

Abstract
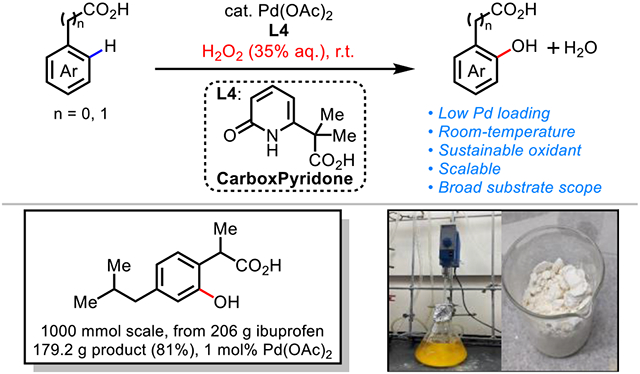
With the large number of Pd(II)-catalyzed C─H activation reactions of native substrates developed in the past decade, the development of catalysts to enable the use of green oxidants under safe and practical conditions has become an increasingly important challenge. Notably, the compatibility of Pd(II) catalysts with sustainable aqueous H2O2 has been a long standing challenge in catalysis including Wacker-type oxidations. We report herein a bifunctional bidentate carboxyl-pyridone (CarboxPyridone) ligand that enables room-temperature Pd-catalyzed C─H hydroxylation of a broad range of benzoic and phenylacetic acids with an industry-compatible oxidant, aqueous hydrogen peroxide (35% H2O2). The scalability of this methodology is demonstrated by a 1000 mmol scale reaction of ibuprofen (206 g) using only a 1 mol% Pd catalyst loading. The utility of this protocol is further illustrated through derivatization of the products and synthesis of polyfluorinated natural product coumasten and pterocarpene from phenol intermediates prepared using this methodology.
Graphical Abstract
1. Introduction
Phenols are not only common motifs in natural products and bioactive compounds (Scheme 1a),1 but also versatile building blocks for organic syntheses.2 Accordingly, a variety of approaches have been developed to access this important motif, including chemical synthesis,3 biochemical synthesis4 and the controlled decomposition of natural compounds,5 such as lignin depolymerization. Modern transition metal catalysis has made significant advances in converting a variety of aryl halides or aryl boronic acids into phenols.6 In principle, the direct hydroxylation of aryl C─H bonds offers an appealing, single-step alternative, but the realization of this strategy has proven challenging. Various strategies ranging from photocatalytic methods7a,7b to nickel,7c manganese,7d copper,7e and iron7f,7g catalysis have been explored to achieve aromatic C─H oxidations. Recently, palladium catalysis has emerged as a powerful strategy for selective oxygenation of C─H bonds.8,9 The C─H hydroxylation reactions of arenes bearing strongly coordinating directing groups such as pyridyl or oxime have been reported using various oxidants,8a-f however, these substrates are not practical for the synthesis. There are a handful of examples on C(sp2)─H oxidation directed by weakly coordinating native directing groups such as free carboxylic acids using hypervalent iodine reagents such as PhI(OAc)2, and an additional hydrolysis step is required to access the final phenolic compound (Scheme 1b).9f,9g
Scheme 1.

Preparation of Phenols through Pd(II) catalyzed C(sp2)─H Oxidations
In order for C─H hydroxylation to be practical and scalable, it is essential that methodologies employ a cheap and environmentally friendly oxidant. Recently we demonstrated the feasibility of using molecular oxygen for the direct hydroxylation of C─H bonds of (hetero)benzoic acids (Scheme 1c),10 however, the efficiency, scope, and safety of pure oxygen are unsuitable for industrial scale applications. Since aqueous H2O2 has been used in several landmark ton-scale industrial processes including the HPPO (converting propene to propylene oxide) and ε-Caprolactam processes,11 we envisioned that Pd-catalyzed C─H hydroxylation with H2O2 could provide a practical solution to the synthesis of phenols. However, hydrogen peroxide tends to decompose under the elevated temperature generally required for previously reported Pd(II)-catalyzed C─H activation reactions. Herein we report the development of a bifunctional bidentate carboxyl-pyridone (CarboxPyridone) ligand that enables room-temperature C(sp2)─H hydroxylation of native carboxylic acid substrates with practical, aqueous H2O2 as the sole oxidant (Scheme 1d). This new protocol provides an efficient synthetic route to access a wide range of phenols, the synthetic utility of which was demonstrated through the synthesis of two polyfluorinated natural products. Importantly, the reaction can be efficiently scaled up, as highlighted by the synthesis of 179 grams of ortho-hydroxylated ibuprofen. Preliminary mechanistic studies demonstrate that ligand is crucial for accelerating C─H cleavage at room temperature, which prevent the decomposition of hydrogen peroxide.
