

Abstract
Heterobifunctional PROTAC degraders are gaining attention as a differentiated therapeutic modality with the potential for oral dosing in the clinic. Belonging to the beyond Rule of Five domain of physicochemical property space, we have sought to understand the determinants of oral absorption for this class of molecules for the rapid development of novel oral agents. We have collected a large data set from PROTAC molecules that have been dosed orally and intravenously in rats to estimate the fraction absorbed from oral dosing. Through this estimation, effects from differential hepatic clearance are normalized, allowing for a better assessment of the absorption. We demonstrate that rats are less permissive to PROTAC absorption than mice. The physicochemical properties of the molecules are then evaluated once compounds are rank-ordered by the fraction absorbed. We derive suggested design constraints on physicochemical properties for PROTAC molecules that are associated with higher probability of being orally absorbed.
Introduction
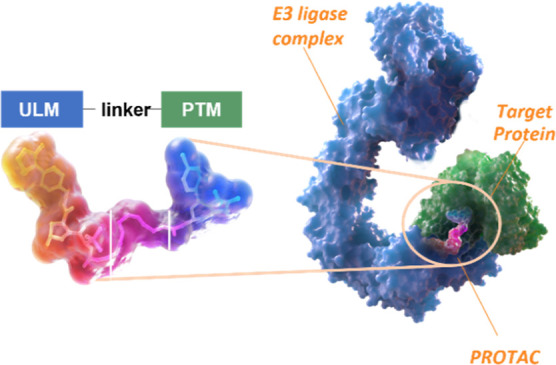
PROteolysis TArgeting Chimera (PROTAC) molecules are heterobifunctional compounds consisting of a protein targeting moiety (PTM) joined by a linker to a ubiquitin ligase moiety (ULM) (Scheme 1). They function by bringing a target protein in physical proximity to an ubiquitin ligase, leading to ubiquitin transfer and subsequent degradation of the target protein by the ubiquitin proteasome system. Over the past decade, there has been explosive growth in interrogating the potential of PROTAC protein degraders as new therapeutic agents, with interest from both academia and the pharmaceutical industry.1,2 Because PROTACs possess inherent functional activity and degrade a target protein rather than simply occupying a binding site, they are well suited for the treatment of diseases where achieving functional activity with small molecules is challenging (classically undruggable targets, proteins with scaffolding functions, or protein aggregates), where mechanisms of drug resistance are driven by overexpression or mutation of a target protein, or where isoform selectivity is desirable. Collectively, we refer to these use cases as the “Tenets of PROTAC targets”.1
Scheme 1. Structures of a PROTAC and Induced Trimer Complex between Target Protein:PROTAC:E3 Ligase.
Perhaps the greatest barrier to harnessing the potential of the PROTAC platform to deliver new drug molecules to patients is achieving suitable oral absorption. By their nature, PROTAC molecules occupy a space beyond that traditionally associated with oral absorption, as enumerated in the Rule of Five (Ro5)3,4 by Lipinski et al., or other analyses that have followed.5 More recently, there has been a renewed interest in understanding the behavior of molecules beyond the borders of Ro5 in extended Rule of Five (eRo5) or beyond Rule of Five (bRo5) space. A series of seminal publications by Kihlberg et al. in particular have laid out determinants of these spaces, where the criteria shift from “probable to be orally absorbed” to “possible to be orally absorbed”.6−8
These challenges notwithstanding, we and others have reported that achievement of oral bioavailability with PROTACs is possible.9 We draw particular attention to two independent analyses conducted by AbbVie10 on bRo5 compounds generally and AstraZeneca11,12 on PROTACs specifically based on proprietary data sets. In the present work, we lay out a comprehensive analysis of PROTAC oral absorption vs common physicochemical properties, discuss where PROTACs sit in the eRo5/bRo5 space, and draw several critical conclusions for the successful design of oral PROTACs.
