- indicating cellular injury and inflammation. (C) Western blot analysis in the quadricep shows elevated content of proinflammatory p-p38 Mapk normalized to p38 Mapk (p-p38 Mapk: p38 Mapk), and p65 NfκB and Stat3 normalized to Gapdh. (D) Staining using Masson’s trichrome for collagen deposition (blue) in the quadricep showing increased fibrosis in Pcyt2+/−. Data are derived from both male and female mice and are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Elevated Lipogenesis With DAG and TAG Accumulation
To probe the myocellular accumulation of lipids, we determined TAG and DAG content in the soleus and gastrocnemius muscles. We observed increased TAG and DAG in the whole muscle of Pcyt2+/- by the method of Bligh and Dyer (Figure 3A) and further quantified this accumulation separately in the soleus and gastrocnemius. In the soleus, TAG is increased by 86% and DAG is increased by 87% in Pcyt2+/- relative to Pcyt2+/+. Similarly, in gastrocnemius, TAG is increased by 92% and DAG is increased by 83% in Pcyt2+/− (Figures 3B and C). Fatty acids and their acyl-CoA esters can activate Pkc, and indeed, there is a 95% increase in Pkcα protein content. There is a 53% increase in cytosolic Pkc activity, and a 35% increase in membrane-bound Pkc activity, indicating increased translocation and activation of Pkc in Pcyt2+/− whole muscle (Figures 3D and E). Next, we measured the protein content of the key lipogenic transcription factor Srebp1c and the FA metabolic regulator Acc. In Pcyt2+/− whole muscle, the protein content of Srebp1c is increased by 89% and Acc is increased by 4.24-fold. The ratio of phosphorylated Acc to total Acc (p-Acc: Acc) is reduced by 45%, showing enhanced Acc dephosphorylation and activation (Figure 3F). Accordingly, the levels of malonyl-CoA, the product of the Acc-catalyzed reaction, are increased by 2-fold in the soleus and 56% in the gastrocnemius (Figure 3G). Lastly, we measured the levels of long-chain fatty acids in their activated acyl-CoA form (LCFA-CoA), which is required for further anabolic or catabolic reactions. This measurement provides a more direct index of intramyocellular lipid metabolism than total TAG content because it mitigates the contamination from closely associated intramuscular adipose tissue. In Pcyt2+/−, LCFA-CoAs are increased by 54% in the soleus and 90% in the gastrocnemius relative to Pcyt2+/+, showing increased lipid availability (Figure 3H). The acyl-CoA form of the most abundant saturated LCFA, palmitoyl-CoA, is well known to associate with the generation of cytotoxic lipid intermediates, inflammation, mitochondrial damage, and cell death. Together, this indicates lipotoxicity as a contributor to Pcyt2+/− skeletal muscle degeneration.
Figure 3.

Increased lipogenesis with TAG and DAG accumulation. (A) Image of a TLC plate showing TAG and DAG accumulation in Pcyt2+/- relative to Pcyt2+/+ by the method of Bligh and Dyer. Quantification of (B) TAG and (C) DAG accumulation showing elevated levels of TAG and DAG across both the soleus and gastrocnemius muscles in Pcyt2+/−. (D) Western blot analysis of Pkcα normalized to β-Tubulin in whole muscle showing increased Pcyt2+/- protein content of Pkcα. (E) Pkc activity in the membrane and cytosolic fractions of whole muscle shows increased membrane-bound and cytosolic Pkc, indicating elevated Pkc activity in Pcyt2+/-. (F) Western blot analysis of lipogenic proteins Srebp1c, p-Acc, and Acc; p-Acc relative to Acc and Srebp1c, and Acc normalized to β -Tubulin, showing increased content of lipogenic proteins in Pcyt2+/−. (G) Elevated malonyl-CoA and (H) LCFA-CoA content in the Pcyt2+/- soleus and gastrocnemius. Together, this shows elevated lipogenesis that leads to DAG and TAG accumulation and the activation of Pkc in Pcyt2+/- skeletal muscle relative to Pcyt2+/+. Data are derived from both male and female mice and are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Reduced Lipolysis and Mitochondrial Fatty Acid Transport and Oxidation
We assessed whole-body lipolysis in Pcyt2+/− by determining the plasma concentrations of FFA and glycerol with and without epinephrine stimulation, which is a primary inducer of adipose and skeletal muscle lipolysis.29 Under basal conditions, no differences are observed between Pcyt2+/+ and Pcyt2+/- plasma FFA and glycerol; however, when stimulated with epinephrine, Pcyt2+/- mice show impaired FA mobilization, evidenced by a 39% lower FFA release and a 38% lower glycerol release than Pcyt2+/+ (Figures 4A and B). In the muscle, activation of the intramuscular lipolysis marker hormone-sensitive lipase (Hsl) is diminished, as shown by a reduction in the protein content of phosphorylated Hsl (p-Hsl: Hsl) by 76% (Figure 4C), indicating a limited liberation FAs from of skeletal muscle TAGs.
Figure 4.

