


Abstract
ATM is a gene mutated in the human disease ataxia telangiectasia with reported homologues in yeast, Drosophila, Xenopus and mouse. Whenever mutants are available they all indicate a role of this gene family in the cellular response to DNA damage. Here, we present the identification and molecular characterisation of the first plant homologue of ATM. The genomic locus of AtATM (Arabidopsis thaliana homologue of ATM) spans over 30 kb and is transcribed into a 12 kb mRNA resulting from the splicing of 79 exons. It is a single copy gene and maps to the long arm of chromosome 3. Transcription of AtATM is ubiquitous and not induced by ionising radiation. The putative protein encoded by AtATM is 3856 amino acids long and contains a phosphatidyl inositol-3 kinase-like (Pi3k-l) domain and a rad3 domain, features shared by other members of the ATM family. The AtAtm protein is highly similar to Atm, with 67 and 45% similarity in the Pi3k-l and rad3 domains respectively. Interestingly, the N-terminal portion of the protein harbours a PWWP domain, which is also present in other proteins involved in DNA metabolism such as human mismatch repair enzyme Msh6 and the mammalian de novo methyl transferases, Dnmt3a/b.
INTRODUCTION
In all living cells, DNA damage induces a variety of responses which contribute to maintaining the integrity of the genome. In eukaryotic cells, these include DNA repair, cell cycle arrest and, for multicellular organisms, apoptosis (1). DNA repair and cell cycle arrest are distinct but complementary genome-preserving responses whereas DNA damage-induced apoptosis prevents the transmission of misrepaired lesions which may have oncogenic potential. Apoptosis may therefore be seen as a genome-preserving response at the level of the organism.
Recent advances in the understanding of the signalling pathways leading to these responses have stemmed from studies of the human disease ataxia telangiectasia (AT). AT patients present a number of symptoms, some of which—e.g. impaired fertility, proneness to develop certain cancers, hypersensitivity to ionising radiation (IR)—suggest a relation to DNA damage (2,3). AT cells are hypersensitive to IR and defective in G1/S and S-phase DNA damage checkpoints, the latter resulting in radio-resistant DNA synthesis (4,5). These defects probably contribute to the radiation sensitive phenotype of AT cells, but there is also evidence for a defect in DNA repair (6,7)
The cloning of the gene responsible for the AT disease, ataxia telangiectasia mutated (ATM), revealed that Atm is a large protein with a Pi-3 kinase-like (Pi3k-l) domain at its C-terminus (8,9), a feature shared by the cell cycle regulators Tor1 and Tor2 from yeast and their mammalian counterpart Frap/Raft (10–13). Whereas the Tor family of proteins are not involved in the control of DNA damage responses, other proteins with a Pi3k-l domain and more closely related to Atm were found to play a role in DNA damage responses. These include Tel1 and Mec1 from budding yeast (14,15), rad3 from fission yeast (16), mei-41 from Drosophila (17), and the mammalian proteins DNA-PKcs (18) and Atr (16,19). They are all very large proteins of over 2500 amino acids with little homology outside the Pi3k-l domain except in a poorly defined region of approximately 1000 amino acids which includes the rad3 domain.
Despite the presence of a Pi3k-l domain, Atm and other members of the family appear to be protein rather than lipid kinases. For example, p53 was shown to be phosphorylated in vitro on Ser15 by Atm (20,21). Because Ser15 phosphorylation is involved in p53 stabilisation, this may explain the delayed accumulation of p53 in AT cells in response to IR, as well as the G1/S checkpoint defect (22). Other potential phosphorylation targets include Replication Protein A (RPA) and the oncogene protein c-abl (23–25). In budding yeast, Mec1 is necessary for the phosphorylation of Rad53, a DNA damage checkpoint kinase (26) and overexpression of Tel1 partially rescues mec1 mutants, including by restoring the phosphorylation of Rad53 (14,27). This indicates that both yeast Atm homologues have partially overlapping roles. In addition, phosphorylation of Rpa and Rad9 after DNA damage requires the presence of active Mec1 (28,29). Also, in fission yeast, activation of the checkpoint kinases cds1 (a RAD53 homologue) and chk1 depends on rad3 (30). The identification of chk1 and cds1 structural and functional homologues in mammals further underlines the conservation of the DNA damage checkpoint pathways among eukaryotic organisms (31,32).
This conservation led us to postulate the existence of similar mechanisms in plants. We describe here the cloning an Arabidopsis thaliana homologue of ATM.
MATERIALS AND METHODS
New sequence
EMBL accession number AJ250248. Arabidopsis thaliana homologue of ATM.
Plant material, cell culture and irradiation experiments
Arabidopsis thaliana var. Colombia plants were cultivated in a growth chamber on a 16/8 h light/dark cycle. Arabidopsis thaliana var. Colombia cell suspension (33) was grown in liquid medium described by Chandler (34) at 28°C in an orbital shaker (120 r.p.m.) and under continuous light (60 µmol photons/m2/s). Cells were collected in the exponential phase of growth and irradiated with various doses of γ rays using a 60Co source at a dose rate of 30 Gy/min. For time course analysis, cells were put back in the shaker for the required time period, filtered, dried and flash-frozen in liquid nitrogen. Samples were stored at –80°C or immediately ground for RNA extraction.
