

Abstract
Canine RPGRIP1-cone-rod dystrophy (CRD), a model for human inherited retinal diseases (IRDs), was originally identified as autosomal recessive early-onset blindness. However, later studies revealed extensive phenotypic variability among RPGRIP1 mutants. This led to the identification of a homozygous MAP9 variant as a modifier associated with early-onset disease. Based on further phenotypic variation affecting cone photoreceptor function, we report mapping of L3 as an additional modifier locus, within a 4.1-Mb locus on canine chromosome 30. We establish the natural disease history of RPGRIP1-CRD based on up to 9-year long-term functional and structural retinal data from 58 dogs including 44 RPGRIP1 mutants grouped according to the modifier status. RPGRIP1 mutants affected by both MAP9 and L3 modifiers exhibited the most severe phenotypes with rapid disease progression. MAP9 alone was found to act as an overall accelerator of rod and cone diseases, while L3 had a cone-specific effect. Ultrastructural analysis of photoreceptors revealed varying degrees of rod and cone damage, while the connecting cilia appeared structurally preserved in all groups. We conclude that RPGRIP1-CRD is an oligogenic disease with at least three loci contributing to the pathogenesis. While the RPGRIP1 variant is required for developing the disease, MAP9 and L3 modifiers exacerbate the phenotype, individually and cumulatively. Oligogenic canine RPGRIP1-CRD illustrates the impact of multiple genetic modifiers on disease phenotype and thus has the potential to reveal new targets for broad-spectrum therapies for oligogenic or polygenic forms of human IRDs.
Introduction
Phenotypic variabilities have been widely observed among inherited retinal disease (IRD) patients who share the same disease-causing genetic variant (reviewed by Schneider et al. 1). The basis for the phenotypic variability in IRDs has not always been established while both genetic modifiers and environmental factors have been considered as a potential contributor. Examples of modifiers most of which utilized small families segregating variable phenotype, include an ABCA4 variant exacerbating PROM1-related retinal disease (2), PDZD7 variants modifying USH2A-Usher syndrome (3), AHI1 variants exacerbating CEP290-Leber congenital amaurosis (LCA) (4), CCDC28B variants exacerbating Bardet-Biedl syndrome phenotypes (5), and CNOT3 attenuating PRPF31-autosomal dominant retinitis pigmentosa (RP) phenotype (6), among others. Due to the often mild and conditional nature of genetic modifiers, it has been extremely challenging to tease out such factors among the human patient population (7). Hence, the examples of established genetic modifiers are limited, but it is likely that its role inherited diseases with variable disease expression or penetrance is underestimated.
Cone-rod dystrophies (CRDs) are a heterogeneous group of IRDs characterized by initial and primary effects on cone photoreceptors followed by rod degeneration, ultimately leading to blindness. Currently, there are at least 23 genes associated with CRDs that affect people worldwide (RetNet, accessed 19 February 2023, https://web.sph.uth.edu/RetNet/). Curtis and Barnett first identified a novel form of early-onset IRD (8) in miniature longhaired dachshunds (MLHDs), which was subsequently determined to be a form of CRD and termed cone-rod dystrophy 1 (cord1) (9). Using a research colony of MLHDs with moderate genetic homogeneity, Mellersh et al. conducted a whole-genome scan and identified a homozygous exonic 44 bp insertion in RPGRIP1 (retinitis pigmentosa GTPase regulator-interacting protein 1; RPGRIP1ins/ins) in all dogs clinically afflicted with early-onset blindness (10). Further examination of the broader pet MHLD population revealed extensive phenotypic heterogeneity and genotype–phenotype discordance, suggesting that additional genetic modifiers could influence the impact of the RPGRIP1 variant on disease expression (10,11). This is not surprising as RPGRIP1 variants have been associated with multiple human IRD clinical phenotypes (12) including autosomal recessive LCA (13–17), CRD (18,19) and RP (20,21). While the search for disease modifiers in human populations is often hindered by their genetic complexity (22,23), we utilized RPGRIP1 mutant dogs selected from the MLHD pet population, which due to its purebred nature represents less genetic heterogeneity than the general canine population, and far less than that of the human population. Mutant dogs were categorized as either early or delayed disease onset in a genome-wide association study (GWAS) (24). A homozygous 22 kb deletion subsequently was identified in MAP9, encompassing its tandem duplication, resulting in an alternative version of MAP9. Double homozygosity of both variants (RPGRIP1ins/insMAP9del/del) was found to correlate with early onset blindness (25). However, the molecular basis for the primary cone dysfunction of cord1 remained elusive, as neither RPGRIP1 nor MAP9 has established cone-specific expression or function.
To further investigate the mechanistic basis for the variable phenotypic effects observed among RPGRIP1 mutant dogs, we developed an outcrossed research colony of dogs harboring the RPGRIP1 variant (26). Interestingly, among the mutant puppies, we observed a bimodal cone electroretinography (ERG) function distribution at the first recording, in which dogs with diminished signals could be distinguished from those with normal signals (27). Importantly, this cone ERG phenotype did not segregate with the RPGRIP1 or MAP9 variant, indicating that the previously established digenic model was insufficient to fully account for the dynamic phenotype of cord1 (27). Herein, we provide the details on the mapping of an additional modifier associated with increased loss of cone function, and termed ‘L3’ as the third locus involved in cord1. L3 is defined as a 4.1-Mb SNP chip haplotype due to the extensive linkage disequilibrium and the complicated nature in pinpointing modifier variants with possibly modest molecular changes. Furthermore, we present a comprehensive longitudinal descriptive data of the oligogenic RPGRIP1-CRD dogs. Establishment of the genotype–phenotype correlation allows us to better understand the effect of each locus, leading to improved understanding and prediction of the disease course. Moreover, characterization of the oligogenic RPGRIP1-CRD dog model establishes a unique platform for teasing out the effect of multiple disease loci in the canine population whose limited genetic complexity compared to the human populations is advantageous for such endeavor (28). Canine RPGRIP1-CRD is the first large animal model of oligogenic IRD and has the potential to inform therapeutic targets to restore vision in both canine and human IRD patients.
Results
Mapping of L3 and lack of discernible pathogenic variant within the interval
To map the loci associated with the observed phenotypic differences in cone ERG in RPGRIP1ins/ins dogs of our colony, we carried out a GWAS in RPGRIP1ins/ins dogs with either diminished/absent or normal cone ERG signals in the first year of life. Mutant dogs with diminished cone ERG were termed ‘cases’, while mutants with normal cone ERG signals were termed ‘controls’. The abnormal cone ERG was observed in dogs of all possible genotypes of the known modifier MAP9. A total of 21 cases (MAP9 wild-type (n = 7), heterozygote (n = 10), and affected (n = 4)) and 19 controls (MAP9 wild-type (n = 7) and heterozygote (n = 12)) were genotyped on an SNP genotyping array (Illumina CanineHD BeadChip; 230 k evenly spaced SNP, Canfam 3.0 assembly; Illumina, Inc. San Diego, CA). After filtering 105 385 non-informative markers, 114 615 SNPs were used for GWAS mapping. The estimated lambda was 1.03, showing a low degree of stratification. The resulting Manhattan plot shows the top 254 most associated SNPs falling in the same CFA30 region (highest P-value 6.46, BICF2S23623902) and maintaining association after 100 000 permutations (Fig. 1A). Parametric linkage analysis was carried out using an incomplete penetrance and recessive inheritance model revealing an interval of LOD > 0 from position 1 to 13.983 cM (heterogeneity LOD (HLOD) > 6 between 8.722 and 8.994 cM) (Fig. 1B). Subsequent haplotype analysis confirmed a 4.1 Mb (CFA30: 9 059 844–13 159 098, CanFam3.1) interval of homozygosity in 18/21 (86%) of cases, but only in 1/19 (5%) of controls (Fig. 1C). The consequent hit on the linkage analysis and the fact that so many of the top hits fall within the same interval brought strong evidence to the validity of the mapping. We refer to this 4.1-Mb interval as L3, as it is the third locus associated with cord1.
