

Abstract
Ferroptosis is a new form of nonapoptotic cell death closely associated with glutathione (GSH) peroxidase 4 inhibition and/or GSH depletion, resulting in the accumulation of cellular iron and lipid peroxides. The exact mechanism by which GSH depletion causes the accumulation of reactive oxygen species (ROS) and lipid-ROS and subsequent ferroptotic cell death in neuronal cells remains unclear. In the present study, using immortalized HT22 mouse hippocampal neuronal cells as a model, we show that nitric oxide (NO) accumulation via protein disulfide isomerase (PDI)-mediated neuronal nitric oxide synthase (nNOS) activation plays a critical role in chemically-induced ferroptosis. Mechanistically, we find that erastin-induced GSH depletion leads to activation of PDI, which then mediates ferroptosis by catalyzing nNOS dimerization, followed by accumulation of cellular NO, ROS and lipid ROS and ultimately ferroptotic cell death. Pharmacological inhibition of PDI enzymatic activity or selective PDI knockdown can effectively abrogate erastin-induced ferroptosis in HT22 cells. The results of this study reveal an important role of PDI in mediating chemically induced ferroptosis in a neuronal cell model, and PDI may serve as a potential drug target for protection against GSH depletion-associated ferroptotic neuronal cell death.
Keywords: protein disulfide isomerase, neuronal nitric oxide synthase, nitric oxide, reactive oxygen species, lipid reactive oxygen species
Introduction
Ferroptosis is a form of oxidative cell death [ 1, 2] and is morphologically distinct from apoptosis-associated characteristics such as cellular shrinkage and plasma membrane blistering [ 3– 5] . Mechanistically, ferroptosis is closely associated with inhibition of glutathione (GSH) peroxidase 4 (GPX4, an enzyme involved in the reduction of lipid peroxides) and/or GSH depletion [ 1, 4, 6, 7] . While RSL-3 is a prototypical inhibitor of GPX4 that induces ferroptosis, erastin is a widely used inducer of cellular GSH depletion through inhibition of the amino acid antiporter system Xc ‒ and thus blocking the influx of extracellular cystine [ 3, 6] . Depletion of intracellular cysteine leads to reduced GSH biosynthesis and ultimately GSH depletion [ 3, 6] . GSH serves as a reducing cosubstrate for many cellular proteins, including GPX4, which plays an important role in detoxification of intracellular lipid reactive oxygen species (ROS) [ 6, 8, 9] . Inhibition of GPX4 leads to accumulation of lipid ROS and ultimately ferroptotic cell death [ 6, 7] . Although a growing body of literature suggests that cellular ROS and lipid-ROS are key factors in ferroptotic cell death, the exact mechanism by which GSH depletion causes ROS/lipid-ROS accumulation and subsequent ferroptotic cell death in neuronal cells remains unclear, which is the focus of our present study [ 10– 12] .
Protein disulfide isomerase (PDI or PDIA1) is the prototype of the PDI family proteins, which are ubiquitous dithiol/disulfide oxidoreductases of the thioredoxin superfamily [ 13– 15] . PDI is primarily localized in the endoplasmic reticulum of mammalian cells, although a small fraction of this protein is also found in the nucleus, cytosol, plasma membrane and extracellular space [ 16– 18] . PDI is involved in protein processing by catalyzing the formation of intra- and intermolecular disulfide bridges in proteins [16]. PDI in its reductive state can be oxidized to its oxidative state ( i. e., with a disulfide bond formed in its catalytic site) by ER oxidoreductin 1 (ERO1) [ 19– 21] . Our earlier study found that PDI plays an important role in mediating glutamate-induced oxidative cytotoxicity in HT22 cells, an immortalized mouse hippocampal neuronal cell line [22].
In the present study, we sought to evaluate the role of PDI in mediating erastin-induced ferroptosis in HT22 neuronal cells. Experimental evidence is provided to show that PDI plays a critical role in mediating erastin-induced ferroptotic cell death in these cells through its ability to catalyze nNOS dimerization, which is followed by cellular accumulation of NO, ROS and lipid-ROS and ultimately ferroptotic cell death. Pharmacological inhibition of PDI enzymatic activity or selective PDI knockdown can effectively abrogate erastin-induced ferroptosis.
Materials and Methods
Reagents
DAF-FM-DA and DCF-DA probes were purchased from Beyotime Biotechnology (Shanghai, China). Anti-nNOS antibody (#610308) was obtained from BD Biosciences (San Jose, USA). Anti-PDI (#P7372) and anti-β-actin antibodies were purchased from Sigma-Aldrich (St. Louis, USA). Mouse anti-nNOS and rabbit anti-PDI antibodies were obtained from Solarbio (Beijing, China). The siRNAs targeting mouse PDI, mouse nNOS and negative control siRNA (#siN0000001-1-5) were purchased from RiboBio (Guangzhou, China). Most of the other chemicals, unless specified, were also obtained from Sigma-Aldrich.
Cell culture and siRNA tranfection
The HT22 murine hippocampal neuronal cells were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China), and cultured in DMEM supplemented with 10% ( v/ v) fetal bovine serum and 1% penicillin and streptomycin and incubated at 37°C under 5% CO 2. Cells were subcultured when they reached ~80% confluence. For siRNA transfection, the siRNAs (at a final concentration of 60 nM) for targeted genes ( PDI and nNOS) were transfected into HT22 cells at 24 h after seeding with Lipofectamine RNA iMAX (Invitrogen). Twenty-four h following siRNA transfection, cells were treated with respective drugs, and subsequently processed for cell viability measurement, fluorescence imaging and immunoblot analysis. The siRNA sequences targeting the mouse PDI (siPDI) are CCAAGTACCAGCTGGACAA (#1), GAACGGTCATTGATTACAA (#2), and TGCTAAGATGGACTCAACA (#3). The siRNA sequences targeting the mouse nNOS (sinNOS) are GCTGCCATCCCATCACATA (#1), CCTCGTGAATGCACTCATT (#2), and GCGACAATTTGACATCCAA (#3).