Physical mapping and sequencing of the BAC clone
The insert of EST number T43304 was hybridised to the TAMU BAC library filter. Six positive clones were identified (T1F6, T3G4, T23E4, T24C20, T25D10, T28I7). Corresponding IGF BAC clones were found using the BAC fingerprint database (http://genome.wustl.edu/gsc/arab/arabidopsis.html ). The physical map position was then determined with the IGF mapping table (http://www.mpimp-golm.mpg.de/101/mpi_mp_map/bac.html ).
Sequencing of BAC clone T24C20 was done at the Genoscope, as part of the European contribution to the Arabidopsis Genome Initiative. 20 µg of DNA were sheared using a HydroShear apparatus (Genemachines) and the resulting fragments were repaired using T4 DNA polymerase. The fragments were ligated to BstXI adaptors and separated on a preparative 0.4% LMP agarose gel (FMC). The DNA fractions of 3 kb were eluted on Qiaquick (Qiagen) and then ligated to a BstXI-digested pcDNA2.1 plasmid (Invitrogen) in a 10× insert/1× vector ratio before transformation of electrocompetent Escherichia coli strain DH10B (Gibco BRL). We isolated 800 subclones (seven genome equivalents) and sequenced the end clones on a LICOR 4200 sequencer and assembled the sequences using Phred and Phrap software, which resulted in the construction of a scaffold composed of several contigs. The gaps and poor quality regions were sequenced by primer walking with dye terminators on an automated ABI 377 sequencer (PE-Applied Biosystems). The resulting sequence was then analysed by comparing restriction digest using three different restriction enzymes to the restriction pattern deduced from the sequence.
RNA isolation
Total RNA was prepared from 2 g of frozen plant tissues (20 day-old plants) and from cultured cells according to Kloppstech et al. (35). Poly(A)+ mRNA was prepared as described by Montané et al. (36).
Reverse transcription (RT)–PCR and determination of transcript structure
RT–PCR was performed on poly(A)+ mRNA (1 µg) with ExpandTM Reverse Transcriptase and specific primers according to the manufacturer’s instructions (Boehringer Mannheim, Germany). Based on genomic sequence and structure prediction (GENSCAN software) 86 PCR primers were designed. The sequence of these primers is available from the authors on request. The first-strand cDNA synthesis reaction products were amplified by PCR using various primer combinations. The initial denaturation was done at 94°C for 2 min, then amplification was performed for at least 30 cycles with a denaturation time of 30 s at 94°C, followed by annealing at different temperatures (depending on primers) and extension for different times at 72°C.
For rapid amplification of cDNA ends (5′ RACE) experiments, first-strand cDNA synthesis was performed as described above, with primer AtATM63 (5′-TCTGTTCGCTGAACGA-CAACGCGAAG-3′), and followed by a phenol/chloroform extraction. A string of deoxyguanosine was added at the 3′ end of the cDNA with DNA Terminal Transferase (New England Biolabs) according to the manufacturer’s instructions and a first PCR was performed with 250 pmol of primer AtATM61 (5′-TGCTGAGTGTATCTGCCGTCATAGC-3′) and primer AtATM83 [5′-(GA)12GCTCACTAGT(C)14-3′] using Taq DNA polymerase (Sigma). The initial denaturation was done at 94°C for 2 min, then amplification was performed for 30 cycles with a denaturation time of 30 s at 94°C, followed by annealing at 55°C for the first 5 cycles and at 63°C for the remaining 25 cycles, and extension for 2 min at 72°C. Following the first PCR, 1 µl of a 10-fold dilution of the reaction mixture was added to the second PCR reaction mixture containing primers AtATM83 and AtATM84 (5′-CGAAGAAGCGAACCACGAAACAAGATGG-3′). The initial denaturation was done at 94°C for 2 min, then amplification was performed for 30 cycles with denaturation time of 30 s at 94°C, followed by annealing at 62°C for 30 s and extension for 90 s at 72°C.
PCR products were sequenced with the BigDye terminator kit (PE-Applied Biosystems) and analysed on an ABI Prism 310 sequencing machine (PE-Applied Biosystems).
Semi-quantitative RT–PCR
For semi-quantitative RT–PCR, 1 µg of total RNA from tissues was reverse transcribed as described above using 5 µM of primer 5′-VN-(T)11-3′, and cDNA were diluted 2-fold. Aliquots of each dilution (1 µl) were used for PCR with either 5 µM AtATM (Atatm89 5′-CTACCTTTCTATTGGTATCCTCTCT-CCTT-3′ and Atatm14 5′-TCTGCATTGGTTTCGCTTATC-3′) or ACT8 (act-8.3 5′-GCACTTTCCAGCAGGTATGGATCT-CTAAGGCA-3′ and act-8.4 5′-CCGGAAAGTTTCTCACA-TAGTGCAC-3′) specific primers, in the presence of 250 µM of each dNTP and 1 U of Taq polymerase (Sigma) in 10 mM Tris–HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2 and 0.001% gelatin. Cycling conditions were identical to those set out above for cDNA cloning except that the number of cycles was reduced (25 for act-8 primers and 30 for AtATM primers). Reaction products were separated on 1% agarose gels, transferred to a Nylon membrane (Hybond N+, Amersham Life Sciences), and hybridized with 32P-labelled fragments. Hybridisation signals were quantified using a PhosphorImager (Molecular Dynamics)
Northern blot analysis
For northern blot analysis, poly(A)+ RNA (5 µg/lane) were separated on formaldehyde gels in 1× MOPS buffer and transferred to a Nylon membrane (Hybond N+, Amersham Life Sciences) as described by Sambrook et al. (37). Gels were stained with ethidium bromide to ensure that equal amounts had been loaded. Hybridisation was performed overnight at 65°C with 32P-labelled fragments in a solution containing 0.5 M NaH2PO4 pH 7.2, 5% SDS, 5 mM EDTA. Labelling of probes was performed with the Megaprime DNA labelling system (Amersham Life Sciences) using 50 µCi of [α-32P]dCTP. Filters were washed twice at 65°C for 10 min in 1× SSC, 0.1% SDS, and 10 min in 0.2× SSC, 0.1% SDS. Filters were exposed to X-ray film (Kodak) or to an imaging plate.