Reduced lipolysis and mitochondrial FA transport and oxidation. Reduced (A) FFA and (B) glycerol levels in plasma under basal and epinephrine stimulation by I.P. showing an impairment in systemic lipolysis in Pcyt2+/- relative to Pcyt2+/+. (C) Western blot analysis of the intramuscular lipolysis marker p-Hsl relative to Hsl shows reduced whole muscle protein content, indicating decreased activation and liberation of FAs from TAG stores in skeletal muscle. This suggests reduced skeletal muscle lipolysis as a contributing factor to the whole-body reduction in lipolysis Pcyt2+/−. (D) Western blot analysis of Sirt1, p-Ampkα, and Ampkα; p-Ampkα relative to Ampkα, Sirt1 and Ampk are normalized to β-Tubulin, showing decreased whole muscle content of lipolytic proteins in Pcyt2+/−. (E) Reduced mRNA expression of the LCFA mitochondrial transport protein Cpt1, normalized to Gapdh in Pcyt2+/- whole muscle. (F) Fatty acid oxidation is decreased, and (G) ATP content is increased in Pcyt2+/- whole muscle. Together, this shows how, relative to Pcyt2+/+, Pcyt2+/- muscle exhibits impaired FA mobilization and oxidation. Data are derived from both male and female mice and are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We have previously shown by indirect calorimetry that fasted whole-body FA oxidation is reduced in adult Pcyt2+/− relative to Pcyt2+/+ mice2 and here, we determined skeletal muscle FA oxidation. In Pcyt2+/− whole muscle, Sirt1 and Ampk proteins, which are activated under lipolytic and FA oxidative conditions, are reduced by 46% and 60%, respectively, and activation of Ampk by phosphorylation (p-Ampk: Ampk) is reduced by 43% (Figure 4D). In line with our found increase in malonyl-CoA, which inhibits the mitochondrial LCFA transport enzyme Cpt1, the mRNA expression of Cpt1 is reduced by 74% in Pcyt2+/− (Figure 4E). As predicted by the above evidence, rates of FA oxidation are decreased by 36% in Pcyt2+/− whole muscle (Figure 4F). Interestingly, despite the decreased FA oxidation, there is a 52% increase in ATP content in Pcyt2+/- muscle relative to Pcyt2+/+ (Figure 4G). This suggests a potential shift in energy substrate oxidation to favor glycolysis, which can occur under conditions of defective FA oxidation in order to meet energetic demands.30 Cumulatively, these findings show that Pcyt2+/- mice exhibit major perturbations to FA metabolism, both through augmented FA synthesis and impaired FA mobilization and oxidation, showing a reduced reliance on FAs as a substrate for ATP production.
Impaired Glucose Metabolism With Glycogen Accumulation
Skeletal muscle is the primary site of insulin-stimulated glucose disposal, making it a critical regulator of systemic glucose homeostasis, therefore, we first determined whole-body insulin sensitivity by GTT (Figure 5A). Pcyt2+/− mice show an elevation in basal plasma glucose concentration and at all measured time points following the administration of an intraperitoneal glucose load. The time required to clear plasma glucose is extended, and the area under the curve is elevated by 43% in Pcyt2+/- compared to Pcyt2+/+. This confirms our previous reports of both elevated plasma glucose levels and insulin levels in response to a GTT, showing hyperglycemia and impaired systemic insulin sensitivity in adult Pcyt2+/- mice.2,3 In skeletal muscle, we measured the levels of insulin signaling proteins and found no significant change in pSer307-Irs1: Irs1; however, Irs1 content was reduced by 44% in Pcyt2+/− whole muscle. p85-Pi3k is reduced by 58%, and both phosphorylated (pSer473-Akt: Akt) and total Akt are reduced by 24% and 31%, respectively (Figure 5B). Insulin induces skeletal muscle glucose transport mainly facilitated by Glut4 translocation to the plasma membrane. In Pcyt2+/− whole muscle, the total pool of Glut4 protein was elevated by 2.43-fold; however, Glut4 plasma membrane content was reduced by 87%, indicating reduced trafficking to the plasma membrane and responsiveness to insulin stimulation (Figure 5C). We evaluated glucose uptake by measuring the incorporation of 14C-2-deoxyglucose into the soleus and gastrocnemius muscles with and without insulin stimulation (Figure 5D). In the soleus, glucose uptake is reduced by 37% under basal conditions and by 23% under insulin stimulation, and in the gastrocnemius is reduced by 35% under basal conditions and by 27% under insulin stimulation (Figure 5E). Finally, periodic acid-Schiff staining revealed altered glycogen storage with an accumulation of muscle glycogen in Pcyt2+/− quadricep (yellow) (Figure 5F), and assay-based quantification of glycogen content shows a 75% increase in whole muscle (Figure 5G). Replete glycogen stores inhibit the continued storage of glucose as glycogen, and indeed, the rate of glycogen synthesis is decreased by 36% in Pcyt2+/− (Figure 5H). These data show impaired glucose handling in Pcyt2+/− with reduced insulin sensitivity, impaired glucose uptake, and glycogen accumulation.
Figure 5.

