

Abstract
Replication fork reversal safeguards genome integrity as a replication stress response. DNA translocases and the RAD51 recombinase catalyze reversal. However, why RAD51 is required and what happens to the replication machinery during reversal remain unknown. Here we find that RAD51 utilizes its strand exchange activity to circumvent the replicative helicase, which remains bound to the stalled fork. RAD51 is not required for fork reversal if the helicase is unloaded. Thus, we propose that RAD51 creates a parental DNA duplex behind the helicase that is utilized as a substrate by the DNA translocases for branch migration to create a reversed fork structure. Our data explains how fork reversal happens while maintaining the helicase in a position poised to restart DNA synthesis and complete genome duplication.
One-Sentence Summary:
Fork reversal requires the RAD51 recombinase to overcome the CMG helicase which remains bound to the stalled fork.
Replication is challenged by various stressors including DNA damage, collisions with transcriptional machineries, and unusual DNA structures that stall replication elongation (1). Often this replication stress uncouples DNA synthesis from unwinding, triggering responses that stabilize the stalled fork and promote genome stability. One of these responses is replication fork reversal (2). Fork reversal is thought to help cells tolerate replication stress by facilitating the repair of DNA lesions, switching DNA templates to allow bypass of obstacles, or stabilizing the fork until a converging replication fork completes DNA synthesis. Reversal involves the coordinated reannealing of the parental DNA template strands combined with displacement and annealing of the nascent DNA strands (2). Previous studies showed several ATP-dependent translocases generate reversed forks, including SMARCAL1, ZRANB3, HLTF, and FBH1 (3–7). In addition, RAD51, a well-studied recombinase in homologous recombination repair of double-strand breaks, is required for reversal but how it acts is unclear (8).
A major unanswered question about fork reversal is the fate of the replisome during the reversal process, especially the CMG complex, which consists of six MCM subunits (MCM2-7) that combine with CDC45 and the GINS hetero-tetramer to form the active helicase. Dissociation of CMG to facilitate reversal would be potentially catastrophic for completing DNA replication and maintaining genome stability since it cannot be reloaded during S-phase (9). Even if another helicase could unwind the parental duplex, it could not easily replace the myriad of other functions mediated by CMG including scaffolding other replication and replication-coupled repair proteins and chaperoning histones to re-establish chromatin (10–12). Current fork reversal models suggest that the DNA fork junction created by the helicase is reversed and converted into a four-way junction. Whether this process can occur in the presence of CMG remains unknown. Indeed, in vitro studies of DNA replication using plasmids in Xenopus egg extracts found that reversal requires unloading the helicase when replisomes converge at an interstrand crosslink (ICL) (13). Confining reversal to situations in which replisomes converge would overcome the need for retaining CMG; however, fork reversal is a common response to fork stalling in human cells even in conditions like hydroxyurea (HU) treatment where fork convergence is prevented. Thus, understanding the fate of the helicase is critical to determining how reversal happens and validating it as a replication stress-tolerance mechanism as opposed to a dead-end pathological event associated with fork collapse.
The CMG replicative helicase remains trapped on the DNA during replication fork reversal
Our previous iPOND proteomics studies indicated that there is little loss of the helicase proteins until at least 8 hours after HU-induced fork stalling even as fork reversal factors like SMARCAL1 are recruited and reversal is detected (Fig. 1A and (8, 14)). When HU is removed, forks rapidly resumed DNA synthesis (Fig. 1B). Fork restart requires CMG since inactivating the MCM2 subunit by proteolysis using an improved auxin-inducible degron (AID2) (15) during the HU treatment prevented restart (Fig. 1B). MCM2 is almost completely lost from chromatin within one hour of addition of 5-ph-IAA (5-phenyl-indole-3-acetic acid) to the MCM2-AID2 degron cells, and this is accompanied by a loss of DNA synthesis and a loss of MCM7 on chromatin suggesting that the entire MCM complex is disassembled and removed (Fig. S1A–S1D). In contrast, MCM7 is not lost from chromatin in HU-treated cells without MCM2 degradation (Fig. S1E).(14). These data confirm that the MCM complex remains bound to stalled forks, poised to promote restart even in conditions that cause fork reversal.
Fig. 1. Fork reversal does not require CMG disassembly.