RESULTS
We first found an Arabidopsis EST (GenBank accession number T43304) with potential homologies to ATM by a word search in the dbEST database. The translated EST sequence showed similarities to a domain just upstream of the Pi3k-l domain. The EST clone was sequenced and found to contain a 1.7 kb insert corresponding to the 3′ end of a mRNA encoding a Pi3K-l domain. Initial BLAST searches (38) with this sequence confirmed a strong similarity with Atm-like proteins and we named it AtATM (A.thaliana homologue of ATM). Preliminary northern hybridisation showed the presence of a 12 kb mRNA (see below). We screened an Arabidopsis cDNA library (39) to obtain larger clones and found one clone with a 3.4 kb chimeric insert of which 2.9 kb corresponded to the 3′ end of the AtATM transcript. After several unsuccessful 5′ RACE attempts, we decided to first characterise the genomic locus of AtATM.
Isolation and sequencing of the genomic locus
The EST fragment was hybridised to the Texas A&M University (TAMU) bacterial artificial chromosome (BAC) library filters (40). Five BAC clones were found to contain the AtATM locus. The Institut für Genbiologische Forschung (IGF) BAC clones with matching restriction fingerprints were found using the fingerprint database. This allowed us to position the gene on chromosome 3 between RFLP markers m409 and I18. All the positive BAC clones mapped to the same chromosomal region suggesting it is a single locus and possibly a single copy gene. This was confirmed by Southern blotting and hybridisation (data not shown). Next, we focused on BAC clone T24C20 and completely sequenced it. The annotated sequence is now available (EMBL accession number AL096856). The genomic region corresponding to the EST was identified and in order to position a possible 5′ end of the gene we performed BLAST searches with 5 kb genomic fragments upstream of the EST. A region with homology to betaine aldehyde dehydrogenase was found ~35 kb upstream of the EST region and in the opposite orientation. This suggested that the AtATM gene is contained within these 35 kb.
Determination of the AtATM mRNA structure by RT–PCR
We then used the GENSCAN software (41) to predict potential translation initiation, donor and acceptor splice sites respectively. This prediction helped us design primers to amplify cDNA fragments by RT–PCR and thus to determine the complete structure of the AtATM gene. The transcript is 11.8 kb long and composed of 79 exons. Several exons were not predicted by GENSCAN or by the annotation from the Munich Information Center for Protein Sequences (MIPS). The exon–intron junctions are presented in Table 1 where they are compared with the annotation of MIPS. The 5′ end of the transcript was determined by 5′ RACE with primers located in the second exon. This exon of 2.8 kb is unusually long relative to the other exons and contains a potential Met initiator codon. The first exon does not contain in-frame Met codons, nor an uninterrupted in-frame ORF, and is thus presumed to be non-coding. Attempts at amplifying cDNA fragments with primers located further upstream failed, confirming the 5′ RACE result. The complete exon–intron structure of AtATM is represented in Figure 1a.
Table 1. Exon–intron organisation of the AtATM gene.

Exons–intron boundaries were determined by RT–PCR and compared with MIPS prediction on T24C20. The numbers indicate the position on BAC T24C20 sequence (EMBL accession number AL096856). Nine exons were absent on the MIPS prediction, and eight have different boundaries. Two exons (numbers 59 and 74) present non-canonical splice site.
Figure 1.

(a) Schematic of the exon–intron structure of the AtATM gene drawn to scale. All coding exons are represented by black boxes; the first exon, which is non-coding, by a white box. (b) Schematic of the primary structure of the AtAtm protein. PWWP, hath or PWWP domain; ATM, similarity to human Atm; rad3, rad3 domain; CC, coiled-coil domain ; Pi3k, phosphatidyl inositol-3 kinase-like domain ; C-ter, C-terminal domain characteristic of the Atm/Tor family of proteins.
The AtAtm protein and sequence similarities
The translated product of the AtATM transcript is a large protein of 3856 amino acids with a predicted molecular weight of 440 kDa (EMBL accession number AJ250258). The overall structure of the protein with the various domains is presented in Figure 1b. The 350 C-terminal residues constitute the Pi3k-l domain with a short tail of 25 amino acids at the very end characteristic of the Atm/Tor family of cell cycle regulators (42). Figure 2a shows an alignment of AtAtm with seven selected members of the family, Atm, X-Atm, Atr, rad3, Mec1, Tel1 and mei-41. Critical residues (Asp3681, Asn3686 and Asp3700) for the catalytic activity of these kinases are conserved in AtAtm. Over this region, AtAtm shares 58% identity and 67% similarity with Atm.
Figure 2.

