

Abstract

Grp94, an ER-localized molecular chaperone, is required for the folding and activation of many membrane and secretory proteins. Client activation by Grp94 is mediated by nucleotide and conformational changes. In this work, we aim to understand how microscopic changes from nucleotide hydrolysis can potentiate large-scale conformational changes of Grp94. We performed all-atom molecular dynamics simulations on the ATP-hydrolysis competent state of the Grp94 dimer in four different nucleotide bound states. We found that Grp94 was the most rigid when ATP was bound. ATP hydrolysis or nucleotide removal enhanced mobility of the N-terminal domain and ATP lid, resulting in suppression of interdomain communication. In an asymmetric conformation with one hydrolyzed nucleotide, we identified a more compact state, similar to experimental observations. We also identified a potential regulatory role of the flexible linker, as it formed electrostatic interactions with the Grp94 M-domain helix near the region where BiP is known to bind. These studies were complemented with normal-mode analysis of an elastic network model to investigate Grp94’s large-scale conformational changes. SPM analysis identified residues that are important in signaling conformational change, many of which have known functional relevance in ATP coordination and catalysis, client binding, and BiP binding. Our findings suggest that ATP hydrolysis in Grp94 alters allosteric wiring and facilitates conformational changes.
1. Introduction
Hsp90s are highly conserved ATP-dependent molecular chaperones important for the maturation and stabilization of proteins, including hormone and immune receptors, transcription factors, and kinases.1,2 Based on interactions with this array of substrates, termed client proteins, perturbations in Hsp90 expression and function have consistently been linked to diseases such as cancers, Alzheimer’s and Parkinson’s diseases, and diabetes, rendering it a useful therapeutic target for the management of many detrimental conditions.3−5 Hsp90s are present in most organisms, from bacteria to eukaryotes, with isoforms localized into different cellular compartments. In mammals, Hsp90α and Hsp90β are located in the cytosol, tumor necrosis factor receptor-associated protein-1 (TRAP1) resides in the mitochondria, and Grp94 is found in the endoplasmic reticulum (ER).1,3 Hsp90s share common structural features, including a nucleotide binding domain (NTD) that binds ATP, a middle domain (MD) that presents residues important for ATP hydrolysis and provides an extended surface for binding to clients and cochaperones, and a C-terminal domain (CTD) that acts as the constitutive dimerization site that is also involved in client binding.6−8 Hsp90s also share a common mechanism that couples ATP induced conformational rearrangements with protein folding and remodeling.7 Despite their sequence, structural, and functional similarities, each Hsp90 homologue is specific in the type of client protein they chaperone.3 Additional structural features distinguish Hsp90s found in different organisms or organelles. Eukaryotic Hsp90s in the cytosol and ER possess a flexible charged linker of variable length between the NTD and MD, and a C-terminal extension following the CTD (Figure 1A). Organelle localized Hsp90s, as well as some cytosolic Hsp90s in higher eukaryotes, also possess an N-terminal extension referred to as the “strap” that is absent in yeast and bacteria.1,6,9 Additional regulation of the Hsp90 conformational and functional cycle is provided by client binding, post-translational modifications, environmental factors, and cochaperones.8,10−12 These “extra” features and factors that regulate Hsp90 activity contribute to the uniqueness of the different Hsp90s in interacting with and activating specific clients. These differences open an avenue for selectively targeting individual Hsp90s, an opportunity that is especially relevant in certain disease conditions when pan-inhibition of all Hsp90s is not desired.
Figure 1.

Grp94 structure. (A) The domain organization of each Grp94 protomer. The ER signal sequence (SS), pre-N domain, and ER retention signal are missing from the structures used in the simulations. (B) The structure of the Grp94 dimer in the ATP-hydrolysis competent conformation, as obtained from the Protein Databank as PDB ID 5ULS.21 This structure is used in the all-atom molecular dynamics simulations. The AMP-PNP from the resolved structure was replaced with ATP, ADP-Pi, or removed completely. (C) The nucleotide binding site and interactions with nucleotide are highlighted. The structures were rendered in PyMOL.
Grp94 is the ER-localized Hsp90 that promotes the proper folding and activation of multiple membrane and secretory proteins, including integrins, Toll-like receptors (TLRs), insulin-like growth factors (IGFs), Her2, and Wnt coreceptor LRP6.3,13,14 Importantly, many Grp94 client proteins are involved in cell growth and maturation pathways and are usually exploited by cancerous cells for survival. Grp94 also interacts with proinsulin and myocilin, implicating it in diseases such as Type 2 diabetes and open angle glaucoma.15−18 Therefore, exploiting the structural and functional differences between Grp94 and other Hsp90s represents a promising approach to target Grp94-specific related diseases.
Grp94 shares a similar ATP-dependent conformational cycle as other Hsp90s that is required for client folding and activation.19−21 Grp94 has been captured in a variety of conformations ranging from an extended open conformation with dimerization at only the CTD to a closed ATP-bound state characterized by additional dimerization at the NTDs.22−26 Like other Hsp90s, nucleotide binding and hydrolysis by Grp94 are weakly coupled to large-scale conformational changes.24,27−29 Recent studies demonstrate that in the presence of ATP, Grp94’s conformational sampling is biased toward more compact closed states.30 The ATP-bound closed conformation that is competent for ATP hydrolysis (Figure 1B) is characterized by ATP lid closure, rotation of the N-M domain to bring catalytic residues from the M-domain close to the ATP binding site (Figure 1C), and transient dimerization of the NTDs.
Certain unique features distinguish Grp94 from its homologues. Grp94 has very low ATP hydrolysis and dimer closure rates compared to its homologues. The barrier to dimer closure is provided in part by the presence of a regulatory pre-N domain in Grp94, which suppresses the rates of ATP hydrolysis.21,31 Additionally, Grp94 has an insertion of five amino acids (QEDGQ) in the ATP lid20,32 that results in an additional hydrophobic pocket specific to Grp94.
A key component in the Grp94 functional cycle that remains largely unexplored is how conformational changes are directly coupled to nucleotide binding and hydrolysis. Allosteric communication may play a critical role in transducing local information from nucleotide binding and hydrolysis to distant sites in multidomain proteins like Grp94.27,33−39 Even in the absence of structural changes, allosteric communication can still influence protein function.40 Physics-based approaches in conjunction with experimental techniques41−43 have been used to identify allosteric communication that is required for protein function. Despite the wealth of information on Grp94, molecular details of the effect of nucleotide hydrolysis on the active site, signal transduction, and dynamics are still elusive. A number of computational studies on Hsp90s have been successful in revealing internal dynamics and communication networks based on nucleotide, cochaperones, and client protein binding.44−57 While prior computational research on Hsp90 has yielded valuable insights into protein dynamics, earlier studies on Grp94 focused on the relaxed structure45,50 that represents the posthydrolysis state. This conformation is nearly identical when either ADP or no nucleotide are bound.19,21 In this study, we used multiscale dynamics to probe the effect of nucleotide hydrolysis and the allosteric wiring within Grp94’s closed, catalytically active conformation. The insights obtained could play an important role in guiding the discovery of allosteric inhibitors.
2. Materials and Methods
2.1. Molecular Dynamics Simulations
One microsecond molecular dynamics simulations were performed for three symmetric Grp94 conformations with either ATP, ADP-Pi, or no nucleotide (apo) bound to both protomers and for one asymmetric Grp94 conformation with one ATP and one ADP-Pi bound. The Grp94 dimer in the closed ATP hydrolysis competent conformation was obtained from PDB ID 5ULS (biological assembly).21 Due to the low rates of ATP hydrolysis in Grp94, the pre-N region of the N domain is commonly truncated in experiments to accelerate the kinetics of ATP hydrolysis.19,21,30 Therefore, we also used this truncated construct, removing residues 48–86 from the resolved structure, in an attempt to accelerate dynamics. The missing regions of a single protomer were modeled using I-TASSER.58−60 The dimer was reconstituted in PyMOL61 by aligning two identical models of each protomer with the biological assembly to produce a full-length homodimer. This approach was used to minimize any potential asymmetry within the protomers of the dimer. The overall RMSD of the modeled full-length dimer with the crystallized dimer was 0.17 Å.
Each unique nucleotide-bound system began with the same Grp94 protein conformation, as the only differences among the systems are in the nucleotide placed in the nucleotide binding pocket. The AMP-PNP structure that was cocrystallized with 5ULS was replaced with ATP from PDB ID1TC0.32 ADP + Pi was introduced into the binding pocket by breaking the ATP γ-phosphate bond to mimic the structure resulting immediately after ATP hydrolysis. The apo structure was derived by removing the ligand from the modeled structure. The unique nucleotide bound structures were then used for molecular dynamics simulations with NAMD 2.1362 using the CHARMM36 force field63 and the explicit TIP3P water model.62 The solution builder module in CHARMM-GUI was used for the preparation and generation of files for the simulations.64−66 The protein–ligand complex was solvated for each system in a rectangular water box with a minimum of 10 Å distance to the edge of the box with periodic boundary conditions in place. Sodium and chloride ions were added to achieve charge neutrality at a physiological concentration of 0.15 M. Then, the system was minimized using the conjugate gradient algorithm for 1,000 steps to remove atomic clashes and optimize atomic coordinates. Before the production run, each of the simulations were equilibrated for 50 ps at 300 K using the NVT ensemble. Finally, a production run was carried out using the NPT ensemble at a temperature (300 K) and pressure (1 atm) controlled by Langevin dynamics and the Langevin piston methods, respectively. The integration time step of the simulations was set to 2.0 fs, and the SHAKE algorithm was used to constrain all chemical bonds involving hydrogen atoms and heavy atoms.67 Electrostatic interactions were calculated with the smooth particle mesh Ewald method.68 The Lennard-Jones potential cutoff was defined at a distance of 12 Å with a switching distance of 10 Å. The simulations were run locally on Miami University’s Redhawk High Performance Computing Cluster or at the Ohio Supercomputing Center.69
2.2. Simulation Analysis Metrics
RMSD
| 1 |
where x, y, z are the coordinates of atom i and the reference coordinates were obtained from the starting structure of the simulations.
RMSF
| 2 |
where xi are coordinates of atom i and ⟨xi⟩ is the average position of residue i.
Dynamic Cross-Correlation
The dynamic cross correlation (DCC) was used to investigate domain motions.70 DCC between two residues i and j is given by
| 3 |
where Δqi = qi – ⟨qi⟩.
Conformational Angle
The conformational angle (θ) is calculated as the dihedral angle between two planes. The planes are defined by the centers of mass of the NTD and CTD of protomer A and the centers of mass of the CTD and NTD of protomer B. The CTD-CTD distance does not significantly change throughout the simulation, making it a good reference point to assess the conformational angle.
Principal Component Analysis
Principal component analysis (PCA) was calculated using ProDy71 and visualized using the NMWiz 1.0 plugin of VMD.72 The principal modes were calculated by diagonalizing the covariance matrix of the 3N fluctuations obtained from the atomic coordinates to produce the eigenvectors.
Perturbation Response Scanning
The Perturbation response scanning analysis (PRS) was calculated using ProDy.71 PRS determines the sensitivity and effectiveness of each residue in transmitting allosteric signals. This method is based on linear response theory73 in which the proteins are represented as network models and are perturbed to evaluate how one residue affects the dynamics of others.
