

SUMMARY
When replication forks encounter template lesions, one result is lesion skipping, where the stalled DNA polymerase transiently stalls, disengages, and then reinitiates downstream to leave the lesion behind in a postreplication gap. Despite considerable attention in the 6 decades since postreplication gaps were discovered, the mechanisms by which postreplication gaps are generated and repaired remain highly enigmatic. This review focuses on postreplication gap generation and repair in the bacterium Escherichia coli. New information to address the frequency and mechanism of gap generation and new mechanisms for their resolution are described. There are a few instances where the formation of postreplication gaps appears to be programmed into particular genomic locations, where they are triggered by novel genomic elements.
KEYWORDS: DNA recombination, DNA repair, postreplication gap, RecA, RecF, RecG, RecO, RecR, mutagenesis, translesion DNA synthesis
INTRODUCTION
DNA damage is common. Nucleated human cells can suffer 100,000 DNA lesions or more daily (1, 2). A bacterial cell growing exponentially in rich medium suffers a few thousand DNA lesions every cell cycle. Most of this damage is repaired via multiple universal pathways, which include but are not limited to nucleotide excision repair (3–10), base excision repair (5, 8–10), mismatch repair (11–16), and direct repair (8). The first three of these processes are predicated on the duplex structure of DNA. A lesion on one strand can be cut out of that strand and replaced, with the replacement being directed by the undamaged complementary strand (Fig. 1A). The vast majority of DNA lesions are corrected rapidly and accurately in this manner. However, encounters of the replisome with lesions where repair is incomplete are inevitable.
FIG 1.

Consequences of DNA lesion repair. (A) Many DNA repair pathways, including nucleotide excision repair, base excision repair, and mismatch repair, rely on the fact that DNA is double stranded. If a lesion occurs in one strand, it can be excised from that strand to leave a DNA gap. DNA synthesis, using the undamaged complementary strand as a template, then completes the repair process in a manner that does not produce mutations. (B) The processes described above for panel A generate a transient discontinuity in one of the DNA strands. If a replication fork encounters this discontinuity before it is eliminated, a double-strand break results. The double-strand break is repaired by RecBCD and RecA in the double-strand break repair pathway.
Nucleotide and base excision repair as well as mismatch repair all generate a transient break in one DNA strand. When the replisome encounters a template discontinuity, a double-strand break (DSB) is produced (17–19). In bacteria, such events elicit RecA- and RecBCD-mediated double-strand break repair (DSBR) (Fig. 1B) (20–22). The broken end is processed by RecBCD. The RecA protein is loaded onto the 3′-ending strand by RecBCD (23, 24), followed by strand invasion. This initiates a replication restart pathway that reconstructs the replication fork structure, allowing the replisome to be reloaded by the repair primosome (25–27). Replication can then continue. As the reconstituted fork replaces the one that collapsed, no overreplication occurs.
DSBR is potentially a significant drain on cellular resources. In processing the broken end, RecBCD may hydrolyze hundreds if not thousands of phosphodiester bonds (28). Replication is ultimately reinitiated upstream of the original break location, forcing the rereplication of a segment of the DNA template.
How often does DSBR happen? It happens often, perhaps up to once every replication cycle. During normal growth in rich medium and using that fluorescently labeled MuGam protein that binds to double-strand ends to detect breaks, Escherichia coli exhibits a detectable double-strand break in about 7.5% of cells (29). Given the 70% detection rate (29), the high rate of replication initiation, and the presence of perhaps 2 to 3 replicating chromosomes at any given moment during growth in rich media, this direct visualization suggests a rate of <0.05 DSBs per replication cycle. However, as these breaks are repaired and, thus, transient, a snapshot of visualized breaks may underestimate their rate of occurrence. Perhaps a better indication may be seen in the fate of cells that lack recB. Here, only approximately 30% of the cells are viable or are able to divide many times. The rest are unable to divide repeatedly (30, 31). More recent estimates of fork breakage using some direct-detection methods suggest that 9% of forks fail to reach the terminus due to encounters with template discontinuities in cells lacking recB (32, 33). As there are two forks per replication cycle, this suggests a rate of about 1 DSB per 5 replication cycles. Viability is further decreased by problems that occur during replication termination in cells lacking recB (32, 33). Given the uncertainties about the detection rates during direct visualization, replisome encounters with template discontinuities and subsequent double-strand break formation may be frequent, with many, if not most, replication forks undergoing such events under aerobic conditions.
