

Abstract
The mTORC2 pathway plays a critical role in promoting tumor progression in human colorectal cancer (CRC). The regulatory mechanisms for this signaling pathway are only partially understood. We previously identified UBXN2A as a novel tumor suppressor protein in CRCs and hypothesized that UBXN2A suppresses the mTORC2 pathway, thereby inhibiting CRC growth and metastasis. We first used murine models to show that haploinsufficiency of UBXN2A significantly increases colon tumorigenesis. Induction of UBXN2A reduces AKT phosphorylation downstream of the mTORC2 pathway, which is essential for a plethora of cellular processes, including cell migration. Meanwhile, mTORC1 activities remain unchanged in the presence of UBXN2A. Mechanistic studies revealed that UBXN2A targets Rictor protein, a key component of the mTORC2 complex, for 26S proteasomal degradation. A set of genetic, pharmacological, and rescue experiments showed that UBXN2A regulates cell proliferation, apoptosis, migration, and colon cancer stem cells (CSCs) in CRC. CRC patients with a high level of UBXN2A have significantly better survival, and high-grade CRC tissues exhibit decreased UBXN2A protein expression. A high level of UBXN2A in patient-derived xenografts and tumor organoids decreases Rictor protein and suppresses the mTORC2 pathway. These findings provide new insights into the functions of an ubiquitin-like protein by inhibiting a dominant oncogenic pathway in CRC.
Keywords: Colon and rectal cancer, mTORC2, Rictor, UBXN2A, mouse
Introduction
Dysregulation of the mTORC2 pathway has been identified as an oncogenic factor in CRC initiation and progression (1). In addition, mTOR complexes develop drug resistance in both colon (2, 3) and rectal cancers (4). With two distinct complexes called mTORC1 and mTORC2, the mTORC pathway contributes to diverse signaling pathways in physiological and pathological conditions (5). The essential roles of mTORC1 in key physiological functions (6, 7) and associated negative feedback loops activated upon its inhibition (8, 9) have limited the benefit of using mTORC1 inhibitors for the treatment of CRC (10–15). However, recent reports have demonstrated a distinct role for mTORC2 in several solid cancers (1, 16–18). Well-supported studies have illustrated that a selective mTORC2 inhibitor can function as an effective anti-tumorigenic agent and spare mTORC1 signaling (1, 16–22). Rictor is a critical member of the mTORC2 complex whose overexpression is associated with tumor progression, metastasis, and poor prognosis (23, 24). The driving role of Rictor protein in epithelial-mesenchymal transition (EMT) and chemosensitivity (25, 26) has turned Rictor protein into a potential targeted therapy for CRC (27, 28).
In the present study, we report that UBXN2A ubiquitinates Rictor protein for 26S proteasomal degradation, resulting in repression of the mTORC2 pathway in colon cancer cells and leaving mTORC1 intact. while mTORC1 remains intact. Gain- and loss-of-function approaches as well as rescue experiments revealed that UBXN2A interferes with the mTORC2’s downstream protein pathways, including apoptosis, epithelial-mesenchymal transition, vascular endothelial growth factor (VEGF), cancer cell migration, and regeneration of colon cancer stem cells (CSCs). Patients with a higher level of UBXN2A have better survival of CRC. On the other hand, the reduction of UBXN2A proteins can be seen in poorly differentiated (high-grade) human tumor tissues. This evidence further confirmed that UBXN2A functions as a dominant tumor suppressor protein in CRC (29–31). Our findings uncovered a novel regulatory pathway capable of suppressing the mTORC2 pathway and its downstream protein targets. Therefore, the enhancement of UBXN2A is a promising targeted therapy to overcome CRC metastasis and drug resistance mediated by the hyperactive mTORC2 pathway.
Results
Haploinsufficiency of the UBXN2A tumor suppressor gene promotes tumorigenesis in the UBXN2A (+/−) mouse
We generated a UBXN2A heterozygous mouse (+/−) that expresses half the amount of endogenous UBXN2A compared with WT littermates (32). UBXN2A−/− is embryonic lethal due to UBXN2A’s role in embryonic development (33). Tumor development by the azoxymethane/dextran sodium sulfate protocol (34) is accelerated in UBXN2A+/− mice (Fig. 1A–E). Figure 1E shows several high-grade dysplasia adenomas that have initiated invading the muscularis mucosa of the colon, the earliest morphologically definable stage of human CRC (35). Figure 1F and supplemental Figure 1A–B show a significant increase in adenomatous areas with low- and high-grade dysplasia in heterozygous mice. Next, proliferating cells in the tumor tissue were stained by immunohistochemistry using Ki-67 staining marker (Fig. 1G). Tumors extracted from UBXN2A +/− mice showed significant nuclear staining of Ki-67 in UBXN2A heterozygous mice versus tumors removed from WT littermates (Supplemental Figure 1C). The rapid progression of pre-malignant high-grade dysplasia in the heterozygous UBXN2A +/− mouse model suggests key anti-proliferative functions for UBXN2A.
Figure 1: Haploinsufficiency of the UBXN2A tumor suppressor gene promotes colon cancer progression.
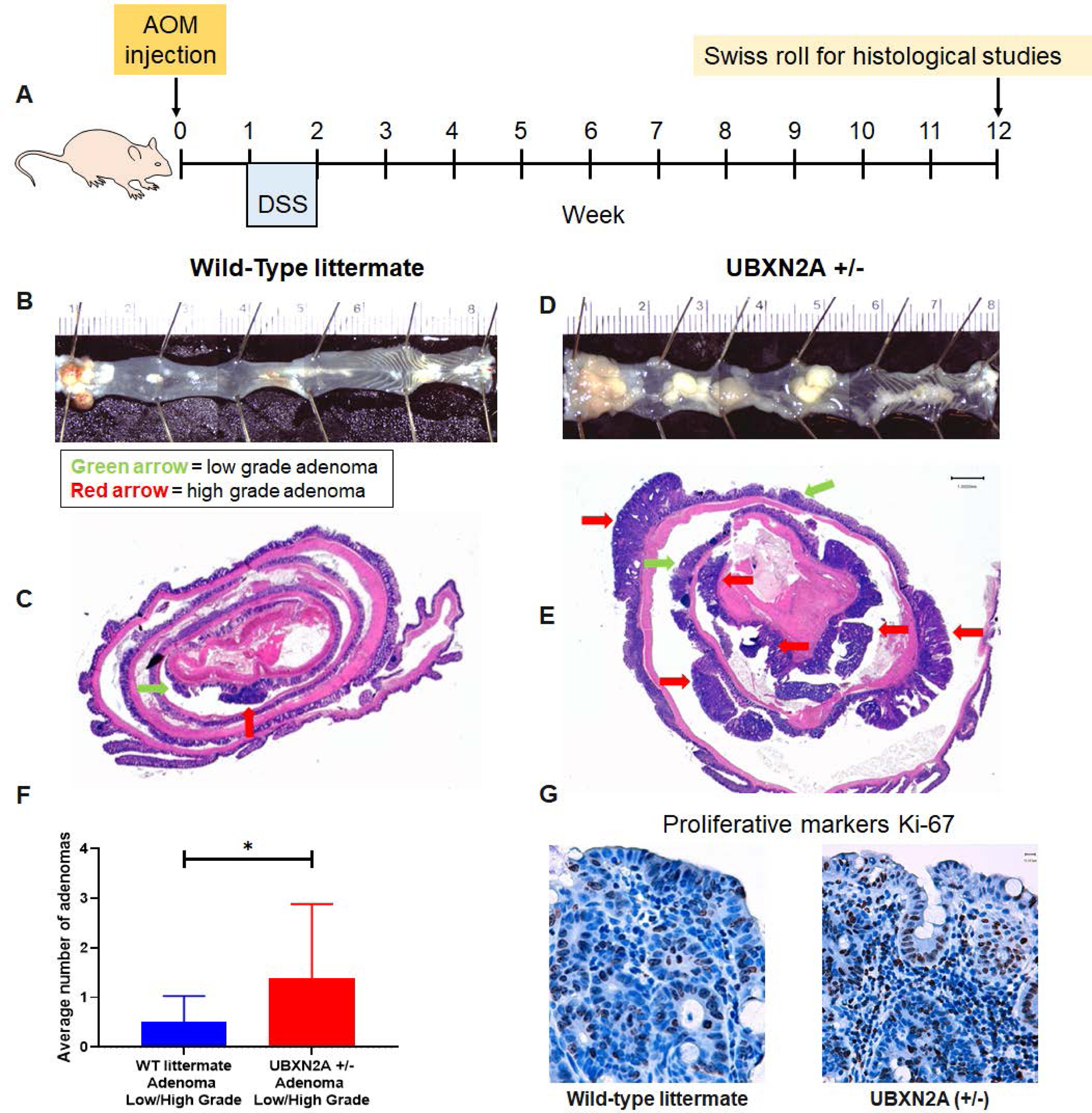
A-D: Colorectal tumors developed in all AOM/DSS treated C57BL/6 mice. E-F: Tumors were stained with hematoxylin and eosin (H & E). The Swiss roll technique followed by IHC did not have access to all those rectal tumors (panels B and D) due to the nature of the technique. Unavoidably, sections are omitted at both ends of the Swiss roll during construction. Low expression of UBXN2A in heterozygote mice (+/−) significantly increased both low- and highgrade adenomas. Green arrow = low grade adenoma, red arrow = high grade adenoma (n = 11 per genotype, male and female, Welch’s t test, *p<0.05, mean ± SD). G: IHC showed a higher level of cell proliferation in UBXN2A +/− (scale bar, 1mm).
Induction of UBXN2A interferes with the mTORC2 pathway without affecting mTORC1
Based on the in vitro anti-growth, anti-migration, and anti-invasion functions of UBXN2A, we hypothesized that UBXN2A potentially interferes with more than one major tumorigenic pathway in CRC (32, 36). A multi-anti-oncoprotein function has been described for several proteins associated with the ubiquitin-proteasome pathway. The nature of these ubiquitin-associated proteins, such as E3 ubiquitin ligases and their adaptors, enable them to target a diverse set of key substrates in different signaling pathways. One example is the FBXW7 (F-box with 7 tandem WD40) protein, which is one of the key players in the Skp1-Cullin1-F-box (SCF) ubiquitin ligase complex. FBXW7 aids in the degradation of several key oncoproteins, such as c-Myc, Notch, cyclin E, c-JUN, and KLF5, via the ubiquitin-proteasome system (UPS) (37). To find the tumorigenic pathway(s) regulated by UBXN2A, we examined the status of several tumorigenic pathways (38) involved in CRC initiation, progression, and metastasis in the presence of induced UBXN2A. Since the PI3K/AKT/MTOR and MAPK pathways are most commonly mutated in CRC in addition to both APC and p53 mutations in patients with CRC (39), we checked key phosphorylated proteins downstream of MAPK and PI3K/AKT pathways. Among studied pathways, we found that UBXN2A significantly interferes with the mTORC2 pathway (40), which has been implicated in CRC cell migration, invasion, and metastasis (41–45). Tet-On inducible HCT-116 cells were incubated with DOX to express GFP or GFP-UBXN2A (Supplemental Figures 2 and 3A) and analyzed by flow cytometry using Alexa Fluor pAKT-Ser473 and pAKT-Thr308. Induction of UBXN2A (48 hours Doxycycline) significantly decreased pAKT-S473 and pAKT-T308 (Fig. 2A–E). Similarly, UBXN2A significantly decreased pAKT-S473 in colon adenocarcinoma SW480 cells. However, UBXN2A showed no significant effect on AKT-T308 in SW480 cells (Fig. 2F). Phosphorylation of AKT at S473 requires the mTORC2 complex containing Rictor protein (46). These results indicate potent inhibition of the pAKT-Ser473 signaling pathway by UBXN2A, irrespective of their cancer cell classifications. As previously reported, phosphorylation at both T308 and S473 can simultaneously be affected in a cell-type-dependent manner (47). In the next set of experiments, xenograft mice carrying Tet-On inducible HCT-116 cells were fed doxycycline as previously reported (48), followed by tumor extraction and western blot (WB) experiments (Fig. 2G–H). WB confirmed that UBXN2A dominantly decreases the level of pAKT-Ser473 in xenograft tumors (Fig. 2I). Supplemental Figure 1D–G confirmed that induced GFP-UBXN2A reduces tumor growth and decreases the protein level of Rictor in tumor tissues. Figure 2J–L and supplemental Figure 1H–I show two sets of patient-derived CRC xenografts (PDXs) with low and high-levels of UBXN2A protein as previously described (48). WB of PDX tissues followed by quantitation of their corresponding bands (Image Studio version 5.0) revealed high-level of UBXN2A in PDXs significantly decreases Rictor protein, and it changes Rictor’s downstream target proteins, including AKT473, VEGF, and E-Cadherin. To determine whether UBXN2A selectively targets the mTORC2 pathway, we checked the effect of induced UBXN2A on phospho-p70 S6 kinase (T389), which is located downstream of mTORC1 (49). ELISA (Fig. 2M) and WB experiments (Fig. 2N–O) indicated that induced UBXN2A has no significant effect on phosphorylation and activation of p70S6K protein. Finally, we used flow-cytometry technology to gain a deeper insight into the levels of phospho-p70 S6 kinase (T389) per cell (10000 events per sample) in the presence and absence of UBXN2A. Panel P shows a small but significant elevation of phospho-p70 S6 kinase (T389) in cells overexpressing UBXN2A due to the overlapping function of mTORC1 and mTORC2 in selected pathways. Results presented in Figure 2 indicate that UBXN2A can selectively inhibit the mTORC2 complex, downregulate Rictor protein, and interfere with Rictor’s downstream target proteins while leaving the mTORC1’s downstream protein targets primarily intact.