(a) Alignment of the C-terminal region spanning the Pi3 kinase-like domain of AtAtm with six closely related members of the Atm family. The numbers on the right indicate the position of the residues for each protein. Amino acids identical to AtAtm are boxed in black. Pale and dark grey boxes highlight amino acid identities between two and three or more proteins respectively, AtAtm excluded. The alignment was performed with the PILEUP program (Genetics Computer Group, v.9.1). Vertical arrows indicate the three conserved residues of the kinase catalytic site, namely Asp 3681, Asn3686 and Asp3700 for AtAtm. (b) Schematic drawing for the alignment of the AtATM translation product to related protein in mammals (Atm, Atr), Drosophila melanogaster (Mei41) and yeast (Rad3, Tel1 and Mec1). Sequences were drawn to scale. Black boxes cover the Pi3-kinase-like region of strong homology, which is shared by all family members; boxes 1–5, the different homology domains between each protein and AtAtm; box 6, the region of similarity shared by AtAtm and Atm only. Thin lines for each sequence indicate region with no similarity (<20% identity). Alignments were performed agains thet AtAtm sequence with each of the family members using the GAP program (Genetics Computer Group, v.9.1). (c) Percentages of amino acid similarity and identity for the domains defined in (b) between the different Atm-related proteins. In each box, the first number represents the similarity and the second number the identity. (d) Alignment of the PWWP domain of the following proteins: AtAtm, Dnmt3b (accession number AAF04015), hMsh6 (accession number AAB39212), MMSET type I (accession number AAF23369), Caenorhabditis elegans similar to Sir2 (accession number U97193), and HDGF (accession number P51858). The alignment was generated as in (a).
Outside the Pi3k-l domain, there is a large region of overall weaker sequence similarity between the various proteins of the selected group, with the exception of X-Atm for which the complete sequence is not yet available. The length of this similarity is variable depending on which proteins are compared. A comparison was performed with each possible pair of the selected proteins. This led us to subdivide this region into six domains common to respectively all, six, five, four, three or two members of the group (Fig. 2). Over this region, the AtAtm protein is more similar to Atm than to any other member of the group.
Several regions outside the Pi3k-l domain have been identified as potentially important for Atm function. The overexpression of a fragment containing a leucine zipper motif between residues 1217 and 1238 confers a dominant negative phenotype in human tumour cells (43). A two-hybrid search with the same fragment identified β-adaptin as a potential partner (44). The fragment sufficient for interaction however did not contain the leucine zipper. A proline-rich motif between residues 1374 and 1382 was also found to be critical in the binding of Atm to c-Abl (24). Finally, an N-terminal fragment of 246 residues was found to selectively bind p53 (45). None of these motifs seem to be conserved in AtAtm. A search for structural motifs, however, identified residues 3249–3288 as potentially forming a coiled-coil domain which does not appear to be present in the human Atm protein.
Blast searches with smaller portions of the AtAtm protein helped us detect the presence of a recently described domain of unknown function. This domain was first identified in a gene family with homology to the hepatoma-derived growth factor (HDGF) and thus called the hath (homologous to the amino terminus of HGDF) domain (46), and was also designated PWWP (for proline–tryptophan–tryptophan–proline) domain by others (47). In AtAtm, the hath domain is located between amino acids 100 and 155. A multiple alignment over this domain is shown in Figure 2d.
Expression analysis
Expression was studied at the level of transcription by northern hybridisation. A transcript of ~12 kb was detected from poly(A)+ mRNA of Arabidopsis cell suspension cultures (Fig. 3). The size of the transcript is compatible with the predicted size based on our RT–PCR analysis (see above). Also, the steady state level of the transcript does not vary over 6 h after treatment of the cells with 100 Gy of γ rays, nor 1 h after irradiation with doses ranging from 0 to 200 Gy.
Figure 3.
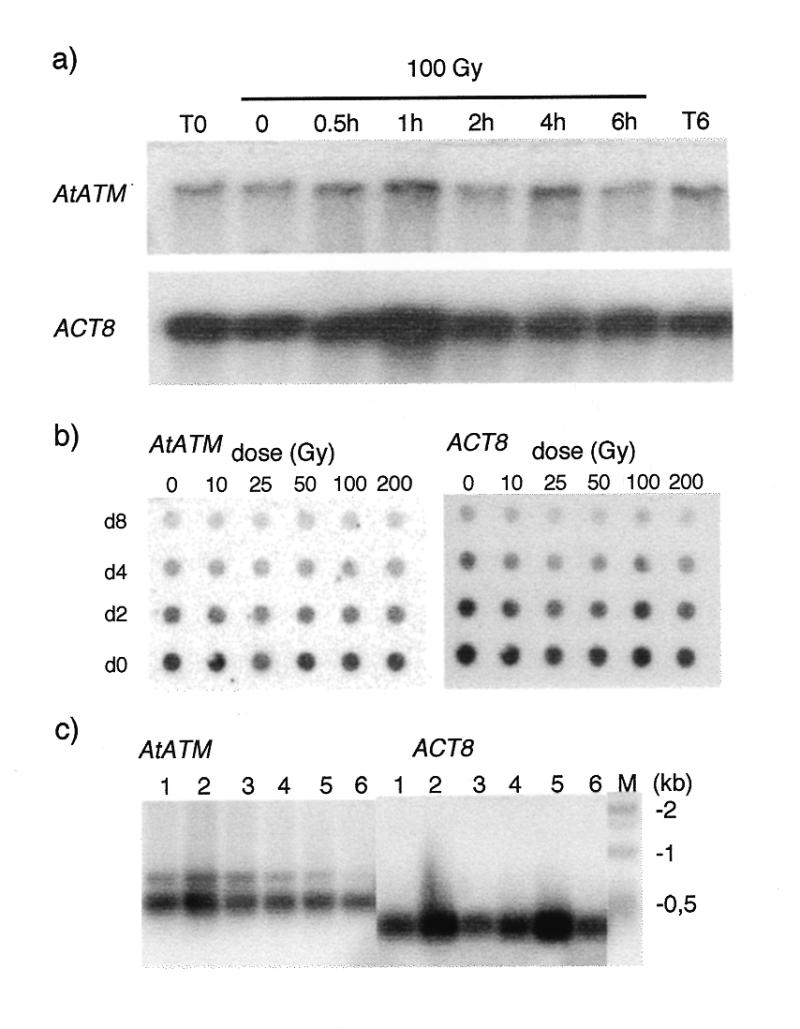
