


Abstract
The cis–syn dimer is the major DNA photoproduct produced by UV irradiation. In order to determine the origin of the mutagenic property of the cis–syn dimer, we used NMR restraints and molecular dynamics to determine the solution structure of a DNA decamer duplex containing a wobble pair between the 3′-T of the cis–syn dimer and the opposite T residue (CS/TA duplex). The solution structure of the CS/TA duplex revealed that the 3′-T·T base pair of the cis–syn dimer had base pair geometry that was significantly different from the canonical Watson–Crick base pair and caused destabilization and conformational distortion of its 3′-region. However, a 3′-T·A base pair at the cis–syn dimer within this related DNA decamer maintains the normal Watson–Crick base pair geometry and causes little distortion in the conformation of its 3′-side. Our results show that in spite of its stable hydrogen bonding, the insertion of a T residue opposite the 3′-T of the cis–syn dimer is inhibited by structural distortion caused by the 3′-T·T base pair. This may explain why the frequency of the 3′-T→A transversion, which is the major mutation produced by the cis–syn dimer, is only 4%.
INTRODUCTION
The cis–syn cyclobutane pyrimidine dimer (cis–syn dimer) (Fig. 1A) and the pyrimidine(6-4)pyrimidone photoproduct [(6-4) adduct] are the two major classes of cytotoxic, mutagenic and carcinogenic DNA photoproducts induced by the UV portion of the solar spectrum (1–3). The cis–syn dimer is the most abundant DNA photoproduct and is repaired by various DNA repair enzymes (4–6). T4 endonuclease V (endo V) is a DNA repair enzyme that catalyzes the first step of the cis–syn dimer-specific base excision repair pathway (6). The crystal structure of the cis–syn dimer–endo V complex reveals that the DNA duplex exhibits a sharp kink at the central cis–syn dimer. This kink causes the adenine base opposite the 5′-T of the cis–syn dimer to flip out of the DNA duplex and be trapped in a pocket of the endo V enzyme (6). The results of a structural study of a DNA dodecamer duplex that contained a cis–syn dimer suggest that the unique BII type backbone linkage that flanks the 3′-side of the dimer might be an important structural element for recognition of the dimer by the cis–syn dimer-specific photolyase and endo V repair enzyme (7). In an in vitro assay, the (6-4) adduct is repaired about nine times faster than the cis–syn dimer by the non-specific Escherichia coli uvr(A)BC excinuclease (4). The binding affinities of the DNA damage binding proteins for the cis–syn dimer are also much lower than those of the (6-4) adduct (8,9). It has been suggested that the binding affinities of the DNA damage recognition proteins and repair enzymes for damaged DNA depend on the degree of DNA unwinding or bending caused by the lesions (6,10). The cis–syn dimer is produced directly by sunlight at a 5–10-fold higher rate than the (6-4) adduct (2,3). Thus, because of its high abundance and low repair rate, the cis–syn dimer might be the most mutagenic photoproduct in the cell.
Figure 1.

(A) Chemical structures of the cis–syn dimer. (B) DNA sequence context of the CS/TA duplex.
DNA photoproducts produced by UV irradiation interfere strongly with DNA replication and transcription if left unrepaired. The cis–syn dimer blocks DNA polymerization by E.coli DNA polymerase I (11). When an SOS response is induced, damaged regions of the DNA are bypassed by DNA polymerase and give rise to mutations caused by the insertion of an incorrect nucleotide opposite the lesion (12–15). This process is termed translesion replication (TR) and is responsible for mutagenesis in prokaryotes and eukaryotes (16). In SOS-induced E.coli cells, adenines were found to be incorporated into the site opposite 5′-T and 3′-T of the cis–syn dimer with frequencies of 98 and 94%, respectively (12). The frequency of mutations caused by the cis–syn dimer during TR is only 7%, and all targeted mutations are single nucleotide substitutions (12,17). About 90% of these mutations are targeted at the 3′-T of the cis–syn dimer, the first of these two thymine residues encountered by DNA polymerase (12). The most common mutation is the 3′-T→A transversion, which results from the misincorporation of a thymine residue opposite the 3′-T of the cis–syn dimer (12).
The most important properties in the determination of base insertion specificity during replication is the Gibbs free energy of base pairing between the template base and the incoming dNTP (16). In addition, as suggested by Echols and Goodman, similarity to the geometry of Watson–Crick base pairs is a second property that determines base selectivity (16). Echols and Goodman also suggest that this geometric mechanism is useful in considering the response of a DNA polymerase to a damaged base in a template. When the base pair between a damaged base and an incoming dNTP deviates from the Watson–Crick geometry, the insertion frequency of this dNTP is decreased during TR. Recent structural studies of photoproduct-containing DNA duplexes suggest that the conformational distortion of the duplex region formed between template and primer as well as hydrogen bonding between the template base and incoming dNTP are important components in the determination of an incoming dNTP opposite a DNA lesion during TR (18). Thus, structural studies of a DNA duplex that contains a cis–syn dimer in a mutated DNA sequence context are required to understand completely the mutagenic property of the cis–syn dimer. In this report, we have determined the three-dimensional solution structure of a DNA decamer duplex that contains a wobble base pair between the 3′-T of the cis–syn dimer and an opposed T residue (CS/TA duplex) (Fig. 1B). The conformational influence of the 3′-T·T base pair in the CS/TA duplex was compared with that of a 3′-T·A base pair in the cis–syn dimer-containing native CS/AA duplex (7,19). This structural comparison provides insight into the mechanism that determines base selection by DNA polymerase in the context of a cis–syn dimer-containing template.