2. Results and Discussion
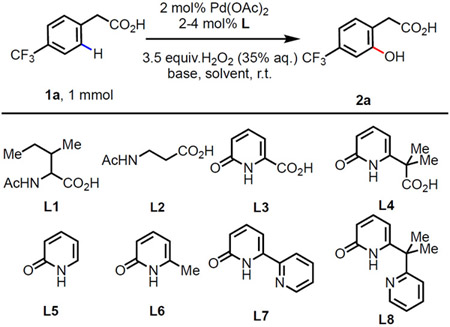
Itoh et al. has reported Pd(II) catalyzed the strongly coordinating pyridine directed C(sp2)─H hydroxylation using hydrogen peroxide.8e Given our goal of developing a practical C─H hydroxylation reaction, we were interested in developing C─H hydroxylation directed by common, weakly coordinating native directing groups. We were particularly interested in carboxylic acid directing groups due to the abundant sources and versatile conversions. We began our investigation of C(sp2)─H hydroxylation using 4-trifluorometylphenylacetic acid (1a) as a model substrate with 2 mol% Pd(OAc)2 loading at a 1.0 mmol scale (Table 1). Mono N-protected amino acid ligand (MPAA) L1 was chosen for preliminary screening as this ligand is known to accelerate C─H bond cleavage with phenyl acetic acid substrates by direct participation of the acetylamino motif (NHAc) as an internal base during the concerted metalation-deprotonation (CMD) step.12 To our delight, L1 enabled the formation of the desired ortho-hydroxylated product 2a in 37% yield when using N,N-dimethylacetamide (DMA) as the solvent and H2O2 (35% aq.) as the oxidant at 90 °C (entry 1). Experiments show that Pd(II) salts decompose 85% of the H2O2 at 90 °C within 20 min (see Supporting Information). We reasoned that the key to improve this reaction is to find a ligand that can ensure Pd(II) catalyst activates C─H preferentially than H2O2. Although the decomposition of H2O2 was not observed under the reaction conditions at room temperature, MPAA ligands L1 and L2 proved incapable of enabling C─H hydroxylation at room temperature (entries 2-3). Therefore, it was crucial to develop a new ligand that would enable C─H activation at room temperature. Recently, 2-pyridones have emerged as particularly effective ligands that play analogous role to NHAc for challenging Pd(II) catalyzed C─H activation reactions.10,13 We hypothesized that incorporation of the 2-pyridone group into the MPAA scaffold in place of NHAc might enable the C─H activation at mild reaction temperatures. Consequently, we designed and tested the pyridone-containing MPAA analogs L3 and L4 (CarboxPyridone). While the five membered chelate L3 did not afford the desired product (entry 4); surprisingly, the six-membered chelate L4 afforded 2a in a 65% yield under the room temperature conditions (entry 5). We propose that the increased flexibility of the six-membered chelate may compensate for the rigidity of the planar 2-pyridone motif, allowing the ligand to adopt a favorable conformation for pyridone-assisted C─H cleavage step. When monodentate 2-pyridone ligand L5 and L6 are applied, only trace amount of product was observed, demonstrating the importance of bidentate nature of the ligand (entries 6-7). Interestingly, L7 and L8, two recently developed effective bidentate pyridone-pyridine ligands for C─H activation reactions were not effective (entries 8-9),13 indicating the importance of retaining a carboxylic acid motif in the bidentate ligand. A control experiment without ligand clearly indicates that ligand plays a key role for this reaction (entry 10). Further optimization with L4 revealed that the base and solvent combinations of KHCO3 with DMA (entry 11) and K2HPO4 with CH3CN (entry 12) afforded the product in excellent yields (see Supporting Information for screening details).
Table 1.
Optimization of the C(sp2)─H Hydroxylation Using Hydrogen Peroxidea
| ||||
|---|---|---|---|---|
| Entry | Ligand | Base | Solvent | Yield (%) |
| 1b | L1 | K2HPO4 | DMA | 37 |
| 2 | L1 | K2HPO4 | DMA | 0 |
| 3 | L2 | K2HPO4 | DMA | 0 |
| 4 | L3 | K2HPO4 | DMA | 0 |
| 5 | L4 | K2HPO4 | DMA | 65 |
| 6 | L5 | K2HPO4 | DMA | trace |
| 7 | L6 | K2HPO4 | DMA | trace |
| 8c | L7 | K2HPO4 | DMA | <5 |
| 9c | L8 | K2HPO4 | DMA | 0 |
| 10 | No L | K2HPO4 | DMA | 0 |
| 11 | L4 | KHCO3 | DMA | 80 |
| 12 | L4 | K2HPO4 | CH3CN | 86 |
Conditions: 4-Trifluoro-phenylacetic acid (1.0 mmol), Pd(OAc)2 (2 mol%), ligand (4 mol%), H2O2 (35% aqueous solution, 3.5 equiv.), base (1.5 equiv.) in solvent (3.0 mL) r.t., 24 h. Yields were determined by 1H NMR using CH3NO2 as the internal standard.
90°C.
2 mol% ligand.
