

Summary
Although vast numbers of putative gene regulatory elements have been cataloged, the sequence motifs and individual bases that underlie their functions remain largely unknown. Here, we combine epigenetic perturbations, base editing, and deep learning to dissect regulatory sequences within the exemplar immune locus encoding CD69. We converge on a ∼170 base interval within a differentially accessible and acetylated enhancer critical for CD69 induction in stimulated Jurkat T cells. Individual C-to-T base edits within the interval markedly reduce element accessibility and acetylation, with corresponding reduction of CD69 expression. The most potent base edits may be explained by their effect on regulatory interactions between the transcriptional activators GATA3 and TAL1 and the repressor BHLHE40. Systematic analysis suggests that the interplay between GATA3 and BHLHE40 plays a general role in rapid T cell transcriptional responses. Our study provides a framework for parsing regulatory elements in their endogenous chromatin contexts and identifying operative artificial variants.
Keywords: regulatory elements, CRISPRi, base editing, transcription factor, immune response
Graphical abstract
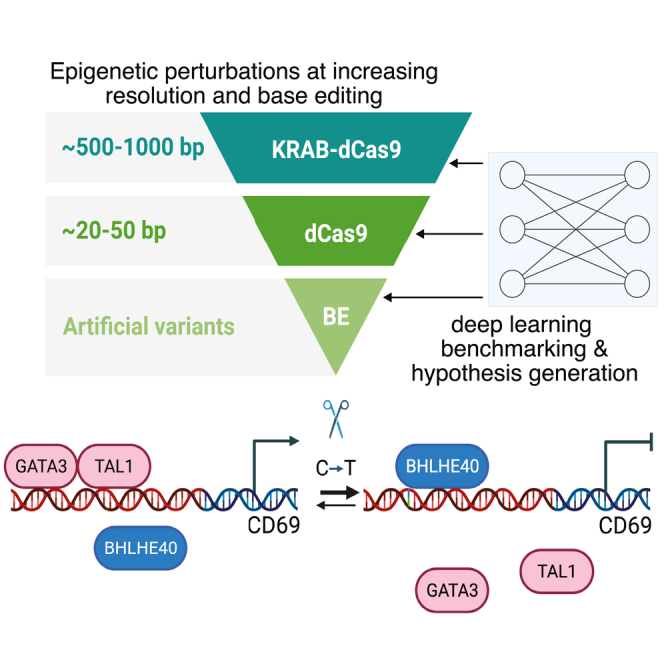
Highlights
-
•
Base editing and deep learning pinpoint individual bases that alter gene expression
-
•
An artificial C-to-T variant in a regulatory element suppresses CD69 expression
-
•
The artificial C-to-T alters the balance of transcription factor binding
-
•
Global interplay between GATA3 and BHLHE40 regulates immune genes and T cell states
Chen and Javed et al. integrated epigenetic perturbations, base editing, and deep learning to dissect a CD69 enhancer. They identified a single C-to-T mutation that impacts gene expression by altering binding of GATA3, TAL1, and BHLHE40. Extending their analysis, they find evidence of a broader interaction between GATA3 and BHLHE40 during T cell activation.
Introduction
Genome-wide maps of chromatin state and transcription factor (TF) binding have nominated more than a million cell type-specific regulatory elements (REs) in the human genome as potential context-specific regulators of gene expression.1,2,3 A critical next step is to determine their functions and sequence determinants. Computational tools that predict functional bases and/or gene targets are rapidly evolving but require systematic benchmarking against perturbational data.4,5 Massively parallel reporter assays (MPRAs) enable high-throughput analysis of sequence determinants within REs but are based on exogenously introduced constructs that do not recapitulate the native chromatin contexts.6,7,8,9 CRISPR interference (CRISPRi) with fusions between dCas9 and the KRAB repressor provides a means to suppress an RE in its native context and evaluate consequent transcriptional changes.10,11,12,13,14 Traditional CRISPR-based genetic perturbations offer increased resolution15,16 but incur variable sequence changes due to heterogeneity of insertions or deletions (indels) after DNA repair. Base editors fused to a nickase Cas9 (hereafter referred to as base editors) introduce single base variants, often without frameshifts or indels. They have been used to characterize coding variants17,18,19,20,21 and are valuable when systematically applied to noncoding REs.
In this study, we integrated CRISPRi, dCas9, and base editing with computational predictions to parse noncoding regulatory sequences in the CD69 locus. We identified a ∼170 bp interval within a ∼1,500 bp upstream enhancer that plays a key role in regulating gene expression. Within this interval, base editing and deep learning converge upon a critical C at chr12:9,764,948 (hg38), where a C-to-T transition reduces element accessibility and CD69 expression. We show that this C-to-T base edit ablates a GATA3 binding site, thereby displacing a GATA3-TAL1 activating complex and increasing the association of the BHLHE40 repressor across the element and CD69 promoter. Systematic analysis of chromatin accessibility and TF binding during T cell activation supports a global role for interplay between GATA3 and BHLHE40 in immune gene responses and T cell polarization.
Results
Resolving functional bases within immune REs
To dissect functional sequences within REs, we established a workflow combining chromatin profiling, deep learning, CRISPRi, dCas9, and base editing (Figure 1A). We combined assay for transposase-accessible chromatin using sequencing (ATAC-seq) accessibility maps with deep learning models to predict REs and functional sequences that regulate inducible gene expression in T cells. We then incorporated CRISPRi, dCas9 interference, and base editing to directly test the regulatory functions of sequences and individual bases (Figure 1A).
Figure 1.

Integrative analysis of the CD69 regulatory landscape
(A) Schematic depicting characterization of the CD69 locus using successive functional perturbations and deep learning.
(B) Genomic tracks depict accessibility of the CD69 locus in primary CD4+ T cells and Jurkat cells, without or with stimulation (PMA/ionomycin). Enformer signal track (summed model gradient) shows the predicted contribution of underlying sequence to CD69 transcriptional output in Jurkat cells. Gray bars depict differentially accessible ATAC peaks in stimulated Jurkat cells relative to resting (FDR < 0.25). CRISPRi sgRNA positions are also indicated. ATAC signal corresponds to reads per genomic content (RPGCs).
(C) Flow cytometry of CD69 expression in Jurkat cells targeted with the indicated CRISPRi sgRNA following a stimulation time course. Samples gated on the live lentiviral transduced population post-puromycin selection.
(D) Expanded view of the absolute value of the Enformer signal (gradient) as described in (B) at single base resolution over RE-4.
(E) Enrichment of dCas9 sgRNAs in CD69+ Jurkat cells relative to CD69− cells (y axis; log2 odds ratio of normalized sgRNA reads). sgRNA positions are plotted along the x axis according to their 5′ starting position on the positive strand. Each data point represents mean ± SEM.
(F) Enrichment/depletion plot of cytidine base editor (CBE) sgRNAs in CD69+ Jurkat cells relative to CD69− cells (as in E). The CBE can edit Cs at base positions 2–11 opposite the NGG PAM, with a strong preference for positions in the central 2–8 base window. sgRNA positions are plotted along the x axis according to their 5′ starting position on the positive strand. Each data point represents mean ± SEM.
For (C), (E), and (F), data represent 2–3 biological independent experiments. A 170 bp region critical for CD69 activation is denoted (D–F, light red).
We focused on the CD69 locus, which encodes a key molecule for T cell signal transduction and tissue residency.22,23 CD69 expression is rapidly induced upon stimulation by T cell receptor crosslinking or PMA/ionomycin in both CD4+ T cells and the Jurkat T cell line (Figures S1A and S1B). Chromatin accessibility maps nominated putative regulatory sites that gain accessibility upon stimulation in primary T cells and Jurkat cells (Figure 1B). We complemented these data with predictions from the Enformer model5 trained on chromatin maps and cap analysis of gene expression (CAGE)-seq data.1,24 We also refined the predictions by fine-tuning the Enformer model to predict differential accessibility between resting and stimulated Jurkat cells (Methods). Genomic intervals corresponding to the promoter, an intronic region, the 3′ UTR, and an interval located ∼4 kb upstream of the transcription start site (TSS) were predicted to have a strong impact on CD69 transcriptional induction (Figure 1B).
We used CRISPRi to test the functional impact of seven candidate REs across the CD69 locus, including the promoter (sgProm), the intronic element (sgInt), the predicted upstream element (RE-4), and four other sites in the locus that also gained accessibility upon T cell activation (RE-1, RE-2, RE-3, and RE-5) (Figure 1B; Table S1). We infected Jurkat cells with lentiviral constructs containing KRAB-dCas9 and sgRNAs, selected positive cells with puromycin, applied PMA/ionomycin stimulation, and measured CD69 surface protein expression by flow cytometry. We found that sgRNAs targeting the promoter and RE-4 had the strongest suppressive effects on CD69 induction (Figures 1C and S1C), while sgRNAs targeting the TSS-proximal RE-3 had a weaker effect (Figures 1C and S1C). The impact of the respective CRISPRi perturbations largely correlated with the Enformer predictions (Figure S1D). RE-4 corresponds to a DNase hypersensitive site bound by multiple TFs that has scored in a luciferase reporter assay and a CRISPR activation screen.1,25,26 Whereas chromatin accessibility over RE-4 spans ∼1.4 kb, the Enformer predictions highlighted a specific ∼170 bp sequence interval within RE-4 as most critical for RE-4 accessibility and CD69 expression (Figures 1D and S1E).