Figure 1.
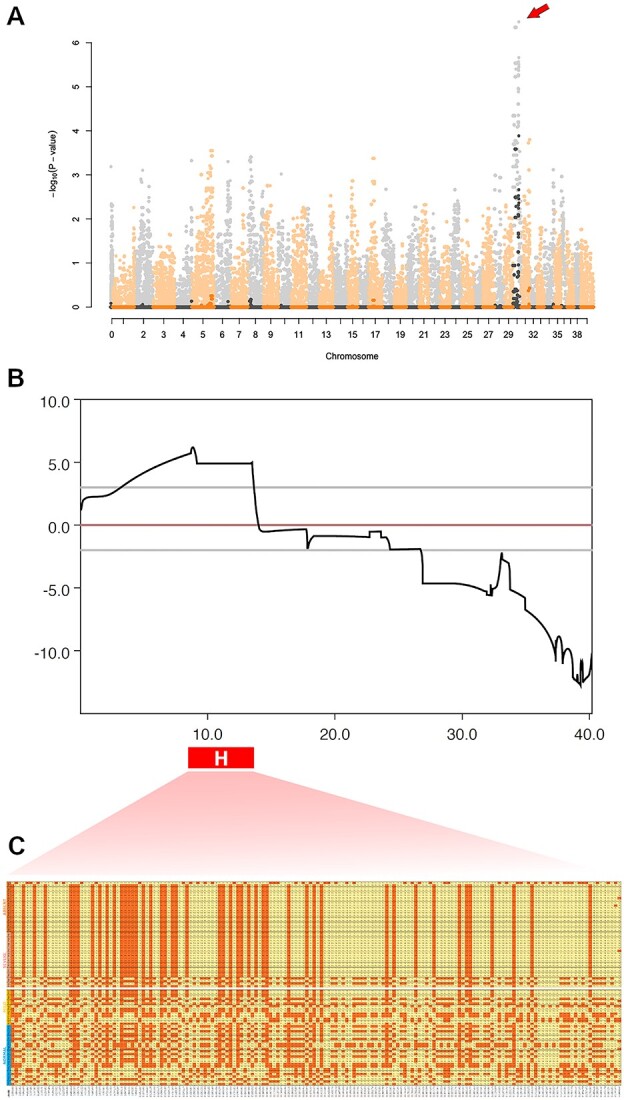
Mapping of L3. (A) Manhattan plot representing a genome-wide association study using RPGRIP1ins/ins dogs with and without significant cone ERG abnormality as cases (n = 21) and controls (n = 19), respectively. A significant peak of association is observed on canine chromosome 30 (−logP = 6.46, arrow). Negative log of P-values are shaded, while permuted ones (100 000 permutations) are plotted in their original colors (black and orange). (B) Parametric (recessive) linkage results for the genotyped severe/absent (cases) and mild/normal (controls) family members. Multiple sub-families ran in parallel. In red, bottom, the candidate haplotype is shown marked with ‘H’. Observe the overlap with the highest LOD score. (C) Haplotype analysis revealing a 4.1-Mb block of homozygosity that is enriched (86%) in the cases but rare (5%) in the controls.
Furthermore, we carried out whole genome sequencing (WGS) of dogs homozygous (n = 2) and heterozygous (n = 2) for L3. The variants that fell within the candidate interval on CFA30 (13 231 variants) were filtered based on selecting variants homozygous for the L3 candidate locus in the severe cases and heterozygous in the moderate, and then further selected using the following criteria: (1) expression in retina and (2) changes to protein coding. Retinal expression was informed by archived canine retinal RNA-seq data produced by our group, identifying 38 genes transcribed out of total 49 predicted genes located in L3. Ultimately, 12 coding variants were found to meet the above criteria (Supplementary Material, Table S1). The frequency of each of these variants was calculated in the WGS database of 816 dogs obtained from Dog Biomedical Variant Database Consortium (DBVDC) (29), identifying four rare variants with MAF < 0.1 in genes ZNF106, STARD9, and SERINC4 (Supplementary Material, Table S1). The MAF < 0.1 was a preliminary filtering implemented to avoid false negatives due to the fact that the database is not homogeneous—in the successive steps, the precise frequencies were calculated for each AA substitution.
Since the variants we focused on were all AA substitutions, the impact on the coded peptide were predicted to be ‘MODERATE’ for all 11 variants by SnpEff (as opposed to ‘HIGH’ impact like premature stop codons). Six different AA change impact predictors were used to estimate which variant could be the most damaging: Polyphen2, Provean, MutPred2, SIFT, FATHMM and VestScore. The output of the tools was not completely uniform and some, especially FATHMM and VestScore, have shown a higher tolerance score for the submitted variants. Nonetheless, the variants occurring on UBR1, WDR76, STARD9 and C15orf43 have shown a higher incidence of possibly damaging predictions (Supplementary Material, Table S1). The c.1300G > A; p.Ala434Thr substitution in SERINC4 is even more egregious, predicted to be damaging in four tools and the only one with a score higher than 0.8 in MutPred2 (5% of FDR). The SERINC4 variant is also remarkable because is the only variant that was both rare and predicted to be potentially deleterious.
Due to the availability of new canine references and of the Dog10K dataset (consisting of more than 2000 dogs, mostly purebred (https://kiddlabshare.med.umich.edu/dog10K/, accessed: 9 February 2023), we decided to re-map the dogs sequenced for the study along with all the unrelated genomes generated in our lab (70 in total), to the same reference the Dog10k dataset was mapped against (canFam4). We re-mapped the candidate interval using the NCBI remapping service and re-calculated the total frequencies of the coding variants detected, and filtered for exclusive variants against such larger dataset. Of the 11 coding variants detected in the previous iteration, six passed the MAF < 0.1 filter: four were under 0.05 (Supplementary Material, Table S1). One additional predicted coding variant was detected in the new-called dataset, in PLA2G4F (c.377C > T, p.Ser126Phe). Filtering against the whole dataset for variants exclusive to our dogs (using the same criteria, homozygous in the severe and heterozygous in the moderate) we failed to detect an actual exclusive variant.
While identification of the variant representing L3 requires further comprehensive in vitro and ex vivo functional validation, the current study aimed to characterize the effect of L3 on the retinal phenotype. As such, we grouped the dogs according to the status of each of the three cord1-associated loci, RPGRIP1, MAP9 and L3, and used the following titles to depict the affected loci of each group (Table 1). ‘Rpgrip1/Map9/L3’ represents triple homozygotes (RPGRIP1ins/insMAP9del/del L3aff/aff), ‘Rpgrip1/Map9’ and ‘Rpgrip1/L3’ each mark RPGRIP1 mutants with only one of the modifiers (RPGRIP1ins/insMAP9deldel and RPGRIP1ins/ins L3aff/aff, respectively), and ‘Rpgrip1’ signifies RPGRIP1 mutants unaffected by either of the modifiers in a homozygous state (RPGRIP1ins/ins).
Table 1.
Genetic subgroups and their contribution to structural and functional database
| Groups | # of dogs | Locus homozygosity | Structural analysis | Functional analysis | |||||
|---|---|---|---|---|---|---|---|---|---|
| RPGRIP1 insertion | MAP9 deletion | L3 haplotype | Fundus score | OCT | LM & TEM | ERG | Visual navigation | ||
| Rpgrip1/Map9/L3 | 5 | Y | Y | Y | 5 | 5 | 1 | 5 | 1 |
| Rpgrip1/Map9 | 3 | Y | Y | N | 1 | 1 | 1 | 3 | 0 |
| Rpgrip1/L3 | 16 | Y | N | Y | 12 | 10 | 1 | 16 | 3 |
| Rpgrip1 | 20 | Y | N | N | 11 | 10 | 0 | 20 | 2 |
| Control | 14 | N |
Y (2) N (12) |
N | 3 | 3 | 0 | 14 | 1 |
| Total # of dogs | 58 | - | - | - | 32 | 29 | 3 | 57 | 7 |
OCT, Optical Coherence Tomography; LM, Light Microscopy; TEM, Transmission Electron Microscopy; ERG, Electroretinography. Y, homozygous with the indicated variation; N, heterozygous with the indicated variant or wild-type homozygous.