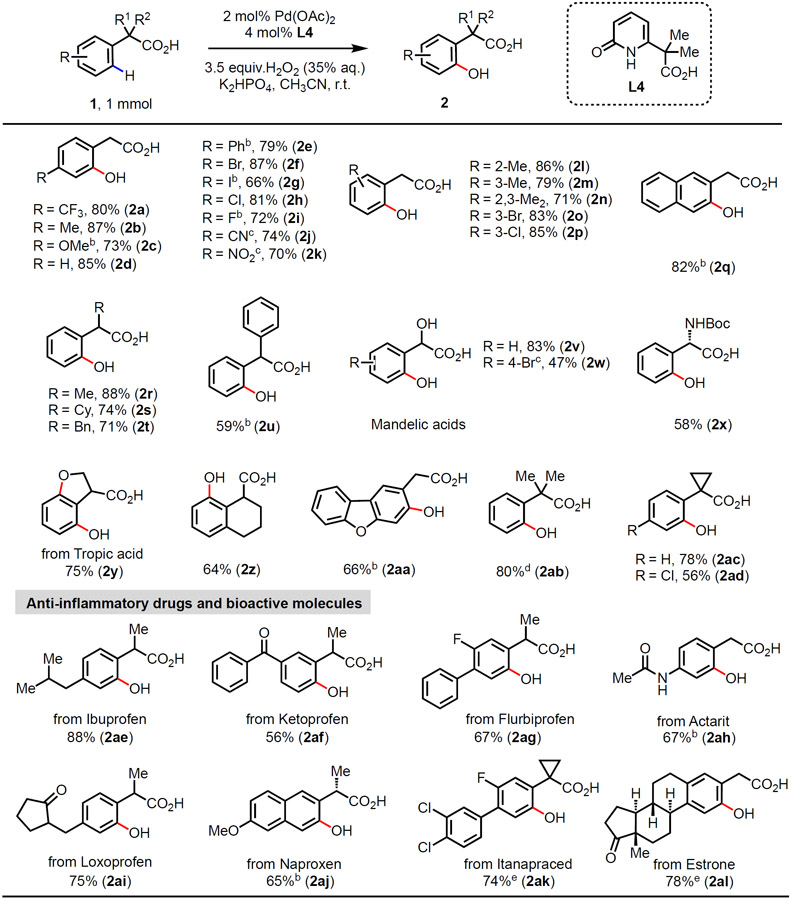
Having determined optimal conditions, we subjected a wide range of phenylacetic acids to the hydroxylation reaction (Table 2). Phenylacetic acids containing electron-withdrawing (1a, 1e-1k) or electron-donating (1b, 1c)para-substituents, as well as un-substituted (1d) all provided the corresponding products in high yields. Substrates with various substitution patterns (1l-1q) were also smoothly hydroxylated in high yields, affording the hydroxylated products at the less hindered positions. In addition, the α-substituents of carboxylic acids (1r-1u) were also tolerated, generating the corresponding hydroxylated products. It is noteworthy that hydroxylations of biologically active molecules such as mandelic acids (1v, 1w), protected phenylglycine (1x) and tropic acid (1y) were feasible providing expedient access to phenol derivatives. The phenylacetic acids with tetralin skeleton (1z), dibenzofuran (1aa) and α-quaternary centers (1ab-1ad) were also compatible. Interestingly, the reactions are highly selective for C(sp2)─H bonds, leaving potentially reactive cyclopropyl β-C(sp3)─H bonds intact (2ac, 2ad). Late-stage modification of the existing drug molecules is a powerful approach to rapidly optimize bioactivity of lead compounds. Various anti-inflammatory drugs such as ibuprofen (1ae), ketoprofen (1af), flurbiprofen (1ag), loxoprofen (1ai), and naproxen (1aj) were all successfully hydroxylated with the new practical method. Other pharmaceuticals, including actarit (1ah) and itanapraced (1ak), were also hydroxylated in high yields and with high regioselectivity. Likewise, the complex phenylacetic acid derived from estrone (1al) was hydroxylated at the ortho-position in good yield.
Table 2.
Ligand enabled C(sp2)─H hydroxylation of phenylacetic acidsa
|
Conditions: Carboxylic acid 1 (1 mmol), Pd(OAc)2 (2 mol%), L4 (4 mol%), H2O2 (35% aqueous solution, 3.5 equiv.), and K2HPO4 (1.5 equiv.) in CH3CN (3.0 mL), r.t., 24 h. Isolated yields.
KHCO3 (2 mmol) instead of K2HPO4, DMA instead of CH3CN.
60 °C.
Isolated yield based on the corresponding lactone. (See SI for details)
0.5 mmol scale.
