

Summary
By a survey of metagenome-wide association studies (MWAS), we found a robust depletion of Bacteroides cellulosilyticus, Faecalibacterium prausnitzii, and Roseburia intestinalis in individuals with atherosclerotic cardiovascular disease (ACVD). From an established collection of bacteria isolated from healthy Chinese individuals, we selected B. cellulosilyticus, R. intestinalis, and Faecalibacterium longum, a bacterium related to F. prausnitzii, and tested the effects of these bacteria in an Apoe/− atherosclerosis mouse model. We show that administration of these three bacterial species to Apoe−/− mice robustly improves cardiac function, reduces plasma lipid levels, and attenuates the formation of atherosclerotic plaques. Comprehensive analysis of gut microbiota, plasma metabolome, and liver transcriptome revealed that the beneficial effects are associated with a modulation of the gut microbiota linked to a 7α-dehydroxylation–lithocholic acid (LCA)–farnesoid X receptor (FXR) pathway. Our study provides insights into transcriptional and metabolic impact whereby specific bacteria may hold promises for prevention/treatment of ACVD.
Subject areas: Human metabolism, Microbiology, Bacteriology, Microbial metabolism
Graphical abstract
Highlights
-
•
Metagenome-wide association studies reveal depletion of three bacterial species
-
•
Gavage of Apoe−/− mice with functionally equivalent bacteria curbs atherosclerosis
-
•
Gavage increases level of bacterially derived LCA in circulation
-
•
Increased LCA, an FXR agonist, links to marked changes in the liver transcriptome
Human metabolism; Microbiology; Bacteriology; Microbial metabolism
Introduction
Atherosclerotic cardiovascular disease (ACVD) is a metabolic disease characterized by dyslipidemia with high morbidity and mortality worldwide.1,2 Increasing understanding of the gut microbiota and its effect on host metabolism has spurred a strong interest in using the information on gut bacteria for diagnostics purposes and using specific gut bacteria for therapeutic interventions in relation to ACVD.3 Fromentin et al. reported that intestinal bacteria play an important role in the regulation of lipid homeostasis and atherosclerosis.4,5,6,7,8 Noteworthy, previous studies have indicated that specific intestinal bacteria may improve lipid homeostasis via production of bacterial metabolites.9,10 Lithocholic acid (LCA), a secondary bile acid produced by the gut microbiota, has been shown to inhibit the biosynthesis and storage of lipids via downregulation of genes involved in lipogenesis.11
Our previous metagenome-wide association studies (MWAS) revealed significant differences in the gut microbiota between patients with ACVD and healthy controls,5 demonstrating a depletion of Bacteroides cellulosilyticus, Faecalibacterium prausnitzii, and Roseburia intestinalis in patients with ACVD.5 The three bacteria were associated with distinct ecological network structures which may represent ACVD depletion groups.5 B. cellulosilyticus was significantly associated with decreased serum triglycerides (TG), total cholesterol (CHOL), and low-density lipoprotein cholesterol (LDLC), and F. prausnitzii was negatively correlated with markers of insulin resistance in healthy individuals, which further suggested important impact on cardiovascular health.12 Colonization with R. intestinalis was reported to improved diabetes and ameliorate intestinal inflammation by producing butyric acid in gnotobiotic mice fed a diet with high content of plant polysaccharides.13 However, the impact of B. cellulosilyticus and F. prausnitzii on atherogenesis has not been experimentally verified. Furthermore, it is still unclear whether supplementation with members of both the Bacteroidota and the Firmicutes phylum would be superior to supplementation with only a single bacterium belonging to the Firmicutes phylum for improving lipid metabolism and preventing atherosclerosis.
Recent developments in anaerobic culturomics have greatly increased the number of available bacteria and genomes of members of the Bacteroidota and Firmicutes phyla from the human gut microbiota, but because of the difficulties associated with cultivation of such anaerobic bacteria and regulatory issues, previous studies have largely investigated the effect of traditional probiotics.14
Here, we first confirmed the significant associations between B. cellulosilyticus, F. prausnitzii, and R. intestinalis and cardiovascular risk factors in a large human cohort. Then, we investigated the possible protective effects of B. cellulosilyticus, Faecalibacterium longum, a novel member of the genus Faecalibacterium closely related to F. prausnitzii,15 and R. intestinalis by intragastric administration of each bacterium individually or in combination to an Apoe−/− atherosclerosis mouse model. Our study showed that a consortium of all three bacteria may exert a persistent beneficial effect improving several risk factors for the development of ACVD.
Results
Depletion of B. cellulosilyticus, F. prausnitzii, and R. intestinalis characterizes individuals with ACVD
We performed a reanalysis of the data obtained by analyzing a cohort consisting of 218 individuals with ACVD and 187 healthy controls5 to confirm the depletion of B. cellulosilyticus, F. prausnitzii, and R. intestinalis in the gut microbiota of individuals with ACVD compared to heathy individuals (Figure 1A). Taking advantage of the large SZ-4D cohort comprising 2,183 deeply phenotyped healthy adults,16 we identified distinct network structures of these three bacterial species in healthy individuals, with especially B. cellulosilyticus being involved in numerous positive interactions with other gut bacterial species (Figure 1B). Genome-scale metabolic models also showed distinct distribution and abundances of potential functions in the three species, including an increased abundance of pathways involved in pectin degradation in B. cellulosilyticus, and biosynthesis of phosphoribosylpyrophosphate, tryptophan, and trehalose in R. intestinalis (Table S1).17 Using the SZ-4D cohort, we further identified several associations between the three species and cardiometabolic risk factors at a false discovery rate (FDR) of 0.05 after adjusting for confounding factors. This analysis revealed that F. prausnitzii was negatively correlated with serum levels of TG, LDLC, and CHOL (Figure 1C). B. cellulosilyticus correlated negatively with blood pressure and serum TG, and positively with high-density lipoprotein cholesterol (HDLC), and R. intestinalis correlated inversely with the obesity-related phenotypes body fat and waist-to-hip ratio. These findings and the results of previous studies indicated that these three bacterial species might exert positive effects in relation to preventing or ameliorating obesity and obesity-associated diseases.
Figure 1.

B. cellulosilyticus, F. prausnitzii, and R. intestinalis are depleted in patients with ACVD
(A) The distribution of B. cellulosilyticus, F. prausnitzii, and R. intestinalis in ACVD cohorts. The reported p values represent Wilcox rank-sum test between patients with ACVD and healthy controls.
(B) Network of three bacteria and other species in the large SZ-4D cohort comprising 2183 healthy individuals. Three bacteria formed distinct ecology groups with dense sub network for B. cellulosilyticus.
(C) Three bacterial species were negatively correlated with cardiovascular risk factors in the SZ-4D cohort. Here shown as the partial Spearman correlation between the three taxa and cardiovascular risk factors after adjusting for factors that potentially might influence the gut microbiome at FDR < 0.1.
Gavage with F. longum, B. cellulosilyticus, and R. intestinalis individually or together lowers plasma lipids levels and attenuates the formation of atherosclerotic plaque in Apoe−/− mice
We have previously reported on a large culture collection of bacterial strains isolated from human stool samples.14 We mapped the metagenomic linkage groups (MLGs) classified as F. prausnitzii, B. cellulosilyticus, and R. intestinalis to the sequenced member of this collection and found that these MLGs exhibited significant matches to the strains F. longum CM04-06, closely related to F. prausnitzii,15 B. cellulosilyticus AF17-25, and R. intestinalis AM43-11.14,15 We designed a mouse experiment involving gavage of atherosclerosis-prone mice with a single strain or a consortium of all three strains to examine to what extent this might prevent/ameliorate the development of atherosclerosis combined with an attempt to explain the underlying mechanisms (Figure 2A). Eight-week-old female apolipoprotein-E-deficient (Apoe−/−) mice on a C57BL/6J background were fed a Western diet and gavaged orally daily with each individual strain or a combination of all strains for a period of 127 days (∼18 weeks). Wild-type C57BL/6J and Apoe−/− mice gavaged with vehicle (PBS), following the same protocol, served as controls. At 25 weeks of age, the mice were euthanized, and plasma was obtained for biochemical analyses and selected tissues were dissected out for atherosclerosis assessment (Figure 2A). As expected, we observed characteristic significant differences (p < 0.05) in plasma TG, LDLC, HDLC, and CHOL as well as the extent of atherosclerotic lesions between wild-type C57BL/6J and Apoe−/− mice (Figures 2B–2J, Table S2).
Figure 2.

Gavage of Western diet-fed Apoe−/− mice with live bacteria reduces plasma lipids and attenuates atherosclerosis
(A) Diagram of the animal study design. Eight-weeks-old animals were fed a Western diet (WD) and randomly divided into six groups: mice in the C57BL/6J and Apoe−/− control group were gavaged with an equivalent volume of vehicle (PBS), the other 4 group were respectively gavaged with live B. cellulosilyticus, F. longum, R. intestinalis and the consortium of the three bacteria, at a daily dose of 1 × 109 CFU in PBS. When the mice were 25 weeks old, gavage was terminated and the mice were sacrificed.
(B–F) Comparison of plasma glucose and lipid profiles (6–7 samples per group).
(G) Representative photomicrographs of oil red O staining and quantitative analysis of atherosclerotic plaque area in the aortas (6–8 samples per group).
(H) Representative photomicrographs of oil red O staining and quantitative analysis of area percentage of atherosclerotic lesion in the aortic sinus (4–8 samples per group). The black bar represents 500 μm.
(I) Representative photomicrographs of hematoxylin and eosin staining and quantitative analysis of atherosclerotic lesion size in the aortic sinus (4–8 samples per group). The black bar represents 500 μm.
(J) Representative photomicrographs of Masson staining and quantitative analysis of area percentage of collagen in the aortic sinus (4–8 samples per group). The black bar represents 500 μm. TG, triglycerides; LDLC, low-density lipoprotein cholesterol; HDLC, high-density lipoprotein cholesterol; CHOL, total cholesterol. Two-tailed Student’s t test or Mann-Whitney U-test was used for comparison between two groups. Data are shown as mean ± standard error of the mean.