Figure. 2: Induction of UBXN2A interferes with the mTORC2 pathway but not the mTORC1 signaling pathway.
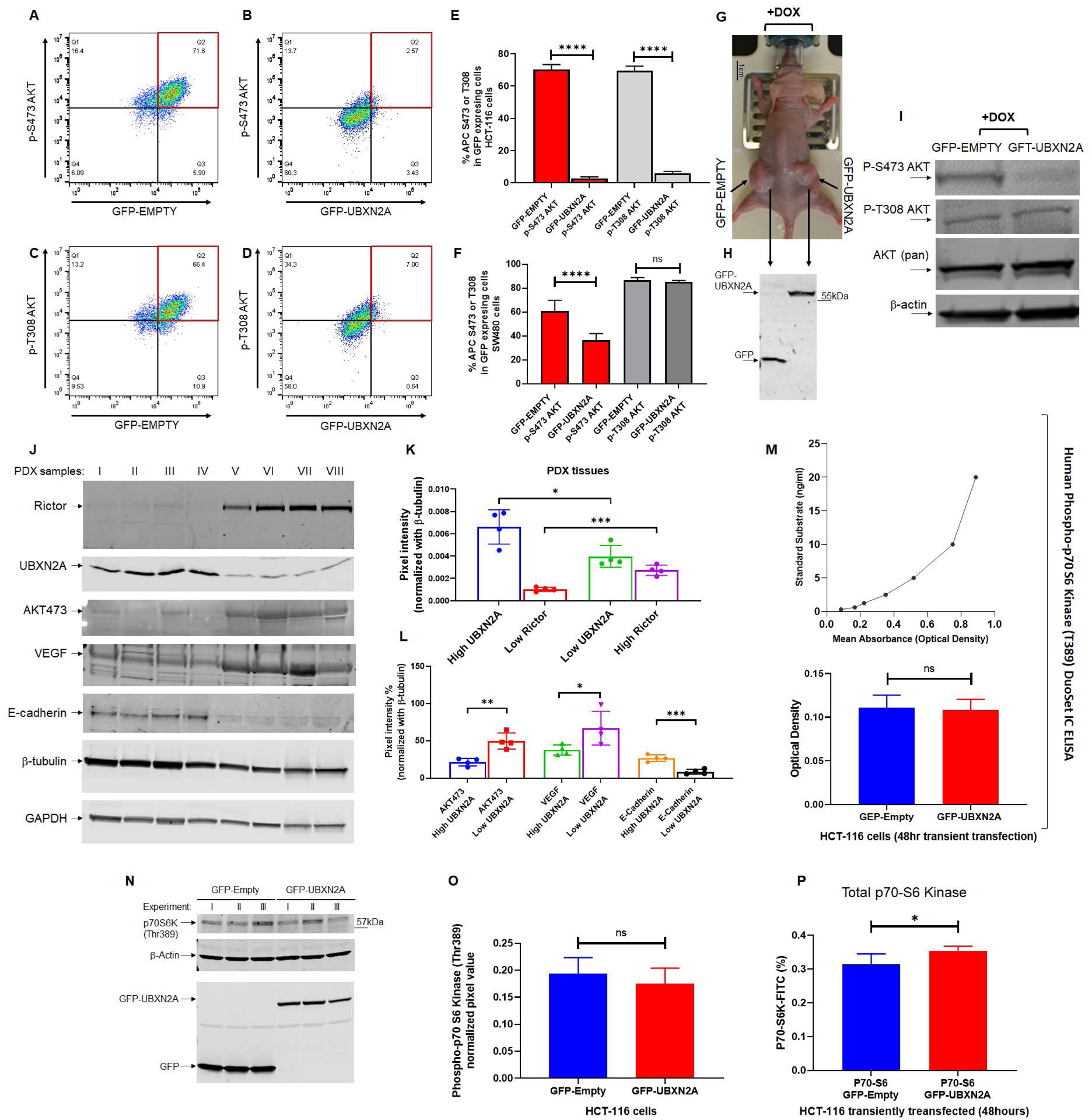
Tet-On inducible HCT-116 cells were incubated with DOX to express GFP or GFP-UBXN2A and analyzed by flow cytometry using Alexa Fluor pAKT-Ser473 and pAKT-T308. Induction of UBXN2A significantly decreased pAKT-Ser473 and pAKT-Thr308 (A-E). Similarly, UBXN2A significantly decreased pAKT-Ser473 in SW480 cells. However, UBXN2A induction showed no effect on pAKT-Thr308 in SW480, suggesting UBXN2A affects mTORC2 in a cell-dependent manner (F). Xenograft mice carrying Tet-On inducible HCT-116 cells were fed doxycycline (G), followed by tumor extraction and WB experiments (H).Representative WB of three mice with similar results (one GFP-empty tumor and one GFP-UBXN32A tumor per mouse) confirmed UBXN2A decreases the level of pAKT-S473/T308 in xenograft tumors (I). PDX tissues were subjected to WB followed by quantitation of their corresponding bands. Results revealed that the high level of UBXN2A in PDXs significantly decreases Rictor protein (J-K), and it changes Rictor’s downstream target proteins, including AKT-473, VEGF, and E-Cadherin (L). ELISA assays (M) and WB of phospho-p70 S6 kinase (T389) in HCT-116 cells (N and O) combined with flow-cytometry analysis of the total level of p70 S6 kinase (P) revealed that elevated UBXN2A has no significant effect on phosphorylation and activation of phospho-p70 S6 kinase (T389), which is located downstream of mTORC1 (n=3, * p< 0.05, **** p< 0.0001, mean +/− SD).
Induction of UBXN2A targets the mTORC2 pathway
Phosphorylation of AKT at S473 is dominantly regulated by Rictor protein, a key member in the mTORC2 complex (46). In the next experiment, we used flow cytometry analysis to determine the effect of UBXN2A on Rictor in HCT-116 cells. Figure 3A–C shows that UBXN2A overexpression leads to a significant reduction of Rictor protein. A set of WB experiments confirmed that induced GFP-UBXN2A significantly decreases the protein level of Rictor (supplemental figure 3A–B). A similar Rictor reduction was observed in SW48 and SW480 cells transiently transfected with GFP-UBXN2A (Fig. 3D and supplemental figure 3C–D). Based on previous reports regarding the role of UBX domain-containing protein in protein turnover (32, 50, 51), we examined the turnover of Rictor protein in Tet-On UBXN2A inducible HCT-116 cells in the presence of the bortezomib, a selective proteasome inhibitor. Figure 3E–F shows that UBXN2A induction leads to a significant reduction of Rictor protein while the presence of bortezomib preserves the level of Rictor protein in the presence of UBXN2A. A set of immunoprecipitation experiments further verified that UBXN2A binds to Rictor protein [Figure. 3G (UBXN2A pull-down) and supplemental Figure 3E–F (Rictor pull-down)], suggesting UBXN2A binds to and increases the turnover of Rictor protein. This effect is blocked by inhibition of the proteasome complex. Next, we looked at the K-48-linked ubiquitin chain of Rictor in the presence of GFP-empty or GFP-UBXN2A overexpression in transiently transfected SW480 and SW620 colon cancer cells (Fig. 3H). We used the K48 Tandem Ubiquitin Binding Entities (TUBEs) magnetic beads kit (32). Panel I in Figure 3 shows that UBXN2A increases K48-linked chain ubiquitination of Rictor protein in both colon cancer cell lines, particularly metastatic SW620 colon cancer cells. Rictor is a heavy protein with a molecular weight of 220 kDa. While we used gradient 4–20% SDS-glycine gel to separate these heavy ubiquitinated Rictor proteins, they still inevitably made dense ladders. Lidia Wrobel et al. showed similar dense ladders of ubiquitinated Rictor (K48 and K63 ubiquitin chain) in their 2021 Cell Report publication (52).
Figure 3: UBXN2A binds to Rictor protein and promotes its proteasomal degradation.