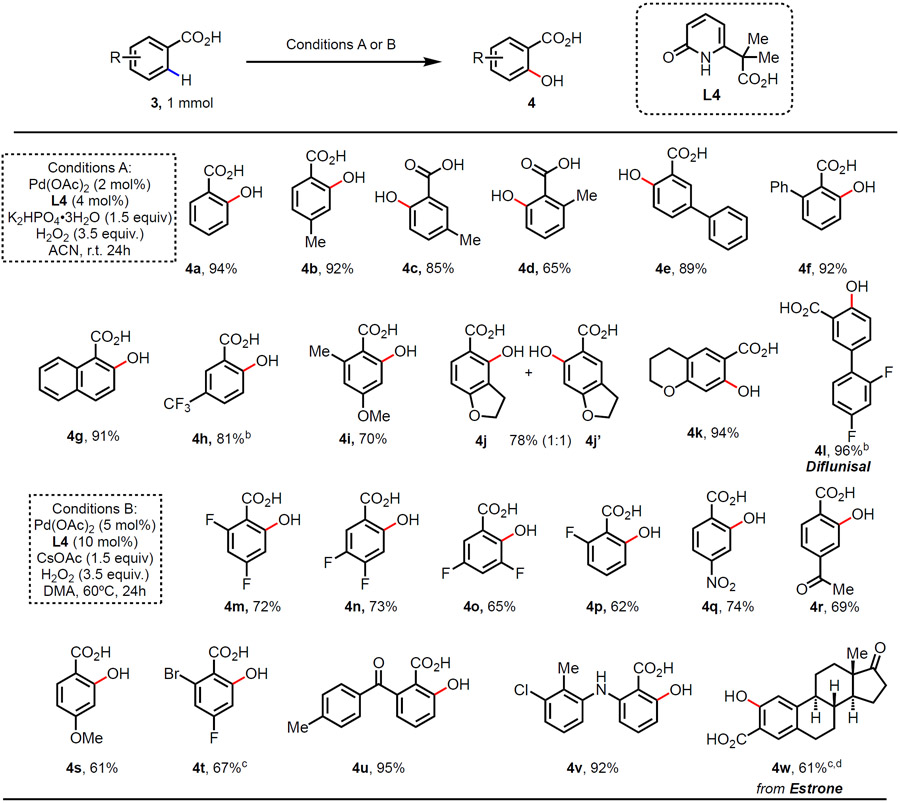
Our discovery of ligand-enabled C(sp2)─H hydroxylation of phenylacetic acid substrates led us to test whether this methodology is also applicable to another important class of substrates, benzoic acids (Table 3). It is worth noting that a single catalyst is often not compatible with both phenylacetic acid and benzoic acid scaffolds. To our delight, salicylic acid (4a) was obtained in high yield when benzoic acid was subjected to the similar conditions, differing only in the use of potassium phosphate dibasic trihydrate (K2HPO4•3H2O) as the base. Alkyl or aryl substituted benzoic acids (3b-3f) were effective substrates, affording the corresponding mono-hydroxylated products in good to excellent yields. The reaction was selective for C(sp2)─H hydroxylation with substrate 3d, for which monodentate 2-pyridone ligands have previously been reported to effect benzylic C(sp3)─H activation.14 1-Naphthoic acid (3g) was successfully hydroxylated on 2-position under the reaction conditions without decarboxylation.8g A higher temperature of 60 °C was required for substrate with electron-withdrawing trifluoromethyl group to achieve good yield (3h). Apparently, no significant H2O2 decomposition occurred at this relatively mild temperature. Methoxy and cyclic ether substituted benzoic acids (3i-3k) were also compatible substrates. For dihydrobenzofuran carboxylic acid substrate 3j, both regioisomers were obtained in 1:1 ratio (4j and 4j'). NSAID compound diflunisal (4l) could be synthesized from corresponding benzoic acid (3l). However, only trace amount of product was observed for the attempted reaction of 2,4-difluorobenzoic acid (3m). With modified conditions, 2,4-difluorobenzoic acid (3m) was hydroxylated in 72% yield by increasing the Pd(OAc)2 loading to 5%, and switching solvent to DMA. These new reaction conditions (Conditions B) were then applied to more substrates. We performed high yielding ortho-hydroxylations of benzoic acids with various substituents such as difluoro (3m-3o), fluoro (3p), nitro (3q), acetyl (3r), methoxy (3s) and 4-toluoyl (3u) groups. An ortho-bromo group remained intact during the selective C(sp2)─H hydroxylation of substrate 3t. A hydroxy group was introduced successfully to NSAID drug tolfenamic acid (3v). Estrone derived benzoic acid compound 3w was also hydroxylated in 61% yield.
Table 3.
Ligand enabled C(sp2)─H hydroxylation of benzoic acidsa
|
Conditions A: Carboxylic acid 3 (1 mmol), Pd(OAc)2 (2 mol%), L4 (4 mol%), H2O2 (35% aqueous solution, 3.5 equiv.), and K2HPO4•3H2O (1.5 equiv.) in CH3CN (3.0 mL), r.t. 24 h.; Conditions B: Carboxylic acid 3 (1 mmol), Pd(OAc)2 (5 mol%), 14 (10 mol%), H2O2 (35% aqueous solution, 3.5 equiv.), and CsOAc (1.5 equiv.) in DMA (3.0 mL), 60 °C. 24 h. Isolated yields.
60 °C
K2HPO4•3H2O instead of CsOAc, 48h.
0.5 mmol scale, r.t.