The experiment revealed that mice gavaged with the live bacteria had better outcomes than those in the Apoe−/− control group with respect to the mean (±SD) change of blood biochemical parameters (Figures 2B–2F, Table S2). Compared with the control Apoe−/− mice, Apoe−/− mice gavaged with B. cellulosilyticus exhibited significantly decreased levels of plasma glucose (p = 0.0411; Figure 2B), TG (p = 0.0016; Figure 2C), and LDLC (p = 0.0293; Figure 2D) corresponding to a decrease of 30.7%, 37.7%, and 23.3%, respectively. Gavage with F. longum significantly decreased TG (p = 0.0372, Figure 2C) and LDLC (p = 0.0472, Figure 2D) corresponding to a decrease of 23.1% and 17.2%, respectively. Gavage with R. intestinalis significantly decreased TG (p = 0.0038, Figure 2C) and increased HDLC (p = 0.0363; Figure 2E) corresponding to changes of 34.8% and 19.0%, respectively, Gavage with the consortium of all three strains significantly decreased TG (p = 0.0053, Figure 2C) and LDLC (p = 0.0464, Figure 2D) corresponding to a decrease of 31.9% and 23.3%, respectively, but without significant differences in CHOL (Figure 2F) and body weight (Figure S1A, Table S3). Oil-red staining of longitudinally opened aortas (Figure 2G) and aortic sinuses (Figure 2H), and hematoxylin and eosin staining of aortic sinuses (Figure 2I), revealed a significantly reduced formation of atherosclerotic lesions in Apoe−/− mice gavaged with B. cellulosilyticus and the three bacterial strains in combination compared with the PBS gavaged Apoe−/− mice (p < 0.05, Table S2). Gavage with B. cellulosilyticus resulted in a significant (p < 0.05) decrease of 24.4%, 67.7%, and 25.1%, respectively, for these three parameters, whereas gavage with all three strains resulted in a 32.1%, 47.4%, and 29.2% decrease (p < 0.05), respectively. Masson staining of aortic sinuses (Figure 2J) revealed that mice gavaged with live R. intestinalis and the combination of all three strains exhibited significantly attenuated collagen deposition corresponding to a reduction of 21.6% and 35.8% (p < 0.05), respectively. Whereas, gavage with individual bacteria or all three strains together improved a number of cardiovascular risk factors in the Apoe−/− mice, gavaging with the three strains was unable to fully reestablish the values observed in the wild-type C57BL/6J mice. The experiments also demonstrated that the combination of all three strains with a dosage of each strain reduced to one-third of the dosage used for gavage with single strains resulted in improvements to at least the same levels for all these metabolic and histological parameters as were observed for gavage with the single strains. Interestingly, compared to supplementation with only a single bacterium, we observed that a number of parameters associated with cardiac performance were improved in mice gavaged with the combination of all three bacterial strains, including decreased left ventricular volume and diameter at end-systole and diastole, and increased left ventricular ejection fraction and fractional shortening (p < 0.05; Figures S1B–S1I, Table S2).
Gavage with F. longum, B. cellulosilyticus, and R. intestinalis individually or together regulates expression of the farnesoid X receptor and genes involved in cholesterol biosynthesis and circadian rhythm
To examine how gavage with each bacterial strain or the combination of bacterial strains might affect lipid metabolism in Apoe−/− mice, expression of genes in the liver of mice at the end of the intervention was examined by RNA-seq. Compared to Apoe−/− mice gavaged with PBS, Apoe−/− mice gavaged with the combination of the three bacterial strains exhibited the greatest number of differentially expressed genes with 247 being downregulated and 640 being upregulated (Figures S2A and 3A–3D).
Figure 3.

Gavage of Western diet-fed Apoe−/− mice with live bacteria downregulates expression of genes involved in lipogenic pathways and upregulates expression of genes involved in circadian rhythm
(A–D) Volcano plots showing transcriptome-wide changes in mRNA levels. The significantly upregulated or downregulated genes (FDR < 0.05 and the absolute value of fold change >1) are marked in red and blue, respectively.
(E) Heatmap of genes related to biosynthesis and metabolism of lipids in mice treated with vehicle or live bacteria.
(F) Heatmap of the genes related to the regulation of circadian rhythm in mice treated with vehicle or live bacteria. The red areas represent highly expressed genes and blue areas represent lowly expressed genes. Apoe−/− control, western diet-fed Apoe−/− mice treated with vehicle (n = 6); B, Western diet-fed Apoe−/− mice treated with B. cellulosilyticus (n = 7); F, Western diet-fed Apoe−/− mice treated with F. longum (n = 8); R, Western diet-fed Apoe−/− mice treated with R. intestinalis (n = 7); B,F,R, Western diet-fed Apoe−/− mice treated with the consortium of the three bacteria (n = 5).
Gene set enrichment analysis based on Gene Ontology revealed that genes that were downregulated in mice receiving gavage with bacteria were significantly enriched (Q < 0.05) for genes involved in lipogenic processes (lipid, fatty acids and cholesterol biosynthesis, and triglyceride metabolic processes), cholesterol synthesis and metabolism (such as Hmgcs1, Fdps, Sc5d, and Pcsk9, FDR < 0.05), and bile acid transport (including Aqp8, Insig2, Slc45a3, and Abcb11, FDR < 0.05) (Figures S2B and S2C, Table S4. Differentially expressed genes in liver, related to Figures 3 and S2, Table S5. Gene Set Enrichment Analysis of differentially expressed genes between B and Apoe−/− control, related to Figure 3 and Table S4, Table S6. Gene Set Enrichment Analysis of differentially expressed genes between F and Apoe−/− control, related to Figure 3 and Table S4, Table S7. Gene Set Enrichment Analysis of differentially expressed genes between R and Apoe−/− control, related to Figure 3 and Table S4, Table S8. Gene Set Enrichment Analysis of differentially expressed genes between B,F,R and Apoe−/− control, related to Figure 3 and Table S4). More specifically, the expression levels of mRNAs encoding enzymes involved in lipogenesis such as Elovl5, Pnpla3, Abca2, Fasn, Acss2, Acly, and Acacb (log2 FC < -1, FDR < 0.05) were all downregulated in response to gavage with either a single bacterium or the combination of the three bacterial species (Figure 3E).
By contrast, genes for which expression was upregulated in mice gavaged with the bacterial strains were enriched for genes involved in lipolysis and oxidation of fatty acids (e.g. Lipe, Cpt1a, and Pnpla2, key enzymes for TG catabolism; Figure 3F), and surprisingly genes involved in the regulation of circadian rhythm (Ciart, Usp2, Per3, Per2, Clock, Arntl, Noct, Dbp, Nr1d1, and Nr1d2; Figure 3G) both in mice gavaged with a single bacterial species or a combination of all three species (Figures S2B and S2C). Together, these results indicated that apart from genes involved in general lipid metabolism, gavage with these bacterial strains may also regulate metabolism by modulating expression of genes involved in the regulation of circadian rhythm. However, more detailed analyses during the dark and the light cycle are needed to determine the physiological effect of these changes in expression of the genes involved in regulation of circadian rhythm.
The majority of the genes being differentially affected by gavage with the bacterial species are controlled by nuclear receptors, such as Rev-erb-alpha regulating the expression of Arntl (also called Bmal1), and in addition, both Rev-erb alpha (encoded by Nr1d1) and Rev-erb beta (encoded by Nr1d2) are important regulator of genes involved in lipid metabolism. Expression of both Nr1d1 and Nr1d2 was also significantly increased in response to gavage (Figure S2D). Expression of nuclear bile acid receptor, farnesoid X receptor (Fxr; Nr1h4), was upregulated in mice gavaged with B. cellulosilyticus or R. intestinalis. Expression of Nr1c1 (Ppara) was upregulated in mice gavaged with R intestinalis, or the combination of all three bacterial species (FDR < 0.5, Table S4). Finally, we observed that expression of Nr4a1 (Nur77) was upregulated both in response to gavage with each individual bacterial strain or in response to gavage with all three bacterial strains (Figure S2D). In conclusion, these results indicated that the improvement in metabolic performance elicited by gavage with the three bacterial strains independently, especially B. cellulosilyticus, or combined improved lipid metabolism by inducing expression of genes reducing the synthesis and increasing the catabolism of lipids in atherosclerosis-prone mice.
Gavage with F. longum, B. cellulosilyticus, and R. intestinalis individually or together lowers plasma long-chain fatty acid and increases the level of lithocholic acid
Plasma metabolites may serve as a ligand for nuclear receptors. To identify metabolite features that were altered by gavage with bacteria improving atherosclerosis, we quantified 617 metabolites in plasma samples collected from all mice using targeted metabolomic profiling. A total of 187, 51, 184, and 169 metabolites exhibited significant different abundances comparing mice gavaged with B. cellulosilyticus, F. longum, R. intestinalis, and the combination of the three species to Apoe−/− mice gavaged with PBS, respectively (Figure 4A, p < 0.05; VIP >1; Table S9). The analysis revealed that of the metabolites that exhibited increased levels after B. cellulosilyticus supplementation, bile acids (LCA and allocholic acid), pantothenate, and methylguanosine were elevated (FDR < 0.01, VIP>1; Figures 4B, 4C, and S3A). On the contrary, multiple lipids especially Lyso-PCs (lysophosphatidylcholines) were reduced. In mice gavaged with F. longum, levels of a number of other lipids (e.g. carnitines, oxidized lipids, and free fatty acids) were reduced (Figures 4C, 4D, and S3B). Similar results were obtained analyzing mice gavaged with R. intestinalis. Moreover, the data revealed that 3-hydroxy-3-methyl butyric acid (HMB) was significantly increased in mice gavaged with R. intestinalis (FDR = 6.52 × 10-3, VIP = 1.35; Figures 4C and 4E). For mice gavaged with the combination of the three bacterial species, a significant enrichment of bile acids (LCA and allocholic acid), HMB, and methylguanosine (FDR < 0.05) and decreasing levels of lipids (e.g. Lyso-PCs and free fatty acids) were observed (Figures 4C, 4F, and S3D). Interestingly, similar trends in changes of plasma metabolites were observed by comparing wild-type mice to Apoe−/− mice (Figure S4A). Thus, the changes in 64 overlapping metabolites reflected a reduction in biosynthesis of unsaturated fatty acids and Western diet-enriched metabolites such as lecithin, choline, and carnitine (Figures S4B and S4C).