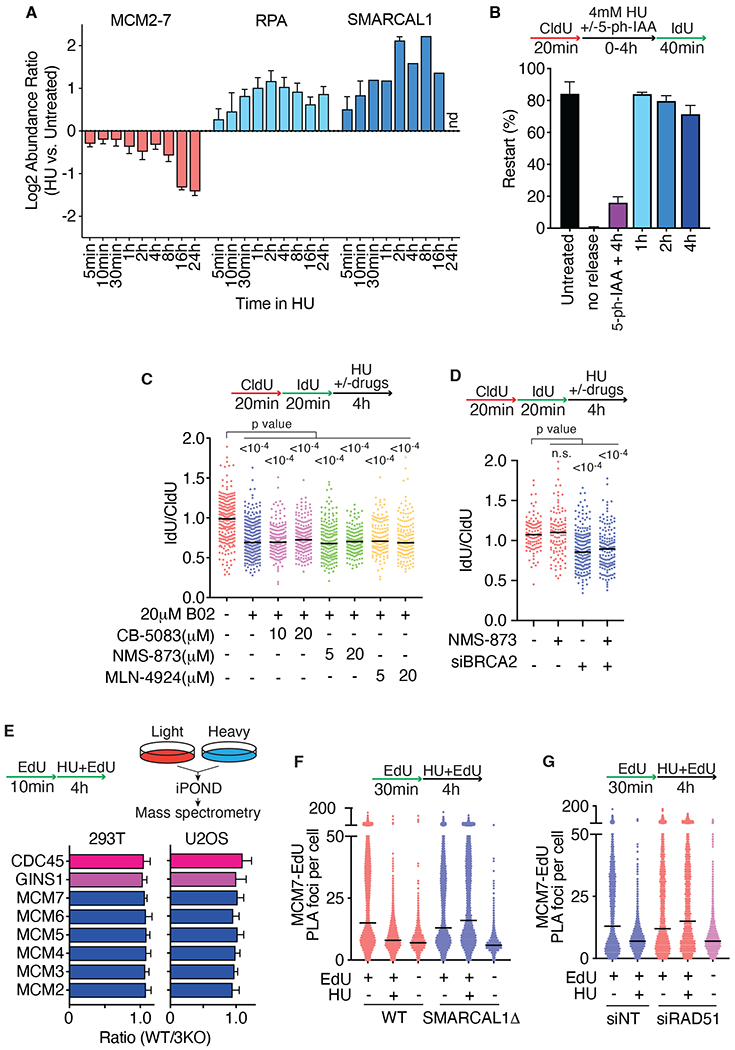
(A) iPOND-SILAC mass spectrometry measured abundance changes of selected proteins or complexes comparing HU vs. untreated cells (generated from original data in (14)). nd, not detected. (B) MCM2-AID2 HCT116 cells were labeled with CldU and IdU and treated with 4mM HU for 0-4 hours. Where indicated, 2 μM 5-ph-IAA was added to degrade MCM2 during the HU treatment. Restart efficiency was calculated as the percentage of continuous red and green fibers compared to the total imaged by DNA combing. Mean and SD of three experiments is shown. (C and D) Fork protection assays were completed as indicated. U2OS cells were treated with the inhibitors during the HU treatment time. All graphs are representative of at least three experiments. siNT, non-targeting siRNA. P values were calculated using a Kruskal-Wallis test. (E) iPOND-SILAC-mass spectrometry was used to measure the abundance of proteins at stalled replication forks in HU-treated wild type (WT) and SMARCAL1, ZRANB3, and HLTF triple knockout (3KO) cells. (F) PLA assay for EdU and MCM7 in wild-type or SMARCAL1Δ U2OS cells. (G) PLA of EdU and MCM7 in cells transfected with RAD51 siRNA.
Since CMG is not removed from most replication forks in response to persistent stalling, we asked whether it needs to be removed to allow reversal. Reversed forks are the substrates for nascent strand degradation in the absence of fork protection factors (7, 16–18), so we used degradation to test that reversal is operational. As previously described, treating cells with the selective RAD51 inhibitor B02 or silencing the fork protection factor BRCA2 caused nascent strand degradation (Fig. 1C and 1D) (19, 20). The known pathways for CMG removal require ubiquitylation followed by extraction by the p97 segregase (21, 22). Suppressing these activities with p97 inhibitors (CB-5083 and NMS-873) or a neddylation inhibitor that blocks MCM ubiquitylation (MLN-4924) (21, 22) did not affect nascent strand degradation (Figs. 1C, 1D, S2A, and S2B)). These results confirm that the presence of the CMG complex at the stalled fork does not prevent nascent strand degradation and, by inference, fork reversal in human cells.