The robustness and scalability of the hydroxylation reaction was demonstrated through the large-scale reactions of several substrates (Scheme 2a). A mole-scale (206 g) reaction of ibuprofen at room temperature resulted in the formation of 179.2 g of 2ae (81% yield) even with lower loading of 1 mol% Pd(OAc)2. Similarly, large-scale reactions of 1a and 1d exhibited good yields. The hydroxylation of benzoic acids also proved to be highly scalable, with an 100 mmol scale reaction of benzoic acid (3a) affording 10.6 g of product 4a (77% yield). These results show the potential of this methodology in industrial processes. To showcase the synthetic utility of the reaction, the carboxyl acid directing group of the hydroxylated products were derivatized to various functional groups (Scheme 2b). 2-Hydroxy phenylacetic acids (2a and 2d) could be converted to the corresponding lactones (5a, 5b) and hydrobenzofurans (5c, 5d) through cyclization. Carboxylic acid directing groups were transformed to heterocycles such as the oxazoline (5e) and the benzimidazole (5f). In addition, we sought to demonstrate the utility of the hydroxylation reaction through the synthesis of analogs of natural products. The coumestans and pterocarpans are found in nature, some of which have significant biological activities.15 Since the presence of fluorine substituent can uniquely impact the biological and physical properties of compounds, we embarked on the synthesis of unnatural fluorinated coumestan and pterocarpan using building blocks prepared from the title C(sp2)─H hydroxylation reaction (Scheme 2c). The fluorinated precursors 2i and 4m were synthesized in one step from the commercially available carboxylic acids. Salicyl aldehyde 6 was synthesized from 4m in 2 steps. A Perkin reaction of 2h and 6 followed by oxidation afforded trifluorinated coumestan derivative 7 in good yield. We were also able to prepare the trifluorinated version of pterocarpene 8 by reduction of 7 followed by ring closure.
Scheme 2.

Synthetic Applications
aConditions: (i) LiAlH4, THF, 50 °C. (ii) PCC, DCM, r.t.. (iii) Ac2O, NaOAc, AcOH, 110 °C. (iv) DDQ, toluene, 120 °C. (v) LiAlH4, THF, 50 °C. (vi) I2, imidazole, PPh3, CH3CN/Et2O r.t.. (See supporting information for detailed procedures)
Preliminary mechanistic studies were conducted to gain further insight into the roles of the reaction components. A deuterium labeling experiments of 1d with D2O in the presence of L4 or L8 resulted in deuterium incorporation into the ortho-positions. However, in the presence of MPAA L1 or in the absence of ligand, H/D exchange was not observed (Scheme 3a). These results suggest that the CMD active 2-pyridone motif in bidentate ligands is critical for enabling room temperature C─H activation. The kinetic isotope effect (KIE) was measured through parallel experiments of 1d and 1d-d5 (see Supporting Information for details). The measured kH/kD value of 1.08 suggests that C─H cleavage is fast and not the rate-limiting step (Scheme 3b). The palladacycle intermediate (9) prepared from 2-methyl benzoic acid was subjected to the hydroxylation conditions and provided the product in 51% yield, consistent with the proposal that palladacycles such as 9 are active intermediates in the reaction (Scheme 3c). The kinetic experiments of the oxidation step with 14 and without ligand shows that PyriCarbox ligand can stabilize the C─H activation intermediate to ensure the smooth oxidation step with H2O2 (Figure S10). Based on these observations and previous reports,16 a Pd(II)/Pd(IV) catalytic cycle is proposed (Scheme 3d). Following ligand enabled C─H cleavage, oxidative addition of H2O2 to Pd(II) forms the high valent Pd(IV) species which subsequently undergoes reductive elimination to form the hydroxylated product. Ligand exchange with 2HX forms water as the sole byproduct and regenerates the catalyst PdX2.
Scheme 3.

Mechanistic Studies and Proposed Catalytic Cycle
3. Conclusion
In summary, we have developed a practical C(sp2)─H hydroxylation of carboxylic acid substrates using hydrogen peroxide aqueous solution as the hydroxylating reagent, enabled by a newly developed bifunctional carboxyl-pyridone (CarboxPyridone) ligand. Mechanistic studies indicate that this ligand scaffold play a crucial role in achieving room temperature C─H activation, thus permitting the development of mild reaction conditions which preclude the decomposition of hydrogen peroxide. With this new protocol, a wide range of phenylacetic acids and benzoic acids were successfully hydroxylated, providing the corresponding phenols. The practicality and scalability of the reaction was highlighted by large scale examples including a 1000 mmol scale reaction of ibuprofen, suggesting the transformation may have the potential to be applied to industrial scale processes. Furthermore, the derivatizations of phenol products and synthesis of trifluorinated coumestan and pterocarpene were achieved.
Supplementary Material
Acknowledgements
We gratefully acknowledge The Scripps Research Institute, the NIH (National Institute of General Medical Sciences grant R01GM102265) and Bristol Myers Squibb. We thank D. A. Strassfeld for editorial assistance. We also thank the Scripps Automated Synthesis Facility (ASF) and Scripps Center for Metabolomics and Mass Spectrometry for guidance on analytical methods. H.S.P thanks the Korea Foundation of Advanced Science for the predoctoral fellowship.
Footnotes
Supporting Information The Supporting Information is available free of charge on the ACS Publications website. Full experimental details and characterization of new compounds (PDF)
Notes The authors declare no competing interests.
