

Abstract
Tumor necrosis factor (TNF) plays an important role in the pathogenesis of inflammatory and autoimmune diseases such as rheumatoid arthritis and Crohn’s disease. The biological effects of TNF are mediated by binding to TNF receptors, TNF receptor 1 (TNFR1), or TNF receptor 2 (TNFR2), and this coupling makes TNFR1-specific inhibition by small-molecule therapies essential to avoid deleterious side effects. Recently, we engineered a time-resolved fluorescence resonance energy transfer biosensor for high-throughput screening of small molecules that modulate TNFR1 conformational states and identified zafirlukast as a compound that inhibits receptor activation, albeit at low potency. Here, we synthesized 16 analogues of zafirlukast and tested their potency and specificity for TNFR1 signaling. Using cell-based functional assays, we identified three analogues with significantly improved efficacy and potency, each of which induces a conformational change in the receptor (as measured by fluorescence resonance energy transfer (FRET) in cells). The best analogue decreased NF-κB activation by 2.2-fold, IκBα efficiency by 3.3-fold, and relative potency by two orders of magnitude. Importantly, we showed that the analogues do not block TNF binding to TNFR1 and that binding to the receptor’s extracellular domain is strongly cooperative. Despite these improvements, the best candidate’s maximum inhibition of NF-κB is only 63%, leaving room for further improvements to the zafirlukast scaffold to achieve full inhibition and prove its potential as a therapeutic lead. Interestingly, while we find that the analogues also bind to TNFR2 in vitro, they do not inhibit TNFR2 function in cells or cause any conformational changes upon binding. Thus, these lead compounds should also be used as reagents to study conformational-dependent activation of TNF receptors.
Keywords: ligand-independent interactions, TNF, TNFR1, NF-κB activation, Inflammatory diseases
Introduction
Tumor necrosis factor receptor 1 (TNFR1) plays a pivotal role in signaling the inflammatory pathway.1 Binding of the tumor necrosis factor (TNF) to the extracellular domain of TNFR1 leads to IκBα degradation and NF-κB activation.2 Upregulation of TNF levels has been associated with several inflammatory and autoimmune diseases, including rheumatoid arthritis (RA), multiple sclerosis, and Crohn’s disease.3 The prevalence of these autoimmune diseases affects approximately 23.5 million people in the USA based on 24 autoimmune diseases with available epidemiologic studies4 and up to 50 million people when considering all autoimmune diseases.5 Hence, TNFR1 is a high-value target and therapeutic intervention of its receptor signaling is a billion-dollar industry of high interest to pharmaceutical companies.
Current clinical strategies to attenuate the interaction of TNF-TNFR1 involve several anti-TNF biologic agents including anti-TNF monoclonal antibodies (infliximab, adalimumab, certolizumab pegol, golimumab) and a soluble TNF receptor (etanercept) that functions by sequestering TNF and blocking ligand binding to the receptor.6−9 These biological agents can be expensive, cause injection site reactions or infusion reactions, and often fail to cross the blood–brain barrier.7,10,11 Recently, several small-molecule anti-TNFs have also been reported.12−14 They are typically allosteric inhibitors that disrupt the trimeric structure of TNF, resulting in asymmetric trimer or dimer and the binding of only two monomer units to the receptors instead of three and completely inhibit the functions of TNF in vitro and in vivo.12 However, there are a few disadvantages of allosteric inhibitors. Generally, allosteric modulators have lower binding affinities than orthosteric modulators due to shallower and narrower binding sites15 and have lower water solubility.16 These characteristics may make it difficult to advance allosteric modulators as therapeutic candidates for clinical testing. Furthermore, allosteric sites are frequently unknown or challenging to discover because they are only accessible in specific protein conformations that may not have an associated resolved structure.17 Finally, allosteric sites complicate testing in various species homologs, such as human vs rodent receptors, because they are less evolutionarily conserved among subspecies than orthosteric sites are across protein families.18,19 Despite high potency—the most potent TNF inhibitors UCB-9260 and TNF IN-42, which inhibit NF-κB with EC50 of 250 and 27 nM, respectively—these TNF targeting therapeutics have several drawbacks stemming from the global blockade of TNF signaling.12,13 These include increased rates of infections and lymphomas due to reduced TNFR1 signaling as well as lupus-like symptoms associated with the lack of TNFR2 stimulation. Low rates of disease remission and the generation of antibodies against biologic anti-TNFs have also been observed in clinical studies.9,20 New receptor-specific treatments that directly target TNFR1 rather than the ligands could overcome these limitations.
Recently, in a fluorescence resonance energy transfer (FRET)-based high-throughput screening study using the National Institutes of Health (NIH) clinical collection library, we have identified a small-molecule compound zafirlukast that directly binds to TNFR1 and inhibits TNF-induced NF-κB activation.21 To the best of our knowledge, zafirlukast is the first and only small-molecule inhibitor of TNFR1 that does not block ligand binding.21 We were intrigued by the potential of using zafirlukast scaffold (Scheme 1) as a template to explore its structure–activity relationships and screen for more potent TNFR1 inhibitors.22−24 In this study, we synthesized 16 analogues and tested their potency and specificity for TNF-induced TNFR1 signaling. Using cell-based functional assays, we identified three zafirlukast analogues with significantly improved activity. Importantly, we showed that the analogues do not block TNF binding to TNFR1. We note that analogues bind to TNFR2, but they do not inhibit TNFR2 function or cause any conformational changes. Thus, these lead compounds can be used not only for therapy development but also as new candidates to learn more about how these receptors’ activation and inhibition differ from one another.
Scheme 1. Synthesis and Schematic of Variations in R1 and R2 Groups.

Reagents and conditions: (a) Ag2O, dioxane, RT, 20 h, (b) NaH, MeI, THF, RT, 1 h, (c) Pd/C, H2, THF, RT, 20 h, (d) chloroformate, Et3N, DCM, 0 °C to RT, 1 h, (e) LiOH, H2O, MeOH, RT,16 h, (f) sulfonamides, EDC, DMAP, DCM, RT, 16 h.
Materials and Methods
Cell Cultures and Reagents
HEK293 cells were cultured in phenol red–free Dulbecco’s modified Eagle medium supplemented with 2 mM l-glutamine, heat-inactivated 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 μg/mL). HUVEC cells were cultured on 0.2% gelatin-coated dishes in EGM-2 medium supplemented with heat-inactivated 2% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 μg/mL). Cell cultures were incubated at 37 °C in a humid atmosphere of 5% CO2.
NF-κB Luciferase Reporter Gene Assay
HEK293 cells were transfected with the NF-κB–luciferase reporter genes in a 10 cm plate with Lipofectamine 3000. Next day, cells were lifted with TrypLE and resuspended in phenol red–free DMEM. Transfected cells (7500 cells/well) were dispensed in 96-well white, solid-bottom plates and incubated with drugs (0.01–500 nM) or DMSO (negative control) in the presence (10 ng/mL) and absence of TNF for 24 h at 37 °C. After incubation, 70 μL of Dual-Glo Luciferase Reagent (Promega, Madison, WI) was added and incubated at room temperature for 15 min, and firefly luminescence was measured using a Cytation 3 Cell Imaging Multi-Mode Reader luminometer. Next, 70 μL of Dual-Glo Stop & Glo Reagent was added and incubated at room temperature for 15 min, and Renilla luminescence was measured using a luminometer.
IkB Degradation Assay
HEK293 cells were cultured into six-well plates at 0.4 million/mL and incubated overnight. Next day, cells were treated with DMSO (negative control) and respective doses of drug compounds (0.01–500 nM) for 2 h, followed by 30 min of TNF (10 ng/mL). Cells were lysed for 30 min on ice with native lysis buffer containing 1% protease inhibitor and centrifuged at 13,000 rpm at 4 °C for 15 min. The total protein concentration of lysates was determined by bicinchoninic acid assay (BCA), and equal amounts of total protein (80 μg) were mixed with 4× Bio-Rad sample buffer and boiled for 5 min. Next, protein samples were resolved by SDS–PAGE and immunoblotted with anti-IκBα and β-actin.
MTT Assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to measure cytotoxicity of zafirlukast and its analogues. To determine the cytotoxicity of zafirlukast and its analogues, HEK293 cells were seeded in 96-well plates at a density of 7500 cells/well and incubated for 24 h at 37 °C and 5% CO2. After incubation, cells were treated separately with increasing concentrations of zafirlukast or analogues (0.0001 nM–10 μM), followed by 24 h of incubation at 37 °C. Cell viability was assessed with a Cytation 3 Cell Imaging Multi-Mode Reader luminometer (BioTek).
TRADD-Induced NF-κB Activation Assay
For TRADD-induced NF-κB activation in HEK293 cells, cells (0.8 million/well, in a six-well plate) were transfected with 1 μg of NF-κB firefly luciferase reporter gene, 0.1 μg of Renilla luciferase reporter gene, 1 μg of TRADD plasmid, and 0.4 μg of empty plasmid for a total of 2.5 μg of DNA. In the control cells, the TRADD plasmid was replaced with an empty plasmid. After 3 h of transfection, cells were harvested and plated (7500 cells/well: total volume, 70 μL) into 96-well white solid-bottom assay plates. Drug treatment (0.01 to 500 nM) or DMSO treatment (negative control) was performed 5 h after cell plating. Luciferase activities were determined 24 h after treatments. Briefly, 70 μL of Dual-Glo Luciferase Reagent was added and incubated at room temperature for 15 min and firefly luminescence was measured using a Cytation 3 Cell Imaging Multi-Mode Reader luminometer. Next, 70 μL of Dual-Glo Stop & Glo Reagent was added and incubated at room temperature for 15 min and measured Renilla luminescence using a luminometer.