Cell viability
The MTT reduction assay was used to determine cell viability. HT22 cells were seeded in 96-well plates at a density of 2000 cells/well 24 h prior to receiving various experimental treatments. To test the effect of an inhibitor, cells were jointly treated with erastin and the inhibitor for 24 h. Afterwards, MTT (Macklin, Shanghai, China) at a final concentration of 0.5 mg/mL was added to each well and incubated for 2.5 h at 37°C under 5% CO 2. After incubation, the MTT-containing medium was removed, and 100 μL of DMSO was added to each well to dissolve the MTT formazan. The absorbance of the MTT formazan was measured with a UVmax microplate reader (Molecular Device, Palo Alto, USA) at 560 nm.
Measurement of ROS and NO
HT22 cells were seeded in 24-well plates at a density of 5×10 4 cells/well 24 h before treatment. Afterwards, cells were treated with erastin and various inhibitors for 8 h. Prior to fluorescent probe staining, cells were washed with HBSS twice. Afterwards, intracellular accumulation of ROS and NO was detected by adding the cell-permeant fluorescent probes DCF-DA (10 μM) and DAF-FM-DA (10 μM) in 200 μL phenol red- and serum-free DMEM to each well. The cells were incubated at 37°C for an additional 20 and 30 min and washed twice with HBSS. Fluorescence images were captured using an AXIO fluorescence microscope (Carl Zeiss Corporation, Oberkochen, Germany).
Intracellular ROS and lipid-ROS detection by flow cytometry
HT22 cells were seeded in 6-well plates at a density of 10×10 4 cells/well 24 h prior to erastin treatment. After erastin treatment for 8 h, cells were collected and resuspended in 10 μM DCF-DA and 10 μM C11-BODIPY (581/591) in 200 μL phenol red- and serum-free DMEM and incubated for 20 or 30 min at 37°C, respectively. Afterward, the cells were washed three times with HBSS to remove the probes. The fluorescence intensity reflecting the intracellular accumulation of ROS and lipid-ROS was detected with a flow cytometer (Beckman Coulter, Brea, USA). Three or more experiments were conducted, and the average fluorescence intensity (mean±SE) was calculated for each experimental group using FlowJo software (FlowJo LLC, Ashland, USA).
Purification of PDI
The cDNA for human PDI protein was cloned into the pET-28a vector. The expressed PDI protein was purified using Ni-NTA agarose (QIAGEN, Dusseldorf, Germany) and separated using the ÄKTA FPLC system (GE Healthcare, Waukesha, USA) as described previously [23]. The purified PDI protein was analysed by western blot analysis, and its protein concentration was determined by Bio-Rad Protein Assay using BSA as a standard.
Western blot analysis
After treatment with erastin for the indicated lengths of time, HT22 cells were collected by trypsinization and centrifugation and then lysed in RIPA buffer containing 1% protease inhibitor cocktail on ice for 30 min. Protein concentrations were determined using a BCA assay kit (Thermo Fisher Scientific, Waltham, USA). For total nNOS analysis (including both monomeric and dimeric forms of nNOS), samples were heated at 95°C for 5 min with a reducing buffer before loading onto the gel. To analyse the monomeric and dimeric forms of nNOS, samples were prepared with a nonreducing buffer and were not heated, and the temperature of the gel was maintained below 15°C during electrophoresis. Proteins were then separated using 10% agarose gel (for total nNOS analysis) or 6% agarose gel (for monomeric and dimeric nNOS analysis) and transferred to PVDF membranes. The membranes were then incubated for 1 h in 5% skim milk at room temperature and incubated with the primary antibody overnight. Afterwards, the membranes were washed three times with TBST at room temperature (10 min each time). The membranes were then incubated with the secondary antibody for 1 h at room temperature and washed with TBST three times before visualization. Desired protein bands were visualized using the Tanon 5200 chemiluminescence imaging system (Tanon, Shanghai, China).
Statistical analysis
All quantitative experiments were repeated at least three times. Data are presented as the mean±SE of multiple measurements. Statistical analysis and comparisons were performed with one-way ANOVA using GraphPad Prism 7 (GraphPad, La Jolla, USA). P<0.05 is used to denote as statistically significant or statistically very significant, respectively. For western blot quantification, one representative data point is shown.
Results
Erastin induces ferroptotic cell death by increasing cellular NO and ROS levels
Treatment of HT22 cells with erastin induced cell death in a dose- and time-dependent manner ( Figure 1A,B). To probe whether erastin-induced ferroptosis in HT22 cells is related to NO and ROS accumulation, we determined the time- and dose-dependent changes in the cellular levels of NO, ROS and lipid-ROS following erastin treatment ( Figure 1C‒S). Based on fluorescence microscopic and flow cytometric analyses, we found that treatment of HT22 cells with erastin elicited a time- and dose-dependent increase in cellular NO levels ( Figure 1C‒F). Similarly, the cellular levels of ROS and lipid ROS were also increased in a time- and dose-dependent manner by erastin treatment ( Figure 1G‒N for ROS and Figure 1O‒S for lipid ROS). Notably, NO accumulation occurred approximately 2 h before the onset of ROS accumulation, whereas accumulation of ROS and lipid-ROS occurred at approximately the same time.
Figure 1 .

Time- and dose-dependent induction of ferroptotic cell death in erastin-treated HT22 cells
(A,B) Time- and dose-dependent effect of erastin treatment on cell viability. (C‒F) Time- and dose-dependent induction of NO accumulation following erastin treatment: fluorescence microscopy images (C,E) and respective quantitative intensity values (D,F). Note that the y-axis is the mean fluorescence intensity. Scale bar: 60 μm. (G‒J) Time- and dose-dependent induction of ROS accumulation following erastin treatment: fluorescence microscopy images (G,I) and quantitative intensity values (H,J). Scale bar: 60 μm. (K‒N) Time- and dose-dependent induction of ROS accumulation following erastin treatment: flow cytometry data (K,M) and quantitative intensity values (L,N). (O‒S) Time- and dose-dependent induction of lipid ROS accumulation following erastin treatment: confocal microscopy images (O), flow cytometry data (P,R) and quantitative intensity values (Q,S). Scale bar: 50 μm. The selected time points for the time-dependent erastin treatment in A‒S are 2, 4, 6 and 8 h, and the concentrations for the dose-dependent erastin treatment in A‒S are 0.2, 0.4, 0.8 and 1 μM. Data are presented as the mean±SE. n=3. * P<0.05, ** P<0.01 vs the control group.