References
- 1.(a) Quideau S; Deffieux D; Douat-Casassus C; Pouysegu L Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chem., Int. Ed 2011, 50, 586–621. [DOI] [PubMed] [Google Scholar]; (b) Albuquerque BR; Heleno SA; Oliveira MBPP; Barros L; Ferreira ICFR Phenolic Compounds: Current Industrial Applications, Limitations and Future Challenges. Food Funct. 2021, 12, 14–29. [DOI] [PubMed] [Google Scholar]; (c) Scott KA; Cox PB; Njardarson JT Phenols in Pharmaceuticals: Analysis of a Recurring Motif. J. Med. Chem 2022, 65, 7044–7072. [DOI] [PubMed] [Google Scholar]
- 2.(a) Wu W-T; Zhang L; You S-L Catalytic Asymmetric Dearomatization (CADA) Reactions of Phenol and Aniline Derivatives. Chem. Soc. Rev 2016, 45, 1570–1580. [DOI] [PubMed] [Google Scholar]; (b) Qiu ZH; Li CJ Transformations of Less-Activated Phenols and Phenol Derivatives via C–O Cleavage. Chem. Rev 2020, 120, 10454–10515. [DOI] [PubMed] [Google Scholar]
- 3.(a) The Chemistry of Phenols; Rappoport Z, Ed.; John Wiley & Sons: Chichester, U.K., 2003; pp 395–489. [Google Scholar]; (b) Liu Y; Liu S; Xiao Y Transition-Metal-Catalyzed Synthesis of Phenols and Aryl Thiols. Beilstein J. Org. Chem 2017, 13, 589–611. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Iqbal Z; Joshi A; Ranjan De S Recent Advancements on Transition-Metal-Catalyzed, Chelation-Induced ortho-Hydroxylation of Arenes. Adv. Synth. Catal 2020, 362, 5301–5351 [Google Scholar]
- 4.(a) Ullrich R; Hofrichter M Enzymatic Hydroxylation of Aromatic Compounds. Cell. Mol. Life Sci 2007, 64, 271–293. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lewis JC; Coelho PS; Arnold FH Enzymatic Functionalization of Carbon-Hydrogen Bonds. Chem. Soc. Rev 2011, 40, 2003–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dong J; Fernández-Fueyo E;Hollmann F; Paul CE; Pesic M; Schmidt S; Wang Y; Younes S; Zhang W Biocatalytic Oxidation Reactions: A Chemist’s Perspective. Angew. Chem., Int. Ed 2018, 57, 9238–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Sun ZH; Fridrich B; de Santi A; Elangovan S; Barta K Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev 2018, 118, 614–678. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schutyser W; Renders T; Van den Bosch S; Koelewijn S-F; Beckham GT; Sels BF Chemicals from Lignin: An Interplay of Lignocellulose Fractionation, Depolymerisation, and Upgrading. Chem. Soc. Rev 2018, 47, 852–908. [DOI] [PubMed] [Google Scholar]
- 6.(a) Willis MC Palladium-Catalyzed Coupling of Ammonia and Hydroxide with Aryl Halides: The Direct Synthesis of Primary Anilines and Phenols. Angew. Chem., Int. Ed 2007, 46, 3402–3404. [DOI] [PubMed] [Google Scholar]; (b) Enthaler S; Company A Palladium-Catalysed Hydroxylation and Alkoxylation. Chem. Soc. Rev 2011, 40, 4912–4924. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ohkubo K; Fujimoto A; Fukuzumi S Visible-Light-Induced Oxygenation of Benzene by the Triplet Excited State of 2,3-Dichloro-5,6-dicyano-p-Benzoquinone. J. Am. Chem. Soc 2013, 135, 5368–5371. [DOI] [PubMed] [Google Scholar]; (b) Zheng Y-W; Chen B;Ye P; Feng K;Wang W; Meng Q-Y; Wu L-Z; Tung C-H Photocatalytic Hydrogen Evolution Cross-Couplings: Benzene C─H Amination and Hydroxylation. J. Am. Chem. Soc 2016, 138, 10080–10083. [DOI] [PubMed] [Google Scholar]; (c) Morimoto Y; Bunno S; Fujieda N; Sugimoto H; Itoh S Direct Hydroxylation of Benzene to Phenol Using Hydrogen Peroxide Catalyzed by Nickel Complexes Supported by Pyridylalkylamine Ligands. J. Am. Chem. Soc 2015, 137, 5867–5870. [DOI] [PubMed] [Google Scholar]; (d) Masferrer-Rius E; Borrell M; Lutz M; Costas M; Gebbink RJMK Aromatic C─H hydroxylation reactions with hydrogen peroxide catalyzed by bulky Manganese complexes. Adv. Synth. Catal 2021, 363, 3783–3795. [Google Scholar]; (e) Tsuji T; Zaoputra AA; Hitomi Y; Mieda K; Ogura T; Shiota Y; Yoshizawa K; Sato H; Kodera