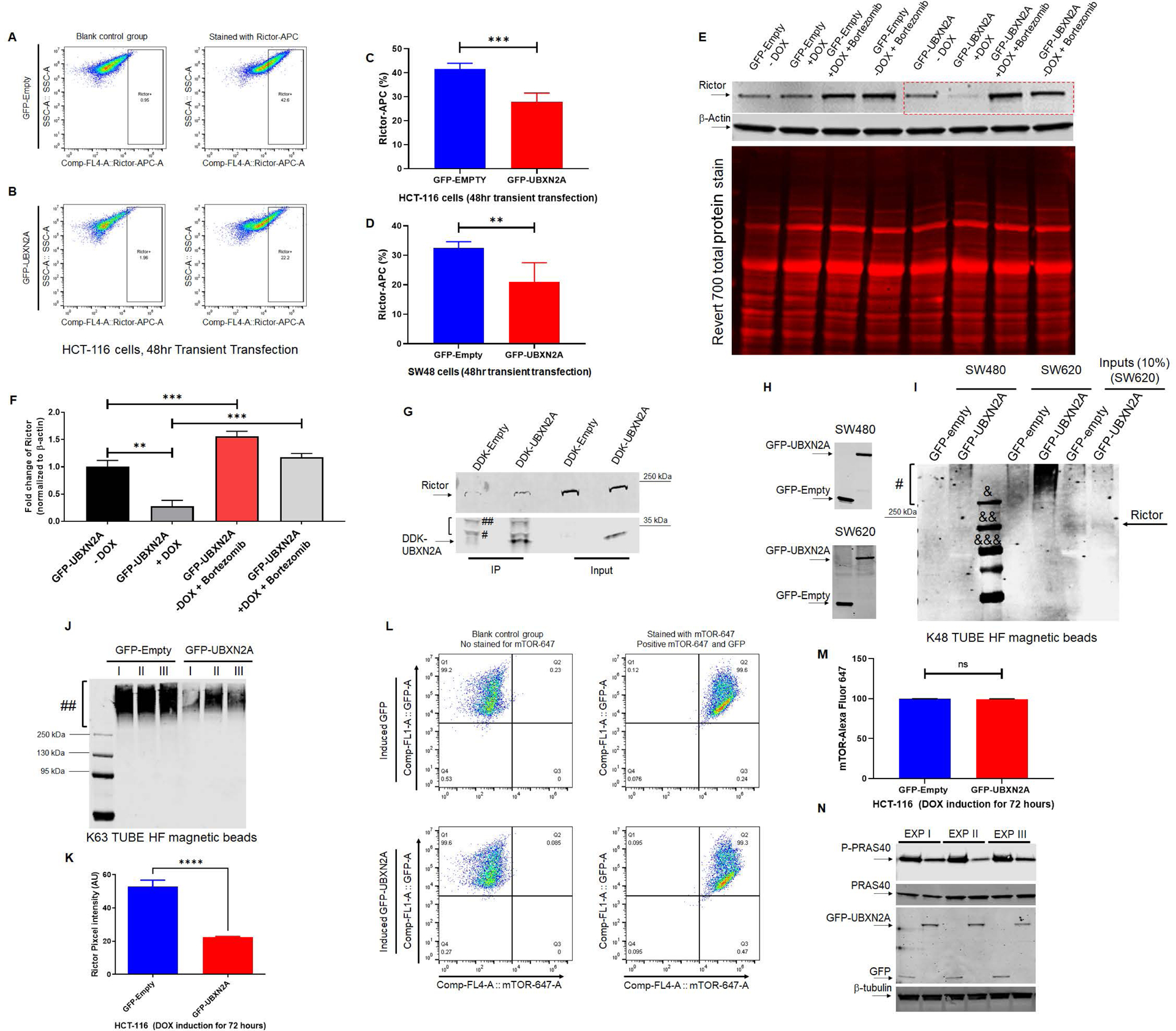
Flowcytometry analysis of HCT-116 cells transiently transfected with GFP-empty or GFP-UBXN2A vectors for 48 hours revealed UBXN2A overexpression significantly decreases Rictor protein (A-C). Similar to the HCT-116 cell line, transient transfection of SW48 cells with GFP-empty or GFP-UBXN2A vectors for 48 hours followed by flow-cytometry analysis showed UBXN2A can significantly decrease the protein level of Rictor (D). Induction of UBXN2A in HCT-116 treated with DOX for 72 hours decreased the half-life of Rictor, while the presence of a proteasome inhibitor (bortezomib, 50nM) rescues rapid turnover of Rictor (red box in panel E; quantitated signals in panel F). A β-actin antibody and Revert 700 total protein stain were used as the loading control. HCT-116 cells were transfected with DDK-Empty or DDK-UBXN2A plasmid for 48 hours followed by immunoprecipitation using anti-DDK antibody immobilized on magnetic IgG beads. DDK-UBXN2A pulled down Rictor protein, indicating UBXN2A binds to Rictor protein. Two bands tagged with one # and two ## are light and heavy chains, respectively (G). Due to the high molecular weight of Rictor protein (220 kDa), we observed a low non-specific affinity of Rictor protein to magnetic beads in control. In another set of experiments, K48 TUBE HF magnetic beads were used to pull down ubiquitinated Rictor in SW480 and SW620 (a metastatic colon cancer cell line) transiently transfected with GFP-empty or GFP-UBXN2A vectors for 48 hours (H). GFP-UBXN2A increases the K48-linked ubiquitination ladder (#) of Rictor protein (I), particularly in SW620. The K48 pull-down experiment was repeated with the same results trend. &, &&, and &&& are protein molecular markers for 250 kDa, 130, kDa, and 95 kDa, respectively. In contrast, K63 magnetic beads revealed induced UBXN2A decreases pulled down K63 linked chain of Rictor protein (n=3, Panels J) due to the reduction of total Rictor protein in the presence of UBXN2A measured by immunofluorescent microscopy in HCT-116 cells (n=150 cells-Panel K). A set of flow-cytometry analyses in HCT-116 cells transiently overexpressing GFP-empty of GFP-UBXN2A proteins showed the presence of UBXN2A has no significant effect on mTOR protein, another protein member of the mTORC2 complex (L and M). Finally, the Tet-on inducible GFP-empty and GFP-UBXN2A were treated with DOX for 72 hours. Cell lysates were subjected to WB using anti-PRAS40 (total protein and P-PRAS40, a phosphorylated form of protein) as well as a GFP antibody. A triplicate WB revealed that UBXN2A decreases the protein level P-PRAS40, activated by the Rictor-mTORC2 signaling pathway (N). Together, these flow-cytometry, K48/K63 linked chains, and WB results indicate UBXN2A selectively targets and degrades Rictor proteins via ubiquitin-proteasome pathway in colon cancer cells (** p< 0.01, *** p< 0.001, mean +/− SD).
In contrast, UBXN2A induction led to the reduction of K63-linked ubiquitinated Rictor protein, which is a proactive form of Rictor protein (52), pulled down by K63 TUBE magnetic beads (Fig. 3J). The reduction of p63-chain ubiquitinated Rictor protein could be due to the reduction of total Rictor in the presence of UBXN2A (Fig. 3K). Interestingly, the turnover of mTOR protein, another key member of the mTORC2 complex (1), remained stable in the presence of induced UBXN2A (Fig. 3L–M). Finally, figure 3N shows that induced GFP-UBXN2A decreases phosphorylation of PRAS40 (proline-rich AKT substrate) at Thr246, which is a key target of the phosphorylated AKT pathway (53, 54). This indicates that UBXN2A-dependent suppression of the Rictor-AKT pathway leads to efficient inhibition of downstream mTORC2-AKT activity.
The absence of UBXN2A leads to the overactivation of the mTORC2 pathway
To further confirm that the presence of UBXN2A is essential for the regulation of Rictor-mTORC2AKT activities, we generated two stably expressed knockout (KO) UBXN2A HCT-116 colon cancer cell lines (Clone 3 and Clone 9) using CRISPR technology. Clones #3 and #9 are heterozygote cells, and we were unable to establish a single clone despite several optimizations. While the QC report shows 85% editing efficiency (supplementary materials), we still observed a weak band for UBXN2A. This issue could be due to the necessity of UBXN2A as a key component of the P97 complex at the ER site, since we previously had a similar issue with shRNA against UBXN2A (55). This could be one of the limitations associated with CRISPR technology in certain cell lines, which needs to be addressed and solved by optimization of Cas systems per cell line (56). Alternatively, it could be due to the potential alternative splice form of UBXN2A (two isoforms produced by alternative splicing) enhanced in the absence of full-length UBXN2A. WT and KO cells were subjected to WB and flow cytometry analysis using an anti-Rictor antibody. Figure 4A shows the elevation of Rictor protein in the absence of UBXN2A in clones 3 and 9. A set of flow-cytometry analyses further confirmed that the absence of UBXN2A leads to a significant elevation of mean fluorescent intensity (MFI) of Rictor protein in HCT-116 cells (Figure 4B–D). A set of immunocytochemistry experiments confirmed that the absence of UBXN2A can significantly increase the level of Rictor protein (Fig. 4E–G-upper panel). Alexa Fluor 546 secondary fluorescent antibody was used to capture and measure corresponding Rictor signals (Figure 4H). Alexa Fluor 488 secondary antibody combined with Z stack imaging, which captured all Rictor signals per cell, further demonstrated the elevated level of total Rictor protein (free and associated with mTORC2 complex) in cytoplasmic compartments in the absence of UBXN2A (Fig. 4E–G, lower panels). As previously described for CRISPR-Cas9 technology (57), Rictor signals per individual cell revealed heterogeneity among cells in terms of elevated Rictor levels in UBXN2A KO cells. The ubiquitination and proteasomal-dependent turnover of Rictor in the presence of UBXN2A (Fig. 3) and its elevation in UBXN2A KO cells encouraged us to examine Rictor’s ubiquitination level in UBXN2A KO cells versus WT cells. Cell lysates were subjected to K48-magnetic bead pull-down experiments. Panel I in Figure 4 revealed that the K48-linked chain ubiquitinated ladder of Rictor protein decreases in the absence of UBXN2A, confirming that UBXN2A binds and ubiquitinates Rictor protein for proteasomal degradation. As previously shown for UBX domain-containing proteins, the total level of ubiquitinated proteins showed a reduction in UBXN2A KO cells (Fig. 4J). In the next step, we decided to examine whether the elevated level of Rictor in UBXN2A KO cells can lead to activation of the downstream pathway of the mTORC2 pathway. The mTORC2 signaling pathway regulates several pathways, including cell apoptosis and proliferation. A set of colony formation assays (Fig. 4K–N) and caspase-3 flow cytometry assays (Fig. 4O) confirmed the elevated Rictor in the absence of UBXN2A leads to significantly higher cell proliferation (Fig. 4K) and less cell death (significant in UBXN2A KO clone 9, Fig. 4O), respectively. On the other hand, overexpression of UBXN2A in HCT-116 cells led to a significant elevation of early and late apoptosis (supplemental figure 4A–F). As previously shown by a dual mTORC1/2 inhibitor (58), we observed elevation of PUMA and BAX in the presence of overexpressed UBXN2A (supplemental figure 4G). Figure 4P confirms that the elevated level of Rictor in UBXN2A KO cells can increase pAKT-473, which propagates a wide range of downstream signaling events. Finally, we used a set of WB experiments to reconfirm that the absence of UBXN2A alters the protein level of pAKT1, p-PRAS40, and cleaved PARP (Fig. 4Q). pAKT1, p-PRAS40, and cleaved PARP are downstream protein targets of the mTORC2-Rictor complex (17).
Fig. 4: Absence of UBXN2A leads to elevation of Rictor, resulting in inhibition of mTORC2’s downstream protein targets.
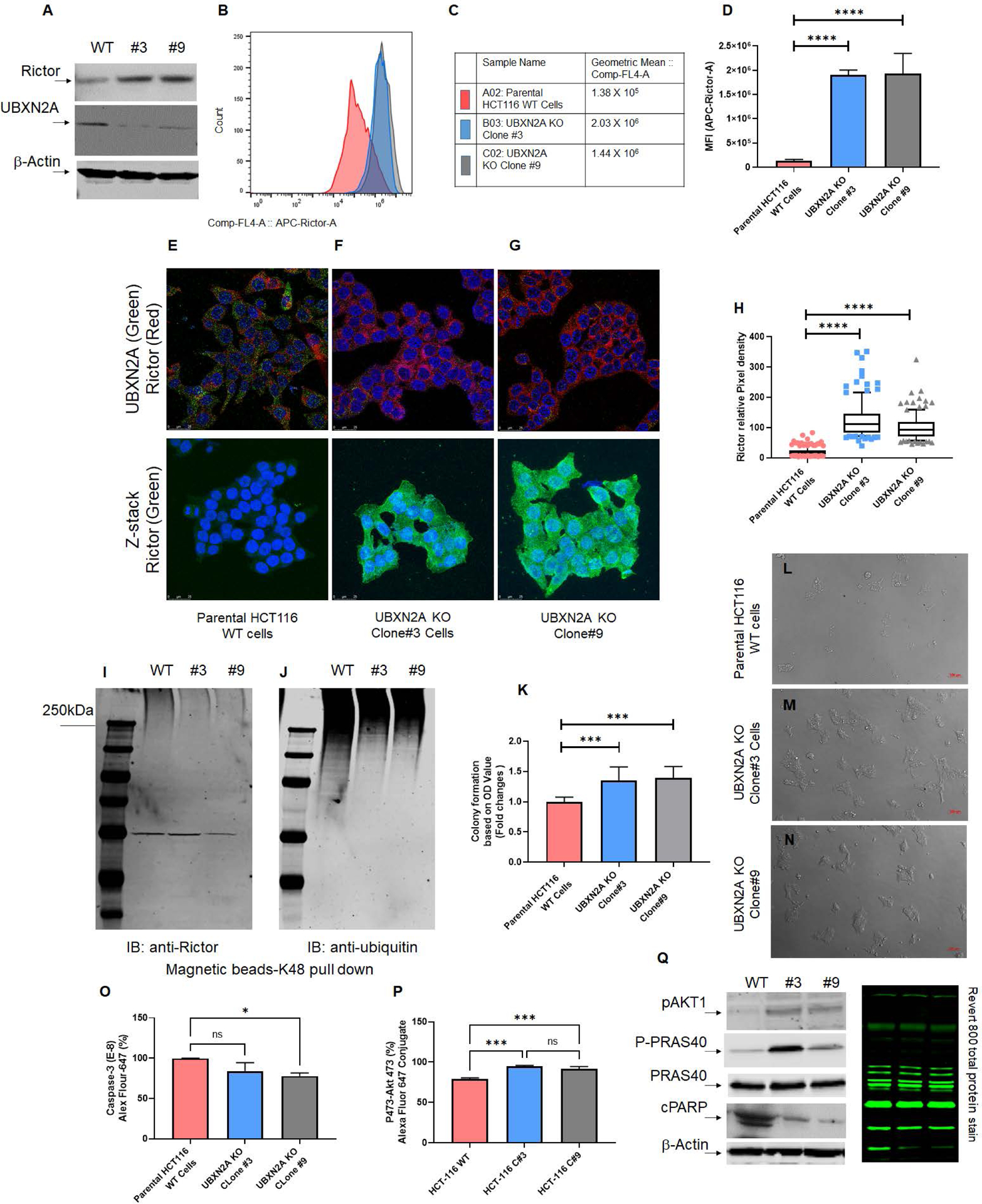
Two stable CRISPR UBXN2A KO HCT-116 cells generated by CRISPR/Cas9 genome editing were validated by WB (A) and flow-cytometry analysis (B-D). Clone 3 and clone 9 UBXN2A KO were subjected to confocal microscopy study (E-H), TUBE K48-linked ubiquitin chain magnetic beads pull-down (I-J), and crystal violet cell viability assay (K-N). These data demonstrate that the absence of UBXN2A in HCT-116 (clones 3 and 9) significantly elevates the level of Rictor protein measured by flow- cytometry and immunocytochemistry (D and H). Furthermore, the absence of UBXN2A decreases the K48 ubiquitinated form of Rictor in HCT-116 cells (I-J). The absence of UBXN2A and simultaneous elevation of Rictor leads to higher cell proliferation and less apoptosis stained and quantitated by crystal violet assay (K-N). Flow-cytometry analysis of Caspase-3 (O) and the elevation of pAKT473 (P) further confirmed the inverse relationship between UBXN2A and Rictor protein levels and Rictor’s downstream pathways responses. A set of WB experiments revealed that the absence of UBXN2A (clones 3 and 9) leads to the elevation of pAKT1 and P-PRAS40, as well as the reduction of cleaved PARP (cPARP), which are regulated by the activated mTORC2 pathway (Q). A β-actin antibody and Revert 800 total protein stain were used as the loading control (* n≥3, p<0.05, ***p<0.001, ****p<0.0001, mean ± SD).