If the CMG complex remains present at the replication fork, it is unclear how fork reversal can happen since DNA footprinting and binding studies suggest that the fork reversal enzymes like SMARCAL1 would need to bind the DNA in a partially overlapping position with CMG (23–25). Thus, we asked if CMG is repositioned during reversal. We first used iPOND proteomics to examine CMG abundance near nascent DNA in cells lacking fork reversal enzymes compared to wild-type cells. We found no change in any of the detected CMG subunits in either U2OS or HEK293T cells lacking SMARCAL1, ZRANB3, and HLTF (Fig. 1E). We next utilized a proximity ligation assay (PLA) assay which has higher spatial resolution than iPOND to ask if the CMG complex is still intimately associated with nascent DNA. The PLA signal between MCM7 and EdU was reduced after HU treatment in a SMARCAL1- and RAD51-dependent manner (Figs. 1F and 1G). This result suggests that the helicase is not pushed backward during fork reversal since then it should be associated with the nascent strands. Instead, it suggests that CMG is trapped in a single-stranded DNA (ssDNA) bubble within the parental DNA ahead of the reversed fork (Fig. S2C). It is possible that the helicase could switch to a double-stranded DNA (dsDNA) binding mode during fork reversal to move it onto the parental DNA away from the fork junction (26). However, it is unclear how CMG encircling dsDNA would avoid the normal unloading process triggered by this transition (27).
RAD51 strand exchange activity promotes fork reversal
If fork reversal happens behind the CMG helicase with it continuing to encircle ssDNA, then the DNA fork that is reversed is not the one that CMG creates by unwinding the parental DNA duplex (Fig. S2C, panel ii). RAD51 is recruited within 15 minutes to stalled forks (14), and we hypothesized that it could use its strand exchange activity to generate a new substrate behind the CMG for the DNA translocases (Fig. S2D). To test this hypothesis, we examined RAD51 mutants to test if its strand exchange activity is required for reversal. Since RAD51 is essential for cell viability, we utilized an siRNA complementation approach in which endogenous RAD51 is silenced in cell lines stably expressing siRNA-resistant, exogenous wild-type or mutant RAD51. We started with fork protection assays in cells depleted of BRCA2 as an indirect readout for fork reversal (Fig. S3A). To accurately measure normal RAD51 function, we utilized microRNA silencing-mediated fine-tuners (miSFITs) vectors to generate cells expressing near endogenous levels of RAD51 (28). Failure to control RAD51 levels in this way leads to overexpression and prevents nascent strand degradation even when BRCA2 is silenced as previously described (Fig. S3B, S3C)(19, 29). The reason this is the case is not known but could be because overexpression interferes with the generation of the degradation substrate (30) or causes protection of the reversed fork without requiring BRCA2 stabilization. Inserting the “8A” miRNA-17 target sequence into the 3’ UTR of the RAD51 expression vector provided near the endogenous expression of wild-type RAD51 (Fig. S3B). Silencing endogenous RAD51 and BRCA2 in these cells caused SMARCAL1-dependent nascent strand degradation indicating this complementation system faithfully restores RAD51 function and can be used to examine RAD51 mutants (Figs. S3C and S3D).
We applied this approach to test the activity of seven RAD51 mutant proteins (summarized in Table S1). Three of these RAD51 proteins, I287T, K133R, and G151D retain strand exchange or D-loop formation activity (31–37). Four of the RAD51 proteins, T131P, A293T, II3A, and Y232A, have decreased or inactive strand exchange or D-loop formation activity (38–43). In addition to selecting the optimal miSFIT vector for each, we also monitored protein expression over time since some of the RAD51 proteins changed expression with increasing cell passages (Figs. S3E–S3I). We also ensured that the system maintains near physiological cell-to-cell heterogeneity in RAD51 protein levels (Figs. S4A–S4B). All analyses were performed when the cells expressed levels of the mutant proteins comparable to endogenous RAD51 unless otherwise noted.
The three RAD51 mutants that retain strand exchange activity, I287T, K133R, and G151D, complemented the loss of endogenous RAD51 to allow nascent strand degradation in BRCA2-deficient cells when expressed at near endogenous levels (Fig. 2A) even though overexpression of I287T and K133R block degradation (Fig S5A and S5B). In contrast, nascent strand degradation was not observed in cells expressing the strand exchange/D-loop formation defective proteins, T131P, A293T, II3A, and Y232A. (Fig. 2A). T131P, A293T, and Y232A have significant defects in DNA binding. However, II3A has only a modest change in DNA binding affinity and retains the ability to form nucleoprotein filaments (42, 44). The II3A mutant was previously reported to be capable of generating a degradation substrate (42); however, that experiment was done in cells significantly overexpressing II3A, and degradation was monitored only 8 hours after HU treatment when degradation happens irrespective of the presence of fork protection factors (45).
Fig. 2. RAD51 recombinase activity promotes fork reversal.