To determine whether NO accumulation causes ROS accumulation and ferroptotic cell death, we determined the effect of 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO), an NO scavenger, on erastin-induced ferroptosis [24]. cPTIO effectively suppressed erastin-induced accumulation of NO ( Figure 2A,B), ROS ( Figure 2C‒F), and lipid ROS in HT22 cells ( Figure 2G‒I). In addition, cPTIO attenuated erastin-induced cell death ( Figure 2J). Notably, sodium nitroprusside (SNP), which can directly release NO, sensitized HT22 cells to erastin-induced ferroptotic cell death and ROS accumulation ( Figure 3A‒C). These results indicate that erastin-induced NO accumulation leads to cellular ROS accumulation and subsequent ferroptotic cell death.
Figure 2 .

NO and ROS accumulation in erastin-treated HT22 cells
(A‒D) Levels of NO (A,B) and ROS (C,D) after treatment with 1 μM erastin± 200 μM cPTIO for 8 h: fluorescence microscopy images (A,C) and fluorescence intensity values (B,D). Scale bar: 60 μm. (E‒I) Levels of ROS (E,F) and lipid ROS (G,H) after treatment with 1 μM erastin± 200 μM cPTIO for 8 h: flow cytometry data (E,G), fluorescence intensity values (F,H), and confocal microscopy data (I). Scale bar: 50 μm. (J) Cell viability change following treatment with 1 μM erastin± 200 μM cPTIO for 24 h. Data are presented as the mean±SE. n=3. * P<0.05, ** P<0.01 vs the control group.
Figure 3 .

Effect of NO and ROS scavengers on erastin-treated HT22 cells
(A) Cell viability change after treatment with 200 μM SNP and different concentrations of erastin. (B,C) Levels of ROS after treatment with 1 μM erastin± 200 μM SNP for different time intervals (2, 4 and 6 h): fluorescence microscopy images (B) and fluorescence intensity values (C). Scale bar: 45 μm. (D‒G) Levels of NO (D,E) and ROS (F,G) after treatment with 1 μM erastin± 200 μM Trolox: fluorescence microscopy images (D,F) and fluorescence intensity values (E,G). Scale bar: 45 μm. (H‒K) Levels of lipid ROS after treatment with 1 μM erastin± 200 μM Trolox or 200 μM SNAP for 8 h: flow cytometry data (H,J), fluorescence intensity values (I,K). (L) Cell viability change following treatment with 1 μM erastin± 200 μM Trolox for 24 h. (M,N) Levels of ROS after treatment with 1 μM erastin± 100 μM NAC for 8 h: fluorescence microscopy images (M) and fluorescence intensity values (N). Scale bar: 50 μm. (O) Cell viability change following treatment with 1 μM erastin and different concentrations of NAC. Data are presented as the mean±SE. n=3. * P<0.05, ** P<0.01 vs the control group.
6-Hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox), which has both NO and ROS scavenging activity [25], had a protective effect against erastin-induced accumulation of NO ( Figure 3D,E), ROS ( Figure 3F‒I) and lipid-ROS ( Figure 3J,K), and it also abrogated erastin-induced ferroptotic cell death ( Figure 3L). Similarly, NAC ( N-acetyl- L-cysteine), another commonly used antioxidant, also showed strong protection against erastin-induced ROS accumulation ( Figure 3M,N) and cell death ( Figure 3O). Together, these results indicate that erastin-induced ferroptosis is jointly mediated by the accumulation of NO, ROS and lipid ROS in HT22 cells.
Formation of cellular NO is catalyzed by NOS using L-arginine as a substrate [ 26– 28] . Three NOS isoforms have been reported in the brain, namely, neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS) [27]. Our earlier study showed that only nNOS (but not eNOS or iNOS) is expressed in HT22 cells [22]. Treatment of HT22 cells with erastin for 8 h significantly increased the levels of total nNOS proteins (which includes both monomer and dimer forms of nNOS) in a dose- and time-dependent manner, whereas the cellular levels of total PDI were not significantly different ( Figure 4A,B). We found that chloropromazine (CPZ), an nNOS inhibitor [29], decreased erastin-induced accumulation of cellular NO ( Figure 4C,D), ROS ( Figure 4E,F) and lipid-ROS ( Figure 4G,H), along with strong protection against erastin-induced ferroptotic cell death ( Figure 4I). Similarly, selective nNOS knockdown also showed a partial protective effect against erastin-induced cytotoxicity ( Figure 4J). The efficiency of nNOS knockdown was confirmed by changes in cellular nNOS protein levels ( Figure 4K). Together, these results indicate that NO accumulation via nNOS activation is involved in mediating erastin-induced ferroptosis in HT22 cells.
Figure 4 .

Erastin induces nNOS activation in HT22 cells
(A,B) Total nNOS levels after treatment with increasing concentrations of erastin for 8 h (A) or after treatment with 1 μM erastin for different time intervals (B). (C‒F) Levels of NO (C,D) and ROS (E,F) after treatment with 1 μM erastin± 20 μM CPZ for 8 h: fluorescence microscopy images (C,E) and fluorescence intensity values (D,F). Scale bar: 50 μm. (G,H) Levels of lipid ROS after treatment with 1 μM erastin± 20 μM CPZ for 8 h: flow cytometry data (G) and fluorescence intensity values (H). (I) Cell viability change following treatment with 1 μM erastin± 20 μM CPZ for 24 h. (J) Effect of nNOS knockdown on erastin-induced cell viability change. Cells were transfected with nNOS-siRNA for 24 h prior to treatment with 1 μM erastin for an additional 24 h. (K) Effectiveness of nNOS-siRNAs in reducing cellular nNOS protein levels. Cells were transfected with nNOS-siRNAs for 48 h, and cellular nNOS protein levels were determined by western blot analysis. Data are presented as the mean±SE. n=3. * P<0.05, ** P<0.01 vs the control group.
