

Abstract

The merger of photocatalysis and transition-metal catalysis has recently emerged as an adaptable platform for the development of innovative and environmentally benign synthetic methodologies. In contrast to classical transformation by Pd complexes, photoredox Pd catalysis operates through a radical pathway in the absence of a radical initiator. Using the synergistic merger of photoredox and Pd catalysis, we have developed a highly efficient, regioselective, and general meta-oxygenation protocol for diverse arenes under mild reaction conditions. The protocol showcases the meta-oxygenation of phenylacetic acids and biphenyl carboxylic acids/alcohols and is also amenable for a series of sulfonyls and phosphonyl-tethered arenes, irrespective of the nature and position of the substituents. Unlike thermal C–H acetoxylation which operates through the PdII/PdIV catalytic cycle, this metallaphotocatalytic C–H activation involves PdII/PdIII/PdIV intermediacy. The radical nature of the protocol is established through radical quenching experiments and EPR analysis of the reaction mixture. Furthermore, the catalytic path of this photoinduced transformation is established through control reactions, absorption spectroscopy, luminescence quenching, and kinetic studies.
Keywords: distal C, H activation, Pd-photoredox catalysis, phenylacetic acid drugs, acetoxylation, radical mechanism
In recent years, transition-metal-catalyzed C–H bond functionalization gains tremendous expansion due to the easy accessibility of C–H bonds in various molecular systems.1−5 Among several C–H functionalization reactions, the construction of the C–O bond attracted much attention in organic synthesis due to its ability to induce polar nature and a unique H-bond donor-accepting capacity. In particular, the installation of the acetoxy group enhances antimicrobial and herbicidal activities in many molecules and is also found in many marketed drugs and natural products.6−11 As a consequence, a number of acetoxylation reactions ranging from proximal to distal C(sp2)–H bonds were developed (Scheme 1a,b).12−25 However, most of these protocols suffer from pitfalls such as the use of excess acetoxylating reagent, high temperature, and use of acids (acetic acid or acetic anhydride) as additives or solvents, which limit their application in the substrates bearing sensitive functional groups and complex moieties. Furthermore, distal C(sp2)–H acetoxylation reactions are limited to the substrates with toluene, benzoic acid, and aniline backbones (Scheme 1b). To overcome these limitations, a sustainable protocol is required, which operates under milder conditions and avoids superstoichiometric amounts of acetoxylating reagents and is applicable for the substrates which are unexplored under thermal conditions. The recent resurgence in photochemistry has aided the development of numerous transformations in the arena of organic chemistry which were previously inaccessible.26−43 Very recently, a unique class of photocatalysis termed “photoredox catalysis” has gained momentum and has been applied by the organic chemistry community worldwide. Photoredox catalysis upon merger with a transition metal (Ni, Pd, Cu, Co, and Au) has led to the accomplishment of prior elusive transformations and received broad attention from organic chemists.31,44−55 To be precise, the ability of a Pd-catalyzed photoredox reaction to reroute the reaction mechanism via alternative Pd/radical-mediated pathways led to improvement in the reaction rate, substrate scope, and functional group compatibility.47,51,56,57 In general, Pd-photocatalyzed reactions work in two different ways (Scheme 1c). The first one is the synergetic cooperativity between a Pd catalyst and a photosensitizer, where an external photocatalyst is a sole light-absorbing species (Scheme 1c, left) and recently gained substantial attention to functionalize a plethora of C–H bonds under ambient conditions.26,44,51,58−68 In the second mode of reaction, the Pd catalyst itself absorbs the photon energy and catalyzes the reaction via a traditional or new type of mechanism without the requirement of an exogenous photocatalyst (Scheme 1c, right).52,69−72 This dual activity of Pd was well documented in the literature by various transformations.73−76 This catalytic transformation usually follows a single catalytic cycle in contrast to photoredox catalysis.77
Scheme 1. Realm of Pd-Catalyzed Proximal and Distal C–H Activation Reactions.
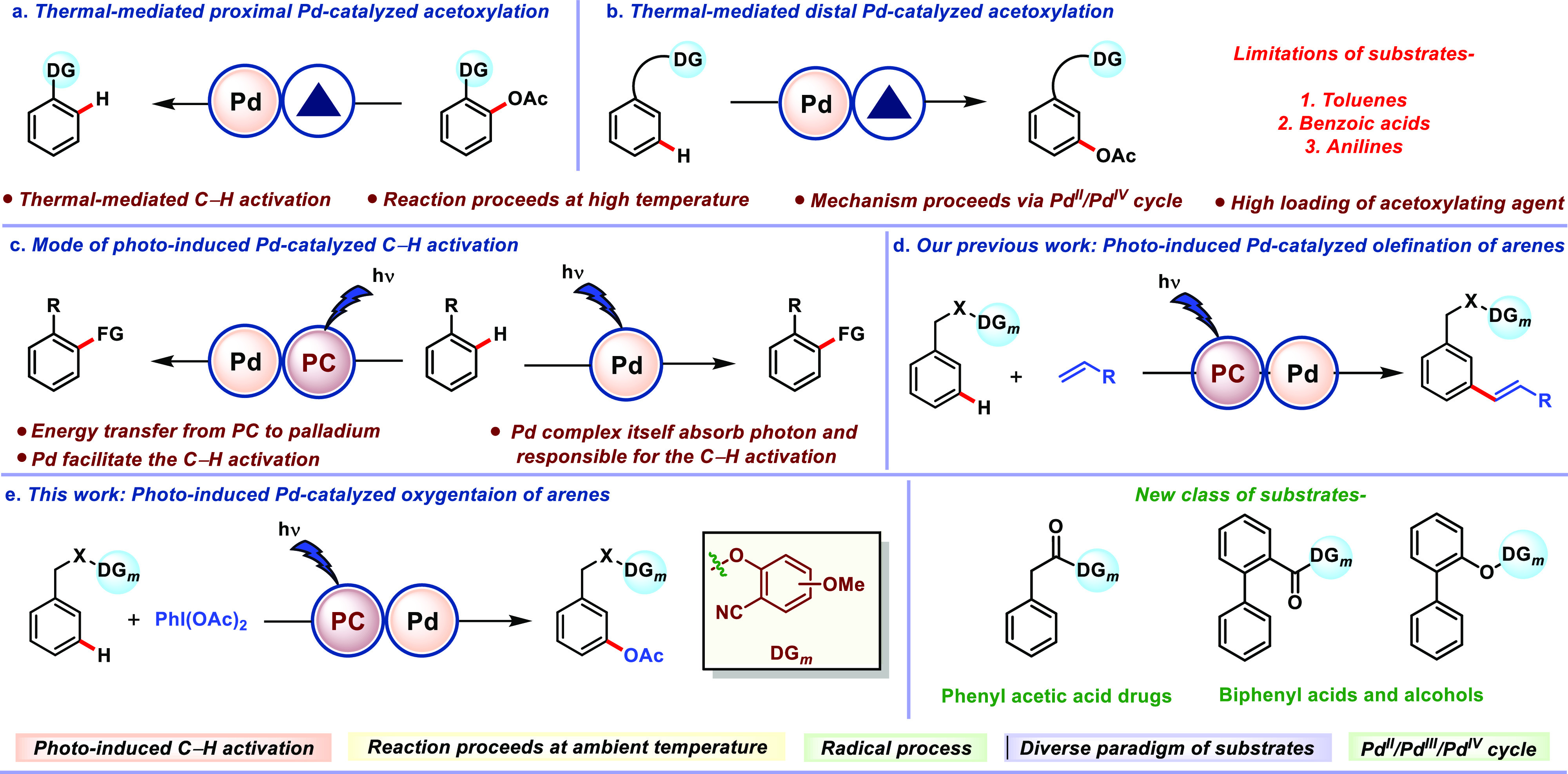
Converging this alluring property of Pd catalysis and photocatalysis, recently, our group reported a highly regioselective C(sp2)–H olefination of arenes and heteroarenes, including distal C(sp2)–H olefination through the directing group assistance (Scheme 1d).78 Inspired by our earlier success, herein, we demonstrated photoredox Pd catalysis for the meta-oxygenation of arenes (Scheme 1e). This synergetic effect of photoredox and Pd catalysis aids to perceive a new class of substrates like phenyl acetic acids, biaryl acids, and alcohols which are not reported earlier. To demonstrate the superiority of our protocol over thermal conditions, parallel experiments under thermal conditions (at 80 °C) were performed, and results are tabulated in the Supporting Information (see Section 11 and Table S8). Notably, the present catalytic system obviates the use of elevated temperature and a superstoichiometric acetylating agent, which is known to play a pivotal role in the reaction under thermal conditions, and afforded superior selectivity.