Progression of ophthalmoscopic changes in RPGRIP1 mutants is impacted by modifiers
Multiple generations of animals resulting from outcross, backcross and intercross breedings (Supplementary Material, Fig. S1) were studied over a 12-year period. The ages of the dogs at the endpoint of data collection ranged from 1 month to 8½ years. The characteristics of the fundus phenotype in the RPGRIP1ins/ins dogs included in the current study was consistent with our previous report (27). Retinal atrophy started in the periphery and extended centrally over time in the tapetal (Fig. 2A) and non-tapetal (Supplementary Material, Fig. S2) regions. Based on the extent of retinal atrophy (e.g. hypo- and hyper-reflectivity) in the tapetal fundus, we scored the disease from one (incipient) through five (end-stage) (Supplementary Material, Table S2 and Fig. 2). Analysis of the scores revealed rapid progression of fundus changes in the ‘Rpgrip1/Map9/L3’ group affected by all three loci, reaching end-stage as early as 3 years of age in 67% of the dogs (Fig. 2B). In contrast, groups ‘Rpgrip1/Map9’ and ‘Rpgrip1/L3’, which were affected by only one of the modifiers, had slower progression rates (Fig. 2B) and did not reach advanced or end-stage even by 8 years of age. A comparison of Rpgrip1/Map9 and Rpgrip1/L3 mutants revealed that initial pathologic changes manifested sooner in the former, but that progression in subsequent years was more rapid in the latter (Fig. 2B). Fundus features in the ‘Rpgrip1’ group with no modifier effects remained between the incipient and early stage throughout the 8½-year evaluation period (Fig. 2B).
Figure 2.

Illustration of the tapetal fundus changes of RPGRIP1ins/ins dogs showing progression of characteristic retinal atrophy with advancing disease stage. (A0) The earliest indication of fundus abnormality characterized by subtle dark discoloration in the far periphery of the tapetal fundus may be indistinguishable from normal fundus. (A1) The early distinct change at Stage one (‘incipient’) is mild mottling dark discoloration of the tapetal fundus developing at the far peripheral tapetum along the superior non-tapetal border (arrow). (A2) Stage two (‘early’) is characterized by inferior expansion of dark mottled lesions, reaching up to superior 1/2 of the tapetal fundus (arrows). (A3) With Stage three (‘mid’), dark discoloration expands to ~2/3 of the tapetal fundus (arrows) and mild tapetal hyper-reflectivity becomes increasingly evident (asterisk; at this stage, changes in reflectivity are not easily photographed). (A4) By Stage four (‘advanced’), the leading edge (arrows) of the atrophic lesions appearing both hypo- and hyper-reflective (asterisk) further extends inferiorly, encompassing nearly all the tapetal fundus. (A5) At Stage five (‘end-stage’), the changes are severe and generalized, mimicking late stages of other forms of retinal degeneration (with vascular attenuation, generalized hyper-reflectivity and optic nerve atrophy). (B) Aggregated longitudinal fundus scoring representing all evaluated dogs per genotype group. The result score per time-point is represented as the mode score for all the animals evaluated in that timeframe. The percentage on each bar represents the proportion of animals that contributed to the mode score.
Oligogenic RPGRIP1-CRD affected dogs undergo uneven outer nuclear layer (ONL) thinning
Longitudinal structural changes of the retina were documented by optical coherence tomography (OCT) and analyzed in a representative dog per genotype. Consistent with the ophthalmoscopic observations, OCT revealed that the Rpgrip1/Map9/L3 dog exhibited the fastest rate of progression (Fig. 3A), with slow and consistent ONL thinning until 2 years of age, followed by rapid degeneration by the third year of life. In this animal, ONL was better preserved centrally with a gradual thinning over time, whereas peripheral thinning rapidly progressed at 3 years of age. Notably, there was a significant difference between the Rpgrip1/Map9 and Rpgrip1/L3 animal analyzed, with the former showing earlier onset and more rapid progression over time than the latter (Fig. 3B and C). In addition, while the Rpgrip1/Map9 dog exhibited marked ONL thinning in the peripheral retina between 5 and 6 years of age, the Rpgrip1/L3 dog exhibited very slow yet steady ONL thinning across the vertical meridian, until the first peripheral changes were observed at age 6 years (Fig. 3C). Interestingly, the difference in ONL thinning between Rpgrip1/Map9 and Rpgrip1/L3 contrasts with the aggregated ophthalmoscopic findings above. This may be attributed to the small sample size of Rpgrip1/Map9 (n = 1) as well as differences in sensitivity between the quantitative and qualitative data. Lastly, the Rpgrip1 animal unaffected by modifiers showed only mild ONL thinning starting around 7 years of age (Fig. 3D). In all cases, retinal thinning was not uniform across the vertical meridian, but initiated in the periphery, and progressively extended towards the central retina. All dogs affected with one or two modifiers had a thinner ONL by the time of the first scan at 9 months of age compared to Rpgrip1 at 3 years of age or to unaffected control.
Figure 3.

Longitudinal evaluation of ONL thickness and retinal histology in canine RPGRIP1 mutants of different modifier combinations. Graphs of ONL thickness (μm) measured on OCT along the superior–inferior meridians show the most rapid and severe progression of ONL thinning in the triple homozygote (A). The RPGRIP1ins/ins mutants with one modifier are less affected, although ONL thinning starts sooner in the animal with the MAP9 modifier (B) compared to the animal with the L3 modifier (C). A related RPGRIP1ins/ins animal without modifiers (D) and a non-RPGRIP1ins/ins animal (E) are illustrated for comparison. Fundus photographs (A1, B1, C1) and photomicrographs of toluidine blue-stained semi-thin sections (A2, B2, C2) from age-matched (6 years) canine mutants are shown. The arrows in the fundus photographs indicate the area centralis (a.k.a. canine macula) from which the semi-thin sections shown in the panel below had been obtained. Histologically, the triple homozygote (Rpgrip1/Map9/L3) had the most advanced ONL atrophy (A1, A2: R62). The animal affected with the MAP9 modifier (Rpgrip1/Map9), had an intermediated ONL phenotype with moderate ONL reduction (B1, B2: R67). The animal affected with the L3 (Rpgrip1/L3) modifier had near-normal level of ONL preservation (C1, C2: R63). Scale bar, 50 μm.
Connecting cilia microstructure is preserved in all RPGRIP1 mutants regardless of modifiers
Using immunohistochemistry (IHC), we previously examined the connecting cilia of RPGRIP1 mutant canine retinas unaffected by MAP9 (RPGRIP1ins/insMAP9+/+(WT)) and of unknown L3 haplotype (27). Of the retinas studied, cilia length was shorter in the cone ERG-absent animal compared to cone ERG-normal. Recent genotyping of these dogs confirmed the affected L3 haplotype (L3aff/aff) of the cone ERG-absent dog, while the cone ERG-normal dog was unaffected. However, the effect of the MAP9 modifier on cilia structure remains unknown, as does the ultrastructure of the connecting cilia in any RPGRIP1 mutant canine retina.
To study the effect of modifiers on retinal cell morphology in the RPGRIP1 mutants, age-matched retinas of different modifier status were examined. Histologic findings were in accordance with those seen by OCT. At 6 years of age, there was ONL thinning down to a single row of nuclei in the Rpgrip1/Map9/L3 retina (Fig. 3A1, A2). In contrast, the Rpgrip1/Map9 retina that exhibited moderate ONL attenuation was reduced to five to six rows of nuclei (Fig. 3B1, B2), while the Rpgrip1/L3 retina had an ONL thickness within normal range at 8–9 nuclei (Fig. 3C1, C2). The preserved thickness of the Rpgrip1/L3 retina based on histology was consistent with the in vivo OCT findings above and suggests that the dark discoloration observed qualitatively by ophthalmoscopy (Fig. 3C1) does not correspond with retinal atrophy but is more likely due to complicating factors, such as outer segment disorganization.