Mutational Analysis
Mutational analysis of Grp94 was carried out by BeAtMuSiC.74−76 This is a coarse grained knowledge-based approach that relies on statistical potentials describing pairwise inter-residue distances, backbone torsion angles, and solvent accessibility. The effect of mutation is evaluated by changes in the stability of the complex and changes in the strength of the interfacial residues interaction.74,76 Free energy changes are computed as ΔΔG = ΔGmutant – ΔGWT. Values of ΔΔG > 0 are destabilizing, while values of ΔΔG < 0 are stabilizing. The calculation was carried out on structures at 500, 750, and 1000 ns, and the values were averaged to represent the equilibrated conformation.
2.3. Elastic Network Model
Using the low frequency modes predicted by elastic network models (ENM) has been shown to successfully describe global motions of proteins and complexes, including structural transitions that connect two allosteric states.77−82 We have modeled Grp94 as an elastic network composed of N nodes where N is the number of amino acids in the structure.71 The full-length ATP structure generated for the all-atom simulations was used as the starting structure for these calculations. The ADP-bound conformation of Grp94 was modeled similarly, beginning with atomic coordinates from PDB ID 2O1U.19 Each system was minimized for 50 steps of steepest descent minimization and 50 steps of Adopted Basis Newton–Raphson minimization and then briefly equilibrated in implicit solvent for 0.2 ps. The first 86 residues and the flexible loops (residues 287–328) were removed. Each system was reduced to a single node at the α-carbon position of each amino acid residue; the nodes that are within a cutoff distance Rc = 9 Å in the PDB structure, are connected via harmonic potential with the energy function:
| 4 |
where γ is the spring constant that defines the energy scale, dij is the dynamic distance between residues i and j, and dij0 is the corresponding PDB distance. The dynamics of the system are obtained by calculating the normal modes of the mass-spring system with potential energies given by eq 4. The normal mode calculation yields a set of 3N-dimensional eigenvectors, qM and corresponding eigenvalues ωM for each mode M. The cutoff distance (Rc = 9 Å) was chosen using a comparison of the normal mode-based calculated B-factors to the B-factors reported in PDB structures, where available.83
2.4. Measured Quantities from Elastic Network Model Calculations
Structural Overlap
The overlap function is used to describe how closely a single mode matches the allosteric change of dynamic proteins with multiple conformational states. The calculation for the structural transition is given by:
| 5 |
where M is the mode index, Δri is the difference in the locations of the ith amino acid α-carbon atom in the two structures that correspond to the starting and end conformations of a structure, and qiM are the corresponding eigenvectors. The sums are over all nodes and thus include 3N terms. Based on the definition in eq 5, 0 ≤ IM ≤ 1. The closer the value is to 1, the more accurately a given mode describes the structural transition between the two states.
To quantify the pairwise correlations of amino acid vibrations and highlight domain motions, we calculate the covariance matrix as
| 6 |
where the sums are over the modes, M. Since −1 ≤ Cij ≤ 1, the regions where Cij values are close to +1 correspond to concerted vibrations, negative Cij values indicate motions that are in opposite directions.
The relative displacement of a node i in mode M is calculated from the normalized eigenvectors qiM as
| 7 |
where 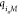 denotes the displacement of the site i in the u direction.
denotes the displacement of the site i in the u direction.
Structural Perturbation Method (SPM)
The structural perturbation method (SPM) assesses the dynamic role of individual amino acids in a structural transition84,85 and quantifies how a point mutation alters the allosteric dynamics of an entire protein. We calculate the response to a mutation at the site i as a perturbation:
| 8 |
where δγ is the perturbed spring constant. While the sum only includes amino acids within the cutoff Rc of the mutation site, the resulting changes in the eigenvectors encompass the entire elastic network. Thus, the greater the response δωiM, the more dynamically significant a specific residue is to a given mode. High δωiM nodes trace a network of residues that can be considered an allostery wiring diagram for a transition. We highlight the nodes that have a top 5% of δωiM values.
3. Results
3.1. Ligand Binding Affects the Stability of Grp94
The objective of this study was to determine the microscopic details of Grp94’s internal dynamics and how nucleotide affects Grp94’s catalytic conformation. All atom molecular dynamics simulations enable the investigation of how subtle changes in fluctuations and local structure can result in long-range allosteric signaling. To this end, we ran 1 μs simulations using the ATP-hydrolysis competent state of Grp94 in the presence of explicit solvent. Three symmetric conformations of Grp94s closed state were studied, including the pre-hydrolysis state containing 2 ATP, the post-hydrolysis state containing 2 ADP-Pi, and the apo state with no nucleotide. We considered the apo state, because the conformation of Grp94 in the absence of nucleotide or with ADP bound is nearly identical. In addition, one asymmetric Grp94 conformation was studied, containing one ATP and one ADP-Pi. This state was designed to mimic the sequential hydrolysis mechanism of mitochondrial TRAP186 and the asymmetric ATP/ADP conformation of Grp94 that has been observed experimentally.30
We began by measuring the structural differences between Grp94’s initial conformation to each position throughout the simulation by calculating the root mean square deviation (RMSD). Smaller RMSD values correspond to more stable protein structures. We calculated the RMSD of the backbone atoms for each ligand-bound structure with respect to the dimer (Figure 2A, Figure S1 (blue trace)). While the backbone deviations of the full protein appear high, Grp94 contains a 41 residue unstructured charged linker between the N- and M- domains. Fluctuations in this region contributed to the RMSD. By comparison, omitting the charged linker in the RMSD calculation resulted in increased stability of Grp94 in all nucleotide bound conditions (Figure 2B, Figure S1 (green)). The distribution of the RMSD values in the protein lacking the charged linker appeared to be the most stable for the ATP state, with broader distributions shifted toward higher RMSD values for the other structures (Figure 2B). Overall, the systems were stable after 100 ns, suggesting they were well equilibrated (Figure S1), which was confirmed by block averaging analysis (Figure S2). In contrast, the ATP/ADP-Pi system appeared to be the least stable. When the RMSD was separated into individual chains lacking the charged linker (Figure S3), all systems showed stability, suggesting the changes in this asymmetric state can be attributed to the quaternary structure. This observation is discussed in more detail in a later section.
Figure 2.

Probability distributions of the Root Mean Square Deviations of the Grp94 catalytically active conformation in the presence of different nucleotides. The probability distributions are shown from the time traces (Figure S1) for the (A) protein backbone, (B) protein backbone lacking the charged linker region, (C) N-terminal domain, (D) middle domain, and (E) C-terminal domain.
The contribution of individual Grp94 domains to the RMSD was also assessed, including the N-, M-, and C-domains, in the presence of different ligands. The largest difference was observed in the N-domain, which exhibited the highest deviations in the absence of nucleotide (Figure 2C, Figure S1 (yellow)). In contrast, the N-domain of the ATP bound Grp94 conformation exhibited the most stability, with the highest probability of RMSD centered around 5 Å. Differences in NTD interaction with nucleotide were observed for different Grp94 configurations. ATP bound protomers maintained an average of 5 hydrogen bonds, which was reduced to an average of 2–3 hydrogen bonds in ADP-Pi containing protomers (Figure S4A–C). In ATP bound protomers, R448 (78–99%) occupancy, N194 (77–86% occupancy), G198 (43–83% occupancy), and D149 (61–65% occupancy) were the most likely to form hydrogen bonds with nucleotide. Once ATP is hydrolyzed, the hydrogen bonds between the γ-phosphate and V197, G198, N194, and R448 were lost, while new bonds were formed. In protomers harboring ADP-Pi, residues E103 (64–98% occupancy) and N107 (60–69% occupancy) form hydrogen bonds with the ADP. E103, D149, and R448 have been previously identified as catalytic residues and are highly conserved throughout the Hsp90 family.19,87 Since the N-terminal domain harbors the nucleotide binding site, conformational changes within this domain are expected. In comparison, the Grp94 M-domain (Figure 2D, Figure S1 (gray)) and the C-domain (Figure 2E, Figure S1 (purple)) exhibited more stability compared to the N-domain. The fluctuations in the different nucleotide-bound states overlapped and suggested no major differences, except for the asymmetric ATP/ADP-Pi structure. In agreement with this result, analysis of the salt bridges indicated that most interactions were formed within the MD and CTD; all states showed similar occupancy, suggesting that only subtle changes occur beyond the NTD (Figure S5 and Table S1).
Strikingly, in all of the RMSD calculations, the asymmetric structure with one ATP and one ADP-Pi had a very broadened distribution and sampled a variety of RMSD values (Figure 2 and Figure S1). These results show that the asymmetric structure is less stable than symmetric structures, with differences in RMSD in all three domains. During the simulation of the asymmetric state with ATP/ADP-Pi bound, the lid covering the ATP pocket (residues 169–194) was observed to open, exposing the bound nucleotide in the protomer containing ADP-Pi. The opening of the lid allowed the escape of inorganic phosphate from the nucleotide binding site. Once the lid no longer covered the nucleotide binding pocket, it became very mobile and moved toward helix 1 (residues 78–93), adopting a conformation similar to the previously observed extended open lid configuration.32Figure 3 shows snapshots of the ATP lid movement from the beginning (red), middle (gray), and end (blue) of the simulations, while the graphs below indicate the lid RMSD for each protomer. In the asymmetric structure (Figure 3B), we were unable to observe the fully open state of the lid in the ADP-bound protomer, characterized by restricted mobility and interactions with helix 1, since the first nine residues of helix 1 were truncated in our starting structures. The fluctuations of the lid in ATP bound protomers in either symmetric or asymmetric structures are relatively small and the lid remains in the ”closed” conformation21 (Figure 3A (both protomers) and 3B (protomer A)). In the apo and symmetric ADP-Pi bound conformations, the lid exhibits some fluctuations and populates open intermediate conformations (Figure 3C,D). The lid flexibility was also apparent in the root mean square fluctuation (RMSF), which calculates the movement of individual residues in each ligand-bound structure (Figure 4). The ATP lid was the most mobile in the ADP-Pi bound protomer of the asymmetric ATP/ADP-Pi bound state. Throughout the simulations, hydrogen bonds with occupancy of >40% involving the ATP lid and NTD were not observed. Weak ATP lid interactions with the NTD could enable higher flexibility to sample extended-open lid conformations observed for Grp94.19,32,88 Nonetheless, our simulations captured significant fluctuations of the lid region and the local conformational transitions that occurred as a result of ATP hydrolysis in one protomer.
Figure 3.

Movement of the ATP lid in simulations. The dynamic position of the ATP lid is shown for (A) one ATP protomer, (B) the ADP-Pi protomer from the asymmetric ATP/ADP-Pi conformation, (C) one ADP-Pi protomer, and (D) one apo protomer. Snapshots are captured throughout the simulation with dark red indicating the position at the start of the simulation and dark blue for the position at the end of the simulation. For clarity, only the NTD of one protomer is shown in the rendering. The NTD is colored in gold and shown in transparency. The helical structure in green indicates the fragment of helix 1 present in the initial Grp94 structure, corresponding to residues 87–93. The nucleotide is shown in yellow van der Waals representation. Structures were rendered in VMD. The time series of the ATP lid RMSD for each protomer are shown below each image. Movement of the lid in protomer A is indicated in blue and in orange for protomer B.
Figure 4.