MATERIALS AND METHODS
Sample preparation
The cis–syn dimer-containing DNA decamer was prepared by direct 254 nm UV irradiation of DNA oligomer in aqueous solution and purified as described (19,20). The CS/TA duplex (Fig. 1B) was prepared by dissolving the lesion-containing strand and the complementary strand at a 1:1 stoichiometric ratio in an aqueous solution containing 10 mM sodium phosphate (pH 6.6) and 200 mM NaCl.
NMR experiments
All NMR data sets generated with the CS/TA duplex were collected with a Bruker DMX-600 spectrometer (Korea Basic Science Institute, Taejon). Details of the NMR experiments and data processing can be found in our earlier published studies of a photoproduct-containing DNA duplex (19,20). NOE distance restraints from non-exchangeable protons were obtained from 2D-NOESY experiments with mixing times of 50, 80 and 160 ms in a D2O buffer solution. Exchangeable proton NOEs were determined using NOESY spectra in H2O buffer with 120 and 400 ms mixing times. Watson–Crick-type hydrogen bonding restraints were imposed on each base pair. The T6·T15 wobble pair was restrained by the hydrogen bonds between the T15-O4 and T6-imino proton and the T6-O2 and T15-imino proton.
Structure calculation
The structure of the CS/TA duplex was calculated using the program X-PLOR 3.1 (21) with restrained molecular dynamics (RMD) simulation. We initially generated the normal A- and B-form starting structures with modification of the cis–syn dimer at the T5-T6 position. These structures were subjected to the RMD and simulated annealing protocol. The first stage of computation began with energy minimization, followed by 10 ps of molecular dynamics (MD) at 1000 K. The force constants for the distance restraints were increased gradually over 10 cycles of 1 ps dynamics. The final values of the force constants were 130 kcal/mol Å2. The system was subsequently cooled to 300 K over 10 cycles of 0.5 ps dynamics followed by energy minimization. The final stage involved 20 ps of RMD at 300 K. The final structure was obtained by averaging the last 6 ps trajectories and subsequent energy minimization. Twenty-one structures (14 from the B-form and seven from the A-form initial structures) were chosen on the basis of the lowest NOE violations and total energies.
The mean structure of the RMD-refined structures was next optimized using full relaxation matrix refinement (based on NOE intensity) with X-PLOR. NOE volumes from 738 cross peaks of the three mixing times of 50, 80 and 160 ms were used as restraints. The first stage of computation began with 10 ps of MD at 500 K. The force constants for the distance restraints were decreased gradually to zero, and those of the intensity restraints were increased to 400 kcal/mol over 10 cycles of 0.5 ps dynamics. The system was subsequently cooled to 300 K over five cycles of 0.5 ps dynamics, followed by energy minimization. The final stage involved 10 ps of the MD at 300 K. The final structure was obtained by averaging the last 6 ps trajectories and subsequent energy minimization. Eight structures were chosen on the basis of the lowest total energies. The helical parameters of the refined structures were calculated using the program CURVES (22).
RESULTS AND DISCUSSION
NMR resonance assignment of the CS/TA duplex
Two-dimensional homonuclear 1H-NOESY spectra of the CS/TA duplex in D2O buffer were acquired at 1°C with 50, 80 and 160 ms mixing times. The non-exchangeable base and sugar protons were assigned according to their intraresidue and sequential nuclear Overhauser effect (NOE) connectivities. NOE spectroscopy (NOESY) data enabled assignment of all non-exchangeable protons except some H5′ and H5′′ protons that are not reported herein. The sequential NOE connectivities between base protons and their own and 5′-flanking sugar H1′ protons is shown for a NOESY in Figure 2A and B for the cis–syn dimer-containing strand and complementary strand, respectively. No sequential NOE cross peak was observed between A4-H1′ and T5-H6 resonances (boxed in Fig. 2A), indicating conformational deviation of the A4 or T5 residues from standard B-form helix. The H6 and 5-methyl resonances of the T5 and T6 residues were assigned from a unique NOE feature produced by four substituents of the cyclobutane ring of the cis–syn dimer (Fig. 3A). Because of the loss of aromaticity of the two thymine bases (T5, T6) upon formation of the cis–syn dimer, their H6 proton resonances were significantly upfield-shifted, and the sequential NOE cross peaks involving the cis–syn dimer occur in a different part of the spectrum (Fig. 2A, right panel). The A-H2 protons were assigned from NOE cross peaks of H2 protons to own and 3′-flanking H1′ protons.