M Specific Enhancement of Catalytic Activity by a Dicopper Core: Selective Hydroxylation of Benzene to Phenol with Hydrogen Peroxide. Angew. Chem., Int. Ed 2017, 56, 7779–7782. [DOI] [PubMed] [Google Scholar]; (f) Shoji O; Kunimatsu T; Kawakami N; Watanabe Y Highly selective hydroxylation of benzene to phenol by wild-type cytochrome P450BM3 assisted by decoy molecules. Angew. Chem., Int. Ed 2013, 52, 6606–6610. [DOI] [PubMed] [Google Scholar]; (g) Cheng L; Wang H; Cai H; Zhang J; Gong X; Han W Iron-Catalyzed Arene C─H Hydroxylation. Science 2021, 374, 77–81. [DOI] [PubMed] [Google Scholar]
- 8.(a) For selected examples of palladium catalyzed hydroxylation of arenes, see: Kim SH; Lee HS; Kim SH; Kim JN Regioselective ortho-Hydroxylation of Aryl Moiety of 2-Arylpyridines using Pd(OAc)2/Oxone in PEG-3400/tert-BuOH. Tetrahedron Lett. 2008, 49, 5863–5866. [Google Scholar]; (b) Yan Y; Feng P; Zheng Q-Z; Liang Y-F; Lu J-F; Cui Y; Jiao N PdCl2 and N-hydroxyphthalimide Co-Catalyzed C(sp2)─H Hydroxylation by Dioxygen Activation. Angew. Chem., Int. Ed 2013, 52, 5827–5831. [DOI] [PubMed] [Google Scholar]; (c) Liang Y-F; Wang X; Yuan Y; Liang Y; Li X; Jiao N Ligand-Promoted Pd-catalyzed Oxime Ether Directed C─H Hydroxylation of Arenes. ACS Catal. 2015, 5, 6148–6152. [Google Scholar]; (d) Dong J; Liu P; Sun P Palladium-Catalyzed Aryl C(sp2)─H Bond Hydroxylation of 2-Arylpyridine Using TBHP as Oxidant. J. Org. Chem 2015, 80, 2925–2929. [DOI] [PubMed] [Google Scholar]; (e) Yamaguchi T; Yamaguchi E; Tada N; Itoh A Direct Ortho-Hydroxylation of 2-Phenylpyridines Using Palladium(II) Chloride and Hydrogen Peroxide. Adv. Synth. Catal 2015, 357, 2017–2021. [Google Scholar]; (f) Chen X-YY; Ozturk S; Sorensen EJ Pd-Catalyzed Ortho C─H Hydroxylation of Benzaldehydes Using a Transient Directing Group. Org. Lett 2017, 19, 6280–6283. [DOI] [PubMed] [Google Scholar]; (g) Zhang Y-H; Yu J-Q Pd(II)-Catalyzed Hydroxylation of Arenes with 1 Atm of O2 or Air. J. Am. Chem. Soc 2009, 131, 14654–14655. [DOI] [PubMed] [Google Scholar]; (h) Huang CH; Ghavtadze N; Chattopadhyay B; Gevorgyan V Synthesis of Catechols from Phenols via Pd-Catalyzed Silanol-Directed C─H Oxygenation. J. Am. Chem. Soc 2011, 133, 17630–17633. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Shan G; Yang X; Ma L; Rao Y Pd-Catalyzed C─H Oxygenation with TFA/TFAA: Expedient Access to Oxygen-Containing Heterocycles and Late-Stage Drug Modification. Angew. Chem., Int. Ed 2012, 51, 13070–13074. [DOI] [PubMed] [Google Scholar]; (j) Mo F; Trzepkowski LJ; Dong G Synthesis of Ortho-Acylphenols through the Palladium-Catalyzed Ketone-Directed Hydroxylation of Arenes. Angew. Chem., Int. Ed 2012, 51, 13075–13079. [DOI] [PubMed] [Google Scholar]; (k) For review on this topic, see: Saha D; Das P; Biswas P; Guin J Synthesis of Phenolic Compounds via Palladium Catalyzed C─H Functionalization of Arenes. Chem. Asian J 2019, 14, 4534–4548. [DOI] [PubMed] [Google Scholar]
- 9.(a) For selected examples of palladium catalyzed acetoxylation of arenes, see: Dick AR; Hull KF; Sanford MS A Highly Selective Catalytic Method for the Oxidative Functionalization of C─H Bonds. J. Am. Chem. Soc 2004, 126, 2300–2301. [DOI] [PubMed] [Google Scholar]; (b) Wang GW; Yuan TT; Wu XL Direct Ortho-Acetoxylation of Anilides via Palladium-Catalyzed sp2 C─H Bond Oxidative Activation. J. Org. Chem 2008, 73, 4717–4720. [DOI] [PubMed] [Google Scholar]; (c) Vickers CJ; Mei TS; Yu J-Q Pd(II)-Catalyzed o-C─H Acetoxylation of Phenylalanine and Ephedrine Derivatives with MeCOOOtBu/Ac2O. Org. Lett 2010, 12, 2511–2513. [DOI] [PubMed] [Google Scholar]; (d) Yang G; Lindovska P;Zhu D; Kim J;Wang P; Tang RY; Movassaghi M;Yu J-Q Pd(II)-Catalyzed meta-C─H Olefination, Arylation, and Acetoxylation of Indolines Using a U-Shaped Template. J. Am. Chem. Soc 2014, 136, 10807–10813. [DOI] [PubMed] [Google