Oral bioavailability (F) is defined in eq 1, wherein fa is the fraction absorbed, fg is the fraction gastrically available (accounting for gut metabolism by CYP enzymes or the microbiome), and fh is the fraction hepatically available. For an orally absorbed drug which enters systemic circulation through the hepatic portal vein and is hepatically cleared, the first-pass effect dictates that fh = 1 – (Cl/Qh), where Cl is the intravenous clearance and Qh is the hepatic blood flow. Experimentally, oral bioavailability (Fobs) is determined as the ratio of the oral area under the concentration–time curve (AUCpo) to the intravenous area under the concentration–time curve (AUCiv) normalized for the oral and intravenous doses.
| 1 |
Although it is difficult to directly determine the value of fa or fg experimentally, the product of these two components can be estimated. In a theoretical scenario where a compound is completely orally absorbed and there is no contributing gut metabolism, fa × fg = 1, and the theoretical maximum possible bioavailability (Fmax) can be determined as in eq 2.
| 2 |
Thus, the product of fa × fg can be approximated as the ratio of the experimentally observed bioavailability and the theoretical maximum bioavailability as shown in eq 3. This equation contains an uncertainty because it assumes that the measured iv clearance is entirely hepatic. A lack of correction for extrahepatic clearance mechanisms, if present, may lead to a misrepresentation of the true fa × fg.
| 3 |
We argue herein that many prior analyses relating physicochemical properties of molecules to oral bioavailability are fundamentally flawed because they inappropriately use Fobs as a surrogate for the true property of interest, fa × fg. To illustrate this point, consider the hypothetical case of two compounds shown in Table 1. Both compounds have an observed bioavailability of 5%, but compound A has high hepatic clearance (90% Qh), whereas compound B has low hepatic clearance (10% Qh). Because Fmax for compound A is only 10%, fa × fg = 0.50, and this compound is relatively well absorbed despite its apparent low bioavailability. In contrast, for compound B, Fmax is 90%, fa × fg = 0.06, and this compound is relatively poorly absorbed. In short, the readily calculated apparent value of fa × fg is a better measure of absorption than Fobs because it appropriately factors in and normalizes for the hepatic clearance of the compound.
Table 1. Hypothetical Compounds and Their PK Parameters, Illustrating Potential Disconnects between Oral Bioavailability and Fraction Absorbed.
| property | compound A | compound B |
|---|---|---|
| Cl (% Qh) | 90 | 10 |
| F (%) | 5 | 5 |
| fa × fg | 0.50 | 0.06 |
| PK diagnosis | high clearance | low absorption |
For our analysis of PROTAC molecules, we considered that to have desirable human pharmacokinetics, a compound should have a minimum oral bioavailability of 20% and low hepatic clearance (<25% Qh), leading to a minimal fa × fg = 0.20/(1 – 0.25) ≈ 0.26.5,11,13 We therefore set fa × fg = 0.25 as a threshold of absorption for PROTACs to be minimally drug-like. It should be noted, however, that this cutoff is arbitrarily chosen based on practical experience, and there is little agreement on metrics constituting drug-likeness.4,14 A key assumption in our analysis is that the degree of absorption in preclinical species, specifically rats, will be predictive of human absorption. In contrast, it has been remarked upon many times in the literature that oral bioavailability in individual preclinical species (or aggregates thereof) is not well correlated with human bioavailability.15 This lack of correlation may be at least partially related to cross-species differences in hepatic clearance, which is better accounted for in the fa × fg term. However, species differences in fg driven by factors such as the complement of gut metabolic enzymes mean that fa × fg is an improved, but imperfect, measure of absorption.16
Results
To analyze the Arvinas PK database, we selected rats as the preclinical species of interest because we have a large (n = 1806), diverse data set spanning multiple programs and chemotypes with the necessary measured intravenous and oral PK data. Rats are also a species that represents a well-established preclinical toxicology model for regulatory agencies. Attainment of exposure multiples above that required for efficacy in rats is thus a critical component for advancement to clinical trials. Furthermore, we have observed when comparing rodent species that rats are less permissive to oral absorption of PROTACs than mice (vide infra). Collectively, attaining oral absorption in rats represents a higher barrier to compound progression than mice.
Although arguably similar analyses in higher species (e.g., dog or monkey) might be more useful or predictive of human outcomes, these data sets were more limited in scope and therefore deprioritized for analysis of physicochemical property trends. Furthermore, because compound progression to higher species PK studies is typically gated on PK performance in rodents, there is an inherent selection bias in the higher species PK data set.