Ultrastructural analysis of the same set of retinas using transmission electron microscopy (TEM) revealed extensive alterations in the outer retina of Rpgrip1/Map9/L3 eye, with a complete lack of outer and inner segments, which were replaced by gliotic structures (Fig. 4A). The Rpgrip1/Map9 retina had extensive outer segment loss, although the inner segment structure was better preserved (Fig. 4B). In the Rpgrip1/L3 retina, both outer and inner segments were preserved (Fig. 4C). Despite the extensive outer retinal destruction in some of these retinas, TEM images suggested that the microstructure of the connecting cilia was preserved in all genotypes along and across the cilia (Fig. 4A2, B2–B4, C2) and the 9 + 0 arrangement of doublet microtubules could be observed consistent with primary cilium (Fig. 4B4).
Figure 4.

Photoreceptor ultrastructure of RPGRIP1 mutant canine retinas with MAP9 and/or L3 modifiers. Transmission electron photomicrographs illustrating photoreceptor-RPE interphase (A1, B1, C1) and connecting cilium structure (A2, B2–B4, C2) taken from age-matched (6 years) canine mutants. (A) The triple homozygote exhibited extreme photoreceptor damage lacking outer segments (OS) and normal inner segment (IS) structure (A1, red arrow). (B) The dog affected with the MAP9 modifier retained some IS structure of rods (B1, red arrows) and cones (B3, purple arrow); both rods and cones frequently lacked normal OS. (C) The animal affected with the L3 modifier largely retained both IS and OS (C1, arrowheads). Evaluation of cilia structures did not reveal a morphologic abnormality in either group of animals (A2, B2–B4, C2). Normal connecting cilia structure was seen in the transverse (B3 insert) and longitudinal (C2 insert) sections. Scale bar, 1 μm.
Temporal aspects of rod and cone ERG functional abnormalities
Evaluation of rod-mediated ERG responses revealed moderate differences in levels and trends according to modifier status (Fig. 5A). Specifically, rod responses were reduced in the Rpgrip1/Map9/L3 group beginning in the first year and continued to decline over time. In the groups affected by one modifier, rod ERG b-wave amplitudes were lower in Rpgrip1/Map9 than in Rpgrip1/L3, the levels of which were comparable to those of Rpgrip1 until 3 years of age. After the third year, the Rpgrip1/Map9 group had a distinct drop in rod response, while Rpgrip1/L3 had a slower decline with better preservation of rod function. The Rpgrip1 group also had generally lower scotopic responses compared to controls, although without significant decrease over time. The mixed rod and cone responses followed similar trends as rod-specific responses (Fig. 5C).
Figure 5.

Longitudinal ERG across all genotype groups and traces from representative animals. Responses to full-field single flash ERGs represented by rod (A) and cone 1 Hz (B) b-wave amplitudes (μV) over time are displayed per age and group. Error bars represent SEM. (C) ERG traces of representative animals of each genotype groups. OD, right eye; OS, left eye.
Cone-mediated ERG responses were significantly attenuated to a variable extent by 6 months of age in all groups of RPGRIP1 mutants with or without modifier(s) (Fig. 5B). Cone responses were undetectable in some Rpgrip1/Map9/L3 dogs at the initial ERG recordings, as early as 6 weeks of age, while others in this group had some detectable albeit minimal cone response, which was rapidly lost during the first year of life. Both Rpgrip1/Map9 and Rpgrip1/L3 groups had significantly reduced cone responses from the earliest ERG recordings, which declined slowly over the 6–8 years of lifetime where data was available. As described above, L3 was mapped based on distinct cone ERG among RPGRIP1 mutants independent of MAP9, suggesting that the Rpgrip1/L3 would more likely show an exaggerated cone phenotype. However, cone levels and ERG trends were surprisingly similar between Rpgrip1/L3 and Rpgrip1/Map9 groups. Still, when analyzed in conjunction with the rod ERG data, the Rpgrip1/L3 group had the highest ratio of rod/cone b-wave amplitude. For example, at 2 years of age, the rod/cone b-wave amplitude ratio was 24 for Rpgrip1/Map9 dogs compared to 119 for Rpgrip1/L3, supporting an effect by L3 on cone function. Cone ERG in the Rpgrip1 group was moderately reduced compared to controls but was more preserved than mutants affected by additional modifier(s), and with only marginal decline up to 8 years of age.
Overall, cone ERG was more attenuated than rod ERG in all RPGRIP1 mutant groups with variable levels of cone functional preservation and trends of progression. Furthermore, the degree of rod functional preservation relative to cone functional loss varied depending on the modifier status.
Variable effects on vision including reasonable photopic navigation despite diminished cone ERG function
A total of seven available animals representing different genotype groups (except Rpgrip1/Map9) were enrolled in longitudinal vision testing to assess their ability to navigate an obstacle course under different light intensities. The progression of scotopic and photopic vision-guided navigation results generally corresponded with the trends observed with ERGs (Supplementary Material, Fig. S3). The first testing of the Rpgrip1/Map9/L3 group was performed at 4 years of age. By this time, both scotopic and photopic vision was impaired as indicated by increased transit times. The Rpgrip1/L3 and Rpgrip1 groups had within normal scotopic and photopic navigation up to 1 year of age. Thereafter, both groups showed a gradual and persistent decline in photopic and, to a lesser degree, scotopic vision, with the Rpgrip1/L3 group marginally more affected, especially under photopic condition. Notably, despite detectable but significantly reduced cone ERG signals in the Rpgrip1/L3 group and to a lesser extent in the Rpgrip1 group across the 6–8 year age range, all these animals retained a reasonable ability to navigate under photopic conditions compared to dogs with achromatopsia in which cone ERG is completely lost (30).
Discussion
We describe the long-term natural history of CRD in a colony of dogs affected with a variant in RPGRIP1, in the presence or absence of two recently identified modifiers, MAP9 (24,25) and L3, whose mapping is described herein. In the original research colony where the RPGRIP1 variant alone was found to completely correspond with early-onset CRD (10), all dogs were affected by the MAP9 variant (MAP9del/del) as confirmed by later typing of the founder dogs (unpublished). Meanwhile, the current outcrossed and expanded canine colony exhibit variable phenotypic trends according to the additional loci involved. Specifically, we found that dogs affected with all three disease-associated loci (Rpgrip1/Map9/L3) consistently resulted in more dramatically affected phenotypes. A lesser effect was observed in mutants with only one of the modifiers (Rpgrip1/Map9 or Rpgrip1/L3), indicating that each modifier can individually exacerbate the phenotype, while together they exert a cumulative effect.
In contrast to OCT, which requires anesthesia in animals, or histology, which is terminal, ophthalmoscopy is a simple, non-invasive method to assess retinal structure in live dogs. Using this approach, characteristic fundus changes consistent with our previous report (27) were observed in this current extended follow-up. Notably, the pattern of ophthalmoscopic changes were comparable across the RPGRIP1 mutant groups. However, the onset of ophthalmoscopic changes and the rate of progression varied and was modifier-dependent, with those affected by both modifiers showing the most severe course. The presence of a single modifier also exacerbated the disease albeit to a lesser degree. At 2–5 years of age, RPGRIP1 mutants affected by L3 exhibited worse ophthalmoscopic disease than those affected by MAP9. However, as discussed below, OCT and histologic findings demonstrated that the MAP9 modifier resulted in more severe disease, highlighting the potential limitations of ophthalmoscopic evaluation whose en face and qualitative features are subjective. Our findings indicate that the RPGRIP1 variant by itself gives rise to the characteristic ophthalmoscopic changes in affected animals at an older age, while the two modifiers independently as well as cumulatively accelerate the pathologic changes.