IL-1α-Induced NF-κB Activation Assay
HEK293 cells were transfected with the NF-κB–luciferase reporter genes in a 10 cm plate with Lipofectamine 3000. Next day, cells were lifted with TrypLE and resuspended in phenol red–free DMEM. Transfected cells (7500 cells/well) were dispensed in 96-well white, solid-bottom plates and incubated with drugs (0.01–500 nM) or DMSO (negative control) in the presence (10 ng/mL) and absence of IL-1α for 24 h at 37 °C. After incubation, 70 μL of Dual-Glo Luciferase Reagent (Promega, Madison, WI) was added and incubated at room temperature for 15 min, and firefly luminescence was measured using a Cytation 3 Cell Imaging Multi-Mode Reader luminometer. Next, 70 μL of Dual-Glo Stop and Glo Reagent was added and incubated at room temperature for 15 min, and Renilla luminescence was measured using a luminometer.
TNF-TNFR1 Pulldown Assay
For TNFR1 pulldown assay, the HEK293 cells were lysed with native lysis buffer and protein concentrations were determined using BCA. Next, 10 μL of anti-FLAG magnetic beads was incubated with 30 μL of 25 μg/mL FLAG-tagged TNF for 2 h at 4 °C. Unbound TNF was then removed using a magnet, and the anti-FLAG beads were washed three times with PBS with 0.5% albumin (PBSA). Next, TNF-coated beads were incubated with 250 μL of HEK293 lysate with (250 nM) and without analogues, and anti-FLAG beads alone were incubated with PBS or HEK293 lysate. After incubation, beads were washed three times with PBSA. 10 μL of 1× loading dye was added to the 10 μL of beads and pipetted up and down 5 times to elute the proteins. Using a magnet, the 10 μL of dye was then removed and placed in a separate tube. This elution step was repeated three more times for each sample. Pulled-down samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotted with anti-Flag and anti-TNFR1 antibodies.
RelB Transcription Factor Activity Assay
The effect of zafirlukast analogues on TNFR2 function was determined using a RelB transcription factor activity assay. Since TNFR2 expression is limited to immune cells such as activated CD4+ and CD8+ lymphocytes, endothelial cells, microglia, and oligodendrocytes,25−28 we performed RelB assay in HUVEC cells. HUVEC cells were plated in a 6-well plate at 1 million cells per well and left in an incubator overnight. Next day, cells were transfected with 0.5 μg of membrane TNF using Lipofectamine 3000. After 6 h, transfecting media was replaced with fresh media and cells were then treated with the zafirlukast analogues and incubated overnight. Next day, cells were lifted and lysed with a native lysis buffer and protein concentrations were determined using BCA. Proteins samples were resolved by SDS–PAGE and immunoblotted with anti-RelB antibody.
Molecular Biology
EGFP and TagRFP vectors were a kind gift from David D. Thomas. cDNAs encoding TNFR1ΔCD or TNFR2ΔCD were inserted at the N-terminus of the EGFP and TagRFP vectors using standard cloning techniques. To prevent the dimerization and aggregation of EGFP, alanine 206 was mutated to lysine (A206K).
Time-Resolved Förster Resonance Energy Transfer Assay
HEK293 cells were transfected using Lipofectamine 3000 with TNFR2ΔCD-GFP and TNFR2ΔCD-GFP–TNFR2ΔCD-RFP (1:6 ratio) for a total of 2.5 μg/well in 6-well plates for 24 h and transfection was confirmed using EVOS fluorescence microscopy. For lifetime measurements, transfected cells were detached with trypsin, washed thrice with PBS, treated with analogues (10 μM), and incubated for 1–2 h. After incubation, cells were dispensed (50 μL/well) into a 96-well glass-bottom plate. The donor lifetime in the presence and absence of the acceptor was measured by using a fluorescence lifetime plate reader (Fluorescence Innovations, Inc., Minneapolis, MN). GFP fluorescence was excited with a 473 nm microchip laser, and emission was filtered with 488 nm long-pass and 517 nm/20 nm band-pass filters. Time-resolved fluorescence waveforms for each well were fitted to single-exponential decays using least-squares minimization global analysis software (Fluorescence Innovations, Inc.) to give the donor lifetime (τD) and donor–acceptor lifetime (τDA). The FRET efficiency (E) was then calculated based on eq 1:
| 1 |
One-Dimensional 19F Ligand-Observed NMR
Zafirlukast analogues were dissolved in DMSO to a concentration of 10 mM. Commercial recombinant TNFR1-ECD and TNFR2-ECD protein (Sino Biological) and death receptor 5 (DR5) (Abcam) were reconstituted following manufacturer’s recommendations. NMR samples were then prepared to give the following final sample conditions: 25 μM compounds with 0, 0.5, 1.25, 2.5, 5, 10, and 20 μM TNFR1 protein or 1.25 μM DR5/TNFR2 supplemented with 5% D2O and 0.0025% TFA in PBS, pH 7.4. NMR data were collected on a Bruker 600 MHz Avance NEO, equipped with a 5 mm triple resonance cryoprobe at 298 K using a standard one-dimensional 19F pulse sequence with 128 scans. The acquisition time is 0.498 s. Spectra were referenced to trifluoroacetic acid (d = −75.20 ppm). The change in chemical shift was plotted against protein concentration to obtain KD values using eq 2.
| 2 |
Results
Synthesis of Zafirlukast Analogues
The derivatives of zafirlukast were synthesized using an adaptation of the route established by Matassa et al.,29 as shown in Scheme 1 and the Supporting Information. The sequence begins with the construction of the zafirlukast benzyl-indole core using a silver-mediated Friedel-Crafts reaction of 5-nitroindole (1) with the aptly-substituted bromobenzyl ester (2) yielding the desired product 3 in 40% yield. The reduced yield noted is most likely due to the 5-nitro group that deactivates the C3 position of the indole to electrophilic substitution by the activated benzyl group. All attempts to improve this yield using alternative Lewis acids and modified reaction conditions failed to improve the percent conversion of the nitroindole starting material to the desired alkylated product. N-methylation was accomplished using sodium hydride in anhydrous THF followed by the addition of methyliodide to obtain 4 in 70% yield. Indole substitutions were subsequently completed in a two-step process involving the palladium on carbon reduction of the 5-nitro group to the amine followed by base-catalyzed acylation of 5 using cyclopentyl chloroformate to obtain the desired carbamate 6 in 50% yield. The final compounds were obtained by simple base-catalyzed saponification of the benzoic ester to the corresponding carboxylic acid (7) followed by EDC activation and coupling with a series of sulfonamides to produce acylsulfonamides 8a–j in 30 to 83% yields. Compounds 9a–f were synthesized using the same set of reactions by first installing the trifluorophenyl sulfonamide moiety followed by diversification of the carbamate using a series of alkylformates.
Subset of Zafirlukast Analogues Partially Inhibits TNFR1-Induced NF-κB Activation with High Potency
The effect of zafirlukast analogues on TNF-induced NF-κB activation was determined using a cell-based functional assay. To do this, we first checked the surface expression TNFR1 and TNFR2 in HEK293 cells using flow cytometry. These results suggest that HEK293 cells only express TNFR1 (Figure 1A) but not TNFR2 (Figure 1B). We then tested the effect of 10 acylsulfonamide (8a–j) on TNFR1 antagonism at two concentrations (0.5 and 5 μM). One out of 10 analogues blocked up to 20% of the NF-κB activity at 0.5 μM treatment (Figure 1C). To further explore the effect of the carbamate moiety on TNFR1 antagonism, we screened a set of 6 compounds (9a–f) (Scheme 1). Two additional potent hits (9a-MeCF3 and 9c-MeOEtCF3) emerged with NF-κB activity blocked up to 32–40% (Figure 1E). Interestingly, 5 μM treatment increased the NF-κB activity when compared with 0.5 μM treatment (Figure 1D,F). It is possible that our compounds triggered a compensatory mechanism at higher concentrations of analogues, or it could be due to compound solubility. Therefore, we tested the solubility of compounds using a dynamic light scattering (DLS) experiment. All of our functional assays are done at ≤10 μM concentration of compounds. Consequently, we tested the solubility of compounds at 10 and 200 μM. We did not see any aggregation of compounds at 10 μM concentration in phosphate buffer saline at physiological pH 7.4. However, aggregation of cpCF3 and zafirlukast was observed at 200 μM (Supplementary Figure 1). Moreover, acyl sulfonamide is a key functional group in zafirlukast and all of the analogues and is regarded as an isostere or surrogate of carboxylic acids as they have comparable pKa’s. The acidic NH of acyl sulfonamide remains deprotonated under pH 7.4 and the ionic nature of the molecule makes it well soluble up to 100 μM concentration. Other functional groups such as carbamate also add polarity and H-bonding sites to the molecule and could contribute to its aqueous solubility as well.30 These results suggest that increased NF-κB activity at 5 μM treatment is not due to insolubility of compounds.
Figure 1.