To resolve the functional sequences within these elements and test the Enformer prediction, we designed a library of 101 sgRNAs that target sequences across RE-3 and RE-4 (Figures S2A and S2B; Table S2). We began with a pooled dCas9 assay, reasoning that dCas9 would specifically occlude TFs overlapping target sites and thus affect a narrower interval than KRAB-dCas9.27 We infected Jurkat cells with a pooled lentiviral CRISPR library composed of dCas9 and the 101 sgRNAs, selected for puromycin resistance and stimulated with PMA/ionomycin for 5 h. We then isolated genomic DNA from pre-sorted and sorted CD69− and CD69+ subsets (Figure S2C) and amplified the sgRNA cassettes for sequencing. The relative effect of each sgRNA on CD69 expression was calculated based on its enrichment/depletion in CD69+ relative to CD69− libraries. Multiple sgRNAs within the ∼1.7 kb tiled region suppressed CD69 activation (Figures 1E and S2D).
To pinpoint individual functional bases in these REs, we complemented the dCas9 tiling with cytidine base editor (CBE) and adenine base editor (ABE) screens. We infected Jurkat cells with lentiviral constructs containing CBE or ABE and the same pool of 101 sgRNAs (Figures S2A and S2B; Table S2). We stimulated and sorted the cells and then sequenced the sgRNA cassettes from pre-sorted, CD69−, and CD69+ subsets (Figure S2C). Multiple sgRNAs scored in these screens as reducing CD69 activation (Figures 1F and S2E–S2G). Notably, the CBE and dCas9 perturbations both pinpointed an ∼150 bp interval within RE-4 that closely corresponded to the interval highlighted by Enformer as critical for CD69 expression (Figures 2A and 1D). Several ABE hits in or near this interval also affected CD69 induction but with lower fold enrichment, indicative of a lower signal-to-noise ratio (Figure S2G). In addition to pinpointing this key functional interval in RE-4, the individual bases nominated by Enformer frequently coincided with sgRNAs that scored in the CBE screen as regulators of CD69 expression (Figures S2H and S2I). The predictive value of Enformer was further supported at a genome-wide level by an enrichment of sequence motifs recognized by TFs with established roles in T cell biology (Figure S2J).
Figure 2.

A critical sequence interval within RE-4 influences CD69 expression
(A) Enrichment and genomic position of sgRNAs in dCas9 and CBE tiling screens as in Figures 1E and 1F limited to the central portion of RE-4. Dashed gray lines correspond to expected C-to-T edit positions for CBE-sgRNAs sg#70 and sg#48.
(B) Transcription factor motif locations (grouped by broad motif class) for key immune regulators shown across the same interval as in (A) (STAR Methods). Dark gray areas represent overlapping motifs. Representative PWM logo plots for each motif class are provided on the right-hand side.
(C) Zoomed-out view of the CD69 locus shows CBE sgRNA depletion (red boxes correspond to top-scoring guides in the sg#70 and sg#48 intervals), common SNPs (black vertical stripes), eQTLs (blue vertical stripes),28 and PhastCon100 conservation score (green stripes).
Further analysis of the implicated RE-4 sequence interval revealed multiple TF motifs relevant to immune function, including GATA, bHLH/e-box, TCF, ETS, and STAT (Figure 2B). Notably, a second top-scoring interval from the CBE and dCas9 screens, centered at sg#48, showed similar TF motif enrichments (Figure 2A; chr12:9,765,200-9,765,310). We also scanned the locus for annotated expression quantitative trait loci (eQTLs). However, the implicated RE-4 intervals are highly conserved evolutionarily and devoid of natural variation in the human population and thus are invisible to eQTL analysis (Figure 2C).28 These findings highlight the importance of engineered variants for parsing highly conserved REs, which tend to be depleted of natural variants.
A single artificial variant alters TF binding patterns and suppresses CD69
We next sought to validate individual base edits and their transcriptional consequences. We infected Jurkat cells with a CBE vector containing either the top-ranked sgRNA (sg#70), a highly ranked sgRNA in an adjacent interval with several scoring sgRNAs (sg#48), or a control sgRNA (sgCtrl) (Figures 1E, 1F, and 2A). We confirmed that CBE-sg#70 strongly suppressed CD69 induction upon stimulation, while sg#48 had a lower but still significant effect (unpaired t test, p < 0.0001), consistent with our tiling data (Figures 3A and S3A–S3C). We next amplified and sequenced the target region from genomic DNA isolated from Jurkat cells infected with CBE-sg#70.29 CBE-sg#70 is predicted to incur C-to-T transitions at positions 948 and/or 952 within RE-4 (chr12: 9,764,948 and 9,764,952). In unsorted cells, C-948 was replaced by T on ∼57% of alleles. The proportion of C-948-edited alleles was higher in sorted CD69− Jurkat cells (67%) and lower in the CD69+ population (53.6%), consistent with a suppressive effect on CD69 induction (Figure 3B). In contrast, edits to the other candidate site, C-952, were less frequent (14.4% at baseline, 16.6% in CD69−, 13.9% in CD69+; Figure 3B). These results indicate that the single C-948-to-T edit strongly impacts transcriptional induction of CD69 in response to stimulation.
Figure 3.

Top-scoring base edits target competitive transcription factor binding sites
(A) Flow cytometry plots of CD69 signal for CBE-sgCtrl and CBE-sg#70 Jurkat cells under resting or stimulated conditions. Bar plot depicts the proportion of CD69+ cells in CBE-sgCtrl (gray) and CBE-sg#70 (red) after stimulation. P value based on unpaired t test, ∗∗p < 0.01. Data are from 4 independent experiments each with 2–3 technical replicates, mean ± SEM.
(B) Table depicts frequency of incurred base edits in CBE-sg#70-infected Jurkat cells. PCR amplicons from unsorted, CD69−, and CD69+ populations were sequenced. Consensus sequence is shown along with stacked bars that depict the proportions of C and T bases in the sequencing data (numbers indicate the percentage of alleles with a C-to-T edit). Shaded boxes indicate the sg#70 target sequence.
(C) Chromatin accessibility shown over the CD69 locus for stimulated CBE-sg#70 (red) and CBE-sgCtrl (gray) Jurkat cells. Bar plot depicts the mean ATAC-seq signal over RE-4 (TMM normalized counts per million; CPM). P value based on unpaired t test, ∗p < 0.05. Data are from 3 replicates, mean ± SEM.
(D) Enformer signal (letter height) for the sg#70 target region corresponds to the model gradient with respect to predicted RE-4 accessibility in Jurkat cells and indicates the predicted impact of each base on RE-4 accessibility. The sgRNA directly coincides with a GATA motif and two e-box/bHLH sites and incurs an edit that disrupts the former (vertical dashed line).
(E) Volcano plot depicts gene expression fold change (x axis) and significance (y axis) for transcription factor (TF) genes in stimulated Jurkat cells relative to resting cells. Labels identify differential GATA (red) and bHLH/e-box (blue) family members with significant differential expression at FDR < 0.05.
(F) Genomic tracks for the CD69 locus depict chromatin accessibility (ATAC), H3K27 acetylation (H3K27ac), GATA3 binding, TAL1 binding, and BHLHE40 binding in CBE-sgCtrl (gray) and CBE-sg#70 (red) Jurkat cells. For ChIP-seq, the y axis represents the −log10(p value) relative to input controls. For ATAC-seq, the y axis represents the RPGC normalized signal.
Jurkat cells in (A)–(C) and (F) were stimulated with PMA/ionomycin for 2 h.
We also examined the impact of the C-948 edit on chromatin accessibility. ATAC-seq profiles revealed reduced RE-4 accessibility in cells harboring the CBE-sg#70 construct relative to CBE controls (Figures 3C and S3D). The effect was most significant for RE-4 (Figure S3E; false discovery rate [FDR] < 0.05; STAR Methods) in the CD69 locus, and we did not observe changes in the vicinity of other activation associated genes such as CD28 and NR4A1 (Figure S3F). Hence, the single base substitution at position C-948 reduces RE-4 accessibility and suppresses CD69 induction in stimulated Jurkat cells.
We next considered the mechanism that underlies the potent effect of this single base mutation. C-948 directly overlaps a GATA motif predicted by the Enformer model to impact element accessibility in Jurkat cells (Figures 3D and S3G). The C-948-to-T edit disrupts a critical position in this motif. Two adjacent e-box/bHLH motifs were also highlighted by the fine-tuned model gradients, one of which is located at ideal spacing to complete a GATA:TAL1 binding site. Among cognate TFs, GATA3 is highly expressed in Jurkat cells (Figure 3E), up-regulated upon stimulation, and broadly implicated in T cell lineage commitment.30
We therefore used chromatin immunoprecipitation sequencing (ChIP-seq) to map GATA3, TAL1, and the enhancer-associated chromatin mark H3K27 acetylation (H3K27ac). This confirmed strong binding of GATA3 and TAL1 to RE-4 (Figure 3F). Remarkably, binding of both TFs was entirely lost in sg#70-edited Jurkat cells, consistent with the disruption of the GATA motif. Loss of TF binding was accompanied by reduced accessibility and acetylation in the edited cells. We also confirmed that GATA3 knockout suppressed CD69 induction in stimulated Jurkat cells (Figure S4A), while overexpression of GATA3 or TAL1 increased CD69 induction (Figures S4B and S4C). These data support key roles for the GATA:TAL1 motif and corresponding TFs in CD69 induction, as is consistent with prior studies that have associated these TFs with transcriptional activation in T cells or other hematopoietic lineages.30,31,32,33
The coordinated changes in chromatin state and transcription suggested that the displacement of GATA3/TAL1 may be accompanied by additional regulatory events, potentially including recruitment of transcriptional repressors. Since the critical interval contains multiple e-box/bHLH motifs (Figure 2B), we considered BHLHE40, which is highly expressed in Jurkat cells and strongly induced upon stimulation (Figure 3E). BHLHE40 is an established T cell regulator that can function as a transcriptional repressor.34,35,36,37
We mapped BHLHE40 by ChIP-seq in wild-type and sg#70-edited Jurkat cells (Figure 3F). In wild-type cells, BHLHE40 binding was largely confined to a punctate site over RE-4. However, in the edited cells, diffuse binding was evident across an ∼8 kb region encompassing RE-4 and the CD69 promoter. Thus, in addition to displacing the GATA3-TAL1 activating complex, the C-948-to-T edit promotes the association of a key T cell repressor across the locus. To further investigate, we evaluated the impact of BHLHE40 perturbations. BHLHE40 overexpression suppressed CD69 induction in both control and CBE-sg#70-edited Jurkat cells, while knockdown increased induction (Figures 4A and 4B). Overexpression of BHLHE41, a homologous TF of BHLHE40, had no effect on CD69 expression (Figure S4D). Consistent with its impact on CD69 transcription, overexpression increased BHLHE40 binding over RE-4 while reducing accessibility and H3K27ac over the element (Figures 4C and 4D). Interestingly, BHLHE40 overexpression also resulted in a peak of the repressive chromatin mark H3K27 trimethylation (H3K27me3), consistent with a direct repressive impact on the element (Figure 4D).