We commenced our study with the meta-acetoxylation of the phenylacetic acid-bearing nitrile-based template under Pd-photoredox catalysis. To start with our hypothesis, we have taken phenylacetic acids as model substrates, as these variants of acids are well abundant in commercially available drugs. The meta-acetoxylation of phenylacetic acids is yet to be explored, either in thermal or photochemical conditions. Here, we report the meta-acetoxylation of phenylacetic acids under photoredox conditions. Taking the idea from our previous report, we performed sequential optimization of each reaction parameter (see Supporting Information, Section 3) and found that the use of Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), and visible light (23 W) in HFIP solvent at ambient temperature for 36 h provided the highest yield of 79% of the meta-acetoxylated product, with selectivity >25:1 (Table 1, entry 1). Experimentation with diverse directing auxiliaries for the meta-functionalization suggests that only a nitrile-based directing template is compatible and also that the methoxy group-bearing template (at the para or meta position) provides the best yield and selectivity (see Supporting Information, Section 3 and Table S1). To testify the role of Pd catalyst, light, and the photocatalyst in this meta-C(sp2)–H acetoxylation reaction, parallel reactions were investigated in the absence of these components (Table 1, entries 2–4). Complete inhibition of the reaction justifies the role of the Pd catalyst, light, and the photocatalyst in endorsing the transformation. Furthermore, Pd catalysts other than Pd(OAc)2 were found to be inferior (Table 1, entries 5–8). Photocatalysts other than eosin Y provide lower yields (Table 1, entries 9 and10). Among the different N-protected amino acids and 2-hydroxy pyridine tested, N-Cbz-Gly-OH emerged to be the best (Table 1, entries 11–15). Solvents other than HFIP were found to be ineffective or less productive for this transformation (Table 1, entries 16 and 17). Finally, after a series of optimization, it was established that a catalytic Pd(OAc)2/ligand/photocatalyst in HFIP solvent can perform meta-acetoxylation under a household compact fluorescent lamp (CFL) bulb (see Supporting Information, Section 3). It was worth noting that the internal temperature of the reaction was found to lie between 30 and 35 °C throughout the reaction time.
Table 1. Optimization of Photoinduced meta-Selective C–H Oxygenation of Arenes.

| entry | deviation from standard conditions | yield (%)a | selectivity |
|---|---|---|---|
| 1 | none | 79 | >25:1 |
| 2 | no Pd(OAc)2 | NR | |
| 3 | no light | NR | |
| 4 | no eosin Y | NR | |
| 5 | Pd(TFA)2 instead of Pd(OAc)2 | 32 | >25:1 |
| 6 | PdCl2 instead of Pd(OAc)2 | trace | |
| 7 | Pd(PPh3)4 instead of Pd(OAc)2 | NR | |
| 8 | Pd(OAc)2 (20 mol %) | 71 | >25:1 |
| 9 | fluorescein instead of eosin Y | 47 | >25:1 |
| 10 | rhodamin B instead of eosin Y | trace | |
| 11 | N-Ac-Gly-OH instead of CBz-Gly-OH | 41 | 16:1 |
| 12 | Fmoc-Gly-OH instead of CBz-Gly-OH | 40 | 17:1 |
| 13 | N-Ac-4-hyrdoxy-L-proline instead of CBz-Gly-OH | 21 | 3:1 |
| 14 | CBz-Gly-OH (10 mol %) | 58 | 18:1 |
| 15 | 2-hydroxy pyridine instead of CBz-Gly-OH | 18 | 2:1 |
| 16 | DCE instead of HFIP | NR | |
| 17 | TFE instead of HFIP | 14 | 20:1 |
Yield determined by 1HNMR using TMB (trimethoxy benzene) as the internal standard. Yield given is the sum of the isomers.
With the suitable reaction conditions, the protocol was subsequently explored to generalize the scope of the transformation concerning arene substrates (Scheme 2). In general, phenylacetic acid derivatives with different substitution patterns underwent meta-acetoxylation smoothly, affording products in moderate-to-good yield with excellent selectivity (53–86%). Substitution of an electronically differentiated group at the para-position does not hamper the selectivity, although a moderate yield was observed for the electron-withdrawing group (2–5) (Scheme 2). The presence of a methyl group at the meta-position of phenylacetic acid (6) delivered the highest yield and selectivity (86%, >25:1). Furthermore, ortho-substituted phenylacetic acid produced synthetically useful yield and selectivity (8–10) (Scheme 2). Diphenylacetic acids (11 and 12) were also working well under optimized conditions, affording meta-acetoxylated products in good yield and exclusive selectivity (Scheme 2). Tethering of a cyclopentyl group at the α-position of phenylacetic acid (13) was also well tolerated. Inspired by the versatility of the protocol, we desired to expand the protocol for the late-stage functionalization of marketed drugs and agrochemicals (Scheme 2). To this aim, ibuprofen (14), clofibric acid (15), felbinac (16), and ketoprofen (17) were transformed to their meta-acetoxylated derivatives in good yield and selectivity. Increasing the chain length of the directing template decreased the meta-selectivity of the protocol, as evident from the outcome of 4-phenylbutanoic acid (18).
Scheme 2. Scope of the Reaction with Phenylacetic Acid Derivatives.

Reaction conditions: substrate (1 equiv), PhI(OAc)2 (2 equiv), Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), HFIP (1 mL), CFL (23 W), 30–35 °C, 36 h. Isolated yields are reported.
To further diversify the structural motifs which are yet to be utilized for the meta-acetoxylation reaction, biphenyl carboxylic acid and alcohols were tested under the optimized reaction conditions (Scheme 3). It was worth noting that biphenyl carboxylic acid showed similar reactivity as phenylacetic acid, affording meta-acetoxylated products with good yield and selectivity (19–29) (Scheme 3). The positional biases of the substituents resulted in a similar reactivity pattern as phenylacetic acid. Additionally, the incorporation of a functional group at the arene ring tethering directing template (28 and 29) also delivered synthetically acceptable yield and selectivity. Furthermore, biphenyl alcohols were also found as suitable substrates for photoinduced meta-acetoxylation using 2-carboxy benzonitrile as an effective directing template (Scheme 3). Biphenyl alcohols (30–32) matched biphenyl carboxylic acids, affording meta-acetoxylated products in good yield (66–75%), except for a slightly lesser selectivity for unsubstituted (30) and para-chloro (32) substrates. After the successful photoinduced meta-acetoxylation of ester-linked arenes, we turned our focus to check the feasibility of the protocol for the sulfonyl-linked benzyl arenes (Scheme 4). This will clearly set a platform to compare the thermal and photochemical modes of reaction. The results obtained for the various substituted sulfonyl-linked substrates (entries 33–46) were satisfying and gave useful yields of the expected products with excellent selectivity. A range of electron-donating and electron-withdrawing substituents were compatible, irrespective of their position in the arene. It is important to mention that the present photoredox condition delivered superior selectivity for these classes of substrates over the thermal-mediated C–H acetoxylation reaction. Furthermore, the robustness of the protocol was demonstrated by the meta-acetoxylation reaction of the phosphonyl-tethered arenes (47 and 48) (Scheme 4).