Impaired glucose transport and glycogen accumulation. (A) Glucose tolerance test and area under the curve showing whole-body reduced insulin sensitivity in Pcyt2+/- relative to Pcyt2+/+. (B) Western blot analysis of proteins in the insulin signaling cascade: pSer307-Irs1, Irs1, p85-Pi3k, pSer473-Akt, Akt; pSer307-Irs1 relative to Irs1; pSer473-Akt relative to Akt; and Irs1, p85-Pi3k, and Akt normalized to β-Tubulin show reduced expression, indicating impaired insulin signaling in Pcyt2+/- whole muscle. (C) Western blot analysis of plasma membrane Glut4 content and total Glut4 content; Glut4 plasma membrane relative to Glut4 total, and Glut4 total normalized to β-Tubulin. This shows reduced activation and translocation of Glut4 to the plasma membrane in Pcyt2+/- whole muscle. 14C-2-deoxyglucose uptake into (D) soleus and (E) gastrocnemius muscles with and without insulin stimulation also showed diminished glucose uptake in Pcyt2+/−. (F) Periodic acid-Schiff staining of quadricep muscle sections showing glycogen accumulation, and (G) assay-based quantification showing elevated levels of glycogen content in Pcyt2+/- whole muscle. (H) Rate of glycogen synthesis is decreased in Pcyt2+/- whole muscle. Together, this shows impaired glucose handling in Pcyt2+/- with glycogen accumulation that inhibits further synthesis, insulin signaling and glucose update. Data are derived from both male and female mice and are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Discussion
We have previously determined that mice with a global, heterozygous deletion of the Pcyt2 gene develop an age-dependent obesity and NASH.2,3 In this study, we report multiple abnormalities within the skeletal muscle of Pcyt2+/−mice, including disrupted muscle structure with degeneration, myosteatosis, fibrosis, and altered FA and glucose metabolism. The Pcyt2+/− quadricep is increased in size and weight relative to Pcyt2+/+, possibly due to elevated adipose tissue infiltration, as H&E staining reveals a dramatic presence of intramuscular adipose tissue. Myosteatosis is associated with NAFLD severity and is an established prognostic indicator for end-stage liver disease patients.31–34 Another possibility for the increased muscle weight is skeletal muscle cell hypertrophy due to the chronic workload imposed on skeletal muscle by Pcyt2+/- obesity,2 reflecting the human obese condition whereby skeletal muscle mass is increased, yet the function is impaired.35 Future work should quantify intramuscular adipose tissue and fat-free muscle volume to better understand the relationship between skeletal muscle composition, physical function, and Pcyt2+/- NASH development.
Metabolic inflammation emanating both from the fatty liver and skeletal muscle is mutually detrimental and suggested as a key process that contributes to skeletal muscle cell death, dysfunction, and deleterious remodeling.36Pcyt2+/- develop NASH with hepatocellular injury, inflammation, and fibrosis, and here we show the skeletal muscle manifestations of these pathologies. Pcyt2+/- skeletal muscle exhibits intramuscular adipose tissue and LCFA-CoA, TAG, and DAG accumulation. Additionally, Pcyt2+/- skeletal muscle cells show evidence of degeneration with vacuolization, disordered sarcomeres, mitochondria ultrastructure irregularities, and paucity. There is increased protein content of the p65 subunit of NfκB, indicating increased NfκB activation and translocation to the nucleus, where it acts as a central regulator of inflammatory pathways and has been shown to participate in the development of multiple metabolic disorders.37,38 Protein content of pro-inflammatory p-p38, Mapk, and Stat3 is also increased, and together these proteins play critical roles in modulating the cellular response to inflammation, stress, and disease progression within skeletal muscle.39Pcyt2+/- skeletal muscle shows macrophage infiltration, indicating cellular injury and the presence of degenerative muscle fibers, and fibrosis, a major cause of muscle weakness and reduced regenerative capacity and a hallmark of muscular dysfunction and degeneration.40 Lastly, Pcyt2+/- exhibits hindlimb clasping upon tail suspension, indicating muscle weakness and/or neuromuscular dysfunction. Together, this shows myopathic processes in Pcyt2+/- skeletal muscle, including myosteatosis, lipotoxicity, inflammation, and fibrosis causing skeletal muscle cell degeneration that is accompanied by preliminary evidence for impaired muscle function.
Interestingly, hindlimb clasping is a marker of disease severity in several mouse models of neurodegenerative and neuromuscular disorders, including cerebellar ataxia. Recently, a small group of patients has been identified with a loss-of-function mutation in the PCYT2 gene that develop a broad progressive ataxia-spasticity condition. This disorder is characterized by progressive cerebral and cerebellar atrophy, multiple muscular pathologies, and spastic para- or tetraplegia, and indicates disturbed PE metabolism as a key pathological mechanism in neuromuscular function.41–43 Given the essential role of the autonomic nervous system in peripheral energy homeostasis, nutrient sensing, inflammation, and tissue function and repair, the role of Pcyt2 and PE homeostasis within neuromuscular systems represents an interesting route for future investigation.
It is well known that intramuscular and intramyocellular FA accumulation contributes to aberrant glucose metabolism and is strongly associated with insulin resistance, although the exact mechanisms underlying this relationship are not completely clear.44–46Pcyt2+/- mice exhibit impaired systemic glucose handling as shown by the GTT. Whole-body glucose homeostasis is dependent on the conserved ability of skeletal muscle to synthesize and store glycogen in response to insulin stimulation, and impairments in this pathway are a primary defect in metabolic syndrome and T2DM.47Pcyt2+/- muscle shows abnormal glycogen metabolism, with an accumulation of glycogen content and reduced glycogen synthesis. Because Pcyt2+/- glycogen stores are replete, negative feedback inhibits insulin signaling and reduces the flux of circulating glucose into muscle glycogen storage. This is shown in Pcyt2+/− whole muscle by the decreased expression of insulin signaling proteins, diminished Glut4 trafficking to the plasma membrane, reduced glucose uptake in both the soleus and gastrocnemius, and reduced glycogen synthesis. These findings are consistent with reports of an inverse relationship between skeletal muscle glycogen content and insulin sensitivity.48–50 Reduced glucose disposal into glycogen stores forces the redirection of glucose within the skeletal muscle into alternative non-oxidative metabolic pathways, such as lipogenesis. Indeed, in Pcyt2+/- muscle, lipogenesis is upregulated in both the soleus and gastrocnemius muscles, which can further inhibit insulin signaling.51,52 These findings may lend important insight into Pcyt2+/- NASH progression and Pcyt2+/- skeletal muscle insulin resistance can drive hepatic lipogenesis via the metabolic rerouting of circulating glucose from skeletal muscle uptake and glycogen storage to liver TAGs. This mechanism has been validated experimentally with muscle-specific inactivation of the insulin receptor gene53 and in insulin-resistant patients.54 Further, insulin stimulation induces protein synthesis, muscle hypertrophy, and inhibits proteolysis55,56 which directly links skeletal muscle insulin resistance during Pcyt2+/- NASH to impaired muscle function.