PDI mediates nNOS dimerization in erastin-treated cells
It is known that the catalytic activity of nNOS can be activated through the formation of nNOS dimers [ 26– 28] , which are stabilized by an intermolecular disulfide bond and facilitate NO production [30]. We found that nNOS in untreated HT22 cells existed mostly as monomers, and treatment of these cells with erastin increased the formation of nNOS dimers in both time- and dose-dependent manners, whereas the monomer nNOS levels appeared to be not markedly different ( Figure 5A,B).
Figure 5 .

PDI knockdown protects HT22 cells against erastin-induced ferroptosis
(A,B) Levels of dimer and monomer nNOS after treatment with 1 μM erastin for the indicated time intervals (A) or at the indicated concentrations for 8 h (B). (C) Levels of dimer and monomer nNOS after incubation of cell lysates with PDI (1 mg/mL) in vitro at 4°C for 60 min. (D) Effectiveness of PDI-siRNA in reducing cellular PDI protein levels. Cells were transfected with PDI-siRNA for 48 h, and the PDI levels were determined by western blot analysis. (E) Effect of PDI knockdown on erastin-induced nNOS dimerization. Cells were transfected with PDI-siRNA for 24 h prior to treatment with 1 μM erastin for an additional 24 h. nNOS dimer formation was detected by western blot analysis. (F‒J) Effect of PDI knockdown on erastin-induced NO and ROS accumulation. Cells were transfected with PDI-siRNA for 24 h prior to erastin (1 μM) treatment. The levels of NO and ROS after 8 h of erastin treatment were assessed by fluorescence microscopy (F,H), and their quantitative intensity values are shown in G and I. Cell viability changes were analysed after 24 h of erastin treatment (J). Scale bar: 45 μm. Data are presented as the mean±SE. n=3. * P<0.05, ** P<0.01 vs the control group.
Next, we determined whether PDI can catalyze nNOS dimerization in erastin-treated HT22 cells. We first incubated HT22 cell lysates with purified PDI protein and observed a significant increase in nNOS dimerization ( Figure 5C). We then used selective PDI-siRNAs to knock down PDI in HT22 cells and examined whether PDI is involved in erastin-induced nNOS dimerization. The effectiveness of PDI knockdown by PDI-siRNAs was confirmed by assessing changes in cellular protein levels ( Figure 5D). We found that erastin-induced dimerization of nNOS was abrogated by PDI knockdown ( Figure 5E). Additionally, erastin-induced accumulation of NO ( Figure 5F,G) and ROS ( Figure 5H,I) and ferroptotic cell death ( Figure 5J) were all strongly attenuated by PDI knockdown. Together, these results show that PDI is directly involved in catalyzing nNOS dimerization during erastin-induced ferroptotic cell death.
To provide further experimental evidence for the suggestion that PDI enzymatic activity is responsible for nNOS dimerization, we sought to determine the effect of cystamine on erastin-induced ferroptosis. Cystamine is an organic disulfide molecule known to selectively inhibit PDI’s enzymatic activity through covalent modification of the cysteine residue(s) in its catalytic site [ 31, 32] . We found that cystamine could inhibit erastin-induced nNOS dimerization ( Figure 6A,B) in erastin-treated HT22 cells. In addition, treatment of cells with cystamine attenuated erastin-induced accumulation of NO ( Figure 6C,D), ROS ( Figure 6E‒H) and lipid ROS ( Figure 6I‒K). Additionally, cystamine strongly inhibited erastin-induced ferroptotic cell death ( Figure 6L).
Figure 6 .

Cystamine (a PDI inhibitor) protects HT22 cells against erastin-induced ferroptosis
(A,B) Levels of dimer and monomer nNOS (A) and total nNOS level (B) after treatment with 1 μM erastin± 100 μM cystamine for 8 h. (C‒F) Levels of NO (C,D) and ROS (E,F) after treatment with 1 μM erastin± 100 μM cystamine for 8 h: fluorescence microscopy images (C,E) and quantitative intensity values (D,F). Scale bar: 45 μm. (G‒K) Levels of ROS (G,H) and lipid ROS (I,J) after treatment with 1 μM erastin± 100 μM cystamine for 8 h: flow cytometry data (G,I), quantitative intensity values (H,J), and confocal microscopy data (K). Scale bar: 50 μm. (L) Cell viability change after treatment with 1 μM erastin± 100 μM cystamine for 24 h. Data are presented as the mean±SE. n=3. * P<0.05, ** P<0.01 vs the control group.
LOC14, another known inhibitor of PDI [ 33, 34] , also showed a strong inhibitory effect against erastin-induced nNOS dimerization ( Figure 7A,B). Similarly, LOC14 abrogated erastin-induced accumulation of NO ( Figure 7C,D), ROS ( Figure 7E‒H) and lipid ROS ( Figure 7I‒K), and it also prevented erastin-induced ferroptotic cell death ( Figure 7L). Together, these results indicate that PDI activation plays an important role in erastin-induced accumulation of NO, ROS/lipid-ROS and ferroptosis. Notably, unlike cystamine, which decreased total nNOS expression in erastin-treated cells, LOC14 did not have a similar effect on total cellular nNOS levels.
Figure 7 .

LOC14 (a PDI inhibitor) protects cells against erastin-induced ferroptosis
(A,B) Levels of dimer and monomer nNOS (A) and total nNOS (B) after treatment with 1 μM erastin± 0.5 μM LOC14 for 8 h. (C‒F) Levels of NO (C,D) and ROS (E,F) after treatment with 1 μM erastin± 0.5 μM LOC14 for 8 h: fluorescence microscopy images (C,E) and quantitative intensity values (D,F). Scale bar: 45 μm. (G‒K) Levels of ROS (G,H) and lipid ROS (I,J) after treatment with 1 μM erastin± 0.5 μM LOC14 for 8 h: flow cytometry data (G,I), quantitative intensity values (H,J), and confocal microscopy data (K). Scale bar: 50 μm. (L) Cell viability change after treatment with 1 μM erastin± 0.5 μM LOC14 for 24 h. Data are presented as the mean±SE. n=3. * P<0.05, ** P<0.01 vs the control group.