Figure 2.

Expanded NOESY (300 ms mixing time) contour plots of the CS/TA duplex in D2O buffer at 1°C. (A) A typical region (base to H1′ protons) in the sequential NOE connectivity of the cis–syn dimer-containing strand and (B) the complementary strand.
Figure 3.

Topology of the cyclobutane ring of the cis–syn dimer. (A) NOE cross peaks between the H6 and methyl protons of the two T residues of the cis–syn dimer in the expanded NOESY spectrum in a D2O buffer at 1°C. (B) Structure of the cis–syn dimer in the mean structure of the CS/TA duplex. Interresidue distances between the H6 and 5-methyl carbon are indicated in the figure (in Å).
The exchangeable protons were assigned by analyzing NOESY data in an H2O buffer solution. Like the CS/AA duplex (19), the imino proton resonance of the 5′-T (T5) of the cis–syn dimer in the CS/TA duplex is shifted further upfield (11.94 p.p.m.) than that of the normal Watson–Crick A·T base pair. The two imino protons of the T6·T15 mismatch exhibited the strong NOE cross peak with each other. The chemical shift values of the imino protons of T6 (9.97 p.p.m.) and T15 (9.77 p.p.m.) were similar to those previously reported for hydrogen-bonded imino protons of the T·T wobble base pair (23). These include hydrogen bonding either between the T6-O2 and T15-O4 carbonyls or the T6-O4 and T15-O2 carbonyls (24). For the CS/TA duplex, the imino protons of the T6 and T15 residues formed hydrogen bonds to the O4 and O2 carbonyls of the opposite T residues, respectively, as shown by the NOE cross peak between T6-imino and T15 methyl resonances (Fig. 4A). In temperature-dependent imino proton spectra conducted in H2O buffer (Fig. 5), all imino resonances except those of the terminal base pairs were intact over 30°C, indicating that this duplex forms a stable double helix at room temperature. The imino proton resonance of the 3′-flanking A7·T14 base pair was exchanged more rapidly with solvent than that of the 5′-flanking A4·T17 base pair, as shown by the weaker NOE cross peak of the T14 imino proton with the opposite A-H2 to that of the T17 imino proton. This result indicates that the T6·T15 wobble pairing give rise to the conformational disruption of the 3′-side of the cis–syn dimer, thus causing the A7·T14 base pair to be weaker than the A4·T17 base pair. Chemical shifts of all the assigned proton resonances for the CS/TA duplex are listed in Table 1.
Figure 4.

Topology of the T6·T15 base pair. (A) NOE cross peaks between the imino and methyl protons of the T6 and T15 residues in the expanded NOESY spectrum in an H2O buffer at 1°C. (B) Chemical structure of the T6·T15 base pair as deduced by the NOE data.
Figure 5.

Temperature dependence of the imino proton resonances of the 1H-NMR spectra for the CS/TA duplex in an H2O buffer solution. The positions of nucleotides in the decamer duplex that give rise to the resonances are indicated. The experimental temperatures are shown at the right of the figure.
Table 1. Chemical shifts of proton resonances in the CS/TA duplex.
| Base | NH/NH2 | H2/H5/Me | H6/H8 | H1′ | H2′/H2′′ | H3′ | H4′ |
|---|---|---|---|---|---|---|---|
| C1 | 8.23/7.15 | 5.87 | 7.66 | 5.73 | 2.03/2.45 | 4.74 | 4.11 |
| G2 | 13.14 | – | 8.01 | 5.95 | 2.70/2.80 | 5.01 | 4.39 |
| C3 | 8.42/6.66 | 5.49 | 7.44 | 5.63 | 2.14/2.46 | 4.89 | 4.24 |
| A4 | – | 7.87 | 8.38 | 6.41 | 2.73/2.87 | 5.04 | 4.48 |
| T5 | 11.94 | 0.75 | 4.40 | 5.45 | 1.97/2.37 | 4.77 | 4.23 |
| T6 | 9.97 | 1.45 | 4.05 | 5.22 | 1.72/1.84 | 4.72 | 3.84 |
| A7 | – | 7.87 | 8.35 | 6.19 | 2.74/2.82 | 5.01 | 4.37 |
| C8 | 8.28/6.74 | 5.29 | 7.36 | 5.55 | 2.05/2.35 | 4.85 | 4.16 |
| G9 | 13.12 | – | 7.88 | 6.00 | 2.61/2.76 | 5.01 | 4.40 |