Pcyt2+/- mice develop adult-onset obesity and a reduced capacity for whole-body FA oxidation, as determined by indirect calorimetry.2 In the present study, we specifically show modifications to skeletal muscle FA metabolism in Pcyt2+/− mice, namely increased lipogenesis and decreased mitochondrial FA oxidation. Pcyt2 catalyzes the synthesis of CDP-ethanolamine, which is condensed with DAG to form PE in the third and final step of the CDP-ethanolamine Kennedy pathway. As the rate-limiting enzyme, Pcyt2 deficiency reduces flux through the pathway by limiting the availability of CDP-ethanolamine and thus restricts the incorporation of DAG into PE formation, causing DAG to accumulate.2 When dysregulated, DAGs are a potentially dangerous lipid species due to their central role as second messengers in signal transduction for a range of cellular processes, such as Pkc activation. Here, we show elevated DAG content in Pcyt2+/− soleus and gastrocnemius muscles, and increased activity of membrane-bound Pkc. DAG-activated Pkc isoforms are well known to promote skeletal muscle insulin resistance in both animal and human models.46,57–59Pcyt2+/- muscle exhibits an upregulation of key lipogenic proteins Srebp1c and Acc, and elevated malonyl-CoA content, which is the primary carbon source for FA synthesis by Fas. Malonyl-CoA modulates FA oxidation through its capacity to inhibit the transport of LCFA-CoA into the mitochondria for β-oxidation via Cpt1 inhibition. Indeed, Cpt1 expression is decreased, and FA oxidation is diminished Pcyt2+/- muscle. This is supported by a reduction in the FA oxidative proteins Sirt1 and p-Ampk. Accordingly, there is an accumulation of LCFA-CoA, a potentially toxic intermediate, that is not able to be oxidized in the mitochondria and must be directed to TAG synthesis. Lastly, we show that systemic lipolysis is diminished under epinephrine stimulation, and within the muscle, there is a reduction in p-Hsl, which is a major inducer of intramuscular lipolysis,60 indicating decreased Pcyt2+/- TAG catabolism. Together, this results in TAG accumulation in Pcyt2+/-soleus and gastrocnemius muscles. Therefore, elevated FA production in Pcyt2+/- muscle likely occurs, in part, to facilitate the conversion of surfeit DAG into neutral TAG storage, to mitigate the potentially deleterious consequences of excessive DAG signaling. This shows how metabolic adaptations to a deficiency in phospholipid synthesis through the CDP-ethanolamine Kennedy pathway result in intramyocellular and intramuscular lipid accumulation, influence skeletal muscle insulin resistance, and facilitate NASH development.
In this study, we measured lipid accumulation and glucose uptake in the soleus, predominantly composed of oxidative Type I and IIA fibers, and the gastrocnemius, which contains a greater volume of glycolytic Type IIB fibers. One of the central mechanisms hypothesized to be driving the pathologies in Pcyt2+/− skeletal muscle is altered DAG metabolism. Therefore, we anticipated that fiber-type differences in the distribution of Pkc may influence glucose metabolism differently in the soleus and gastrocnemius muscles. Glycolytic fibers exhibit a greater abundance of Pkcθ than oxidative,61,62 which is the major DAG-sensing isoform of Pkc and driver of insulin resistance in skeletal muscle.63 However, insulin-stimulated glucose uptake was similarly reduced in Pcyt2+/− soleus and gastrocnemius. Likewise, we found that LCFA-CoA, DAG, and TAG accumulate similarly in both Pcyt2+/− soleus and gastrocnemius muscles, despite differences in oxidative capacity. The soleus possesses an increased oxidative potential and enrichment in the lipase Hsl,64 which has an ∼10-fold greater affinity toward DAG compared to TAG.65 Hence, we anticipated that the soleus would more readily oxidize DAGs shunted from the CDP-ethanolamine pathway and require less TAG production; however, this was not the case, showing that higher inherent rates of FA oxidation in the soleus are not able to protect against DAG and TAG accumulation during Pcyt2 deficiency.
The explanation for the lack of differences in lipid accumulation and glucose uptake in across soleus and gastrocnemius muscles is unclear. Multiple mechanistic lines of reasoning have been proposed to explain the pathogenesis of skeletal muscle insulin resistance, including intramyocellular and intramuscular lipid accumulation,44,46 inflammation,66 mitochondrial dysfunction,67,68 and oxidative stress,69,70 and it is possible the relative influence of these processes on insulin resistance varies across muscle fiber type. Type I and Type II fibers exhibit differences in autophagic processes71,72 mitochondrial volume density, network dynamics, and respiratory properties,73 metabolism of ROS,74,75 and calcium handling at the mitochondria and SERCA,76,77 allowing them distinct metabolic characteristics and contractile properties. Given that PE plays critical roles in all of these processes, autophagy and mitophagy,78,79 mitochondrial respiration and oxidative stress,80–83 and SERCA function,84–86 it stands to reason that the mechanisms driving insulin resistance and the development of Pcyt2 muscle pathologies are fiber-type specific. For example, glycolytic muscles appear to have a lessened ability to buffer ROS than oxidative muscle,75,87 and thus, may be more susceptible to oxidative stress and inflammation during Pcyt2 deficiency. Therefore, it is plausible that the mechanistic consequences of Pcyt2 deficiency differ according to fiber type, but ultimately result in convergent signals that induce FA accumulation, insulin resistance, and manifest a similarly impaired phenotype.