Expression of AtATM in response to IR and in different tissues. (a) Time course analysis by northern hybridisation of AtATM (top panel) and ACT8 (bottom panel) transcript levels in cell suspensions after 100 Gy of γ irradiation. The AtATM and ACT8 transcripts migrate at 12 and 1.6 kb respectively. (b) Dot blot hybridisation analysis of AtATM and ACT8 transcript levels 1 h after different doses of irradiation. The following amounts of poly(A)+ mRNA were deposited on the membrane: do, 150 pg; d2, 75 pg; d4, 37.5 pg; and d8, 18.6 pg. (c) RT–PCR analysis of AtATM and ACT8 transcript levels in major plant organs. 1, flowers; 2, flower buds; 3, leaves; 4, roots; 5, stems; 6, siliques. Total RNA (1 mg) was reverse transcribed and amplified for 30 and 25 cycles for AtATM and ACT8 respectively. The RT–PCR products were separated on agarose gels and transferred to Nylon membranes which were then hybridised to gene-specific probes.
Expression levels of AtATM in different tissues was determined by semi-quantitative RT–PCR (Fig. 3). It is expressed at low levels in all tissues examined with slightly higher levels in flower buds. This is similar to the situation in mammals, where ATM is expressed ubiquitously at low levels (9).
DISCUSSION
We have identified an Arabidopsis homologue of ATM via sequence homology. The AtATM gene is situated on chromosome 3 between markers m409 and I18, respectively at 64.02 and 64.99 cM on the RI map.
From the genomic sequence we were able to determine by RT–PCR the complete structure of the AtATM mRNA. The transcript is 11.8 kb long and composed of 79 exons. To our knowledge, this is the longest Arabidopsis coding sequence to date. Out of the 79 exons, nine were not predicted by the MIPS annotation, two of which (numbers 59 and 74) have non-canonical splice sites, and eight have different boundaries than predicted by MIPS. This piece of data may be useful to improve current programs used to predict gene structure. The first exon is non-coding and may be involved in the stability of the transcript and/or regulation of transcription. This has been observed for other plant genes, such as AtDMC1 (48). Alternative splicing in the 5′UTR and multiple polyadenylation sites have been described for the human ATM gene although the significance of these observations is currently unknown (49). Except for a putative alternative transcription initiation site, we did not detect any alternative splicing over the whole length of the gene. We cannot exclude the possibility that there is alternative splicing, however, because the RNA used for the RT–PCR analysis came from a single source, namely a cell suspension culture, and alternative splicing is known to reflect tissue specific expression (50–53). RT–PCR analysis on different tissues will have to be performed to determine if this the case for AtATM.
The predicted protein encoded by AtATM is 3856 amino acids long for a total molecular weight of 440 kDa. AtAtm shows very strong homology to Atm and other related members of the family in the C-terminal region corresponding to the pi3K-l domain. This suggests that AtAtm is a true member of this family and, like Mec1, Tel1, mei-41, DNA-PK, Atr and Atm, plays an important role in the cellular response to DNA damage. In addition, over the loosely defined upstream domain, AtAtm is more closely related to human Atm than to the other members of the family, mouse Atm excluded. Although this may be taken as additional evidence that AtAtm is an orthologue of human Atm, we cannot further predict the precise role of AtAtm. For example, the yeast TEL1 gene is more closely related to ATM than MEC1, but the cellular phenotype of AT cells resembles that of mec1 mutants (14,15). The phenotype of mec1 tel1 double mutants, however, suggest they have partially overlapping roles (14,27,29). Thus, it is difficult to assign a particular role based simply on sequence homology. Nonetheless, Keegan et al. suggest that rad3, Mec1, mei-41 and Atr on one hand and Tel1 and Atm on the other hand constitute two distinctive subgroups of the family, based not only on structural but also functional similarities (54).
As in other members of the Atm family, the Pi3k-l domain of AtAtm is preceded by a very long N-terminal fragment. Sequence comparisons over this region again show that AtAtm is more closely related to Atm than to the other members and further underlines its status as a true Atm homologue. In Atm, a putative leucine zipper domain was identified and a fragment comprising this motif overexpressed in human cells has a dominant negative effect (43). AtAtm does not contain a putative leucine zipper. However a coiled-coil domain between residues 3249 and 3288 was predicted by two different programs. Since coiled-coil domains are involved in protein–protein interactions and particularly multimerisation, it is possible that AtAtm interacts with itself or other proteins via this domain.