Although ophthalmoscopy allowed qualitative evaluation of retinal phenotype by subjective scoring, OCT scans with ONL measurement provided objective quantification of photoreceptor loss. Longitudinal evaluation of ONL thickness revealed progressive outer retinal atrophy in RPGRIP1 mutants affected with one or both modifiers. As with the ophthalmoscopic assessment, there was a modifier-dependent difference in the onset and the rate of progression. ONL thinning was most severe and progressed rapidly in the animal affected with both modifiers, while the presence of a single modifier led to mild to moderate disease progression, with MAP9 accelerating the OCT phenotype more than L3. This is in contrast with the ophthalmoscopic phenotype, which appeared to be accelerated by modifier L3 more than by MAP9. This discrepancy may be attributed to intra-group variation. It is also possible that the ophthalmoscopically observed dark discoloration at the initial stages does not directly translate to ONL thickness changes, but is instead associated with disorganization of the outer segments. However, obvious outer segment disorganization was not observed in the TEM images studied in the Rpgrip1/L3 animal, although observation was limited to the area centralis of each animal.
Retinal samples from the same cohort of age-matched RPGRIP1 mutants in the longitudinal OCT study were examined histologically. TEM analysis of the outer retina revealed extreme, severe, and mild photoreceptor destruction in mutants affected with both modifiers, MAP9 only and L3 only, respectively. While the ERG reduction in these animals was distinctly cone-led, functional abnormalities in rods were observed as well, as morphological deterioration involved both rods and cones in the retinas with extreme and severe phenotypes. Similarly, differences in morphologic changes between rods and cones were not evident in the retina with mild phenotype. Importantly, despite varying degrees of outer and inner segment destruction, the ultrastructure of the photoreceptor connecting cilia appeared to be preserved in all RPGRIP1 mutant animals. Both RPGRIP1 and MAP9 localize in the connecting cilia (27,31) with proposed roles in protein trafficking (32). Our finding indicates that variants in RPGRIP1, MAP9 or the gene represented by L3 do not affect ciliogenesis or structural maintenance, but rather, may affect trafficking and signaling of key proteins by altering their distribution within photoreceptors, particularly in cones.
In the current study, we used two methods to assess retinal function, ERG and vision-guided navigation. While both methods captured functional decline by the advanced disease stage, in the early disease stage, vision-guided navigation was less reliable due to lack of sensitivity. In turn, cone-mediated ERG was the most sensitive and informative measure, identifying severe reduction in RPGRIP1 mutant animals of all modifier status from the earliest time point of observation. Interestingly, and consistent with our previous reports (26,27), in the presence of very low but detectable trace cone ERG, photopic vision-guided navigation was not as dramatically affected as in canine achromatopsia (30). However, with further loss of cone ERG amplitudes, marked delay in photopic navigation became evident, which had not been captured in our previous work (27). Rod ERG was much less affected than cone ERG, showing only mild to moderate reduction across the RPGRIP1 mutant groups over time. Of them, rod ERG was generally worse in the two groups with MAP9 involvement. Scotopic navigation seemed to decline in concordance with photopic navigation, despite distinct cone-led deterioration based on ERG. Of note, while the modest decline in navigation is quantifiable using our standardized method (33), the degree of vision loss may be insufficient to affect the animals’ ability to perform basic visual tasks, and thus may not be considered clinically relevant and may even go unnoticed in companion animals.
In a previous study, we further evaluated the role of the RPGRIP1 variant (44 bp insertion) as the primary cause of cord1 based on the apparent lack of disease in some RPGRIP1 mutant animals (26). However, the duration of phenotypic follow-up was limited in that report, with the age at final examination ranging from 2½ to 4 years, leaving open the possibility of late-onset disease. In the current long-term study, we found that the only recognizable phenotype for most of the lifespan in animals with no modifiers (Rpgrip1) was reduced cone ERG. Some deviation in vision, both photopic and scotopic, was detectable as early as 2 years in this group, although it was unlikely to be clinically relevant. Retinal structure as determined by OCT in this group remained normal up to 7 years of age at which time some initial ONL thinning occurred. Age-related physiologic retinal thinning is a normal process (34,35), but previous canine studies found no significant differences between animals at 7 and 11 years of age (36). Therefore, ONL thinning at age 7–8 years in the Rpgrip1 animal is considered to represent late-onset retinal degeneration rather than physiologic thinning.
Taken together, our findings indicate that the RPGRIP1 variant itself is indeed sufficient to cause pathology, although the phenotype is limited and may go unnoticed unless detailed cone ERG studies are performed. However, all RPGRIP1 mutant animals regardless of modifier status have varying degree of cone ERG reduction, even as early as 6 weeks of age. Thus, even by itself, the RPGRIP1 variant appears to disrupt retinal function, particularly cone-mediated physiology. As such, these findings imply that gene augmentation therapy of RPGRIP1 alone could be therapeutic in RPGRIP1 mutant animals regardless of modifier status, as was previously shown (37,38) and that single augmentation of functional MAP9 or the gene represented by L3 are unlikely to completely rescue the disease or its progression. While the L3 modifier has cone-specific features, and thus may have less overall structural impact on the ONL in the rod-dominated canine retina, the MAP9 modifier seems to affect both rods and cones, exacerbating the overall pathology and accelerating scotopic ERG decline and ONL thinning.
Of interest, there were two dogs that were affected just with the MAP9 modifier (R22 and R26, Supplementary Material, Fig. S1) (26). Their ERG and ophthalmoscopic findings were within normal parameters up to 1 year of age, which was the study endpoint for these ‘control’ dogs. This was before the discovery of the MAP9 variant. Unfortunately, no definitive conclusion could be drawn from this small group of n = 2 with short follow-up. In the current study group, there were no animals homozygous for the L3 haplotype but unaffected by the primary RPGRIP1 variant or the MAP9 modifier. It is of great importance to study the long-term phenotype of dogs affected just with the MAP9 variant or L3 haplotype. It can certainly inform on and tease out the individual functional effect of each modifier and will be the scope of future studies.
Following the mapping of L3, RNA-seq and WGS were utilized to identify 12 missense variants in genes within the L3 interval and that are expressed in the retina. Subsequent analyses based on variant screening in a large canine WGS database as well as impact prediction pointed to six variants in SERINC4, ZNF106, STARD9, UBR1 and C15orf43 as possible candidates. None of these genes have previously been indicated in IRDs. While we examined rare coding variants that are more readily validated, this is not to exclude variants in regulatory regions such as promoters and enhances. As L3 is a modifier locus, it is possible that the L3 molecular change alone has only modest effects and does not cause a phenotype unless together with the RPGRIP1 variant. Therefore, future efforts to pinpoint the molecular changes associated with L3 can pose challenges even with high-throughput sequencing approach such as low-pass WGS. Further studies are warranted to functionally validate the exact genetic variant representing L3, to examine the association between RPGRIP1, MAP9 and the third protein at the molecular level, and to determine the role of each protein in photoreceptor function, particularly in connecting cilia physiology. To date, variants in hundreds of genes have been associated with human IRDs, which are typically considered monogenic diseases. However, phenotypic variabilities even among patients harboring the same causal variant are increasingly recognized (18,39). While the presence of disease modifiers, either genetic or environmental, have been suggested in such cases, the extensive genetic and phenotypic heterogeneities in human patient populations have hindered the identification of such modifiers. In contrast, certain canine subpopulations, such as research colonies or registered canine breeds, exhibit moderate to high genomic uniformity, thus facilitating identification of genetic disease modifiers as in another example of phenotypically variability in canine RPGR-retinal degeneration with a shared RPGRorf15 variant (40). The oligogenic RPGRIP1-CRD canine model has indeed been an ideal sample group for isolating multiple genetic modifiers and for studying their effect on the phenotype under a controlled research environment (25). Establishment of modifiers and their molecular mechanisms has the potential for developing new target or agents for broad-spectrum therapy in multiple forms of IRDs resulting from genetic variants in different genes (39).