UBXN2A-dependent suppression of mTORC2 complex impairs its downstream metastatic pathways
It has been shown that mTORC2 inactivation suppresses vascular endothelial growth factor (VEGF), a key element in tumor neovascularization (59, 60). In addition, siRNA silencing of Rictor protein leads to elevation of E-cadherin and reduction of N-cadherin, two key proteins in epithelial-mesenchymal transition (EMT) (61). Flow cytometry experiments indicate that the presence of UBXN2A decreases the level of VEGF in HCT-116 (Fig. 5A and 5C) and LoVo cells (Fig. 5B and 5D). Similarly, transient overexpression of UBXN2A decreases VEGF in SW620, a metastatic colon cancer cell line (Fig. 5E and Supplemental Figure 5). Next, the level of two EMT markers, E-cadherin and N-cadherin, were determined in the UBXN2A Tet-On inducible HCT-116 cell line after 72 hours of UBXN2A-induction with DOX. WB results confirmed the presence of UBXN2A can significantly elevate E-cadherin (Fig. 5F–G). While the protein levels of N-cadherin were not significantly affected by UBXN2A, it does show a reduction in response to UBXN2A. To further verify the inhibitory effect of UBXN2A on EMT, the expression levels and subcellular localization of E-cadherin and N-cadherin were examined by immunofluorescent confocal microscopy. Measurement of individual cells (N=150) in 5 different areas of the HCT-116 cell slide revealed that induction of UBXN2A oppositely switches the levels of E-cadherin (Fig. 5 H–I) and N-cadherin (Fig. 5 J–K), which can weaken EMT. To determine whether UBXN2A has a causal relationship with the Rictor protein, we conducted two rescue experiments using flow cytometry analysis. Supplemental Figure 6A–C shows overexpression of MYC-Rictor in HCT-116 cells with induced GFP-UBXN2A can lead to a significant reduction of early apoptosis enhanced by GFP-UBXN2A (column red versus orange). WB experiments focused on E-cadherin and Vimentin, two proteins downstream of the Rictor-mTORC2 pathway (62). Supplemental Figure 6D revealed that elevated E-cadherin and reduced Vimentin in the presence of induced UBXN2A are reversed by overexpression of MYC-Rictor. In another set of experiments, clones 3 and 9 UBXN2A KO cells were transfected with DDK-empty or DDK-UBXN2A (supplemental Figure 6E). WB experiments in supplemental Figure 6F show the level of E-cadherin increases in clones 3 and 9 overexpressing DDK-UBXN2A (Lanes 4 and 5) versus low level of E-cadherin proteins in clones 3 and 9 UBXN2A KO cells (Lanes 2 and 3).
Figure. 5: UBXN2A expression leads to the reduction of VEGF proteins in metastatic colon cancer cells.
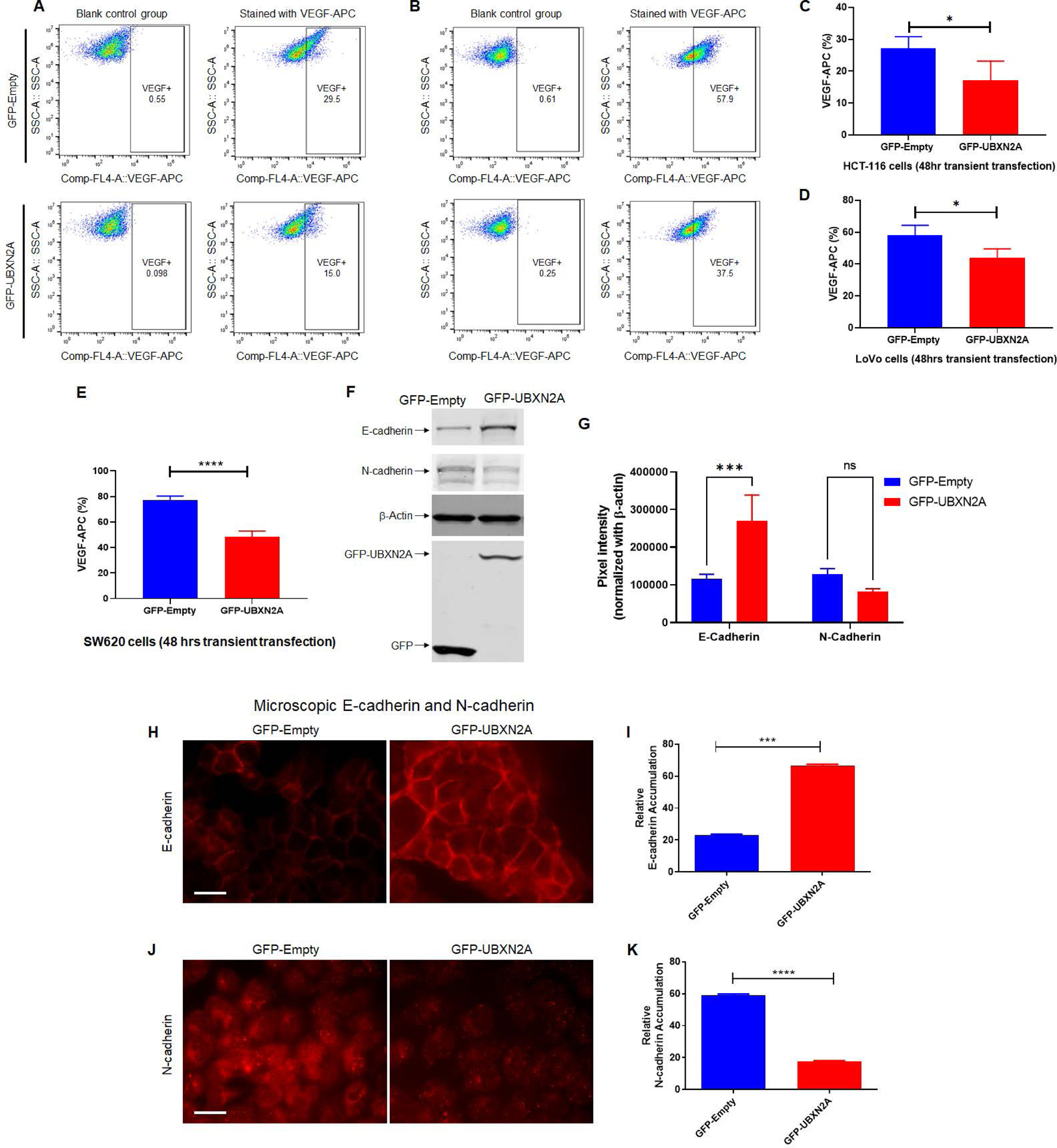
To determine the effects of UBXN2A expression on the VEGF protein level, we transiently transfected HCT-116 (A and C) and LoVo (B and D) colon cancer cells with GFP-empty and GFP-UBXN2A. Flow-cytometry experiments revealed that GFP-UBXN2A and not GFP-Empty decreases the level of VEGF proteins (C and D). A similar significant reduction of VEGF was measured in SW620 metastatic colon cancer cells (n=4, E). Cell lysates of HCT-116 cells transiently transfected with GFP-empty or GFP-UBXN2A were subjected to WB and probed with anti-E-cadherin and anti-N-cadherin antibodies (F). The quantitation of E-cadherin and N-cadherin bands normalized by β-actin in WB results (n=3, G). To confirm that UBXN2A regulates functional E-cadherin and N-Cadherin at the plasma membrane, we conducted a set of immunocytochemistry experiments (H and J). Measured fluorescent signals (n=150 cells per group, I and K) revealed UBXN2A overexpression can switch the E-cadherin and N-cadherin which potentially suppress EMT. These results indicate that UBXN2A expression can directly interfere with the two major metastatic pathways (angiogenesis and EMT) activated by the Rictor-mRORC2 pathway (* p<0.05, ***p<0.001, ****p<0.0001, mean ± SD).
Overall, the above gain of function and rescue experiments indicate a direct interaction between UBXN2A as a regulatory protein and Rictor protein in the mTORC2 signaling pathway in CRC cells.
UBXN2A suppresses cell adhesion and migration of colon cancer cells
It has been shown that disruption of the Rictor-mTORC2 pathway suppresses cell adhesion and migration in cancer cells (63, 64). We determined whether adhesion and migration can be affected by the presence or absence of UBXN2A in colon cancer cells. We first used a 16-well E-Plate format within the xCELLigence RTCA instrument to optimize the number of HCT-116 cells for the migration assay as well as to measure cell adhesion as previously described (65). Figure 6A–B- shows that induction of GFP-UBXN2A, but not GFP alone, significantly decreases cancer cell adhesion monitored by E-Plates. To determine whether UBXN2A expression suppresses colon cancer migration, 16-well CIM plates within the xCELLigence RTCA instrument were used (66, 67). Figure 6C–D shows that the expression of GFP-UBXN2A significantly decreases cell migration. To further verify that UBXN2A functions as a suppressor of colon cancer migration, we used a pharmacological tool, veratridine (48), to increase the level of endogenous UBXN2A in HCT-116 cells expressing GFP-UBXN2A. Results (Fig. 6C–D, blue column) confirmed that induction of endogenous UBXN2A further decreases cell migration, suggesting a dominant role for UBXN2A in colon cancer cell migration. Panel E in Figure 6 confirmed Dox robustly induces expression of GFP or GFP-UBXN2A at similar levels, and cells treated with VTD have elevated levels of endogenous UBXN2A. Finally, we examined the migration of clone 3 and clone 9 CRISPR UBXN2A KO HCT-116 cells versus control cells. Panels F and G in Figure 6 show a significant elevation of migration in the absence of UBXN2A, confirming a significant elevation of cell migration due to elevated stability of Rictor protein in the absence of UBXN2A KO cells.
Figure. 6: Genetic and pharmacological regulation of UBXN2A suppress colon cancer migration.