Root mean square fluctuations of Grp94 domains with different nucleotides bound. Each protomer of the dimer is represented by a blue trace (protomer A) or an orange trace (protomer B). The background is colored according to the domain; NTD (light yellow), charged linker (olive), MD (gray), and CTD (purple). RMSF is calculated as described in the Materials and Methods and shown for the (A) symmetric ATP, (B) asymmetric ATP/ADP-Pi, (C) symmetric ADP-Pi, and (D) symmetric apo (no nucleotide) states. Due to the truncation of the pre-N domain through residue 86 in all structures, amino acid numbering begins at residue 87.
The sequence of the ATP lid is moderately conserved (Figure S6). Additionally, the lid of Grp94 has a 5-residue insertion compared to cytosolic Hsp90s. Isolated structures of the Grp94 NTD have indicated that the position of the lid transitions between open and extended open conformations upon the addition of nucleotide.32,89 In the hydrolysis competent state, the ATP lid is stabilized by NTD dimerization and adopts the closed conformation,21 similar to that observed for yeast Hsp90.26 The orientation of the lid may translate into regulation mechanisms for client interactions. The ATP lid of Hsp90 was proposed to adopt an open conformation when interacting with clients and promote client release during lid closure.11 Previous molecular dynamics simulations of the N-terminal domain of Hsp90 showed that the lid of Hsp90 acts as a nucleotide sensitive conformational switch that is influenced by binding partners; structural rigidity was observed with ATP binding and increased flexibility with inhibitor binding.47 Together, these results highlight the importance of ATP lid dynamics in regulating Hsp90 function.
3.2. The Charged Linker Interacts with All Grp94 Domains
The largest fluctuations in all simulations were observed in the charged linker region (Figure 4), as the linker fluctuated freely in solution in all of the systems. The position of the charged linker is poorly resolved in Hsp90 structures or absent from the Hsp90 constructs used for crystallization due to its disorder and mobility. Therefore, we modeled the linker as an unstructured region (Materials and Methods). In our simulations, we observed electrostatic interactions between the negatively charged linker and positively charged regions in the N-, M-, and C-domains (Figure S7). Interestingly, in the simulations of the ATP/ADP-Pi and the ADP-Pi conformations, the linker of an ADP-bound protomer interacted with the positively charged residues located on the M-domain helix of the opposite protomer (Figure 5) near the region that binds to BiP.90 Importantly, this region is in the same vicinity as the lumenal channel in Grp94 that is implicated in client binding. The charged linker was mostly disordered as it was trapped in this region, and this interaction may mimic Grp94 interactions with unfolded or misfolded client proteins. Electrostatic interactions between Hsp90 and client proteins have been observed for both bacterial and human cytosolic Hsp90s and contribute to Hsp90-client affinity.10,91 Once these interactions were formed during the simulation, the linker remained tightly bound for the remainder of the simulation. The β-strands N8 and N9 (residues 279–285 and 332–337) that flank the linker retained their structure in all simulations. Throughout the ATP/ADP-Pi and ADP-Pi simulations, the linker transiently formed secondary β-strand conformations that extended from N8 and N9 strands (Figure 5A, left protomer) or that were formed within multiple regions of the linker (Figure 5B, left protomer). In Grp94, the charged linker is shorter compared to other Hsp90s (Figure S6). Additionally, it is moderately conserved with a divided E/D rich region responsible for binding 1–2 Ca2+ ions.13 When the Ca2+ sites are occupied, Grp94’s peptide binding activity is enhanced.92 The effect of Ca 2+ in protein remodeling has yet to be demonstrated. In contrast, deletions of the charged linker of eukaryotic Hsp90 resulted in the loss of cochaperone interactions and deficiencies in client remodeling.93 The charged linker of Grp94 is often replaced with a four glycine linker without compromising Grp94 function.21,94,95 Taken together, our results show a potential regulatory mechanism for the charged linker based on nucleotide state. This regulation may be coupled to the Ca2+ dependent activity of Grp94.
Figure 5.

Snapshots of electrostatic interactions between the charged linker and the middle domain. Interactions are observed in (A) the asymmetric ATP/ADP-Pi conformation and in (B) the symmetric ADP-Pi conformation. Grp94 is colored in gray, the charged linker is highlighted in magenta (residues 287 to 327), the β-strands flanking the linker are highlighted in orange, and the M-domain helix (residues 455 to 475) is colored in green. In the inset pictures, the positively charged residues on the M-domain helix and the NTD are shown in van der Waals representation in green and cyan, respectively, while negatively charged residues on the charged linker are shown in van der Waals representation in magenta. Residues forming the lumenal channel are shown in yellow. To the right of the rendering are the interaction energies between the linker and M-domain helix as a function of time. Structures were rendered in VMD.
3.3. Asymmetry Is Observed within the Grp94 Dimer, Independent of the Nucleotide Bound State
One surprising observation was asymmetry in motions when comparing Grp94 protomers, even in the symmetric nucleotide bound states. The resolved crystal structure was captured in a highly symmetric state, with a Cα backbone RMSD of 0.6 Å.21 In preparing the protein for simulations, identical copies of the full-length protomer were used. Comparison of the RMSF analysis for each Grp94 protomer in each nucleotide bound state hinted that the behavior within each protomer may differ since each protomer had a unique fluctuation profile (Figure 4). While the largest differences in mobility were observed in the asymmetric state with one ATP and one ADP-Pi, mobility differences were also observed when comparing protomers in the symmetric ATP, ADP-Pi, and apo states. The ligand RMSD values also demonstrated similar trends when comparing ligand fluctuations within each protomer (Figure S8). To further characterize the effects of nucleotide and asymmetry, we investigated the domain motions by calculating the dynamic cross-correlation as shown in Figure 6. In this calculation, we removed the flexible linker due to its inherent mobility. Residues 1–619 correspond to the first protomer and residues 620–1238 correspond to the second protomer, with the individual domain boundaries indicated at the top and to the right of each matrix. In all of the simulations, the domains moved as rigid bodies, with high positive correlation within each domain (N, M, and C). Within each nucleotide bound state, different correlations were observed within protomer A and protomer B, with the largest differences in the N-M domain of the ATP/ADP-Pi state. In addition to the high positive correlation within individual domains, long-range correlations were also observed. A strong positive correlation was observed between the N- and C-domains within a protomer as well as between the M-domains of opposite protomers. These results are in agreement with observations of long-range networking between the N- and C-domains from computational studies investigating allosteric networking in Hsp90s45,48,55,96,97 and with experimental studies showing that sequences within the C-terminus of Hsp90 regulate ATP hydrolysis at the NTD.98,99
Figure 6.

Dynamic cross-correlation analysis of Grp94. The correlation of motions between atoms are quantified for all residue pairs in the (A) symmetric ATP, (B) asymmetric ATP/ADP-Pi, (C) symmetric ADP-Pi, and (D) symmetric apo configurations. Blue indicates correlated motions, red indicates anticorrelated motions, and white indicates no correlation of motions. The motions for each nucleotide-bound structure are decomposed into the first two principal components, labeled PC1 and PC2. Arrows depicting the motions are drawn onto the structures to the right of the matrix in cyan for PC1 and PC2, and the contributions of each principal component are listed below the structures.
ATP hydrolysis alters the allosteric wiring within Grp94, with the most pronounced effects observed at the NTD (Figure 6, residues ≃1–200 and ≃600–800). The motions of the NTD within ATP bound protomers were strongly correlated, as highlighted by blue coloring. In protomers containing ADP-Pi or no nucleotide, this correlation was reduced and the pattern was more dispersed. Interestingly, in the asymmetric state, the signaling within the N-terminal domain of the ADP-Pi bound protomer was significantly reduced, beyond what was observed in the symmetric ADP-Pi conformation. Long range signaling was also altered upon nucleotide hydrolysis. The highest positive correlation between the N- and C- terminal domains within protomers was observed in the symmetric ATP-bound state and was weakened in all the other nucleotide bound states. This suggests that the sensing of the terminal phosphate by both protomers results in signaling to the C-terminal domain for allostery. In the asymmetric structure with one ATP and one ADP-Pi bound, this communication between the N-terminal domain and C-terminal domain, even in the ATP bound protomer, was lost. Similarly, the positive M-domain correlation between protomers was present in the ATP state, weakened in the ADP-Pi state, and lost when nucleotide was removed. We observed that in the symmetric ATP-bound state, the N-terminal domain acted independently of the M- domain, which moved as a rigid body with the C-domain. Analysis of the relaxed conformation of Grp94 has indicated similar motions and domain correlations, suggesting that the local wiring within Grp94 may be independent of conformation.50
Together, these results indicate that the most correlated motions occur in the symmetric ATP conformation. ATP hydrolysis or removal of nucleotide reduces the inter- and intraprotomer correlation, highlighting how local dynamics are altered by nucleotide. As a result, the global dynamics of the protein were altered and the domain and protomer motions were decoupled. In simulations of the human cytosolic Hsp90α, more relaxed structures, characterized by decoupled NTD-CTD fluctuations, were also observed for ADP-Pi and apo states compared to the ATP conformation.55 Differences in internal dynamics were also observed for TRAP1, dependent on the bound nucleotides;100 though, the MD-CTD interface was the most affected. In contrast, E. coli Hsp90 and yeast Hsp82 show more symmetric fluctuations in some studies46,48 and more asymmetric fluctuations in other studies.50 The uncoupling of domain motions could be a structurally encoded mechanism to allow Hsp90s to more freely transition toward open configurations.
To gain further insight into the internal protein dynamics, we used principal component analysis (PCA) to analyze the motions observed in molecular dynamics simulations. This technique reduces the dimensionality of the motions to describe protein dynamics that correspond to the largest variance. This requires diagonalization of the covariance matrix to provide eigenvectors or principal components. It is important to note that for our system, PCA analysis characterizes the local fluctuations around the catalytically competent closed state, but does not describe motions for global conformational changes toward more relaxed conformations. The fluctuations from the first two principal components are mapped onto Grp94 structures to the right of the corresponding covariance matrix, and the associated variance is listed below each structure (Figure 6 and Methods). PCA1 accounted for ≥30% of the motions in all nucleotide bound states and the combination of PCA1 and PCA2 accounted for ≃50%. Supplementary Movies 1–8 show the motions for the first two principal components for the ATP, ATP/ADP-Pi, ADP-Pi, and apo states. Calculation of the first two principal components showed local fluctuations induced by ATP hydrolysis were largely localized to the N-domain of Grp94 that harbors the nucleotide. This is in agreement with the broadened RMSD distribution observed for the NTDs (Figure 2 and Figure S1). The B-factors for the lid mobility, described for each principal component, were also in agreement with the RMSF and RMSD calculations (Figure 2, Figure 4, and Figure S1), indicating minimal fluctuations in the ATP conformation and higher mobility in the nucleotide altered conformations (Figure S9). The B-factor data suggested that in all of the conformations, similar regions contributed to the dominant fluctuations. Enhanced motions were observed in the ATP lid and the lumenal channel compared to the rest of the protein. After ATP hydrolysis, the N-domain became more mobile, as characterized by larger B-factors. This enhanced mobility in the N-terminal domain may lead to additional rearrangements that are required for large scale conformational changes toward the relaxed conformation. Our results suggest asymmetrical fluctuations within Grp94 protomers.