For curation, all 1806 PROTAC compounds with both rat iv and po PK data were considered and fa × fg calculated according to eq 3. This calculation assumes only hepatic clearance and ignores potential contributions from extrahepatic clearance mechanisms, which are difficult to determine systematically for large compound sets. Any apparent fa × fg values > 1 were set equal to 1 as the theoretical maximum. 75% of our compound collection displayed low or moderate clearance in rats (Figure S1), indicating that clearance is not a major limiting factor in attaining oral bioavailability. A separate analysis of rat clearance vs physicochemical descriptors (Figure S2) did not identify any noteworthy trends. A total of 233 compounds (∼13% of the data set, leaving n = 1573 compounds in the final analysis) with rat Cl ≥ Qh (55 mL/min/kg)17,18 were excluded because the theoretical Fmax = 0 if clearance is entirely hepatic, and thus, no meaningful assessment of fa × fg can be made. Indeed, 79% of the 233 high clearance compounds displayed F < 5%, as expected within a typical experimental error in PK studies; of the remainder, assessing fa × fg is complicated by the potential contribution of extrahepatic clearance mechanisms. It should also be noted that the error in the apparent value of fa × fg becomes large as Cl approaches Qh and F approaches zero, so such compounds should be treated with appropriate caution. Nonetheless, their inclusion in the analysis does not skew the overall interpretation. Where available (n = 1016), the same computations were done on the same compound set using mouse PK data using a standard Qh value of 90 mL/min/kg.17 Although a standard formulation was not used throughout the data set, our general practice is to dose solution formulations rather than suspensions, irrespective of the formulation chosen. Of the 1573 remaining compounds analyzed, there are 2395 unique rat po PK experiments, reflecting multiple experiments on the same compound assessing dose, schedule, vehicle, and the like. 2102/2395 (88%) of those experiments were conducted at a dose ≤ 30 mg/kg, reflecting normal “screening PK” conditions. For simplicity, compounds with >1 unique experiment (n = 352, 22% of the total) are represented in the figures by their average bioavailability. While this simplification may have the effect of depressing the reported success rates in attaining suitable fa × fg, particularly when dose escalations are included, it does not change the overall conclusions of the property analysis.
We began by comparing the calculated value of fa × fg in rats to that in mice. As shown in Figure 1, fa × fg values in rats are lower than those calculated from PK experiments in mice in 861/1016 (85%) of cases where comparator data in both rats and mice was available. All further analyses thus focused on rat data, as it is ultimately the most restrictive to compound advancement.
Figure 1.

Comparison of the fractions absorbed of PROTAC molecules in rats and mice using the fa × fg term.
The results of the analysis are summarized in Figure 2. A clear cutoff was noted for HBD, with HBD ≤ 4 being an apparent upper limit for achieving fa × fg ≥ 0.25. Upon closer inspection of structures, however, nearly all (97%) of the three HBD and all the four HBD compounds with fa × fg ≥ 0.25 had one or two internally satisfied HBD, respectively. The cutoff is thus better stated as unsatisfied HBD ≤ 2. This observation potentially affords the chemist a greater degree of design flexibility with respect to HBD count, provided that an internal acceptor can be provided for additional HBD beyond the first two. A similar trend held for HBA, with ∼15 being an upper limit. Two compounds with HBA ≥ 15 achieved the threshold, but the limited sample size in this space (n = 13) may not be suitably powered to draw a strong conclusion. Previously, Veber et al. defined cutoffs for rat oral bioavailability based on RB (≤ 10) and TPSA (≤ 140 Å2).5 We observed upper limits of 14 RB and TPSA 200 Å2 for fa × fg ≥ 0.25, which represent an expanded range vs the Veber rules. We found MW and c log P/D to have significantly expanded ranges vs the Ro5, with fa × fg ≥ 0.25 achievable up to a MW of 950, a c log P range from 1 to 7, and a c log D range from 0 to 6. The upper limit on NAr was 5, although only a small fraction (9%) of compounds at NAr = 5 were well absorbed and a more realistic cutoff may be NAr = 4. Last, we calculated the previously reported AB-MPS score,10 which is a composite of c log D, NAr, and RB (Figure 3), and found the upper limit to be 20. The AB-MPS score was not more predictive of risk for low oral absorption than any of its individual composite parameters alone.
Figure 2.

Estimated fraction absorbed in rats as a function of the physicochemical properties of the PROTAC molecules in the Arvinas database. The x-axis of discrete variables has been jittered for clarity.