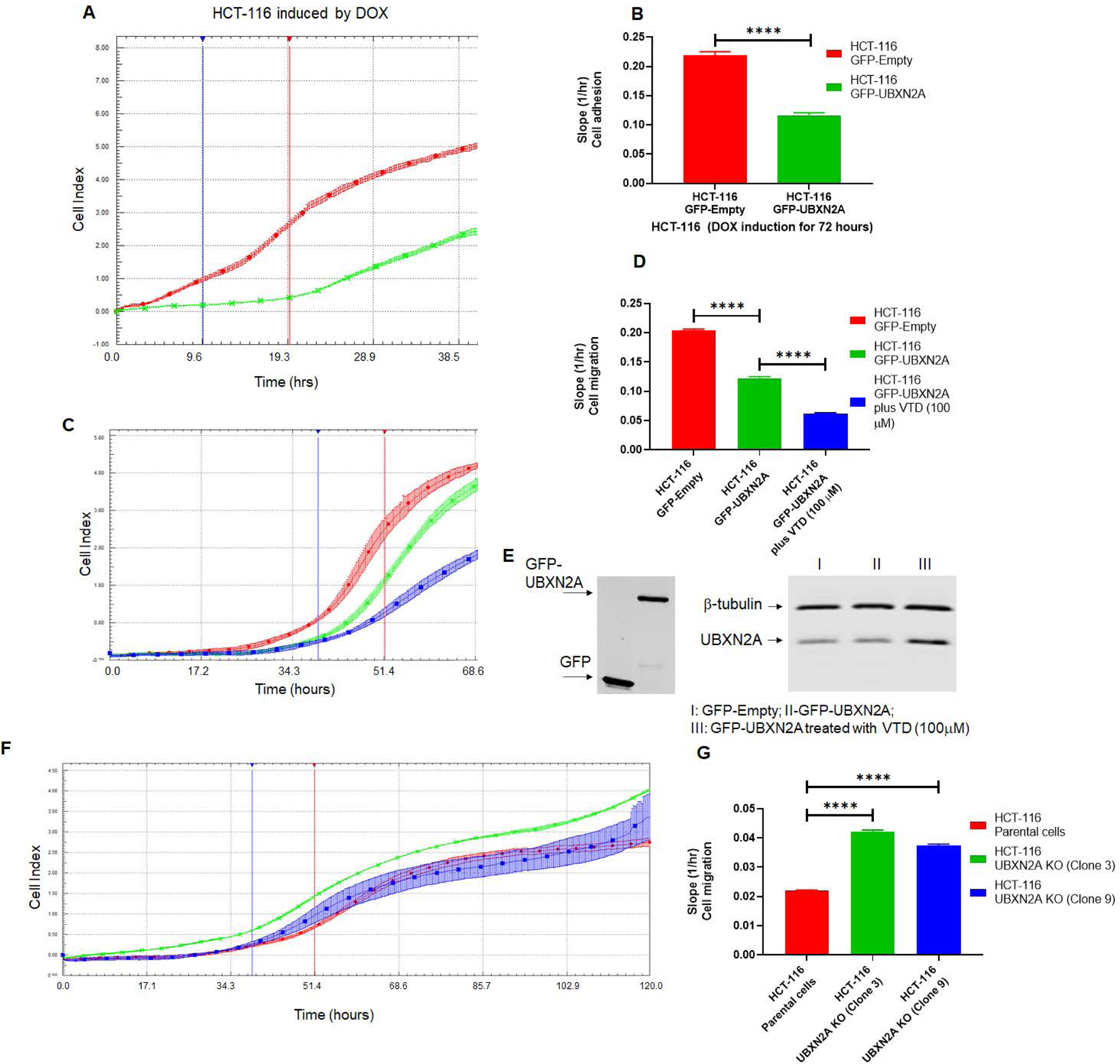
HCT-116 GFP-empty or GFP-UBXN2A treated with DOX for 72 hours, were plated in 16-wells E-plate or CIM-plate (xCELLigence Real-Time technology) and monitored in real-time for cell adhesion (A-B) and migration (C-D). A and C are representative graphs comparing the rate of adhesion and migration using the calculated cell index (see methods section). B and D show calculated slopes for these two events during critical time points marked with blue and red lines in the A and C diagrams. Interestingly, enhancement of UBXN2A endogenous protein by the pharmacological tool, veratridine, further decreases HCT-116 cancer cell migration (D, blue column versus green column). E shows equal overexpression of exogenous GFP and GFP-UBXN2A in HCT-116 with the Tet-on promoter system and elevated endogenous UBXN2A in veratridine-treated cells subjected to xCELLigence analysis. To examine the physiological effect of UBXN2A in cell migration, HCT-116 UBXN2A KO cells (clones 3 and 9) were subjected to a set of xCELLigence migration assays (F). Results shown in G indicate that the UBXN2A’s loss of function leads to significant cell migration indicating that the negative interaction of UBXN2A with Rictor affects downstream signaling pathways regulated by the mTORC2-Rictor pathway. Experiments were repeated two times with N of 4 per cell line per experiment (****P<0.0001, mean ± SD).
UBXN2A decreases positive cancer stem cell populations in human colon cancer cells
It is well accepted that cancer stem cells (CSCs) are the drivers of tumor progression and drug resistance (68). A recent report indicates that the mTORC2 signaling pathway regulates CSCs via the hedgehog pathway (69). We used flow cytometry to determine whether the expression of UBXN2A can decrease CSC populations. We used LGR5, CD44, and CD133 to examine the effect of UBXN2A in CSCs in two colon cancer cell lines. Figure 7A–F and statistical analysis of recorded positive cells (Fig. 7G–H and supplemental figure 7A–F) revealed UBXN2A significantly decreases the level of CD44- and LGR5-positive CSCs in HCT-116 cells. Both CD44+ and LGR5+ are involved in both local and liver metastasis in colorectal cancer (70). As previously reported in HCT-116 cells (71), we had no changes in CD-133 CSCs (Fig. 7I and supplemental figure 7G–L). Figure 7J–L shows overexpression of UBXN2A significantly decreases positive CSCs for CD-44, LGR5, and CD-133 in SW480 cells (Supplemental Figures 8, 9, and 10). It has been reported that the CD-133+ CSC population in SW480 colon cancer cells can generate tumor sphere-forming efficiency in vitro, and they increase tumorigenicity in animal models (72). In addition, colon CSCs are highly tumorigenic, aggressive, and chemoresistant, and they are a critical factor in the metastasis and recurrence of CRC (73). These data suggest that UBXN2A can regulate the stemness of colon cancer cells via the mTORC2-hedgehog axis, potentially resensitizing cancer cells to 5-FU. Therefore, we hypothesized that UBXN2A suppresses mTORC2 in 5-Fluorouracil (5-FU) resistant colon cancer cells enriched in CSCs. We generated 5-FU HCT-116 resistant cells carrying Tet-on GFP-empty and GFP-UBXN2A as previously described (74). Supplemental Figure 11 shows that induction of UBXN2A significantly decreases the pAKT473 in 5-FU-resistant cells. Finally, the UBXN2A Tet-on inducible cells were subjected to a set of rescue experiments using flow-cytometry analysis. Supplemental Figure 12 shows induction of GFP-UBXN2A significantly decreases CD44+ cells in comparison to GFP-empty cells (column blue versus red). However, the level of CD44+ cells reversed when MYC-Rictor was overexpressed in induced GFP-UBXN2A cells (column purple versus red).
Fig. 7: UBXN2A induction decreases cancer stem cell populations in human colon cancer cells.
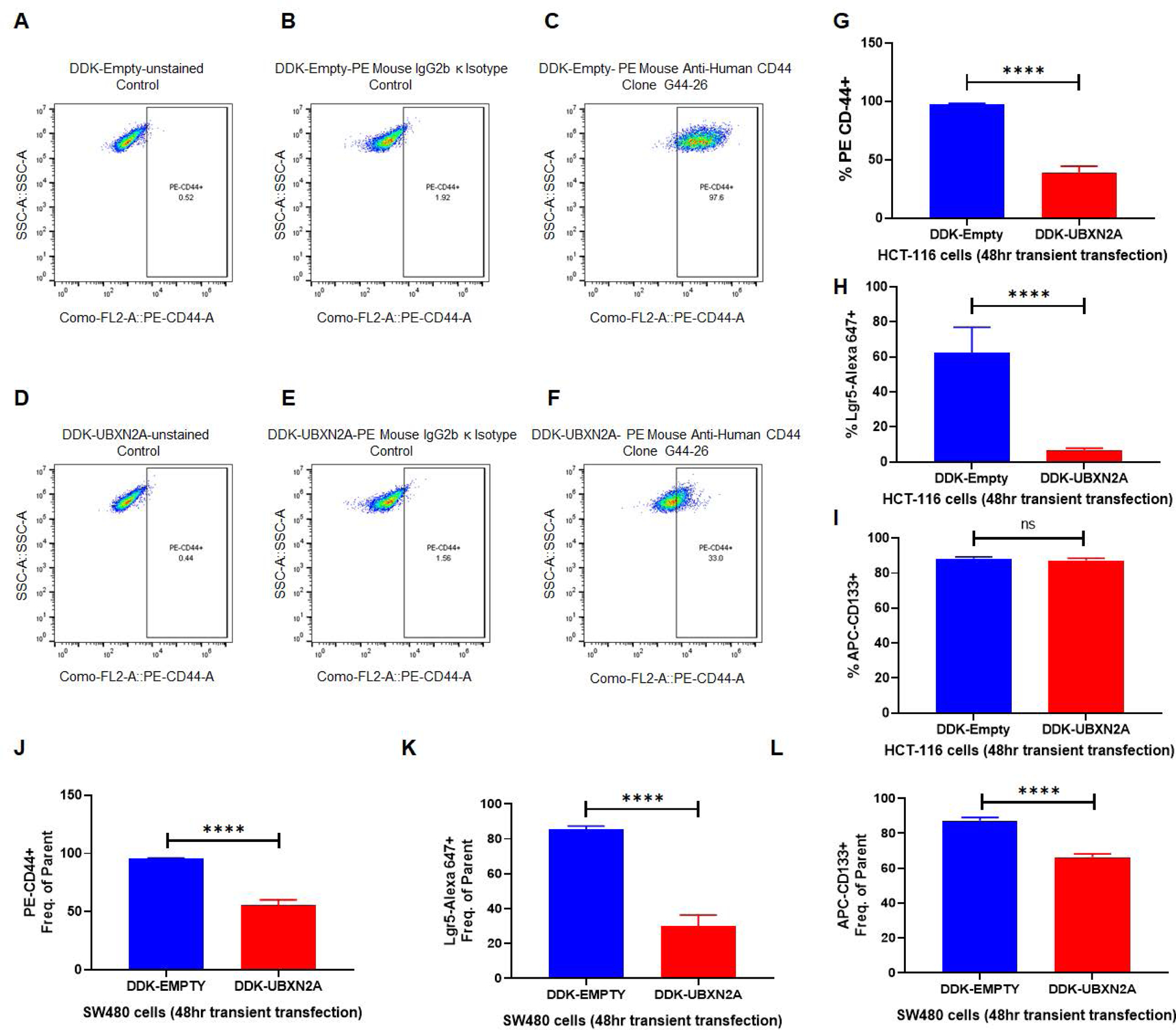
CSCs positive for Lgr5, CD44, and CD133 are regulated by the mTORC2-hedgehog axis in cancer cells and are potentially responsible for the high recurrence rates of CRC. HCT-116 (A-I) and SW480 (J-L) colon cancer cells were transfected with DDK-tag empty vector or DDK-tag UBXN2A. After 48 hours, cells were subjected to flow cytometry analysis using three CSC markers (Lgr5, CD44, and CD133). As previously described for HCT-116 cells, overexpression of UBXN2A has no significant effect on CD133 (I). UBXN2A overexpression led to a significant reduction of CD44 (J) and Lgr5 (K) as well as CD133 (L), which exhibits increased tumor sphere-forming efficiency and increased tumorigenic potential in the SW480 cell line (N=4, **** p< 0.0001, mean +/− SD).