Scholar]; (e) Li Y-Q;Yang Q-F; Fang P; Mei T-S; Zhang D Palladium-Catalyzed C(sp2)─H Acetoxylation via Electrochemical Oxidation. Org. Lett 2017, 19, 2905–2908. [DOI] [PubMed] [Google Scholar]; (f) Dastbaravardeh N; Toba T; Farmer ME; Yu J-Q, Monoselective o-C─H Functionalizations of Mandelic Acid and α-Phenylglycine. J. Am. Chem. Soc 2015, 137, 9877–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wang X; Wang H; Zhou C; Yang L; Fu L; Li G Native Carboxyl Group-assisted C─H Acetoxylation of Hydrocinnamic and Phenylacetic Acids. Chem. Commun 2022, 58, 4993–4996. [DOI] [PubMed] [Google Scholar]
- 10.Li Z; Wang Z; Chekshin N; Qian S; Qiao JX; Cheng PT; Yeung K-S; Ewing WR; Yu J-Q A Tautomeric Ligand Enables Directed C─H Hydroxylation with Molecular Oxygen. Science 2021, 372, 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clerici MG; Kholdeeva OA Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Wiley: Hoboken, NJ, 2013. [Google Scholar]
- 12.(a) Wang D; Engle KM; Shi B; Yu J Ligand-Enabled Reactivity and Selectivity in a Synthetically Versatile Aryl C─H Olefination. Science 2010, 327, 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Engle KM; Wang D-H; Yu J-Q Ligand-Accelerated C─H Activation Reactions: Evidence for A Switch of Mechanism. J. Am. Chem. Soc 2010, 132, 14137–14151. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Engle KM; Thuy-Boun PS; Dang M; Yu J-Q Ligand-Accelerated Cross-Coupling of C(sp2)─H Bonds with Arylboron Reagents. J. Am. Chem. Soc 2011, 133, 18183–18193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Wang P; Verma P; Xia G; Qiao JX; Tao S; Cheng PTW; Poss MA; Farmer ME; Yeung K-S; Yu J-Q Ligand-Accelerated Non-directed C─H Functionalization of Arenes. Nature 2017, 551, 489–404. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) For recent discoveries on bidentate pyridone ligands, see: Wang Z; Hu L; Chekshin N; Zhuang Z; Qian S; Qiao JX; Yu J-Q Ligand-Controlled Divergent Dehydrogenative Reactions of Carboxylic Acids via C─H Activation. Science 2021, 374, 1281–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chan HSS; Yang J-M; Yu J-Q Catalyst-Controlled Site-Selective Methylene C─H Lactonization of Dicarboxylic Acids. Science 2022, 376, 1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sheng T; Zhuang Z; Wang Z; Hu L; Herron AN; Qiao JX; Yu J-Q One-Step Synthesis of β-Alkylidene-γ-lactones via Ligand-Enabled β,γ-Dehydrogenation of Aliphatic Acids. J. Am. Chem. Soc 2022, 144, 12924–12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian SQ; Li Z-Q; Li M; Wisniewski SR; Qiao JX; Richter JM; Ewing WR; Eastgate MD; Chen JS; Yu J-Q Ligand-Enabled Pd(II)-catalyzed C(sp3)─H Lactonization Using Molecular Oxygen as Oxidant. Org. Lett 2020, 22, 3960–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selvam C; Jordan BC; Prakash S; Mutisya D; Thilagavathi R Pterocarpan Scaffold: A Natural Lead Molecule with Diverse Pharmacological Properties. Eur. J. Med. Chem 2017, 128, 219–236. [DOI] [PubMed] [Google Scholar]
- 16.(a) Giri R; Liang J; Lei J-G; Li J-J; Wang D-H; Chen X; Naggar IC; Guo C; Foxman BM; Yu J-Q Pd-Catalyzed Stereoselective Oxidation of Methyl Groups by Inexpensive Oxidants under Mild Conditions: A Dual Role for Carboxylic Anhydrides in Catalytic C─H Bond Oxidation. Angew. Chem., Int. Ed 2005, 44, 7420–7424. [DOI] [PubMed] [Google Scholar]; (b) Oloo W; Zavalij PY; Zhang J; Khaskin E; Vedernikov AN Preparation and C–X Reductive Elimination Reactivity of Monoaryl PdIV–X Complexes in Water (X ═ OH, OH2, Cl, Br). J. Am. Chem. Soc 2010, 132, 14400–14402. [DOI] [PubMed] [Google Scholar]; (c) Zhuang Z; Herron AN; Fan Z; Yu J-Q Ligand-Enabled Monoselective β-C(sp3)─H Acyloxylation of Free Carboxylic Acids Using A Practical Oxidant. J. Am. Chem. Soc 2020, 142, 6769–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.