Materials and Methods
Canine research colony
The pedigree for the study group consisting of 58 research dogs (32 males, 26 females) bred and maintained over 12 years at the Retinal Disease Studies Facility, Kennett Square, PA, is presented in Supplementary Material, Fig. S1. The research colony primarily originates from three purebred MLHDs and one mongrel dog founders. All the procedures were conducted in full compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and with Institutional Animal Care and Use Committee (IACUC, Protocol# 804956) approval. The animals were typed for the RPGRIP1 and MAP9 genetic variants as well as the genetic markers associated with L3 (see below). The origin of this research colony has been described previously (26,27). The animals were categorized into four groups according to the modifier genotypes (Table 1 and Supplementary Material, Fig. S1). One group was used as a control and included those animals that were unaffected (i.e. heterozygous or wild-type) with RPGRIP1, and having variable MAP9 and L3 genotypes (Table 1 and Supplementary Material, Fig. S1). This included one MAP9del/+ and two MAP9+/+ in the fundus score and OCT control dataset, two MAP9del/del, eight MAP9del/+ and four MAP9+/+ in the ERG control group as well as one MAP9+/+ in the control group of the visual behavior studies.
Mapping and survey of modifier locus L3
DNA samples from 42 RPGRIP1ins/ins dogs were genotyped on the Illumina CanineHD BeadChip (230 k SNPchip; Illumina, Inc. San Diego, CA). Genotyping data were pre-processed using Plink v1.9 (41) and analyzed using the GenABEL package (https://cran.r-project.org/src/contrib/Archive/GenABEL/, v 1.8) developed for the R Studio integrated environment (https://rstudio.com/, v 0.99.903) (42). Parametric linkage was carried out using MERLIN (42,43), using a 90% or penetrance in the parameters file. Haplotype phasing was carried out using BEAGLE 3.0 (44). The mapped L3 interval was examined using archived RNA-seq data from selected RPGRIP1 mutants with or without cone ERG loss. RNA-seq reads were aligned to the canine references (CanFam3.1) using STAR v2.7.0 and genes transcribed in the retina were visually identified. Variants unique to the cone ERG phenotype were called using the GATK pipeline (v2.4.9) (45); minimum coverage used was ignored to avoid any chance of false negatives. The biological impact of the variant was assessed using SnpEff (46). In addition, any changes in the exon structure were examined using the Integrative Genome Browser (IGV), using the sashimi plot function of IGV as a visual aid (47).
Four DNA fragment libraries were prepared from samples derived from two severe and two moderate cases for WGS. Libraries of 300 bp insert size were prepared, llumina HiSeq2500 paired-end reads (2 × 100 bp, one lane per sample); Casava was used for the fastq preparation (https://bioweb.pasteur.fr/packages/pack@casava@1.8.2, v 1.8.2). A total of ~20 billion reads (100 bp paired-end reads) were collected, corresponding to an approximate average of 33.4×). The reads were aligned using Burrows-Wheeler Aligner (BWA) (48) against CanFam3.1 and canFam4. The SAM file, which was generated by BWA, was then converted to a BAM file and the reads were sorted by chromosomes using Samtools v 1.1 (49). The PCR duplicates were marked using Picard tools (http://sourceforge.net/projects/picard/). WGS variant calling was also carried out with GATK and preliminary variant impact was predicted with SnpEff (46). AA change impact was predicted with Polyphen2, Provean, MutPred2, SIFT, FATHMM and VestScore. Data extraction of variants from the Dog Biomedical Variant Database Consortium (DBVDC) (29) and Dog10K dataset were done using the software BCFtools (https://samtools.github.io/bcftools/, v 1.11), and MAF was calculated using PLINK. Delly2 (50) was used to detect structural variants falling on coding regions.
Genotyping of variants and loci associated with cord1
All the dogs enrolled in the study were typed for the known RPGRIP1 and MAP9 genetic variants as well as for haplotypes representing the L3 interval. The 44 bp insertion in RPGRIP1 was typed by a modified PCR-gel electrophoresis protocol based on previous reports (10,27). The ~22 kb deletion in MAP9 was typed by a modified duplex PCR–gel electrophoresis protocol based on previous reports (25,27). The status of L3 was determined by typing of four genetic variants (CanFam3.1 CFA30: 10 426 046C > T, 10 426 138T > C, 11 938 924A > G and 11 939 061T > C;) to establish the haplotype spanning the 4.1-Mb L3 interval.
Ophthalmic examination
The animals underwent periodic routine ophthalmic examinations, and wide-field color fundus photographs were acquired using a RetCam® Shuttle retinal camera (Natus Medical Inc. Pleasanton, CA) with a 130-degree field of view lens. Fundus images from 32 animals over multiple time-points (age 1–8½ years) were scored from one to five (Fig. 2, Supplementary Material, Table S2) based on the severity of the retinal degeneration in both eyes. Three investigators blinded to sample identity each analyzed 122 pair of randomized images.
Spectral-domain OCT
In vivo imaging was carried out under general anesthesia using Spectralis® HRA + sdOCT (Spectralis, Heidelberg Engineering Inc., Heidelberg, Germany) as previously described (27). ONL thickness was measured in representative animals using single, high-resolution 30° longitudinal b-scans from the optic nerve head to the periphery in both superior and inferior meridians. Measurements were acquired using the software (HEYEX) provided in the Spectralis unit and segmented manually every 0.5 mm from the optic nerve head. ONL measurements acquired from both eyes were averaged and used to represent linear graphs across the vertical meridian.
Light and TEM
Within 3 min of humane euthanasia followed by enucleation, retinal samples with the adjacent choroid and sclera were collected from the superotemporal fundus, centered at the area centralis. Samples were fixed (2 h, 4°C) in a mixture of 1% paraformaldehyde and 2.5% glutaraldehyde in 0.2 M phosphate buffer (pH 7.4). Samples were then washed, postfixed in 2% osmium tetroxide (2 h, room temperature) and embedded in Epon 812. Semi-thin sections were cut from each tissue block, stained with toluidine blue, and examined to determine the regions to be captured by TEM. Ultrathin sections were contrasted with uranyl acetate and lead citrate and then examined using TEM (Tecnai™ 12 Spirit G2 BioTwin, Field Electron and Ion, Hillsboro, OR).
ERG
Retinal responses elicited under scotopic and photopic stimuli were recorded as previously described (51) in 57 animals at ages ranging from 1 month to 8½ years. Amplitudes of recorded scotopic b-wave, mixed rod and cone b-wave, photopic one Hz b-wave, and the trough-to-peak amplitudes of the 29.4 Hz flicker were measured in the ERG software (Espion V6, Diagnosys, Lowel, MA). Responses from both eyes were averaged per time-point and longitudinal descriptive analysis (mean ± SEM) was performed across groups using GraphPad (GraphPad Software, San Diego, CA).
Visually guided obstacle course navigation
Vision-guided navigation was tested in an obstacle avoidance course as previously described (26,33). The test results from scotopic, mesopic and photopic lightings at intensities, 0.003, 0.2 and 646 lux, translating to rod, mixed rod-cone and cone vision, respectively. Data from three wild-type animals that had been previously published were used as control (52).
Supplementary Material
Acknowledgements
The authors thank Drs. Valerie Dufour, Jose Manuel Guzman, Simone Iwabe, Sara Jastrebski, and Kendra McDaid for assistance in collecting or analyzing part of the clinical data. We also thank staff members of the RDSF including Terry Jordan, Jacqueline Wivel, Nanci Newsom, Mayra Quiroz, among many others, for anesthesia and animal care support, and Dr Leslie King for critical review of the manuscript. We thank the Dog Biomedical Variant Database Consortium for sharing whole-genome sequencing data from control dogs.
Conflict of Interest statement. All authors declare no conflict of interest.