Figure 4.

GATA3-BHLHE40 interaction impacts global T cell transcriptional responses
(A) Flow cytometry plots of CD69 signal for stimulated Jurkat cells transduced with CBE-sg#70 and a BHLHE40 overexpression (OE) construct (BHLHE40-OE) or with corresponding controls (sgCtrl and Ctrl-lentivirus [LV], respectively). Bar plot depicts the proportion of CD69+ cells in each condition. P value based on unpaired t test, ∗∗∗∗p < 0.0001. Data are from 3 independent experiments with 2–3 technical replicates, mean ± SEM.
(B) Flow cytometry plots of CD69 signal for stimulated Jurkat cells transduced with shBHLHE40-1, shBHLHE40-2, or shCtrl RNA LV. Flow plots gated on GFP+ cells. Bar plot depicts the proportion of CD69+ cells in each condition. P value based on unpaired t test, ∗∗∗p < 0.001, ∗p < 0.05.
(C) Chromatin accessibility in the CD69 locus for CBE-sg#70 Jurkat cells transduced with either BHLHE40-OE LV (light blue) or control (gray). Cells were stimulated with PMA/ionomycin. ∗FDR < 0.25. Bar plot data are from 2 replicates, mean ± SEM ATAC-seq signal over RE-4 (TMM normalized CPM).
(D) Genomic tracks for the CD69 locus depict BHLHE40 binding, H3K27ac signal, and H3K27me3 in Ctrl-LV (gray) and BHLHE40-OE (blue) Jurkat cells. The y axis represents the −log10(p value) relative to input controls.
(E) Venn diagram (left) depicts the overlap between GATA3- and BHLHE40-bound sites within 25 kb of a TSS (STAR Methods), while (right) depicts overlap between genes with a GATA3- or BHLHE40-bound site within 25 kb of the annotated TSS. Bound sites were defined based on IDR ChIP-seq peaks for either factor that overlapped an H3K27ac peak in Ctrl-LV Jurkat cells.
(F) Heatmap shows differentially expressed genes with GATA3 or BHLHE40 binding within 25 kb of the TSSBHLHE40-OE Jurkat cells relative to control. Cells were stimulated with PMA/ionomycin. Select immune genes and hits from gene set enrichment analysis are shown on the right.
Further evidence of interplay between these TFs emerged in our examination of the second interval identified in our dCas9 and CBE screens. The top-ranked edit in this interval (sg#48) also incurs a C-to-T edit that disrupts a GATA motif flanked by a bHLH/e-box motif (Figures 2A and 2B). Hence, this second functional edit may similarly affect the interplay among GATA3, TAL1 and BHLHE40 in the regulation of CD69.
Global interplay between BHLHE40 and GATA3 in T cell responses
Finally, we considered whether BHLHE40 may contribute more broadly to immune gene regulation and T cell phenotypes. We found that BHLHE40 overexpression (OE) led to a global reduction in chromatin accessibility (Figure S5A). BHLHE40 OE also altered the expression of multiple immune genes, up-regulating Th1 and effector T cell-related genes and down-regulating Th2 and naive/stemness pathways (Figures S5B and S5C). ChIP-seq analysis further indicated that BHLHE40 binds REs proximal to many of these deregulated genes, suggesting that the regulation is at least in part direct (Figures S5D and S5E). Notably, the global expression programs associated with BHLHE40 OE are in opposition to those reported for GATA3,31,38,39,40 which is instead implicated in Th2 differentiation (e.g., GATA3, interleukin 21 receptor [IL-21R], IL32) and naive/stemness T cell phenotypes (e.g., BACH2, CD28).
We therefore compared the REs and genes targeted by the respective TFs. First, we evaluated the correspondence in H3K27ac-marked REs bound by the respective TFs genome-wide. This revealed 16,323 GATA3 and 21,146 BHLHE40 sites, with a substantial overlap of 10,623 (28%), consistent with extensive interplay between these TFs (Figure 4E; Table S3). We next collated the set of 5,478 candidate target genes whose promoters were located near bound sites for both factors (STAR Methods; Table S3). These refined targets were also enriched for genes that were responsive to BHLHE40 OE (Figure S5F). However, the co-bound genes were relatively biased toward effector T cell pathways over Th1/Th2 differentiation pathways (Figure 4F). Overall, these data clarify connections from GATA3 and BHLHE40 to several T cell transcriptional programs and in particular support direct roles for the opposing regulators in the rapid transcriptional responses associated with effector T cell biology.
Discussion
Resolving functional sequences within the vast numbers of putative REs in the human genome is a critical challenge in human genetics. Here, we integrate chromatin maps, deep learning, epigenetic editing, and base editing to parse sequences that control an exemplar inducible gene in Jurkat T cells. Top-scoring base edits clustered in an evolutionarily conserved interval within a CD69 enhancer that was also highlighted by the deep learning model. Integrating regulatory predictions with TF ChIP-seq revealed that these edits disrupt cooperative TAL1 and GATA3 binding, thereby opening the locus to opposing TFs such as BHLHE40. Genome-wide analysis suggests a broader interaction between GATA3 and BHLHE40 in regulating immune genes and T cell phenotypes.
Our results emphasize the importance of epigenetic perturbations and introducing artificial sequence variants for characterizing regulatory sequences, which tend to be highly conserved and may be invisible to methods that rely on natural genetic variation. The approach utilized here also has implications for future studies. First, there remains a considerable gap between the throughput of current approaches and the eventual goal of deciphering the regulatory code of the entire human genome. Functional perturbations will need to be combined with computational approaches, which can help prioritize regions, such as the deep learning model incorporated here. Second, different epigenetic and genetic editing tools may be combined to resolve functional sequences more effectively than either tool alone. Epigenetic perturbations with KRAB-dCas9 allow sensitive identification of putative TF binding regions, which may then be precisely resolved with dCas9 and base editors.
Our study also highlights the interplay between two key TFs, GATA3 and BHLHE40. Two top-scoring C-to-T base edits that suppress the CD69 response both appear to act by shifting the balance away from coordinate GATA3-TAL1 binding toward increased BHLHE40 association with the regulatory sequences. The lack of natural variation makes this region invisible to eQTL and genetic mapping studies, and hence the artificial variants uniquely offer insight into the TFs and motifs underlying its function. Based on their opposing effects at the CD69 locus, we extended our analysis genome-wide and found that the two factors co-regulate a number of immune genes and exert opposing effects on effector T cell function as well as Th1-Th2 differentiation pathways. We speculate that the opposing regulators may be particularly critical to the rapid responsiveness of co-regulated genes during T cell activation.
In conclusion, we have benchmarked emerging experimental and computational strategies to resolve regulatory genomic sequences with increasing precision. Our study demonstrates, in particular, the potential of base editing screens and artificial variants for identifying critical regulatory motifs and TF interactions that underlie rapid and robust transcriptional responses. Further computational and experimental innovations are needed to scale these approaches and further illuminate the syntax of human regulatory genomics.
Limitations of the study
We also note limitations of our study. Although our pooled screen tested a large number of perturbations, it was limited to a single inducible gene locus in one cell model. Extension of the approach to additional immune loci and primary T cells is an exciting future opportunity. Our approach was also limited by technical aspects of the perturbation tools. The base editor construct is limited by PAM site availability, meaning that we could only edit ∼28% of the Cs or Gs within the targeted REs. Base editors with less restrictive PAM site requirements41 could improve the resolution of future screens. While base editor approaches are mainly focused on C-to-T or A-to-G transitions, prime editors could enable more systematic base changes if they could be applied at scale.42 We also acknowledge that while our study characterizes a set of key regulators acting at the base edit site, it is not exhaustive, and other factors are likely binding the enhancer and impacting CD69 expression.