| C10 | 8.30/6.70 | 5.17 | 7.34 | 6.16 | 2.23/2.23 | 4.54 | 4.04 |
| G11 | 13.11 | – | 8.00 | 5.99 | 2.66/2.82 | 4.88 | 4.31 |
| C12 | 8.50/6.64 | 5.35 | 7.40 | 5.82 | 2.10/2.48 | 4.92 | 4.23 |
| G13 | 13.00 | – | 8.01 | 6.06 | 2.71/2.81 | 5.01 | 4.42 |
| T14 | 14.16 | 1.59 | 7.34 | 6.05 | 2.21/2.53 | 4.85 | 4.31 |
| T15 | 9.77 | 1.61 | 7.20 | 5.72 | 1.80/2.12 | 4.87 | 4.05 |
| A16 | – | 7.72 | 8.34 | 6.19 | 2.75/2.86 | 5.00 | 4.37 |
| T17 | 13.69 | 1.50 | 7.23 | 5.65 | 2.10/2.41 | 4.87 | 4.17 |
| G18 | 12.78 | – | 7.92 | 5.91 | 2.65/2.71 | 5.00 | 4.41 |
| C19 | 8.50/6.73 | 5.45 | 7.40 | 5.78 | 1.96/2.37 | 4.86 | 4.21 |
| G20 | 13.22 | – | 7.98 | 6.18 | 2.39/2.68 | 4.72 | 4.21 |
Overall structure of the CS/TA duplex
The solution structure of the CS/TA duplex was calculated according to the protocols outlined in Materials and Methods. A total of 386 distances were restrained in the RMD calculation; these distance restraints consist of 275 interproton distances derived from NOESY data in D2O buffer, 24 interproton distances obtained from H2O-NOESY cross peaks, and 87 distances for the Watson–Crick base pairs of the flanking residues. A converged subset of 21 structures refined by RMD was identified on the basis of low NOE violations and total energies. These structures exhibited pairwise root-mean-squared deviation (rmsd) values of 1.15 ± 0.28 Å for all heavy atoms. Full relaxation matrix refinements of the mean structure yielded a well-converged subset of eight refined structures. Eight superimposed refined structures of the CS/TA duplex exhibited pairwise rmsd values of 0.87 ± 0.18 Å for all heavy atoms. The R1/6 value for NOE volume intensities was 0.028 ± 0.001. Statistical estimations are shown in Table 2. Figure 6A shows a stereo view of eight individual structures superimposed on the mean structure of the CS/TA duplex. A ribbon representation was applied to the phosphate backbone of the mean structure in order to emphasize the distortion of the phosphate backbone caused by formation of the cyclobutane ring and the T6·T15 wobble pairing. Similarly to the CS/AA duplex (7), the backbone distortion of the CS/TA duplex included minor groove widening of the 3′-side of the cis–syn dimer. However, in contrast to the CS/AA duplex (7), narrowing of the minor groove was not observed at the 5′-side of the cis–syn dimer in the CS/TA duplex. The formation of the cis–syn dimer causes slight unwinding (15°) and bending (9°) of the DNA helix in the CS/AA duplex (19). Our structural calculation showed that the 3′-T·T mismatch of the cis–syn dimer maintained the helical bending (12 ± 7°), but diminished the helical unwinding.
Table 2. Structure refinement statistics and analysis of CS/TA duplex.
| Restrained molecular dynamics | |
| Number of distance restraints | 386 |
| Number of accepted structures | 21 |
| Pairwise rmsd (Å) | 1.15 ± 0.28 |
| NOE violation energy | 69.2 ± 2.4 |
| Number of NOE violations: | |
| Greater than 0.2 Å | 0.0 ± 0.0 |
| Greater than 0.1 Å | 15.9 ± 1.5 |
| Rmsd from restraints | 0.038 ± 0.001 |
| Relaxation matrix refinement | |
| Number of intensity restraints | 684 |
| Number of accepted structures | 8 |
| Pairwise rmsd (Å) | 0.87 ± 0.18 |
| R1/6-factor | 0.028 ± 0.001 |
| X-PLOR energies (kcal/mol): | |
| Total | –773.8 ± 14.0 |
| Bond length | 52.1 ± 1.1 |
| Bond angle | 180.8 ± 2.4 |
| Dihedral angle | 204.3 ± 1.7 |
| Improper angle | 22.1 ± 0.1 |
| Hydrogen bond | –160.8 ± 0.9 |
| Van der Waals | –133.5 ± 0.9 |
| Electrostatic | –1119.8 ± 3.8 |
| Relaxation | 172.2 ± 1.4 |
| Rmsd from ideal geometry: | |
| Bond length | 0.016 ± 0.000 |
| Bond angle | 3.146 ± 0.053 |
| Improper angle | 6.019 ± 0.141 |
Figure 6.