Contributor Information
Ana Ripolles-Garcia, Division of Experimental Retinal Therapies, Department of Clinical Sciences & Advanced Medicine, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Leonardo Murgiano, Division of Experimental Retinal Therapies, Department of Clinical Sciences & Advanced Medicine, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Natalia Ziolkowska, Department of Histology and Embryology, Faculty of Veterinary Medicine, University of Warmia and Mazury, Olsztyn 10-719, Poland.
Felipe Pompeo Marinho, Division of Experimental Retinal Therapies, Department of Clinical Sciences & Advanced Medicine, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Karolina Roszak, Division of Experimental Retinal Therapies, Department of Clinical Sciences & Advanced Medicine, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Sommer Iffrig, Division of Experimental Retinal Therapies, Department of Clinical Sciences & Advanced Medicine, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Gustavo D Aguirre, Division of Experimental Retinal Therapies, Department of Clinical Sciences & Advanced Medicine, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Keiko Miyadera, Division of Experimental Retinal Therapies, Department of Clinical Sciences & Advanced Medicine, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Data availability statement
The RNA-seq and whole-genome sequencing and SNP genotyping datasets generated and analysed as part of this study are available in the Dryad archive (https://doi.org/10.5061/dryad.wdbrv15rj).
Funding
Margaret Q. Landenberger Research Foundation (K.M.), NEI/NIH grant EY-006855 (G.D.A., K.M.), Foundation Fighting Blindness (G.D.A.), the Van Sloun Foundation for Canine Genetic Research (G.D.A., L.M.), and the Sanford and Susan Greenberg End Blindness Outstanding Achievement Prize (G.D.A.).
References
- 1. Schneider, N., Sundaresan, Y., Gopalakrishnan, P., Beryozkin, A., Hanany, M., Levanon, E.Y., Banin, E., Ben-Aroya, S. and Sharon, D. (2022) Inherited retinal diseases: linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res., 89, 101029. [DOI] [PubMed] [Google Scholar]
- 2. Lee, W., Paavo, M., Zernant, J., Stong, N., Laurente, Z., Bearelly, S., Nagasaki, T., Tsang, S.H., Goldstein, D.B. and Allikmets, R. (2019) Modification of the PROM1 disease phenotype by a mutation in ABCA4. Ophthalmic Genet., 40, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ebermann, I., Phillips, J.B., Liebau, M.C., Koenekoop, R.K., Schermer, B., Lopez, I., Schafer, E., Roux, A.F., Dafinger, C., Bernd, A.et al. (2010) PDZD7 is a modifier of retinal disease and a contributor to digenic usher syndrome. J. Clin. Invest., 120, 1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coppieters, F., Casteels, I., Meire, F., De Jaegere, S., Hooghe, S., vanRegemorter, N., Van Esch, H., Matuleviciene, A., Nunes, L., Meersschaut, V.et al. (2010) Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum. Mutat., 31, E1709–E1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Badano, J.L., Leitch, C.C., Ansley, S.J., May-Simera, H., Lawson, S., Lewis, R.A., Beales, P.L., Dietz, H.C., Fisher, S. and Katsanis, N. (2006) Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature, 439, 326–330. [DOI] [PubMed] [Google Scholar]
- 6. Venturini, G., Rose, A.M., Shah, A.Z., Bhattacharya, S.S. and Rivolta, C. (2012) CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet., 8, e1003040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kousi, M. and Katsanis, N. (2015) Genetic modifiers and oligogenic inheritance. Cold Spring Harb Perspect Med, 5, a017145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Curtis, R. and Barnett, K.C. (1993) Progressive retinal atrophy in miniature longhaired dachshund dogs. Br Vet J, 149, 71–85. [DOI] [PubMed] [Google Scholar]
- 9. Turney, C., Chong, N.H., Alexander, R.A., Hogg, C.R., Fleming, L., Flack, D., Barnett, K.C., Bird, A.C., Holder, G.E. and Luthert, P.J. (2007) Pathological and electrophysiological features of a canine cone-rod dystrophy in the miniature longhaired dachshund. Invest. Ophthalmol. Vis. Sci., 48, 4240–4249. [DOI] [PubMed] [Google Scholar]
- 10. Mellersh, C.S., Boursnell, M.E., Pettitt, L., Ryder, E.J., Holmes, N.G., Grafham, D., Forman, O.P., Sampson, J., Barnett, K.C., Blanton, S.et al. (2006) Canine RPGRIP1 mutation establishes cone-rod dystrophy in miniature longhaired dachshunds as a homologue of human Leber congenital amaurosis. Genomics, 88, 293–301. [DOI] [PubMed] [Google Scholar]
- 11. Miyadera, K., Kato, K., Aguirre-Hernandez, J., Tokuriki, T., Morimoto, K., Busse, C., Barnett, K., Holmes, N., Ogawa, H., Sasaki, N.et al. (2009) Phenotypic variation and genotype-phenotype discordance in canine cone-rod dystrophy with an RPGRIP1 mutation. Mol. Vis., 15, 2287–2305. [PMC free article] [PubMed] [Google Scholar]
- 12. Beryozkin, A., Aweidah, H., Carrero Valenzuela, R.D., Berman, M., Iguzquiza, O., Cremers, F.P.M., Khan, M.I., Swaroop, A., Amer, R., Khateb, S.et al. (2021) Retinal degeneration associated with RPGRIP1: a review of natural history, mutation Spectrum, and genotype-phenotype correlation in 228 patients. Front Cell Dev Biol, 9, 746781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dryja, T.P., Adams, S.M., Grimsby, J.L., McGee, T.L., Hong, D.H., Li, T., Andreasson, S. and Berson, E.L. (2001) Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am. J. Hum. Genet., 68, 1295–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gerber, S., Perrault, I., Hanein, S., Barbet, F., Ducroq, D., Ghazi, I., Martin-Coignard, D., Leowski, C., Homfray, T., Dufier, J.L.et al. (2001) Complete exon-intron structure of the RPGR-interacting protein (RPGRIP1) gene allows the identification of mutations underlying Leber congenital amaurosis. Eur. J. Hum. Genet., 9, 561–571. [DOI] [PubMed] [Google Scholar]
- 15. Imani, S., Cheng, J., Mobasher-Jannat, A., Wei, C., Fu, S., Yang, L., Jadidi, K., Khosravi, M.H., Mohazzab-Torabi, S., Shasaltaneh, M.D.et al. (2018) Identification of a novel RPGRIP1 mutation in an Iranian family with leber congenital amaurosis by exome sequencing. J. Cell. Mol. Med., 22, 1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li, T. (2014) Leber congenital amaurosis caused by mutations in RPGRIP1. Cold Spring Harb Perspect Med, 5, a017384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao, Y., Long, Y., Liu, B., Cao, Q., Li, Y., Li, S., Wang, G., Meng, X. and Li, S. (2021) Ocular characteristics of patients with Leber congenital amaurosis 6 caused by pathogenic RPGRIP1 gene variation in a Chinese cohort. J. Ophthalmol., 2021, 9966427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hameed, A., Abid, A., Aziz, A., Ismail, M., Mehdi, S.Q. and Khaliq, S. (2003) Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J. Med. Genet., 40, 616–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang, L., Zhang, Q., Li, S., Guan, L., Xiao, X., Zhang, J., Jia, X., Sun, W., Zhu, Z., Gao, Y.et al. (2013) Exome sequencing of 47 chinese families with cone-rod dystrophy: mutations in 25 known causative genes. PLoS One, 8, e65546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Booij, J.C., Florijn, R.J., tenBrink, J.B., Loves, W., Meire, F., vanSchooneveld, M.J., deJong, P.T. and Bergen, A.A. (2005) Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J. Med. Genet., 42, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang, H., Wang, Y., Chen, H., Chen, Y., Wu, J., Chiang, P.W., Fan, N., Su, Y., Deng, J., Chen, D.et al. (2017) Targeted next generation sequencing identified novel mutations in RPGRIP1 associated with both retinitis pigmentosa and Leber’s congenital amaurosis in unrelated Chinese patients. Oncotarget, 8, 35176–35183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beigi, F., Del Pozo-Valero, M., Martin-Merida, I., Perea-Romero, I., Manaviat, M.R., Ayuso, C. and Ghasemi, N. (2021) Apparent but unconfirmed digenism in an Iranian consanguineous family with syndromic retinal disease. Exp. Eye Res., 207, 108533. [DOI] [PubMed] [Google Scholar]