Figure 3.
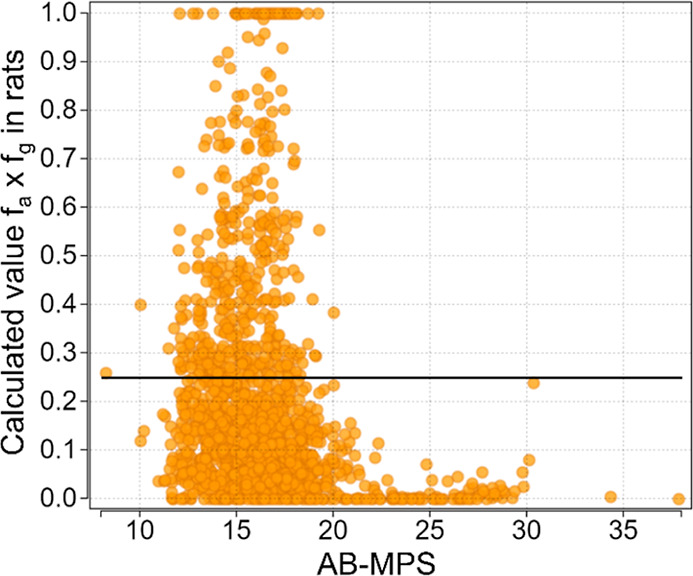
Estimated fraction absorbed in rats as a function of AB-MPS score of the PROTAC molecules in the Arvinas database.
The property space of small-molecule drugs has been classified by Kihlberg into three categories: traditional Ro5, which hews to the Lipinski rules; extended Rule of Five (eRo5) space, which represents the natural tail of the Ro5 distribution; and beyond Rule of Five (bRo5) space, where other factors (e.g., hydrophobic collapse, internally satisfied HBD-HBA pairs, etc.) come into play to enable oral absorption.6 In Table 2, we show where the oral absorption cutoffs sit for PROTACs with respect to these empirically defined regions of chemical space.
Table 2. Upper Limits of Physicochemical Properties for Oral Absorption for Ro5 + Veber, bRo5, and PROTACs.
| property | Ro5/Veber | bRo5 | PROTAC |
|---|---|---|---|
| HBD | 5 | 6 | 2 unsatisfied |
| HBA | 10 | 15 | 15 |
| RB | 10 | 20 | 14 |
| MW | 500 | 1000 | 950 |
| TPSA | 140 | 250 | 200 |
| NAr | ND | ND | 5 |
| c log P | 5 | 7.5 | 7 |
| c log D | ND | ND | 6 |
In general, we have observed that acceptable oral absorption in rats is possible in a window of HBA, RB, TPSA, MW, and c log P that is expanded vs traditional Ro5 guidance, but only MW, HBA, and c log P approach the bRo5 limit. This observation may be in part because these computed physicochemical descriptors are not completely independent variables, e.g., there is a positive correlation between MW and TPSA, MW and RB, etc. It has also been noted that there can be significant discrepancies between calculated and experimental log P/D values.19 Limits may be softer for properties like c log P and TPSA due to the chameleonic nature of larger molecules,20 where they are able to adapt their conformation, and therefore effective physicochemical properties, in response to their environment. In contrast, however, there are hard limits on HBD which hew closely to the Ro5 space. HBD have previously been noted as the main restriction to oral absorption even within the traditional Ro5.4 This latter constraint, therefore, becomes the most restrictive to compound design.
The practical consequences of these constraints can be seen when considering the additive physicochemical properties in the context of a PROTAC molecule. We calculated the properties for representative ULMs for cereblon and VHL (Figure 4), as well as the average properties for a linker derived from the 20 most represented linkers in our PROTAC collection. We then derive a “budget” remaining for a PTM for an orally bioavailable PROTAC degrader (Figure 5) and compare that budget to the properties of an average oral drug molecule (HBD 1.6, HBA 5.3, TPSA 74, RB 4.5, and MW 343).21
Figure 4.

Representative ULMs used in physicochemical property budget analysis.
Figure 5.

Analysis of the physicochemical property budget with respect to the design of orally bioavailable PROTACs based on targeting (A) VHL or (B) CRBN as E3 with representative ULMs depicted in Figure 4.