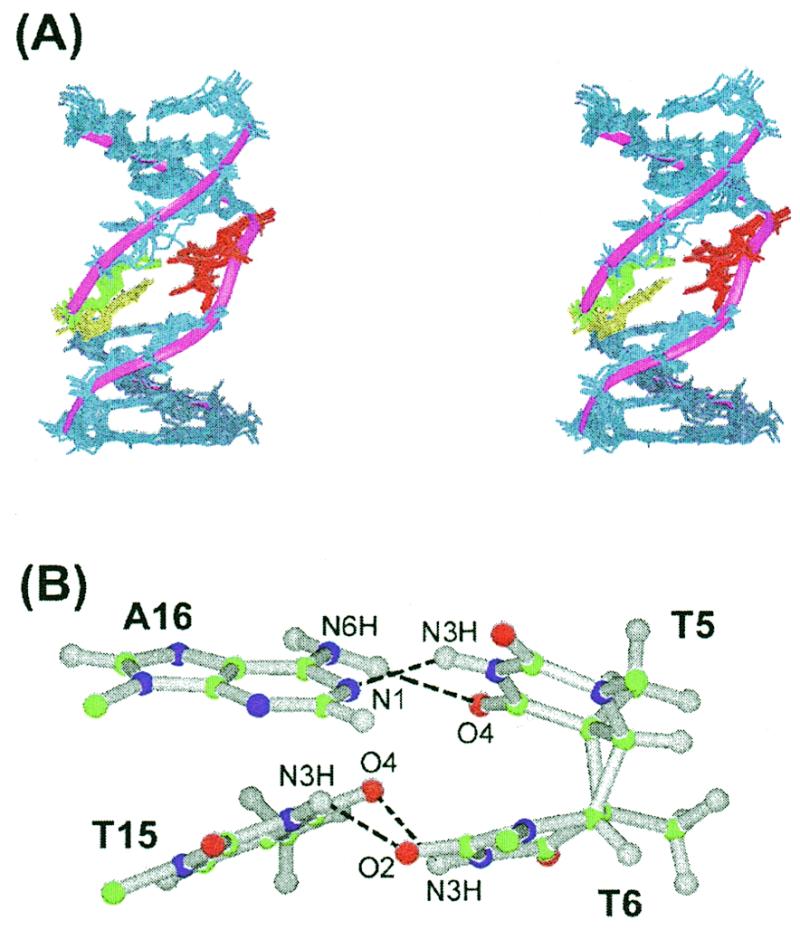
Solution structure of the CS/TA duplex. (A) Superimposed stereo view of eight intensity-refined structures of the CS/TA duplex. The two thymine residues (T5, T6) of the cis–syn dimer are colored red, and their opposite T15 and A16 residues are colored yellow and green, respectively. The other flanking residues are colored dark blue. A ribbon representation is applied to the phosphate backbone of the mean structure. The hydrogen atoms are excluded in this figure. (B) Ball and stick view of the two base pairs of the cis–syn dimer. Balls are colored using the accepted atomic color representation: gray, hydrogen; green, carbon; blue, nitrogen; red, oxygen. The dotted lines indicate the hydrogen bonds determined by the program INSIGHT II.
The cis–syn dimer has a cyclobutane ring formed by the C5–C5 and C6–C6 covalent linkages between the two thymine bases (Fig. 1A). In crystal form, the cyclobutane ring of an isolated cis–syn dimer of a TpT dinucleotide displays a left-handed (CB–) puckering (25), while in solution it has a flexible conformation that interconverts between a right-handed (CB+) and CB– puckering (26). Right-handed CB+ puckering exists only when the cis–syn dimer is within a DNA duplex (19,27). In the CS/TA duplex, the cyclobutane ring of the cis–syn dimer also adopted the CB+ puckering (Fig. 3B), evidenced by that fact that we observed a stronger NOE cross peak for T5-H6↔T6-Me than for T5-Me↔T6-H6 (Fig. 3A).
Base pairing of the cis–syn dimer
The T5·A16 base pair of the cis–syn dimer in the CS/TA duplex showed Watson–Crick base pairing (Fig. 6B), as deduced from the NOE cross peak between the A16-H2 and T5-imino proton. In the CS/TA duplex, the formation of the cis–syn dimer did cause structural distortion of the 5′-T·A base pair as in the CS/AA duplex (19), confirmed by the values of the propeller twist (–45 ± 5°), opening (16 ± 4°) and buckle (12 ± 5°) angles. However, the overall geometry of this base pair is similar to that of the canonical Watson–Crick base pair (Fig. 7).
Figure 7.

Base pairing geometry of (A) a normal A·T base pair, (B) the 5′-T·A (upper) and 3′-T·T (lower) base pairs of the CS/TA duplex and (C) the 5′-T·A (upper) and 3′-T·A (lower) base pairs of the CS/AA duplex (19). The C1′–C1′ distances and C1′-involving angles are shown on the figure.