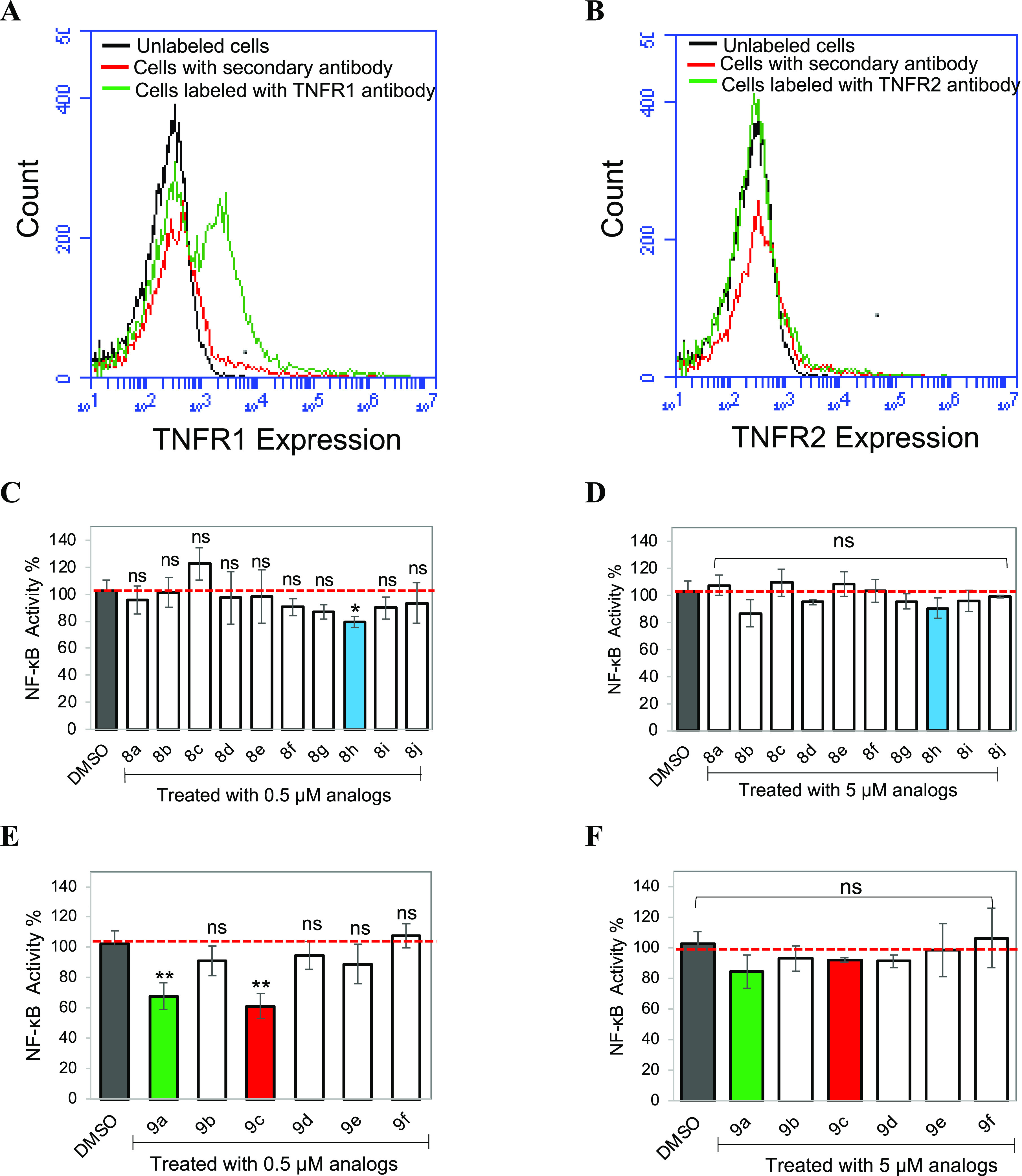
Effect of zafirlukast analogues on TNF-induced NF-κB activation. FACS data demonstrate surface expression of TNFR1 (A) and TNFR2 (B) on HEK293 cells. Cells were incubated with the anti-TNFR1 or TNFR2 antibody and followed by the AlexaFluor647-conjugated secondary antibody. Luciferase assay of NF-κB activation in HEK293 cells transfected with luciferase reporter plasmids and treated with 0.5 μM (C, E) and 5 μM (D, F) zafirlukast analogues for 2 h, followed by the addition of TNF (10 ng/mL) for 24 h. Data are means ± SD from three experiments.
Next, the zafirlukast analogues with the most potent NF-κB inhibition (8h cpCF3, 9a MeCF3, 9c MeOEtCF3) were selected and tested for their dose–response effect on TNF-induced NF-κB activity. While 9a MeCF3 inhibited about 63% of TNF-induced NF-κB activity with a relative IC50 of 0.38 ± 0.17 nM (Figure 2A), 9c MeOEtCF3 and 8h cpCF3 inhibited only about 35–42% of TNF-induced NF-κB activity (Figure 2B,C). MeCF3 showed better potency and efficacy as compared to zafirlukast (Figure 2D). Although 9a MeCF3, 9c MeOEtCF3, and 8h cpCF3 inhibited NF-κB activation in a dose–response fashion from 0.01 nM up to 250 nM and plateaued at around 250 nM, efficacies plummeted at 500 nM of antagonist. Whereas zafirlukast inhibited NF-κB activation to a comparable extent, the curve neither plateaued nor plummeted at the highest concentration tested (Figure 2D).
Figure 2.

Effect of zafirlukast analogues on TNF-induced NF-κB activation. Luciferase assay of NF-κB activation in HEK293 cells transfected with luciferase reporter plasmids and treated with zafirlukast (0.001 nM to 10 μM) or its analogues (0.001 nM to 0.5 μM) in a dose-dependent manner for 2 h, followed by the addition of TNF (10 ng/mL) for 24 h. NF-κB inhibition in HEK293 cells treated with MeCF3 (A), MOEtCF3 (B), cpCF3 (C), and zafirlukast (D). Data are means ± SD from three experiments.
Lead Compounds Inhibited TNFR1-Induced IκBα Degradation More Efficiently than Zafirlukast
Next, the effect of zafirlukast analogues on TNF-induced IκBα degradation was determined by immunoblotting (Figure 3). After TNF treatment, IκBα was rapidly degraded to 10–20% of the basal levels in HEK 293 cells (Figure 3A). IκBα degradation was inhibited in a dose-dependent manner in cells treated with zafirlukast and its analogues (Figure 3A).
Figure 3.
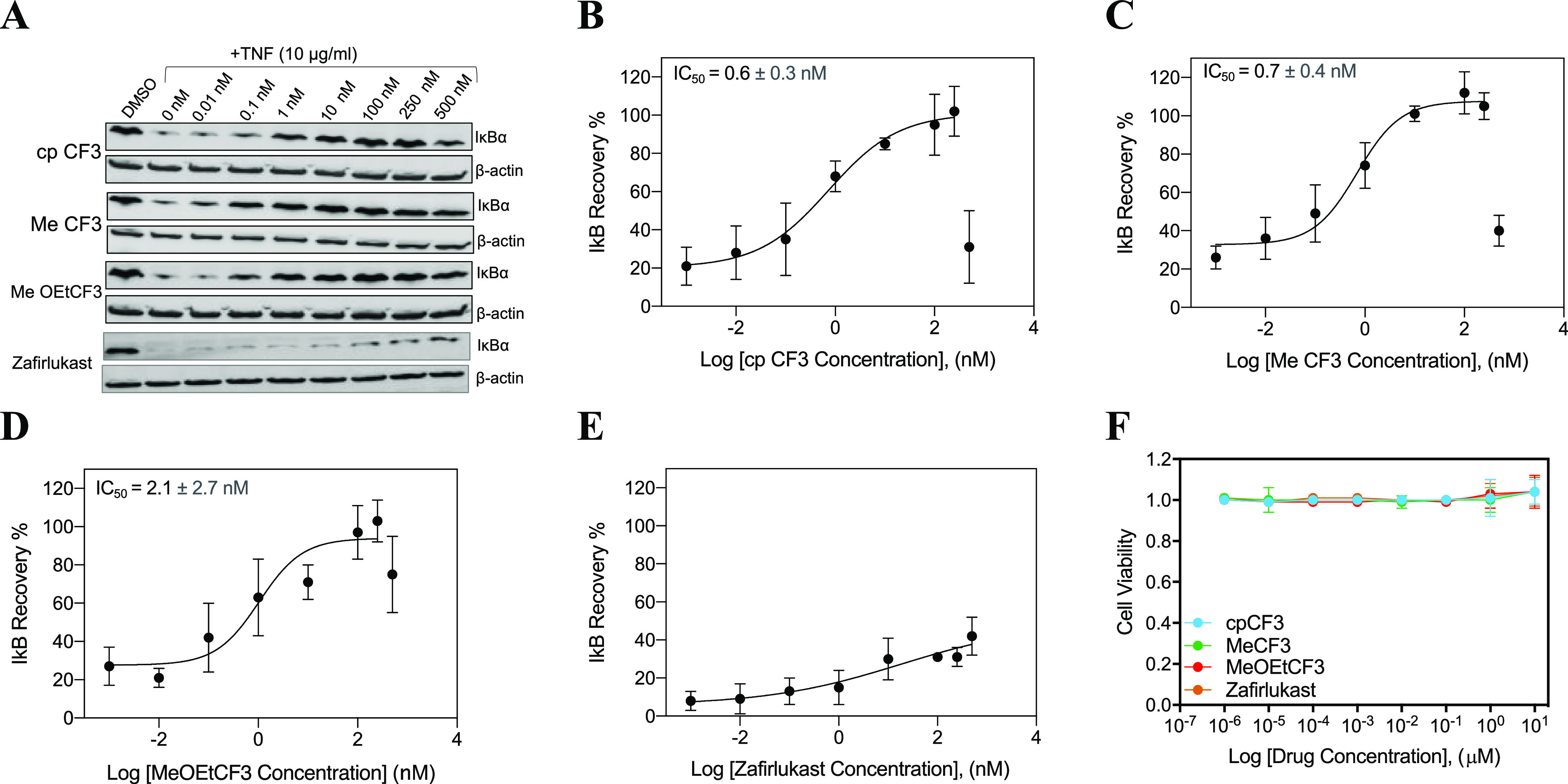
Effect of zafirlukast analogues on TNF-induced IκBα degradation. (A) HEK293 cells with endogenous TNFR1 receptors were treated with DMSO and cpCF3, MeCF3 and MeOEtCF3 in a dose-dependent manner (0.01 to 500 nM) for 2 h, followed by the addition of TNF (10 ng/mL) for 30 min and then analyzed with Western blot. Qualitative dose-dependent inhibition of IκBα degradation was observed from the increase in the intensity of the IκBα protein bands. Quantification of IκBα protein bands in cells treated with cpCF3 (B), MeCF3 (C), MeOEtCF3 (D), and zafirlukast (E) using ImageJ (n = 3). (F) Cytotoxicity of analogues. HEK293 cells were treated with increasing concentrations of zafirlukast or its analogues (0.0001 nM–10 μM). Cytotoxicity of analogues was measured using MTT assay. Data are presented as mean ± standard deviation (n = 3).