Scheme 3. Scope of the Reaction with Biphenyl Acid/Alcohol Derivatives.

Reaction conditions: substrate (1 equiv), PhI(OAc)2 (2 equiv), Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), HFIP (1 mL), CFL (23 W), 30–35 °C, 36 h. Isolated yields are reported. DGm’ = 2-carboxy benzonitrile.
Scheme 4. Scope of the Reaction with Sulfonyl- and Phosphonate-Linked Arenes.

Reaction conditions: substrate (1 equiv), PhI(OAc)2 (2 equiv), Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), HFIP (1 mL), CFL (23 W), 30–35 °C, 36 h. Isolated yields are reported.
In order to check the feasibility of other acetoxylating sources, PhI(TFA)2 was used, and it led to the formation of the meta-hydroxylated compound as the only product (Scheme 5). The formation of hydroxylated products can be rationalized due to the higher electrophilicity of trifluoroacetate ester carbonyl, which is likely hydrolyzed in situ. This observation was further confirmed experimentally, where an independently synthesized trifluoro-acetoxylated substrate afforded the hydroxylated product quantitatively when treated under standard reaction conditions (see Supporting Information, Section 5). A similar reactivity trend for meta-hydroxylation was observed as for meta-acetoxylation (entries 49–54), and it was demonstrated through representative examples of various arenes (Scheme 5). The present photoinduced protocol could also be scaled up (3 mmol) without altering the selectivity and affording the meta-acetoxylation product in acceptable yield (33, see Supporting Information, Section 4.F.).
Scheme 5. Representative Examples for meta-Hydroxylation of Various Arenes.

Reaction conditions: substrate (1 equiv), PhI(CO2CF3)2 (2 equiv), Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), HFIP (1 mL), CFL (23 W), 30–35 °C, 36 h. Isolated yields are reported.
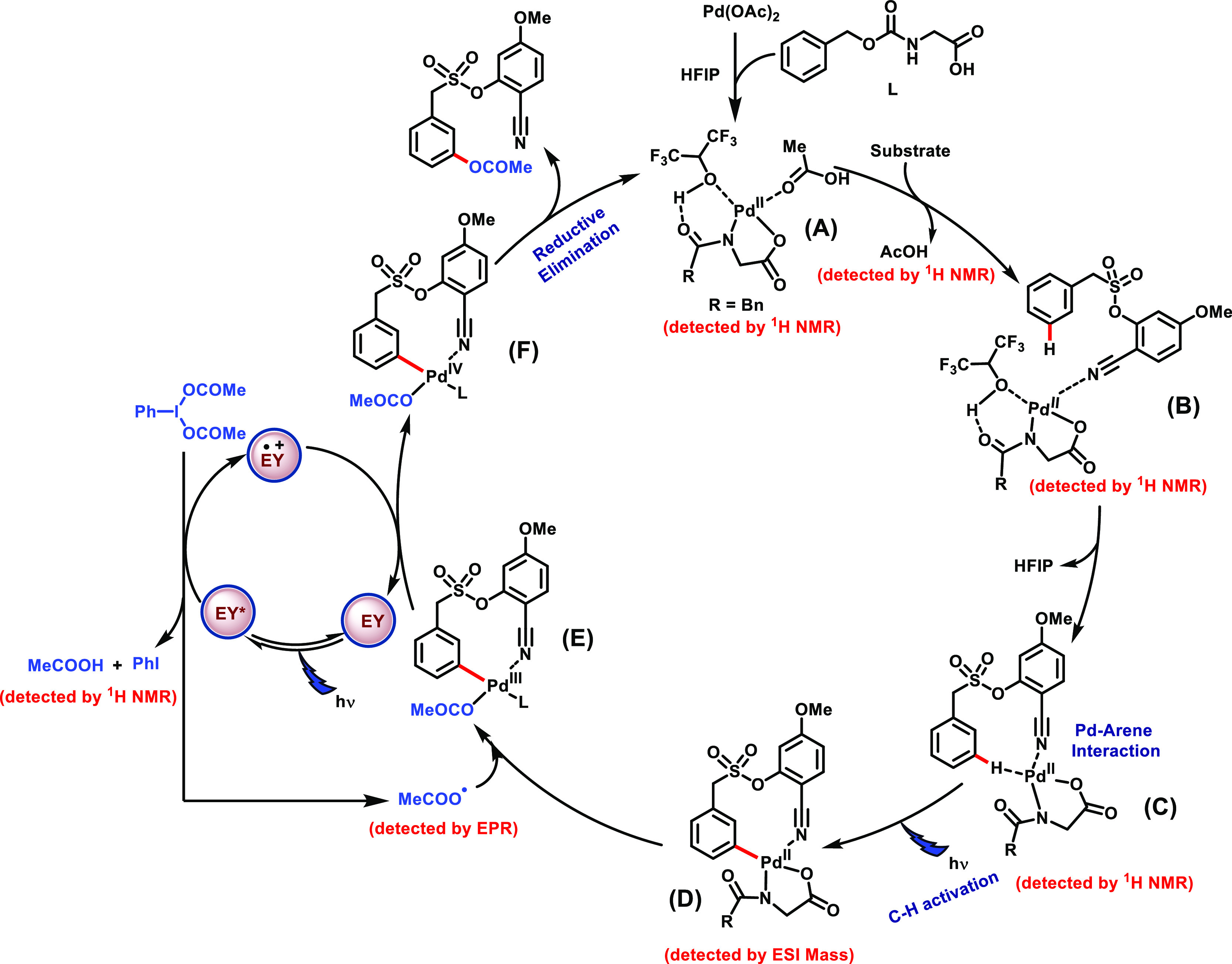
Under thermal conditions, the Pd-catalyzed C(sp2)-H-acetoxylation reaction followed a PdII/PdIV catalytic cycle12−25 in contrast to photoredox catalysis, where two catalytic cycles operate synergistically. The merger of photocatalysis with Pd catalysis can reroute the traditional Pd-catalyzed reactions, thereby enabling the reaction development through an entirely new mechanistic paradigm. To delve into the possible reaction mechanism for this photoinduced Pd-catalyzed meta-acetoxylation reaction, a series of experiments were conducted. As discussed earlier, in the absence of light (dark condition) and a photocatalyst (eosin Y), no desired product formation was observed highlighting the role of light and the photocatalyst in the transformation. In our previous report, we demonstrated that light plays a significant role in the C–H activation process. To further confirm the hypothesis, when a model reaction was set up in the absence of PhI(OAc)2, a homo-dimerized product was obtained through cross-dehydrogenative coupling engaging the meta-C–H bond, which remains unreactive at room temperature in dark conditions (Scheme 6d). The requirement of light irradiation for dimerization indicates that the threshold energy required for the C–H activation step is fulfilled by photons. Apart from this, the conversion profile of the reaction was supervised through on/off experiments (see Supporting Information, Section 6.V.). The meta-acetoxylated product formation ceased when the CFL was switched off, signifying the requirement of constant photoirradiation for an effective outcome. A UV–visible study of individual reaction components and reaction mixture was carried out, and a broad signal between 440 and 480 nm for the reaction mixture that falls in the visible region is expected to be the light-absorbing species to favor the C–H activation step (Scheme 6a). The role of light in the C–H activation step is the key feature of this protocol. In order to understand the photomediated C–H activation via the intermediate C to D, luminescence quenching (Scheme 6b) experiment was performed. Initially, we have taken the palladium catalyst and the meta-scaffold, but the emission profile suggests that there is no emission spectra for the palladium catalyst. Hence, the luminescence quenching experiment between the Pd catalyst and the scaffold cannot be performed. This further strengthens our hypothesis that the overall complex of the ligand, Pd catalyst, and meta-scaffold undergoes C–H activation in the presence of visible light. Therefore, we have performed another experiment, where an equimolar mixture of the Pd catalyst–ligand was taken and quenching experiment was performed in the presence of the meta-scaffold. Interestingly, we observed the quenching phenomenon for the Pd catalyst and ligand mixture (Scheme 6b, I). This observation further corroborated with our hypothesis that light is compulsory