Tumor suppressor protein UBXN2A acts by shutting down the function of the overactivated mTORC2 pathway in CRC tumor tissues
We have previously shown that UBXN2A is elevated in ~50% of human CRC tissues (48). To determine the status of UBXN2A expression in a different stage of CRC, we conducted a set of immunohistochemistry (IHC) experiments on human CRC tissues at three different stages. Figure 8A shows a basal expression of UBXN2A in normal colon tissue. We stained well-differentiated (N=26), moderately differentiated (N=74), and poorly differentiated (N=23) CRC tumors by polyclonal anti-UBXN2A antibodies. Scored signals revealed a significant upregulation of UBXN2A in well-differentiated tumor tissues in comparison to normal colon tissue. However, the level of UBXN2A significantly decreased in moderately and poorly differentiated tumor tissues (Fig. 8B–E). As previously observed in several tumor suppressor proteins (29, 75, 76), UBXN2A protein levels predominantly upgrade during the early stage of tumor development (Fig. 8F). According to IHC results, we further explored the prognostic relevance of UBXN2A expression in CRC using the OncoLnc online tool. From the reanalysis of the available Kaplan–Meier survival analysis data in TCGA, survival analysis shows a consistent effect of UBXN2A on colorectal cancer. However, when analyzing colon and rectal adenocarcinoma, we found inconsistencys in survival rates between adenocarcinoma (COAD) and rectal cancer adenocarcinoma (READ), two subtypes of colon cancer. As in overall colorectal cancer survival analysis, an elevated UBXN2A level in COAD correlated with higher survival rates in patients (log-rank p-value=0.0361, Fig. 8G). This could not be said conclusively about READ, as the survival curve analysis showed a modest survival rate correlation (log-rank p-value=0.811, Fig. 8H). Calculation of the 5-year survival rate for patients with colon cancer (Fig. 8G) and rectal cancer (Fig. 8H) revealed that those who had tumors expressing high levels of UBXN2A experienced better overall survival compared to those with tumors expressing low levels of UBXN2A. To further find the prognostic value of UBNX2A in COAD patients, we downloaded the gene expression and clinical patient data from TCGA for alive patients and processed the level of UBXN2A expression. Analysis of UBXN2A expression in the alive patient cohort shows a prognostic benefit of high UBXN2A with improved overall survival (Fig. 8I). Furthermore, to assess whether UBXN2A is indicative of prognosis in patients with CRC, cox-regression analysis was performed (Supplementary material).
To further confirm that elevation of UBXN2A can target the mTORC2 pathway in human cancer tissues, we used two sets of patient-derived human tumor organoids (see materials and methods) treated with and without the UBXN2A enhancer veratridine (VTD) (77) for 72 hours. As previously reported, VTD increased the level of UBXN2A protein roughly by 2-fold (Fig. 8J). WB analysis of lysates revealed that enhancement of UBXN2A can decrease the level of Rictor, which consequently leads to elevation of E-cadherin and reduction of VEGF and phosphorylated PRAS40 proteins (Fig. 8K). In summary, a hyperactive form of mTORC2 in CRC stimulates and maintains several tumorigenic pathways such as EMT, angiogenesis (VEGF), and CSCs. A high level of UBXN2A observed in 50% of patients with CRC or pharmacological stimulation of UBXN2A can interrupt developing metastatic pathways in CRC patients by targeting and degrading Rictor protein, a key member of the mTORC2 pathway (Fig. 8L).
Discussion
In this study, we demonstrated a negative regulatory interaction between UBXN2A tumor suppressor protein and Rictor protein. Rictor is a key member of the mTORC2 complex. Activation of Rictor plays a critical role in CRC formation and progression (1). A combination of genetic and pharmacological tools revealed that the presence of UBXN2A regulates the proteasomal degradation of Rictor protein in colon cancer cells. UBXN2A-dependent regulation of Rictor protein affects phosphorylation of AKT protein and its downstream pathways, including VEGF, EMT markers, and apoptosis, as well as cancer cell adhesion and migration. Results indicate that the reduction of VEGF was moderate in response to UBXN2A which can be due to the fact that mTORC2 regulates angiogenesis independent of AKT/mTORC1 (59). A moderate reduction of VEGF by UBXN2A can ideally avoid triggering of compensatory mechanisms observed with current VEGF inhibitors (78). This study revealed that suppression of the mTORC2-Rictor pathway by UBXN2A can significantly decrease CSCs in the colon cancer cells that are responsible for tumor metastasis and drug resistance (79).
Analyzing the TCGA dataset with the optimal cutoff value revealed that the survival rate is significantly better for colon cancer patients with high expression of UBXN2A than for those with low expression. Based on this set of patient genomics data, we used IHC to determine the level of UBXN2A in different stages of colon cancer. In this study, we showed that the UBXN2A protein level was highly upregulated in colon tumor tissues with low invasiveness corresponding to the early stages of colon cancer. More importantly, UBXN2A was downregulated in colon tumor cells with high invasiveness (poorly differentiated tumors) corresponding to later stages of colon adenocarcinoma. Similar examples are retinoblastoma and MCIP1 tumor suppressor proteins; their expressions have been shown to increase during the early stages of tumor development and then to be reduced or absent in later stages of cancer (80, 81). As previously reported for selected tumor suppressor proteins (29, 82, 83), UBXN2A may inversely regulate the migration and metastasis of colon adenocarcinoma cells in the early stages of cancer. The loss or downregulation of UBXN2A found in later stages of colon cancer could happen in response to overall genetic alternations developed in late-stage CRC, including the UBXN2A gene. Alternatively, low UBXN2A levels in late-stage CRC tumors could be due to post-translational modification. We believe the adaptor role of UBXN2A and its cross-talk with E3 ligases, including the CHIP E3 ligase (32), allow UBXN2A to target different oncoproteins such as Mortalin and Rictor protein, perhaps in a tissue- or stage-dependent manner.
In conclusion, we have demonstrated UBXN2A as a novel inhibitor of the mTORC2 signaling pathway. UBXN2A suppresses several metastatic signaling pathways downstream of the mTORC2 pathway via proteasomal degradation of Rictor protein. It is highly plausible that the UBXN2A-Rictor-mTORC2 axis may be altered during the progression of colon cancer, determining the prognosis for patients (Fig. 8L). Understanding the physiological and therapeutic potential of UBXN2A in human colon cancer will open a new platform for developing selective anti-mTORC2 drugs that leave the mTORC1 pathways primarily intact.
Materials and methods
Mice
The UBXN2A-null mouse line (Ubxn2atm1(KOMP)Mbp) was engineered by the Knockout Mouse Project (KOMP,www.KOMP.org) in the C57BL/6 background. We used an AOM (Azoxymethane) and DSS (Dextran sodium sulfate, MP Biomedicals LLC, Irvine, CA) mouse model of colon cancer. Mice were first treated with one dose of AOM and subsequently administered DSS (2% in drinking water) for one week, as previously established (34). Our AOM/DSS protocol generated a polypoid growth of adenoma-carcinoma sequence predominantly in the distal colon mimicking human CRC (84). The xenograft mouse model with HCT-116 colon cancer cells expressing DOX TET-inducible GFP-empty or GFP-UBXN2A has previously been described (48)
Histopathological Examination
Isolated colon samples (swiss rolled) were fixed instantly with 10% formalin and kept in 70% ethanol. The paraffin-embedded samples were sectioned to 5 mm thickness and stained with H&E and periodic acid-Schiff (PAS). The degree of hyperplasia for low- and high-grade adenoma were determined by a pathologist, who was blinded to group identity.
Western Blot, Immunoprecipitation, Ubiquitin-pull down, immunofluorescent, and crystal violet staining method assays:
Mouse colon tissues were dissected and cleaned in ice-cold PBS. Following snap freezing in liquid nitrogen, they were stored at −80°C. 60mg colon tissue was subsequently prepared and placed in a digitonin lysis buffer (50 mM Tris/HCl, pH 7.5, 150 mM NaCl, 1% Digitonin (Sigma-Aldrich, St. Louis, MO) plus 1x mammalian complete protease inhibitor (Research Products International Corp). Silicon/Zirconia Beads (2.3mm) were then added to the tube containing the lysis buffer, and cells were mechanically homogenized for 30 seconds with the MiniBead Beater (Biospec Products). Following homogenization (two hours gentle rocking at 4°C), tissue lysates were subjected to centrifugation at 13000RPM for 10min at 4°C, and the supernatant was moved to a fresh Eppendorf tube in preparation for WB. Cell lysates used in WB were normalized for equal loading by NanoDrop using direct absorbance at 280 nm (ThermoFisher Scientific). We loaded the samples onto SDS-PAGE 4–20% gradient gel. We performed protein transfer using an iBlot 2 system for probing with the corresponding antibodies. For a detailed list of antibodies used in this experiment, see the supplemental materials. Immunoprecipitation, Ubiquitin-pull down, and immunofluorescent assays were conducted as previously described (32). The cytotoxicity test protocol using a crystal violet staining method has been described previously (36).
Tissue Specimens, Immunohistochemistry and Immunohistochemical Analysis
We procured tissue microarrays (TMAs) containing colon cancer tissues of various grades and adjacent normal tissues from patients from AccuMax (catalog number A203 [VI]; catalog numbers A203 [III] and A203 [IV] ISU Abxis Co., Ltd., San Diego, CA). In addition, we obtained archived colon cancer samples from Sanford Health. The final number of colon cancer tissue spots suitable for analysis was 123. We processed the slides for immunohistochemistry (IHC) analysis using a polymer-based MACH4 IHC kit (Biocare Medical; Concord, CA) according to the manufacturer’s recommendation (unless indicated, all reagents were purchased from Biocare Medical) (85). All slides were studied using an Olympus BX 41 microscope (Olympus Corporation; Center Valley, PA). We performed quantitative analysis of immunoreactivity by calculating the composite score, which is a function of the percentage of cancer cells positively immunostained multiplied by the UBXN2A staining intensity (range, 0–16). For rating the intensity of the staining, we scored the percentage of cancer cells positively stained for UBXN2A on a scale of 0 to 4 as follows: 1=minimal, 2=moderate, 3=strong, and 0 if there is no staining. In addition, the score for the number of stained cells was as follows: 1 = 1–25% staining, 2 = 26–50% staining, 3 = 51 −75% staining, 4 = 76–100% staining, and 0 if there was no staining. The mean composite score (MCS) of UBXN2A staining was calculated based on total immunoreactivity localized to the cytoplasm or nucleus. All recorded signals was scored by an independent pathologist blinded to the pathology reports. This type of semi-quantitative analysis (i.e., MCS), which takes into account both intensity of staining as well as the extent of tumor cells stained, has been widely used for the quantification of protein expression obtained from IHC (85).
UBXN2A affects survival analysis in COAD, READ, and CRC
We used two interactive survival analysis online tools (Human protein atlas and www.oncolnc.org) to perform UBXN2A survival analysis in COAD, READ, and CRC. The human protein atlas contains 3 sets of datasets: 1) RNA-seq tissue data in mean transcripts per million from HPA, 2) RNA-seq data in median reads per kilobase per million mapped reads from GTEx dataset, and 3) cap analysis gene expression in tags per million data from FANTOM5 dataset. OncoLnc contains data from 8647 patients on 21 cancer studies from the Cancer Genome Atlas (TCGA). The Kaplan-Meier plots were created based on specific gene (UBXN2A) expression levels among the subjects in these datasets
Statistical analysis
We analyzed all statistical values presented in this study with the software GraphPad Prism 9. The difference between groups was analyzed by Student’s t-test or the one-way ANOVA. A p-value of ≤0.05 was used to compare mean values and signify a statistically significant result. Data are presented as the mean ± standard deviation (SD). We used N=3 for Tet-on xenograft experiments and n=4 for patient-derived xenograft experiments (Figure 2).
DATA AVAILABILITY STATEMENT
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Supplementary Material
Fig. 8: UBXN2A protein levels predominantly upgrade during the early stage of tumor development and improve survival rates in colorectal cancer patients.