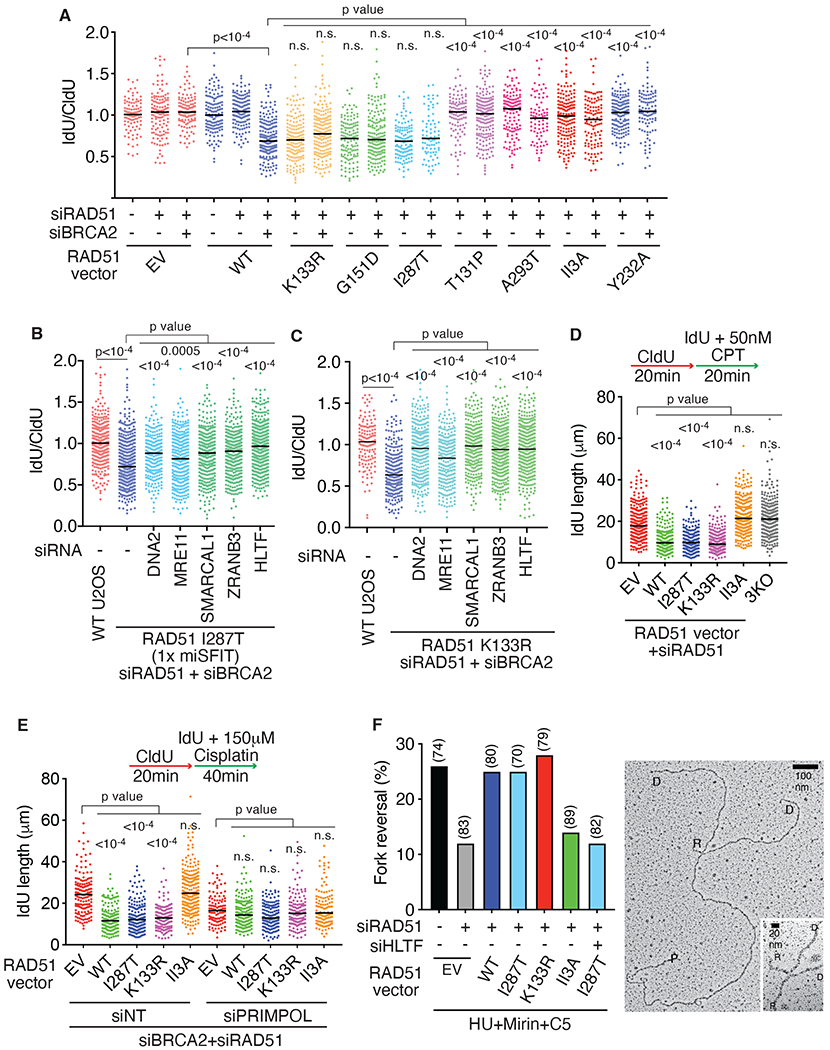
(A-C) Fork protection assays were completed in U2OS cells expressing near endogenous levels of the indicated RAD51 proteins after transfection with the indicated siRNAs. P values were calculated using a Kruskal-Wallis test. (EV, empty vector) (D-E) Replication elongation rate in the presence of 50nM camptothecin (CPT) in D) or 150mM cisplatin in E). P values were calculated using a Kruskal-Wallis test. (F) Percentage of reversed replication forks in U2OS cells expressing endogenous levels of the indicated RAD51 proteins. Cells were transfected with the indicated siRNA and treated 72 hours later with 4mM HU, 20μM MRE11 inhibitor (Mirin) and 25μM DNA2 inhibitor (C5) for 5 hours. The number of replication intermediates analyzed for each condition is indicated in parentheses and a representative image is shown (right panel), P: parental strand, D: daughter strand and R: reversed arm.
As reported previously using the heterozygous Fanconi Anemia patient cells (38), RAD51 T131P has a dominant-negative effect on the fork protection activity of endogenous RAD51 (Fig. S5C). However, nascent strand degradation is prevented once endogenous RAD51 is depleted and only T131P RAD51 is expressed (Fig. 2A). Thus, the T131P mutant itself cannot perform fork reversal, but when co-expressed with wild-type RAD51, there is sufficient RAD51 function to do reversal but not protection. This is consistent with the observations that the T131P RAD51 protein is deficient in strand exchange activity but combining the mutant and wild-type proteins can yield sufficient RAD51 function to perform exchange and promote homologous recombination (38). This situation may also mimic the observation that partial loss of RAD51 function through depletion or chemical inhibition is sufficient to inactivate its fork protection but not fork reversal functions (7, 46).
Nascent strand degradation after BRCA2 silencing in cells expressing only the RAD51 I287T or K133R proteins is dependent on the MRE11 and DNA2 nucleases, and the fork reversal enzymes SMARCAL1, ZRANB3, and HLTF confirming that these three DNA translocases promote the formation of a reversed fork substrate for degradation in these cells (Fig. 2B and 2C). Nascent strand degradation happened in cells expressing endogenous levels of the I287T, K133R, and G151D mutant proteins even when BRCA2 is not silenced (Fig. 2A). SMARCAL1, ZRANB3, HLTF, MRE11, and DNA2 silencing reduced this degradation in the RAD51 I287T or K133R expressing cells (Fig. S5D and S5E). In addition, silencing the structure-specific endonuclease MUS81 and the endonuclease scaffold SLX4 also reduced degradation (Fig. S5D and S5E). Thus, these mutants may generate reversed forks and substrates for the endonucleases, but further studies will be needed to understand why these forks are insensitive to BRCA2-mediated stabilization even though overexpression of either I287T or K133R prevents degradation in BRCA2-deficient cells (Figures S5A and S5B)(19).