Finally, we acknowledge several limitations of the Enformer model and its use within this study. First, although the model predictions aided in variant interpretation and hypothesis generation, the biological findings were primarily underpinned by the experimental perturbations. Second, while the model predictions and experimental data highlighted similar intervals and nucleotides, further innovations and more comprehensive evaluations are needed to improve base-level accuracy of the model. In particular, algorithmic improvements, ideally trained in iterative cycles with experimental tests of artificial variants, may ultimately yield sufficiently accurate predictive models to resolve regulatory sequences across the vast noncoding genome. Given the increasing prevalence and potential of deep learning for regulatory genomics, integrative studies such as ours will be critical moving forward.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies and dyes | ||
| Brilliant Violet 510™ anti-human CD69 Antibody | Biolegend | Cat#310936 RRID:AB_2563834 |
| APC anti-human CD69 Antibody | Biolegend | Cat# 310910 RRID:AB_314845 |
| Zombie NIR™ Fixable Viability Kit | Biolegend | Cat# 423106 |
| H3K27Ac antibody | Active Motif | Cat# 39133 RRID:AB_2561016 |
| H3K27Me3 antibody | Active Motif | Cat# 39155 RRID:AB_2561020 |
| GATA-3 (D13C9) XP Rabbit mAb | Cell Signaling Technology | Cat# 5852, RRID:AB_10835690 |
| Dec1 antibody | Novus Biologicals | Cat#NB100-1800 |
| TAL1 antibody | SantaCruz Biotech | Cat#sc-393287 |
| Cell lines and primary cells | ||
| Jurkat cell line, Clone E6.1 | ATCC | Cat#TIB152, RRID:CVCL_0367 |
| CD4+ T cells | AllCells | N/A |
| Chemicals and buffers | ||
| Phorbol 12-myristate 13-acetate | Sigma-Alrich | Cat#P8139 |
| Ionomycin calcium salt | Sigma-Alrich | Cat# I0634 |
| Chloroquine diphosphate | Millipore Sigma | Cat#C6628 |
| Polybrene | Sigma-Alrich | Cat#107689 |
| Brilliant Staining Buffer | BD | Cat#566349 |
| Recombinant DNA | ||
| KRAB-dCas9-sgRNA-Puro | Broad GPP | pXPR_066 |
| LentiCRISPR v2-dCas9 | Addgene | Cat#112233 RRID:Addgene_112233 |
| rApobec-nCas9-UGI-Puro | Broad GPP | pRDA_256 |
| EFS-ABE8e-V106W-nCas9-puro | Broad GPP | pRDA_426 |
| GATA3-Overexpression-GFP | In this study | N/A |
| BHLHE40-Overexpression-GFP | In this study | N/A |
| BHLHE41-Overexpression-GFP | OriGene | Cat#: RC206882L2 |
| sgCtrl-1 CRISPR-Cas9-GFP | In this study | sgRNA sequence: GGCTAAATTCCTCTTATTCA |
| sgCtrl-2 CRISPR-Cas9-GFP | In this study | sgRNA sequence: GTAACCAAGAGTCAGGACTG |
| sgGATA3-1 CRISPR-Cas9-GFP | In this study | sgRNA sequence: ACCGAGTTTCCGTAGTAGGG |
| sgGATA3-2 CRISPR-Cas9-GFP | In this study | sgRNA sequence: TACGTGCCCGAGTACAGCTC |
| shCtrl(Scamble shRNA-EGFP) | VectorBuilder | Target sequence:CCTAAGGTTAAGTCGCCCTCG |
| shBHLHE40-1(EGFP) | VectorBuilder | Target sequence: AGAAAGGATCGGCGCAATTAA |
| shBHLHE40-2(EGFP) | VectorBuilder | Target sequence: ACCCGAACATCTCAAACTTAC |
| TAL1-Overexpression-Puro | OriGene | Cat#RC222628L3 |
| pLenti-Overexpression-Puro | OriGene | Cat#PS100092 |
| psPAX2 | Addgene | Cat#12260 RRID:Addgene_12260 |
| pMD2.G | Addgene | Cat#12259 RRID:Addgene_12259 |
| Lentivirus packing | ||
| Lipofectamine 3000 Transfection Reagent | ThermoFisher | Cat#L3000001 |
| OptiMEM | ThermoFisher | Cat# 31985070 |
| ATAC-seq reagents | ||
| Illumina tagmentation kit | Illumina | Cat#20034197 |
| Nextera XT Index Kit | Illumina | Cat# FC-131-1001 |
| MinElute Reaction Purification Kit | QIAGEN | Cat#28003 |
| MinElute PCR Purification Kit | QIAGEN | Cat#28004 |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat# M0541 |
| RNA-seq reagents | ||
| QIAGEN RNeasy Micro kit | QIAGEN | Cat# 74004 |
| Dynabeads mRNA Direct Kit | ThermoFisher | Cat# 610.12 |
| RNA Fragmentation Reagents | ThermoFisher | Cat# AM8740 |
| Turbo DNase | ThermoFisher | Cat#AM2238 |
| FastAP enzyme | ThermoFisher | Cat# EF0651 |
| Dynabeads MyOne Silane | ThermoFisher | Cat# 37002D |
| T4 RNA ligase | NEB | Cat#M0204L |
| AffinityScript RT Enzyme | Agilent | Cat#600107 |
| Phusion Master Mix | NEB | Cat# M0531L |
| AMPure XP Beads | Beckman Coulter | Cat# B23318 |
| IDT indexes | IDT | N/A |
| ChIP-seq reagents | ||
| Protein G beads | ThermoFisher | Cat#10003D |
| RNAse | Roche | Cat#11119915001 |
| Proteinase K | Invitrogen | Cat# 25530-015 |
| DNA end-repair kit | Epicenter Biotech | Cat# ER0720 |
| Klenow Fragment | NEB | Cat# M0212L |
| Quick Ligation kit | NEB | Cat# M2200S |
| PFU Ultra II HS 2x Master Mix | Agilent | Cat#600850-51 |
| Amplicon-seq reagents and primers | ||
| QIAamp DNA Micro Kit | QIAGEN | Cat#6304 |
| DNeasy Blood and Tissue Kit | QIAGEN | Cat#69504 |
| Titanium® Taq DNA Polymerase | Takara | Cat# 639208 |
| Agencourt AMPure XP SPRI beads | Beckman Coulter | Cat# A63880 |
| P5 Primer for tiling: AATGATACGGCGAC CACCGAGATCTACACTCTTTCCCTACAC GACGCTCT TCCGATCT TTGTGGAAAGGACGAAACACCG |
IDT | N/A |
| P7 Primer for tiling: CAAGCAGAAGACGG CATACGAGATNNNNNNNNGTGACTGGA GTTCAGAC GTGTGCTCTTCCGATCTCCAATTCCCAC TCCTTTCAAGACCT |
IDT | N/A |
| sg#70 amplicon F-primer: GGTGAGACG TCAGAAAGGAAGT |
IDT | N/A |
| sg#70 amplicon R-primer: AATTCACCC ACTGAAAGGAAAA |
IDT | N/A |
| Software and algorithms | ||
| CRISPResso2 | Clement et al.29 | http://crispresso.pinellolab.org/submission/ |
| EQTLGEN | Võsa et al.28 | https://eqtlgen.org/cis-eqtls.html |
| Python v3.9 | http://www.python.org/downloads/release/python-390/ | |
| R v4.2 | http://www.r-project.org/ | |
| Bioconductor v3.15 | http://www.bioconductor.org/ | |
| DESeq v2 | Anders and Huber43 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| CSAW | Lun et al.44 | https://bioconductor.org/packages/release/bioc/html/csaw.html |
| ComplexHeatmap | Gu et al.45 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Enformer v1 | Avsec et al.5 | https://tfhub.dev/deepmind/enformer/1 |
| Enformer fine-tuning code for this paper | https://doi.org/10.5281/zenodo.7775557, https://github.com/BernsteinLab/BE_CD69_paper_2022 | |
| DeepTools 3.5.0 | Ramirez et al.46 | https://github.com/deeptools/deepTools |
| TFModisco 0.4.2.3 | Shrikumar et al.47 | https://github.com/kundajelab/tfmodisco |
| MEME suite v5.4.1 | Bailey et al.48 | https://meme-suite.org/meme/ |
| ENCODE ATAC-seq pipeline v 2.1.3 | https://github.com/ENCODE-DCC/atac-seq-pipeline | |
| ENCODE ChIP-seq pipeline v 2.1.6 | https://github.com/ENCODE-DCC/chip-seq-pipeline2 | |
| STAR v2.7.9a | Dobin et al.49 | https://github.com/alexdobin/STAR |
| Salmon v1.6 | Patro et. al50 | https://github.com/COMBINE-lab/salmon |
| Bedtools v2.30.0 | Quinlan et al.51 | https://github.com/arq5x/bedtools2 |
| Samtools v1.12 | Li et al.52 | https://github.com/samtools/samtools |
| MOODS v1.9.4.1 | Korhonen et al.53 | https://github.com/jhkorhonen/MOODS |
| Deposited data | ||
| Jurkat ATAC-seq, wild-type | Nasser et al.4 | GEO: GSE155555 |
| CD4+ T-cell ATAC-seq | GEO: GSE124867 | |
| Jurkat RNA-seq, wild-type | Brignall et al.54 | GEO: GSE90718 |
| +/- edited Jurkat ChIP-seq, ATAC-seq | this paper | GEO: GSE206377 |
| +/- BHLHE40-OE Jurkat RNA-seq, ChIP-seq, ATAC-seq | this paper | GEO: GSE206377 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bradley E. Bernstein (bradley_bernstein@dfci.harvard.edu).
Material availability
Base-editor construct will be available on addgene upon publication. sgRNA library requests should be directed to Fadi J. Najm (fadinajm@broadinstitute.org)
Method details
Guide library design and cloning
Pooled libraries for expression of sgRNAs were generated as detailed previously.55 Briefly, DNA oligos were annealed into double stranded fragments with compatible overhangs and ligated into BsmBI sites into vectors. Vector backbones were CRISPRi+guide puro (pXPR_066, Broad GPP), lentiCRISPR v2-dCas9 (gift of Thomas Gilmore, Addgene 112233), rApobec-nCas9-UGI-puro (pRDA_256, Broad GPP) and EFS-ABE8e-V106W-nCas9-puro(pRDA_426, Broad GPP). Libraries were then transformed by electroporation into electrocompetent E-coli (Invitrogen) and spread onto bioassay plates. Bacterial colonies were harvested and isolated using the Plasmid Plus Midi Kit (Qiagen). Peak proximity and acceptable on-target efficacy scores56 determined sgRNA selection for the CRISPRi tests. After RE-3 and RE-4 were identified, sgRNAs in these peak regions were selected and included for screening with dCas9 and base editors and can be found in Table S2.