Of particular interest, we noted the presence of a PWWP domain near the N-terminus of AtAtm. The existence of this conserved domain of approximately 25 amino acids was first described in the HDGF and two related proteins (46). It is present in a number of proteins including a mammalian MutS homologue (hMsh6 or G/T binding protein) (55,56), the mammalian DNA cytosine-5 methyl transferase 3B (Dnmt3b) (57), the multiple myeloma SET domain protein (58), a candidate gene for the Wolf-Hirschhorn syndrome WHSC1 (47), and an adenovirus E1A-associated protein (BS69) (59). These proteins play a role in various aspects of DNA metabolism including DNA repair, regulation of transcription and the cell cycle. Recently, it was found that the gene mutated in the human chromosome instability and immunodefficiency syndrome (ICF) is the DNMT3B gene (60,61). This disorder is due to the absence of methylation in heterochromatic regions, thus it is possible that the role of the PWWP domain is to direct the Dnmt3b proteins to certain chromosomal regions either by binding to other proteins or directly to DNA. The presence of other chromatin-associated domains such as the SET or bromo domains amongst the proteins with a PWWP domain further supports this assumption. Thus the PWWP domain of AtAtm may be of particular interest in a search for interacting partners or to test binding to DNA.
We were able to detect a 12 kb transcript hybridising to a specific AtATM probe in cell suspensions. The steady state levels of this transcript are fairly low and do not rise upon treatment with γ radiation over a 6 h period. In human cells, ATM expression is not induced by IR. Thus, like its human counterpart, AtAtm is likely to play a role very early in the response to genotoxic stress, via activation of its kinase activity upon binding to DNA breaks as was recently shown for human Atm (62).
Based on RT–PCR analysis of the expression in various tissues, it appears that AtATM is transcribed at low levels in all tissues, including tissues where few cell divisions occur like mature leaves. This is similar to the situation in mammals and raises the question of the role of these proteins in non-dividing cells.
In conclusion, we have characterised a plant homologue of ATM. The high degree of sequence conservation, and the fact that all members of this family of proteins from eukaryotic organisms as varied as yeast, Drosophila and mammals, play a critical role in DNA damage cell cycle checkpoints, suggest that AtATM does also play a similar role in Arabidopsis. We plan to confirm this hypothesis by studying insertion mutants of AtATM obtained by a reverse genetics strategy.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Marie-Hélène Montané for critical reading of the manuscript and for supplying mRNAs, and Olivier Pierrugues for supplying plant tissues and total RNA. V.G. is funded by a predoctoral fellowship ‘Contrat Formation Recherche’ from the Commissariat à l’Energie Atomique.
DDBJ/EMBL/GenBank accession no. AJ250248
REFERENCES
- 1.Wang J.Y. (1998) Curr. Opin. Cell Biol., 10, 240–247. [DOI] [PubMed] [Google Scholar]
- 2.Taylor A., Harnden,D., Arlett,C., Harcourt,S., Lehmann,A., Stevens,S. and Bridges,B. (1975) Nature, 258, 427–429. [DOI] [PubMed] [Google Scholar]
- 3.Shiloh Y. (1997) Annu. Rev. Genet., 31, 635–662. [DOI] [PubMed] [Google Scholar]
- 4.Beamish H., Williams,R., Chen,P. and Lavin,M.F. (1996) J. Biol. Chem., 271, 20486–20493. [DOI] [PubMed] [Google Scholar]
- 5.Houldsworth J. and Lavin,M.F. (1980) Nucleic Acids Res., 8, 3709–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foray N., Priestley,A., Alsbeih,G., Badie,C., Capulas,E.P., Arlett,C.F. and Malaise,E.P. (1997) Int. J. Radiat. Biol., 72, 271–283. [DOI] [PubMed] [Google Scholar]
- 7.Dar M.E., Winters,T.A. and Jorgensen,T.J. (1997) Mutat. Res., 384, 169–179. [DOI] [PubMed] [Google Scholar]
- 8.Savitsky K., Sfez,S., Tagle,D.A., Ziv,Y., Sartiel,A., Collins,F.S., Shiloh,Y. and Rotman,G. (1995) Hum. Mol. Genet., 4, 2025–2032. [DOI] [PubMed] [Google Scholar]