To determine how allosteric signaling was networked in the various structures, we applied perturbation-response scanning (PRS) analysis (Figure 7 and Table S2). This computational tool has been successful in identifying allosterically connected residues of large proteins.33,55,101 In all of the Grp94 structures, residues that bind nucleotide and residues that form the nucleotide binding pocket were highlighted as important for signal transduction. Residue E103, which was shown to be important for ATP binding,19,87,102 was not highlighted in this analysis. However, residues on the same helix containing E103, such as L104, I105, and A108 are indicated in the signaling network. Similarly, residues nearby D149 and G153 that are important for ATP hydrolysis, such as H146, T148, G151, V152, M154, are also implicated in allosteric networking. Residue H146 was previously shown to be important in peptide binding and may also play a role in coordinating allosteric transmission by nucleotide.103,104 In addition to signaling networks at the N-terminal ATP binding pocket, long-range signaling to residues in the C-terminus also occurred. These residues are not directly involved in the dimerization interface, and the majority are not surface exposed with the exception of C645, E686, and N688. Similar crosstalk between the N- and C- domains of cytosolic human Hsp90 has also been observed.105 In cytosolic yeast and human Hsp90, a regulatory point in the C-terminal domain was experimentally identified.106 Mutation of this residue (A577 in yeast Hsp82 or C598 in human Hsp90α) altered Hsp90 ATPase and chaperone activity, as well as the conformational equilibrium. This residue corresponds to A646 in Grp94, which was identified in signaling allosteric changes in all of the states. Furthermore, a second ATP binding site at the Hsp90 C-terminus has been identified.107,108 Studies involving the development of a competitive inhibitors for Hsp90α implicated residues M474-N487, V502–N503, E537, T540, K560, N590–I599, S602-T603, A652-L662, Q682, and N686 in forming the C-terminal allosteric pocket.105,109 Homologous residues in Grp94 are I521–K534, A549-S551, D584, R587, S607, Q638-I647, S650-Q651, E703-L713, K733, and D737. Some of these C-terminal residues were highlighted by PRS Analysis (Table S2), specifically through the regulatory motif comprised of residues 642–647. Computational studies of the relaxed state of Grp94 also implicated this region in allosteric signaling.50 Many of the residues highlighted by PRS analysis are highly conserved in Hsp90s (Figure S6).
Figure 7.

Perturbation response scanning analysis of Grp94. The effectivity profile for each residue in Protomer A (black) or Protomer B (red) is shown for Grp94 in the (A) symmetric ATP, (B) asymmetric ATP/ADP-Pi, (C) symmetric ADP-Pi, and (D) symmetric apo configurations. The charged linker was removed in the PCA calculations due to its flexibility. A gray box was used for this missing region to maintain amino acid numbering. Magenta circles indicate residues involved in nucleotide binding and catalysis, including 102–108, 148–154, and 444–454. Yellow circles indicate residues located in the ATP lid, including residues 172–191. Cyan circles indicate residues involved in the M-domain loop or residues that reside in the lumenal channel, including residues 394–407, 428, 497, 498, 575, 662, and 665–668. The blue circle indicates residue 146 involved in peptide binding.
Overall, the networks of residues implicated in signaling are similar for all of the nucleotide binding states and suggest that long-range signaling occurs between the C- and N-terminal domains. Despite the asymmetry in protomer motions of Grp94, the effectivity profile of both protomers was also similar in the four different nucleotide-bound states (Figure 7). In a previous study with cytosolic Hsp90α, PRS analysis of molecular dynamics simulations revealed residues in the ATP lid and the M-domain catalytic loop were implicated in signaling.55 The different allosteric networks involved in Grp94 signaling could be partially due to differences in structure. Interestingly, no residues with high effectivity were identified in the M-domain of Grp94 (Figure 7 and Table S2). Differences in signaling pathways of Hsp90 homologues and the lack of close signaling networks throughout the Grp94 structure may help to explain the low hydrolysis rate of Grp94. Alternatively, the limited simulation time and sampling was not effective in populating all of the possible states and some allosteric networks may be missed. We explored an alternative coarse grained approach to capture dynamics in a later section.
3.4. Nucleotide Binding Alters the Global Dynamics of Grp94
In all four nucleotide bound states, the N-terminal domain exhibited relatively large fluctuations compared to the middle and C-terminal domains (Figure 2, Figure S1, Figure 4, and Figure 6). To determine whether the dominant motions of the NTDs correspond to the opening of N-domains and relaxation of the protomers toward more open configurations,19,20 we calculated the distance between the center of mass of the NTD’s of each Grp94 dimer as a function of time (Figure 8A). The distances were similar when comparing the starting and ending configurations, suggesting that the NTD’s have not largely separated for the symmetric ATP, ADP-Pi, and apo configurations. However, the asymmetric ATP/ADP-Pi structure indicated a separation of the N-terminal domains. Visual inspection of the trajectories suggested a rotational motion of the NTD’s. Since the C-terminal domains did not undergo significant movement in our simulations, we calculated the conformational angle (θ) between the N–C domains of protomer A and the N–C domains of protomer B, measured as a dihedral angle of the centers of mass of the four domains (Figure 8B and Materials and Methods). In the active conformation of Grp94 when the NTD’s are dimerized, the NTDs and CTDs of both protomers criss-cross to form a twisted state. The conformational angle gives insight into the ”twistedness” of the structure. For the symmetric structures with no nucleotide or ADP-Pi, this angle slowly increased indicating that the N-domains are becoming less twisted (Figure 8C), but remained similarly separated based on the distances from Figure 8A. The ATP structure showed no significant changes and remained in a similar twisted state throughout the simulation, suggesting that ATP stabilizes this conformation. In contrast, the torsional angle decreased in the asymmetric ATP/ADP-Pi structure (Figure 8D), suggesting the structure assumed a more twisted conformation. This additional twisting resulted in the separation of the N-terminal domains as they rotated outward (Figure 8A). This compaction is further supported by the decrease in radius of gyration for the asymmetric ATP/ADP-Pi conformation (Figure S10).
Figure 8.

Characterization of Grp94 domain movements. (A) Distances between the centers of mass of Grp94 N-terminal domains as a function of time. The probability distributions of the distances are shown on the right. Since the ATP lid opened and exhibited greater flexibility in some simulations, it was omitted in the center of mass calculations. (B) The conformational angle between the N and C domains of both protomers was calculated as described in the Materials and Methods. (C) Illustration of untwisting at the NTD observed in symmetric apo and ADP conformations. (D) Illustration of additional twisting at the NTD of the asymmetric conformation. The NTD and CTD are indicated for protomers A (blue) and B (green). Movements depicted in the illustrations are exaggerated for clarity.
Since the dimer interface appeared to be affected by nucleotide, we used the ensemble structures from the all-atom molecular dynamics simulations and performed mutational analysis to assess the stability of the Grp94 dimer. The BeAtMuSic approach was used to determine the binding interactions between protomers and predict energetic changes for all four Grp94 systems. We began by using in silico alanine scanning to identify hot spots of Grp94 that are sensitive to mutation. Destabilizing substitutions with ΔΔG > 2.5 kcal/mol were selected for all Grp94 conformations (Figure 9A) and mapped onto the catalytically active conformation (Figure 9B). The hot spot regions correspond to the NTD dimer interface, including the nontruncated portion of helix 1 associated with the ATP lid, and the CTD dimer interface. Additionally, the lumenal channel residues important for client binding were also implicated, though they do not form a tight dimerization interface like the NTD or CTD. We carried out full mutational analysis of these sites to get a better picture of the effect of nucleotide on Grp94’s stability (Figure 9C–F). The lumenal channel residues F398, Y401, and Y667 were predicted to be destabilized by mutation in nucleotide-bound configurations, but mutation insensitive in apo configurations. Of these three residues, F398 is highly conserved in cytosolic Hsp90s and not TRAP1, though Y401 or Y667 are not conserved. Another interesting residue highlighted by this analysis was V446, which is nearby the M-domain catalytic residue R448 that is required for ATP hydrolysis.19 This residue was very sensitive in the symmetric ATP bound configuration but no longer sensitive once one ATP hydrolysis event occurred, which is in agreement with structural rearrangements in this region post-hydrolysis.19 This region of the dimerization interface into the M-domain is highly conserved (Figure S6). In Figure 3, the relationship between nucleotide hydrolysis and ATP lid dynamics is highlighted. In order for reopening of the Grp94 dimer post-hydrolysis, destabilization of the NTD dimerization interface must occur. Mutational analysis links alteration by nucleotide to the NTD dimerization site.
Figure 9.

In silico mutational analysis of Grp94. (A) Changes in binding free energy at the Grp94 dimer interface resulting from alanine scanning. The effect on the binding free energy is quantified for each structure and shown in a stacked representation. The most sensitive residues (ΔΔG values >2.5 in at least one of the structures) are shown. The residues identified in (A) are highlighted on the structure in (B) in cyan. (C–F) Full mutational analysis of the most sensitive residues for all Grp94 conformations; (C) symmetric ATP, (D) asymmetric ATP/ADPPi, (E) symmetric ADP-Pi, and (F) symmetric apo. Residues sensitive to mutation are indicated in blue, while substitutions that result in stabilization are shown in red.
Our findings suggest that nucleotide influences structural changes at the NTD. In the case of asymmetric nucleotide, this resulted in a more compact conformation. No significant changes in dynamics were observed in the client binding region, though mutational analysis revealed sensitivity at this interface. These results are in agreement with findings for other Hsp90s. The mitochondrial Hsp90, TRAP1, has been characterized to function by a sequential hydrolysis mechanism and populate an intermediate asymmetric conformation.86 One protomer of the TRAP1 dimer remains in a straight conformation while the second protomer assumes a “buckled” conformation. The buckle originates at the interface between the MD and CTD, as measured by ≃20 Å, changes in distances between residues K439C and D684C. Importantly, the conformational changes associated with changes in symmetry were localized to regions in TRAP1 important for client binding, providing a direct relationship between ATP hydrolysis-induced conformational rearrangement and client activation.9,86 Simulations of the asymmetric TRAP1 conformation revealed asymmetric water dynamics near the β and γ phosphates, with more water molecules surrounding the nucleotide in the straight protomer and fewer surrounding the buckled protomer.86 We calculated the distances of the corresponding Grp94 residues, T483 and P730, to assess for a potential buckled structure. This region did not demonstrate large changes in distance in the time scales sampled, nor did we observe large scale conformational rearrangements within the client binding region (Figure S11A). We also monitored the number of water molecules within 5 Å of the α and β phosphates in the asymmetric structure, which revealed similar water dynamics (Figure S11B). The number of water molecules around the ADP-Pi containing protomer of Grp94 was skewed toward a higher number of water molecules, likely due to the exposure of the nucleotide binding pocket when the ATP lid opens. It is possible that Grp94 does not sample such an asymmetric state with a buckled protomer, as described for TRAP1.
Our findings are in agreement with simulations and single-molecule FRET experiments of yeast Hsp90 that reported that the asymmetric ATP/ADP structure populated a compacted conformation,44 while symmetric ATP conformations of yeast Hsp90 remained in a state resembling the crystal structure.26 The structural asymmetry in the ATP/ADP state led to the collapse of the M-domain catalytic loop. Arg380 (Arg448 in Grp94), which is conserved in all Hsp90s, is hypothesized to be responsible for conveying allosteric information from the nucleotide to the client binding domain. In our simulation, we discovered the loss of interaction of Arg448 with the nucleotide after ATP hydrolysis but did not observe structural changes in the client binding domain. However, this could be due to the limited time scales we were able to sample, since mutational analysis linked dimer stability to the homologous region of Grp94 (Figure 9). Single molecule FRET experiments have also reported that Grp94 populates a more compact closed conformation when asymmetric nucleotide (ATP/ADP) is present and cycles between two closed conformations.30 This conformation was suggested to represent an intermediate state during the ATP hydrolysis reaction, possibly constituting a newly observed coiled-coil conformation that was previously reported for TRAP1,110 though the exact structural details are not currently known. Together, the results suggest a compaction of the twisted state for Grp94’s ATP/ADP-Pi conformation, which may be a conserved feature for Hsp90s.