In the CS/TA duplex, the T6·T15 wobble pair of the cis–syn dimer forms hydrogen bonds between the T6-imino and T15-O4 and between the T15-imino and T6-O2 (Fig. 6B). This hydrogen bonding feature is consistent with the NMR results described above. The calculated structure of the CS/TA duplex showed that the T6·T15 base pair was distorted, confirmed by the values of the shear (2.4 ± 0.1 Å), stretch (–1.3 ± 0.1 Å) and stagger (2.1 ± 0.1 Å) values and the geometry of this base pair differed significantly from the canonical Watson–Crick base pairing geometry. This can be confirmed by comparing the C1′–C1′ distances and C1′-involving angles. The T6-C1′ to T15-C1′ distance, and the T6-N1, T6-C1′, to T15-C1′ and T15-N1, T15-C1′, to T6-C1′ angles are 8.8 ± 0.1 Å, 76 ± 2° and 45 ± 2°, respectively, compared to 11.1 Å, 50° and 51° for the canonical Watson–Crick T·A base pair (Fig. 7). However, although the structure of the 3′-T·A base pair in the CS/AA duplex is distorted (19), the geometry of this base pair is similar to that of a canonical Watson–Crick base pair. The T6-C1′ to A15-C1′ distance, and the T6-N1, T6-C1′, to A15-C1′ and the A15-N9, A15-C1′, to T6-C1′ angles in the CS/AA duplex (19) are 10.8 Å, 54° and 48°, respectively (Fig. 7).
Conformational feature of the cis–syn dimer and its 3′-flanking side
The glycosidic bond torsion angles of both T residues (T5: χ = –84 ± 4°; T6: χ = –94 ± 3°) of the cis–syn dimer were similar to those in the CS/AA duplex (7). In the CS/AA duplex (7), the backbone conformation involving the phosphorous atom between the 3′-T of the cis–syn dimer and its 3′-neighboring A residue is significantly distorted. The gauche– orientation (–80°) of the ɛ angle and the increased rise (4.7 Å) at this step are related to the BII conformation of DNA (7). However, in the CS/TA duplex the ɛ angle exhibited the trans orientation (–142 ± 4°) at the corresponding step, and the value (4.0 ± 0.2 Å) of the rise at this step was less than that of the CS/AA duplex (7). The structural features indicate that the 3′-T·T mispairing of the cis–syn dimer diminished the BII conformation at its 3′-side. The interphosphate distance of the cis–syn dimer is similar in both the CS/TA (6.4 ± 0.1 Å) and CS/AA duplexes (6.2 Å), although the backbone conformations of the 3′-T residue in two duplexes are in contrast with each other. In the crystal structure of the cis–syn dimer–endo V complex (6), this interphosphate distance (6.3 Å) is shortened as compared with the normal B-DNA (7.8 Å). It was suggested that the hydrogen bonds between these two phosphates and the side chain of Arg-3 of the endo V may play the key role in the cis–syn dimer recognition (6). In the crystal structure, the A residue opposite the 5′-T is completely flipped out from the interior of the DNA duplex and is accommodated into the cavity on the surface of the endo V. This binding feature of the cis–syn dimer by the endo V may be correlated to the structural distortion of the 5′-T·A base pair in both the CS/TA and CS/AA duplexes.
The formation of the four-membered ring of the cis–syn dimer causes narrowing of the minor groove at the cis–syn dimer site when the two thymine bases are base paired with adenine residues (7). In contrast, when the DNA duplex contained the 3′ T·T mispairing of the cis–syn dimer, distortion of the phosphate backbone at the T-T step of the cis–syn dimer diminished this narrowing of the minor groove at the dimer site, as confirmed by comparing some distances. For the CS/TA duplex, the T5-C1′ to T6-C1′, T5-C2′ to T6-C4′, and T5-C3′ to A7-C5′ distances are 4.0 ± 0.1, 4.7 ± 0.1 and 4.6 ± 0.1 Å, respectively, compared to 3.6, 3.8 and 3.8 Å for the corresponding distances in the CS/AA duplex (7).
The two base pairs of the cis–syn dimer show distinct stacking features with respect to their adjacent base pairs (Fig. 8). The stacking interaction of T6·T15/A7·T14 base pair step was disrupted, as evidenced by its positive tilt (10 ± 2°) and negative roll (–17 ± 4°), while the A4·T17/T5·A16 step was well stacked. The non-planar nature of the T6 base caused a significant disruption of the A7·T14 base pair. This destabilization was confirmed by the observed temperature dependence of the imino resonance (Fig. 5) and the weakness of NOE cross peak between the T14 imino proton and A7-H2 in the NOESY spectrum in H2O buffer. Taken together, these results indicate that the 3′-T·T wobble pairing of the cis–syn dimer disrupts the 3′-flanking base pair, rather than the cis–syn dimer itself or the 5′-flanking base pair.
Figure 8.