Cell culture and stimulation
The Jurkat cell line (ATCC, Clone E6.1, TIB152) was cultured in complete RPMI (RPMI Medium 1640,Gibco, 11875085, 1% Penicillin-Streptomycin, Gibco, 15140122, 10% Heat Inactivate Fetal Bovine Serum, Peak Serum, 20mM HEPES,Gibco,15630080,1% Sodium Pyruvate, Gibco, 11360070, and 1% NEAA,Gibco, 11140050) at a maximum density of 2 x 106 cells/ml in 25 cm or 75 cm cell culture dishes. Stimulation of Jurkat cells for 2-7 hour experiments was achieved with 50ng/ml Phorbol 12-myristate 13-acetate (PMA, Sigma-Alrich, P8139) and 500ng/ml ionomycin calcium salt from Streptomyces conglobatus (ionomycin, Sigma-Alrich, I0634).
Cryopreserved CD4+ T cells isolated from healthy donors were obtained from AllCells. On the day of stimulation, cells were thawed in RPMI 1640 medium supplemented with 2mM L-glutamine and 50% FBS, counted and resuspended in TexMACS medium (Miltenyi Biotec) supplemented with 20 IU/mL human Interleukin-2 (IL-2) and 1% penicillin-streptomycin. Cells were seeded at 1 million cells per well in a 48-well plate. Cells were either left untreated or stimulated with 10 μL T Cell TransAct™, human (Miltenyi Biotec) via CD3 and CD28 for 24hrs.
Lentivirus production
293T cells approaching 70-80% confluency in 10 cm cell culture dishes were used for packaging. Cells were pre-treated with 25 uM chloroquine diphosphate (Millipore Sigma, C6628) in 3 ml of complete DMEM (Gibco DMEM with 1% Penicillin-Streptomycin and 10% Heat Inactivate Fetal Bovine Serum) and incubate in the 37°C and 5% CO2 incubator for more than 30 minutes. Lipofectamine 3000 Transfection Reagent (ThermoFisher, L3000001) was used to deliver plasmids into 293T cells. Briefly, 15 ug lentiviral vector plasmid, 15 ug of psPAX2 and 5 ug pMD.G plasmid were vortexed with 40 ul P3000 reagent in 1.5 ml OptiMEM (ThermoFisher, 31985070). Then 40 ul Lipofectamine was added to 1.5 ml OptiMEM and briefly vortexed. The two OptiMEM solutions were combined and mixed well by vortexing for 30s and incubated at room temperature for at least 20 minutes. Carefully, the OptiMEM mixture was added dropwise to 293T cells and incubated in a 37°C and 5% CO2 incubator for 6 hours. Media were aspirated and replaced with 5ml of fresh complete RPMI. Lentiviral supernatant was harvested between 24 hours and 48 hours after transfection.
Lentivirus tranduction
Jurkat cells were resuspended in 1ml media and seeded at a density of 2-5 x 105 cells per well of a 12-well plate. Lentiviral supernatant was supplemented with 8 ug/ml polybrene (Sigma-Alrich) added to the Jurkat cells. The plate was then centrifuged at 2000xg, 32°C for 60mins. Cells were then incubated at 37°C and 5% CO2 overnight and changed into complete RPMI on the next day. For GFP+ marked lentivirus, cells were sorted or analyzed 4-5 days after transfection via flow cytometry(Note: too long overexpression or KD for TFs completely changed the cell status, thus keeping experiments in 4-5 days window is critical). For puromycin selection, 5 ug/ml of puromycin was added to the transduced cells and selected for 2 days.
Flow cytometry and sorting
Suspended cells were centrifuged down at 300xg, room temperature for 5 minutes. The cells were stained with the antibody cocktail in the staining buffer of a 1:1 mix of PBS and Brilliant Staining Buffer (BD, 566349), at room temperature for 20 mins or at 4°C for 30-40 mins. Cells were washed once in PBS with 1% FBS and then resuspended in the same buffer. Flow cytometry or FACS was processed on either BD LSRFortessa X-20 or SONY SH800 following the manufacturing instructions. Antibodies and dyes used from Biolegend: Brilliant Violet 510™ anti-human CD69 Antibody (310936);APC anti-human CD69 Antibody (310910); Zombie NIR™ Fixable Viability Kit (423106).
At least 2 x 105 CRISPR library infected Jurkat cells were collected as a pre-sorted baseline. 2-4 x 106 CRISPR library infected Jurkat cells were resuspended in 2 ml of complete RPMI and stimulated with 50 ng/ml PMA and 500 ng/ml ionomycin for 5 hours, and then processed for FACS as described above. Sorted CD69- and CD69+ populations were collected for genomic DNA isolation.
Genomic DNA isolation and sequencing
Genomic DNA (gDNA) was isolated using QIAamp DNA Micro Kit (QIAGEN, 6304) or DNeasy Blood and Tissue Kit (QIAGEN, 69504) according to the manufacturer’s protocol. The gDNA concentrations were quantified by Qubit. For PCR amplification, at least 330 ng of gDNA was used per reaction for greater than 500-fold library coverage. Each reaction contained 1.5 ul Titanium Taq (Takara), 10 μl of 10× Titanium Taq buffer, 8 μl deoxyribonucleotide triphosphate provided with the enzyme, 5 μl DMSO, 0.5 μl P5 stagger primer mix (stock at 100 μM concentration), 10 μl of a uniquely barcoded P7 primer (stock at 5 μM concentration), and water up to 100ul.
P5 Primer: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCT TCCGATCT
TTGTGGAAAGGACGAAACACCG
P7 Primer: CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGAC
GTGTGCTCTTCCGATCTCCAATTCCCACTCCTTTCAAGACCT
PCR cycling conditions included: an initial 5 min at 95°C; followed by 30 s at 54°C, 30 s at 53°C, 20 s at 72°C, for 28 cycles; and a final 10-min extension at 72°C. PCR primers were synthesized at Integrated DNA Technologies. PCR products were purified with Agencourt AMPure XP SPRI beads according to the manufacturer’s instructions (Beckman Coulter, A63880). Samples were sequenced on a MiSeq (Illumina). Reads were counted by alignment to a reference file of all possible guide RNAs present in the library. The read was then assigned to a condition on the basis of the 8-nt index included in the P7 primer.
Amplicon sequencing
To assess base editing frequency of the sg#70 locus, we designed primers flanking this region resulting in a 214bp product. Forward primer: GGTGAGACGTCAGAAAGGAAGT and reverse primer: AATTCACCCACTGAAAGGAAAA. Amplicons were next ligated with Illumina Truseq adaptors, cleaned and size selected with AMPure XP SPRI beads, and sequenced on a MiSeq paired end run. FASTQ files were processed with CRISPResso2 v2 with standard settings for base editor29.
ATAC-seq experimental processing
ATAC-seq lysis buffer contains 10mM Tris-HCl (pH=7.4), 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 0.1% NP40, 0.1% Digitonin, 1% BSA and topped up with ddH2O. ATAC-seq washing buffer contains 10mM Tris-HCl (pH=7.4), 10mM NaCl, 3mM MgCl2, 1% BSA and topped up with ddH2O.
5 x 10ˆ4 cells were centrifuged down with the resuspension buffer (PBS with 1%BSA) in a low-binding eppendorf tube at 4°C, 500xg for 5 mins. Each pellet is resuspended with 50 ul of lysis buffer and incubated on ice for 5 minutes. 50 ul of wash buffer was added to the lysis buffer containing nuclei and centrifuged down at 4°C, 500xg for 5 minutes. The supernatant is then removed and 50ul resuspension buffer is added to the tube without disturbing the pellet. Nucleus are then centrifuged down at 4°C, 500xg for 5minutes. Tagmentation of the genome DNA is processed using the Illumina tagmentation kit (20034197) for 30 mins in 37°C. Fragmented products are then isolated via MinElute Reaction Purification Kit (QIAGEN, 28003) according to the manufacturer's instructions. Illumina Nextera XT SetA indexes and NEBNext High-Fidelity 2X PCR Master mix (NEB, M0541) are used to amplify the fragmented products of each sample, with 12 PCR cycles of 98°C-10s, 63°C-30s and 72°C-1min. PCR products are then isolated via MinElute PCR Purification Kit (QIAGEN, 28004) following manufacturer’s instructions.
RNA-seq experimental processing
Whole RNA was extracted from over 1 × 10ˆ5 cells using the QIAGEN RNeasy Micro kit (QIAGEN, 74004) according to the manufacturer's instructions. 1ug RNA was then used to prepare the RNA-seq library. Poly-A+ RNA is enriched using Dynabeads mRNA Direct Kit (ThermoFisher, 610.12) according to the manufacturer's instructions and eluted in 18ul Tris-HCl buffer(pH=7.4). Zinc fragmentation is processed using RNA Fragmentation Reagents(ThermoFisher, AM8740), followed by Turbo DNase (ThermoFisher, AM2238) and FastAP enzyme (EF0651) treatment. Then the fragmented RNA are cleaned-up using Dynabeads MyOne Silane (ThermoFisher, 37002D) and eluted in 7ul of nuclease-free water. Next, RNA-adaptors are ligased to eluted RNA using T4 RNA ligase (NEB, M0204L) at 23°C for 1 hour and adaptor-ligated RNA was cleaned-up using Dynabeads MyOne Silane and eluted in 13.5ul of nuclease-free water. First strand of cDNA is synthesized using AffinityScript RT Enzyme (Agilent, 600107) according to the manufacturer's instructions at 54°C for 1 hour. First-strand cDNA was cleaned-up using Dynabeads MyOne Silane and eluted in 5.5ul of nuclease-free water, followed by cDNA adaptor ligation. After another round of clean-up, the adaptor-ligated cDNA was processed to library PCR amplification using Phusion Master Mix (NEB, M0531L) with IDT adaptor indexes. The final library was cleaned-up with AMPure XP Beads (Beckman Coulter, B23318) to a final size around 280bps.