Double-strand breaks are perhaps the most deleterious DNA damage event that can be inflicted on a genome, at a minimum halting replication during the time required for fork reconstitution and proceeding at a substantial cost in deoxynucleoside triphosphates (dNTPs) (34–36). Minimizing such events may be an evolutionary imperative.
When excision repair has not yet been initiated, replisome collision with a template lesion has at least three possible outcomes (Fig. 2). First, the replisome can stall and regress, allowing normal nucleotide/base excision repair processes to operate before replication continues (33, 37–41). Second, the situation may be resolved quickly by translesion DNA synthesis (TLS). In this scenario, a translesion DNA polymerase such as DNA polymerase II (Pol II) or Pol IV replaces part of the DNA Pol III replisome, synthesizes DNA over the lesion, and is then replaced in kind by DNA Pol III (42–48). This strategy carries with it the potential for introducing a mutation.
FIG 2.

Three possible outcomes of replisome collisions with template lesions. The outcomes are, from left to right, fork stalling and reversal, translesion DNA synthesis, and lesion skipping. Lesion skipping occurs often in bacteria and is the focus of this review.
The final strategy and the primary focus of this review is lesion skipping, whereby the replisome halts replication (stalls), disengages, and reinitiates replication downstream (33, 49). The process of lesion skipping must vary to a degree on the leading- and lagging-strand templates. In both cases, the replisome must stall at the lesion and then disengage from the template. On the lagging strand, reinitiation can take advantage of the mechanisms organic to lagging-strand DNA synthesis. On the leading strand, the reinitiation mechanism is less well defined. In vitro, reinitiation dependent on DnaG and DnaB has been documented (39, 50–54).
In either case, the lesion is left behind in a single-strand gap, often called a postreplication gap (55–59) or a daughter strand gap (60, 61). The term postreplication gap is used in this article. Lesion skipping has a major advantage in that replisome progress is interrupted minimally if at all. In principle, replication can proceed unhindered, and the lesion can be repaired behind the fork.
However, the presence of a lesion in a region of single-stranded DNA (ssDNA) imposes a daunting set of problems for repair. If a trailing replisome encounters the gap prior to any repair, a double-strand break would be created, triggering an energetically expensive repair process that delays replication. This would eliminate the replicative advantage gained in the first place by lesion skipping. Cleavage of the lesion-containing single strand in the gap to remove the lesion is not a viable option. This would introduce a double-strand break, not at the replication fork but behind the fork that originally skipped over the lesion. Repair by the normal double-strand break repair pathway could ultimately create an unscheduled replication fork that would overreplicate a potentially significant segment of the genome. The repair of postreplication gaps must thus be efficient, keep the damaged single DNA strand intact, and be rapid enough that it can be completed prior to the arrival of the next replisome.
As described below, lesion skipping to generate lesion-containing postreplication gaps occurs up to several times per replication cycle in bacterial cells grown in rich media. The repair of these gaps requires the acquisition of an undamaged strand complementary to the gap DNA so that nucleotide excision repair can eventually proceed normally. This undamaged strand can be provided via recombination with the undamaged daughter chromosome or via replication using a translesion DNA polymerase. Based on the defects observed when key components are missing, recombinational DNA repair via RecA, which is loaded onto the ssDNA in the gap that is coated by the ssDNA binding protein (SSB) by the RecFOR targeting system, is the predominant pathway for the repair of postreplication gaps, at least under normal growth conditions (62–64). A RecA-independent template switch pathway plays an apparently secondary role that might be essential in particular situations (65, 66). Under normal growth conditions where the SOS response is not induced, DNA polymerases II, IV, and V provide the potential for translesion synthesis (45, 49, 67, 68). DNA polymerase V adds to the TLS repertoire primarily when DNA damage is extensive enough to induce the SOS response. All of the pathways have potential genomic drawbacks. In principle, recombinational DNA repair via the RecFOR system or by template switching does not introduce mutations, although it has a limited potential to introduce genetic rearrangements. More importantly, the recombination pathways create a transient joint molecule linking daughter chromosomes behind the fork. These joint molecules can be resolved to form either separate monomeric chromosome products or a dimeric chromosome. Dimeric chromosomes can be resolved during chromosome segregation at cell division (69). If the resolution is incomplete, the replicated genome products remain joined and cannot be segregated to daughter cells. Translesion DNA synthesis, in contrast, introduces a greater risk of mutagenesis.