Figure 4.

Gavage of Western diet-fed Apoe−/− mice with live bacteria changes the levels of plasma metabolites
(A) Principal component analysis of plasma metabolite profiles in mice treated with vehicle or live bacteria.
(B and D–F) Heatmap of differentially affected metabolites screened by OPLS-DA. The grids filled with graduated color indicate the relative abundance of metabolites, and the grids filled with pure red and blue represent the correlation between metabolites and lipids. Differential metabolites were defined by a p value smaller than 0.05 and a VIP value larger than 1. Two-tailed Student’s t test was used for comparison between two groups.
(C) Boxplots of LCA and HMB in mice treated with vehicle or bacteria.
(G) The scatterplots of linear regression of LCA with TG, LDLC, and CHOL. WT, Western diet-fed normal C57BL/6J mice treated with vehicle (n = 7); Apoe−/− control, Western diet-fed Apoe−/− mice treated with vehicle (n = 6); B, Western diet-fed Apoe−/− mice treated with B. cellulosilyticus (n = 7); F, Western diet-fed Apoe−/− mice treated with F. longum (n = 8); R, Western diet-fed Apoe−/− mice treated with R. intestinalis (n = 7); B,F,R, Western diet-fed Apoe−/− mice treated with the cocktail of the three bacteria (n = 5).
LCA is one the most abundant secondary bile acids in humans, and is a major component of the recirculating bile acid pool.18 LCA has important roles in the prevention of Clostridium difficile outgrowth,19 and controlling host metabolic and immune responses.20,21,22,23 HMB (also named 3-hydroxyisovaleric acid) is a byproduct of the branched-chain amino acid (BCAA) degradation pathway in R. intestinalis.24 Moreover, we observed significant negative correlations (FDR < 0.05) among LCA, HMB, and metabolic parameters (Figure 4G, Table S10). The effects of LCA on lipid homeostasis were further confirmed in a causal network edge-oriented model (Figure S5). Thus, these results suggested that LCA and HMB represent metabolites that may decrease plasma lipids and atherosclerosis in mice in response to supplementation with the three bacterial strains.
We also examined metabolites that can be produced by the microbiota and have been reported to be associated with cardiovascular disease (Figure S6). Among these, we observed that the levels of circulating phenylacetylglutamine,25 isoleucine,26 glutamate,27,28 tyrosine,29 and succinic acid30,31 were significantly diminished (FDR < 0.05, VIP >1), while the level of indole-3-acetic acid32 was increased. Overall, these results suggested that colonization with three strains also modulated the level of metabolites that directly or indirectly may play a role in adverse cardiovascular events.
Gavage with F. longum, B. cellulosilyticus, and R. intestinalis individually or together modulates the fecal microbiota composition
To determine the effects of supplementation on the gut microbiota, we collected high-quality metagenomic data (∼15 Gb/sample) from 476 fecal samples at baseline and during supplementation (Figure 5A). We were unable to detect the three bacterial species at baseline for the four intervention groups and in the Apoe−/− control mice during the experiment. Notably, there was no significant differences of the fecal microbiota between the bacterial intervention groups and the Apoe−/− control group at baseline (post hoc permutational multivariate analysis of variance (PERMANOVA), R2 = 0.118–0.286, Bonferroni adjusted p = 0.008–0.775; Figure 5A). Gavage with each of the individual bacterial strains significantly (p < 1 × 10−8) increased the relative abundance of each of the three bacteria in the feces of the gavaged Apoe−/− mice (Figure S7). Interestingly, following gavage with all three bacteria combined, only the abundance of B. cellulosilyticus exhibited a highly significant increase in abundance (p < 1 × 10−8) in Apoe−/− mice, and R. intestinalis had a slightly higher (p = 0.025) relative abundance in mice gavaged with the combination of bacterial species compared to Apoe−/− control mice. At the end of the intervention, we detected significant differences in gut microbiota composition between the groups of Apoe−/− mice (R2 = 0.285, p = 1 × 10−4; Figure 5A). Of these groups, mice gavaged with R. intestinalis and the combination of all strains had shifted to the greatest extent. A general overview of bacterial taxonomic development in each time point is provided in Figure S8. The microbiome from mice gavaged with each individual bacterial species or the combination of strains developed over time into stages that differed from that of Apoe−/− control mice, following different and distinct trajectories according to dirichlet multinomial mixtures clustering. By the end of the intervention, two major clusters were present in the control Apoe−/− control mice. By contrast, all Apoe−/− mice gavaged with either a single bacterium or the combination with all three bacteria followed trajectories that converged into one dominant cluster, with cluster 3 characterizing mice gavaged with B. cellulosilyticus, cluster 4 characterizing mice gavaged with F. longum, and cluster 2 characterizing mice gavaged with R. intestinalis or all three bacteria. Overall, these results indicated that supplementation with each individual strain or the combination of strains in a distinct manner affected the overall structure of the gut microbiome.
Figure 5.

The interventions change the fecal microbiome composition and the changes link to lipids
(A) Visualization of the fecal microbiota composition at baseline and at the endpoint of the intervention in mice receiving each individual bacterium or the consortium of all three bacteria. Microbiota composition is represented by multidimensional scaling (genus level Aitchison distance). Before represents baseline. After represents the end of the study. Intervention effects are symbolized by the colored arrows, with direction and length corresponding to the shift in group centroid coordinates from each intervention group to control group. Fecal composition of all groups was not different at baseline while microbiota composition of intervention group was different from that of control group at the end of study.
(B) Lipid-associated taxa. The β (the linear model coefficients, dot) ±SE for each taxon that was significantly associated with the lipid parameter. An asterisk before the label indicates a taxon that has been reported as having the ability for transforming bile acid and a circle before the label represents a taxon producing acetic acid.
(C) Treatment effect on lipid-associated taxa. The coefficient β±SE represents the treatment effect on lipid associated taxa in b compared to the control group at the end of the study. A comparison of the effects of treatment and control on growth rates of each taxon was performed using a mixed effects linear model by interaction between weeks in the intervention and treatment (coefficient β3±SE) and a random intercept for each mouse.
We then used linear models to determine the relationships between the levels of bacterial taxa and lipid levels for each mouse. We showed that among the 97 species that were above 10% prevalence, 18 were significantly associated with lipids (termed lipid-associated taxa, q value <0.05; Figure 5B, Table S11). Of these taxa, 11 were negatively associated, including closely related species of the Lactobacillus johnsonii cluster33 (e.g., L. johnsonii34,35,36,37,38 and Lactobacillus taiwanensis), Bacteroides caecimuris,39,40,41,42 Bilophila wadsworthia,43,44,45 Alistipes inops,46 Staphylococcus nepalensis,47,48 and B. cellulosilyticus; the 7 taxa that were positively associated included Aerococcus viridans,49 Bordetella pseudohinzii, Oscillibacter sp 1 3, and Turicimonas muris. Of the taxa associated inversely with lipid metabolism, 5 taxa have been identified to be involved in bile acid metabolism, 5 taxa produce acetic acid, reported to modulated adiposity, and 1 taxon was experimental shown to lower lipid content. An analysis of covariance between plasma metabolites and gut microbiota revealed that the levels of these 11 taxa correlated with the 69 plasma metabolites including lecithin, choline, carnitine, BCAA, and LCA that have been shown to be associated with whole body metabolism (Table S12).
Both the relative levels and growth rate of the taxa associated inversely with lipid anabolism increased significantly more in the gut microbiota of mice gavage with live bacteria compared to the Apoe−/− control mice in a linear mixed-effects model (q value < 0.05; Figure 5C, Table S13). Of the taxa associated positively with lipid anabolism, the pathogen Aerococcus viridans50 exhibited the greatest decrease in mice gavaged with the three bacteria.
To explore unmeasured microbiota-derived metabolites previously reported to play important role in cardiovascular disease, we determined the functional potential of the taxa from the shotgun sequencing data. Consistent with independent blood metabolomics data, we found that the BCAA biosynthesis potential was decreased both in mice gavaged with each bacterial strain and the combination of all strains (FDR < 0.1; Figure S9, Table S14). B. cellulosilyticus, F. longum, and R. intestinalis are all main short-chain fatty acid (SCFA) producers. Here, we observed that SCFA pathways, tetrapyrrole biosynthesis I (from glutamate), pyruvate fermentation to propanoate I, phosphopantothenate biosynthesis I, and phytate degradation I were increased in mice gavaged with each bacterial strain and the combination of all strains (p < 0.05; Figure S9).
To get more insight into the role of the microbiota in relation to the increase in the levels of the important secondary bile acid, LCA, we determined the per kilobase million reads of the bai operon, known to contribute to the conversion of primary to secondary bile acids via the 7α-dehydroxylation pathway (Figure S10A). Quantification of blood LCA levels by MS was found to be in agreement with baiG metagenomic transcript abundances (Figure S10B). We found that the bai operon (A-I) was overall highly abundant in mice gavage with live bacteria compared to the Apoe−/− control mice (Kruskal-Wallis p < 0.05; Figure S10C). We confirmed the presence of the bai operon in lipid-associated taxa and taxa that increased in response to treatment using homology sequence alignment (phmmer) and correlation analysis (Figure S10D).