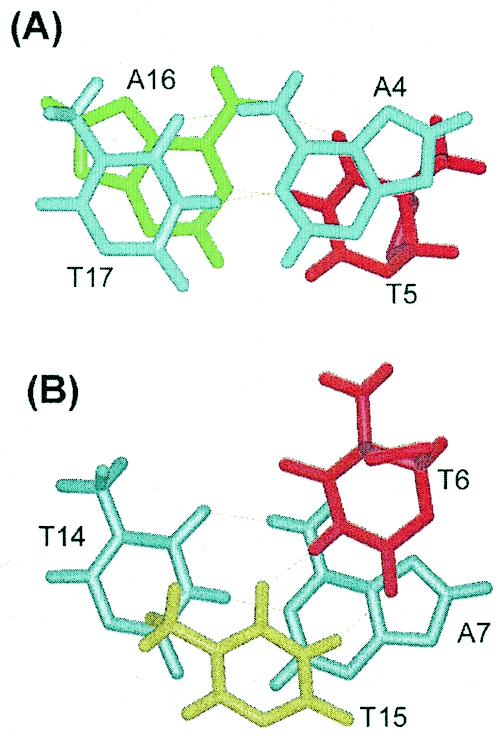
Comparative stick view of the stacking interactions between (A) the A4·T17 and T5·A16 base pairs and between (B) the T6·T15 and A7·T14 base pairs.
Structural basis for the mutagenic property of the cis–syn dimer
When an SOS response is induced, the cis–syn dimer is bypassed by DNA polymerase and produces base substitution mutations (12,17). The most abundant mutation, the 3′-T→A transversion, is caused by mispairing between the 3′-T of the cis–syn dimer and an incoming T residue. Our study reveals that the incoming T residue is able to form hydrogen bonds with the opposite 3′-T of the cis–syn dimer. This finding can account for why the 3′-T→A transversion mutation is sometimes caused by the cis–syn dimer during TR. However, an A residue has been shown to be inserted into the site opposite the 3′-T of the cis–syn dimer with a frequency of 94% (12). For a non-instructional DNA lesion (such as an abasic site), an A residue is incorporated with a frequency of ∼70% during TR in E.coli, in accordance with the ‘A rule’ (28). Thus, the observed accurate replication at the site opposite the 3′-T of the cis–syn dimer may not be the result of the non-instructive insertion of adenine at this position by DNA polymerase.
A comparison of the solution structure of the CS/TA duplex with those of the CS/AA duplexes (7,19) can explain the much higher insertion frequency for A residues than for T residues opposite the 3′-T site of the cis–syn dimer. First, although formation of the cis–syn dimer distorts the conformation of its 3′-T residue, the 3′-T·A base pair of the cis–syn dimer maintains the Watson–Crick base pair, and its geometry is also similar to that of the canonical Watson–Crick T·A base pair. Echols and Goodman suggest that base pair geometry that is equivalent to the canonical Watson–Crick base pair is an important component in the determination of the fidelity of TR as well as normal replication (16). However, the geometry of the 3′-T·T base pair of the cis–syn dimer deviated from the canonical Watson–Crick geometry more severely than did the 3′-T·A base pair. This base pairing geometry may explain why during TR DNA polymerase prefers to insert an A residue opposite the 3′-T of the cis–syn dimer rather than a T residue (the frequency of insertion is 94% for A and 4% for T), although both A and T residues are able to form hydrogen bonds with the opposite 3′-T of the cis–syn dimer.
Secondly, the stacking interaction between the 3′-T·T base pair of the cis–syn dimer and 3′-flanking A·T base pairs in the CS/TA duplex was disrupted more than the corresponding step in the CS/AA duplex (19). The 3′-flanking A·T base pair was unstable only in the CS/TA duplex and not in the CS/AA duplex. Therefore, the 3′-T·T base pair of the cis–syn dimer caused the destabilization of the 3′-flanking base pair. The stability of the 3′-flanking base pair of the lesion may be an important physical property in the selection of the dNTP to be incorporated opposite the lesion by DNA polymerase. This disruption of the 3′-side of the cis–syn dimer in the CS/TA duplex can explain inhibition of insertion of a T residue opposite the 3′-T of the cis–syn dimer during TR.
In summary, using experimental NMR restraints and MD, we have demonstrated the solution structural features unique to the 3′-T·T base pair of the cis–syn dimer in the CS/TA duplex. The structural comparison between the 3′-T·T and 3′-T·A base pairs of the cis–syn dimer offers an explanation of its mutagenic property. The T·T base pair on the 3′-side of the cis–syn dimer had base pair geometry that was significantly different from the canonical Watson–Crick base pair and caused destabilization as well as conformational distortion of its 3′-region. In contrast, a 3′-T·A base pair at the cis–syn dimer within the CS/AA duplex maintains the normal Watson–Crick base pair geometry and causes little distortion in the conformation of its 3′-side. Our results show that in spite of its hydrogen bonding, the insertion of a T residue opposite the 3′-T of the cis–syn dimer is inhibited by structural distortion caused by the 3′-T·T base pair. This may explain why the frequency of the 3′-T→A transversion, which is the major mutation produced by the cis–syn dimer, is only 4% during TR.
Coordinate
The coordinate for the minimized mean structure of the CS/TA duplex has been deposited in the Protein Data Bank under accession code 1ql5.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by the Korea Science and Engineering Foundation through the Center for Molecular Catalysis at Seoul National University and by the Molecular Medicine Research Group Program (MOST, 98-J03-01-01-A-01).