and has an integral role in the C–H activation step. Additionally, we have performed the quenching experiment for the photocatalyst and PhI(OAc)2. The result fits with our mechanistic postulate as the interaction of the photocatalyst and PhI(OAc)2 was observed by the quenching experiment (Scheme 6b, II). To understand the nature of the reaction mechanism, radical quenching experiments were performed with 3 equiv of 2,2,6,6- tetramethylpiperidin-1-oxyl (TEMPO), phenyl N-tert-butylnitrone (PNB), and butylated hydroxytoluene (BHT), respectively (see Supporting Information, Section 6.VIII.). A omplete inhibition of the product formation suggests the generation of radical species in the reaction. The photo-irradiated generation of radical species from PhI(OAc)2 is documented in the literature;79−81 however, the intriguing aspect of the protocol is how the Pd substrate cooperativity enables the meta-oxygenation of arenes by engaging the in situ-generated acetoxy radical. In order to gain an insight into the aforesaid aspect, EPR study of the eosin Y and PhI(OAc)2 mixture under light at different time intervals was conducted and was found to be active. On the contrary, in the absence of either eosin Y or light, it was found to be EPR-silent, amplifying their role in the generation of acetoxy radicals. Furthermore, a prominent EPR signal was also observed for the standard reaction, indicating the formation of a radical species in the reaction medium (Scheme 6c). Also, quantitative iodobenzene formation was observed when an equimolar mixture of eosin Y and PhI(OAc)2 was irradiated with light for 6 h (see Supporting Information, Section 6.VI.). To get further insights into the complexity of the reaction mechanism, the coordination affinity of Pd with the ligand and substrate was monitored through 1H NMR. It was observed that the acetic acid (-CH3) peak intensified when the substrate was added to the stoichiometric Pd(OAc)2–ligand mixture (see Supporting Information, Section 10.A.). The phenomenon of the photoinduced C–H palladation step was also monitored through the 1H NMR of reaction mixtures at room temperature in dark and under visible light. When the reaction mixture was kept under dark conditions, no shift in the peak of the 1H NMR signal was observed, while the reaction kept under visible light showed a significant shift in signal (see Supporting Information, Section 10.B.). This observation further strengthens our hypothesis on the role of visible light in the C–H activation step. The presence of the C–H-palladated intermediate was also supported by the ESI-MS studies of the reaction mixture in the absence of PhI(OAc)2, while in dark condition, no C–H-activated complex mass was observed (see Supporting Information, Section 10.C.). All these observations further strengthen the hypothesis on the role of visible light in the C–H activation step. The isotope-labeling experiment carried out between nondeuterated and deuterated substrates showed a high value of the kinetic isotope effect (kH/kD = 2.5) (see Supporting Information, Section 7) (Scheme 6e). The high value of KIE explicitly establishes that the C–H activation step is likely to be the rate-limiting step in the overall transformation. Furthermore, the kinetic study showed the first-order rate dependency for the substrate, ligand, and palladium catalyst, confirming their participation in the rate-limiting step (see Supporting Information, Section 8). Relying on these observations and literature precedence, an intuitive mechanistic cycle is proposed involving PdII/PdIII/PdIV intermediates (Scheme 7). The weak coordination of palladium with the nitrile group brings the Pd–ligand complex in close proximity to the meta-C–H bond, which activates the C–H bond through photoexcitation to form a large palladacyclic intermediate D. Next, the acetoxy radical formed through eosin Y-assisted reduction of PhI(OAc)2 coordinates with intermediate D to afford the PdIII intermediate E (Scheme 7). One electron oxidation of intermediate E by oxidized eosin Y results in the regeneration of eosin Y and PdIV intermediate F, which undergoes reductive elimination to deliver the meta-acetoxylated product and restore the PdII catalyst.
Scheme 6. Understanding the Mechanism for Pd-Catalyzed Photoredox meta-Acetoxylation of Arenes.
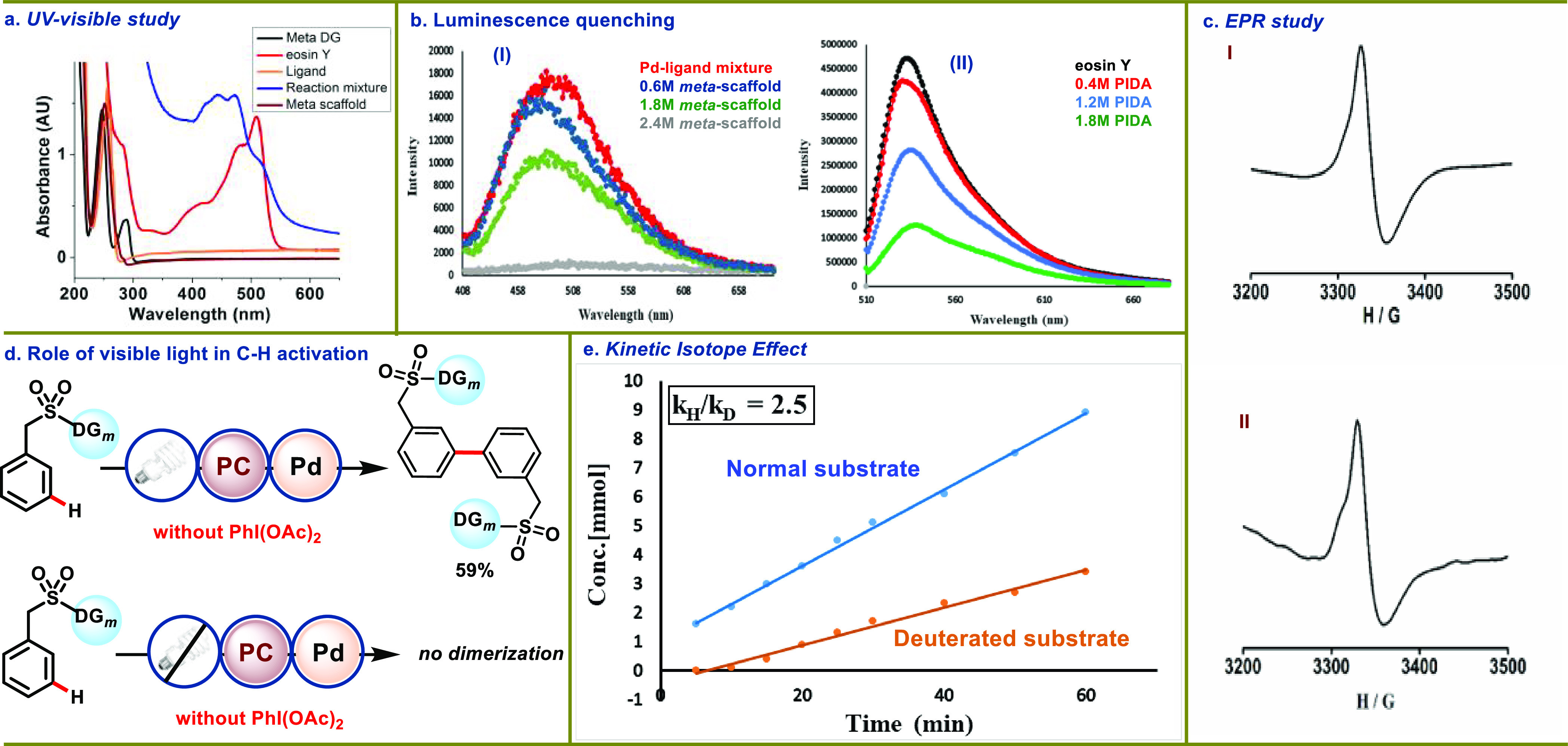
Mechanistic investigations: (a) UV–vis absorption spectroscopy studies of the reaction components and reaction mixture. (b) Luminescence quenching experiments. (c) EPR spectroscopy for the detection of radical species. (I) eosin Y, PhI(OAc)2 in HFIP at 100 K (g = 1.993), (II) scaffold, ligand, Pd(OAc)2, eosin Y, PhI(OAc)2 in HFIP at 100 K (g = 2.001). (d) Role of visible light in C–H activation. (e) Kinetic isotope effect studies.