While zafirlukast partially inhibited TNF-induced IκB degradation (Figure 3E), 8h cpCF3 (Figure 3B), 9a MeCF3 (Figure 3C), and 9c MeOEtCF3 (Figure 3D) completely inhibited IκB activity with IC50s of 0.6 ± 0.3, 0.71 ± 0.35, and 2.1 ± 2.6 nM, respectively. Taken together, these results confirm that three zafirlukast analogues enhanced potency and efficacy of TNFR1 inhibition. Next, we investigate the specificity and mode of action of these lead analogues.
Next, to rule out the possibility that the inhibitory effect of zafirlukast analogues on TNF-induced biological responses was not caused by their cytotoxic effect, we investigated the cytotoxicity of analogues in HEK293 cells using the MTT assay. No appreciable cytotoxicity is observed, which suggests that the inhibitory effect of zafirlukast and its analogues is not due to cell death (Figure 3F).
Specificity of Zafirlukast Analogues
We aimed to determine whether the functional effects of the analogues were specifically due to the mitigation of TNFR1 mediated NF-κB activation rather than through the inhibition of other proteins in alternative signaling pathways. The specificity of lead compounds was determined in three ways: (1) the NF-κB activation induced by TNFR1-associated death domain (TRADD) overexpression, (2) IL1α-induced NF-κB activation, and (3) TNFR2 function.
Effect of Zafirlukast Analogues on NF-κB Activation Induced by TRADD Overexpression
TRADD is the essential adaptor protein recruited to TNFR1 upon TNFα binding, and TRADD overexpression is independent of TNF-induced NF-kB activation.31 Therefore, testing the effect of the analogues on NF-κB activation induced by TRADD overexpression,31 without TNF, represents the important control experiment for specificity. As shown in Figure 4A, overexpression of TRADD significantly increased NF-κB activation in HEK293 cells. Zafirlukast and its three lead analogues had little effect on TRADD-induced NF-κB activation, even at concentrations well above the IC50s (Figure 4B). These results indicated that the reduced NF-κB activation was not resulted from direct inhibition of TRADD and ruled it out as a possible target of our compounds and zafirlukast.
Figure 4.

Effect of zafirlukast analogues on TRADD-induced NF-κB activation. HEK293 cells transfected with luciferase reporter plasmids and TRADD plasmids and treated with increasing concentrations of zafirlukast analogues (0.01 to 500 nM). (A) NF-κB activity in TRADD expressing cells and (B) TRADD expressing cells treated with analogues. Data are means ± SD of three experiments.
Effect of Zafirlukast Analogues on IL-1α Induced NF-κB Activation
In addition to TNF, IL1α is another cytokine that promotes the activation of NF-κB upon binding to its receptor IL1R.32,33 Therefore, we tested the effect of analogues on IL-1α induced NF-κB activation in HEK293 cells. As shown in Figure 5A, IL1α treatment significantly increased NF-κB activation in HEK293 cells, and zafirlukast and its analogues failed to mitigate the IL-1α-induced NF-κB activation (Figure 5B). This experiment further confirmed that these antagonists act independent of the IL1 pathway.
Figure 5.

Effect of zafirlukast analogues on IL-1α-induced NF-κB activation. NF-κB activation in HEK293 cells transfected with luciferase reporter plasmids and treated with IL1-α (10 ng/mL) only (A), and zafirlukast analogues (0.01 to 500 nM) and IL1-α for 24 h (B). Data are means ± SD from three experiments.
Effect of Zafirlukast on TNFR2-Induced Activation of the Noncanonical Pathway
It has been shown previously that the noncanonical NF-κB pathway selectively responds to a subset of TNFR superfamily members.34−36 The noncanonical pathway depends on ligand-induced processing of NF-κB2 precursor protein, p100, as opposed to the degradation of IκBα in the canonical NF-κB pathway.34−36 This p100 protein mediates activation of the p52/RelB complex.34−36 Recently, it has been reported that the membrane TNF induces p100 processing via TNFR2.37 Therefore, we sought to study the effect of the analogues on membrane TNF-induced activation of RelB in HUVEC cells using immunoblotting (Figure 6A). Densitometry analysis of protein bands showed zafirlukast and its analogues have a minimal effect on membrane TNF-TNFR2-induced activation of RelB (Figure 6B).
Figure 6.

Specificity of zafirlukast analogues. (A) Effect of zafirlukast analogues on TNF-induced RelB activation (TNFR2 noncanonical pathway). HUVEC cells transfected with membrane TNF for 6 h and then treated with zafirlukast analogues (250 nM), and then analyzed with Western blot. (B) Quantification of RelB protein bands by densitometry using ImageJ.
Effect of Zafirlukast Analogues on TNF-TNFR1 Interactions
It has been shown previously that blocking TNF binding to TNFR1 inhibits NF-κB activation.38,39 We therefore investigated if the anti-inflammatory function of zafirlukast analogues resulted from blocking TNF binding. We tested the effect of analogues on TNF–TNFR1 interactions using a pulldown assay (Figure 7). For this assay, we have used recombinant FLAG-tagged TNF and anti-FLAG beads. It has been shown that in the native state, recombinant TNF exists mainly as a trimer with a threefold symmetry, resulting in three identical receptor binding sites.40−42 Trimeric TNF binds to three TNFR1 and forms a basic unit of signaling. First, we examined whether TNFR1 in HEK293 lysate nonspecifically binds to anti-FLAG beads alone. As shown in Figure 7A, the TNFR1 band did not appear when beads were incubated with just HEK293 lysate, which indicates that TNFR1 did not bind to anti-FLAG beads. Densitometry analysis of protein bands showed no significant difference in the binding of TNF to TNFR1 in the presence and absence of analogues (Figure 7B,C). To further support this observation, we tested the effect of zafirlukast analogues on ligand binding in TNFR1ΔCD-GFP-expressing HEK293 cells using flow cytometry. No significant difference was observed in the binding of TNF to TNFR1ΔCD-GFP in the presence and absence of analogues (Figure 7D). These results further confirm that the analogues do not block TNF binding.
Figure 7.