Discussion and Conclusions
Several conclusions are immediate from this analysis. First, only a judiciously chosen ULM (e.g., a lenalidomide-derived compound targeting CRBN) that is relatively compact will be compatible with rat oral absorption. Barring major changes, the opportunity for rat oral absorption sufficient to enable advancement toward preclinical development of PROTACs derived from larger ULMs, e.g., the prototypical VHL ligand described in Figure 4 and other reported ubiquitin ligase binders (IAP, MDM2, etc.), is limited.11 In our data set, few VHL PROTACs demonstrated mouse or rat fa × fg > 0.25.
Second, even with an efficient ULM, significant constraints are still in operation on the PTM. Particularly in the era of molecular property inflation,21−23 finding suitable PTM starting points with 1 unsatisfied HBD, 9 HBA, 120 TPSA, 10 RB, and 488 MW can be a challenging exercise. These guidelines should influence the selection of PTMs for oral PROTAC design. Furthermore, chemists working in the PROTAC space would be well served to commit part of their medicinal chemistry program to making PTM modifications to bring these parts of a PROTAC molecule in line with the properties needed for oral absorption. Historical PTMs may be suitable starting points, but in many cases, they need further modification. Our analysis also assumes an optimized, more druglike linker, and the derived property budget will be further shrunk with unoptimized PEG- or alkyl-type linkers.
Last, we have shown that PROTAC oral absorption in rats is generally more restricted than in mice. There is a dearth of published pharmacokinetic data for PROTACs in species other than mice, but mice may provide an incomplete picture of PROTAC absorption properties. Indeed, a few studies have reported useful levels of oral exposure in mice for challenging classes of PROTACs such as those based on recruitment of the VHL E3 ligase.24,25 Based on the relative permissiveness of mice toward oral absorption, such studies merit caution in their interpretation. Greater integration of pharmacokinetic data across multiple species is needed to support eventual clinical progression.
A potential limitation of this analysis is that the threshold for acceptable rat oral absorption—and by inference human oral absorption—is empirical. Oral absorption must ultimately be viewed through the lens of pharmacodynamics: “acceptable” is sufficient human in vivo free drug exposure at the site of action relative to potency on the target. Indeed, the original formulation of the Rule of 5 is derived in such fashion rather than prescribing an absorption or bioavailability floor.3 The lack of larger numbers of clinically advanced PROTAC agents precludes such an analysis for PROTACs, but the strong bias toward CRBN-derived heterobifunctional degraders in clinical trials9 and the alignment of our physicochemical property limits with a recent IQ consortium survey26 supports the trends we report here. It is possible that a sufficiently potent PROTAC could have clinical exposure suitable to drive efficacy despite having rat fa × fg < 0.25. Nonetheless, such compounds may encounter other challenges to their preclinical and clinical progression, such as achieving efficacious exposure multiples in preclinical toxicology studies, which require dose escalation beyond the projected efficacious dose.
Looking to the future, several needs are apparent to expand the potential of PROTACs as a platform technology. First, there is a strong need for more screening work, both to identify new, compact ULMs, but also new PTMs. Indeed, an entirely new definition of a PTM may be required. Modern screening campaigns to discover PTMs that have functional activity, coupled with more challenging target space such as protein–protein interactions, have contributed significantly to molecular property inflation over the last two decades. An advantage of PROTAC degraders, however, is that the downstream functional pharmacology is now event driven rather than occupancy driven, and because of this change, PTMs no longer need to have function—they simply need to bind their target, in some cases with only micromolar affinity.27 The heterobifunctional PROTAC mechanism engages the ubiquitin proteasome system and supplies the missing function. The built-in functionality of the PROTAC mechanism invites reinvestigation of old screening hits that were discarded because they were silent target binders that lacked functional activity. In addition, the physicochemical properties of compounds in a PTM screening library also merit reconsideration, given that PTMs derived from screening are now no longer the end point structures, but rather beginning points of a PROTAC medicinal chemistry campaign. Second, there is an ongoing need for more predictive in vitro ADME assays for PROTACs. It has been noted as a general trend across multiple companies,12,26 as well as at Arvinas, that such assays as configured for more traditional small molecules are often poorly predictive of in vivo PROTAC behavior. Third, greater attention to the potential of parenterally administered PROTACs is warranted. While the early clinical stage degraders have been biased toward oral administration, there may be cases where an intravenous or subcutaneous agent may be appropriate based on the target, its resynthesis rate, and the needs of the patient population.28
The final and greatest need is for more detailed analysis of the determinants of successful oral absorption for prospective use. The present analysis is retrospective and furthermore is exclusive rather than inclusive. Our work lays out the boundaries of the PROTAC oral absorption space, beyond which the probability of failure is high. Comparing an early year of our data set to the most recent one, awareness of these limits coupled with other workflow enhancements has enriched our success in obtaining viable oral absorption in rats by ∼6-fold. Further enrichment of this success rate is desirable for the continued growth of the oral PROTAC platform.