Scheme 7. Plausible Mechanism for meta-Acetoxylation of Arenes.
The meta-acetoxylated product (33) was utilized for further functionalization to deliver synthetically versatile compounds (Scheme 8). The olefination reaction of meta-acetoxylated product delivered the meta-selective olefinated compound (55) in good yield (75%) and excellent selectivity. Similarly, meta-selective hydroxylation of the product (33) resulted in the formation of a potential resveratrol precursor (56) in moderate yield and excellent selectivity. Furthermore, the sulfonyl linker of the benzylsulfonyl ester scaffold can be easily cleaved through modified Julia olefination conditions (Scheme 8). When meta-acetoxylated products reacted with aldehydes under modified Julia olefination conditions, meta-hydroxylated alkenes (57 and 58) with a phase II “quinone reductase” (QR) activity inducer (58) were obtained in good yields. Subsequently, the isolated meta-hydroxylated alkene (57) was easily masked with triflate (59), which was then transformed into synthetically useful molecules via alkynylation (60) and arylation (61) reactions (Scheme 8).
Scheme 8. Application of the meta-Acetoxylated Product.

In summary, using the synergistic merger of photocatalysis and Pd catalysis, we have developed a highly efficient and selective photoinduced meta-oxygenation of a variety of arene systems. The reaction features a high level of regioselectivity at ambient temperature and is compatible for a series of functional groups. In the present protocol, light energy is involved in C–H activation as well as acetoxy radical generation. The control experiments suggest the formation of radical species in the reaction and the involvement of PdII/PdIII/PdIV species in the catalytic cycle. The significance of the present transformation has been illustrated through the synthesis of the resveratrol precursor and the phase II QR activity inducer.
Methods
General Procedure for meta-Acetoxylation of Phenylacetic Acid Derivatives
In an oven-dried screw-capped reaction tube charged with a magnetic stir bar, the corresponding ester of phenyl acetic acid (0.1 mmol), Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), and PhI(OAc)2 (0.2 mmol, 2 equiv) in 1 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) were added. The reaction tube was capped and placed 3 cm away from four 23 W household CFL bulbs under stirring (1500 rpm) at room temperature for 36 h. The temperature was maintained at approximately 30–35 °C through cooling with a fan. Upon completion, the mixture was diluted with ethyl acetate and filtered through a celite pad. The filtrate was evaporated under reduced pressure, and the crude mixture was purified by column chromatography using silica (100–200 mesh size) and petroleum ether/ethyl acetate as the eluent.
General Procedure for meta-Acetoxylation of Biphenyl Ester/Alcohol Derivatives
In an oven-dried screw-capped reaction tube charged with a magnetic stir bar, the corresponding biphenyl ester/alcohol (0.1 mmol), Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), and PhI(OAc)2 (0.2 mmol, 2 equiv) in 1 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) were added. The reaction tube was capped and placed 3 cm away from four 23 W household CFL bulbs under stirring (1500 rpm) at room temperature for 36 h. The temperature was maintained at approximately 30–35 °C through cooling with a fan. Upon completion, the mixture was diluted with ethyl acetate and filtered through a celite pad. The filtrate was evaporated under reduced pressure, and the crude mixture was purified by column chromatography using silica (100–200 mesh size) and petroleum ether/ethyl acetate as the eluent.
General Procedure for meta-Acetoxylation of Sulfonyl/Phosphonyl Ester Derivatives
In an oven-dried screw-capped reaction tube charged with a magnetic stir bar, the corresponding sulfonyl/phosphonyl ester (0.1 mmol), Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), and PhI(OAc)2 (0.2 mmol, 2 equiv) in 1 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) were added. The reaction tube was capped and placed 3 cm away from four 23 W household CFL bulbs under stirring (1500 rpm) at room temperature for 36 h. The temperature was maintained at approximately 30–35 °C through cooling with a fan. Upon completion, the mixture was diluted with ethyl acetate and filtered through a celite pad. The filtrate was evaporated under reduced pressure, and the crude mixture was purified by column chromatography using silica (100–200 mesh size) and petroleum ether/ethyl acetate as the eluent.
General Procedure for meta-Hydroxylation of Various Arene Derivatives
In an oven-dried screw-capped reaction tube charged with a magnetic stir bar, the corresponding sulfonyl/phosphonyl ester (0.1 mmol), Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), and PhI(OCOCF3)2 (0.2 mmol, 2 equiv) in 1 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) were added. The reaction tube was capped and placed 3 cm away from four 23 W household CFL bulbs under stirring (1500 rpm) at room temperature for 36 h. The temperature was maintained at approximately 30–35 °C through cooling with a fan. Upon completion, the mixture was diluted with ethyl acetate and filtered through a celite pad. The filtrate was evaporated under reduced pressure, and the crude mixture was purified by column chromatography using silica (100–200 mesh size) and petroleum ether/ethyl acetate as the eluent.
General Procedure for meta–meta Homocoupling
In an oven-dried screw-capped reaction tube charged with a magnetic stir bar, the corresponding sulfonic ester (0.1 mmol), Pd(OAc)2 (10 mol %), N-CBz-Gly-OH (20 mol %), and eosin Y (3 mol %) in 1 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) were added. The reaction tube was capped and placed 3 cm away from four 23 W household CFL bulbs under stirring (1500 rpm) at room temperature for 36 h. The temperature was maintained at approximately 30–35 °C through cooling with a fan. Upon completion, the mixture was diluted with ethyl acetate and filtered through a celite pad. The filtrate was evaporated under reduced pressure, and the yield was monitored using the 1H NMR signal in the presence of 1,3,5-trimethoxybenzene as the internal standard.