REFERENCES
- 1.Patrick M.H. and Rahn,R.O. (1976) In Wang,S.Y. (ed.), Photochemistry and Photobiology of Nucleic Acids. Vol. II. Academic Press, New York, pp. 35–95.
- 2.Mitchell D.L. (1988) Photochem. Photobiol., 48, 51–57. [DOI] [PubMed] [Google Scholar]
- 3.Brash D.E. (1988) Photochem. Photobiol., 48, 59–66. [DOI] [PubMed] [Google Scholar]
- 4.Svoboda D.L., Smith,C.A., Taylor,J.-S. and Sancar,A. (1993) J. Biol. Chem., 268, 10694–10700. [PubMed] [Google Scholar]
- 5.Sancar A. (1994) Biochemistry, 33, 2–9. [DOI] [PubMed] [Google Scholar]
- 6.Vassylyev D.G., Kashiwagi,T., Mikami,Y., Ariyoshi,M., Iwai,S., Ohtsuka,E. and Morikawa,K. (1995) Cell, 83, 773–782. [DOI] [PubMed] [Google Scholar]
- 7.McAteer K., Jing,Y., Kao,J., Taylor,J.-S. and Kennedy,M.A. (1998) J. Mol. Biol., 282, 1013–1032. [DOI] [PubMed] [Google Scholar]
- 8.Reardon J.T., Nichols,A.F., Keeney,S., Smith,C.A., Taylor,J.-S., Linn,S. and Sancar,A. (1993) J. Biol. Chem., 268, 21301–21308. [PubMed] [Google Scholar]
- 9.Burns J.L., Guzder,S.N., Sung,P., Prakash,S. and Prakash,L. (1996) J. Biol. Chem., 271, 11607–11610. [DOI] [PubMed] [Google Scholar]
- 10.Oh E.Y. and Grossma,L. (1986) Nucleic Acids Res., 14, 8557–8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan G.L., Deotsch,P.W. and Haseline,W.A. (1985) Biochemistry, 24, 5723–5728. [DOI] [PubMed] [Google Scholar]
- 12.Banerjee S.K., Christensen,R.B., Lawrence,C.W. and LeClerc,J.E. (1988) Proc. Natl Acad. Sci. USA, 85, 8141–8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LeClerc J.E., Borden,A. and Lawrence,C.W. (1991) Proc. Natl Acad. Sci. USA, 88, 9685–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuchs R.P.P. and Napolitano,R.L. (1998) Proc. Natl Acad. Sci. USA, 95, 13114–13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomer G., Reuven,N.B. and Livneh,Z. (1998) Proc. Natl Acad. Sci. USA, 95, 14106–14111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Echols H. and Goodman,M.F. (1991) Annu. Rev. Biochem., 60, 477–511. [DOI] [PubMed] [Google Scholar]
- 17.Smith C.A., Wang,M., Jiang,N., Che,L., Zhao,X., Taylor,J.-S. (1996) Biochemistry, 35, 4146–4154. [DOI] [PubMed] [Google Scholar]
- 18.Lee J.-H., Hwang,G.-S. and Choi,B.-S. (1999) Proc. Natl Acad. Sci. USA, 96, 6632–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J.-K., Patel,D.J. and Choi,B.-S. (1995) Photochem. Photobiol., 62, 44–50. [DOI] [PubMed] [Google Scholar]
- 20.Lee J.-H., Hwang,G.-S., Kim,J.-K. and Choi,B.-S. (1998) FEBS Lett., 428, 269–274. [DOI] [PubMed] [Google Scholar]
- 21.Brünger A.T. (1992) X-PLOR Version 3.1: A System for X-ray Crystallography and NMR. Yale University Press, New Haven, CT.
- 22.Lavery R. and Sklenar,H. (1988) J. Biomol. Struct. Dyn., 6, 63–91. [DOI] [PubMed] [Google Scholar]
- 23.Trotta E. and Paci,M. (1998) Nucleic Acids Res., 26, 4706–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kouchakdjian M., Li,B.F.L., Swann,P.F. and Patel,D.J. (1988) J. Mol. Biol., 202, 139–155. [DOI] [PubMed] [Google Scholar]
- 25.Cadet J., Voituriez,L., Hruska,F.E. and Grand,A. (1985) Biopolymers, 24, 897–903. [DOI] [PubMed] [Google Scholar]
- 26.Kim J.-K. and Alderfer,J.L. (1992) J. Biomol. Struct. Dyn., 9, 706–718. [Google Scholar]
- 27.Taylor J.-S., Garrett,D.S., Brockie,I.R., Svoboda,D.L. and Telser,J. (1990) Biochemistry, 29, 8858–8866. [DOI] [PubMed] [Google Scholar]
- 28.Jing Y., Kao,J.F.-L. and Taylor,J.-S. (1998) Nucleic Acids Res., 26, 3845–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]