- 9.Savitsky K., Bar-Shira,A., Gilad,S., Rotman,G., Ziv,Y., Vanagaite,L., Tagle,D.A., Smith,S., Uziel,T., Sfez,S., Ashkenazi,M., Pecker,I., Frydman,M., Harnik,R., Patanjali,S.R., Simmons,A., Clines,G.A., Sartiel,A., Gatti,R.A., Chessa,L., Sanal,O., Lavin,M.F., Jaspers,N.G.J., Taylor,A.M.R., Arlett,C.F., Miki,T., Weissman,S.M., Lovett,M., Collins,F.S. and Shiloh,Y. (1995) Science, 268, 1749–1753. [DOI] [PubMed] [Google Scholar]
- 10.Helliwell S.B., Wagner,P., Kunz,J., Deuter-Reinhard,M., Henriquez,R. and Hall,M.N. (1994) Mol. Biol. Cell, 5, 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunz J., Henriquez,R., Schneider,U., Deuter-Reinhard,M., Movva,N.R. and Hall,M.N. (1993) Cell, 73, 585–596. [DOI] [PubMed] [Google Scholar]
- 12.Brown E.J., Albers,M.W., Shin,T.B., Ichikawa,K., Keith,C.T., Lane,W.S. and Schreiber,S.L. (1994) Nature, 369, 756–758. [DOI] [PubMed] [Google Scholar]
- 13.Sabatini D.M., Erdjument-Bromage,H., Lui,M., Tempst,P. and Snyder,S.H. (1994) Cell, 78, 35–43. [DOI] [PubMed] [Google Scholar]
- 14.Morrow D.M., Tagle,D.A., Shiloh,Y., Collins,F.S. and Hieter,P. (1995) Cell, 82, 831–840. [DOI] [PubMed] [Google Scholar]
- 15.Paulovitch A.G. and Hartwell,L.H. (1995) Cell, 82, 841–847. [DOI] [PubMed] [Google Scholar]
- 16.Bentley N.J., Holtzman,D.A., Flaggs,G., Keegan,K.S., DeMaggio,A., Ford,J.C., Hoekstra,M. and Carr,A.M. (1996) EMBO J., 15, 6641–6651. [PMC free article] [PubMed] [Google Scholar]
- 17.Hari K.L., Santerre,A., Sekelsky,J.J., McKim,K.S., Boyd,J.B. and Hawley,S.R. (1995) Cell, 82, 815–821. [DOI] [PubMed] [Google Scholar]
- 18.Hartley K.O., Gell,D., Smith,G.C., Zhang,H., Divecha,N., Connelly,M.A., Admon,A., Lees-Miller,S.P., Anderson,C.W. and Jackson,S.P. (1995) Cell, 82, 849–856. [DOI] [PubMed] [Google Scholar]
- 19.Cliby W.A., Roberts,C.J., Cimprich,K.A., Stringer,C.M., Lamb,J.R., Schreiber,S.L. and Friend,S.H. (1998) EMBO J., 17, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canman C.E., Lim,D.S., Cimprich,K.A., Taya,Y., Tamai,K., Sakaguchi,K., Appella,E., Kastan,M.B. and Siliciano,J.D. (1998) Science, 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- 21.Banin S., Moyal,L., Shieh,S., Taya,Y., Anderson,C.W., Chessa,L., Smorodinsky,N.I., Prives,C., Reiss,Y., Shiloh,Y. and Ziv,Y. (1998) Science, 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- 22.Kastan M.B., Zhan,Q., el-Deiry,W.S., Carrier,F., Jacks,T., Walsh,W.V., Plunkett,B.S., Vogelstein,B. and Fornace,A.J.,Jr (1992) Cell, 71, 587–597. [DOI] [PubMed] [Google Scholar]
- 23.Gately D.P., Hittle,J.C., Chan,G.K. and Yen,T.J. (1998) Mol. Biol. Cell, 9, 2361–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shafman T., Khanna,K.K., Kedar,P., Spring,K., Kozlov,S., Yen,T., Hobson,K., Gatei,M., Zhang,N., Watters,D., Egerton,M., Shiloh,Y., Kharbanda,S., Kufe,D. and Lavin,M.F. (1997) Nature, 387, 520–523. [DOI] [PubMed] [Google Scholar]
- 25.Baskaran R., Wood,L.D., Whitaker,L.L., Canman,C.E., Morgan,S.E., Xu,Y., Barlow,C., Baltimore,D., Wynshaw-Boris,A., Kastan,M.B. and Wang,J.Y. (1997) Nature, 387, 516–519. [DOI] [PubMed] [Google Scholar]
- 26.Sun Z., Fay,D.S., Marini,F., Foiani,M. and Stern,D.F. (1996) Genes Dev., 10, 395–406. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez Y., Desany,B.A., Jones,W.J., Liu,Q., Wang,B. and Elledge,S.J. (1996) Science, 271, 357–360. [DOI] [PubMed] [Google Scholar]
- 28.Brush G.S., Morrow,D.M., Hieter,P. and Kelly,T.J. (1996) Proc. Natl Acad. Sci. USA, 93, 15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vialard J.E., Gilbert,C.S., Green,C.M. and Lowndes,N.F. (1998) EMBO J., 17, 5679–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinho R.G., Lindsay,H.D., Flaggs,G., DeMaggio,A.J., Hoekstra,M.F., Carr,A.M. and Bentley,N.J. (1998) EMBO J., 17, 7239–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanchez Y., Wong,C., Thoma,R.S., Richman,R., Wu,Z., Piwnica-Worms,H. and Elledge,S.J. (1997) Science, 277, 1497–1501. [DOI] [PubMed] [Google Scholar]
- 32.Brown A.L., Lee,C.H., Schwarz,J.K., Mitiku,N., Piwnica-Worms,H. and Chung,J.H. (1999) Proc. Natl Acad. Sci. USA, 96, 3745–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Axelos M., Curie,C., Mazzolini,L., Bardet,C. and Lescure,B. (1992) Plant Physiol. Biochem., 30, 123–128. [Google Scholar]