General Procedure for Scale-Up Reaction
In an oven-dried screw-capped reaction tube charged with a magnetic stir bar, the corresponding sulfonyl ester (3.0 mmol), Pd(OAc)2 (10 mol %), N-Cbz-Gly-OH (20 mol %), eosin Y (3 mol %), and PhI(OAc)2 (6.0 mmol, 2 equiv) in 10 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) were added. The reaction tube was capped and placed 3 cm away from four 23 W household CFL bulbs under stirring (1500 rpm) at room temperature for 36 h. The temperature was maintained at approximately 30–35 °C through cooling with a fan. Upon completion, the mixture was diluted with ethyl acetate and filtered through a celite pad. The filtrate was evaporated under reduced pressure, and the crude mixture was purified by column chromatography using silica (100–200 mesh size) and petroleum ether/ethyl acetate as the eluent.
Acknowledgments
Financial support received from SERB CRG is gratefully acknowledged (CRG/2022/004197). Financial support received from IIT Bombay (W.A.) and CSIR-India (fellowship to A.S.) is gratefully acknowledged. H.G. acknowledges NSF (CHE-2029932) and Robert A. Welch Foundation (D-2034-20200401) for the financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00231.
Experimental details including starting material synthesis, optimization details, mechanistic study, characterization data, and 1H and 13C NMR spectra of all the isolated compounds (PDF)
Author Contributions
⊥ W.A. and A.S. contributed equally
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript. W.A. and A.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- McMurray L.; O’Hara F.; Gaunt M. J. Recent developments in natural product synthesis using metal-catalysed C–H bond functionalization. Chem. Soc. Rev. 2011, 40, 1885–1898. 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]
- Abrams D. J.; Provencher P. A.; Sorensen E. J. Recent applications of C–H functionalization in complex natural product synthesis. Chem. Soc. Rev. 2018, 47, 8925–8967. 10.1039/C8CS00716K. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ge H. Site-selective C–H arylation of primary aliphatic amines enabled by a catalytic transient directing group. Nat. Chem. 2017, 9, 26–32. 10.1038/nchem.2606. [DOI] [Google Scholar]
- Wencel-Delord J.; Glorius F. C–H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem. 2013, 5, 369–375. 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]
- Cernak T.; Dykstra K. D.; Tyagarajan S.; Vachal P.; Krska S. W. The medicinal chemist’s toolbox for late-stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- Patterson L. D.; Miller M. J. Enzymatic deprotection of the cephalosporin 3′-acetoxy group using candida antarctica Lipase B. J. Org. Chem. 2010, 75, 1289–1292. 10.1021/jo902406b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerst E. P.; Arntzen C. J.; Pfister K.; Penner D. Herbicide cross-resistance in triazine-resistant biotypes of four species. Weed Sci. 1986, 34, 344–353. 10.1017/S0043174500066960. [DOI] [Google Scholar]
- May J. A.; Ratan H.; Glenn J. R.; Losche W.; Spangenberg P.; Heptinstall S. GPIIb-IIIa antagonists cause rapid disaggregation of platelets pre-treated with cytochalasin D. Evidence that the stability of platelet aggregates depends on normal cytoskeletal assembly. Platelets 1998, 9, 227–232. 10.1080/09537109876744. [DOI] [PubMed] [Google Scholar]
- O’Connor S. E.; Grosset A. The pharmacological basis and pathophysiological significance of the heart rate-lowering property of diltiazem. Fundam. Clin. Pharmacol. 1999, 13, 145–153. 10.1111/j.1472-8206.1999.tb00333.x. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Guo R.; Hu Y.; Dong X.; Lin N.; Dai X.; Wu H.; Ma S.; Yang B. Design, synthesis and biological evaluation of valepotriate derivatives as novel antitumor agents. RSC Adv. 2017, 7, 31899. 10.1039/C6RA27478A. [DOI] [Google Scholar]
- Murakami A.; Kitazono Y.; Jiwajinda S.; Koshimizu K.; Ohigashi H. Niaziminin, a thiocarbamate from the leaves of Moringa oleifera, holds a strict structural requirement for inhibition of tumor-promoter-induced Epstein-Barr virus activation. Planta Med. 1998, 64, 319–323. 10.1055/s-2006-957442. [DOI] [PubMed] [Google Scholar]
- Lyons T. W.; Sanford M. S. Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick A. R.; Hull K. R.; Sanford M. S. A highly selective catalytic method for the oxidative functionalization of C-H bonds. J. Am. Chem. Soc. 2004, 126, 2300–2301. 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]
- Yadav M. R.; Rit R. K.; Sahoo A. K. Sulfoximines: A reusable directing group for chemo- and regioselective ortho C-H oxidation of arenes. Chem. – Eur. J. 2012, 18, 5541–5545. 10.1002/chem.201200092. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Wang Y.; Yang T.; Li L.; Li D. Palladium catalyzed ortho-C–H-benzoxylation of 2-arylpyridines using iodobenzene dibenzoates. Tetrahedron Lett. 2015, 56, 6136–6141. 10.1016/j.tetlet.2015.09.097. [DOI] [Google Scholar]
- Chen K.; Wang D.; Li Z.-W.; Liu Z.; Pan F.; Zhang Y.-F.; Shi Z.-J. Palladium catalyzed C(sp3)–H acetoxylation of aliphatic primary amines to γ-amino alcohol derivatives. Org. Chem. Front. 2017, 4, 2097–2101. 10.1039/C7QO00432J. [DOI] [Google Scholar]
- Li S.; Cai L.; Ji H.; Yang L.; Li G. Pd(II)-catalysed meta-C–H functionalizations of benzoic acid derivatives. Nat. Commun. 2016, 7, 10443. 10.1038/ncomms10443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Fu L.; Li G. Incorporation of carbon dioxide into carbamate directing groups: Palladium-catalyzed meta-C–H olefination and acetoxylation of aniline derivatives. Adv. Synth. Catal. 2017, 359, 2235–2240. 10.1002/adsc.201700261. [DOI] [Google Scholar]
- Tang R.-Y.; Li G.; Yu J. Q. Conformation-induced remote meta-C–H activation of amines. Nature 2014, 507, 215. 10.1038/nature12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Lindovska P.; Zhu D.; Kim J.; Wang P.; Tang R.-Y.; Movassaghi M.; Yu J.-Q. Pd(II)-Catalyzed meta-C–H olefination, arylation, and acetoxylation of indolines using a U-shaped template. J. Am. Chem. Soc. 2014, 136, 10807–10813. 10.1021/ja505737x. [DOI] [PubMed] [Google Scholar]
- Maji A.; Bhaskararao B.; Singha S.; Sunoj R. B.; Maiti D. Directing group assisted meta-hydroxylation by C–H activation. Chem. Sci. 2016, 7, 3147–3153. 10.1039/C5SC04060D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta U.; Modak A.; Bhaskararao B.; Bera M.; Bag S.; Mondal A.; Lupton D. W.; Sunoj R. B.; Maiti D. Catalytic arene meta-C–H functionalization exploiting a quinoline-based template. ACS Catal. 2017, 7, 3162–3168. 10.1021/acscatal.7b00247. [DOI] [Google Scholar]