Discussion
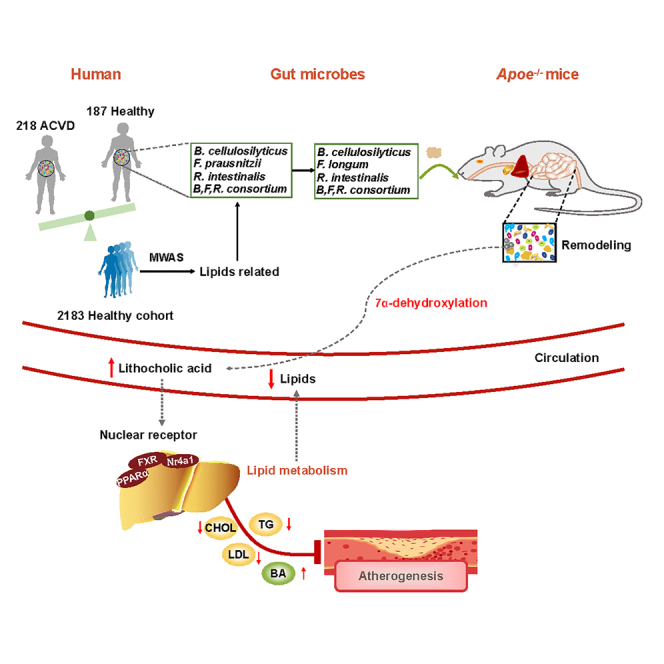
In this study, we exploited MWAS biomarkers discovered in human cohorts and conducted a systematic survey of the literature on microbiome studies of ACVD including our previous work. This survey demonstrated a consistent depletion of B. cellulosilyticus, F. prausnitzii, and R. intestinalis in individuals with ACVD compared to healthy individuals. These species were also frequently reported to be enriched in healthy individuals in other cross-sectional studies of metabolic disorders.51 Of note, the abundances of these three bacteria correlated inversely with plasma lipid levels in 3,587 young healthy individuals.52 The robust depletion of these three bacteria indicated that administration of these bacteria might hold a potential for treatment of ACVD. In our collection of cultivated gut bacterial species from young healthy individuals,14 isolates of B. cellulosilyticus and R. intestinalis were available, whereas F. prausnitzii was missing. However, as a related species with similar metabolic competences F. longum was available. We used these three bacterial species to investigate a possible beneficial effect of supplementation using an Apoe−/− atherosclerosis mouse model. Our results revealed that administration beneficially affected the metabolism of the Apoe−/− mice and provided information on potential modes of actions. A schematic summary illustrating how the bacterial produced LCA may affect hepatic gene expression and metabolism and thereby influence atherogenesis is presented in Figure 6.
Figure 6.

Schematics illustrating how bacterially produced lithocholic acid may affect hepatic gene expression and lipid metabolism
Administration of B. cellulosilyticus, F. longum, and R. intestinalis leads to an increase in the relative abundance of LCA-producing bacteria increasing LCA levels through the 7α-dehydroxylation pathway. The increased levels of LCA in circulation downregulate expression of genes involved in lipid biosynthesis and upregulates genes involved in of lipid catabolism via activation of FXR.
Major risk factors for ACVD/atherosclerosis are hypercholesterolemia and dyslipidemia, indicating that the improvement of lipid metabolism may play an important role in the prevention and treatment of cardiovascular disease.53,54 F. longum treatment may improve hepatic functions by reducing fat content in the liver,55 and colonization with R. intestinalis has been reported to inhibit the development of atherosclerotic plaque.13 Here, we found that the levels of atherosclerotic plaque and lipid were diminished in Apoe−/− mice after gavage with the three strains of bacteria alone or in combination, indicating a potential advantage by administration of members of both the Bacteroidota and the Firmicutes phylum. Moreover, combination of all three bacterial strains brings superior cardiac performance such as decreased left ventricular volume and diameter at end-systole and diastole, and increased left ventricular ejection fraction and fractional shortening.
We observed that treatment with B. cellulosilyticus, F. longum, and R. intestinalis alone or in combination remodeled the balance of species in the gut microbiota. We identified bacteria that associated positively or negatively with lipid levels. Of note, administration of B. cellulosilyticus, F. longum, and R. intestinalis resulted in a transition of the gut microbiota from communities comprising taxa associated with high levels of lipids including Aerococcus viridans, which may induce endocarditis49 to communities comprising taxa inversely associated with lipid levels including Lactobacillus (L. taiwanensis, L. johnsonii), Bacteroides (A. inops, B. caecimuris, and B. cellulosilyticus), and Eubacterium (Eubacterium plexicaudatum), some of which have been extensively studied for their probiotic properties.34,35,36,37,38
Administration of B. cellulosilyticus, F. longum, and R. intestinalis exerted a marked effect on secondary bile acid synthesis leading to an increase in the plasma level of LCA accompanied by an increase in taxa with a potential for biosynthesis of LCA such as E. plexicaudatum harboring a 7α-dehydroxylation gene cluster. The combined results of the analyses of the liver transcriptome and the plasma metabolome indicated that activation of FXR mediated by the bacterial metabolite LCA reduced the expression of genes involved in lipogenesis (such as Srebf1 and Fasn) and increased the expression of target genes involved in lipolysis (such as Pnpla2 and Lipe) suggesting an additional mechanism for lowering lipids.11,56,57 Interesting, the level of HMB was increasing greatly by administration of R. intestinalis in which HMB is produced via degradation of BCAA.24 HMB has been observed to decrease plasma cholesterol via activation of LXR and PPARγ and lowering expression of Acc and Srebf1.58 HMB supplementation in humans has been reported to result in a net decrease in CHOL and LDLC, thereby reducing the risk of stroke and heart attack.59 For trimethylamine N-oxide (TMAO) associated with atherosclerosis,60 we did not observe significant effects of administration of the three bacteria individually or as a consortium on the levels of TMAO (Figure S6). However, we observed that levels of circulating phenylacetylglutamine,61 and hydroxyphenyllactic acid (4-hydroxyphenyllactate),62 all in the phenylalanine metabolism pathway, were significantly reduced (FDR < 0.05, VIP >1) after B. cellulosilyticus gavage (Figure S6, Table S9). We conclude that B. cellulosilyticus plays an important role in microbiota-host phenylalanine co-metabolism.
In conclusion, this study demonstrates a protective effect of administration of B. cellulosilyticus, F. longum, and R. intestinalis to Apoe−/− mice suggesting a potential for use of these bacteria to prevent/ameliorate ACVD. Obviously, more studies using other animal models are needed to determine the potential beneficial effects of supplementation B. cellulosilyticus, F. longum, and R. intestinalis in relation to ACVD.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial strains | ||
| Bacteroides cellulosilyticus | Isolated from fecal samples of healthy donors as reported previously | https://doi.org/10.1038/s41587-018-0008-8 |
| Faecalibacterium longum | Isolated from fecal samples of healthy donors as reported previously | https://doi.org/10.1038/s41587-018-0008-8 |
| Roseburia intestinalis | Isolated from fecal samples of healthy donors as reported previously | https://doi.org/10.1038/s41587-018-0008-8 |
| Critical commercial assays | ||
| Oil red O staining | Servicebio, Wuhan, China | #G1016 |
| Oil red O staining | Servicebio, Wuhan, China | #G1004 |
| Hematoxylin and eosin staining | Servicebio, Wuhan, China | #G1003 |
| Masson staining | Servicebio, Wuhan, China | #G1006 |
| Total cholesterol | Rayto, ShenZhen, China | #S03042 |
| Triglycerides | Rayto, ShenZhen, China | #S03027 |
| Low-density lipoprotein cholesterol | Rayto, ShenZhen, China | #S03029 |
| High-density lipoprotein cholesterol | Rayto, ShenZhen, China | #S03025 |
| Glucose | Rayto, ShenZhen, China | #S03039 |
| Deposited data | ||
| Raw and analyzed data | This paper | Available at CNSA (https://db.cngb.org/cnsa/) of (CNGB) database under the accession code CNP0002026 |
| Raw and analyzed data from the ACVD cohort and large scale healthy cohort | (Jie et al.5), (Jie et al., 2022)63 | Available at CNSA (https://db.cngb.org/cnsa/) of (CNGB) database under the accession code CNP0000426, CNP0000289 |
| Software and algorithms | ||
| ImageJ® | https://doi.org/10.1038/nmeth.2089 | https://ImageJ.nih.gov/ij/ |
| R Studio 4.1.0 | RStudio: Integrated Development for R | https://posit.co/products/open-source/rstudio/ |
| Network edge orienting (NEO) network | ||
| cOMG | This paper | https://github.com/jiezhuye/cOMG |
| Bowtie2(v2.3.564) | Langmead and Salzberg64 | http://bowtie-bio.sourceforge.net/bowtie2 |
| MetaPhlAn3 | (Francesco et al., 2021)65 | https://huttenhower.sph.harvard.edu/metaphlan3/ |
| HUMAnN2 | (Franzosa et al., 2018)66 | https://huttenhower.sph.harvard.edu/humann |
| ggplot2 | (Wickham, 2016)67 | https://ggplot2.tidyverse.org/ |
| PERMANOVA | Anderson68 | https://doi.org/10.1002/9781118445112.stat07841 |
| DMM | (Holmes, et al., 2012)69 | http://code.google.com/p/microbedmm/ |
| ShortBRED | (Kaminski et al., 2015)70 | https://huttenhower.sph.harvard.edu/shortbred |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J Mice | Beijing Vitalstar Biotechnology Co., Ltd | N/A |
| Mouse: Apolipoprotein E-deficient (Apoe−/−) mice on C57BL/6J background | Beijing Vitalstar Biotechnology Co., Ltd | N/A |
Resource availability
Lead contact
Requests for further information and/or reagents and resources should be directed to and will be fulfilled by the lead contact, Shilong Zhong (zhongsl@hotmail.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Animal model
Apolipoprotein E-deficient (Apoe−/−) mice were on a C57BL/6J background. All mice were purchased from the Beijing Vitalstar Biotechnology Co., Ltd and housed at the Guangzhou General Biomedical Technology Co., Ltd. Eight-week-old female mice were fed Western diet (Harlan TD.88137) and water ad libitum under a strict 12-h light cycle for 18 weeks. Mice were randomly divided into six groups: mice in the wild-type (WT) and Apoe−/− control group were gavaged with vehicle (PBS), those in the bacteria groups were gavaged daily with live B. cellulosilyticus, F. longum, R. intestinalis or the consortium of the three bacteria, respectively, with a final total dose of 1 × 109 CFU in PBS. All animal experiments were approved by the Committee of Guangdong Provincial People’s Hospital.
Method details
Atherosclerotic lesion assessment
After the mice were anesthetized, the aorta segment was perfused with saline and dissected from the proximal ascending aorta arch to the celiac artery and fixed in 4% paraformaldehyde. For enface lesion analysis of aortic, the adventitial tissue was carefully removed and the aorta was opened longitudinally, stained with oil red O (#G1016; Servicebio, Wuhan, China), pinned on a black surface and images were captured using a digital camera.