3.5. Fluctuations Observed in All-Atom Molecular Dynamics Simulations Agree with Fluctuations Observed in Normal Mode Analysis Calculations of Coarse-Grained Grp94
Due to the limited time scales that all-atom molecular dynamics can sample given large system sizes, we compared the motions sampled in all-atom simulations with those of methods that sample large-scale conformational changes. This method efficiently investigates large amplitude movements using a harmonic model. One significant advantage of this methodology is its ability to study slow structural dynamics with reduced computational cost. We created an elastic network model of Grp94 in the ATP-hydrolysis competent conformation (Materials and Methods). The collective protein motions of the hydrolysis competent state were compared with the relaxed conformation to identify the top modes involved in the conformational change (ATP → ADP). Modes that had a significant structural overlap with the relaxed structure (Im ≥ 0.20) were considered (Figure 10A). Three dominant modes were identified. The cross-correlation was calculated for the top modes (Figure S12), scaled by the structural overlap contribution of each mode respective to the total, and summed for comparison to the dynamic cross-correlation from all-atom simulations. Comparison of the normalized fluctuations of the first three modes in the ATP → ADP conformational change (Figure 10B) with the all-atom molecular dynamics simulations with symmetric ADP-Pi bound (Figure 6) revealed a similar cross-correlation pattern of internal motion for each protomer. This suggests that the all-atom simulations reproduce the collective internal dynamics involved in large scale conformational changes wired by nucleotide. This observation is consistent with earlier reports from Mishra and Jernigan111 suggesting that all-atom molecular dynamics simulations and elastic network models can capture similar dynamic communities. The fluctuations were mapped onto the Grp94 structure (Figure 10C). The largest fluctuations were observed in a region in the CTD (residues 695–710) and the NTD, specifically in the ATP lid residues (residues 172–191). Other mobile regions included the NTDs and MD1 domains. These results are in agreement with those from all-atom molecular dynamics simulations (Figure 2, Figure 3, Figure 4, Figure 7). In contrast, regions with the most stability included a region of the CTD particularly the dimer interface, the client binding region, the lumenal channel, and a region of the NTD (Figure 10C).
Figure 10.

Normal modes of the ATP-bound Grp94 conformation associated with the conformational change toward the ADP state. (A) Structural overlap with the relaxed Grp94 conformation to identify the modes that contribute to the biological movement. The closed circles represent the overlap for individual modes. Lines are drawn for clarity. Nonzero modes with the highest structural overlap are considered. (B) The correlation matrix of the top three modes with the highest structural overlap with the relaxed conformation. The individual modes from Figure S12 were scaled based on their contribution (Im value) and summed. Blue indicates high correlation red indicates anticorrelation, and white indicates no correlation. (C) The B-factors of the top 3 modes Figure S12 were summed and mapped onto the Grp94 structure. Regions in red are the most mobile, while regions in blue are rigid.
The motion from the ATP bound state to the relaxed conformation was decomposed into three dominant motions. In the first dominant mode, mode 13, the N-domains undergo a bending motion that results in the straightening (or decreasing the twistedness) of both protomers, namely at the N-M interfaces (Supplementary movie 9). The decrease in conformational angle for symmetric post-hydrolysis states in all-atom simulations agrees with this finding (Figure 8B). To further understand the movement, the collective motions of the amino acids are quantified by the correlated motion of pairs of amino acids in each mode. Figure S12A shows the correlation matrix for Mode 13 of Grp94. One protomer consists of 641 residues, and the second protomer consists of residues 642–1284, as the pre-N domain and the charged linker were not present in these calculations. For clarity, the domain boundaries are indicated on the top X-axis, since regions with inherent flexibility are removed and the system is renumbered. Mode 13 is characterized by correlated motions in the N- and the M-C domains. The N- and M-C domain motions were anticorrelated within a protomer. Similarly, M-domain motions between protomers were anticorrelated. Larger displacements occurred in the NTD and M1 domains (Figure S12D, black). The second dominant mode, mode 14, is characterized by a scissoring open → close motion that results in a bending of the protomers to separate the M-domains by bringing the N-domains closer (Supplementary movie 10). In this mode, each domain acts independently, as connected by hinges (Figure S12B). Motions of N- and M-domains from opposite protomers were highly correlated, with the largest displacements at the NTD (Figure S12D, red). In the third dominant mode, mode 20, a shearing motion occurs primarily at the M2-C domain and the N-domains (Figure S12C and Supplementary movie 11). This resulted in large fluctuations at the M2- and C-domains (Figure S12D, green). Together, the largest fluctuations occurred in the NTD and CTD, as shown by the combined fluctuations in Figure S12D (blue).
To determine the network of residues involved in the conformational changes, we carried out Structural Perturbation Method (SPM) Analysis. The top 5% of residues involved in allosteric communication are highlighted in Table 1. SPM analysis reveals that residues in the lumenal channel (394–407, 428, 497, 498, 575, 662, 665, 666, 667, 668) and residues in the client binding region (653–678)21,112,113 are important for signaling allosteric changes. These residues were found to be relatively rigid (Figure 10C). This finding agrees with identified signaling networks in the relaxed structure of Grp94.50 A similar region in bacterial Hsp90, corresponding to residues 653–682, demonstrated high mobility and transient interactions with the ATP lid.50 Additionally, in mitochondrial TRAP1 the nucleotide binding site was shown to be allosterically linked to the client binding site.56 Changes in Grp94’s lumenal channel and client binding regions during the closed → open transition could invoke client release or client remodeling mechanisms. Residues in Grp94’s ATP lid were also identified in allosteric signaling. This is in agreement with our all-atom molecular dynamics simulations that show higher fluctuations in the lid when nucleotide is changed from ATP to ADP-Pi or removed (Figure 2, Figure S1, Figure 3). Interestingly, residues in the N-terminal ATP binding site were not involved in these collective motions, unlike the observations from the PRS analysis (Figure 7 and Table S2). Several residues in the M-domain regulatory motif (444–454) important for ATP hydrolysis were identified through SPM analysis, which is in agreement with the involvement of this region in signaling when Grp94 is in the relaxed conformation.50 The salt bridge between R448, the main catalytic residue in this regulatory motif, and the nucleotide was broken when ATP is hydrolyzed in the all-atom simulations. Additionally, residue K467 and other residues along the M-domain helix were identified in signaling networks by SPM analysis. This region is important in BiP binding and this Hsp70-Hsp90 direct interaction is conserved in cytosolic chaperones.114−117 It is not surprising that this region is networked in the conformational change, as BiP binding accelerates Grp94 closure to the catalytically competent state.90 Residue K364 and surrounding residues were also identified. Mutation of this residue accelerates Grp94 ATPase activity 4-fold.118 This residue was also shown to directly contact the nucleotide in the relaxed conformation,119 though this contact was not observed in the ATP structure, nor was it formed in any of our simulations. In many modes, residues in helix 1 near the pre-N domain were identified in signaling. While the pre-N region was truncated in these studies to accelerate conformational changes, these regions may be important for signaling allosteric changes leading to dimer opening. This region of the ATP lid also forms a dimer interface. For instance, residues 73–78 of Grp94 are important for the pre-N domain swap between the N-terminal domains.21 Lastly, several identified residues are also post-translationally modified (https://www.phosphosite.org). Together, this analysis highlighted many residues with functional importance.
Table 1. Structural Perturbation Method Analysis of Grp94 from ENM Calculations.

Residues important for signaling allosteric changes are highlighted for each normal mode contributing to the transition to the relaxed state. Residues in the ATP loop are highlighted in blue, residues in the lumenal channel or client binding region are highlighted in red, residues in the M-domain catalytic loop essential for ATP hydrolysis are highlighted in bold font, residues along the M-domain helix implicated in BiP binding are highlighted in orange, and residues with asterisks were identified to undergo post-translational modifications (https://www.phosphosite.org).
4. Discussion
In this work, we show that minor alterations in the nucleotide binding pocket of Grp94 can have far-reaching effects on other domains or regions far from the active site. While ATP stabilized the closed conformation and enabled the ATP lid to adopt a closed conformation, hydrolysis of the γ-phosphate of ATP or the absence of a nucleotide destabilizes the N-terminal domain and the active site, including the ATP lid. In the asymmetric conformation, ATP lid opening toward an extended open conformation was observed, allowing the release of inorganic phosphate from the ADP bound protomer. In Grp94’s relaxed conformation where the NTDs are not dimerized, the lid region (helix 4/5 and helix 1), which forms the secondary dimer interface in the catalytically active closed state, assembles as an integral structural unit whose movements are highly coordinated. The closure of the lid releases helix 1, allowing it to stabilize the secondary dimerization interface in the closed dimer. While the transition from the open to closed lid dimer has been observed experimentally, our results capture the reverse transition from the closed to the open lid conformation. Nucleotide release occurs in the extended lid conformation, though we did not observe a collapse of the dimerization interface as a result of lid opening. This may be a limitation of our methods given the time scales sampled. Alternatively, this may suggest that further structural perturbations are required to destabilize the dimer interface, leading to reopening of the closed dimer.
Allosteric signaling was detected from the NTD to the CTD in all Grp94 conformations. As nucleotide was hydrolyzed or removed, the global dynamics of the protein were altered and the domain and protomer motions were decoupled. The least stable structure in all of the simulations was the asymmetric ATP/ADP-Pi state that populated a more compact conformation, characterized by a more twisted structure. The high degree of domain decoupling in this structure may provide a direct pathway for conformational changes. The compact asymmetric structure in the ATP/ADP-Pi state is in agreement with experimental observations for Grp94 and cytosolic yeast Hsp90 identifying alternative closed states that are more compact.30,44 While Grp94 was shown to undergo enhanced conformational cycling between closed states when one protomer is hydrolysis defective,30 it remains unclear if this sequential hydrolysis mechanism extends to client activation and remodeling. Structural asymmetry has also been observed for cytosolic Hsp90s, both in the apo form and in complex with clients and cochaperones.120 In contrast, the more compact Grp94 structure observed in our simulations is not characteristic of the asymmetric structure observed for TRAP1 that is composed of one buckled protomer and one straight protomer.9,86 Together, these results suggest that asymmetry may be a conserved feature of Hsp90s, though the structural details may be specific to each Hsp90 member.
We observed a new novel role for the charged linker of Grp94 in simulations of the ADP-Pi and ATP/ADP-Pi conformations, where the linker formed interactions with the M-domain helix and NTD of Grp94. In single molecule experiments with Grp94, the charged linker contributed a large energy difference between folded and unfolded conformations, unlike other Hsp90 members.121 In our simulations, we observed strong interactions between the linker and all domains that could contribute to this energy barrier. In eukaryotic cytosolic systems, Hsp90s have likely evolved to also use the linker to modulate chaperone activity; transient linker-NTD interactions facilitate client binding to p53,11 the charged linker sequence is important for modulating Hsp90 interactions with clients and cochaperones and some Hsp90 functions,122 coordination between ATP and peptide binding is regulated by the charged linker,123 and several post-translational modifications are localized to the linker region (https://www.phosphosite.org). Our results indicate that the linker may interact with the same region of Grp94 as BiP, which accelerates ATP hydrolysis rates and dimer closure of Grp94, and suggest a potentially important role of the charged linker in mediating Grp94 conformational changes. Since Grp94 does not have general cochaperones to modulate ATPase activity like cytosolic Hsp90s, the charged linker could add a similar layer of functional regulation.