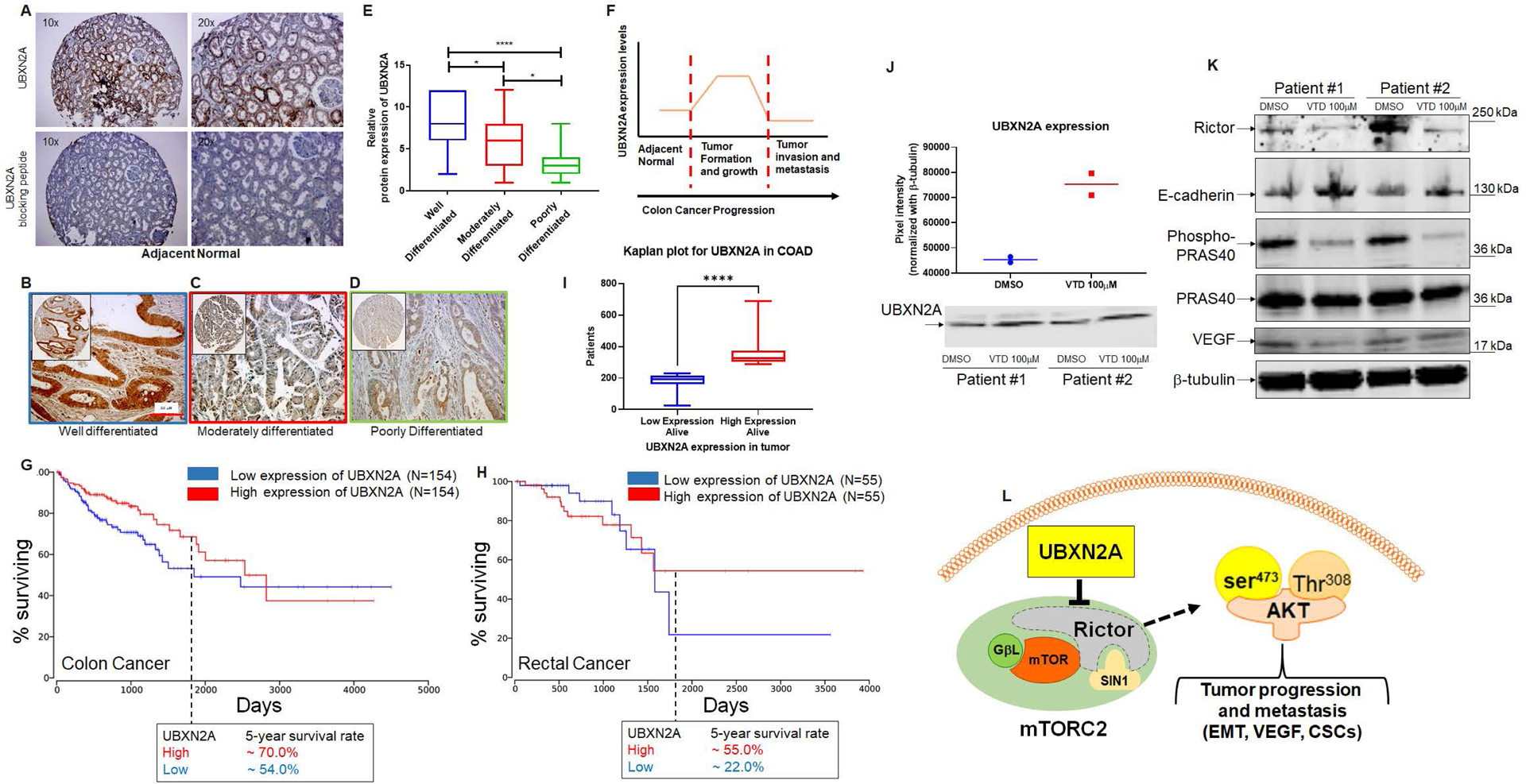
A shows UBXN2A’s medium protein expression level in normal colon tissues. A peptide-blocking assay confirmed the specificity of the antibody against the UBXN2A protein in the IHC study. B-D: IHC staining was used to stain cytoplasmic and nuclear UBXN2A in well-differentiated (n=26), moderately (n=74), and poorly differentiated (n=24 tumor tissues) human colon tumor tissues. E: UBXN2A expression levels of colon cancer tissues were manually scored. The quantitative scoring system revealed that UBXN2A, as a tumor suppressor protein, significantly upregulates in the early stage of colon cancer and shows a significant reduction in a higher stage of colon cancer. The reduction of UBXN2A is associated with a poorer prognosis in higher stages of CRC. (F). Kaplan–Meier’s analysis of extracted survival data from TCGA shows COAD, a subtype of colorectal cancer, indicates a correlation of higher survival rate with higher UBXN2A expression. The 5-year survival rate is ~20% higher than patients with low expression of UBXN2A in tumors (G). READ, a subtype of colorectal cancer Kaplan–Meier’s analysis, shows a favorable survival outcome in high UBXN2A expression. Patients with rectal cancer with a high level of UBXN2A show ~30% elevation of 5-year survival (H). Panel G and H were produced using the Human Protein Atlas and ONCL tools. For ONCL, we used 33 low and 33 high percentiles parameters. Further analysis shows a significantly larger portion of alive patients with COAD had higher UBXN2A expression compared to lower expression of UBXN2A, indicating a longer progression-free survival than those with low UBXN2A in the TCGA (I). PDOs generated from surgically removed CRC tumors (n=2) were treated with the UBXN2A enhancer Veratridine (VTD, 100μM) for 72 hours. Two individual PDOs showed VTD elevates the level of UBXN2A protein expression by approximately two-fold (J) and simultaneously decreases Rictor resulting in alteration of mTORC2 protein targets, including reduction of P-PRAS40 and VEGF proteins as well as elevation of E-cadherin (K). Schematic diagram showing the mechanistic inhibitory action of UBXN2A on mTORC2 tumorigenic pathway through selective proteasomal degradation of Rictor protein. The absence of a fully functional mTORC2 complex leads to the inhibition of several downstream metastatic pathways (L).
ACKNOWLEDGEMENT
The DaCCoTA funding is supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number U54GM128729 and the National Cancer Institute of the National Institutes of Health under award number 1R03CA223935–01. ARB and the organoid core at UNMC were supported by Nebraska Center for Molecular Target Discovery and Development (P20GM121316) and Fred & Pamela Buffett Cancer Center Support Grant (CA036727). The bioinformatic studies (EGZ) were partially funded by the National Science Foundation/Experimental Program to Stimulate Competitive Research (EPSCoR) Grant OIA-1849206 awarded to Gilbert Ustad and the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health P20GM103443 (V.C.Huber).
Footnotes
COMPETING INTRESTS
The authors declare no competing financial interests.
ETHICAL APPROVAL
The use of archived clinical samples in this study was approved by the institutional review board (IRB) at Sanford Research/USD, Sioux Falls, South Dakota. All animal procedures were approved by the Institutional Animal Care and Use Committee by federal guidelines (the University of South Dakota- IACUC#04-03-20-23E).
Supplementary information
Additional materials and methods are detailed in Supplementary Materials. The information on antibodies is listed in Table S2.
References:
- 1.Roulin D, Cerantola Y, Dormond-Meuwly A, Demartines N, Dormond O. Targeting mTORC2 inhibits colon cancer cell proliferation in vitro and tumor formation in vivo. Molecular Cancer. 2010;9:57-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong CK, Lambert AW, Ozturk S, Papageorgis P, Lopez D, Shen N, et al. Targeting RICTOR Sensitizes SMAD4-Negative Colon Cancer to Irinotecan. Mol Cancer Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ponnurangam S, Standing D, Rangarajan P, Subramaniam D. Tandutinib inhibits the Akt/mTOR signaling pathway to inhibit colon cancer growth. Mol Cancer Ther. 2013;12(5):598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shiratori H, Kawai K, Okada M, Nozawa H, Hata K, Tanaka T, et al. Metastatic role of mTOR signaling activation by chemoradiotherapy in advanced rectal cancer. Cancer Sci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jhanwar-Uniyal M, Wainwright JV, Mohan AL, Tobias ME, Murali R, Gandhi CD, et al. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Advances in Biological Regulation. 2019;72:51–62. [DOI] [PubMed] [Google Scholar]
- 6.Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell. 2015;162(2):259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reilly KE, Rojo F, She Q-B, Solit D, Mills GB, Smith D, et al. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Research. 2006;66(3):1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of Feedback Loops Mediated by PI3K/mTOR Induces Multiple Overactivation of Compensatory Pathways: An Unintended Consequence Leading to Drug Resistance. Molecular Cancer Therapeutics. 2014;13(11):2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. The Journal of Clinical Investigation. 2008;118(9):3065–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf. 2013;12(2):177–86. [DOI] [PubMed] [Google Scholar]
- 11.Le Tourneau C, Faivre S, Serova M, Raymond E. mTORC1 inhibitors: is temsirolimus in renal cancer telling us how they really work? British Journal Of Cancer. 2008;99:1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spindler K- LG, Sorensen MM, Pallisgaard N, Andersen RF, Havelund BM, Ploen J, et al. Phase II trial of temsirolimus alone and in combination with irinotecan for KRAS mutant metastatic colorectal cancer: Outcome and results of KRAS mutational analysis in plasma. Acta Oncologica. 2013;52(5):963–70. [DOI] [PubMed] [Google Scholar]
- 13.Sharma S, Becerra CR, Matrana MR, Alistar AT, Chiorean EG, Schmid AN, et al. A phase I/II multicenter study of ABI-009 (nab-sirolimus) combined with FOLFOX and bevacizumab as first-line (1L) therapy in patients (pts) with metastatic colorectal cancer (mCRC) with or without PTEN loss. Journal of Clinical Oncology. 2019;37(4_suppl):TPS730–TPS. [Google Scholar]
- 14.Kawata T, Tada K, Kobayashi M, Sakamoto T, Takiuchi Y, Iwai F, et al. Dual inhibition of the mTORC1 and mTORC2 signaling pathways is a promising therapeutic target for adult T-cell leukemia. Cancer science. 2018;109(1):103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altomare I, Hurwitz H. Everolimus in colorectal cancer. Expert Opin Pharmacother. 2013;14(4):505–13. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Lin J, Wang X, Yao G, Wang L, Zheng H, et al. Targeting of mTORC2 prevents cell migration and promotes apoptosis in breast cancer. Breast Cancer Res Treat. 2012;134(3):1057–66. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Lai P, Zhang Z, Huang M, Wang L, Yin M, et al. Targeted inhibition of mTORC2 prevents osteosarcoma cell migration and promotes apoptosis. Oncol Rep. 2014;32(1):382–8. [DOI] [PubMed] [Google Scholar]
- 18.Werfel TA, Wang S, Jackson MA, Kavanaugh TE, Joly MM, Lee LH, et al. Selective mTORC2 Inhibitor Therapeutically Blocks Breast Cancer Cell Growth and Survival. Cancer Research. 2018;78(7):1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benavides-Serrato A, Lee J, Holmes B, Landon KA, Bashir T, Jung ME, et al. Specific blockade of Rictor-mTOR association inhibits mTORC2 activity and is cytotoxic in glioblastoma. PLOS ONE. 2017;12(4):e0176599. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Murray ER, Cameron AJM. Towards specific inhibition of mTORC2. Aging. 2017;9(12):2461–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Wang X, Xu T, Zhong S, Shen Z. Targeting of mTORC2 may have advantages over selective targeting of mTORC1 in the treatment of malignant pheochromocytoma. Tumour Biol. 2015;36(7):5273–81. [DOI] [PubMed] [Google Scholar]
- 22.Zou Z, Chen J, Yang J, Bai X. Targeted Inhibition of Rictor/mTORC2 in Cancer Treatment: A New Era after Rapamycin. Curr Cancer Drug Targets. 2016;16(4):288–304. [DOI] [PubMed] [Google Scholar]
- 23.Aimbetov R, Chen CH, Bulgakova O, Abetov D, Bissenbaev AK, Bersimbaev RI, et al. Integrity of mTORC2 is dependent on the rictor Gly-934 site. Oncogene. 2012;31(16):2115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Qi J, Yu J, Chen H, Zou Z, Lin X, & Guo L Overexpression of Rictor protein in colorectal cancer is correlated with tumor progression and prognosis. ONCOLOGY LETTERS. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruicci KM, Plantinga P, Pinto N, Khan MI, Stecho W, Dhaliwal SS, et al. Disruption of the RICTOR/mTORC2 complex enhances the response of head and neck squamous cell carcinoma cells to PI3K inhibition. Molecular oncology. 2019;13(10):2160–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jebali A, Dumaz N. The role of RICTOR downstream of receptor tyrosine kinase in cancers. Mol Cancer. 2018;17(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao D, Jiang M, Zhang X, Hou H. The role of RICTOR amplification in targeted therapy and drug resistance. Molecular medicine (Cambridge, Mass). 2020;26(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang F, Zhang X, Li M, Chen P, Zhang B, Guo H, et al. mTOR complex component Rictor interacts with PKCzeta and regulates cancer cell metastasis. Cancer Res. 2010;70(22):9360–70. [DOI] [PubMed] [Google Scholar]
- 29.Hsu TI, Wang MC, Chen SY, Yeh YM, Su WC, Chang WC, et al. Sp1 expression regulates lung tumor progression. Oncogene. 2012;31(35):3973–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HS, Park CK, Oh E, Erkin OC, Jung HS, Cho MH, et al. Low SP1 expression differentially affects intestinal-type compared with diffuse-type gastric adenocarcinoma. PLoS One. 2013;8(2):e55522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864–70. [DOI] [PubMed] [Google Scholar]