- Bera M.; Sahoo S. K.; Maiti D. Room-temperature meta-functionalization: Pd(II)-catalyzed synthesis of 1,3,5-trialkenyl arene and meta-hydroxylated olefin. ACS Catal. 2016, 6, 3575–3579. 10.1021/acscatal.6b00675. [DOI] [Google Scholar]
- Jayarajan R.; Das J.; Bag S.; Chowdhury R.; Maiti D. Diverse meta-C-H functionalization of arenes across different linker lengths. Angew. Chem., Int. Ed. 2018, 57, 7659–7663. 10.1002/anie.201804043. [DOI] [PubMed] [Google Scholar]
- Giri R.; Liang J.; Lei J.-G.; Li J.-J.; Wang D.-H.; Chen X.; Naggar I. C.; Guo C.; Foxman B. M.; Yu J.-Q. Pd-Catalyzed stereoselective oxidation of methyl groups by inexpensive oxidants under mild conditions: A dual role for carboxylic anhydrides in catalytic C-H bond oxidation. Angew. Chem., Int. Ed. 2005, 44, 7420–7424. 10.1002/anie.200502767. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker J. W.; Stephenson C. R. Shining light on photoredox catalysis: theory and synthetic applications. J. Org. Chem. 2012, 77, 1617–1622. 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- Beatty J. W.; Stephenson C. R. J. Amine functionalization via oxidative photoredox catalysis: methodology development and complex molecule synthesis. Acc. Chem. Res. 2015, 48, 1474–1484. 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q.; Jiang H.; Hu Z.; Ren D.; Yu S. Functionalization of C-H bonds by photoredox catalysis. Chem. Rec. 2017, 17, 754–774. 10.1002/tcr.201600125. [DOI] [PubMed] [Google Scholar]
- Crisenza G. E. M.; Melchiorre P. Chemistry glows green with photoredox catalysis. Nat. Commun. 2020, 11, 803. 10.1038/s41467-019-13887-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A. Y.; Perry I. B.; Bissonnette N. B.; Buksh B. F.; Edwards G.; Frye A. L. I.; Garry O. L.; Lavagnino M. N.; Li Y. B.; Liang X.; Mao E.; Millet A.; Oakley J. V.; Reed N. L.; Sakai H. A.; Seath C. P.; MacMillan D. W. C. Metallaphotoredox: The merger of photoredox and transition metal catalysis. Chem. Rev. 2022, 122, 1485–1542. 10.1021/acs.chemrev.1c00383. [DOI] [PubMed] [Google Scholar]
- Douglas N. H.; Nicewicz D. A. Photoredox-catalyzed C–H functionalization reactions. Chem. Rev. 2022, 122, 1925–2016. 10.1021/acs.chemrev.1c00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärkäs M. D. Jr.; Porco J. A.; Stephenson C. R. J. Photochemical approaches to complex chemotypes: Applications in natural product synthesis. Chem. Rev. 2016, 116, 9638–9747. 10.1021/acs.chemrev.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach T.; Hehn J. P. Photochemical reactions as key Steps in natural product synthesis. Angew. Chem., Int. Ed. 2011, 50, 1000–1045. 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]
- Hoffmann N. Photochemical reactions as key steps in organic synthesis. Chem. Rev. 2008, 108, 1052–1103. 10.1021/cr0680336. [DOI] [PubMed] [Google Scholar]
- Jeong D. Y.; Lee D. S.; Lee H. L.; Nah S.; Lee J. Y.; Cho E. J.; You Y. Evidence and governing factors of the radical-ion photoredox catalysis. ACS Catal. 2022, 12, 6047–6059. 10.1021/acscatal.2c00763. [DOI] [Google Scholar]
- Chandrashekar H. B.; Maji A.; Halder G.; Banerjee S.; Bhattacharyya S.; Mait D. Photocatalyzed borylation using water-soluble quantum dots. Chem. Commun. 2019, 55, 6201–6204. 10.1039/C9CC01737B. [DOI] [PubMed] [Google Scholar]
- Ghosh I.; Marzo L.; Das A.; Shaikh R.; König B. Visible light mediated photoredox catalytic arylation reactions. Acc. Chem. Res. 2016, 49, 1566–1577. 10.1021/acs.accounts.6b00229. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Organic photoredox catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- Shaw M. H.; Twilton J.; MacMillan D. W. C. Photoredox catalysis in organic chemistry. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noël T.; Zysman-Colman E. The promise and pitfalls of photocatalysis for organic synthesis. Chem Catal. 2022, 2, 468–476. 10.1016/j.checat.2021.12.015. [DOI] [Google Scholar]
- Govaerts S.; Nyuchev A.; Noël T. Pushing the boundaries of C–H bond functionalization chemistry using flow technology. J. Flow Chem. 2020, 10, 13–71. 10.1007/s41981-020-00077-7. [DOI] [Google Scholar]
- Wei X.-J.; Abdiaj I.; Sambiagio C.; Li C.; Zysman-Colman E.; Alcázar J.; Noël T. Visible-light-promoted iron-catalyzed C(sp2)–C(sp3) Kumada cross-coupling in flow. Angew. Chem., Int. Ed. 2019, 58, 13030–13034. 10.1002/anie.201906462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twilton J.; Le C. C.; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 2017, 1, 0052 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- Buzzetti L.; Crisenza G. E. M.; Melchiorre P. Mechanistic studies in photocatalysis. Angew. Chem., Int. Ed. 2019, 58, 3730–3747. 10.1002/anie.201809984. [DOI] [PubMed] [Google Scholar]
- Ravelli D.; Protti S.; Fagnoni M. Carbon–Carbon bond forming reactions via photogenerated intermediates. Chem. Rev. 2016, 116, 9850–9913. 10.1021/acs.chemrev.5b00662. [DOI] [PubMed] [Google Scholar]
- Wang C. S.; Dixneuf P. H.; Soulé J. F. Photoredox catalysis for building C-C bonds from C(sp2)-H bonds. Chem. Rev. 2018, 118, 7532–7585. 10.1021/acs.chemrev.8b00077. [DOI] [PubMed] [Google Scholar]
- Hopkinson M. N.; Sahoo B.; Li J. L.; Glorius F. Dual catalysis sees the light: combining photoredox with organo-acid, and transition-metal catalysis. Chem. – Eur. J. 2014, 20, 3874–3886. 10.1002/chem.201304823. [DOI] [PubMed] [Google Scholar]
- Lipp A.; Badir S. O.; Molander G. Stereoinduction in metallaphotoredox catalysis. Angew. Chem., Int. Ed. 2021, 60, 1714–1726. 10.1002/anie.202007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. H.; Chen H.; Zhu C.; Yu S. A review of enantioselective dual transition metal/photoredox catalysis. Sci. Chi. Chem. 2020, 63, 637. 10.1007/s11426-019-9701-5. [DOI] [Google Scholar]
- Shee M.; Singh N. D. P. Cooperative photoredox and palladium catalysis: recent advances in various functionalization reactions. Catal. Sci. Technol. 2021, 11, 742–767. 10.1039/D0CY02071K. [DOI] [Google Scholar]
- Cheung K. P. S.; Sarkar S.; Gevorgyan V. Visible light-induced transition metal catalysis. Chem. Rev. 2022, 122, 1543–1625. 10.1021/acs.chemrev.1c00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti S.; Roy S.; Ghosh P.; Kasera A.; Maiti D. Photo-excited nickel-catalyzed silyl-radical-mediated direct activation of carbamoyl chlorides to access (Hetero)aryl carbamides. Angew. Chem., Int. Ed. 2022, 61, e202207472 10.1002/anie.202207472. [DOI] [PubMed] [Google Scholar]