RAD54 and the RAD51AP1-UAF1 complex are required to assist RAD51 to form D-loops (47, 48). If fork reversal involves strand invasion and D-loop formation, we might expect these proteins to also be required for reversal and nascent strand degradation. As predicted, silencing RAD54 or RAD51AP1-UAF1 prevented nascent strand degradation consistent with a requirement for strand invasion and D-loop formation in the reversal process (Fig. S5F).
We next examined fork elongation rates in cisplatin- or camptothecin-treated cells as a second measure of fork reversal since reversal slows elongation in these conditions (6, 8, 49). Indeed, fork speeds were significantly faster in camptothecin- or cisplatin-treated cells lacking RAD51 or expressing the RAD51 II3A mutant compared to wild-type, I287T, or K133R RAD51 as predicted if RAD51 strand exchange is required for fork reversal (Figs. 2D and 2E). Faster elongation in cells that lack fork reversal is due to PRIMPOL-dependent repriming to tolerate the replication stress (4, 49). S1 nuclease digestion of DNA fibers from cells lacking RAD51 or expressing only the II3A mutant shortened the fibers, and PRIMPOL depletion slowed elongation in these circumstances suggesting that PRIMPOL-dependent repriming that leaves ssDNA gaps is active in these cells as an alternative to reversal (Figs. 2E and S5G). In contrast, fibers in the wild-type, I287T or K133R RAD51 expressing cells were unaffected by S1 nuclease or PRIMPOL depletion.
We examined replication intermediates by electron microscopy as a final test to determine if fork reversal is only operable in cells expressing strand-exchange proficient RAD51 proteins. Consistent with the nascent strand degradation and fork elongation assays, WT, I287T, and K133R RAD51 supported fork reversal, but the II3A RAD51 protein expressing cells showed the same reduction in reversal as RAD51-deficient cells (Figs. 2F and S6). Silencing HLTF in the I287T mutant cells reduced fork reversal as expected.
RAD51 is not required for fork reversal if the CMG helicase is removed from replication forks
Altogether, these results suggest that fork reversal requires the strand exchange activity of RAD51. One possibility is that RAD51-dependent strand exchange generates a paranemic DNA duplex behind the CMG complex. Paranemic joints are formed by RAD51 when there is not a free DNA end(50). This would create a substrate for fork reversal enzymes without requiring the removal of CMG. This model predicts that RAD51 may not be required for fork reversal if the CMG complex is removed. To test this prediction, we degraded MCM2 using the auxin-inducible degron during the HU treatment period of the fork protection assay and asked if RAD51 is still needed to generate a substrate for nascent strand degradation. As predicted, the destruction of MCM2 and disassembly of the MCM complex allowed nascent strand degradation even when RAD51 is silenced and unable to promote reversal (Fig. 3A). This degradation remained dependent on MRE11, DNA2, SMARCAL1, ZRANB3, and HLTF indicating that it occurs downstream of a RAD51-independent fork reversal process (Fig. 3B). Furthermore, nascent strand degradation is also observed after MCM2 degradation in cells only expressing the II3A RAD51 mutant indicating the RAD51 strand exchange function is needed to overcome the presence of the MCM complex (Fig. S7A).
Fig. 3. RAD51 is not required for fork reversal when CMG is disassembled from the stalled replication fork.