In the discussion below, the RecFOR pathway for the recombinational repair of postreplication gaps is a significant focus. Although the term RecFOR is used to describe the pathway or system, it is important to note that a stable complex of the three proteins has never been observed. Thus, this term is always used in the context of a system or pathway. The individual proteins function in distinct ways within this system or pathway.
As a brief introduction, this pathway is outlined in Fig. 3, and many of the key proteins involved are introduced in Table 1. The gap is generated when a replisome encounters an unrepaired template lesion and stalls. The replisome then disengages from the template and reinitiates DNA synthesis downstream, generating a lesion-containing postreplication gap. This gap is specifically targeted by the RecF protein, interacting with the DnaN β-clamp component of the replisome and likely deposited at one gap end when the replisome disengages. The gap is initially coated with SSB. The formation of a RecA filament in the gap, a step often referred to as presynapsis, occurs with the aid of the RecF, RecO, and RecR proteins, in a variety of complexes that serve both to target RecA to the appropriate gaps and to overcome the SSB barrier to RecA filament nucleation. The RecJ nuclease is also involved in enlarging the gap prior to RecA binding. The RecA protein then promotes DNA pairing or synapsis, creating a branched joint molecule that links the gapped chromosome with its undamaged sister chromosome behind the fork. The joint molecule is resolved in the postsynaptic phase and is facilitated by a host of proteins, including but not limited to Uup, RecG, RuvA, RuvB, RuvC, RecQ, RarA, RadA, and RadD. Detailed citations for these proteins and the processes that they promote are provided below as the pathway is presented in more detail.
FIG 3.

Recombinational DNA repair of a postreplication gap mediated by the RecFOR system. The RecF protein, in a complex with RecR, targets the system to the appropriate postreplication gaps. RecO is recruited and activated, most likely via a handoff of RecR from RecF to RecO. RecOR loads a RecA filament into the gap. RecA promotes the formation of a joint molecule, which is then resolved by one of multiple pathways. Topo I, topoisomerase I.
TABLE 1.
Proteins involved in recombinational postreplication gap repair in E. coli
| Protein | Mol wta | Functional oligomer | Known interaction(s) | Activity(ies) | Gene(s) |
|---|---|---|---|---|---|
| All | |||||
| SSB | 18,843 | Tetramer | ~20 proteins, including RadD, RarA, RecG, RecJ, RecO, and RecQ | Binding to ssDNA and many repair proteins | ssb |
| Generation of postreplication gaps and targeting repair to them | |||||
| RecF | 40,519 | Monomer or dimer | RecR, DnaN, DnaG, RecX | ATPase, targeting, RecA loading | recF |
| DnaN | 40,587 | Dimer | DNA polymerase III subunits; RecF, many others | DNA β-clamp and replisome processivity factor | dnaN |
| RecR | 21,965 | Dimer or tetramer | RecF, RecO | Gap targeting and RecA loading | recR |
| RecJ | 63,389 | Monomer | 5′→3′ exonuclease | recJ | |
| Presynapsis | |||||
| RecFORJ | 37,842 | ||||
| RecO | 27,260 | Monomer | RecR, SSB | RecA loading | recO |
| RecR | 21,965 | Dimer or tetramer | RecF, RecO | Gap targeting and RecA loading | recR |
| UvrD | 81,990 | RecA | 3′→5′ helicase and RecA removal when necessary | uvrD | |
| RecA | 37,842 | Nucleoprotein filament | LexA, RecOR, UvrD, RadD, DinI, DNA polymerase V, RecX, PsiB | ATPase, filament formation, DNA pairing, polymerase V subunit, LexA cleavage | recA |
| Synapsis | |||||
| RecA | 37,842 | recA | |||
| Topoisomerase I or III | Type I topoisomerase | topA, topB | |||
| RecX | Monomer | RecA | |||
| Postsynapsis | |||||
| UvrD | 81,990 | RecA | 3′→5′ helicase and RecA removal | uvrD | |
| RarA | 49,594 | Tetramer | SSB | ATPase, DNA binding? | rarA |
| RuvA | 22,086 | Tetramer | RuvB, RuvC | Binds Holliday junctions | ruvA |
| RuvB | 37,174 | 2 hexameric rings | RuvA, RuvC | Holliday junction translocase | ruvB |
| RuvC | 18,747 | Dimer | RuvA, RuvB | Holliday junction cleavage | ruvC |
| Uup | 72,067 | Monomer | RecG? | DNA scanning, Holliday junction binding | uup |
| RadD | 66,413 | Monomer | SSB, RecA | RecA-dependent helicase, ATPase | radD |
| RadA | 49,472 | Monomer | Helicase, facilitates RecA strand exchange | ||
| RecG | 76,430 | Monomer | Uup?, SSB | ATPase, helicase | recG |
| RecQ | 68,364 | Monomer, multimer | SSB | recQ |
The molecular weight includes the N-terminal Met residue except for RecA, RecF, RecO, and Rec.