ChIP-seq experimental processing
Jurkat cells were pelleted and fixed using 1% formaldehyde at 37°C for 10 mins then quenched by glycine. Samples were next washed with cold PBS+proteinase inhibitor (ThermoFisher, 78429), resuspended in lysis buffer (1% SDS, 0.25% DOC, 50mM Tris-HCl, pH=7.4), and incubated on ice for 10 mins. Samples were diluted up to 1ml in eppendorf using ChIP dilution buffer (0.01% SDS, 150mM NaCl, 0.25% Triton, 50mM Tris-HCl, pH=7.4) and sonicated using a Covaris E220, with the following settings: 24 mins with 5% duty factor, 140W max power and 200 cycles/burst. For Figure 4F, each sample(2.5 x 10ˆ7 cells) was then split into 4 eppendorf tubes: 1) 20ul, top up to 200ul for input; 2)180ul, top up to 1ml for H3K27Ac ChIP (2.5ul, Active Motif, 39133); 3) 400ul, top up to 1ml for GATA3 ChIP (10ul, CST-D13C9, 5852); 4) 400ul, top up to 1ml for BHLHE40 ChIP (10ul, Novus Biological, NB100-1800); or 1 x 10ˆ7 cells per sample was used for TAL1 ChIP(10ul, Santa Cruz Biotechnology, # SC-393287). For Figure 4D, each sample (1 x 10ˆ7 cells) was then split into 3 eppendorf tubes: 1) 200ul, top up to 1ml for H3K27Ac ChIP (2.5ul, Active Motif, 39133); 2) 200ul, top up to 1ml for H3K27Me3 ChIP(2.5ul, Active Motif, 39155); 3) 600ul, top up to 1ml for BHLHE40 ChIP (10ul, Novus Biological, NB100-1800). The tubes were incubated overnight at 4°C on a rotator.
On the next day, Protein G beads (ThermoFisher, 10003D) were washed and added to the antibody-containing suspension and rotated at 4°C for 2 hours. The beads were then washed with an ice-cold RIPA wash buffer: RIPA-500, LiCl, and 10mM Tris-HCl buffer (pH=8.5). The beads were eluted in a wash buffer (10mM Tris-HCl, pH=8.0, 0.1% SDS, 150mM NaCl, 5mM DTT) and incubated at 65°C on a shaker for 1 hour. Samples were then treated with RNAse (Roche, 11119915001) at 37°C for 30 mins and then with proteinase K (Invitrogen, 25530-015) at 63°C for 3 hours. AMPure XP Beads (Beckman Coulter, B23318) were used to purify the DNA fragments from the samples. Eluted fragments were then processed for DNA end-repair (Epicenter Biotech, ER0720), Klenow A base adding (Klenow from NEB, M0212L), adaptor ligation (Ligase from NEB, M2200S) and PCR amplification (PFU Ultra II HS 2x master mix from Agilent, 600850-51) according to manufacturer’s protocols. Index primers were ordered from Integrative DNA Technology. PCR was set up with the following conditions: 2 mins for 95°C; 30 sec at 95°C, 30 sec at 55°C,30 sec at 72°C for 16 cycles; 1 min at 72°C. PCR products were purified using AMPure XP Beads with a final size of around 300 bps.
Enformer predictions and fine-tuning
Fine tuning
The published Enformer model without any modifications was downloaded from https://tfhub.dev/deepmind/enformer/1. For model fine-tuning, we loaded the model checkpoint made available by the authors at gs://dm-enformer/models/enformer/sonnet_weights/. The cell-type/organism specific heads in the original model were then replaced with three new dense layers, corresponding to ATAC-seq from resting and stimulated Jurkat T-cells not in the original training data as well as the difference between the two. Training data for these output heads were obtained by downsampling bam files for resting and stimulated Jurkat T-cells to 20 million reads, and generating normalized bigwigs using the bam_cov function in the basenji suite available at https://github.com/calico/basenji. The difference track between the two was created using bigwigCompare from DeepTools. Bigwigs were then converted into the required input format using the basenji_data script. The modified model was then trained on a Google Cloud TPU-VM v3-64 pod-slice using a multi-learning rate scheme. For the differential accessibility prediction, all negative values in the target track were set to 0 in order to be able to keep the original softplus activation used in the final dense layer. The original model trunk, consisting of all convolutional and transformer layers shared for all organisms/tracks was trained using the AdamW optimizer from the tensorflow addons library at a learning rate of 1.0e-05 and weight decay of 5.0e-07. The three added output heads were trained at a higher learning rate of 1.0e-03 and weight decay of 5.0e-07. The model was trained for 32 epochs, with checkpointing every 4 epochs, and training was stopped when the validation loss did not decrease by 1.0e-03 relative to the lowest recorded validation loss for 30 epochs. The best checkpointed model was chosen at epoch 32 which reached a validation pearson’s correlation of 0.7625, 0.7318 and 0.6419 for stimulated,resting, and the differential ATAC-seq profiles for Jurkat cells respectively.
Gradient score calculation
Gradient based model interpretation was conducted as described at https://github.com/deepmind/deepmind-research/blob/master/enformer/enformer-usage.ipynb. For CAGE-seq interpretation, we calculated the gradient of the model for unstimulated Jurkat T-cells with respect to the predicted CAGE-seq signal at the CD69 promoter. This was achieved by centering a 393216 bp genomic window within the CD69 promoter(chr12:9760820-9760903) and computing the gradient for human output head # 4831 with respect to output bins 446-450(corresponding to the approximate promoter width). The absolute value of the gradients were then summed in 128bp bins for coarse grain resolution (Figure 1D). A similar approach to nominate bases contributing to RE-4 accessibility was adopted to obtain the base resolution contribution scores for the fine-tuned model corresponding to Figure S2 and 3. For this analysis, the window was centered around RE-4 (chr12:9764300-9765900) and the gradient was computed with respect to output bins 442–454 (Figures S1D and 3D), which corresponds to the approximate width of RE-4 (∼1.5kb).
Enhancer score calculation
For calculating the enhancer score, we computed the model gradient with respect to the predicted CAGE-seq output at the CD69 TSS. The absolute value of the gradient score was summed over a 2kb window centered at the ATAC-seq narrowpeak at each candidate RE, with the exception of RE-3 for which we manually selected the region chr12: 9762300-9764300 based on the small peak in accessibility in stimulated Jurkat cells and CD4-T cells at this position.
Variant effect prediction
For predicting the results of each BE guide, we started with a 196608 bp sequence centered at the CD69 TSS (the same used to compute the model gradient as described above). We then mutated each C->T lying within 2-8 bp opposite the NGG PAM site for each guide, and ran two forward passes of the model using the mutant sequence and its reverse complement. For each forward pass, the predicted CAGE-seq output was computed as the summed signal over the TSS bins (446-450). We then computed the % difference in predicted CD69 output for each mutation relative to WT, and averaged across the two predictions (forward and reverse strand).
ATAC-seq data processing
All ATAC-seq data were aligned and processed using the ENCODE uniform ATAC-seq processing pipeline v2.1.3 available at https://github.com/ENCODE-DCC/atac-seq-pipeline. The pipeline was configured to use default parameters, adapter auto-detection, the bowtie2 aligner,57 and MACS258 for peak calling. GRCh38 V29 and associated mitochondrial genomes and blacklists were obtained from https://www.encodeproject.org/references/ENCSR938RZZ/.
RNA-seq processing
RNA-seq datasets were processed using a custom pipeline utilizing fastp v0.23.259 for automatic adapter trimming with default settings for paired-end datasetsGene quantifications were obtained using Salmon v1.650 and the GENCODE V38 annotation60 with the seqBias, gcBias, posBias, and validateMappings flags enabled.
ChIP-seq data processing
ChIP-seq read alignment, quality filtering, duplicate marking and removal, peak calling, signal generation, and quality-control was conducted using the ENCODE ChIP-seq pipeline v2.1.6 available at https://github.com/ENCODE-DCC/chip-seq-pipeline2. GRCh38 V29 and blacklists were obtained from https://www.encodeproject.org/references/ENCSR938RZZ/. In brief, reads were aligned to the GRCh38 genome using bowtie2(-X2000), filtered to remove poor quality reads (Samtools) and de-duplicated (Picard MarkDuplicates). We provided matched input controls for each TF ChIP sample (GATA3, BHLHE40, TAL1) when running the pipeline (further details provided with the data submission at GSE206377). With the exception of sg70_P260 (edited), BHLHE40 ChIP p value bigwigs shown for each sample represent the MACS2 signal track output for pooled replicates (corresponding to call-macs2_signal_track_pooled p value output in the above pipeline). For sg70_P260 BHLHE40, we used only replicate 1 due to poor quality of replicate 2.
Other software
Figures and graphical abstract were assembled into panels using Adobe Illustrator and BioRender.
Quantification and statistical analysis
RNA-seq analysis
Differential expression analysis between conditions was conducted using DESeq V243 with default settings. Significance of differential expression for Figures 3E, 4F, and S5B, and S5E was determined using an FDR cutoff of 0.05. Log fold change values were corrected with the lfcShrink option using the apeglm method. For BHLHE40-OE gene expression analysis, BHLHE40-OE was compared to wild-type only in for stimulated Jurkat cells. Differential expression results can be found in Table S3.
Heatmap was constructed using the Complex heatmap package v 2.12.0.45 Genes were subsetted to only keep those differentially expressed between BHLHE40-OE and BHLHE40-WT at FDR < 0.05, and further subsetted to those with BHLHE40 or GATA3 binding events (see methods ChIP-seq processing) within 25kb of the gene TSS as described in the further legends.
Gene set enrichment analysis was conducted using the FGsea package v 1.22.061 and the ImmuneSigDB subset of the C7 immunologic gene set62 from http://www.gsea-msigdb.org/gsea/msigdb/collections.jsp. The product of -log10(p value) and log2FoldChange was used as the ranking metric for input to gene set enrichment. Significant pathways were collapsed using the collapsePathways function from FGsea with default settings.