Experimental Section
ChemAxon JWS version 21.9 was used to calculate HBD, HBA, RB, NAr, MW, TPSA, c log P, and c log D. Data analysis and visualization were performed in D360 version 22.5.5 (Certara).
Acknowledgments
We thank Jordan Clark for assistance with the property calculations and Bradley Pearce, Song Yang, Larry Snyder, Andy Crew, and the Arvinas medicinal chemistry team for helpful discussions.
Glossary
Abbreviations
- c log D
calculated log octanol–water partition coefficient at pH 7.4
- c log P
calculated log octanol–water partition coefficient
- CRBN
cereblon
- F
bioavailability
- HBA
hydrogen bond acceptors
- HBD
hydrogen bond donors
- MW
molecular weight
- NAr
number of aromatic rings
- PK
pharmacokinetics
- PTM
protein targeting moiety
- RB
rotatable bonds
- TPSA
topological polar surface area
- ULM
ubiquitin ligase moiety
- VHL
von Hippel–Lindau
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00740.
Author Contributions
The manuscript was written through contributions of all authors. The authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Research was conducted at Arvinas Operations, Inc. where the authors are employees and shareholders.
Supplementary Material
References
- Békés M.; Langley D. R.; Crews C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discovery 2022, 21, 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; Crews C. M. PROTACs: past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. 10.1039/d2cs00193d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. 10.1016/s0169-409x(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Hartung I. V.; Huck B. R.; Crespo A. Rules were made to be broken. Nat. Rev. Chem. 2023, 7, 3–4. 10.1038/s41570-022-00451-0. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Doak B. C.; Over B.; Giordanetto F.; Kihlberg J. Oral druggable space beyond the rule of 5: insights from the drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Doak B. C.; Zheng J.; Dobritzsch D.; Kihlberg J. How beyond Rule of 5 drugs and clinical candidates bind to their targets. J. Med. Chem. 2016, 59, 2312–2327. 10.1021/acs.jmedchem.5b01286. [DOI] [PubMed] [Google Scholar]
- Poongavanam V.; Doak B. C.; Kihlberg J. Opportunities and guidelines for discovery of orally absorbed drugs in beyond rule of 5 space. Curr. Opin. Chem. Biol. 2018, 44, 23–29. 10.1016/j.cbpa.2018.05.010. [DOI] [PubMed] [Google Scholar]
- Chirnomas D.; Hornberger K. R.; Crews C. M. Protein degraders enter the clinic a new approach to cancer therapy. Nat. Rev. Clin. Oncol. 2023, 20, 265–278. 10.1038/s41571-023-00736-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGoey D. A.; Chen H.-J.; Cox P. B.; Wendt M. D. Beyond the rule of 5: lessons learned from AbbVie’s drugs and compound collection. J. Med. Chem. 2018, 61, 2636–2651. 10.1021/acs.jmedchem.7b00717. [DOI] [PubMed] [Google Scholar]
- Edmondson S. D.; Yang B.; Fallan C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- Pike A.; Williamson B.; Harlfinger S.; Martin S.; McGinnity D. F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov. Today 2020, 25, 1793–1800. 10.1016/j.drudis.2020.07.013. [DOI] [PubMed] [Google Scholar]
- AstraZeneca (ref (12)) uses a 30% F cutoff but their analysis is based on mouse PK, vs our analysis based on rat PK. We have determined mouse to be a more permissive species than rat for PROTAC oral absorption (Figure 1).