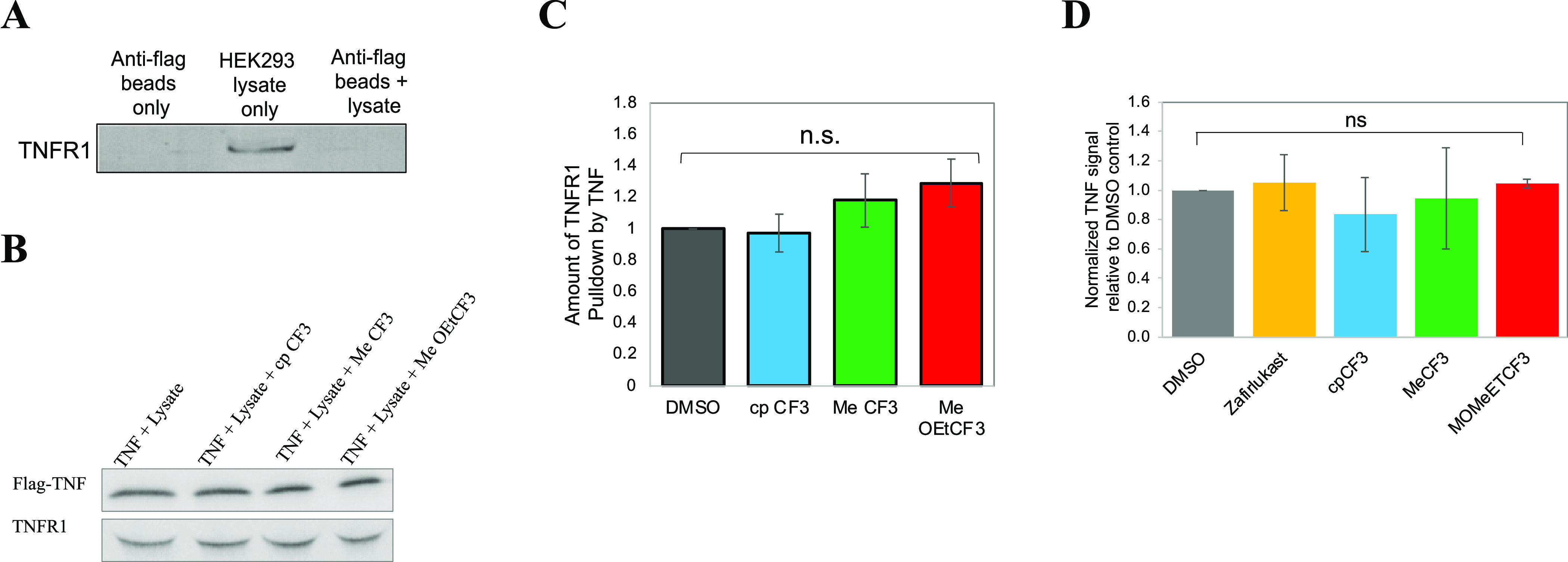
Effects of analogues on TNF-TNFR1 interactions. (A) Anti-FLAG beads were treated separately with PBS and HEK293 lysate and incubated at 4 °C for 2 h. Next, beads were washed thrice and resolved by SDS–PAGE and immunoblotted with the anti-TNFR1 antibody. A total of 100 μg of lysate (total input) was loaded. (B) TNFR1–TNF binding was assessed by a pulldown assay with anti-FLAG magnetic beads. Flag-TNF was mixed with anti-FLAG beads and incubated at 4 °C for 2 h. Next, beads were washed thrice to remove the unbound TNF. HEK293 lysates with and without zafirlukast analogues (250 nM) were added to TNF-coated magnetic beads and rotated at 4 °C for 2 h. Beads were washed three times and pulled-down proteins were resolved by SDS–PAGE and immunoblotted with anti-Flag and TNFR1 antibodies. (C) TNFR1 and TNF bands were quantified using ImageJ. The TNFR1 recovery for each condition was normalized to the TNF content (n = 3). TNFR1 band intensities in presence of analogues were compared to TNF + DMSO control by one-way ANOVA with Dunnett’s multiple comparison test using Graph Pad Prism. (D) HEK293 cells with stable expression of TNFR1ΔCD-GFP were incubated with Flag-TNF (20 μg/mL) and Flag-TNF with analogues (250 nM). Flag-TNF binding was detected with the AF647-conjugated anti-Flag antibody, as measured by flow cytometry (n = 3).
Effect of Analogues on Ligand-Independent Interactions of TNFR1
Next, we evaluated the effect of zafirlukast analogues on ligand-independent TNFR1-TNFR1 interactions. Others and our group have previously reported that several members of the TNF receptor superfamily, including TNFR1, TNFR2, DR5, and FAS, exist as ligand-independent, homophilic, and heterophilic oligomers, which are important for the function of TNF receptors.21,43−47 In our previous work, we established that it is possible to inhibit TNF-induced TNFR1 signaling by disrupting the TNFR1–TNFR1 interaction.21 Thus, we hypothesized that the anti-TNFR1 effect of analogues might be due to disruption of ligand-independent interactions of TNFR1 receptors. We therefore investigated the effect of zafirlukast analogues on ligand-independent TNFR1–TNFR1 interactions using live-cell time-resolved Förster resonance energy transfer (TR-FRET). We previously showed that TR-FRET could directly report on TNFR1-TNFR1 interactions (basal FRET), increase in TNFR1 oligomerization (increase in basal FRET), disruption of TNFR1–TNFR1 interactions (decrease in basal FRET), and conformational changes (decrease or increase in basal FRET).21,48−50 TR-FRET experiments were carried out in HEK293 cells transiently expressing the TNFR1 or TNFR2 without a cytoplasmic domain (TNFRΔCD) fused to GFP and co-expressing TNFRΔCD fused to GFP and RFP (TNFR biosensor) just downstream of the transmembrane domain of the receptors. The TNFRΔCD-GFP (donor) lifetime in the presence and absence of the acceptor (TNFRΔCD-RFP) was measured and then used to calculate the FRET efficiency using eq 1.
For lifetime measurements, we first checked the expression levels of the TNFR1ΔCD-GFP and TNFR2ΔCD-GFP using fluorescence microscopy. Cells transfected with TNFR1ΔCD-GFP (Supplementary Figure 2A) or TNFR2ΔCD-GFP (Supplementary Figure 2B) showed similar transfection efficiencies. Lifetime measurements showed a significant decrease in the fluorescence lifetime of the donor (2.186 ± 0.04 ns) in the presence of the acceptor compared with the donor only (2.598 ± 0.04 ns), which confirms efficient energy transfer between the FRET pairs (Figure 8A). These results confirm that TNFR1 exists as ligand-independent oligomers. We then evaluated the effect of analogues on ligand-independent interactions of TNFR1 or TNFR2. Interestingly, TNFR1 biosensor expressing cells that are treated with analogues showed lower FRET compared with DMSO-treated cells (Figure 8A). These results suggest that analogues are breaking TNFR1-TNFR1 interactions or causing conformational changes. However, analogues have no significant effect on FRET efficiency of the TNFR2 biosensor when compared to DMSO control (Figure 8B). No change in FRET indicates that analogues are not breaking ligand-independent TNFR2-TNFR2 interactions or causing conformational changes or conformational rearrangements are too small to change the distance between the cytoplasmic ends of preassembled TNFR2 receptors.
Figure 8.

Effect of analogues on ligand-independent interactions of TNF receptors determined using live-cell TR-FRET measurements. For lifetime measurements, transfected cells were detached with trypsin, washed thrice with PBS, treated with analogues (10 μM) or DMSO alone, and incubated for 1–2 h. After being incubated, the donor lifetime was measured using a fluorescence lifetime plate reader. (A) Effect of analogues on TNFR1 ligand-independent interaction. (B) Effect of analogues on TNFR2 ligand-independent interactions. Data are means ± SD (n = 3).
Direct Binding of Analogues
19F ligand-observed NMR was utilized to demonstrate that the zafirlukast analogues directly bind to the soluble extracellular domain (ECD) of TNFR1 as all three lead zafirlukast analogues contain trifluoromethyl groups (−CF3). Upon titration of increasing amounts of TNFR1 into solutions of each analogue, the −CF3 peak undergoes line broadening and a downfield shift, indicating that the compounds are binding to TNFR1 (Figure 9A–C). The change in chemical shift was plotted against protein concentration to obtain KD values.
Figure 9.
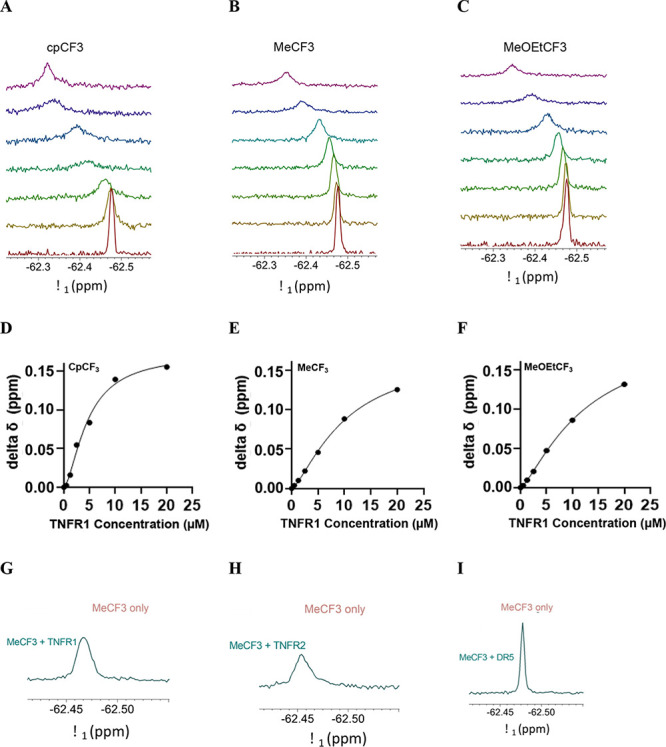
19F-NMR ligand-observed NMR experiments for 25 μM zafirlukast analogues in PBS with TNFR1. Soluble ECD of TNFR1 was titrated into solutions of 25 μM (A) cpCF3, (B) MeCF3, and (C) MeOEtCF3. Titrations were carried out with 0, 0.5, 1.25, 2.5, 5, 10, and 20 μM TNFR1-ECD (increasing in concentration from the bottom to top). Resonances were referenced to an internal TFA control. The change in chemical shift was plotted versus compound concentration to determine KD values for (D) cpCF3, (E) MeCF3, and (F) MeOEtCF3. To investigate whether there is selectivity between TNFR1 and TNFR2, MeCF3 tested with 1.25 μM TNFR1 (G), 1.25 μM TNFR2 (H), and 1.25 μM DR5 (I). Compound only is shown in brown while compound with 1.25 μM protein is shown in teal for all plots. MeCF3 exhibits line broadening and a downfield chemical shift in the presence of TNFR1 and TNFR2 while little to no change is observed for the chemical shift of the compounds in the presence of DR5 at the same concentration.
Of the three analogues, cpCF3 appears to have the highest affinity (KD = 4.6 μM) for TNFR1 (Figure 9D) when compared to MeCF3 (KD = 10 μM) (Figure 9E) and MeOEtCF3 (KD = 12 μM) (Figure 9F). All three analogues showed positive cooperativity with a Hill coefficient greater than 1 (nH = 1.593 for cpCF3, 1.389 for MeCF3, and 1.325 for MeOEtCF3).