ATAC-seq analysis
Differential accessibility analysis was conducted with the CSAW package v 1.28.44 Briefly, a consensus set of peaks was obtained from the union of peaks across all input samples/replicates. Peaks lying within blacklist regions and with low signal(less than −3 log10CPM) were removed. Reads were counted in 300 bp windows genome-wide and merged to a maximum width of 5 kb. Finally, counts were TMM normalized and significant differentially accessible peaks were identified based on a genome-wide FDR cutoff of 0.05 unless otherwise indicated.
For assessing accessibility changes near RE-4 in the edited cells in Figure S3E, we calculated a less conservative FDR/BH correction by restricting the analysis to peaks within a 1Mb window of RE-4 (using the p.adjust R command on the smaller set of nearby peaks).
ATAC-seq tracks in all figures were computed by pooling replicates where applicable using samtools52 and creating signal tracks using DeepTools bamcoverage.46 Signal tracks were normalized using the reads per genomic bin normalization (RPGC) options in DeepTools with the pre-computed effective genome size for GRCh38 (2747877777) in order to create coverage bigwigs.
ChIP-seq analysis
For Ctrl_LV GATA3 and BHLHE40 ChIP, we further processed the data by calling peaks using MACS2 with the default parameters in the above pipeline (p value < 0.01, max # peaks 500,000, and matched input control for GATA3/BHLHE40). Consensus peaks across replicates for Ctrl-LV GATA3 and BHLHE40 ChIP were obtained using the included IDR analysis step (IDR cutoff at 0.05). For H3K27ac, a less conservative set of consensus peaks were obtained by pooling both replicates and calling peaks using MACS2 (same parameters as above). For GATA3 and BHLHE40, we defined the binding event as the 500 bp window centered at the called narrowPeak summit. For H3K27ac, the enhancer was defined as the 2000 bp window centered at the narrowPeak summit. De-novo motif analysis using the XSTREME package (Grant and Bailey, 2021) from the MEME-suite yielded the expected GATA and BHLHE40 motifs among the top discovered motifs.
For the analyses in Figures 4E and 4F, binding events for BHLHE40/GATA3 were obtained by intersecting the centered IDR peaks with H3K27ac peaks in Ctrl-LV Jurkats. BHLHE40 or GATA3 regulated genes were identified by using the bedtools closest51 command to identify gene TSS that were <25kb away from a GATA3 or BHLHE40 binding event. Co-regulated genes were defined as the intersection between BHLHE40 and GATA3 regulated genes. Gene TSS locations were defined using the GENCODE V38 annotation, and defining the TSS as the starting point of the first exon for the longest isoform of each gene, extended to 500 bp by +125/+375 bp in the 3’/5′ direction respectively. GATA3 and BHLHE40 binding events and target genes (based on the distance cutoff) are available in Table S3.
Motif analysis
Motif scan of the RE-4 region corresponding to chr12: 9764556–9765505 was conducted by extracting the regions genomic sequence using bedtools getfasta51 and scanned using the MOODs motif scanner v1.9.4.153 and the HOCOMOCO v11 database, with a p value cutoff of 0.0001 and background base probabilities of 2.977e-01 2.023e-01 2.023e-01 2.977e-01. We first grouped motif matches with the same annotated motif class and start position, keeping only the match with the highest MOODs score. We then removed motif matches with a MOODs score <0, and further filtered to keep the top 25% of matches, corresponding to a score cutoff of 2.78. We then clustered motifs based on whether the motif cluster name contained GATA, bHLH, TCF, ETS, NFKB, NFAT, CREB, or STAT. Representative PWMs plotted in Figure 2 were obtained from the HOCOMOCO web portal.
Common SNP, eQTL and conservation score analysis
Common SNPs with MAF 1% were downloaded from Ensembl GRCh38.63 Expression quantitative trait loci(eQTL) were downloaded from eQTLGEN,28 and filtered to keep only cis-eQTLs with FDR < 0.05 (https://eqtlgen.org/cis-eqtls.html, gene locus for CD69). Conservation scores in Figure 2C correspond to phastCons100way score (0–1, clear to dark green).64
Enformer based motif identification
For identifying TF motifs using Enformer base importance scores (Figure S3), we used the TFModisco suite.47 This tool clusters short stretches of bases using base importance scores to discover motifs that can then be matched to known databases. First we centered 393216 bp genomic windows as above at the promoter of each of 2195 genes that were differentially expressed (FDR < 0.01, see Methods, RNA-seq analysis) between resting and stimulated Jurkat cells. Then, we computed the gradient of the model at each base within the window for output head 4831 as above with respect to the CAGE-seq signal at the promoter, corresponding to bins 446–450. For each window, we also computed the model gradient on a dinucleotide shuffled version of the sequence which was averaged across all genes in order to obtain an empirical null distribution of gradients. In order to reduce computing time, we extracted model gradients, sequence, and null gradients for the 750 centered bp window centered at each ATAC-seq peak detected from unstimulated Jurkat cells. Predictions were run in parallel across all genes simultaneously using a custom WDL/Google Cloud script. Finally, hypothetical contribution scores at each position within the 750 bp input window were computed as the model gradient corresponding to each non-reference base. TFmodisco was then used with default settings in order to identify putative regulatory motifs. Candidate seqlets were then matched to HOCOMOCO v11 motifs65 using Tomtom from the MEME-suite V5.4.148 with a q-value cutoff of 0.05.
Other statistical analysis
Unpaired t-test was used for Figure 1C, S2I, 3A, 3C, S3B, S3C, 4A–4C, and S4A–S4D using Graphpad Prism Version 9.5.0(525). Data points represent mean ± s.e.m, with 2–4 sample replicates per experimental group as described in the figure legends.
Each dot of mean ± s.e.m in Figures 1E, 1F, and S2D–S2H represents triplicates from 3 individual screening experiments, using Graphpad Prism Version 9.5.0(525).
Pearson’s correlation test was performed in Figure S2H using the cor.test command in R version 4.2. Pearson’s correlation and p value are indicated in the figure legend.
Acknowledgments
We thank S.M. Bevil, S. Battaglia, G. Rahme, K. Macias, and J. Verga for technical assistance. We thank the Google TPU Research Cloud for providing TPU access and support. This work was supported by funds from the NCI/NIH Director’s Fund (DP1CA216873 to B.E.B.) and the Gene Regulation Observatory and the Variant-to-Function Initiative at the Broad Institute. Z.C. is supported by NCI-CA-234842. L.P. is supported by the National Human Genome Research Institute (NHGRI) (R35HG010717 and UM1HG012010). J.W. is supported by a postdoctoral fellowship from the Damon Runyon Cancer Research Foundation. M.V. is supported by the National Cancer Institute (NCI) of the National Institutes of Health under the Ruth L. Kirschstein National Research Service Award (F31CA257625). B.E.B. is the Richard and Nancy Lubin Family Endowed Chair at the Dana-Farber Cancer Institute and an American Cancer Society Research Professor.
Author contributions
Z.C., N.J., F.J.N., and B.E.B. conceived the study. Z.C., F.J.N., and B.E.B. designed the experiments. Z.C., M.M., G.S., and A.C. performed the experiments. M.V. and L.P. provided computational assistance. Z.C., N.J., and J.W. analyzed the data. Z.C., N.J., F.J.N., and B.E.B. interpreted the data and wrote the manuscript.
Declaration of interests
B.E.B. declares outside interests in Fulcrum Therapeutics, HiFiBio, Arsenal Biosciences, Design Pharmaceuticals, Cell Signaling Technologies, and Chroma Medicine.
Published: April 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100318.
Contributor Information
Fadi J. Najm, Email: fadinajm@broadinstitute.org.
Bradley E. Bernstein, Email: bradley_bernstein@dfci.harvard.edu.