- Shultz M. D. Two decades under the influence of the rule of five and the changing properties of approved oral drugs. J. Med. Chem. 2019, 62, 1701–1714. 10.1021/acs.jmedchem.8b00686. [DOI] [PubMed] [Google Scholar]
- Musther H.; Olivares-Morales A.; Hatley O. J.; Liu B.; Rostami Hodjegan A. Animal versus human oral drug bioavailability: do they correlate?. Eur. J. Pharm. Sci. 2014, 57, 280–291. 10.1016/j.ejps.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau E.; Petersson C.; Dolgos H.; Peters S. A. A comparative evaluation of models to predict human intestinal metabolism from nonclinical data. Biopharm. Drug Dispos. 2017, 38, 163–186. 10.1002/bdd.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B.; Morris T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- A more recent analysis has suggested a higher value for hepatic blood flow in rats:Gibson C. R.; Gleason A.; Messina E. Measurement of total liver blood flow in intact anesthetized rats using ultrasound imaging. Pharmacol. Res. Perspect. 2021, 9, e00731 10.1002/prp2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannhold R.; Poda G. I.; Ostermann C.; Tetko I. V. Calculation of molecular lipophilicity: State-of-the-art and comparison of log P methonds on more than 96,000 compounds. J. Pharm. Sci. 2009, 98, 861–893. 10.1002/jps.21494. [DOI] [PubMed] [Google Scholar]
- Rossi Sebastiano M.; Doak B. C.; Backlund M.; Poongavanam V.; Over B.; Ermondi G.; Caron G.; Matsson P.; Kihlberg J. Impact of dynamically exposed polarity on permeability and solubility of chameleonic drugs beyond the Rule of 5. J. Med. Chem. 2018, 61, 4189–4202. 10.1021/acs.jmedchem.8b00347. [DOI] [PubMed] [Google Scholar]
- Leeson P. D. Molecular inflation, attrition, and the rule of five. Adv. Drug Deliv. Rev. 2016, 101, 22–33. 10.1016/j.addr.2016.01.018. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Young R. J. Molecular property design: does everyone get it?. ACS Med. Chem. Lett. 2015, 6, 722–725. 10.1021/acsmedchemlett.5b00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann M. M. Molecular obesity, potency and other addictions in drug discovery. MedChemComm 2011, 2, 349–355. 10.1039/c1md00017a. [DOI] [Google Scholar]
- Kofink C.; Trainor N.; Mair B.; Wöhrle S.; Wurm M.; Mischerikow N.; Roy M. J.; Bader G.; Greb P.; Garavel G.; Diers E.; McLennan R.; Whitworth C.; Vetma V.; Rumpel K.; Scharnweber M.; Fuchs J. E.; Gerstberger T.; Cui Y.; Gremel G.; Chetta P.; Hopf S.; Budano N.; Rinnenthal J.; Gmaschitz G.; Mayer M.; Koegl M.; Ciulli A.; Weinstabl H.; et al. A selective and orally bioavailable VHL-recruiting PROTAC achieves SMARCA2 degradation in vivo. Nat. Commun. 2022, 13, 5969. 10.1038/s41467-022-33430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Kalogeropulou A. F.; Domingos S.; Makukhin N.; Nirujogi R. S.; Singh F.; Shpiro N.; Saalfrank A.; Sammler E.; Ganley I. G.; Moreira R.; Alessi D. R.; Ciulli A. Discovery of XL01126: A potent, fast, cooperative, selective, orally bioavailable, and blood-brain barrier penetrant PROTAC degraders of leucine-rich repeat kinase 2. J. Am. Chem. Soc. 2022, 144, 16930–16952. 10.1021/jacs.2c05499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volak L. P.; Duevel H. M.; Humphreys S.; Nettleton D.; Phipps C.; Pike A.; Rynn C.; Scott-Stevens P.; Zhang D.; Zientek M. Industry perspective on the pharmacokinetic and ADME characterization of heterobifunctional protein degraders. Drug Metab. Dispos. 2023, 51, DMD-AR-2022-001154. 10.1124/dmd.122.001154. [DOI] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018, 25, 78–87.e5. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien Laramy M. N.; Luthra S.; Brown M. F.; Bartlett D. W. Delivering on the promise of protein degraders. Nat. Rev. Drug Discovery 2023, 22, 410–427. 10.1038/s41573-023-00652-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.