- Le C.; Chen T. Q.; Liang T.; Zhang P.; MacMillan D. W. C. A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 2018, 360, 1010–1014. 10.1126/science.aat4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-M.; Bellotti P.; Erchinger J. E.; Paulisch T. O.; Glorius F. Radical carbonyl umpolung arylation via dual nickel catalysis. J. Am. Chem. Soc. 2022, 144, 1899–1909. 10.1021/jacs.1c12199. [DOI] [PubMed] [Google Scholar]
- Guilleard L.; Wencel-Delord J. When metal-catalyzed C-H functionalization meets visible-light photocatalysis. Beilstein J. Org. Chem. 2020, 16, 1754–1804. 10.3762/bjoc.16.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabry D. C.; Rueping M. Merging visible light photoredox catalysis with metal-catalyzed C–H activations: on the role of oxygen and superoxide ions as oxidants. Acc. Chem. Res. 2016, 49, 1969–1979. 10.1021/acs.accounts.6b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Xun X.; Song H.; Liu Y.; Wang Q. Palladium metallaphotoredox-catalyzed 2-arylation of indole derivatives. Org. Lett. 2022, 24, 4580–4585. 10.1021/acs.orglett.2c01674. [DOI] [PubMed] [Google Scholar]
- Wang X.; Yu M.; Song H.; Liu Y.; Wang Q. Palladium metallaphotoredox-catalyzed 3-acylation of indole derivatives. Chem. Commun. 2022, 58, 9492–9495. 10.1039/D2CC03658D. [DOI] [PubMed] [Google Scholar]
- Lang S. B.; O’Nele K. M.; Tunge J. A. Decarboxylative allylation of amino alkanoic acids and esters via dual catalysis. J. Am. Chem. Soc. 2014, 136, 13606–13609. 10.1021/ja508317j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller J.; Fabry D. C.; Ronge M. A.; Rueping M. Synthesis of indoles using visible light: photoredox catalysis for palladium-catalyzed C-H Activation. Angew. Chem., Int. Ed. 2014, 53, 13264–13268. 10.1002/anie.201405478. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Li P.; Zhu X.; Wang L. Merging photoredox with palladium catalysis: Decarboxylative ortho-acylation of acetanilides with α-oxocarboxylic acids under mild reaction conditions. Org. Lett. 2015, 17, 6198–6201. 10.1021/acs.orglett.5b03192. [DOI] [PubMed] [Google Scholar]
- Song C.; Zhang H.-H.; Yu S. Regio- and enantioselective decarboxylative allylic benzylation enabled by dual palladium/photoredox catalysis. ACS Catal. 2022, 12, 1428–1432. 10.1021/acscatal.1c05461. [DOI] [Google Scholar]
- Wang H.; Li T.; Hu D.; Tong X.; Zheng L.; Xia C. Acylation of arenes with aldehydes through dual C–H activations by merging photocatalysis and palladium catalysis. Org. Lett. 2021, 23, 3772–3776. 10.1021/acs.orglett.1c01184. [DOI] [PubMed] [Google Scholar]
- Narayanam J. M. R.; Stephenson C. R. J. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]
- Schultz D. M.; Yoon T. P. Solar synthesis: prospects in visible light photocatalysis. Science 2014, 343, 1239176 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischay M. A.; Anzovino M. E.; Du J.; Yoon T. P. Efficient visible light photocatalysis of [2+2] enone cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- Narayanam J. M. R.; Tucker J. W.; Stephenson C. R. J. Electron-transfer photoredox catalysis: Development of a tin-free reductive dehalogenation reaction. J. Am. Chem. Soc. 2009, 131, 8756–8757. 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]
- Parasram M.; Gevorgyan V. Visible light-induced transition metal-catalyzed transformations: beyond conventional photosensitizers. Chem. Soc. Rev. 2017, 46, 6227–6240. 10.1039/C7CS00226B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuentragool P.; Kurandina D.; Gevorgyan V. Catalysis with palladium complexes photoexcited by visible light. Angew. Chem., Int. Ed. 2019, 58, 11586–11598. 10.1002/anie.201813523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuribara T.; Nakajima M.; Nemoto T. A. A visible-light activated secondary phosphine oxide ligand enabling Pd-catalyzed radical cross-couplings. Nat. Commun. 2022, 13, 4052. 10.1038/s41467-022-31613-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G.; Yao W.; Mauro J. N.; Ngai M.-Y. Excited-state palladium-catalyzed 1,2-spin-center shift enables selective C-2 reduction, deuteration, and iodination of carbohydrates. J. Am. Chem. Soc. 2021, 143, 1728–1734. 10.1021/jacs.0c11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K. P. S.; Kurandina D.; Yata T.; Gevorgyan V. Photoinduced palladium-catalyzed carbo-functionalization of conjugated dienes proceeding via radical-polar crossover scenario: 1,2-aminoalkylation and beyond. J. Am. Chem. Soc. 2020, 142, 9932–9937. 10.1021/jacs.0c03993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvasovs N.; Iziumchenko V.; Palchykov V.; Gevorgyan V. Visible light-induced Pd-catalyzed alkyl-Heck reaction of oximes. ACS Catal. 2021, 11, 3749–3754. 10.1021/acscatal.1c00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasram M.; Chuentragool P.; Sarkar D.; Gevorgyan V. Photoinduced formation of hybrid aryl Pd-radical species capable of 1,5-HAT: Selective catalytic oxidation of silyl ethers into silyl enol ethers. J. Am. Chem. Soc. 2016, 138, 6340–6343. 10.1021/jacs.6b01628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratushnyy M.; Parasram M.; Wang Y.; Gevorgyan V. Palladium-catalyzed atom-transfer radical cyclization at remote unactivated C(sp3)–H Sites: Hydrogen-atom transfer of hybrid vinyl palladium radical intermediates. Angew. Chem., Int. Ed. 2018, 57, 2712–2715. 10.1002/anie.201712775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. Room-temperature C–H arylation: Merger of Pd-catalyzed C–H functionalization and visible-light photocatalysis. J. Am. Chem. Soc. 2011, 133, 18566–18569. 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A.; Guin S.; Ali W.; Bhattacharya T.; Sasmal S.; Goswami N.; Prakash G.; Sinha S. K.; Chandrashekar H. B.; Panda S.; Anjana S. S.; Maiti D. Photoinduced regioselective olefination of arenes at proximal and distal sites. J. Am. Chem. Soc. 2022, 144, 1929–1940. 10.1021/jacs.1c12311. [DOI] [PubMed] [Google Scholar]
- He Z.; Bae M.; Wu J.; Jamison T. F. Synthesis of highly functionalized polycyclic quinoxaline derivatives using visible-light photoredox catalysis. Angew. Chem., Int. Ed. 2014, 53, 14451–14455. 10.1002/anie.201408522. [DOI] [PubMed] [Google Scholar]
- Xie J.; Xu P.; Li H.; Xue Q.; Jin H.; Chenga Y.; Zhu C. A room temperature decarboxylation/C–H functionalization cascade by visible-light photoredox catalysis. Chem. Commun. 2013, 49, 5672–5674. 10.1039/c3cc42672f. [DOI] [PubMed] [Google Scholar]
- Paul A.; Chatterjee D. R.; Halder T.; Banerjee S.; Yadav S. Metal free visible light photoredox activation of PhI(OAc)2 for the conversion of arylboronic acids to phenols. Tetrahedron Lett. 2015, 56, 2496–2499. 10.1016/j.tetlet.2015.03.107. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.