Supplemental information
Data and code availability
Datasets generated as part of this study have been deposited at GSE206377. All code for data processing, analysis, and Enformer model tuning/analysis are available on Zenodo at https://doi.org/10.5281/zenodo.7775557 and github at https://github.com/BernsteinLab/BE_CD69_paper_2022.git. Accession numbers and original study references for the publicly available ATAC-seq data in resting and stimulated Jurkat and CD4+ T cell ATAC-seq, as well as wild-type Jurkat RNA-seq, are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.ENCODE Project Consortium. Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A., et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. doi: 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stunnenberg H.G., Hirst M., International Human Epigenome Consortium the international human epigenome consortium: a blueprint for scientific collaboration and discovery. Cell. 2016;167:1897. doi: 10.1016/j.cell.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Andersson R., Sandelin A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020;21:71–87. doi: 10.1038/s41576-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 4.Nasser J., Bergman D.T., Fulco C.P., Guckelberger P., Doughty B.R., Patwardhan T.A., Jones T.R., Nguyen T.H., Ulirsch J.C., Lekschas F., et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593:238–243. doi: 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avsec Ž., Agarwal V., Visentin D., Ledsam J.R., Grabska-Barwinska A., Taylor K.R., Assael Y., Jumper J., Kohli P., Kelley D.R. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods. 2021;18:1196–1203. doi: 10.1038/s41592-021-01252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melnikov A., Murugan A., Zhang X., Tesileanu T., Wang L., Rogov P., Feizi S., Gnirke A., Callan C.G., Jr., Kinney J.B., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein J.C., Agarwal V., Inoue F., Keith A., Martin B., Kircher M., Ahituv N., Shendure J. A systematic evaluation of the design and context dependencies of massively parallel reporter assays. Nat. Methods. 2020;17:1083–1091. doi: 10.1038/s41592-020-0965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kheradpour P., Ernst J., Melnikov A., Rogov P., Wang L., Zhang X., Alston J., Mikkelsen T.S., Kellis M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013;23:800–811. doi: 10.1101/gr.144899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maricque B.B., Chaudhari H.G., Cohen B.A. A massively parallel reporter assay dissects the influence of chromatin structure on cis-regulatory activity. Nat. Biotechnol. 2018;37:90–95. doi: 10.1038/nbt.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fulco C.P., Munschauer M., Anyoha R., Munson G., Grossman S.R., Perez E.M., Kane M., Cleary B., Lander E.S., Engreitz J.M. Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science. 2016;354:769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canver M.C., Smith E.C., Sher F., Pinello L., Sanjana N.E., Shalem O., Chen D.D., Schupp P.G., Vinjamur D.S., Garcia S.P., et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korkmaz G., Lopes R., Ugalde A.P., Nevedomskaya E., Han R., Myacheva K., Zwart W., Elkon R., Agami R. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016;34:192–198. doi: 10.1038/nbt.3450. [DOI] [PubMed] [Google Scholar]
- 14.Sanjana N.E., Wright J., Zheng K., Shalem O., Fontanillas P., Joung J., Cheng C., Regev A., Zhang F. High-resolution interrogation of functional elements in the noncoding genome. Science. 2016;353:1545–1549. doi: 10.1126/science.aaf7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajagopal N., Srinivasan S., Kooshesh K., Guo Y., Edwards M.D., Banerjee B., Syed T., Emons B.J.M., Gifford D.K., Sherwood R.I. High-throughput mapping of regulatory DNA. Nat. Biotechnol. 2016;34:167–174. doi: 10.1038/nbt.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diao Y., Fang R., Li B., Meng Z., Yu J., Qiu Y., Lin K.C., Huang H., Liu T., Marina R.J., et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods. 2017;14:629–635. doi: 10.1038/nmeth.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim Y.B., Komor A.C., Levy J.M., Packer M.S., Zhao K.T., Liu D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017;35:371–376. doi: 10.1038/nbt.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cuella-Martin R., Hayward S.B., Fan X., Chen X., Huang J.-W., Taglialatela A., Leuzzi G., Zhao J., Rabadan R., Lu C., et al. Functional interrogation of DNA damage response variants with base editing screens. Cell. 2021;184:1081–1097.e19. doi: 10.1016/j.cell.2021.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanna R.E., Hegde M., Fagre C.R., DeWeirdt P.C., Sangree A.K., Szegletes Z., Griffith A., Feeley M.N., Sanson K.R., Baidi Y., et al. Massively parallel assessment of human variants with base editor screens. Cell. 2021;184:1064–1080.e20. doi: 10.1016/j.cell.2021.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Sathaliyawala T., Kubota M., Yudanin N., Turner D., Camp P., Thome J.J.C., Bickham K.L., Lerner H., Goldstein M., Sykes M., et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38:187–197. doi: 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cibrián D., Sánchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017;47:946–953. doi: 10.1002/eji.201646837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.FANTOM Consortium and the RIKEN PMI and CLST DGT. Forrest A.R.R., Kawaji H., Rehli M., Baillie J.K., de Hoon M.J.L., Haberle V., Lassmann T., Kulakovskiy I.V., Lizio M., Itoh M., et al. A promoter-level mammalian expression atlas. Nature. 2014;507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mumbach M.R., Satpathy A.T., Boyle E.A., Dai C., Gowen B.G., Cho S.W., Nguyen M.L., Rubin A.J., Granja J.M., Kazane K.R., et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 2017;49:1602–1612. doi: 10.1038/ng.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laguna T., Notario L., Pippa R., Fontela M.G., Vázquez B.N., Maicas M., Aguilera-Montilla N., Corbí Á.L., Odero M.D., Lauzurica P. New insights on the transcriptional regulation of CD69 gene through a potent enhancer located in the conserved non-coding sequence 2. Mol. Immunol. 2015;66:171–179. doi: 10.1016/j.molimm.2015.02.031. [DOI] [PubMed] [Google Scholar]
- 27.Dominguez A.A., Lim W.A., Qi L.S. Beyond editing: repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016;17:5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Võsa U., Claringbould A., Westra H.-J., Bonder M.J., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Yazar S., et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021;53:1300–1310. doi: 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho I.-C., Tai T.-S., Pai S.-Y. GATA3 and the T-cell lineage: essential functions before and after T-helper-2-cell differentiation. Nat. Rev. Immunol. 2009;9:125–135. doi: 10.1038/nri2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei G., Abraham B.J., Yagi R., Jothi R., Cui K., Sharma S., Narlikar L., Northrup D.L., Tang Q., Paul W.E., et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. 2011;35:299–311. doi: 10.1016/j.immuni.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu W., Morrissey C.S., Keller C.A., Mishra T., Pimkin M., Blobel G.A., Weiss M.J., Hardison R.C. Dynamic shifts in occupancy by TAL1 are guided by GATA factors and drive large-scale reprogramming of gene expression during hematopoiesis. Genome Res. 2014;24:1945–1962. doi: 10.1101/gr.164830.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porcher C., Chagraoui H., Kristiansen M.S. SCL/TAL1: a multifaceted regulator from blood development to disease. Blood. 2017;129:2051–2060. doi: 10.1182/blood-2016-12-754051. [DOI] [PubMed] [Google Scholar]
- 34.Cook M.E., Jarjour N.N., Lin C.-C., Edelson B.T. Transcription factor Bhlhe40 in immunity and autoimmunity. Trends Immunol. 2020;41:1023–1036. doi: 10.1016/j.it.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asanoma K., Liu G., Yamane T., Miyanari Y., Takao T., Yagi H., Ohgami T., Ichinoe A., Sonoda K., Wake N., Kato K. Regulation of the mechanism of TWIST1 transcription by BHLHE40 and BHLHE41 in cancer cells. Mol. Cell Biol. 2015;35:4096–4109. doi: 10.1128/MCB.00678-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zawel L., Yu J., Torrance C.J., Markowitz S., Kinzler K.W., Vogelstein B., Zhou S. DEC1 is a downstream target of TGF-β with sequence-specific transcriptional repressor activities. Proc. Natl. Acad. Sci. USA. 2002;99:2848–2853. doi: 10.1073/pnas.261714999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Honma S., Kawamoto T., Takagi Y., Fujimoto K., Sato F., Noshiro M., Kato Y., Honma K.-I. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. 2002;419:841–844. doi: 10.1038/nature01123. [DOI] [PubMed] [Google Scholar]
- 38.Zhu J., Min B., Hu-Li J., Watson C.J., Grinberg A., Wang Q., Killeen N., Urban J.F., Guo L., Paul W.E. Conditional deletion of Gata3 shows its essential function in TH1-TH2 responses. Nat. Immunol. 2004;5:1157–1165. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 39.Ouyang W., Ranganath S.H., Weindel K., Bhattacharya D., Murphy T.L., Sha W.C., Murphy K.M. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 1998;9:745–755. doi: 10.1016/s1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- 40.Singer M., Wang C., Cong L., Marjanovic N.D., Kowalczyk M.S., Zhang H., Nyman J., Sakuishi K., Kurtulus S., Gennert D., et al. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell. 2017;171:1221–1223. doi: 10.1016/j.cell.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290–296. doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lun A.T.L., Smyth G.K. csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 2016;44:e45. doi: 10.1093/nar/gkv1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 46.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shrikumar A., Tian K., Avsec Ž. Technical note on transcription factor motif discovery from importance scores (TF-MoDISco) arXiv. 2018 doi: 10.48550/arXiv.1811.00416. Preprint at. [DOI] [Google Scholar]
- 48.Bailey T.L., Johnson J., Grant C.E., Noble W.S. The MEME suite. Nucleic Acids Res. 2015;43:W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup Genome project data processing subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korhonen J., Martinmäki P., Pizzi C., Rastas P., Ukkonen E. MOODS: fast search for position weight matrix matches in DNA sequences. Bioinformatics. 2009;25:3181–3182. doi: 10.1093/bioinformatics/btp554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brignall R., Cauchy P., Bevington S.L., Gorman B., Pisco A.O., Bagnall J., Boddington C., Rowe W., England H., Rich K., et al. Integration of kinase and calcium signaling at the level of chromatin underlies inducible gene activation in T cells. J. Immunol. 2017;199:2652–2667. doi: 10.4049/jimmunol.1602033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joung J., Konermann S., Gootenberg J.S., Abudayyeh O.O., Platt R.J., Brigham M.D., Sanjana N.E., Zhang F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frankish A., Diekhans M., Jungreis I., Lagarde J., Loveland J.E., Mudge J.M., Sisu C., Wright J.C., Armstrong J., Barnes I., et al. Gencode 2021. Nucleic Acids Res. 2021;49:D916–D923. doi: 10.1093/nar/gkaa1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 62.Godec J., Tan Y., Liberzon A., Tamayo P., Bhattacharya S., Butte A.J., Mesirov J.P., Haining W.N. Compendium of immune signatures identifies conserved and species-specific biology in response to inflammation. Immunity. 2016;44:194–206. doi: 10.1016/j.immuni.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cunningham F., Allen J.E., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Austine-Orimoloye O., Azov A.G., Barnes I., Bennett R., et al. Ensembl 2022. Nucleic Acids Res. 2022;50:D988–D995. doi: 10.1093/nar/gkab1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A., et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018;46:D252–D259. doi: 10.1093/nar/gkx1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets generated as part of this study have been deposited at GSE206377. All code for data processing, analysis, and Enformer model tuning/analysis are available on Zenodo at https://doi.org/10.5281/zenodo.7775557 and github at https://github.com/BernsteinLab/BE_CD69_paper_2022.git. Accession numbers and original study references for the publicly available ATAC-seq data in resting and stimulated Jurkat and CD4+ T cell ATAC-seq, as well as wild-type Jurkat RNA-seq, are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.