PDI is activated in erastin-treated cells through denitrosylation and ERO1
Our earlier study showed that PDI in untreated HT22 cells is mostly present in the S-nitrosylated form, and it becomes denitrosylated following GSH depletion [22]. To probe the involvement of PDI S-nitrosylation in mediating erastin-induced cytotoxicity in HT22 cells, we studied the effect of S-nitroso- N-acetylpenicillamine (SNAP), an S-nitrosylating agent [35]. We found that joint treatment of HT22 cells with SNAP abrogated erastin-induced nNOS dimerization ( Figure 8A,B), accumulation of NO ( Figure 8C,D), ROS ( Figures 3H,I and 8E,F) and lipid-ROS ( Figure 3J,K), and ultimately, ferroptotic cell death ( Figure 8G). This result indicates that S-nitrosylation of PDI is associated with inhibition of PDI activity and reduction in nNOS dimer formation, which then results in decreased levels of NO, ROS and lipid-ROS as well as protection from ferroptosis.
Figure 8 .

SNAP (an S-nitrosylating agent) protects cells from erastin-induced ferroptosis
(A,B) Levels of dimer and monomer nNOS (A) and total nNOS (B) after treatment with 1 μM erastin± 200 μM SNAP for 8 h. (C‒F) Levels of NO (C,D) and ROS (E,F) after treatment with 1 μM erastin± 200 μM SNAP for 8 h: fluorescence microscopy images (C,E) and quantitative intensity values (D,F). Scale bar: 50 μm. (G) Cell viability change after treatment with 1 μM erastin± 200 μM SNAP for 24 h. Data are presented as the mean±SE. n=3. * P<0.05, ** P<0.01 vs the control group.
ERO1 is an important cellular enzyme that catalyzes the oxidation of PDI’s disulfide bonds in its catalytic site [ 19, 21] . When the intracellular GSH/GSSG ratio is markedly reduced by erastin treatment, PDI is easily oxidized by ERO1, and the oxidative form of PDI then catalyzes nNOS dimerization. In this study, we tested the effect of EN460, a known ERO1 inhibitor [36], on erastin-induced ferroptotic cell death. In line with our prediction, EN460 showed a protective effect against erastin-induced accumulation of NO ( Figure 9A,B), ROS ( Figure 9C,D), lipid ROS ( Figure 9E,F) and subsequent cytotoxicity ( Figure 9G). These results indicate that ERO1-mediated PDI oxidation is an important step that contributes to PDI-mediated nNOS dimerization, which subsequently leads to NO accumulation and ferroptotic cell death.
Figure 9 .

EN460 (an ERO1 inhibitor) protects cells from erastin-induced ferroptosis
(A‒D) Levels of NO (A,B) and ROS (C,D) after treatment with 1 μM erastin± 40 μM EN460 for 8 h: fluorescence microscopy images (A,C) and quantitative intensity values (B,D). Scale bar: 45 μm. (E,F) Levels of lipid ROS after treatment with 1 μM erastin± 40 μM EN460 for 8 h: flow cytometry data (E), quantitative intensity values (F). (G) Cell viability change after treatment with 1 μM erastin± 40 μM EN460 for 24 h. Data are presented as the mean±SE. n=3. * P<0.05, ** P<0.01 vs the control group.
Discussion
GSH depletion is closely associated with ROS accumulation and oxidative cytotoxicity (ferroptosis) [ 3, 4, 6] , but the precise mechanism underlying this association remains unclear. Erastin is a prototypical inducer of GSH depletion-associated ferroptosis [3]. We found that during the development of erastin-induced ferroptosis in HT22 neuronal cells, NO accumulation is an early event that subsequently leads to the formation and accumulation of ROS and lipid-ROS and ultimately the development of ferroptotic cell death. The experimental evidence in support of this suggestion includes the following: First, NO formation was increased in a time- and dose-dependent manner in HT22 cells following erastin treatment ( Figure 1). NO accumulation occurred approximately 2 h before ROS and lipid ROS accumulation ( Figure 1). Second, SNP, which directly releases NO, sensitized HT22 cells to erastin-induced cell death ( Figure 3). Third, cPTIO, an NO scavenger, effectively abrogated erastin-induced accumulation of ROS and lipid ROS as well as cell death in HT22 cells ( Figure 2). Similarly, joint treatment of cells with erastin plus Trolox, which has both NO and ROS-scavenging activity, also effectively abrogated erastin-induced accumulation of NO, ROS and lipid-ROS, and these effects were accompanied by strong protection against erastin-induced cytotoxicity ( Figure 3). Fourth, NOS is the major enzyme that catalyzes NO formation [ 26– 28] . CPZ, which is an nNOS inhibitor, showed a protective effect against erastin-induced NO and ROS accumulation as well as cytotoxicity ( Figure 4). Similarly, nNOS knockdown with siRNAs also showed a protective effect against erastin-induced cytotoxicity ( Figure 4). Together, these experimental observations demonstrated that following erastin-induced GSH depletion, NO production is an important early event that subsequently leads to the accumulation of ROS and lipid ROS and ultimately the induction of ferroptotic cell death.
In cells, NO synthase (NOS) is a major enzyme that catalyzes NO formation [ 26– 28] . While nNOS in its monomer form is catalytically inactive, its dimer form is active for catalyzing NO formation [ 27, 28] . In this study, we found that erastin treatment induced the formation of nNOS dimers in a time- and dose-dependent manner in HT22 cells. In addition, we demonstrated that PDI is involved in catalyzing nNOS dimer formation in erastin-treated HT22 cells on the basis of the following lines of evidence. First, transient PDI knockdown by siRNAs efficiently abrogated erastin-induced nNOS dimerization, and this effect was associated with reduced accumulation of NO and ROS as well as strong protection against erastin-induced oxidative cell death ( Figure 5). Second, joint treatment of HT22 cells with erastin plus cystamine or LOC14 (two known PDI inhibitors) strongly reduced erastin-induced formation of nNOS dimers, accumulation of NO, ROS and lipid-ROS, and oxidative cytotoxicity ( Figures 6 and 7). Third, in vitro biochemical analysis showed that incubation of purified PDI protein with HT22 whole cell lysates increased the formation of nNOS dimers ( Figure 6). Together, these experimental observations supported the notion that PDI is directly involved in catalyzing nNOS dimerization in erastin-treated HT22 cells, which subsequently results in NO accumulation, followed by ROS and lipid-ROS accumulation, and ultimately ferroptotic cell death.