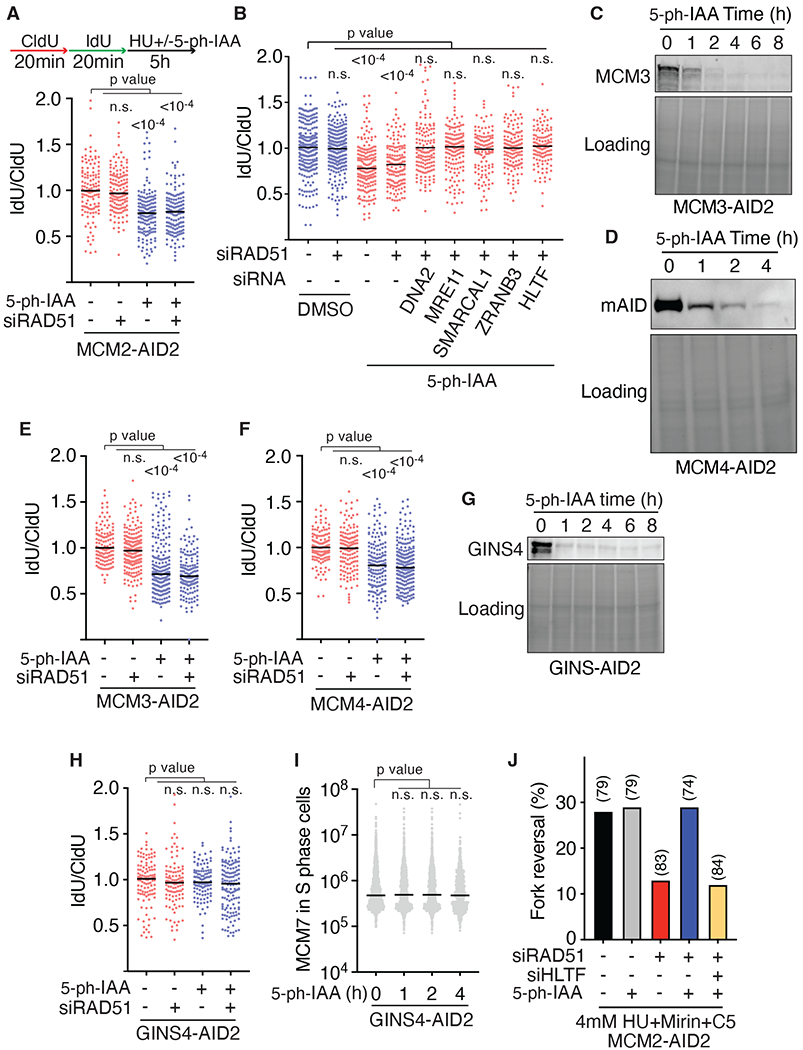
(A-B) Fork protection assays were completed in MCM2-AID2 degron cells after transfection with siRNAs. 2μM 5-ph-IAA was added to induce MCM2 degradation. (C-D) Immunoblots of MCM3-AID2 and MCM4-AID2 degron cells. (E-F) Fork protection assays in the MCM3-AID2 and MCM4-AID2 degron cells. (G) Immunoblot of GINS4 degron cells. (H) Fork protection assay in the GINS4-AID2 degron cells. (I) MCM7 integrated intensity in the nucleus of GINS4-AID2 degron cells was measured by immunofluorescence. All graphs are representative of at least three experiments. P values were calculated using a Kruskal-Wallis test. (J) Percentage of reversed replication forks in MCM2-AID2 cells transfected with the indicated siRNA and treated 72 hours later with DMSO or 2μM 5-ph-IAA together with 4mM HU, Mirin, and C5 for 5 hours. The number of replication intermediates analyzed for each condition is indicated in parentheses.
MCM2 degradation causes nascent strand degradation even when RAD51 is not silenced (Fig. 3A). Again, this degradation depended on the same fork reversal and nuclease enzymes (Fig. S7B). To better understand why nascent strand degradation happens after MCM2 destruction even when wild-type RAD51 is present to protect the reversed fork, we examined the fork proteome in these conditions using iPOND. Degradation of MCM2 caused the loss of the entire CMG complex along with other replisome components (Fig. S7C). ssDNA binding proteins like RPA were enriched after MCM degradation, as were RAD51 and SMARCAL1. In contrast, FANCD2 and FANCI were lost. FANCD2 was one of the first fork protection factors identified (51). It directly interacts with and inhibits DNA2 and MRE11 nucleases (52). Since FANCD2 binds MCM2-7 (53), we hypothesized that the loss of MCMs reduces FANCD2 accumulation at the stalled fork leading to DNA2 and MRE11 mediated degradation. Consistent with this interpretation, overexpression of FANCD2 in the MCM2-degron cells prevented nascent strand degradation (Fig. S7D).
We further confirmed that RAD51 is no longer required to generate a nascent strand degradation substrate if the helicase is removed using MCM3 and MCM4 degron cells. Like MCM2, the destruction of either MCM3 or MCM4 caused a rapid reduction in DNA synthesis and disassembly of the entire MCM complex as evidenced by the loss of MCM7 on chromatin (Figs. 3C, 3D, S7E–S7H). Removing MCM3 or MCM4 during the HU treatment allowed nascent strand degradation irrespective of whether RAD51 was depleted (Fig. 3E and 3F). In contrast, degrading GINS4 did not remove the MCM complex from the chromatin and did not allow nascent strand degradation in the absence of RAD51 suggesting that the presence of the MCM ring at the fork and not helicase activity itself is why RAD51 is needed (Fig. 3G–3I).
To directly monitor if replication forks can reverse after MCM destruction when RAD51 is depleted, we examined the frequency of reversed fork structures by electron microscopy. As previously reported, silencing RAD51 reduced fork reversal in response to replication stress (8) (Fig. 3J and S7I). However, removing the MCM complex largely restored the frequency of reversed forks in RAD51-deficient cells, and this reversal remained dependent on the fork reversal enzyme HLTF (Fig. 3J and S7I).