For analysis of atherosclerotic lesion in the aortic root, samples were obtained from the portion of the ascending aorta proximal to the aortic sinus. Five serial 10-μm-thick sections of the aortic sinus were collected from each mouse using a cryotome (#CryoStar NX50; Thermo Fisher Scientific, Waltham, MA, USA) and stored at - 80 °C. The frozen sections of each mice were stained with oil red O (#G1016, #G1004, Servicebio, Wuhan, China) or hematoxylin and eosin (#G1003, Servicebio, Wuhan, China), masson (#G1006, Servicebio, Wuhan, China). The stained sections were digitally captured using an all-in-one fluorescence microscope (#Eclipse Ci; Nikon, Tokyo, Japan). The quantification of lesion area size was performed with the ImageJ software.
Biochemical parameters assays
The animals were fasted overnight and blood was collected in EDTA containing tubes from the mice by removing the eyeball using the eyeball removal method under anesthesia. The blood samples were centrifuged at 4 °C, 3,000 rpm for 10 min, and stored at −80 °C before measurement. The plasma levels of total cholesterol (#S03042), triglycerides (#S03027), low-density lipoprotein cholesterol (#S03029), high-density lipoprotein cholesterol (#S03025), and glucose (#S03039) were measured enzymatically with commercial kits (Rayto, ShenZhen, China) using an automated chemistry analyzer (#Chemray 800; Rayto, ShenZhen, China) according to the manufacturer’s instructions.
Mice fecal sample DNA extraction and metagenomics shotgun sequencing
Fecal samples from mice were collected at 6 and 7 weeks of age before gavage and the first and fourth day after gavage performed when the mice were 8 weeks old, and when the mice were 9, 11, 13, 16 and 25 weeks of age. Fecal samples were frozen at −80 °C. DNA extraction of the stored fecal samples was performed as previously described.73 This manual extraction protocol has been described in detail,74 and used in many studies.5,75,76,77,78 Briefly, a frozen aliquot of fecal sample was suspended in 250 μL of guanidine thiocyanate, 0.1 M Tris (pH 7.5) and 40 μL of 10% N-lauroyl sarcosine. Then 500 μL 5% N-lauroyl sarcosine was added. After 1h incubation, 500 μL of glass beads (0.1 mm) and 500 μL of TENP were added to the tube for vortexing, followed by centrifugation. The supernatant was transferred to a new tube, and DNA was precipitated by isopropanol.79 Metagenomic sequencing was performed on the BGISEQ-500 platform (PCR-free without size selection, 100 bp of paired-end reads for fecal samples, and four libraries were constructed for each lane), and quality-controlled as previously reported.80
Bacterial culture
B. cellulosilyticus, F. longum, and R. intestinalis were original isolated from fecal samples of healthy donors as reported previously.14 In brief, the fecal sample was firstly placed into a sterile tube and transferred into an anaerobic chamber (Bactron Anaerobic Chamber, Bactron IV-2, Shellab, USA) within 30 min after collection. Next, the sample was preincubated at 37 °C for 1 day using sterile sheep blood and then resuspended in pre-reduced phosphate buffered saline (PBS) containing 0.1% cysteine. The resuspended sample was plated on modified peptone-yeast extract-glucose (MPYG) agar plates supplemented with 5% sterile sheep blood by 6–8 serial 10-fold dilutions and then incubated in an anaerobic atmosphere at 37 °C for 2–3 days. The isolated colonies were re-streaked onto MPYG blood agar for purification and then frozen in a glycerol suspension (20%, v/v) supplemented with 0.1% cysteine at −80 °C. Lastly, the species of isolates were identified by mapping 16S rRNA gene sequences with the sequences of type strains retrieved from the EzBioCloud database (https://www.ezbiocloud.net/).
Three successfully identified bacteria were all incubated using MPYG medium under anaerobic condition at 37 °C for 24h. After incubation, the fresh bacterial cultures were centrifuged at 4 °C in 8000 rpm for 5 min, and then washed with sterile anaerobic PBS (supplemented with resazurin 1 mg/L). The bacteria were resuspended in sterile anaerobic PBS supplemented with 10% glycerol, aliquoted and stored at - 80 °C until use. Bacterial cells for gavage were prepared by suspending them in PBS to a cell density of 5 × 109 CFU/mL in an anaerobic atmosphere. For a combination of the three species, each species was mixed in a ratio of the concentration by 1:1:1 and with a final total density of 5 × 109 CFU/mL. Mice were gavaged daily for 127days with 200 μL PBS containing live B. cellulosilyticus, F. longum, R. intestinalis or a combination of the three bacterial species.
RNA preparation and sequencing
Total RNA was extracted from the liver tissues of each mouse using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. About 60 mg of tissues were ground into powder under liquid nitrogen in a 2 mL tube, mixed with 1.5 mL TRIzol and allowed to rest horizontally for 5 min. The tissue homogenate was centrifuged at 12000×g at 4°C for 5 min, then the supernatant was transferred into 300 μL chloroform/isoamyl alcohol (24:1). The sample was mixed upside down and shaken vigorously for 15s, and then centrifuged at 12000×g at 4°C for 10 min. After centrifugation, the supernatant was transferred into a centrifuge tube with 600 μL of isopropyl alcohol to precipitate RNA and then centrifuged at 13600 rpm for 20 min at 4°C. The RNA pellet was washed twice with 1 mL 75% ethanol, centrifuged at 13600 rpm for 3 min at 4°C, and the pellet was left to air dry for 5–10 min in a biosafety cabinet. The RNA pellet was dissolved in 25 μL–100 μL of DEPC-treated water and analyzed using a Nano Drop and an Agilent 2100 bioanalyzer (Thermo Fisher Scientific, MA, USA).
mRNA was isolated using oligo(dT) magnetic beads, fragmented, and cDNA was prepared by random hexamer-primed reverse transcription, followed by end repair, 3′ adenylation, and adapters were ligated to the ends of 3′ adenylated cDNA fragments followed by PCR amplification. The PCR products were purified using Ampure XP Beads (AGENCOURT) and dissolved in EB solution. An Agilent Technologies 2100 bioanalyzer was used to detect the concentration and length of the library. The double-strand PCR products were denatured at high temperature and amplified to single strand circle DNA in the cyclization reaction reagent. The library was amplified with phi29 (Thermo Fisher Scientific, MA, USA) to make DNA nanoball (DNB) which had more than 300 copies of a single molecule. The DNBs were load into the patterned nanoarray and pair end 150 bases reads were generated on MGISEQ2000 platform (BGI-Shenzhen, China).
FastQC (https://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc) was used to check the quality of the raw data, and AdapterRemoval (https://adapterremoval.readthedocs.io/en/latest/) was used to trim the adaptors, N bases and low-quality ends. Sequencing reads was aligned to the mouse genome (mm10) using STAR applying default settings. Then, the qualified results of alignment were used to estimate gene expression and read counts by Cufflinks (http://cole-trapnell-lab.github.io/cufflinks/) and HTSeq-count (https://htseq.readthedocs.io/en/master/), respectively.
The identification of differentially expressed genes (DEGs)
The “edgeR” package was used to identify differentially expressed genes. Prior to downstream analysis, the only genes having a count-per-million (CPM) values above 1 in at least six libraries were kept. The raw counts on each gene level were then normalized by the algorithm of trimmed mean of M (TMM) values. An adjusted p value of less than 0.05 and log2 fold change greater than 1 were used to indicate genes that were significantly differentially expressed.
Pathway enrichment analysis of DEG
Kyoto Encyclopedia of Genes and Genomes databases (KEGG) enrichment analysis and Gene Ontology (GO) enrichment analysis were performed using the R package “clusterProfiler”. KEGG terms or GO terms with adjusted p values < 0.05 were considered significantly enriched by DEGs.
Targeted metabolomic profiling
Widely targeted metabolomic profiling of plasma from each mouse was performed using an ultra-performance liquid chromatography mass spectrometry (UPLC-MS/MS) system (UPLC, ExionLC AD, https://sciex.com.cn/; MS, QTRAP System, https://sciex.com/) at Wuhan Metware Biotechnology. In total, 617 plasma metabolites comprising 15 classes were targeted. Briefly, the samples were thawed on ice, followed by vortexing for 10s. Second, 300 μL pure methanol were added to 50 μL of plasma. Then, the mixture was vortexed for 3 min and centrifuged at 12000 r/min at 4 °C for 10 min. About 200 μL supernatant were transferred to a clean centrifuge tube and centrifuged at 12000 rpm at 4 °C for 5 min, and then left in a refrigerator at −20 °C for 30 min. The tube was centrifuged at 12000 r/min at 4 °C for 3 min, and 150 μL of supernatant were used for analysis.
The identification of differential plasma metabolites
The raw values of metabolites were log10-transformed and then employed to determine the metabolites exhibiting differential abundances by orthogonal projections to latent structures discriminant analysis (OPLS-DA). Variable important in projection (VIP), which reflects both the loading weights for each component and the variability of the response explained by this component, was used for feature selection. The statistical significance between two groups were conducted using a t test. A p value of <0.05 and VIP >1 were used as the screening criteria for significant differential metabolites.
Network edge orienting network
We used the network edge orienting (NEO) network81 to test the association between bacteria intervention and metabolic markers and plasma lipids. The local-structure edge orienting (LEO) score based on the likelihood of the structural equation model was used to evaluate the oriented relationship between quantitative traits and markers. The candidate pleiotropic anchor (CPA) model was used to test single marker edge orienting and the orthogonal causal anchor (OCA) model was used to test multiple markers. The likelihood-based CPA score assessed whether the chosen model would yield a higher likelihood than the alternative models. We used a threshold of 0.8, as the software suggested, which implies that the model likelihood score of the causal model was 100.8 = 6.3-folds higher than that of the next best model. For the OCA score, we used a threshold of 0.3, as suggested, which implies that the model likelihood score of the causal model was 100.3 = 2-folds higher than that of the next best model.
A modified NEO was used to build a local structure equation model and to obtain edge-oriented scores. The orthogonal causal anchors (LEO.NB.OCA) (A → B) > 0.3 and candidate pleiotropic anchor (LEO.NB.CPA) (A → B) > 0.8 indicated the regulation direction was A to B.