S-Nitrosylation of the cysteine residues in PDI is an important posttranslational regulatory mechanism of its function, which inactivates its enzymatic activity [ 33, 37] . Our earlier study showed that PDI in untreated HT22 cells is mostly present in the S-nitrosylated form, but following glutamate treatment (which induces cellular GSH depletion), PDI becomes denitrosylated [22]. Notably, only the oxidized form of PDI ( i. e., with a disulfide bond formed in its catalytic site) is capable of catalyzing nNOS dimer formation, and during the catalytic process, the disulfide bond in PDI’s active site is being reduced [ 17, 22] (depicted in Figure 10). ERO1 is an enzyme that oxidizes PDI and thus keeps it in the catalytically active state [ 19– 21] . The ERO1-mediated reaction is favored under oxidative conditions when the cellular GSH/GSSG ratio is decreased [ 38, 39] . In the present study, we found that EN460, which is an ERO1 inhibitor, exerted approximately 80% protection against erastin-induced cytotoxicity. This observation suggests that ERO1 helps to keep PDI in a catalytically active state ( i. e., the oxidative state) for the formation of nNOS dimers. In comparison, EN460 at the same concentration showed a very weak protective effect against RSL3-induced ferroptotic cell death (data not shown). RSL3 induces ferroptotic cytotoxicity through direct inhibition of GPX4 [40] and subsequently causes lipid ROS accumulation and cytotoxicity [ 7, 40, 41] . Based on previous reports, although the cellular GSH levels were reduced in RSL3-treated cells, the reduction was mild compared to GSH reduction in erastin-treated cells [ 7, 42] . Therefore, it is possible that in RSL3-induced ferroptosis in which GSH depletion is not strongly depleted, ERO1-mediated PDI oxidation would be less favored, thus reducing the fraction of oxidized PDI. This may explain why the ERO1 inhibitor showed significantly weaker protection in RSL3-treated cells than in erastin-treated cells.
Figure 10 .
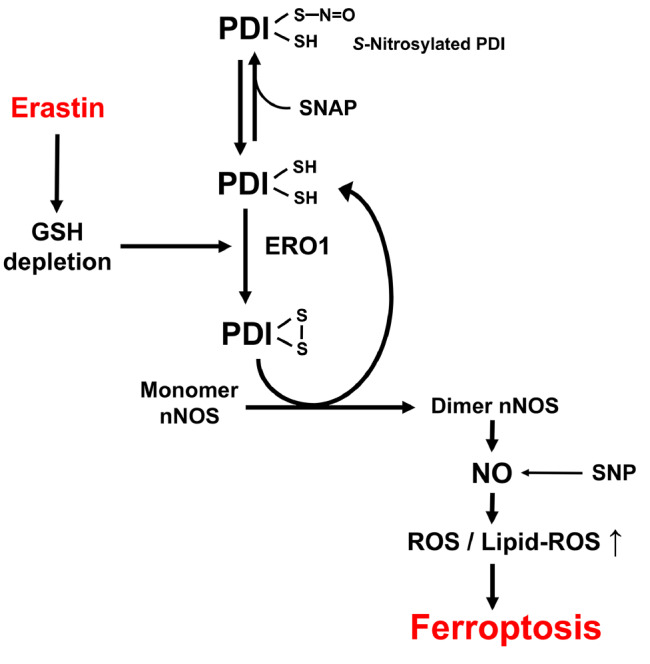
A schematic depiction of the proposed role of PDI in mediating chemically induced ferroptosis
Our earlier study showed that PDI in untreated HT22 cells is mostly present in its S-nitrosylated form, and GSH depletion is associated with S-denitrosylation of PDI [22]. Under conditions of cellular GSH depletion ( i. e., with an increased GSSG/GSH ratio), the reduced form of PDI is facilely converted to its oxidized form by ERO1. Oxidized PDI is the active form for catalyzing the conversion of the nNOS monomer to its dimer form, which then leads to increased formation of NO, followed by accumulation of cellular ROS and lipid ROS and ultimately ferroptotic cell death.
Here, it is also worth noting that there are seven known PDI proteins in the PDI gene family, which include PDIA1 (commonly referred to as PDI) [ 13, 14] , PDIA2 (also called PDIp, a pancreas-specific PDI) [43], PDIA3 [44], PDIA4 [45], PDIA5 [46], PDIA6 [46] and PDILT [47]. Earlier studies have reported that PDI, PDIA3, PDIA4 and PDIA6 are expressed in neuronal cells [ 48– 54] , and the expressions of PDI and PDIA3 are often dysregulated in disease states of the nervous system [ 48– 50, 55] . While we have demonstrated in the present study that PDI plays an important role in mediating erastin-induced ferroptotic cell death in a neuronal cell line, it is not known whether other PDI family proteins share a similar role in mediating chemically induced ferroptotic cytotoxicity. Since our results showed that selective knockdown of PDI only grants partial (~75%) protection against erastin-induced cytotoxicity, this possibility cannot be completely ruled out, and therefore, it will be of interest to explore this possibility in the future.
Last, it is of note that while we demonstrated in this study an important role of PDI in ferroptosis induction using an immortalized neuronal cell line, further establishment of the impact of PDI in ferroptosis animal models would be challenging, as PDI knockout in mice would cause embryonic lethality [ 42, 56] . Additionally, conditional knockout of PDI may also affect other physiological processes beyond ferroptosis due to the involvement of PDI in a variety of biological functions with different interacting partners. Consequently, it may be necessary to collect strong correlations for multiple ferroptosis-associated biochemical changes in the future to draw inferences from in vivo experiments on PDI or its inhibitors.