Another explanation for the lack of observed differences in the soleus and gastrocnemius muscles is the possibility that oxidative fibers undergo a fiber-type metabolic switch toward a profile that resembles glycolytic fibers, which can occur as an adaptive response to conditions of impaired FA oxidation.30Pcyt2+/− exhibit elevated whole muscle ATP content with reduced FA oxidation, indicating a transition to glucose as the primary fuel source for ATP production. In support, glycogen content is increased in Pcyt2+/− muscle, a characteristic of glycolytic fibers. Further, a well-known factor that can induce fiber-type switching is alterations in neurological input and activity.88 Disruptions in phospholipid genes are increasingly associated with motor neuron diseases,89 and specifically, Pcyt2 deficiency in humans causes progressive ataxia-spasticity41,42 implicating Pcyt2 in neuronal homeostasis. Spasticity has been associated with a Type II fiber-type switch,90 suggesting the propensity for Pcyt2+/- mice to undergo a similar shift. Additionally, we showed that Pcyt2+/− mice exhibit hindlimb clasping, which is a key finding in mouse models of neuromuscular disorders. Future work should distinguish fiber-type-specific mechanisms governing the development of Pcyt2+/-skeletal muscle pathologies to improving the understanding of how PE homeostasis influences the metabolic and functional properties of skeletal muscle within the context of NASH pathogenesis.
Conclusion
In summary, these data support a hypothesis whereby diminished phospholipid synthesis through the CDP-ethanolamine Kennedy pathway due to Pcyt2 deficiency results in deleterious metabolic adaptations to accommodate reduced DAG usage. Pcyt2+/- skeletal muscle exhibits impaired glucose handling and storage, increased de novo lipogenesis, and reduced FA oxidation that leads to insulin resistance, myosteatosis, skeletal muscle inflammation, fibrosis, and degeneration, and these processes are linked to metabolic and functional decline in skeletal muscle. A limitation of this study is due to the global, and not muscle-specific, heterozygous deletion of Pcyt2, making it not possible to conclude whether skeletal muscle dysfunction is a primary cause of the overall Pcyt2+/- pathological phenotype or a secondary consequence of the metabolic disease. Future studies should address this by determining the role of phospholipid homeostasis in the increasingly appreciated bidirectional muscle-liver crosstalk during metabolic disease pathogenesis.
Supplementary Material
Contributor Information
Sophie Grapentine, Department of Human Health and Nutritional Sciences, University of Guelph, Guelph N1G 2W1, Canada.
Rathnesh K Singh, Department of Human Health and Nutritional Sciences, University of Guelph, Guelph N1G 2W1, Canada.
Marica Bakovic, Department of Human Health and Nutritional Sciences, University of Guelph, Guelph N1G 2W1, Canada.
Author Contributions
S.G. wrote the manuscript and collected data. R.K.S. collected data. M.B. supervised and designed the study and reviewed/edited the manuscript.
Funding
This work was supported by the Canadian Institutes of Health Research Grant # CIHR-450137 (M.B.) and Ontario Graduate Scholarship (SG). Graphical abstract was created with BioRender.com.
Conflict of interest statement
The authors declare no competing interests.
Data Availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1. Van Der Veen JN, Kennelly JP, Wan S, Vance JE, Vance D E, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr. 2017;1859(9):1558–1572. [DOI] [PubMed] [Google Scholar]
- 2. Fullerton MD, Hakimuddin F, Bonen A, Bakovic M. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J Biol Chem. 2009;284(38):25704–25713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grapentine S, Singh RK, Basu Pet al. Pcyt2 deficiency causes age-dependant development of nonalcoholic steatohepatitis and insulin resistance that could be attenuated with phosphoethanolamine. Sci Rep. 2022;12(1):1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Der Veen JN, Lingrell S, Mccloskey Net al. A role for phosphatidylcholine and phosphatidylethanolamine in hepatic insulin signaling. FASEB J. 2019;33(4):5045–5057. [DOI] [PubMed] [Google Scholar]
- 5. Nachit M, Leclercq IA. Emerging awareness on the importance of skeletal muscle in liver diseases: time to dig deeper into mechanisms!. Clin Sci (Lond). 2019;133(3):465–481. [DOI] [PubMed] [Google Scholar]
- 6. Fullerton MD, Hakimuddin F, Bakovic M. Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2). Mol Cell Biol. 2007;27(9):3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grapentine S, Bakovic M. Significance of bilayer-forming phospholipids for skeletal muscle insulin sensitivity and mitochondrial function. J Biomed Res. 2020;34(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zurlo F, Larson K, Bogardus C, Ravussin E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest. 1990;86(5):1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313(22):2263–2273. [DOI] [PubMed] [Google Scholar]
- 10. Defronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(Suppl 2):S157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Addison O, Marcus RL, Lastayo PC, Ryan AS. Intermuscular fat: a review of the consequences and causes. Int J Endocrinol. 2014;2014:309570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bhanji RA, Moctezuma-Velazquez C, Duarte-Rojo Aet al. Myosteatosis and sarcopenia are associated with hepatic encephalopathy in patients with cirrhosis. Hepatol Int. 2018;12(4):377–386. [DOI] [PubMed] [Google Scholar]