Metagenomic data quality control, taxonomic abundance calculation and function profile computation
The sequencing reads were quality-controlled as described previously80 and implementation of the pipeline is available at https://github.com/jiezhuye/cOMG. Briefly, adapter trimming/filtering was automatically processed by the BGISEQ-500 sequencing platform. The raw sequences with low quality were filtered and trimmed by overall accuracy (OA) control The raw sequences were quality filtered and trimmed by with overall accuracy (OA) control strategy80 using OAs1 (-Qsys = 33, -minLen = 30, -Scut = 0.9, -Qcut = 0.8). , Then the high-quality reads were aligned to mice reference genome GRCm39 (Genebank assembly accession: GCF_000001635.27) by bowtie2 with default parameter. The taxonomic profile was computed via MetaPhlAn382 (default parameter) with the retained high-quality clean reads. Taxa with more than 10% occurrence were kept for further analysis, yielding a filtered table containing 97 species. Functional profiling was performed with HUMAnN266 with default parameter. Human metagenomic data was processed using metagenomic linkage group (MLG) method. Detailed of MLG profiles based on the 9.9M gene set was performed as described.5 Briefly, it clustered genes into sub-cluster based on that genes from same specie tended to co-vary in many samples. We then estimate the relative abundance of an MLG in all samples by using the relative abundance values of genes from this MLG.
Functional analysis of the 7α-dehydroxylation pathway
To investigate the possible link between microbial function and LCA, we constructed a Hidden Markov Chain Models (HMM) for 8 bai gene from the 7α-dehydroxylation pathway. HMM was built based on multiple sequence alignments generated by ClustalW283 with default parameter, containing the respective reference sequences and close homologs identified using HMMsearch.84 All reference gene sequences were available at https://www.pathofunctions.com. Screening for bai was performed by mapping reference sequences against downloaded genomes shown in MetaPhlAn3 profile (>10% occurrence taxa) using phmmer (v. 3.1b2); only top hits were recorded. Score extracted from phmmer was ploted against mice taxa with R package pheatmap. We used ShortBRED70 to obtain gene RPKM abundances using the above reference after removing redundancy. Function group abundance was computed as described previously.85 Briefly, we clustered genes based on their co-variance abundance across samples (Pearson correlation) using the Ward algorithm in the ‘hclust’ function in R. We got two gene clusters and we kept the complete one (that is, how many genes of the operon were present in the cluster). For statistical analysis, the genes in the selected gene clusters were summed within each group or all together for the overall analysis.
Microbiome composition analysis
Multidimensional scaling was performed at the genus level based on Aitchison distance with R (base function, cmdscale) to remove composition effect. Aitchison distance is a Euclidian distance based on center log ratio transformation of relative abundance profiles with R package compositions (clr function). PERMANOVA test was performed between genus proofing (Aitchison distance) of groups at baseline and end of the studies with R package vegan (adonis function, 9999 times permutation). Post hoc compositions were performed between treatment group and control group at FDR <0.05. Dirichlet multinomial mixtures (DMM) modeling was applied to detect microbiome development during intervention. DMM bins samples on the basis of microbial community structure.69 All samples from −2 weeks to 18weeks of intervention were included, and species profiles formed nine clusters (based on lowest Laplace approximation).
Identification of lipid associated taxa and treatment associated taxa
Simple regression between level of metabolic parameter (such as LDL, HDL, TG, CHOL) and taxa abundance were used to define ‘Lipid-associated’ taxa. Because metabolic parameters were only measured at the last time point, we also used the taxa profile at last time point.
The regression model form:
For the shifting of each taxa, we computed two metrics for the effect of treatment. One was the abundance difference between groups at last time point. Taxa were used for regression against the treatment and Apoe−/− control group was treated as reference group using model:
Growth rate of taxa. A comparison of the effects of the live bacteria group and Apoe−/− control group on growth rates was tested with a mixed effects linear model (lme function in nlme package) predicting taxa abundance from the interaction between months in the intervention and treatment, controlling for treatment, months in the intervention, and a random intercept for each mouse. Benjamini-Hochberg adjusted p values are reported.
The model:
The same model was applied to the functional level to determine pathway shifting between treatments.
Covariance between features of plasma metabolites and members of the gut microbiota
Simple regression between level of plasma metabolites (such as LCA) and taxa abundance were used to qualify the associations between metabolites and taxa. Only the last time point taxa profile was used since plasma metabolites were only measured at the last time point.
Quantification and statistical analysis
Statistical analyses were performed using Prism version 7.0 (GraphPad Software; San Diego, CA). Data are presented as mean ± SD. A two-tailed Student’s t test was used for comparison between two groups with normally distributed data, Mann-Whitney U-test was used for non-normally distributed data. A value of p < 0.05 was considered to indicate statistical significance.
Acknowledgments
This study was funded by the National Natural Science Foundation of China (No. 81872934, 81673514 [Principle investigator: Shilong Zhong]), Science and Technology Program of Guangzhou (No. 2023B03J1251 [Principle investigator: Shilong Zhong]), the Key Area Research and Development Program of Guangdong Province, China (No. 2019B020229003 [Principle investigator: Shilong Zhong]), Science and Technology Development Projects of Guangdong Province (2019A050510025 [Principle investigator: Yong Li]), Excellent Young Talent Program of GDPH (KY012021187 [Principle investigator: Xiao Xiao]). The authors are very grateful to colleagues at BGI-Shenzhen and China National Genebank (CNGB), Shenzhen, for sample collection, DNA extraction, library construction, sequencing, and discussions.
Author contributions
S.Z. was the principal investigator of this study and designed the study. Z.J. performed the data analysis and drafted the manuscript. Q.Z. and Y.Z. performed the experiment and drafted the manuscript. Q.W. performed the experiment and revised the manuscript. M.Q. performed data analysis and revised the manuscript. D.H., X.L., X.T., J.Z., Zhu Jie, W.L., X.X., S.C., Y.W., G.G., and Shufen Zheng were assisted in data acquisition and experiment. Y.L., Weihua Lai, H.Y., J.W., L.X., J.C., and T.Z. assisted in data curation and revised the manuscript. K.K. and H.J. provided resources and critically revised the manuscript. All authors reviewed and approved the final manuscript.
Declaration of interests
The authors declare no competing financial interest.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106960.
Contributor Information
Karsten Kristiansen, Email: kk@bio.ku.dk.
Huijue Jia, Email: jiahuijue@genomics.cn.
Shilong Zhong, Email: zhongsl@hotmail.com.
Supplemental information
Data and code availability
-
•
The public metagenomic shotgun-sequencing data for the ACVD cohort (Acvd, n = 218; control, n = 186) have been deposited in the European Bioinformatics Institute (EBI) database under the accession code ERP023788. The public metagenomic shotgun-sequencing data for large scale healthy cohort is available at CNSA (https://db.cngb.org/cnsa/) of (CNGB) database under the accession code CNP0000426, CNP0000289.71,72 Metagenomic sequencing data for all samples in this study “have been deposited to the CNSA (https://db.cngb.org/cnsa/) of (CNGB) database under the accession code CNP0002026. The accession numbers and DOI are listed in the key resources table.
-
•
All original code has been deposited on GitHub (https://github.com/xxx) and is available up on request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Timmis A., Townsend N., Gale C.P., Torbica A., Lettino M., Petersen S.E., Mossialos E.A., Maggioni A.P., Kazakiewicz D., May H.T., et al. European society of cardiology: cardiovascular disease statistics 2019. Eur. Heart J. 2020;41:12–85. doi: 10.1093/eurheartj/ehz859. [DOI] [PubMed] [Google Scholar]
- 2.Zhou M., Wang H., Zeng X., Yin P., Zhu J., Chen W., Li X., Wang L., Wang L., Liu Y., et al. Mortality, morbidity, and risk factors in China and its provinces, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2019;394:1145–1158. doi: 10.1016/S0140-6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakaroun R.M., Olsson L.M., Bäckhed F. The potential of tailoring the gut microbiome to prevent and treat cardiometabolic disease. Nat. Rev. Cardiol. 2022;20:217–235. doi: 10.1038/s41569-022-00771-0. [DOI] [PubMed] [Google Scholar]
- 4.Fromentin S., Forslund S.K., Chechi K., Aron-Wisnewsky J., Chakaroun R., Nielsen T., Tremaroli V., Ji B., Prifti E., Myridakis A., et al. Microbiome and metabolome features of the cardiometabolic disease spectrum. Nat. Med. 2022;28:303–314. doi: 10.1038/s41591-022-01688-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jie Z., Xia H., Zhong S.L., Feng Q., Li S., Liang S., Zhong H., Liu Z., Gao Y., Zhao H., et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017;8:845. doi: 10.1038/s41467-017-00900-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang W.H.W., Bäckhed F., Landmesser U., Hazen S.L. Intestinal microbiota in cardiovascular health and disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2019;73:2089–2105. doi: 10.1016/j.jacc.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Witkowski M., Weeks T.L., Hazen S.L. Gut microbiota and cardiovascular disease. Circ. Res. 2020;127:553–570. doi: 10.1161/CIRCRESAHA.120.316242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao X., Oduro P.K., Tong W., Wang Y., Gao X., Wang Q. Therapeutic potential of natural products against atherosclerosis: targeting on gut microbiota. Pharmacol. Res. 2021;163:105362. doi: 10.1016/j.phrs.2020.105362. [DOI] [PubMed] [Google Scholar]