To evaluate the specificity of the most efficient analogue MeCF3, we examined its binding to extracellular domains of TNFR2 and death receptor 5 (DR5), homologous members of the TNFR superfamily. There was little to no change in the chemical shifts of MeCF3 in the presence of 1.25 μM DR5 (Figure 9I). Conversely, in the presence of soluble TNFR2-ECD (Figure 9H), the peaks undergo appreciable line broadening and downfield shifts similar to the effect seen in the presence of TNFR1-ECD (Figure 9G). It is interesting to note that MeCF3 binds to TNFR2, but it does not inhibit TNFR2 function or cause any conformational changes in the FRET studies. It is also important to note that our original screening platform did not discriminate TNFR1 or TNFR2 binding, and given the sequence and structure similarity (52% similar and 30% identical to TNFR1, determined using the European Bioinformatics Institute’s LALIGN software), it is not surprising to find binding. Taken together, these results indicate that the zafirlukast analogues appear to have selectivity for the TNF receptors. We note the variation in functional IC50s and KDs, which may be due to differences in the experimental conditions between these two assays: analogue IC50 are derived for cell-based studies where receptors are expressed on plasma membranes and concentration of endogenous proteins may differ from those of NMR studies. The NMR studies were done with soluble TNFR1 in PBS.
Next, we investigated whether zafirlukast analogues also bind at the PLAD interface of TNFR1 like zafirlukast. Using 19F ligand-observed NMR, we investigated the direct binding of cpCF3 (the compound with the highest affinity among all three analogues) to soluble PLAD. When increasing amounts of PLAD are titrated into cpCF3 solution, the −CF3 peak broadens and shifts downfield, indicating that the compound is binding to PLAD (Supplementary Figure 3A). However, cpCF3 does not have the same affinity for PLAD as TNFR1-ECD because we could not obtain a full binding curve even at 20 μM. Since we do not have a saturation point, we did not fit the data to obtain a KD value (Supplementary Figure 3B).
Discussion
Selective inhibition of TNFR1 has the potential to prevent the detrimental effects associated with total inhibition of TNF. Most of the known TNFR1 receptor-specific inhibitors are small molecules51,52 or antibodies53−56 that competitively block receptor-ligand interactions. Due to the avidity effect and very high affinity for ligand binding to TNFR1 (KD = 0.4 nM),57 small molecules that work by competitively eliminating ligand binding may not be effective.58 In addition, soluble forms of TNFR1 function as native decoys of membrane-bound counterparts to neutralize TNF in the circulation, and this beneficial effect may be reduced by such TNFR1 antagonists.59−61 Consequently, approaches that do not involve eliminating ligand binding are highly pursued.
In this study, we identified three analogues that were two orders of magnitude more potent than zafirlukast as an inhibitor of TNFR1. We do note that this first limited set of analogues did not achieve the goal of completely knocking out activity, although MeCF3 did somewhat reduce the observed maximal effect. Whether additional medicinal chemistry could be used to further augment this enhanced efficacy is the focus of ongoing efforts. Furthermore, while we have shown that all three lead analogues inhibit TNFR1 activation without interfering with TNF binding, the exact mode of action of these analogues remains unknown. Importantly, the FRET data confirm that analogues perturb TNFR1 but not TNFR2 (Figure 8). The decrease in TNFR1 FRET in the presence of the analogues could be due to disruption of receptor-receptor interactions or structural rearrangements in the TNFR1 backbone, which favors the nonfunctional conformational states of TNFR1.62 Nonetheless, further studies are required to identify modes of action of these functional analogues. Regarding the binding to TNFR2, we note that our original FRET-based screen for small molecules that induced conformational changes in TNFR121 and that identified zafirlukast did not counter-screen against TNFR2 binding. Given the high sequence and structural homology, we are not surprised to find binding to TNFR2 nor that we do not see binding to the DR5 control. However, we are pleased to report that the analogues do not inhibit TNFR2 activity in cells (Figure 6). While this is a more complex result than if the binding mode was selective, the fact that the analogues bind but do not alter activity of TNFR2 confirms that there are critical differences in the structure-functional relationship of the two receptors, even if there are similar small-molecule binding sites. This is a novel and important finding which has not been previously reported in the TNF receptor literature. We have focused on structural dynamics of TNF receptors over the past several years,50,62 and these findings further support our novel discovery that receptor conformational states, which can be selectively modified by small molecules, dictate TNF receptor activity.
We note that the relative IC50 for the zafirlukast analogues is lower than their KD. It remains uncertain whether this difference reflects a secondary mode of action or whether it can be consistent with specific targeting of TNFR1. Generally, IC50 and KD values can be similar when the response is directly proportional to the concentration of the drug. However, in more complex systems, there can be amplification between receptor occupancy and effect, in which case, the IC50 can be lower than the KD.63−65 Moreover, the IC50 can be influenced by factors such as receptor expression levels and cell densities.66,67 Therefore, while IC50 and KD can provide complementary information about drug-receptor interactions, they may not be necessarily comparable or interchangeable.
Zafirlukast was originally designed as an antagonist for leukotriene receptors but stood out in our previous screening against TNFR1. In the crystal structure of zafirlukast in leukotriene receptor CysLT1R,68 the methylene bridge between the indole core and the adjacent benzene ring gave two sides of the molecule certain extent of flexibility to each make contacts with different parts of the pocket. Both the acyl sulfonamide and the carbamate moieties have strong hydrogen bonding interactions with the surrounding polar residues via the carbonyl oxygens, carbamate NH, and sulfone oxygens. The toluene group appending the sulfone is projected into a hydrophobic pocket. Without a previously known or well-defined binding pocket in the preligand binding assembly domain of TNFR1, we suspect that these features may still be essential to the binding conformation and account for superior antagonistic activity of zafirlukast. Therefore, we decided to retain these key functional groups in our analogue design and only vary the sulfonamide and carbamate moieties. In our SAR campaign, compound 8h, with aryl CF3 substituent para to the sulfonamide, appeared as the only hit at the concentrations tested in the cell-based reporter assay. Its CH3 counterpart 8b did not produce such activity. The CF3 group is roughly the same in size as CH3 but offers improved lipophilicity and electron-withdrawing effect.69 These factors combined likely contributed to its potent NF-κB inhibition. Bulkier electron-withdrawing group SF5 with greater lipophilicity70,71 or other electron-withdrawing groups that are less lipophilic like Cl or CN did not achieve such an effect. Analogues with electron-donating alkyl substituents did not produce such activity nor did the ones with N-heterocycles. As we fixed the CF3 benzenesulfonamide moiety and moved on to optimize the carbamate group, methyl and methoxy ethyl carbamate downregulated the NF-κB activity further to 60%. While all six carbamate derivatives we explored in addition to 8h showed reduced NF-κB activation, the smaller or linear alkyl groups seemed to be tolerated better among other bulkier groups. It is not clear why compound 9b with an ethyl group did not follow this trend. Such results warrant more extensive SAR campaigns in future studies.
In summary, we identified three zafirlukast analogues that can be employed to develop therapeutics for RA as well as novel candidates to learn more about how these receptors’ activation and inhibition differ from one another. Further efforts will be required to improve the efficacy of these three lead compounds and investigate their pharmacological properties and effects of the compounds in animal models of inflammation prior to preclinical assessment of this class of compounds.
Statistical Analysis
Acknowledgments
We thank Professor Courtney Aldrich for his guidance in the analogue synthesis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.2c00048.
Synthetic procedures for zafirlukast analogues; solubility of zafirlukast and its analogues; expression of TNFR1ΔCD-GFP or TNFR2ΔCD-GFP on transfected HEK293T cells; and binding cpCF3 to TNFR1-PLAD (PDF)
Author Contributions
# N.V., M.Y., and C.H.L. contributed equally to this work. CRediT: William D Fiers methodology (supporting); .
Author Contributions
CRediT: Nagamani Vunnam investigation (lead), methodology (lead), resources (lead), writing-original draft (lead), writing-review & editing (lead); Mu Yang investigation (lead), methodology (lead), writing-original draft (lead); Chih Hung Lo conceptualization (lead), investigation (supporting), methodology (supporting), writing-original draft (supporting), writing-review & editing (supporting); Carolyn Paulson data curation (supporting); William D Fiers methodology (supporting); Evan Huber methodology (supporting); MaryJane Olivia Been methodology (supporting); David M. Ferguson conceptualization (lead), supervision (supporting), writing-review & editing (lead); Jonathan N. Sachs conceptualization (lead), funding acquisition (lead), project administration (lead), resources (lead), supervision (lead), validation (lead), writing-review & editing (lead).
This work was supported by the U.S. National Institutes of Health (NIH) grants to J.N.S. (R35GM131814).
The authors declare no competing financial interest.
Notes
Experimental data in this study were analyzed using Graph Pad Prism 8.0 software and P-values were calculated using a two-tailed unpaired t-test or one-way ANOVA. *p-value <0.05; **p-value <0.01; ***p-value <0.001; and ****p-value <0.0001. All quantitative variables are shown as means ± SD from three experiments.
Supplementary Material
References
- Ashkenazi A.; Dixit V. M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Wajant H.; Scheurich P. TNFR1-induced activation of the classical NF-kappaB pathway. FEBS J. 2011, 278, 862–876. 10.1111/j.1742-4658.2011.08015.x. [DOI] [PubMed] [Google Scholar]
- Brenner D.; Blaser H.; Mak T. W. Regulation of tumour necrosis factor signalling: live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- NIH , Progress in Autoimmune Diseases Research, Report to Congress; National Institutes of Health, The Autoimmune Diseases Coordinating Committee, March 2005, forward and pages i, 1, 2, 16, 17, 28, 29, 30, 32, 52. 2005. [Google Scholar]
- AARDA Autoimmune Disease Statistics, September 30, 2019. https://www.aarda.org/news-information/statistics/.