As summarized in Figure 10, the results of our present study demonstrated that PDI plays a critical role in mediating erastin-induced ferroptotic cell death in HT22 cells by catalyzing nNOS dimer formation, followed by cellular accumulation of NO, ROS and lipid-ROS, and ultimately ferroptotic cell death. PDI is activated in erastin-treated cells by S-denitrosylation followed by ERO1-mediated oxidation. The discovery of the role of PDI in ferroptosis offers the possibility that PDI might serve as a potential drug target for protection against ferroptosis-related neuronal cell death.
COMPETING INTERESTS
The authors declare that they have no conflict of interest.
Funding Statement
This work was supported in part by the grants from the National Natural Science Foundation of China (No. 81630096), the Shenzhen Key Laboratory of Steroid Drug Development and Discovery (No. ZDSYS20190902093417963), the Shenzhen Peacock Plan (No. KQTD2016053117035204), and the Shenzhen Bay Laboratory (No. SZBL2019062801007).
References
- 1.Stockwell B R. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. . 2022;185:2401–2421. doi: 10.1016/j.cell.2022.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. . 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. . 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, et al. Ferroptosis: process and function. Cell Death Differ. . 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. . 2007;447:865–869. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radical Biol Med. . 2020;152:175–185. doi: 10.1016/j.freeradbiomed.2020.02.027. [DOI] [PubMed] [Google Scholar]
- 7.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. . 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. . 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. . 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan S, Wood M, Maher P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J Neurochem. . 1998;71:95–105. doi: 10.1046/j.1471-4159.1998.71010095.x. [DOI] [PubMed] [Google Scholar]
- 11.Meister A. Glutathione metabolism. Methods Enzymol. 1995, 251: 3–7 . [DOI] [PubMed]
- 12.Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017, 403: 143–170 . [DOI] [PubMed]
- 13.Tasanen K, Parkkonen T, Chow LT, Kivirikko KI, Pihlajaniemi T. Characterization of the human gene for a polypeptide that acts both as the beta subunit of prolyl 4-hydroxylase and as protein disulfide isomerase. J Biol Chem. . 1988;263:16218–16224. doi: 10.1016/S0021-9258(18)37581-1. [DOI] [PubMed] [Google Scholar]
- 14.Pihlajaniemi T, Helaakoski T, Tasanen K, Myllylä R, Huhtala ML, Koivu J, Kivirikko KI. Molecular cloning of the beta-subunit of human prolyl 4-hydroxylase. This subunit and protein disulphide isomerase are products of the same gene. EMBO J. . 1987;6:643–649. doi: 10.1002/j.1460-2075.1987.tb04803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freedman RB, Hirst TR, Tuite MF. Protein disulphide isomerase: building bridges in protein folding. Trends Biochem Sci. . 1994;19:331–336. doi: 10.1016/0968-0004(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 16.Cai H, Wang CC, Tsou CL. Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J Biol Chem. . 1994;269:24550–24552. doi: 10.1016/S0021-9258(17)31426-6. [DOI] [PubMed] [Google Scholar]
- 17.Wilkinson B, Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta. . 2004;1699:35–44. doi: 10.1016/S1570-9639(04)00063-9. [DOI] [PubMed] [Google Scholar]
- 18.Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol. . 2002;193:154–163. doi: 10.1002/jcp.10172. [DOI] [PubMed] [Google Scholar]
- 19.Sevier CS, Kaiser CA. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta. . 2008;1783:549–556. doi: 10.1016/j.bbamcr.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 20.Benham AM, van Lith M, Sitia R, Braakman I. Ero1–PDI interactions, the response to redox flux and the implications for disulfide bond formation in the mammalian endoplasmic reticulum. Phil Trans R Soc B. . 2013;368:20110403. doi: 10.1098/rstb.2011.0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araki K, Nagata K. Functional in vitro analysis of the ERO1 protein and protein-disulfide isomerase pathway. J Biol Chem. . 2011;286:32705–32712. doi: 10.1074/jbc.M111.227181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada K, Fukui M, Zhu BT. Protein disulfide isomerase mediates glutathione depletion-induced cytotoxicity. Biochem Biophys Res Commun. . 2016;477:495–502. doi: 10.1016/j.bbrc.2016.06.066. [DOI] [PubMed] [Google Scholar]
- 23.Choi HJ, Chen T, Hou MJ, Song JH, Li P, Liu C, Wang P, et al. Protection against glutathione depletion-associated oxidative neuronal death by neurotransmitters norepinephrine and dopamine: protein disulfide isomerase as a mechanistic target for neuroprotection. Acta Pharmacol Sin. . 2022;43:2527–2541. doi: 10.1038/s41401-022-00891-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maeda H, Akaike T, Yoshida M, Suga M. Multiple functions of nitric oxide in pathophysiology and microbiology: analysis by a new nitric oxide scavenger. J Leukocyte Biol. . 1994;56:588–592. doi: 10.1002/jlb.56.5.588. [DOI] [PubMed] [Google Scholar]
- 25.Castro IA, Rogero MM, Junqueira RM, Carrapeiro MM. Free radical scavenger and antioxidant capacity correlation of α-tocopherol and Trolox measured by three in vitro methodologies . Int J Food Sci Nutr. . 2006;57:75–82. doi: 10.1080/09637480600656199. [DOI] [PubMed] [Google Scholar]
- 26.Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res. . 1999;43:521–531. doi: 10.1016/S0008-6363(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 27.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. . 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. . 2001;357:593–615. doi: 10.1042/bj3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palacios M, Padron J, Glaria L, Rojas A, Delgado R, Knowles R, Moncada S. Chlorpromazine inhibits both the constitutive nitric oxide synthase and the induction of nitric oxide synthase after LPS challenge. Biochem Biophys Res Commun. . 1993;196:280–286. doi: 10.1006/bbrc.1993.2246. [DOI] [PubMed] [Google Scholar]