Lastly, we identified residues involved in nucleotide signaling. The networked residues were predominantly found in the NTD surrounding the site where nucleotide binds, as well as near the nucleotide binding site in the C-terminal domain. The all-atom molecular dynamics study was complemented with a coarse grained approach utilizing normal-mode analysis of an elastic network model to probe networking involved in the conformational changes between open and closed states. The results were consistent with the all atom molecular dynamics simulations in reproducing the collective dynamics. However, differences emerged in the networks of residues identified. The residues involved in the conformational change included residues in the client binding region, the M-domain catalytic loop, and the M-domain helix. One explanation for the differences observed in networked residues may be attributed to the investigation of equilibrium dynamics by all-atom molecular dynamics simulation versus the investigation of large scale conformational changes by the coarse grained approach. However, an alternative explanation is that nucleotide hydrolysis alone is not sufficient to induce large scale conformational changes, as Hsp90s are highly flexible, adopt numerous conformations, and may have different preferences in conformation despite the presence of nucleotide. Conformational plasticity appears to be an inherent property of individual Hsp90 members. Thermal fluctuations have been hypothesized to be the driving force in conformational dynamics of more flexible Hsp90s.124 Client binding, cochaperone binding, and post-translational modifications may also play a pivotal role in providing thermodynamic and kinetic contributions that result in large-scale conformational changes.10,11,46,90,125−129 The transmission of allostery may occur through multiple predefined pathways, while perturbations to the system by binding events influence the pathway utilized.130 Perhaps in the case of Grp94, nucleotide hydrolysis alters the interdomain and intraprotomer crosstalk to allow more flexibility within the Grp94 structure, while Brownian motion, cellular environment, or cofactor binding alters the predominant signaling network utilized to confer large scale structural rearrangements.
Together, these studies have been successful in identifying allosteric wiring and the local effects of nucleotide hydrolysis. While the simulations produced details in atomistic resolution, the sampling was limited to one conformational state. Conversely, the coarse grained approach was able to investigate large conformational changes and signal transduction. These results highlighted low frequency modes that contribute to dynamics, though the exact pathway of the conformational changes and intermediate conformations were not captured. Thus, advanced sampling methods are required to bridge the gap between the time and length scales of simulations. We expect this information will begin to provide a more complete picture of the structure–function relationship of Grp94.
5. Conclusions
Our work demonstrates how nucleotides facilitate conformational changes and affect allosteric wiring of Grp94. Alteration of nucleotide in Grp94’s nucleotide binding pocket destabilized the structure, specifically the NTD. On the microscopic level, nucleotide alteration resulted in decoupling intradomain and interdomain fluctuations. We identified residues that are involved in short-range allosteric communication within the NTD and residues that are involved in long-range allosteric communication in the CTD. Our simulation results also revealed a potential regulatory role for the flexible linker, in forming strong electrostatic interactions with Grp94 domains, specifically a region in the middle domain. The complementary coarse grained approach investigating Grp94 allostery reproduced the collective dynamics observed in the all-atom molecular dynamics simulations. Catalytic residues, residues in the ATP lid, client binding residues, and BiP binding residues were implicated in signaling allosteric changes. Together, this work provides the groundwork for the development of rationally designed allosteric modulators of Grp94. Such modulators have the potential to have a significant impact on clinical practice.
Acknowledgments
This research was supported by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number R35GM146963. The authors are grateful for computational support by Dr. Jens Mueller and the Redhawk High Performance Computing Center at Miami University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.3c00260.
Details of the RMSD, Hydrogen bond analysis of ligand interaction, interaction energy between Grp94 domains and linker, salt-bridge pairs, B-factors from PCA, movies of PCA fluctuations for each nucleotide-bound state, Hsp90 sequence alignment, radius of Gyration, comparison to TRAP1, normal modes and associated movies indicating fluctuations and perturbation response scanning analysis (PDF)
PCA1 for all atom simulations of the symmetric ATP Grp94 conformation (MPG)
PCA2 for all atom simulations of the symmetric ATP Grp94 conformation (MPG)
PCA1 for all atom simulations of the asymmetric ATP/ADP-Pi Grp94 conformation (MPG)
PCA2 for all atom simulations of the asymmetric ATP/ADP-Pi Grp94 conformation (MPG)
PCA1 for all atom simulations of the symmetric ADP-Pi Grp94 conformation (MPG)
PCA2 for all atom simulations of the symmetric ADP-Pi Grp94 conformation (MPG)
PCA1 for all atom simulations of the symmetric apo Grp94 conformation (MPG)
PCA2 for all atom simulations of the symmetric apo Grp94 conformation (MPG)
Fluctuations from first dominant mode (mode 13) for the conformational change of the Grp94 closed conformation to the relaxed conformation (MPG)
Fluctuations from second dominant mode (mode 14) for the conformational change of the Grp94 closed conformation to the relaxed conformation (MPG)
Fluctuations from third dominant mode (mode 20) for the conformational change of the Grp94 closed conformation to the relaxed conformation (MPG)
The authors declare no competing financial interest.
Special Issue
Published as part of The Journal of Physical Chemistry virtual special issue “Early-Career and Emerging Researchers in Physical Chemistry Volume 2”.
Supplementary Material
References
- Genest O.; Wickner S.; Doyle S. M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. 10.1074/jbc.REV118.002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biebl M. M.; Buchner J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harbor Perspectives in Biology 2019, 11, a034017. 10.1101/cshperspect.a034017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoter A.; El-Sabban M. E.; Naim H. Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. International Journal of Molecular Sciences 2018, 19, 2560. 10.3390/ijms19092560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackie R. E.; Maciejewski A.; Ostapchenko V. G.; Marques-Lopes J.; Choy W. Y.; Duennwald M. L.; Prado V. F.; Prado M. A. The Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases. Frontiers in Neuroscience 2017, 11, 254. 10.3389/fnins.2017.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopf F. H.; Biebl M. M.; Buchner J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- Krukenberg K. A.; Street T. O.; Lavery L. A.; Agard D. A. Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys. 2011, 44, 229–255. 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl L. H. Review: The HSP90 molecular chaperone - An enigmatic ATPase. Biopolymers 2016, 105, 594–607. 10.1002/bip.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Soroka J.; Buchner J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2012, 1823, 624–635. 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Lavery L. A.; Partridge J. R.; Ramelot T. A.; Elnatan D.; Kennedy M. A.; Agard D. A. Structural asymmetry in the closed state of mitochondrial Hsp90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Mol. Cell 2014, 53, 330–343. 10.1016/j.molcel.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street T. O.; Lavery L. A.; Agard D. A. Substrate binding drives large-scale conformational changes in the Hsp90 molecular chaperone. Mol. Cell 2011, 42, 96–105. 10.1016/j.molcel.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez A.; Dahiya V.; Delhommel F.; Freiburger L.; Stehle R.; Asami S.; Rutz D.; Blair L.; Buchner J.; Sattler M. Client binding shifts the populations of dynamic Hsp90 conformations through an allosteric network. Science Advances 2021, 7, 7295. 10.1126/sciadv.abl7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukenberg K. A.; Southworth D. R.; Street T. O.; Agard D. A. pH-Dependent Conformational Changes in Bacterial Hsp90 Reveal a Grp94-Like Conformation at pH 6 That Is Highly Active in Suppression of Citrate Synthase Aggregation. J. Mol. Biol. 2009, 390, 278–291. 10.1016/j.jmb.2009.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzec M.; Eletto D.; Argon Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 2012, 1823, 774–787. 10.1016/j.bbamcr.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eletto D.; Dersh D.; Argon Y. GRP94 in ER quality control and stress responses. Seminars in Cell & Developmental Biology 2010, 21, 479–485. 10.1016/j.semcdb.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khilji M. S.; Bresson S. E.; Verstappen D.; Pihl C.; Andersen P. A. K.; Agergaard J. B.; Dahlby T.; Bryde T. H.; Klindt K.; Nielsen C. K.; Walentinsson A.; Zivkovic D.; Bousquet M.-P.; Tyrberg B.; Richardson S. J.; Morgan N. G.; Mandrup-Poulsen T.; Marzec M. T. The inducible β5i proteasome subunit contributes to proinsulin degradation in GRP94-deficient β-cells and is overexpressed in type 2 diabetes pancreatic islets. American Journal of Physiology - Endocrinology and Metabolism 2020, 318, 892–900. 10.1152/ajpendo.00372.2019. [DOI] [PubMed] [Google Scholar]