- 13. Altajar S, Baffy G. Skeletal muscle dysfunction in the development and progression of nonalcoholic fatty liver disease. J Clin Transl Hepatol. 2020;8(4):414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Post JA, Bijvelt JJ, Verkleij AJ. Phosphatidylethanolamine and sarcolemmal damage during ischemia or metabolic inhibition of heart myocytes. Am J Physiol. 1995;268(2 Pt 2):H773–780. [DOI] [PubMed] [Google Scholar]
- 15. Funai K, Lodhi IJ, Spears LDet al. Skeletal muscle phospholipid metabolism regulates insulin sensitivity and contractile function. Diabetes. 2016;65(2):358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Newsom SA, Brozinick JT, Kiseljak-Vassiliades Ket al. Skeletal muscle phosphatidylcholine and phosphatidylethanolamine are related to insulin sensitivity and respond to acute exercise in humans. J Appl Physiol (1985). 2016;120(11):1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee S, Norheim F, Gulseth HLet al. Skeletal muscle phosphatidylcholine and phosphatidylethanolamine respond to exercise and influence insulin sensitivity in men. Sci Rep. 2018;8(1):6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paran CW, Verkerke ARP, Heden TDet al. Reduced efficiency of sarcolipin-dependent respiration in myocytes from humans with severe obesity. Obesity (Silver Spring). 2015;23(7):1440–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leonardi R, Frank MW, Jackson PD, Rock CO, Jackowski S. Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis. J Biol Chem. 2009;284(40):27077–27089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp. 2010;(39):1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Preiss J, Loomis CR, Bishop WR, Stein R, Niedel JE, Bell RM. Quantitative measurement of sn-1,2-diacylglycerols present in platelets, hepatocytes, and ras- and sis-transformed normal rat kidney cells. J Biol Chem. 1986;261(19):8597–8600. [PubMed] [Google Scholar]
- 22. Pandey GN, Ren X, Dwivedi Y, Pavuluri MN. Decreased protein kinase C (PKC) in platelets of pediatric bipolar patients: effect of treatment with mood stabilizing drugs. J Psychiatr Res. 2008;42(2):106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Antinozzi PA, Segall L, Prentki M, Mcgarry JD, Newgard CB. Molecular or pharmacologic perturbation of the link between glucose and lipid metabolism is without effect on glucose-stimulated insulin secretion. A re-evaluation of the long-chain acyl-CoA hypothesis. J Biol Chem. 1998;273(26):16146–16154. [DOI] [PubMed] [Google Scholar]
- 24. Kim J-Y, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279(5):E1039–E1044. [DOI] [PubMed] [Google Scholar]
- 25. Lo S, Russell JC, Taylor AW. Determination of glycogen in small tissue samples. J Appl Physiol. 1970;28(2):234–236. [DOI] [PubMed] [Google Scholar]
- 26. Ceddia R, William W, Curi R. Comparing effects of leptin and insulin on glucose metabolism in skeletal muscle: evidence for an effect of leptin on glucose uptake and decarboxylation. Int J Obes. 1999;23(1):75–82. [DOI] [PubMed] [Google Scholar]
- 27. Boido M, De Amicis E, Valsecchi Vet al. Increasing agrin function antagonizes muscle atrophy and motor impairment in spinal muscular atrophy. Front Cell Neurosci. 2018;12;1–14. article 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hayward LJ, Kim JS, Lee M-Yet al. Targeted mutation of mouse skeletal muscle sodium channel produces myotonia and potassium-sensitive weakness. J Clin Invest. 2008;118(4):1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gorski J. Muscle triglyceride metabolism during exercise. Can J Physiol Pharmacol. 1992;70(1):123–131. [DOI] [PubMed] [Google Scholar]
- 30. Pereyra AS, Lin C-T, Sanchez DMet al. Skeletal muscle undergoes fiber type metabolic switch without myosin heavy chain switch in response to defective fatty acid oxidation. Mol Metab. 2022;59:101456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tanaka M, Okada H, Hashimoto Yet al. Relationship between nonalcoholic fatty liver disease and muscle quality as well as quantity evaluated by computed tomography. Liver Int. 2020;40(1):120–130. [DOI] [PubMed] [Google Scholar]
- 32. Chen VL, Wright AP, Halligan Bet al. Body composition and genetic lipodystrophy risk score associate with nonalcoholic fatty liver disease and liver fibrosis. Hepatol Commun. 2019;3(8):1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kitajima Y, Hyogo H, Sumida Yet al. Severity of non-alcoholic steatohepatitis is associated with substitution of adipose tissue in skeletal muscle. J Gastroenterol Hepatol. 2013;28(9):1507–1514. [DOI] [PubMed] [Google Scholar]
- 34. Montano-Loza AJ, Angulo P, Meza-Junco Jet al. Sarcopenic obesity and myosteatosis are associated with higher mortality in patients with cirrhosis. J Cachexia Sarcopenia Muscle. 2016;7(2):126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morgan PT, Smeuninx B, Breen L. Exploring the impact of obesity on skeletal muscle function in older age. Front Nutr. 2020;7;56904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gehrke N, Schattenberg JM. Metabolic inflammation—a role for hepatic inflammatory pathways as drivers of comorbidities in nonalcoholic fatty liver disease?. Gastroenterology. 2020;158(7):1929–1947.e6. [DOI] [PubMed] [Google Scholar]