- 30.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. . 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 31.Fujita I, Nobunaga M, Seki T, Kurauchi Y, Hisatsune A, Katsuki H. Cystamine-mediated inhibition of protein disulfide isomerase triggers aggregation of misfolded orexin-A in the Golgi apparatus and prevents extracellular secretion of orexin-A. Biochem Biophys Res Commun. . 2017;489:164–170. doi: 10.1016/j.bbrc.2017.05.118. [DOI] [PubMed] [Google Scholar]
- 32.Hoffstrom BG, Kaplan A, Letso R, Schmid RS, Turmel GJ, Lo DC, Stockwell BR. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol. . 2010;6:900–906. doi: 10.1038/nchembio.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplan A, Gaschler MM, Dunn DE, Colligan R, Brown LM, Palmer 3rd AG, Lo DC, et al. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc Natl Acad Sci USA. . 2015;112:E2245–E2252. doi: 10.1073/pnas.1500439112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chamberlain N, Korwin-Mihavics BR, Nakada EM, Bruno SR, Heppner DE, Chapman DG, Hoffman SM, et al. Lung epithelial protein disulfide isomerase A3 (PDIA3) plays an important role in influenza infection, inflammation, and airway mechanics. Redox Biol. . 2019;22:101129. doi: 10.1016/j.redox.2019.101129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mallis RJ, Thomas JA. Effect of S-nitrosothiols on cellular glutathione and reactive protein sulfhydryls. Arch Biochem Biophys. . 2000;383:60–69. doi: 10.1006/abbi.2000.2048. [DOI] [PubMed] [Google Scholar]
- 36.Blais JD, Chin KT, Zito E, Zhang Y, Heldman N, Harding HP, Fass D, et al. A small molecule inhibitor of endoplasmic reticulum oxidation 1 (ERO1) with selectively reversible thiol reactivity. J Biol Chem. . 2010;285:20993–21003. doi: 10.1074/jbc.M110.126599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, et al. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. . 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 38.Ponsero AJ, Igbaria A, Darch MA, Miled S, Outten CE, Winther JR, Palais G, et al. Endoplasmic reticulum transport of glutathione by Sec61 is regulated by Ero1 and Bip. Mol Cell. . 2017;67:962–973.e5. doi: 10.1016/j.molcel.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeeshan H, Lee G, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci. . 2016;17:327. doi: 10.3390/ijms17030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. . 2008;15:234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, Han X, et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol. . 2018;9:1371. doi: 10.3389/fphar.2018.01371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. . 2021;31:107–125. doi: 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Desilva MG, Lu J, Donadel G, Modi WS, Xie H, Notkins AL, Lan MS. Characterization and chromosomal localization of a new protein disulfide isomerase, PDIp, highly expressed in human pancreas. DNA Cell Biol. . 1996;15:9–16. doi: 10.1089/dna.1996.15.9. [DOI] [PubMed] [Google Scholar]
- 44.Koivunen P, Horelli-Kuitunen N, Helaakoski T, Karvonen P, Jaakkola M, Palotie A, Kivirikko KI. Structures of the human gene for the protein disulfide isomerase-related polypeptide ERp60 and a processed gene and assignment of these genes to 15q15 and 1q21. Genomics. . 1997;42:397–404. doi: 10.1006/geno.1997.4750. [DOI] [PubMed] [Google Scholar]
- 45.Marcus N, Shaffer D, Farrar P, Green M. Tissue distribution of three members of the murine protein disulfide isomerase (PDI) family. Biochim Biophys Acta. . 1996;1309:253–260. doi: 10.1016/S0167-4781(96)00133-9. [DOI] [PubMed] [Google Scholar]
- 46.Ferrari DM, Soling HD. The protein disulphide-isomerase family: unravelling a string of folds. Biochem J. . 1999;339:1–10. doi: 10.1042/bj3390001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Lith M, Hartigan N, Hatch J, Benham AM. PDILT, a divergent testis-specific protein disulfide isomerase with a non-classical SXXC motif that engages in disulfide-dependent interactions in the endoplasmic reticulum. J Biol Chem. . 2005;280:1376–1383. doi: 10.1074/jbc.M408651200. [DOI] [PubMed] [Google Scholar]
- 48.Conway ME, Harris M. S-nitrosylation of the thioredoxin-like domains of protein disulfide isomerase and its role in neurodegenerative conditions. Front Chem. . 2015;3:27. doi: 10.3389/fchem.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andreu CI, Woehlbier U, Torres M, Hetz C. Protein disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett. . 2012;586:2826–2834. doi: 10.1016/j.febslet.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 50.Castillo V, Oñate M, Woehlbier U, Rozas P, Andreu C, Medinas D, Valdés P, et al. Functional role of the disulfide isomerase ERp57 in axonal regeneration. PLoS One. 2015, 10: e0136620 . [DOI] [PMC free article] [PubMed]
- 51.Di Risola D, Ricci D, Marrocco I, Giamogante F, Grieco M, Francioso A, Vasco‐Vidal A, et al. ERp57 chaperon protein protects neuronal cells from Aβ‐induced toxicity. J Neurochem. . 2022;162:322–336. doi: 10.1111/jnc.15655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paschen W, Gissel C, Linden T, Doutheil J. Erp72 expression activated by transient cerebral ischemia or disturbance of neuronal endoplasmic reticulum calcium stores. Metab Brain Dis. . 1998;13:55–68. doi: 10.1023/A:1020631029168. [DOI] [PubMed] [Google Scholar]
- 53.Linden T, Doutheil J, Paschen W. Role of calcium in the activation of erp72 and heme oxygenase-1 expression on depletion of endoplasmic reticulum calcium stores in rat neuronal cell culture. Neurosci Lett. . 1998;247:103–106. doi: 10.1016/S0304-3940(98)00278-X. [DOI] [PubMed] [Google Scholar]
- 54.Honjo Y, Ito H, Horibe T, Takahashi R, Kawakami K. Protein disulfide isomerase-immunopositive inclusions in patients with Alzheimer disease. Brain Res. . 2010;1349:90–96. doi: 10.1016/j.brainres.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 55.Chichiarelli S, Altieri F, Paglia G, Rubini E, Minacori M, Eufemi M. ERp57/PDIA3: new insight. Cell Mol Biol Lett. . 2022;27:12. doi: 10.1186/s11658-022-00315-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu S, Sankar S, Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today. . 2014;19:222–240. doi: 10.1016/j.drudis.2013.10.017. [DOI] [PubMed] [Google Scholar]