- 32.Sane S, Hafner A, Srinivasan R, Masood D, Slunecka JL, Noldner CJ, et al. UBXN2A enhances CHIPmediated proteasomal degradation of oncoprotein mortalin-2 in cancer cells. Molecular oncology. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerovska D, Araúzo-Bravo MJ. Does mouse embryo primordial germ cell activation start before implantation as suggested by single-cell transcriptomics dynamics? Mol Hum Reprod. 2016;22(3):208–25. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94(11):965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plentz RR, Wiemann SU, Flemming P, Meier PN, Kubicka S, Kreipe H, et al. Telomere shortening of epithelial cells characterises the adenoma-carcinoma transition of human colorectal cancer. Gut. 2003;52(9):1304–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sane S, Abdullah A, Boudreau DA, Autenried RK, Gupta BK, Wang X, et al. Ubiquitin-like (UBX)-domain-containing protein, UBXN2A, promotes cell death by interfering with the p53-Mortalin interactions in colon cancer cells. Cell Death Dis. 2014;5:e1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sailo BL, Banik K, Girisa S, Bordoloi D, Fan L, Halim CE, et al. FBXW7 in Cancer: What Has Been Unraveled Thus Far? Cancers (Basel). 2019;11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmad R, Singh JK, Wunnava A, Al-Obeed O, Abdulla M, Srivastava SK. Emerging trends in colorectal cancer: Dysregulated signaling pathways (Review). Int J Mol Med. 2021;47(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Atanasova VS, Riedl A, Strobl M, Flandorfer J, Unterleuthner D, Weindorfer C, et al. Selective Eradication of Colon Cancer Cells Harboring PI3K and/or MAPK Pathway Mutations in 3D Culture by Combined PI3K/AKT/mTOR Pathway and MEK Inhibition. Int J Mol Sci. 2023;24(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koveitypour Z, Panahi F, Vakilian M, Peymani M, Seyed Forootan F, Nasr Esfahani MH, et al. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019;9:97-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malinowsky K, Nitsche U, Janssen KP, Bader FG, Spath C, Drecoll E, et al. Activation of the PI3K/AKT pathway correlates with prognosis in stage II colon cancer. Br J Cancer. 2014;110(8):2081–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pandurangan AK. Potential targets for prevention of colorectal cancer: a focus on PI3K/Akt/mTOR and Wnt pathways. Asian Pac J Cancer Prev. 2013;14(4):2201–5. [DOI] [PubMed] [Google Scholar]
- 43.Roy HK, Olusola BF, Clemens DL, Karolski WJ, Ratashak A, Lynch HT, et al. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis. 2002;23(1):201–5. [DOI] [PubMed] [Google Scholar]
- 44.Saturno G, Valenti M, De Haven Brandon A, Thomas GV, Eccles S, Clarke PA, et al. Combining trail with PI3 kinase or HSP90 inhibitors enhances apoptosis in colorectal cancer cells via suppression of survival signaling. Oncotarget. 2013;4(8):1185–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agarwal E, Robb CM, Smith LM, Brattain MG, Wang J, Black JD, et al. Role of Akt2 in regulation of metastasis suppressor 1 expression and colorectal cancer metastasis. Oncogene. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Breuleux M, Klopfenstein M, Stephan C, Doughty CA, Barys L, Maira S-M, et al. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Molecular Cancer Therapeutics. 2009;8(4):742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang W, Shen T, Dong B, Creighton CJ, Meng Y, Zhou W, et al. MAPK4 overexpression promotes tumor progression via noncanonical activation of AKT/mTOR signaling. J Clin Invest. 2019;129(3):1015–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdullah A, Sane S, Branick KA, Freeling JL, Wang H, Zhang D, et al. A plant alkaloid, veratridine, potentiates cancer chemosensitivity by UBXN2A-dependent inhibition of an oncoprotein, mortalin-2. Oncotarget. 2015;16(27):23561–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biever A, Valjent E, Puighermanal E. Ribosomal Protein S6 Phosphorylation in the Nervous System: From Regulation to Function. 2015;8(75). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song EJ, Yim SH, Kim E, Kim NS, Lee KJ. Human Fas-associated factor 1, interacting with ubiquitinated proteins and valosin-containing protein, is involved in the ubiquitin-proteasome pathway. Mol Cell Biol. 2005;25(6):2511–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu-Baer F, Ludwig T, Baer R. The UBXN1 protein associates with autoubiquitinated forms of the BRCA1 tumor suppressor and inhibits its enzymatic function. Mol Cell Biol. 2010;11:2787–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wrobel L, Siddiqi FH, Hill SM, Son SM, Karabiyik C, Kim H, et al. mTORC2 Assembly Is Regulated by USP9X-Mediated Deubiquitination of RICTOR. Cell Rep. 2020;33(13):108564. [DOI] [PubMed] [Google Scholar]
- 53.Yang G, Murashige Danielle S, Humphrey Sean J, James David E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Reports. 2015;12(6):937–43. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Zhang Q, Wen Q, Zheng Y, Lazarovici P, Jiang H, et al. Proline-rich Akt substrate of 40kDa (PRAS40): a novel downstream target of PI3k/Akt signaling pathway. Cell Signal. 2012;24(1):17–24. [DOI] [PubMed] [Google Scholar]
- 55.Abdullah A, Sane S, Freeling JL, Wang H, Zhang D, Rezvani K. Nucleocytoplasmic Translocation of UBXN2A Is Required for Apoptosis during DNA Damage Stresses in Colon Cancer Cells. J Cancer. 2015;6(11):1066–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Y, Xu J, Ge S, Lai L. CRISPR/Cas: Advances, Limitations, and Applications for Precision Cancer Research. Frontiers in medicine. 2021;8:649896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olive JF, Qin Y, DeCristo MJ, Laszewski T, Greathouse F, McAllister SS. Accounting for tumor heterogeneity when using CRISPR-Cas9 for cancer progression and drug sensitivity studies. PLoS One. 2018;13(6):e0198790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gupta M, Hendrickson AE, Yun SS, Han JJ, Schneider PA, Koh BD, et al. Dual mTORC1/mTORC2 inhibition diminishes Akt activation and induces Puma-dependent apoptosis in lymphoid malignancies. Blood. 2012;119(2):476–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Farhan MA, Carmine-Simmen K, Lewis JD, Moore RB, Murray AG. Endothelial Cell mTOR Complex2 Regulates Sprouting Angiogenesis. PLoS One. 2015;10(8):e0135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Amato KR, Song W, Youngblood V, Lee K, Boothby M, et al. Regulation of endothelial cell proliferation and vascular assembly through distinct mTORC2 signaling pathways. Mol Cell Biol. 2015;35(7):1299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R. TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J Cell Sci. 2012;125(Pt 5):1259–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, et al. mTORC1 and mTORC2 Regulate EMT, Motility, and Metastasis of Colorectal Cancer via RhoA and Rac1 Signaling Pathways. Cancer Research. 2011;71(9):3246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morrison Joly M, Williams MM, Hicks DJ, Jones B, Sanchez V, Young CD, et al. Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast Cancer Research. 2017;19(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen L, Xu B, Liu L, Liu C, Luo Y, Chen X, et al. Both mTORC1 and mTORC2 are involved in the regulation of cell adhesion. Oncotarget. 2015;6(9):7136–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hamidi H, Lilja J, Ivaska J. Using xCELLigence RTCA Instrument to Measure Cell Adhesion. Bio Protoc. 2017;7(24):e2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roshan Moniri M, Young A, Reinheimer K, Rayat J, Dai LJ, Warnock GL. Dynamic assessment of cell viability, proliferation and migration using real time cell analyzer system (RTCA). Cytotechnology. 2015;67(2):379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edwards G, Campbell T, Henderson V, Danaher A, Wu D, Srinivasan R, et al. SNAIL Transctiption factor in prostate cancer cells promotes neurite outgrowth. Biochimie. 2021;180:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang C, Xie J, Guo J, Manning HC, Gore JC, Guo N. Evaluation of CD44 and CD133 as cancer stem cell markers for colorectal cancer. Oncol Rep. 2012;28(4):1301–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maiti S, Mondal S, Satyavarapu EM, Mandal C. mTORC2 regulates hedgehog pathway activity by promoting stability to Gli2 protein and its nuclear translocation. Cell Death Dis. 2017;8(7):e2926. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Wang XF, Zhang XL, Xu LP, Shi GG, Zheng HY, Sun BC. [Expression of stem cell markers CD44 and Lgr5 in colorectal cancer and its relationship with lymph node and liver metastasis]. Zhonghua yi xue za zhi. 2018;98(36):2899–904. [DOI] [PubMed] [Google Scholar]
- 71.Dittfeld C, Dietrich A, Peickert S, Hering S, Baumann M, Grade M, et al. CD133 expression is not selective for tumor-initiating or radioresistant cell populations in the CRC cell line HCT-116. Radiother Oncol. 2010;94(3):375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y, Zhou L, Qing Q, Li Y, Li L, Dong X, et al. Gene expression profile of cancer stem-like cells in the SW480 colon adenocarcinoma cell line. Oncol Rep. 2019;42(1):386–98. [DOI] [PubMed] [Google Scholar]
- 73.Ma Y-S, Li W, Liu Y, Shi Y, Lin Q-L, Fu D. Targeting Colorectal Cancer Stem Cells as an Effective Treatment for Colorectal Cancer. Technology in Cancer Research & Treatment. 2020;19:1533033819892261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boyer J, McLean EG, Aroori S, Wilson P, McCulla A, Carey PD, et al. Characterization of p53 wild-type and null isogenic colorectal cancer cell lines resistant to 5-fluorouracil, oxaliplatin, and irinotecan. Clin Cancer Res. 2004;10(6):2158–67. [DOI] [PubMed] [Google Scholar]
- 75.Qi L, Ding Y. Screening of Tumor Suppressor Genes in Metastatic Colorectal Cancer. BioMed Research International. 2017;2017:2769140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sugimura-Nagata A, Koshino A, Nagao K, Nagano A, Komura M, Ueki A, et al. SPATA18 Expression Predicts Favorable Clinical Outcome in Colorectal Cancer. Int J Mol Sci. 2022;23(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Freeling JL, Scholl JL, Eikanger M, Knoblich C, Potts RA, Anderson DJ, et al. Pre-clinical safety and therapeutic efficacy of a plant-based alkaloid in a human colon cancer xenograft model. Cell Death Discov. 2022;8(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loges S, Mazzone M, Hohensinner P, Carmeliet P. Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell. 2009;15(3):167–70. [DOI] [PubMed] [Google Scholar]
- 79.Kozovska Z, Gabrisova V, Kucerova L. Colon cancer: cancer stem cells markers, drug resistance and treatment. Biomed Pharmacother. 2014;68(8):911–6. [DOI] [PubMed] [Google Scholar]
- 80.Xu HJ, Hu SX, Cagle PT, Moore GE, Benedict WF. Absence of retinoblastoma protein expression in primary non-small cell lung carcinomas. Cancer Res. 1991;51(10):2735–9. [PubMed] [Google Scholar]
- 81.Stathatos N, Bourdeau I, Espinosa AV, Saji M, Vasko VV, Burman KD, et al. KiSS-1/G Protein-Coupled Receptor 54 Metastasis Suppressor Pathway Increases Myocyte-Enriched Calcineurin Interacting Protein 1 Expression and Chronically Inhibits Calcineurin Activity. The Journal of Clinical Endocrinology & Metabolism. 2005;90(9):5432–40. [DOI] [PubMed] [Google Scholar]
- 82.Wu D Epithelial protein lost in neoplasm (EPLIN): Beyond a tumor suppressor. Genes & diseases. 2017;4(2):100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang LH, Wu CF, Rajasekaran N, Shin YK. Loss of Tumor Suppressor Gene Function in Human Cancer: An Overview. Cell Physiol Biochem. 2018;51(6):2647–93. [DOI] [PubMed] [Google Scholar]
- 84.Nascimento-Gonçalves E, Mendes BAL, Silva-Reis R, Faustino-Rocha AI, Gama A, Oliveira PA. Animal Models of Colorectal Cancer: From Spontaneous to Genetically Engineered Models and Their Applications. Veterinary sciences. 2021;8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gupta BK, Maher DM, Ebeling MC, Sundram V, Koch MD, Lynch DW, et al. Increased expression and aberrant localization of mucin 13 in metastatic colon cancer. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2012;60(11):822–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.