- 37. Li Y-P, Reid MB. NF-κB mediates the protein loss induced by TNF-α in differentiated skeletal muscle myotubes. Am J Physiol Regul Integr Comp Physiol. 2000;279(4):R1165–R1170. [DOI] [PubMed] [Google Scholar]
- 38. Baker RG, Hayden MS, Ghosh S. Inflammation, and metabolic disease. Cell Metab. 2011;13(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brennan CM, Emerson CP, Owens J, Christoforou N. p38 MAPKs—roles in skeletal muscle physiology, disease mechanisms, and as potential therapeutic targets. JCI Insight. 6(12):e149915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mahdy MAA. Skeletal muscle fibrosis: an overview. Cell Tissue Res. 2019;375(3):575–588. [DOI] [PubMed] [Google Scholar]
- 41. Vaz FM, Mcdermott JH, Alders Met al. Mutations in PCYT2 disrupt etherlipid biosynthesis and cause a complex hereditary spastic paraplegia. Brain. 2019;142(11):3382–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vélez-Santamaría V, Verdura E, Macmurdo Cet al. Expanding the clinical and genetic spectrum of PCYT2-related disorders. Brain. 2020;143(9):e76. [DOI] [PubMed] [Google Scholar]
- 43. Kaiyrzhanov R, Wortmann S, Reid Tet al. Defective phosphatidylethanolamine biosynthesis leads to a broad ataxia-spasticity spectrum. Brain. 2021;144(3):e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miljkovic I, Zmuda JM. Epidemiology of myosteatosis. Curr Opin Clin Nutr Metab Care. 2010;13(3):260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Correa-De-Araujo R, Addison O, Miljkovic Iet al. Myosteatosis in the context of skeletal muscle function deficit: an interdisciplinary workshop at the national institute on aging. Front Physio. 2020;11;963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abdul-Ghani MA, DeFronzo RA. Pathogenesis of insulin resistance in skeletal muscle. Biomed Res Int. 2010;2010:476279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jensen J, Rustad PI, Kolnes AJ, Lai Y-C. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front Physiol. 2011;2:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jensen J, Jebens E, Brennesvik EOet al. Muscle glycogen inharmoniously regulates glycogen synthase activity, glucose uptake, and proximal insulin signaling. Am J Physiol Endocrinol Metab. 2006;290(1):E154–E162. [DOI] [PubMed] [Google Scholar]
- 49. Nielsen JN, Derave W, Kristiansen S, Ralston E, Ploug T, Richter EA. Glycogen synthase localization and activity in rat skeletal muscle is strongly dependent on glycogen content. J Physiol. 2001;531(Pt 3):757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Laurent D, Hundal RS, Dresner Aet al. Mechanism of muscle glycogen autoregulation in humans. Am J Physiol Endocrinol Metab. 2000;278(4):E663–668. [DOI] [PubMed] [Google Scholar]
- 51. Solini A, Di Virgilio F, Chiozzi P, Fioretto P, Passaro A, Fellin R. A defect in glycogen synthesis characterizes insulin resistance in hypertensive patients with type 2 diabetes. Hypertension. 2001;37(6):1492–1496. [DOI] [PubMed] [Google Scholar]
- 52. Litherland GJ, Morris NJ, Walker M, Yeaman SJ. Role of glycogen content in insulin resistance in human muscle cells. J Cell Physiol. 2007;211(2):344–352. [DOI] [PubMed] [Google Scholar]
- 53. Kim JK, Michael MD, Previs SFet al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest. 2000;105(12):1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Petersen KF, Dufour S, Savage DBet al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci. 2007;104(31):12587–12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fujita S, Rasmussen BB, Cadenas JGet al. Aerobic exercise overcomes the age-related insulin resistance of muscle protein metabolism by improving endothelial function and Akt/mammalian target of rapamycin signaling. Diabetes. 2007;56(6):1615–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Castellino P, Luzi L, Simonson DC, Haymond M, Defronzo RA. Effect of insulin and plasma amino acid concentrations on leucine metabolism in man. Role of substrate availability on estimates of whole body protein synthesis. J Clin Invest. 1987;80(6):1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Heydrick SJ, Ruderman NB, Kurowski TG, Adams HB, Chen KS. Enhanced stimulation of diacylglycerol and lipid synthesis by insulin in denervated muscle: altered protein kinase C activity and possible link to insulin resistance. Diabetes. 1991;40(12):1707–1711. [DOI] [PubMed] [Google Scholar]
- 58. Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes. 2002;51(7):2005–2011. [DOI] [PubMed] [Google Scholar]
- 59. Schmitz-Peiffer C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann N Y Acad Sci. 2002;967(1):146–157. [DOI] [PubMed] [Google Scholar]
- 60. Oscai LB, Essig DA, Palmer WK. Lipase regulation of muscle triglyceride hydrolysis. J Appl Physiol. 1990;69(5):1571–1577. [DOI] [PubMed] [Google Scholar]
- 61. Meller N, Altman A, Isakov N. New perspectives on PKCtheta, a member of the novel subfamily of protein kinase C. Stem Cells. 1998;16(3):178–192. [DOI] [PubMed] [Google Scholar]
- 62. Donnelly R, Reed MJ, Azhar S, Reaven GM. Expression of the major isoenzyme of protein kinase-C in skeletal muscle, nPKC theta, varies with muscle type and in response to fructose-induced insulin resistance. Endocrinology. 1994;135(6):2369–2374. [DOI] [PubMed] [Google Scholar]
- 63. Szendroedi J, Yoshimura T, Phielix Eet al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci. 2014;111(26):9597–9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Donsmark M,Langfort J,Plough Tet al. Hormone-sensitive lipase (HSL) expression and regulation by epinephrine and exercise in skeletal muscle. Eur J Sport Sci. 2010;2(6):1–10. [Google Scholar]