- Li P.; Zheng Y.; Chen X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. 10.3389/fphar.2017.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedger L. M.; McDermott M. F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants – past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. 10.1016/j.cytogfr.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Lis K.; Kuzawińska O.; Bałkowiec-Iskra E. Tumor necrosis factor inhibitors – state of knowledge. Arch. Med. Sci. 2014, 6, 1175–1185. 10.5114/aoms.2014.47827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliolias G. D.; Ivashkiv L. B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schabert V. F.; Watson C.; Joseph G. J.; Iversen P.; Burudpakdee C.; Harrison D. J. Costs of Tumor Necrosis Factor Blockers Per Treated Patient Using Real-World Drug Data in a Managed Care Population. J. Manag. Care Pharm. 2013, 19, 621–630. 10.18553/jmcp.2013.19.8.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltsonoudis E.; Voulgari P. V.; Konitsiotis S.; Drosos A. A. Demyelination and other neurological adverse events after anti-TNF therapy. Autoimmun. Rev. 2014, 13, 54–58. 10.1016/j.autrev.2013.09.002. [DOI] [PubMed] [Google Scholar]
- O’Connell J.; Porter J.; Kroeplien B.; Norman T.; Rapecki S.; Davis R.; McMillan D.; Arakaki T.; Burgin A.; Fox D. III; Ceska T.; Lecomte F.; Maloney A.; Vugler A.; Carrington B.; Cossins B. P.; Bourne T.; Lawson A. Small molecules that inhibit TNF signalling by stabilising an asymmetric form of the trimer. Nat. Commun. 2019, 10, 5795. 10.1038/s41467-019-13616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H. Y.; Li N.; Duan J. J.; Jiang B.; Lu Z.; Ngu K.; Tino J.; Kopcho L. M.; Lu H.; Chen J.; Tebben A. J.; Sheriff S.; Chang C. Y.; Yanchunas J. Jr.; Calambur D.; Gao M.; Shuster D. J.; Susulic V.; Xie J. H.; Guarino V. R.; Wu D. R.; Gregor K. R.; Goldstine C. B.; Hynes J. Jr.; Macor J. E.; Salter-Cid L.; Burke J. R.; Shaw P. J.; Dhar T. G. M. Biologic-like In Vivo Efficacy with Small Molecule Inhibitors of TNFalpha Identified Using Scaffold Hopping and Structure-Based Drug Design Approaches. J. Med. Chem. 2020, 63, 15050–15071. 10.1021/acs.jmedchem.0c01732. [DOI] [PubMed] [Google Scholar]
- Sun W.; Wu Y.; Zheng M.; Yang Y.; Liu Y.; Wu C.; Zhou Y.; Zhang Y.; Chen L.; Li H. Discovery of an Orally Active Small-Molecule Tumor Necrosis Factor-alpha Inhibitor. J. Med. Chem. 2020, 63, 8146–8156. 10.1021/acs.jmedchem.0c00377. [DOI] [PubMed] [Google Scholar]
- van Westen G. J. P.; Gaulton A.; Overington J. P. Chemical, target, and bioactive properties of allosteric modulation. PLoS Comput. Biol. 2014, 10, e1003559 10.1371/journal.pcbi.1003559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthur C. J.; Gentry P. R.; Mathews T. P.; Lindsley C. W. Drugs for allosteric sites on receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 165–184. 10.1146/annurev-pharmtox-010611-134525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.; Ji M.; Ni D.; Zhang J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov. Today 2018, 23, 359–365. 10.1016/j.drudis.2017.10.001. [DOI] [PubMed] [Google Scholar]
- Urwyler S. Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacol. Rev. 2011, 63, 59–126. 10.1124/pr.109.002501. [DOI] [PubMed] [Google Scholar]
- Wagner J. R.; Lee C. T.; Durrant J. D.; Malmstrom R. D.; Feher V. A.; Amaro R. E. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev. 2016, 116, 6370–6390. 10.1021/acs.chemrev.5b00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M.; Maini R. N. Perspectives From Masters in Rheumatology and Autoimmunity: Can We Get Closer to a Cure for Rheumatoid Arthritis?. Arthritis Rheumatol. 2015, 67, 2283–2291. 10.1002/art.39269. [DOI] [PubMed] [Google Scholar]
- Lo C. H.; Vunnam N.; Lewis A. K.; Chiu T. L.; Brummel B. E.; Schaaf T. M.; Grant B. D.; Bawaskar P.; Thomas D. D.; Sachs J. N. An Innovative High-Throughput Screening Approach for Discovery of Small Molecules That Inhibit TNF Receptors. SLAS Discov. 2017, 22, 950–961. 10.1177/2472555217706478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaagstra M. E.; Schoenmakers S. H. H. F.; Nederkoorn P. H. J.; Gelens E.; Timmerman H.; Zhang M.-Q. Development of a Three-Dimensional CysLT1 (LTD4) Antagonist Model with an Incorporated Amino Acid Residue from the Receptor. J. Med. Chem. 1998, 41, 1439–1445. 10.1021/jm970180w. [DOI] [PubMed] [Google Scholar]
- Bernstein P. R. Chemistry and structure--activity relationships of leukotriene receptor antagonists. Am. J. Respir. Crit. Care Med. 1998, 157, S220–S226. 10.1164/ajrccm.157.6.mar-3. [DOI] [PubMed] [Google Scholar]; discussion S225–6, S247–8
- Brown M. F.; Marfat A.; Antognoli G.; Chambers R. J.; Cheng J. B.; Damon D. B.; Liston T. E.; McGlynn M. A.; O’Sullivan S. P.; Owens B. S.; Pillar J. S.; Shirley J. T.; Watson J. W. N-carbamoyl analogs of zafirlukast: Potent receptor antagonists of leukotriene D4. Bioorg. Med. Chem. Lett. 1998, 8, 2451–2456. 10.1016/S0960-894X(98)00442-9. [DOI] [PubMed] [Google Scholar]
- Yang S.; Wang J.; Brand D. D.; Zheng S. G. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784. 10.3389/fimmu.2018.00784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustman D.; Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. 10.1038/nrd3030. [DOI] [PubMed] [Google Scholar]
- Dopp J. M.; Sarafian T. A.; Spinella F. M.; Kahn M. A.; Shau H.; de Vellis J. Expression of the p75 TNF receptor is linked to TNF-induced NFkappaB translocation and oxyradical neutralization in glial cells. Neurochem. Res. 2002, 27, 1535–1542. 10.1023/A:1021608724117. [DOI] [PubMed] [Google Scholar]
- Madsen P. M.; Motti D.; Karmally S.; Szymkowski D. E.; Lambertsen K. L.; Bethea J. R.; Brambilla R. Oligodendroglial TNFR2 Mediates Membrane TNF-Dependent Repair in Experimental Autoimmune Encephalomyelitis by Promoting Oligodendrocyte Differentiation and Remyelination. J. Neurosci. 2016, 36, 5128–5143. 10.1523/JNEUROSCI.0211-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matassa V. G.; Maduskuie T. P.; Shapiro H. S.; Hesp B.; Snyder D. W.; Aharony D.; Krell R. D.; Keith R. A. Evolution of a series of peptidoleukotriene antagonists: synthesis and structure/activity relationships of 1,3,5-substituted indoles and indazoles. J. Med. Chem. 1990, 33, 1781–1790. 10.1021/jm00168a037. [DOI] [PubMed] [Google Scholar]
- Ballatore C.; Huryn D. M.; Smith A. B. 3rd Carboxylic acid (bio)isosteres in drug design. ChemMedChem 2013, 8, 385–395. 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H.; Xiong J.; Goeddel D. V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Dinarello C. A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. 10.1111/imr.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik A.; Kanneganti T. D. Function and regulation of IL-1alpha in inflammatory diseases and cancer. Immunol. Rev. 2018, 281, 124–137. 10.1111/imr.12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. C. The noncanonical NF-kappaB pathway. Immunol. Rev. 2012, 246, 125–140. 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. 10.1038/nri.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauert H.; Wicovsky A.; Muller N.; Siegmund D.; Spindler V.; Waschke J.; Kneitz C.; Wajant H. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 2010, 285, 7394–7404. 10.1074/jbc.M109.037341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey D.; Klareskog L.; Sasso E. H.; Salfeld J. G.; Tak P. P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Pisetsky D. S. Tumor necrosis factor blockers in rheumatoid arthritis. N. Engl. J. Med. 2000, 342, 810–811. 10.1056/NEJM200003163421110. [DOI] [PubMed] [Google Scholar]
- McMillan D.; Martinez-Fleites C.; Porter J.; Fox D. 3rd; Davis R.; Mori P.; Ceska T.; Carrington B.; Lawson A.; Bourne T.; O’Connell J. Structural insights into the disruption of TNF-TNFR1 signalling by small molecules stabilising a distorted TNF. Nat. Commun. 2021, 12, 582. 10.1038/s41467-020-20828-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. A.; Baglioni C. The active form of tumor necrosis factor is a trimer. J. Biol. Chem. 1987, 262, 6951–6954. 10.1016/S0021-9258(18)48183-5. [DOI] [PubMed] [Google Scholar]
- Vanamee E. S.; Faustman D. L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 11, eaao4910 10.1126/scisignal.aao4910. [DOI] [PubMed] [Google Scholar]