- 9.Wang Z., Klipfell E., Bennett B.J., Koeth R., Levison B.S., Dugar B., Feldstein A.E., Britt E.B., Fu X., Chung Y.M., et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang W.H.W., Wang Z., Levison B.S., Koeth R.A., Britt E.B., Fu X., Wu Y., Hazen S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luu T.H., Bard J.M., Carbonnelle D., Chaillou C., Huvelin J.M., Bobin-Dubigeon C., Nazih H. Lithocholic bile acid inhibits lipogenesis and induces apoptosis in breast cancer cells. Cell. Oncol. 2018;41:13–24. doi: 10.1007/s13402-017-0353-5. [DOI] [PubMed] [Google Scholar]
- 12.Brahe L.K., Le Chatelier E., Prifti E., Pons N., Kennedy S., Hansen T., Pedersen O., Astrup A., Ehrlich S.D., Larsen L.H. Specific gut microbiota features and metabolic markers in postmenopausal women with obesity. Nutr. Diabetes. 2015;5:e159. doi: 10.1038/nutd.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasahara K., Krautkramer K.A., Org E., Romano K.A., Kerby R.L., Vivas E.I., Mehrabian M., Denu J.M., Bäckhed F., Lusis A.J., Rey F.E. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat. Microbiol. 2018;3:1461–1471. doi: 10.1038/s41564-018-0272-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou Y., Xue W., Luo G., Deng Z., Qin P., Guo R., Sun H., Xia Y., Liang S., Dai Y., et al. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2019;37:179–185. doi: 10.1038/s41587-018-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou Y., Lin X., Xue W., Tuo L., Chen M.S., Chen X.H., Sun C.H., Li F., Liu S.W., Dai Y., et al. Characterization and description of Faecalibacterium butyricigenerans sp. nov. and F. longum sp. nov., isolated from human faeces. Sci. Rep. 2021;11:11340. doi: 10.1038/s41598-021-90786-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jie Z., Liang S., Ding Q., Li F., Tang S., Wang D., Lin Y., Chen P., Cai K., Qiu X., et al. 2021. A Transomic Cohort as a Reference Point for Promoting a Healthy Human Gut Microbiome. [Google Scholar]
- 17.McNulty N.P., Wu M., Erickson A.R., Pan C., Erickson B.K., Martens E.C., Pudlo N.A., Muegge B.D., Henrissat B., Hettich R.L., Gordon J.I. Effects of diet on resource utilization by a model human gut microbiota containing Bacteroides cellulosilyticus WH2, a symbiont with an extensive glycobiome. PLoS Biol. 2013;11:e1001637. doi: 10.1371/journal.pbio.1001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ridlon J.M., Kang D.J., Hylemon P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Buffie C.G., Bucci V., Stein R.R., McKenney P.T., Ling L., Gobourne A., No D., Liu H., Kinnebrew M., Viale A., et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Aguiar Vallim T.Q., Tarling E.J., Edwards P.A. Pleiotropic roles of bile acids in metabolism. Cell Metabol. 2013;17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wahlström A., Sayin S.I., Marschall H.U., Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metabol. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Brestoff J.R., Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013;14:676–684. doi: 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo C., Xie S., Chi Z., Zhang J., Liu Y., Zhang L., Zheng M., Zhang X., Xia D., Ke Y., et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45:802–816. doi: 10.1016/j.immuni.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 24.Hillman E.T., Kozik A.J., Hooker C.A., Burnett J.L., Heo Y., Kiesel V.A., Nevins C.J., Oshiro Jordan M.K.I., Robins M.M., Thakkar R.D., et al. Comparative genomics of the genus Roseburia reveals divergent biosynthetic pathways that may influence colonic competition among species. Microb. Genom. 2020;6:mgen000399. doi: 10.1099/mgen.0.000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nemet I., Saha P.P., Gupta N., Zhu W., Romano K.A., Skye S.M., Cajka T., Mohan M.L., Li L., Wu Y., et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell. 2020;180:862–877.e22. doi: 10.1016/j.cell.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White P.J., McGarrah R.W., Herman M.A., Bain J.R., Shah S.H., Newgard C.B. Insulin action, type 2 diabetes, and branched-chain amino acids: a two-way street. Mol. Metabol. 2021;52:101261. doi: 10.1016/j.molmet.2021.101261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X., Yang R., Zhang W., Wang S., Mu H., Li H., Dong J., Chen W., Yu X., Ji F. Serum glutamate and glutamine-to-glutamate ratio are associated with coronary angiography defined coronary artery disease. Nutr. Metabol. Cardiovasc. Dis. 2022;32:186–194. doi: 10.1016/j.numecd.2021.09.021. [DOI] [PubMed] [Google Scholar]
- 28.Liu R., Hong J., Xu X., Feng Q., Zhang D., Gu Y., Shi J., Zhao S., Liu W., Wang X., et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017;23:859–868. doi: 10.1038/nm.4358. [DOI] [PubMed] [Google Scholar]
- 29.Jauhiainen R., Vangipurapu J., Laakso A., Kuulasmaa T., Kuusisto J., Laakso M. The association of 9 amino acids with cardiovascular events in Finnish men in a 12-year follow-up study. J. Clin. Endocrinol. Metab. 2021;106:3448–3454. doi: 10.1210/clinem/dgab562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shan W., Cui H., Xu Y., Xue J., Zheng L. Vol. 1. 2022. Succinate Metabolism in Cardiovascular Diseases. [DOI] [Google Scholar]
- 31.Zhang S., Liang Y., Li L., Chen Y., Wu P., Wei D. Succinate: a novel mediator to promote atherosclerotic lesion progression. DNA Cell Biol. 2022;41:285–291. doi: 10.1089/dna.2021.0345. [DOI] [PubMed] [Google Scholar]
- 32.Krishnan S., Ding Y., Saedi N., Choi M., Sridharan G.V., Sherr D.H., Yarmush M.L., Alaniz R.C., Jayaraman A., Lee K. Gut microbiota-derived tryptophan metabolites modulate inflammatory response in hepatocytes and macrophages. Cell Rep. 2018;23:1099–1111. doi: 10.1016/j.celrep.2018.03.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin X.B., Wang T., Stothard P., Corander J., Wang J., Baines J.F., Knowles S.C.L., Baltrūnaitė L., Tasseva G., Schmaltz R., et al. The evolution of ecological facilitation within mixed-species biofilms in the mouse gastrointestinal tract. ISME J. 2018;12:2770–2784. doi: 10.1038/s41396-018-0211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DiMarzio M., Rusconi B., Yennawar N.H., Eppinger M., Patterson A.D., Dudley E.G. Identification of a mouse Lactobacillus johnsonii strain with deconjugase activity against the FXR antagonist T-beta-MCA. PLoS One. 2017;12:e0183564. doi: 10.1371/journal.pone.0183564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elkins C.A., Moser S.A., Savage D.C. Genes encoding bile salt hydrolases and conjugated bile salt transporters in Lactobacillus johnsonii 100-100 and other Lactobacillus species. Microbiology (Read.) 2001;147:3403–3412. doi: 10.1099/00221287-147-12-3403. [DOI] [PubMed] [Google Scholar]
- 36.Wang M., Zhang B., Hu J., Nie S., Xiong T., Xie M. Intervention of five strains of Lactobacillus on obesity in mice induced by high-fat diet. J. Funct.Foods. 2020;72:104078. [Google Scholar]
- 37.Wang H., Ni X., Qing X., Zeng D., Luo M., Liu L., Li G., Pan K., Jing B. Live probiotic Lactobacillus johnsonii BS15 promotes growth performance and lowers fat deposition by improving lipid metabolism, intestinal development, and gut microflora in broilers. Front. Microbiol. 2017;8:1073. doi: 10.3389/fmicb.2017.01073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foley M.H., O'Flaherty S., Allen G., Rivera A.J., Stewart A.K., Barrangou R., Theriot C.M. Lactobacillus bile salt hydrolase substrate specificity governs bacterial fitness and host colonization. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2017709118. e2017709118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukunaga M., Suriki K., Kuda T., Shikano A., Toyama A., Takahashi H., Kimura B. Typical indigenous bacteria in the cecum of ddY mice fed a casein-beef tallow diet or whole-egg diet. J. Food Biochem. 2019;43:e13064. doi: 10.1111/jfbc.13064. [DOI] [PubMed] [Google Scholar]
- 40.Marion S., Desharnais L., Studer N., Dong Y., Notter M.D., Poudel S., Menin L., Janowczyk A., Hettich R.L., Hapfelmeier S., Bernier-Latmani R. Biogeography of microbial bile acid transformations along the murine gut. J. Lipid Res. 2020;61:1450–1463. doi: 10.1194/jlr.RA120001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Behary J., Amorim N., Jiang X.T., Raposo A., Gong L., McGovern E., Ibrahim R., Chu F., Stephens C., Jebeili H., et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021;12:187. doi: 10.1038/s41467-020-20422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lagkouvardos I., Pukall R., Abt B., Foesel B.U., Meier-Kolthoff J.P., Kumar N., Bresciani A., Martínez I., Just S., Ziegler C., et al. The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat. Microbiol. 2016;1:16131. doi: 10.1038/nmicrobiol.2016.131. [DOI] [PubMed] [Google Scholar]