- 34.Chandler M.T., Tandeau de Marsac,N. and de Kouchkovsky,Y. (1972) Can. J. Bot., 50, 2265–2270. [Google Scholar]
- 35.Kloppstech K., Otto,B. and Sierralta,W. (1991) Mol. Gen. Genet., 225, 468–473. [DOI] [PubMed] [Google Scholar]
- 36.Montané M.H., Dreyer,S., Triantaphylides,C. and Kloppstech,K. (1997) Planta, 202, 293–302. [Google Scholar]
- 37.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 38.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minet M., Dufour,M.E. and Lacroute,F. (1992) Plant J., 2, 417–422. [DOI] [PubMed] [Google Scholar]
- 40.Choi S.D., Creelman,R., Mullet,J. and Wing,R.A. (1995) Weeds World, 2, 17–20. [Google Scholar]
- 41.Burge C. and Karlin,S. (1997) J. Mol. Biol., 268, 78–94. [DOI] [PubMed] [Google Scholar]
- 42.Keith C.T. and Schreiber,S.L. (1995) Science, 270, 50–51. [DOI] [PubMed] [Google Scholar]
- 43.Morgan S.E., Lovly,C., Pandita,T.K., Shiloh,Y. and Kastan,M.B. (1997) Mol. Cell. Biol., 17, 2020–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim D.S., Kirsch,D.G., Canman,C.E., Ahn,J.H., Ziv,Y., Newman,L.S., Darnell,R.B., Shiloh,Y. and Kastan,M.B. (1998) Proc. Natl Acad. Sci. USA, 95, 10146–10151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khanna K.K., Keating,K.E., Kozlov,S., Scott,S., Gatei,M., Hobson,K., Taya,Y., Gabrielli,B., Chan,D., Lees-Miller,S.P. and Lavin,M.F. (1998) Nature Genet., 20, 398–400. [DOI] [PubMed] [Google Scholar]
- 46.Izumoto Y., Kuroda,T., Harada,H., Kishimoto,T. and Nakamura,H. (1997) Biochem. Biophys. Res. Commun., 238, 26–32. [DOI] [PubMed] [Google Scholar]
- 47.Stec I., Wright,T.J., van Ommen,G.J., de Boer,P.A., van Haeringen,A., Moorman,A.F., Altherr,M.R. and den Dunnen,J.T. (1998) Hum. Mol. Genet., 7, 1071–1082. [DOI] [PubMed] [Google Scholar]
- 48.Klimyuk V.I. and Jones,J.D. (1997) Plant J., 11, 1–14. [DOI] [PubMed] [Google Scholar]
- 49.Savitsky K., Platzer,M., Uziel,T., Gilad,S., Sartiel,A., Rosenthal,A., Elroy-Stein,O., Shiloh,Y. and Rotman,G. (1997) Nucleic Acids Res., 25, 1678–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valcarcel J. and Gebauer,F. (1997) Curr. Biol., 7, R705–R708. [DOI] [PubMed] [Google Scholar]
- 51.Lou H. and Gagel,R.F. (1998) J. Endocrinol., 156, 401–405. [DOI] [PubMed] [Google Scholar]
- 52.Edwalds-Gilbert G., Veraldi,K.L. and Milcarek,C. (1997) Nucleic Acids Res., 25, 2547–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chabot B. (1996) Trends Genet., 12, 472–478. [DOI] [PubMed] [Google Scholar]
- 54.Keegan K.S., Holtzman,D.A., Plug,A.W., Christenson,E.R., Brainerd,E.E., Flaggs,G., Bentley,N.J., Taylor,E.M., Meyn,M.S., Moss,S.B., Carr,A.M., Ashley,T. and Hoekstra,M.F. (1996) Genes Dev., 10, 2423–2437. [DOI] [PubMed] [Google Scholar]
- 55.Edelmann W., Yang,K., Umar,A., Heyer,J., Lau,K., Fan,K., Liedtke,W., Cohen,P.E., Kane,M.F., Lipford,J.R., Yu,N., Crouse,G.F., Pollard,J.W., Kunkel,T., Lipkin,M., Kolodner,R. and Kucherlapati,R. (1997) Cell, 91, 467–477. [DOI] [PubMed] [Google Scholar]
- 56.Miyaki M., Konishi,M., Tanaka,K., Kikuchi-Yanoshita,R., Muraoka,M., Yasuno,M., Igari,T., Koike,M., Chiba,M. and Mori,T. (1997) Nature Genet., 17, 271–272. [DOI] [PubMed] [Google Scholar]
- 57.Okano M., Xie,S. and Li,E. (1998) Nature Genet., 19, 219–220. [DOI] [PubMed] [Google Scholar]
- 58.Chesi M., Nardini,E., Lim,R.S., Smith,K.D., Kuehl,W.M. and Bergsagel,P.L. (1998) Blood, 92, 3025–3034. [PubMed] [Google Scholar]
- 59.Hateboer G., Gennissen,A., Ramos,Y.F., Kerkhoven,R.M., Sonntag-Buck,V., Stunnenberg,H.G. and Bernards,R. (1995) EMBO J., 14, 3159–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hansen R.S., Wijmenga,C., Luo,P., Stanek,A.M., Canfield,T.K., Weemaes,C.M. and Gartler,S.M. (1999) Proc. Natl Acad. Sci. USA, 96, 14412–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu G.-L., Bestor,T.H., Bourc’his,D., Hsieh,C.-L., Tommerup,N., Bugge,M., Hulten,M., Qu,X., Russo,J.J. and Viegas-Péquignot,E. (1999) Nature, 402, 187–191. [DOI] [PubMed] [Google Scholar]
- 62.Smith G.C., Cary,R.B., Lakin,N.D., Hann,B.C., Teo,S.H., Chen,D.J. and Jackson,S.P. (1999) Proc. Natl Acad. Sci. USA, 96, 11134–11139. [DOI] [PMC free article] [PubMed] [Google Scholar]