- Ghiasi S. M.; et al. Endoplasmic reticulum chaperone glucose-regulated protein 94 is essential for proinsulin handling. Diabetes 2019, 68, 747–760. 10.2337/db18-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothert A. R.; Suntharalingam A.; Huard D. J.; Fontaine S. N.; Crowley V. M.; Mishra S.; Blagg B. S.; Lieberman R. L.; Dickey C. A. Exploiting the interaction between Grp94 and aggregated myocilin to treat glaucoma. Hum. Mol. Genet. 2014, 23, 6470–6480. 10.1093/hmg/ddu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard D. J.; Jonke A. P.; Torres M. P.; Lieberman R. L. Different Grp94 components interact transiently with the myocilin olfactomedin domain in vitro to enhance or retard its amyloid aggregation. Sci. Rep. 2019, 9, 12769. 10.1038/s41598-019-48751-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollins D. E.; Warren J. J.; Immormino R. M.; Gewirth D. T. Structures of GRP94-Nucleotide Complexes Reveal Mechanistic Differences between the hsp90 Chaperones. Mol. Cell 2007, 28, 41–56. 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukenberg K. A.; Böttcher U. M.; Southworth D. R.; Agard D. A. Grp94, the endoplasmic reticulum Hsp90, has a similar solution conformation to cytosolic Hsp90 in the absence of nucleotide. Protein Sci. 2009, 18, 1815–1827. 10.1002/pro.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huck J. D.; Que N. L.; Hong F.; Li Z.; Gewirth D. T. Structural and Functional Analysis of GRP94 in the Closed State Reveals an Essential Role for the Pre-N Domain and a Potential Client-Binding Site. Cell Reports 2017, 20, 2800–2809. 10.1016/j.celrep.2017.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf C.; Stankiewicz M.; Kramer G.; Mayer M. P. Spatially and kinetically resolved changes in the conformational dynamics of the Hsp90 chaperone machine. EMBO J. 2009, 28, 602–613. 10.1038/emboj.2008.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessling M.; Richter K.; Buchner J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nature Structural & Molecular Biology 2009, 16, 287–293. 10.1038/nsmb.1565. [DOI] [PubMed] [Google Scholar]
- Krukenberg K. A.; Förster F.; Rice L. M.; Sali A.; Agard D. A. Multiple Conformations of E. coli Hsp90 in Solution: Insights into the Conformational Dynamics of Hsp90. Structure 2008, 16, 755–765. 10.1016/j.str.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau A. K.; Harris S. F.; Southworth D. R.; Agard D. A. Structural Analysis of E. coli hsp90 Reveals Dramatic Nucleotide-Dependent Conformational Rearrangements. Cell 2006, 127, 329–340. 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Ali M. M.; Roe S. M.; Vaughan C. K.; Meyer P.; Panaretou B.; Piper P. W.; Prodromou C.; Pearl L. H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C. N.; Krukenberg K. A.; Agard D. A. Intra- and intermonomer interactions are required to synergistically facilitate ATP hydrolysis in Hsp90. J. Biol. Chem. 2008, 283, 21170–21178. 10.1074/jbc.M800046200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron P.; Giudice E.; Rolland J.-P.; Buey R. M.; Barbier P.; Diaz J. F.; Peyrot V.; Thomas D.; Garnier C. Apo-Hsp90 coexists in two open conformational states in solution. Biology of the Cell 2008, 100, 413–425. 10.1042/BC20070149. [DOI] [PubMed] [Google Scholar]
- Ye W.; Gotz M.; Celiksoy S.; Tuting L.; Ratzke C.; Prasad J.; Ricken J.; Wegner S. V.; Ahijado-Guzman R.; Hugel T.; Sonnichsen C. Conformational Dynamics of a Single Protein Monitored for 24 h at Video Rate. Nano Lett. 2018, 18, 6633. 10.1021/acs.nanolett.8b03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Friedman L. J.; Sun M.; Gelles J.; Street T. O. Conformational Cycling within the Closed State of Grp94, an Hsp90-Family Chaperone. J. Mol. Biol. 2019, 431, 3312–3323. 10.1016/j.jmb.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge J. R.; Lavery L. A.; Elnatan D.; Naber N.; Cooke R.; Agard D. A. A novel N-terminal extension in mitochondrial TRAP1 serves as a thermal regulator of chaperone activity. eLife 2014, 3, e03487 10.7554/eLife.03487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immormino R. M.; Dollins D. E.; Shaffer P. L.; Soldano K. L.; Walker M. A.; Gewirth D. T. Ligand-induced conformational shift in the N-terminal domain of GRP94, an Hsp90 chaperone. J. Biol. Chem. 2004, 279, 46162–46171. 10.1074/jbc.M405253200. [DOI] [PubMed] [Google Scholar]
- General I. J.; Liu Y.; Blackburn M. E.; Mao W.; Gierasch L. M.; Bahar I. ATPase Subdomain IA Is a Mediator of Interdomain Allostery in Hsp70 Molecular Chaperones. PLOS Computational Biology 2014, 10, e1003624 10.1371/journal.pcbi.1003624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodey N. M.; Benkovic S. J. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008, 4, 474–482. 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- Tsai C. J.; Del Sol A.; Nussinov R. Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms. Molecular Biosystems 2009, 5, 207–216. 10.1039/b819720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Tekpinar M. Large-scale evaluation of dynamically important residues in proteins predicted by the perturbation analysis of a coarse-grained elastic model. BMC Structural Biology 2009, 9, 45–640. 10.1186/1472-6807-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Song G.; Jernigan R. L. Protein elastic network models and the ranges of cooperativity. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 12347–12352. 10.1073/pnas.0902159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chennubhotla C.; Bahar I. Signal Propagation in Proteins and Relation to Equilibrium Fluctuations. PLOS Computational Biology 2007, 3, e172. 10.1371/journal.pcbi.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I.; Chennubhotla C.; Tobi D. Intrinsic Enzyme Dynamics in the Unbound State and Relation to Allosteric Regulation. Curr. Opin. Struct. Biol. 2007, 17, 633. 10.1016/j.sbi.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C. J.; del Sol A.; Nussinov R. Allostery: Absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 2008, 378, 1–11. 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokholyan N. V. Controlling Allosteric Networks in Proteins. Chem. Rev. 2016, 116, 6463–6487. 10.1021/acs.chemrev.5b00544. [DOI] [PubMed] [Google Scholar]
- Wodak S. J.; et al. Allostery in Its Many Disguises: From Theory to Applications. Structure 2019, 27, 566–578. 10.1016/j.str.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Zhou H. X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 116, 6503–6515. 10.1021/acs.chemrev.5b00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S.; Sohmen B.; Hellenkamp B.; Thurn J.; Stock G.; Hugel T. Hierarchical dynamics in allostery following ATP hydrolysis monitored by single molecule FRET measurements and MD simulations †. Chemical Science 2021, 12, 3350–3359. 10.1039/D0SC06134D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra G.; Potestio R.; Micheletti C.; Colombo G. Corresponding functional dynamics across the Hsp90 chaperone family: Insights from a multiscale analysis of MD simulations. PLOS Computational Biology 2012, 8, e1002433 10.1371/journal.pcbi.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindle M. P.; Carter B.; Alao J. P.; Connors K.; Tehver R.; Kravats A. N. Structural Communication between the E. coli Chaperones DnaK and Hsp90. International Journal of Molecular Sciences 2021, 22, 2200. 10.3390/ijms22042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo G.; Morra G.; Meli M.; Verkhivker G. Understanding ligand-based modulation of the Hsp90 molecular chaperone dynamics at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 7976–2781. 10.1073/pnas.0802879105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra G.; Verkhivker G.; Colombo G. Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full-length dimer. PLOS Computational Biology 2009, 5, e1000323 10.1371/journal.pcbi.1000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetz G.; Tse A.; Verkhivker G. M. Dissecting Structure-Encoded Determinants of Allosteric Cross-Talk between Post-Translational Modification Sites in the Hsp90 Chaperones. Sci. Rep. 2018, 8, 6899. 10.1038/s41598-018-25329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A.; Verkhivker G. M. Probing Molecular Mechanisms of the Hsp90 Chaperone: Biophysical Modeling Identifies Key Regulators of Functional Dynamics. PLoS One 2012, 7, e37605. 10.1371/journal.pone.0037605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacklock K.; Verkhivker G. M. Differential Modulation of Functional Dynamics and Allosteric Interactions in the Hsp90-Cochaperone Complexes with p23 and Aha1: A Computational Study. PLoS One 2013, 8, e71936. 10.1371/journal.pone.0071936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacklock K.; Verkhivker G. M. Allosteric Regulation of the Hsp90 Dynamics and Stability by Client Recruiter Cochaperones: Protein Structure Network Modeling. PLoS One 2014, 9, e86547. 10.1371/journal.pone.0086547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astl L.; Verkhivker G. M. Dynamic View of Allosteric Regulation in the Hsp70 Chaperones by J-Domain Cochaperone and Post-Translational Modifications: Computational Analysis of Hsp70 Mechanisms by Exploring Conformational Landscapes and Residue Interaction Networks. J. Chem. Inf. Model. 2020, 60, 1614–1631. 10.1021/acs.jcim.9b01045. [DOI] [PubMed] [Google Scholar]
- Seifert C.; Gräter F. Force Distribution Reveals Signal Transduction in E. coli Hsp90. Biophys. J. 2012, 103, 2195–2202. 10.1016/j.bpj.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkler D. L.; Atilgan C.; Tastan Bishop O. Allosteric Modulation of Human Hsp90α Conformational Dynamics. J. Chem. Inf. Model. 2018, 58, 383–404. 10.1021/acs.jcim.7b00630. [DOI] [PubMed] [Google Scholar]
- D’Annessa I.; Moroni E.; Colombo G. Visualizing the Dynamics of a Protein Folding Machinery: The Mechanism of Asymmetric ATP Processing in Hsp90 and its Implications for Client Remodelling. J. Mol. Biol. 2021, 433, 166728. 10.1016/j.jmb.2020.166728. [DOI] [PubMed] [Google Scholar]
- Czemeres J.; Buse K.; Verkhivker G. M. Atomistic simulations and network-based modeling of the Hsp90-Cdc37 chaperone binding with Cdk4 client protein: A mechanism of chaperoning kinase clients by exploiting weak spots of intrinsically dynamic kinase domains. PLoS One 2017, 12, e0190267 10.1371/journal.pone.0190267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Yan R.; Roy A.; Xu D.; Poisson J.; Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 2015, 12, 7–8. 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008, 9, 1–8. 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A.; Kucukural A.; Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols 2010, 5, 725–738. 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger, LLC, The PyMOL Molecular Graphics System, Version 1.8. 2015.
- Acun B.; Hardy D. J.; Kale L. V.; Li K.; Phillips J. C.; Stone J. E. Scalable Molecular Dynamics with NAMD on the Summit System. IBM J. Res. Dev. 2018, 62, 4:1–4:9. 10.1147/JRD.2018.2888986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Mackerell A. D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Ohio Supercomputer Center. Ohio Supercomputer Center. 1987; http://osc.edu/ark:/19495/f5s1ph73.