- 23. Phelps, I.G., Dempsey, J.C., Grout, M.E., Isabella, C.R., Tully, H.M., Doherty, D. and Bachmann-Gagescu, R. (2018) Interpreting the clinical significance of combined variants in multiple recessive disease genes: systematic investigation of Joubert syndrome yields little support for oligogenicity. Genet Med, 20, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyadera, K., Kato, K., Boursnell, M., Mellersh, C.S. and Sargan, D.R. (2012) Genome-wide association study in RPGRIP1(−/−) dogs identifies a modifier locus that determines the onset of retinal degeneration. Mamm. Genome, 23, 212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Forman, O.P., Hitti, R.J., Boursnell, M., Miyadera, K., Sargan, D. and Mellersh, C. (2016) Canine genome assembly correction facilitates identification of a MAP9 deletion as a potential age of onset modifier for RPGRIP1-associated canine retinal degeneration. Mamm. Genome, 27, 237–245. [DOI] [PubMed] [Google Scholar]
- 26. Kuznetsova, T., Iwabe, S., Boesze-Battaglia, K., Pearce-Kelling, S., Chang-Min, Y., McDaid, K., Miyadera, K., Komaromy, A. and Aguirre, G.D. (2012) Exclusion of RPGRIP1 ins44 from primary causal association with early-onset cone-rod dystrophy in dogs. Invest. Ophthalmol. Vis. Sci., 53, 5486–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Das, R.G., Marinho, F.P., Iwabe, S., Santana, E., McDaid, K.S., Aguirre, G.D. and Miyadera, K. (2017) Variabilities in retinal function and structure in a canine model of cone-rod dystrophy associated with RPGRIP1 support multigenic etiology. Sci. Rep., 7, 12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyadera, K., Acland, G.M. and Aguirre, G.D. (2012) Genetic and phenotypic variations of inherited retinal diseases in dogs: the power of within- and across-breed studies. Mamm. Genome, 23, 40–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jagannathan, V., Drogemuller, C., Leeb, T. and Dog Biomedical Variant Database, C (2019) A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet., 50, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Komaromy, A.M., Alexander, J.J., Rowlan, J.S., Garcia, M.M., Chiodo, V.A., Kaya, A., Tanaka, J.C., Acland, G.M., Hauswirth, W.W. and Aguirre, G.D. (2010) Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet., 19, 2581–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Downs, L.M., Scott, E.M., Cideciyan, A.V., Iwabe, S., Dufour, V., Gardiner, K.L., Genini, S., Marinho, L.F., Sumaroka, A., Kosyk, M.S.et al. (2016) Overlap of abnormal photoreceptor development and progressive degeneration in Leber congenital amaurosis caused by NPHP5 mutation. Hum. Mol. Genet., 25, 4211–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hong, D.H., Yue, G., Adamian, M. and Li, T. (2001) Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J. Biol. Chem., 276, 12091–12099. [DOI] [PubMed] [Google Scholar]
- 33. Garcia, M.M., Ying, G.S., Cocores, C.A., Tanaka, J.C. and Komaromy, A.M. (2010) Evaluation of a behavioral method for objective vision testing and identification of achromatopsia in dogs. Am. J. Vet. Res., 71, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dufour, V.L., Yu, Y., Pan, W., Ying, G.S., Aguirre, G.D. and Beltran, W.A. (2020) In-vivo longitudinal changes in thickness of the postnatal canine retina. Exp. Eye Res., 192, 107926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Trinh, M., Khou, V., Zangerl, B., Kalloniatis, M. and Nivison-Smith, L. (2021) Modelling normal age-related changes in individual retinal layers using location-specific OCT analysis. Sci. Rep., 11, 558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ofri, R. and Ekesten, B. (2020) Baseline retinal OCT measurements in normal female beagles: the effects of eccentricity, meridian, and age on retinal layer thickness. Vet. Ophthalmol., 23, 52–60. [DOI] [PubMed] [Google Scholar]
- 37. Lheriteau, E., Libeau, L., Stieger, K., Deschamps, J.Y., Mendes-Madeira, A., Provost, N., Lemoine, F., Mellersh, C., Ellinwood, N.M., Cherel, Y.et al. (2009) The RPGRIP1-deficient dog, a promising canine model for gene therapy. Mol. Vis., 15, 349–361. [PMC free article] [PubMed] [Google Scholar]
- 38. Lheriteau, E., Petit, L., Weber, M., Le Meur, G., Deschamps, J.Y., Libeau, L., Mendes-Madeira, A., Guihal, C., Francois, A., Guyon, R.et al. (2014) Successful gene therapy in the RPGRIP1-deficient dog: a large model of cone-rod dystrophy. Mol. Ther., 22, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li, S., Datta, S., Brabbit, E., Love, Z., Woytowicz, V., Flattery, K., Capri, J., Yao, K., Wu, S., Imboden, M.et al. (2021) Nr2e3 is a genetic modifier that rescues retinal degeneration and promotes homeostasis in multiple models of retinitis pigmentosa. Gene Ther., 28, 223–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Appelbaum, T., Murgiano, L., Becker, D., Santana, E. and Aguirre, G.D. (2020) Candidate genetic modifiers for RPGR retinal degeneration. Invest. Ophthalmol. Vis. Sci., 61, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chang, C.C., Chow, C.C., Tellier, L.C.A.M., Vattikuti, S., Purcell, S.M. and Lee, J.J. (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aulchenko, Y.S., Ripke, S., Isaacs, A. and vanDuijn, C.M. (2007) GenABEL: an R library for genome-wide association analysis. Bioinformatics, 23, 1294–1296. [DOI] [PubMed] [Google Scholar]
- 43. Abecasis, G.R., Cherny, S.S., Cookson, W.O. and Cardon, L.R. (2002) Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet., 30, 97–101. [DOI] [PubMed] [Google Scholar]
- 44. Browning, S.R. and Browning, B.L. (2007) Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet., 81, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M.et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cingolani, P., Platts, A., Wang, L.L., Coon, M., Nguyen, T., Wang, L., Land, S.J., Lu, X. and Ruden, D.M. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly, 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robinson, J.T., Thorvaldsdottir, H., Wenger, A.M., Zehir, A. and Mesirov, J.P. (2017) Variant review with the integrative genomics viewer. Cancer Res., 77, e31–e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rausch, T., Zichner, T., Schlattl, A., Stutz, A.M., Benes, V. and Korbel, J.O. (2012) DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics, 28, i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aguirre, G.D., Cideciyan, A.V., Dufour, V.L., Ripolles-Garcia, A., Sudharsan, R., Swider, M., Nikonov, R., Iwabe, S., Boye, S.L., Hauswirth, W.W.et al. (2021) Gene therapy reforms photoreceptor structure and restores vision in NPHP5-associated Leber congenital amaurosis. Mol. Ther., 29, 2456–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beltran, W.A., Cideciyan, A.V., Iwabe, S., Swider, M., Kosyk, M.S., McDaid, K., Martynyuk, I., Ying, G.S., Shaffer, J., Deng, W.T.et al. (2015) Successful arrest of photoreceptor and vision loss expands the therapeutic window of retinal gene therapy to later stages of disease. Proc. Natl. Acad. Sci. U. S. A., 112, E5844–E5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and whole-genome sequencing and SNP genotyping datasets generated and analysed as part of this study are available in the Dryad archive (https://doi.org/10.5061/dryad.wdbrv15rj).