Discussion
Altogether, our data support a model of fork reversal that explains how reversal can happen without CMG unloading, identifies a specific function for RAD51 in the reversal process, and suggests that the fork that is reversed is not the same DNA junction that the helicase creates by unwinding. RAD51 utilizes the same strand invasion activity it uses during homologous recombination to generate a new fork junction behind the helicase, which the ATP-dependent motor proteins can then branch migrate to yield the reversed fork structure observed by electron microscopy (Fig. S8). This model provides an explanation for both what happens to CMG during reversal and why RAD51 is required. While RAD51 could have additional functions in the process such as directly stimulating the fork reversal enzymes (54), by circumventing and trapping CMG within the parental ssDNA, RAD51 allows the helicase to remain poised to resume unwinding to facilitate DNA synthesis after the source of replication stress is resolved.
Supplementary Material
Acknowledgments:
The authors thank the Vanderbilt Proteomics core for assistance with the mass spectrometry. We thank Dr. Judith Campbell lab for the FANCD2 expression plasmid.
Funding:
National Institutes of Health grant R01GM116616 (DC)
Breast Cancer Research Foundation Grant (DC)
National Cancer Institute grant R01CA237263 (AV)
National Cancer Institute grant R01CA248526 (AV)
U.S. Department of Defense (DOD) Breast Cancer Research Program (BRCP) Expansion Award BC191374 (AV)
JSPS KAKENHI grant JP21K15021 (YS)
JSPS KAKENHI grants JP21H04719 and JP22H04703 (MTK)
JST CREST program JPMJCR21E6 (MTK)
Footnotes
Competing interests: Authors declare that they have no competing interests.
Data and materials availability:
All data is provided in the manuscript or supplementary materials. All materials are available upon request. Some plasmids and cell lines may require a material transfer agreement (MTAs).
References and Notes:
- 1.Cortez D, Replication-Coupled DNA Repair. Molecular cell 74, 866–876 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berti M, Cortez D, Lopes M, The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol, (2020). [DOI] [PubMed] [Google Scholar]
- 3.Betous R et al. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev 26, 151–162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bai G et al. HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Molecular cell 78, 1237–1251 e1237 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fugger K et al. FBH1 Catalyzes Regression of Stalled Replication Forks. Cell Rep 10, 1749–1757 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Vujanovic M et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Molecular cell 67, 882–890 e885 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taglialatela A et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Molecular cell 68, 414–430 e418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zellweger R et al. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costa A, Diffley JFX, The Initiation of Eukaryotic DNA Replication. Annu Rev Biochem 91, 107–131 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Cabello-Lobato MJ et al. Physical interactions between MCM and Rad51 facilitate replication fork lesion bypass and ssDNA gap filling by non-recombinogenic functions. Cell Rep 36, 109440 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Huang H et al. A unique binding mode enables MCM2 to chaperone histones H3-H4 at replication forks. Nat Struct Mol Biol 22, 618–626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gambus A et al. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol 8, 358–366 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Amunugama R et al. Replication Fork Reversal during DNA Interstrand Crosslink Repair Requires CMG Unloading. Cell Rep 23, 3419–3428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dungrawala H et al. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Molecular cell 59, 998–1010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yesbolatova A et al. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nature communications 11, 5701 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolinjivadi AM et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Molecular cell 67, 867–881 e867 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu W, Krishnamoorthy A, Zhao R, Cortez D, Two replication fork remodeling pathways generate nuclease substrates for distinct fork protection factors. Science advances 6, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mijic S et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nature communications 8, 859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlacher K et al. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 145, 529–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leuzzi G, Marabitti V, Pichierri P, Franchitto A, WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. The EMBO journal 35, 1437–1451 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maric M, Maculins T, De Piccoli G, Labib K, Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 346, 1253596 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreno SP, Bailey R, Campion N, Herron S, Gambus A, Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 346, 477–481 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Betous R et al. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep 3, 1958–1969 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhat KP, Betous R, Cortez D, High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J Biol Chem 290, 4110–4117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mason AC et al. A structure-specific nucleic acid-binding domain conserved among DNA repair proteins. Proc Natl Acad Sci U S A, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wasserman MR, Schauer GD, O’Donnell ME, Liu S, Replication Fork Activation Is Enabled by a Single-Stranded DNA Gate in CMG Helicase. Cell 178, 600–611 e616 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenkyn-Bedford M et al. A conserved mechanism for regulating replisome disassembly in eukaryotes. Nature 600, 743–747 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michaels YS et al. Precise tuning of gene expression levels in mammalian cells. Nature communications 10, 818 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V, Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305–1311 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krishnamoorthy A et al. RADX prevents genome instability by confining replication fork reversal to stalled forks. Molecular cell 81, 3007–3017 e3005 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fortin GS, Symington LS, Mutations in yeast Rad51 that partially bypass the requirement for Rad55 and Rad57 in DNA repair by increasing the stability of Rad51-DNA complexes. The EMBO journal 21, 3160–3170 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies OR, Pellegrini L, Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat Struct Mol Biol 14, 475–483 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrison C et al. The essential functions of human Rad51 are independent of ATP hydrolysis. Molecular and cellular biology 19, 6891–6897 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stark JM et al. ATP hydrolysis by mammalian RAD51 has a key role during homology-directed DNA repair. J Biol Chem 277, 20185–20194 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Chi P, Van Komen S, Sehorn MG, Sigurdsson S, Sung P, Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair (Amst) 5, 381–391 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Chen J et al. Tumor-associated mutations in a conserved structural motif alter physical and biochemical properties of human RAD51 recombinase. Nucleic acids research 43, 1098–1111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marsden CG et al. The Tumor-Associated Variant RAD51 G151D Induces a Hyper-Recombination Phenotype. PLoS Genet 12, e1006208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang AT et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Molecular cell 59, 478–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prasad TK, Yeykal CC, Greene EC, Visualizing the assembly of human Rad51 filaments on double-stranded DNA. Journal of molecular biology 363, 713–728 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Zadorozhny K et al. Fanconi-Anemia-Associated Mutations Destabilize RAD51 Filaments and Impair Replication Fork Protection. Cell Rep 21, 333–340 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Ameziane N et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nature communications 6, 8829 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mason JM, Chan YL, Weichselbaum RW, Bishop DK, Non-enzymatic roles of human RAD51 at stalled replication forks. Nature communications 10, 4410 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marie L, Symington LS, Mechanism for inverted-repeat recombination induced by a replication fork barrier. Nature communications 13, 32 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cloud V, Chan YL, Grubb J, Budke B, Bishop DK, Rad51 is an accessory factor for Dmc1-mediated joint molecule formation during meiosis. Science 337, 1222–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thangavel S et al. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208, 545–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhat KP et al. RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep 24, 538–545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang F et al. Promotion of RAD51-Mediated Homologous DNA Pairing by the RAD51AP1-UAF1 Complex. Cell Rep 15, 2118–2126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sigurdsson S, Van Komen S, Petukhova G, Sung P, Homologous DNA pairing by human recombination factors Rad51 and Rad54. J Biol Chem 277, 42790–42794 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Quinet A et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Molecular cell 77, 461–474 e469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bianchi M, DasGupta C, Radding CM, Synapsis and the formation of paranemic joints by E. coli RecA protein. Cell 34, 931–939 (1983). [DOI] [PubMed] [Google Scholar]
- 51.Schlacher K, Wu H, Jasin M, A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer cell 22, 106–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu W et al. FANCD2 and RAD51 recombinase directly inhibit DNA2 nuclease at stalled replication forks and FANCD2 acts as a novel RAD51 mediator in strand exchange to promote genome stability. bioRxiv, 2021.2007.2008.450798 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lossaint G et al. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Molecular cell 51, 678–690 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Halder S, Ranjha L, Taglialatela A, Ciccia A, Cejka P, Strand annealing and motor driven activities of SMARCAL1 and ZRANB3 are stimulated by RAD51 and the paralog complex. Nucleic acids research 50, 8008–8022 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saito Y, Kanemaki MT, Targeted Protein Depletion Using the Auxin-Inducible Degron 2 (AID2) System. Curr Protoc 1, e219 (2021). [DOI] [PubMed] [Google Scholar]
- 56.Dungrawala H, Cortez D, Purification of proteins on newly synthesized DNA using iPOND. Methods in molecular biology 1228, 123–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mehta KPM et al. CHK1 phosphorylates PRIMPOL to promote replication stress tolerance. Science advances 8, eabm0314 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jackson J, Vindigni A, Studying Single-Stranded DNA Gaps at Replication Intermediates by Electron Microscopy. Methods in molecular biology 2444, 81–103 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Malik PS, Symington LS, Rad51 gain-of-function mutants that exhibit high affinity DNA binding cause DNA damage sensitivity in the absence of Srs2. Nucleic acids research 36, 6504–6510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prakash R et al. Distinct pathways of homologous recombination controlled by the SWS1-SWSAP1-SPIDR complex. Nature communications 12, 4255 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsuo Y, Sakane I, Takizawa Y, Takahashi M, Kurumizaka H, Roles of the human Rad51 L1 and L2 loops in DNA binding. The FEBS journal 273, 3148–3159 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is provided in the manuscript or supplementary materials. All materials are available upon request. Some plasmids and cell lines may require a material transfer agreement (MTAs).