- Tekpinar M.; Neron B.; Delarue M. Correction to “Extracting Dynamical Correlations and Identifying Key Residues for Allosteric Communication in Proteins by correlationplus. J. Chem. Inf. Model. 2021, 61, 5720. 10.1021/acs.jcim.1c01333. [DOI] [PubMed] [Google Scholar]
- Bakan A.; Meireles L. M.; Bahar I. Structural bioinformatics ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics Applications Note 2011, 27, 1575–1577. 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Atilgan C.; Atilgan A. R. Perturbation-Response Scanning Reveals Ligand Entry-Exit Mechanisms of Ferric Binding Protein. PLOS Computational Biology 2009, 5, e1000544. 10.1371/journal.pcbi.1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehouck Y.; Kwasigroch J. M.; Rooman M.; Gilis D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic acids research 2013, 41, W333–W339. 10.1093/nar/gkt450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehouck Y.; Grosfils A.; Folch B.; Gilis D.; Bogaerts P.; Rooman M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. 10.1093/bioinformatics/btp445. [DOI] [PubMed] [Google Scholar]
- Dehouck Y.; Gilis D.; Rooman M. A New Generation of Statistical Potentials for Proteins. Biophys. J. 2006, 90, 4010–4017. 10.1529/biophysj.105.079434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirion M. M. Large Amplitude Elastic Motions in Proteins from a Single-Parameter, Atomic Analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- Ma J. Usefulness and Limitations of Normal Mode Analysis in Modeling Dynamics of Biomolecular Complexes. Structure 2005, 13, 373–380. 10.1016/j.str.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Doniach S. A comparative study of motor-protein motions by using a simple elastic-network model. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13253–13258. 10.1073/pnas.2235686100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehver R.; Chen J.; Thirumalai D. Allostery Wiring Diagrams in the Transitions that Drive the GroEL Reaction Cycle. J. Mol. Biol. 2009, 387, 390–406. 10.1016/j.jmb.2008.12.032. [DOI] [PubMed] [Google Scholar]
- Bahar I.; Atilgan A. R.; Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Folding and Design 1997, 2, 173–181. 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- Atilgan A. R.; Durell S. R.; Jernigan R. L.; Demirel M. C.; Keskin O.; Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Brooks B. R.; Thirumalai D. Allosteric transitions in the chaperonin GroEL are captured by a dominant normal mode that is most robust to sequence variations. Biophys. J. 2007, 93, 2289–2299. 10.1529/biophysj.107.105270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Brooks B. R.; Doniach S.; Thirumalai D. Network of dynamically important residues in the open/closed transition in polymerases is strongly conserved. Structure 2005, 13, 565–577. 10.1016/j.str.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Tehver R.; Thirumalai D. Rigor to Post-Rigor Transition in Myosin V: Link between the Dynamics and the Supporting Architecture. Structure 2010, 18, 471–481. 10.1016/j.str.2010.01.019. [DOI] [PubMed] [Google Scholar]
- Elnatan D.; Betegon M.; Liu Y.; Ramelot T.; Kennedy M. A.; Agard D. A. Symmetry broken and rebroken during the ATP hydrolysis cycle of the mitochondrial Hsp90 TRAP1. eLife 2017, 6, e25235 10.7554/eLife.25235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F.; Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat. Cell Biol. 2001, 3, 891–896. 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- Dollins D. E.; Immormino R. M.; Gewirth D. T. Structure of Unliganded GRP94, the Endoplasmic Reticulum Hsp90. J. Biol. Chem. 2005, 280, 30438–30447. 10.1074/jbc.M503761200. [DOI] [PubMed] [Google Scholar]
- Soldano K. L.; Jivan A.; Nicchitta C. V.; Gewirth D. T. Structure of the N-terminal Domain of GRP94. J. Biol. Chem. 2003, 278, 48330–48338. 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- Huang B.; Sun M.; Hoxie R.; Kotler J. L.; Friedman L. J.; Gelles J.; Street T. O. The endoplasmic reticulum chaperone BiP is a closure-accelerating cochaperone of Grp94. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2118793119 10.1073/pnas.2118793119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagöz G. E.; et al. Hsp90-Tau Complex Reveals Molecular Basis for Specificity in Chaperone Action. Cell 2014, 156, 963–974. 10.1016/j.cell.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas C.; Ostrovsky O.; Makarewich C. A.; Wanderling S.; Gidalevitz T.; Argon Y. The peptide-binding activity of GRP94 is regulated by calcium. Biochem. J. 2007, 405, 233–241. 10.1042/BJ20061867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainzl O.; Lapina M. C.; Buchner J.; Richter K. The charged linker region is an important regulator of Hsp90 function. J. Biol. Chem. 2009, 284, 22559–22567. 10.1074/jbc.M109.031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler P. M.; Shinsky S. A.; Hong F.; Li Z.; Cosgrove M. S.; Gewirth D. T. Characterization of the Grp94/OS-9 Chaperone–Lectin Complex. J. Mol. Biol. 2014, 426, 3590–3605. 10.1016/j.jmb.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y.; Kotler J. L.; Wang S.; Huang B.; Halpin J. C.; Street T. O. The ER chaperones BiP and Grp94 regulate the formation of insulin-like growth factor 2 (IGF2) oligomers. J. Mol. Biol. 2021, 433, 166963. 10.1016/j.jmb.2021.166963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehn A.; Moroni E.; Zierer B. K.; Tippel F.; Morra G.; John C.; Richter K.; Colombo G.; Buchner J. Allosteric Regulation Points Control the Conformational Dynamics of the Molecular Chaperone Hsp90. J. Mol. Biol. 2016, 428, 4559–4571. 10.1016/j.jmb.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Moroni E.; Zhao H.; Blagg B. S.; Colombo G. Exploiting conformational dynamics in drug discovery: Design of C-terminal inhibitors of Hsp90 with improved activities. J. Chem. Inf. Model. 2014, 54, 195–208. 10.1021/ci4005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan W. P.; Owen B. A.; Toft D. O. The influence of ATP and p23 on the conformation of hsp90. J. Biol. Chem. 2002, 277, 45942–45948. 10.1074/jbc.M207754200. [DOI] [PubMed] [Google Scholar]
- Prodromou C. The ATPase cycle of Hsp90 drives a molecular clamp’ via transient dimerization of the N-terminal domains. EMBO Journal 2000, 19, 4383–4392. 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni E.; Agard D. A.; Colombo G. The Structural Asymmetry of Mitochondrial Hsp90 (Trap1) Determines Fine Tuning of Functional Dynamics. J. Chem. Theory Comput. 2018, 14, 1033–1044. 10.1021/acs.jctc.7b00766. [DOI] [PubMed] [Google Scholar]
- Penkler D.; Sensoy O.; Atilgan C.; Tastan Bishop O. Perturbation-Response Scanning Reveals Key Residues for Allosteric Control in Hsp70. J. Chem. Inf. Model. 2017, 57, 1359–1374. 10.1021/acs.jcim.6b00775. [DOI] [PubMed] [Google Scholar]
- Flynn J. M.; Rossouw A.; Cote-Hammarlof P.; Fragata I.; Mavor D.; Hollins C.; Bank C.; Bolon D. N. Comprehensive fitness maps of hsp90 show widespread environmental dependence. eLife 2020, 9, e53810 10.7554/eLife.53810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C.; Siligardi G.; O’Brien R.; Woolfson D. N.; Regan L.; Panaretou B.; Ladbury J. E.; Piper P. W.; Pearl L. H. Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO Journal 1999, 18, 754–762. 10.1093/emboj/18.3.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidalevitz T.; Biswas C.; Ding H.; Schneidman-Duhovny D.; Wolfson H. J.; Stevens F.; Radford S.; Argon Y. Identification of the N-terminal Peptide Binding Site of Glucose-regulated Protein 94. J. Biol. Chem. 2004, 279, 16543–16552. 10.1074/jbc.M313060200. [DOI] [PubMed] [Google Scholar]
- Morra G.; Neves M. A.; Plescia C. J.; Tsustsumi S.; Neckers L.; Verkhivker G.; Altieri D. C.; Colombo G. Dynamics-based discovery of allosteric inhibitors: Selection of new ligands for the C-terminal domain of Hsp90. J. Chem. Theory Comput. 2010, 6, 2978–2989. 10.1021/ct100334n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retzlaff M.; Stahl M.; Eberl H. C.; Lagleder S.; Beck J.; Kessler H.; Buchner J. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Reports 2009, 10, 1147–1153. 10.1038/embor.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu M. G.; Chadli A.; Bouhouche I.; Catelli M.; Neckers L. M. The Heat Shock Protein 90 Antagonist Novobiocin Interacts with a Previously Unrecognized ATP-binding Domain in the Carboxyl Terminus of the Chaperone. J. Biol. Chem. 2000, 275, 37181–37186. 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- Garnier C.; Lafitte D.; Tsvetkov P. O.; Barbier P.; Leclerc-Devin J.; Millot J.-M.; Briand C.; Makarov A. A.; Catelli M. G.; Peyrot V. Binding of ATP to Heat Shock Protein 90. J. Biol. Chem. 2002, 277, 12208–12214. 10.1074/jbc.M111874200. [DOI] [PubMed] [Google Scholar]
- Matts R. L.; Dixit A.; Peterson L. B.; Sun L.; Voruganti S.; Kalyanaraman P.; Hartson S. D.; Verkhivker G. M.; Blagg B. S. Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem. Biol. 2011, 6, 800–807. 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung N.; Lee J.; Kim J. H.; Chang C.; Joachimiak A.; Lee S.; Tsai F. T. Mitochondrial Hsp90 is a ligand-activated molecular chaperone coupling ATP binding to dimer closure through a coiled-coil intermediate. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 2952–2957. 10.1073/pnas.1516167113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S. K.; Jernigan R. L. Protein dynamic communities from elastic network models align closely to the communities defined by molecular dynamics. PLoS One 2018, 13, e0199225 10.1371/journal.pone.0199225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S. F.; Shiau A. K.; Agard D. A. The crystal structure of the carboxy-terminal dimerization domain of htpG, the Escherichia coli Hsp90, reveals a potential substrate binding site. Structure 2004, 12, 1087–1097. 10.1016/j.str.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Fang L.; Ricketson D.; Getubig L.; Darimont B. Unliganded and hormone-bound glucocorticoid receptors interact with distinct hydrophobic sites in the Hsp90 C-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 18487–18492. 10.1073/pnas.0609163103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravats A. N.; Doyle S. M.; Hoskins J. R.; Genest O.; Doody E.; Wickner S. Interaction of E. coli Hsp90 with DnaK Involves the DnaJ Binding Region of DnaK. J. Mol. Biol. 2017, 429, 858–872. 10.1016/j.jmb.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle S. M.; Hoskins J. R.; Kravats A. N.; Heffner A. L.; Garikapati S.; Wickner S. Intermolecular Interactions between Hsp90 and Hsp70. J. Mol. Biol. 2019, 431, 2729–2746. 10.1016/j.jmb.2019.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M.; Kotler J. L.; Liu S.; Street T. O. The endoplasmic reticulum (ER) chaperones BiP and Grp94 selectively associate when BiP is in the ADP conformation. J. Biol. Chem. 2019, 294, 6387–6396. 10.1074/jbc.RA118.007050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Y. R.; Noddings C. M.; Kirschke E.; Myasnikov A. G.; Johnson J. L.; Agard D. A. Structure of Hsp90–Hsp70–Hop–GR reveals the Hsp90 client-loading mechanism. Nature 2021 601:7893 2022, 601, 460–464. 10.1038/s41586-021-04252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y.; Hoxie R. S.; Street T. O. Molecular mechanism of bacterial Hsp90 pH-dependent ATPase activity. Protein Science: A Publication of the Protein Society 2017, 26, 1206–1213. 10.1002/pro.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin J. C.; Street T. O. Hsp90 Sensitivity to ADP Reveals Hidden Regulation Mechanisms. J. Mol. Biol. 2017, 429, 2918–2930. 10.1016/j.jmb.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J. M.; Mishra P.; Bolon D. N. Mechanistic Asymmetry in Hsp90 Dimers. J. Mol. Biol. 2015, 427, 2904–2911. 10.1016/j.jmb.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn M.; Tych K.; Girstmair H.; Steinmaßl M.; Hugel T.; Buchner J.; Rief M. Folding and Domain Interactions of Three Orthologs of Hsp90 Studied by Single-Molecule Force Spectroscopy. Structure 2018, 26, 96–105. 10.1016/j.str.2017.11.023. [DOI] [PubMed] [Google Scholar]
- Tsutsumi S.; Mollapour M.; Prodromou C.; Lee C. T.; Panaretou B.; Yoshida S.; Mayer M. P.; Neckers L. M. Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 2937–2942. 10.1073/pnas.1114414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibel T.; Siegmund H. I.; Jaenicke R.; Ganz P.; Lilie H.; Buchner J. The charged region of Hsp90 modulates the function of the N-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 1297–1302. 10.1073/pnas.96.4.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratzke C.; Nguyen M. N.; Mayer M. P.; Hugel T. From a Ratchet Mechanism to Random Fluctuations Evolution of Hsp90’s Mechanochemical Cycle. J. Mol. Biol. 2012, 423, 462–471. 10.1016/j.jmb.2012.07.026. [DOI] [PubMed] [Google Scholar]
- Zuehlke A.; Johnson J. L. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 2010, 93, 211–217. 10.1002/bip.21292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickler M.; Hessling M.; Ratzke C.; Buchner J.; Hugel T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nature Structural & Molecular Biology 2009 16:3 2009, 16, 281–286. 10.1038/nsmb.1557. [DOI] [PubMed] [Google Scholar]
- Graf C.; Lee C. T.; Eva Meier-Andrejszki L.; Nguyen M. T.; Mayer M. P. Differences in conformational dynamics within the Hsp90 chaperone family reveal mechanistic insights. Frontiers in Molecular Biosciences 2014, 1, 4. 10.3389/fmolb.2014.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe K.; et al. Posttranslational modification and conformational state of Heat Shock Protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget 2013, 4, 1065–1074. 10.18632/oncotarget.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M.; Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 2012, 1823, 648–655. 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Sol A.; Tsai C. J.; Ma B.; Nussinov R. The Origin of Allosteric Functional Modulation: Multiple Pre-existing Pathways. Structure 2009, 17, 1042–1050. 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.