- Chan F. K.-M.; Chun H. J.; Zheng L.; Siegel R. M.; Bui K. L.; Lenardo M. J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354. 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- Naismith J. H.; Devine T. Q.; Brandhuber B. J.; Sprang S. R. Crystallographic evidence for dimerization of unliganded tumor necrosis factor receptor. J. Biol. Chem. 1995, 270, 13303–13307. 10.1074/jbc.270.22.13303. [DOI] [PubMed] [Google Scholar]
- Papoff G.; Hausler P.; Eramo A.; Pagano M. G.; Di Leve G.; Signore A.; Ruberti G. Identification and characterization of a ligand-independent oligomerization domain in the extracellular region of the CD95 death receptor. J. Biol. Chem. 1999, 274, 38241–38250. 10.1074/jbc.274.53.38241. [DOI] [PubMed] [Google Scholar]
- Lee H. W.; Lee S. H.; Lee H. W.; Ryu Y. W.; Kwon M. H.; Kim Y. S. Homomeric and heteromeric interactions of the extracellular domains of death receptors and death decoy receptors. Biochem. Biophys. Res. Commun. 2005, 330, 1205–1212. 10.1016/j.bbrc.2005.03.101. [DOI] [PubMed] [Google Scholar]
- Valley C. C.; Lewis A. K.; Mudaliar D. J.; Perlmutter J. D.; Braun A. R.; Karim C. B.; Thomas D. D.; Brody J. R.; Sachs J. N. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces death receptor 5 networks that are highly organized. J. Biol. Chem. 2012, 287, 21265–21278. 10.1074/jbc.M111.306480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo C. H.; Schaaf T. M.; Thomas D. D.; Sachs J. N. Fluorescence-Based TNFR1 Biosensor for Monitoring Receptor Structural and Conformational Dynamics and Discovery of Small Molecule Modulators. Methods Mol. Biol. 2021, 2248, 121–137. 10.1007/978-1-0716-1130-2_9. [DOI] [PubMed] [Google Scholar]
- Vunnam N.; Campbell-Bezat C. K.; Lewis A. K.; Sachs J. N. Death Receptor 5 Activation Is Energetically Coupled to Opening of the Transmembrane Domain Dimer. Biophys. J. 2017, 113, 381–392. 10.1016/j.bpj.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo C. H.; Huber E. C.; Sachs J. N. Conformational states of TNFR1 as a molecular switch for receptor function. Protein Sci. 2020, 29, 1401–1415. 10.1002/pro.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter P. H.; Scherle P. A.; Muckelbauer J. K.; Voss M. E.; Liu R. Q.; Thompson L. A.; Tebben A. J.; Solomon K. A.; Lo Y. C.; Li Z.; Strzemienski P.; Yang G.; Falahatpisheh N.; Xu M.; Wu Z.; Farrow N. A.; Ramnarayan K.; Wang J.; Rideout D.; Yalamoori V.; Domaille P.; Underwood D. J.; Trzaskos J. M.; Friedman S. M.; Newton R. C.; Decicco C. P. Photochemically enhanced binding of small molecules to the tumor necrosis factor receptor-1 inhibits the binding of TNF-alpha. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 11879–11884. 10.1073/pnas.211178398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Feng Z.; Wang Y.; Ma S.; Hu Z.; Yang P.; Chai Y.; Xie X. Discovery of Novel Ligands for TNF-α and TNF Receptor-1 through Structure-Based Virtual Screening and Biological Assay. J. Chem. Inf. Model. 2017, 57, 1101–1111. 10.1021/acs.jcim.6b00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata H.; Yoshioka Y.; Ohkawa A.; Abe Y.; Nomura T.; Mukai Y.; Nakagawa S.; Taniai M.; Ohta T.; Mayumi T.; Kamada H.; Tsunoda S.-I.; Tsutsumi Y. The therapeutic effect of TNFR1-selective antagonistic mutant TNF-α in murine hepatitis models. Cytokine 2008, 44, 229–233. 10.1016/j.cyto.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Steeland S.; Puimège L.; Vandenbroucke R. E.; Van Hauwermeiren F.; Haustraete J.; Devoogdt N.; Hulpiau P.; Leroux-Roels G.; Laukens D.; Meuleman P.; De Vos M.; Libert C. Generation and Characterization of Small Single Domain Antibodies Inhibiting Human Tumor Necrosis Factor Receptor 1. J. Biol. Chem. 2015, 290, 4022–4037. 10.1074/jbc.M114.617787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontermann R. E.; Munkel S.; Neumeyer J.; Muller D.; Branschadel M.; Scheurich P.; Pfizenmaier K. A humanized tumor necrosis factor receptor 1 (TNFR1)-specific antagonistic antibody for selective inhibition of tumor necrosis factor (TNF) action. J. Immunother. 2008, 31, 225–234. 10.1097/CJI.0b013e31816a88f9. [DOI] [PubMed] [Google Scholar]
- Zettlitz K. A.; Lorenz V.; Landauer K.; Münkel S.; Herrmann A.; Scheurich P.; Pfizenmaier K.; Kontermann R. E. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. mAbs 2010, 2, 639–647. 10.4161/mabs.2.6.13583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali R.; Cheng X.; Berezov A.; Du X.; Schon A.; Freire E.; Xu X.; Chen Y. H.; Greene M. I. Disabling TNF receptor signaling by induced conformational perturbation of tryptophan-107. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 10970–10975. 10.1073/pnas.0504301102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon A.; Lam S. Y.; Freire E. Thermodynamics-based drug design: strategies for inhibiting protein-protein interactions. Future Med. Chem. 2011, 3, 1129–1137. 10.4155/fmc.11.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer R.; Kontermann R.; Maier O. Targeting sTNF/TNFR1 Signaling as a New Therapeutic Strategy. Antibodies 2015, 4, 48. 10.3390/antib4010048. [DOI] [Google Scholar]
- Gohda T.; Niewczas M. A.; Ficociello L. H.; Walker W. H.; Skupien J.; Rosetti F.; Cullere X.; Johnson A. C.; Crabtree G.; Smiles A. M.; Mayadas T. N.; Warram J. H.; Krolewski A. S. Circulating TNF Receptors 1 and 2 Predict Stage 3 CKD in Type 1 Diabetes. J. Am. Soc. Nephrol. 2012, 23, 516–524. 10.1681/ASN.2011060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M.; Loughran P. A.; Zhang L.; Scott M. J.; Billiar T. R. Shedding of the tumor necrosis factor (TNF) receptor from the surface of hepatocytes during sepsis limits inflammation through cGMP signaling. Sci. Signal. 2015, 8, ra11. 10.1126/scisignal.2005548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo C. H.; Schaaf T. M.; Grant B. D.; Lim C. K.; Bawaskar P.; Aldrich C. C.; Thomas D. D.; Sachs J. N. Noncompetitive inhibitors of TNFR1 probe conformational activation states. Sci. Signaling 2019, 12, eaav5637 10.1126/scisignal.aav5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbet J.; Huclier-Markai S. Equilibrium, affinity, dissociation constants, IC5O: Facts and fantasies. Pharm. Stat. 2019, 18, 513–525. 10.1002/pst.1943. [DOI] [PubMed] [Google Scholar]
- Buchwald P. A three-parameter two-state model of receptor function that incorporates affinity, efficacy, and signal amplification. Pharmacol. Res. Perspect. 2017, 5, e00311 10.1002/prp2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adham N.; Ellerbrock B.; Hartig P.; Weinshank R. L.; Branchek T. Receptor reserve masks partial agonist activity of drugs in a cloned rat 5-hydroxytryptamine1B receptor expression system. Mol. Pharmacol. 1993, 43, 427–433. [PubMed] [Google Scholar]
- Hu J.; Kapoor M.; Zhang W.; Hamilton S. R.; Coombes K. R. Analysis of dose-response effects on gene expression data with comparison of two microarray platforms. Bioinformatics 2005, 21, 3524–3529. 10.1093/bioinformatics/bti592. [DOI] [PubMed] [Google Scholar]
- He Y.; Zhu Q.; Chen M.; Huang Q.; Wang W.; Li Q.; Huang Y.; Di W. The changing 50% inhibitory concentration (IC50) of cisplatin: a pilot study on the artifacts of the MTT assay and the precise measurement of density-dependent chemoresistance in ovarian cancer. Oncotarget 2016, 7, 70803–70821. 10.18632/oncotarget.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luginina A.; Gusach A.; Marin E.; Mishin A.; Brouillette R.; Popov P.; Shiriaeva A.; Besserer-Offroy É.; Longpré J.-M.; Lyapina E.; Ishchenko A.; Patel N.; Polovinkin V.; Safronova N.; Bogorodskiy A.; Edelweiss E.; Hu H.; Weierstall U.; Liu W.; Batyuk A.; Gordeliy V.; Han Gye W.; Sarret P.; Katritch V.; Borshchevskiy V.; Cherezov V. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 2019, 5, eaax2518 10.1126/sciadv.aax2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T.; Nishioka G.; Koyama M.; Ando A.; Miki T.; Kumadaki I. The Steric Effect Of A Trifluoromethyl Group. Chem. Pharm. Bull. 1991, 39, 233–235. 10.1248/cpb.39.233. [DOI] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- True J. E.; Thomas T. D.; Winter R. W.; Gard G. L. Electronegativities from Core-Ionization Energies: Electronegativities of SF5 and CF3. Inorg. Chem. 2003, 42, 4437–4441. 10.1021/ic0343298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.