- 43.Natividad J.M., Lamas B., Pham H.P., Michel M.L., Rainteau D., Bridonneau C., da Costa G., van Hylckama Vlieg J., Sovran B., Chamignon C., et al. Bilophila wadsworthia aggravates high fat diet induced metabolic dysfunctions in mice. Nat. Commun. 2018;9:2802. doi: 10.1038/s41467-018-05249-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Devkota S., Wang Y., Musch M.W., Leone V., Fehlner-Peach H., Nadimpalli A., Antonopoulos D.A., Jabri B., Chang E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peck S.C., Denger K., Burrichter A., Irwin S.M., Balskus E.P., Schleheck D. A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia. Proc. Natl. Acad. Sci. USA. 2019;116:3171–3176. doi: 10.1073/pnas.1815661116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamura M., Hoshi C., Kobori M., Takahashi S., Tomita J., Nishimura M., Nishihira J. Quercetin metabolism by fecal microbiota from healthy elderly human subjects. PLoS One. 2017;12:e0188271. doi: 10.1371/journal.pone.0188271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hosoya S., Kutsuna S., Shiojiri D., Tamura S., Isaka E., Wakimoto Y., Nomoto H., Ohmagari N. Leuconostoc lactis and Staphylococcus nepalensis Bacteremia, Japan. Emerg. Infect. Dis. 2020;26:2283–2285. doi: 10.3201/eid2609.191123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D'Alessandro-Gabazza C.N., Kobayashi T., Yasuma T., Toda M., Kim H., Fujimoto H., Hataji O., Takeshita A., Nishihama K., Okano T., et al. A Staphylococcus pro-apoptotic peptide induces acute exacerbation of pulmonary fibrosis. Nat. Commun. 2020;11:1539. doi: 10.1038/s41467-020-15344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L.Y., Yu W.C., Huang S.H., Lin M.L., Chen T.L., Fung C.P., Liu C.Y. Successful treatment of Aerococcus viridans endocarditis in a patient allergic to penicillin. J. Microbiol. Immunol. Infect. 2012;45:158–160. doi: 10.1016/j.jmii.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 50.Mohan B., Zaman K., Anand N., Taneja N. Aerococcus viridans: a rare pathogen causing urinary tract infection. J. Clin. Diagn. Res. 2017;11:DR01–DR03. doi: 10.7860/JCDR/2017/23997.9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin L., Shi X., Yang J., Zhao Y., Xue L., Xu L., Cai J. Gut microbes in cardiovascular diseases and their potential therapeutic applications. Protein Cell. 2021;12:346–359. doi: 10.1007/s13238-020-00785-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.From the American Association of Neurological Surgeons AANS, American Society of Neuroradiology ASNR, Cardiovascular and Interventional Radiology Society of Europe CIRSE, Canadian Interventional Radiology Association CIRA, Congress of Neurological Surgeons CNS, European Society of Minimally Invasive Neurological Therapy ESMINT, European Society of Neuroradiology ESNR, European Stroke Organization ESO, Society for Cardiovascular Angiography and Interventions SCAI, Society of Interventional Radiology SIR, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int. J. Stroke. 2018;13:612–632. doi: 10.1177/1747493018778713. [DOI] [PubMed] [Google Scholar]
- 53.Saeed A., Feofanova E.V., Yu B., Sun W., Virani S.S., Nambi V., Coresh J., Guild C.S., Boerwinkle E., Ballantyne C.M., Hoogeveen R.C. Remnant-like particle cholesterol, low-density lipoprotein triglycerides, and incident cardiovascular disease. J. Am. Coll. Cardiol. 2018;72:156–169. doi: 10.1016/j.jacc.2018.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deprince A., Haas J.T., Staels B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metabol. 2020;42:101092. doi: 10.1016/j.molmet.2020.101092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munukka E., Rintala A., Toivonen R., Nylund M., Yang B., Takanen A., Hänninen A., Vuopio J., Huovinen P., Jalkanen S., Pekkala S. Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. ISME J. 2017;11:1667–1679. doi: 10.1038/ismej.2017.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martínez-Sena T., Soluyanova P., Guzmán C., Valdivielso J.M., Castell J.V., Jover R. The vitamin D receptor regulates glycerolipid and phospholipid metabolism in human hepatocytes. Biomolecules. 2020;10:493. doi: 10.3390/biom10030493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang K., Liao M., Zhou N., Bao L., Ma K., Zheng Z., Wang Y., Liu C., Wang W., Wang J., et al. Parabacteroides distasonis alleviates obesity and metabolic dysfunctions via production of succinate and secondary bile acids. Cell Rep. 2019;26:222–235.e5. doi: 10.1016/j.celrep.2018.12.028. [DOI] [PubMed] [Google Scholar]
- 58.Duan Y., Zhong Y., Xiao H., Zheng C., Song B., Wang W., Guo Q., Li Y., Han H., Gao J., et al. Gut microbiota mediates the protective effects of dietary beta-hydroxy-beta-methylbutyrate (HMB) against obesity induced by high-fat diets. FASEB J. 2019;33:10019–10033. doi: 10.1096/fj.201900665RR. [DOI] [PubMed] [Google Scholar]
- 59.Nissen S., Sharp R.L., Panton L., Vukovich M., Trappe S., Fuller J.C., Jr. beta-hydroxy-beta-methylbutyrate (HMB) supplementation in humans is safe and may decrease cardiovascular risk factors. J. Nutr. 2000;130:1937–1945. doi: 10.1093/jn/130.8.1937. [DOI] [PubMed] [Google Scholar]
- 60.Bogiatzi C., Gloor G., Allen-Vercoe E., Reid G., Wong R.G., Urquhart B.L., Dinculescu V., Ruetz K.N., Velenosi T.J., Pignanelli M., Spence J.D. Metabolic products of the intestinal microbiome and extremes of atherosclerosis. Atherosclerosis. 2018;273:91–97. doi: 10.1016/j.atherosclerosis.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 61.Poesen R., Claes K., Evenepoel P., de Loor H., Augustijns P., Kuypers D., Meijers B. Microbiota-derived phenylacetylglutamine associates with overall mortality and cardiovascular disease in patients with CKD. J. Am. Soc. Nephrol. 2016;27:3479–3487. doi: 10.1681/ASN.2015121302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lam V., Su J., Hsu A., Gross G.J., Salzman N.H., Baker J.E. Intestinal microbial metabolites are linked to severity of myocardial infarction in rats. PLoS One. 2016;11:e0160840. doi: 10.1371/journal.pone.0160840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jie Z., Liu T., Peishan C., Mo H., Liju S., Xin T., Xiaohuan S., Fangming Y., Zhipeng L., Xing L., et al. Over 50,000 metagenomically assembled draft genomes for the human oral microbiome reveal new taxa. Genomics Proteomics Bioinformatics. 2022;20:246–259. doi: 10.1016/j.gpb.2021.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Francesco B., Lauren J.M., Aitor B.M., Leonard D., Francesco A., Sagun M., Ana M., Paolo M., Matthias S., Andrew M.T., et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife. 2021;10:e65088. doi: 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Franzosa E.A., McIver L.J., Rahnavard G., Thompson L.R., Schirmer M., Weingart G., Lipson K.S., Knight R., Caporaso J.G., Segata N., Huttenhower C. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods. 2018;15:962–968. doi: 10.1038/s41592-018-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wickham H. ggplot2. Springer Cham; 2016. [DOI] [Google Scholar]
- 68.Anderson M.J. Wiley; 2014. Wiley StatsRef: Statistics Reference Online; pp. 1–15. [Google Scholar]
- 69.Holmes I., Harris K., Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One. 2012;7:e30126. doi: 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaminski J., Gibson M.K., Franzosa E.A., Segata N., Dantas G., Huttenhower C. High-Specificity targeted functional profiling in microbial communities with ShortBRED. PLoS Comput. Biol. 2015;11:e1004557. doi: 10.1371/journal.pcbi.1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo X., Chen F., Gao F., Li L., Liu K., You L., Hua C., Yang F., Liu W., Peng C., et al. CNSA: a data repository for archiving omics data. Database. 2020;2020 doi: 10.1093/database/baaa055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen F.Z., You L.J., Yang F., Wang L.N., Guo X.Q., Gao F., Hua C., Tan C., Fang L., Shan R.Q., et al. CNGBdb: China national GeneBank DataBase. Yi Chuan. 2020;42:799–809. doi: 10.16288/j.yczz.20-080. [DOI] [PubMed] [Google Scholar]
- 73.Qin J., Li Y., Cai Z., Li S., Zhu J., Zhang F., Liang S., Zhang W., Guan Y., Shen D., et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 74.Li J., Jia H., Cai X., Zhong H., Feng Q., Sunagawa S., Arumugam M., Kultima J.R., Prifti E., Nielsen T., et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 75.He Q., Gao Y., Jie Z., Yu X., Laursen J.M., Xiao L., Li Y., Li L., Zhang F., Feng Q., et al. Two distinct metacommunities characterize the gut microbiota in Crohn's disease patients. GigaScience. 2017;6:1–11. doi: 10.1093/gigascience/gix050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang X., Zhang D., Jia H., Feng Q., Wang D., Liang D., Wu X., Li J., Tang L., Li Y., et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015;21:895–905. doi: 10.1038/nm.3914. [DOI] [PubMed] [Google Scholar]
- 77.Feng Q., Liang S., Jia H., Stadlmayr A., Tang L., Lan Z., Zhang D., Xia H., Xu X., Jie Z., et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015;6:6528. doi: 10.1038/ncomms7528. [DOI] [PubMed] [Google Scholar]
- 78.Jie Z., Yu X., Liu Y., Sun L., Chen P., Ding Q., Gao Y., Zhang X., Yu M., Liu Y., et al. The baseline gut microbiota directs dieting-induced weight loss trajectories. Gastroenterology. 2021;160:2029–2042.e16. doi: 10.1053/j.gastro.2021.01.029. [DOI] [PubMed] [Google Scholar]
- 79.Han M., Hao L., Lin Y., Li F., Wang J., Yang H., Xiao L., Kristiansen K., Jia H., Li J. A novel affordable reagent for room temperature storage and transport of fecal samples for metagenomic analyses. Microbiome. 2018;6:43. doi: 10.1186/s40168-018-0429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fang C., Zhong H., Lin Y., Chen B., Han M., Ren H., Lu H., Luber J.M., Xia M., Li W., et al. Assessment of the cPAS-based BGISEQ-500 platform for metagenomic sequencing. GigaScience. 2018;7:1–8. doi: 10.1093/gigascience/gix133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aten J.E., Fuller T.F., Lusis A.J., Horvath S. Using genetic markers to orient the edges in quantitative trait networks: the NEO software. BMC Syst. Biol. 2008;2:34. doi: 10.1186/1752-0509-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beghini F., McIver L.J., Blanco-Míguez A., Dubois L., Asnicar F., Maharjan S., Mailyan A., Manghi P., Scholz M., Thomas A.M., et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife. 2021;10:e65088. doi: 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 84.Vital M., Rud T., Rath S., Pieper D.H., Schlüter D. Diversity of bacteria exhibiting bile acid-inducible 7alpha-dehydroxylation genes in the human gut. Comput. Struct. Biotechnol. J. 2019;17:1016–1019. doi: 10.1016/j.csbj.2019.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wirbel J., Pyl P.T., Kartal E., Zych K., Kashani A., Milanese A., Fleck J.S., Voigt A.Y., Palleja A., Ponnudurai R., et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019;25:679–689. doi: 10.1038/s41591-019-0406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The public metagenomic shotgun-sequencing data for the ACVD cohort (Acvd, n = 218; control, n = 186) have been deposited in the European Bioinformatics Institute (EBI) database under the accession code ERP023788. The public metagenomic shotgun-sequencing data for large scale healthy cohort is available at CNSA (https://db.cngb.org/cnsa/) of (CNGB) database under the accession code CNP0000426, CNP0000289.71,72 Metagenomic sequencing data for all samples in this study “have been deposited to the CNSA (https://db.cngb.org/cnsa/) of (CNGB) database under the accession code CNP0002026. The accession numbers and DOI are listed in the key resources table.
-
•
All original code has been deposited on GitHub (https://github.com/xxx) and is available up on request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.