- 65. Moro C, Bajpeyi S, Smith SR. Determinants of intramyocellular triglyceride turnover: implications for insulin sensitivity. Am J Physiol Endocrinol Metab. 2008;294(2):E203–213. [DOI] [PubMed] [Google Scholar]
- 66. Tornatore L, Thotakura AK, Bennett J, Moretti M, Franzoso G. The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends Cell Biol. 2012;22(11):557–566. [DOI] [PubMed] [Google Scholar]
- 67. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350(7):664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mogensen M, Sahlin K, FernströM Met al. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56(6):1592–1599. [DOI] [PubMed] [Google Scholar]
- 69. Fridlyand LE, Philipson LH. Reactive species and early manifestation of insulin resistance in type 2 diabetes. Diabetes Obes Metab. 2006;8(2):136–145. [DOI] [PubMed] [Google Scholar]
- 70. Barazzoni R, Zanetti M,Cappellari GGet al. Fatty acids acutely enhance insulin-induced oxidative stress and cause insulin resistance by increasing mitochondrial reactive oxygen species (ROS) generation and nuclear factor-κB inhibitor (IκB)-nuclear factor-κB (NFκB) activation in rat muscle, in the absence of mitochondrial dysfunction. Diabetologia. 2012;55(3):773–782. [DOI] [PubMed] [Google Scholar]
- 71. Yamada E, Bastie CC, Koga H, Wang Y, Cuervo AM, Pessin JE. Mouse skeletal muscle fiber-type-specific macroautophagy and muscle wasting are regulated by a Fyn/STAT3/Vps34 signaling pathway. Cell Rep. 2012;1(5):557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morales-Scholz MG, Wette SG, Stokie JRet al. Muscle fiber type-specific autophagy responses following an overnight fast and mixed meal ingestion in human skeletal muscle. Am J Physiol Endocrinol Metabol. 2022;323(3):E242–E253. [DOI] [PubMed] [Google Scholar]
- 73. Picard M, Hepple RT, Burelle Y. Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: tailoring the organelle for optimal function. Am J Physiol Cell Physiol. 2012;302(4):C629–C641. [DOI] [PubMed] [Google Scholar]
- 74. Anderson EJ, Neufer PD. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H2O2 generation. Am J Physiol Cell Physiol. 2006;290(3):C844–C851. [DOI] [PubMed] [Google Scholar]
- 75. Pinho RA, Sepa-Kishi DM, Bikopoulos Get al. High-fat diet induces skeletal muscle oxidative stress in a fiber type-dependent manner in rats. Free Radic Biol Med. 2017;110:381–389. [DOI] [PubMed] [Google Scholar]
- 76. Picard M, Csukly K, Robillard M-Eet al. Resistance to Ca2+-induced opening of the permeability transition pore differs in mitochondria from glycolytic and oxidative muscles. Am J Physiol Regul Integr Comp Physiol. 2008;295(2):R659–R668. [DOI] [PubMed] [Google Scholar]
- 77. Baylor SM, Hollingworth S. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J Physiol. 2003;551(Pt 1):125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Girardi JP, Pereira L, Bakovic M. De novo synthesis of phospholipids is coupled with autophagosome formation. Med Hypotheses. 2011;77(6):1083–1087. [DOI] [PubMed] [Google Scholar]
- 79. Rockenfeller P, Koska M, Pietrocola Fet al. Phosphatidylethanolamine positively regulates autophagy and longevity. Cell Death Differ. 2015;22(3):499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem. 2005;280(48):40032–40040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, Vance JE. Phosphatidylethanolamine deficiency in mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem. 2013;288(6):4158–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Baker CD, Basu Ball W, Pryce EN, Gohil VM. Specific requirements of nonbilayer phospholipids in mitochondrial respiratory chain function and formation. Mol Biol Cell. 2016;27(14):2161–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Heden TD, Johnson JM, Ferrara PJet al. Mitochondrial PE potentiates respiratory enzymes to amplify skeletal muscle aerobic capacity. Sci Adv. 2019;5(9):eaax8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Starling AP, Dalton KA, East JM, Oliver S, Lee AG. Effects of phosphatidylethanolamines on the activity of the Ca(2+)-ATPase of sarcoplasmic reticulum. Biochem J. 1996;320(Pt 1):309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hunter GW, Negash S, Squier TC. Phosphatidylethanolamine modulates Ca-ATPase function and dynamics. Biochemistry. 1999;38(4):1356–1364. [DOI] [PubMed] [Google Scholar]
- 86. Fajardo VA, Mikhaeil JS, Leveille CF, Tupling AS, LeBlanc PJ. Elevated whole muscle phosphatidylcholine: phosphatidylethanolamine ratio coincides with reduced SERCA activity in murine overloaded plantaris muscles. Lipids Health Dis. 2018;17(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jackson MJ. Reactive oxygen species and redox-regulation of skeletal muscle adaptations to exercise. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2285–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev. 2011;91(4):1447–1531. [DOI] [PubMed] [Google Scholar]
- 89. Rickman OJ, Baple EL, Crosby AH. Lipid metabolic pathways converge in motor neuron degenerative diseases. Brain. 2020;143(4):1073–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Olsson MC, Krüger M, Meyer L-Het al. Fibre type-specific increase in passive muscle tension in spinal cord-injured subjects with spasticity. J Physiol. 2006;577(Pt 1):339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.