A Multitude of Questions
First documented in the 1960s (60, 70–76), the existence of postreplication gaps has been recognized for more than 5 decades. Despite much work, key questions remain unresolved. How often does lesion skipping occur under different growth conditions? How does it occur? Are there any proteins or factors that help catalyze replisome disengagement during lesion skipping that are not part of what is currently defined as the bacterial replisome? Postreplication gaps with lesions represent a very small fraction of the gaps created during a replication cycle. In an environment with high numbers of lagging-strand gaps and occasional mismatch repair gaps, how does RecF target the RecFOR system specifically to the lesion-containing gaps that result from lesion skipping? The RecOR proteins alone form a complex with the demonstrated capacity to load the RecA protein onto any SSB-coated stretch of ssDNA. In principle, loading RecA onto lagging-strand gaps or mismatch repair gaps at random could cause genomic chaos as the loaded RecA would both block replication on the bound templates (77, 78) and generate unproductive joint molecules. As this presumably does not happen, how is RecOR activity constrained? What other proteins are involved in postreplication gap repair? How are the various pathways and subpathways regulated so that they do not interfere with each other? This review represents an effort to frame these and other questions and summarize current progress in answering them. Another recent review covers some other aspects of this topic, with more of a focus on eukaryotes (61).
History
A brief historical perspective might begin in 1968. Okazaki et al. proposed discontinuous lagging-strand DNA replication in E. coli and bacteriophage T4-infected E. coli (79). The Okazaki model provided a clear and compelling explanation for obligate discontinuous DNA synthesis on the lagging strand in terms of the properties of E. coli and T4 DNA polymerases, which are able to synthesize DNA only in the 5′→3′ direction (80). Okazaki provided evidence for discontinuous DNA synthesis also on the leading strand (81) albeit most probably not in the form of lagging-strand Okazaki fragments (~1,200 to 1,700 nucleotides [nt] in length). More recent work has helped establish that leading-strand synthesis is not entirely continuous, with discontinuities occurring less frequently than on the lagging strand (52, 82). Thus, while the origin of the predominant class of lagging-strand replicative gaps is attributable to Okazaki fragment synthesis, the origin of leading-strand gaps is less clear. This subject is especially relevant today in terms of defining the extent to which the generation of ssDNA gaps in the E. coli chromosome may be caused by the repair of DNA damage, homologous recombination, nonhomologous DNA end joining, replication fork collapse on damaged and undamaged DNA, and replication restart.
Ten years after Okazaki et al.’s paper, papers from B. K. Tye et al. showed that the inactivation of dUTPase (dut null mutant, originally isolated by E. B. Konrad and I. R. Lehman [83]), which results in a large increase in the concentration of the dUTP pool in E. coli, leads to an ~100-fold increase in homologous recombination (“hyper-rec phenotype”) accompanied by the presence of short DNA fragments (~200 to 400 nt), termed short Okazaki fragments (“SOF phenotype”) (84–86). Both phenotypes were attributed to the removal of excess U’s from the DNA. Although both aberrant phenotypes were completely suppressed in an E. coli dut ung double mutant, which incorporates but cannot excise U’s from DNA, full-size Okazaki fragments nevertheless remained. Thus, Okazaki fragments cannot be explained by the removal of U’s. But what about the presence of discontinuities of DNA synthesis on the leading strand and on the lagging strand that are not Okazaki fragments? In 2019, Cronan and coworkers inactivated essentially all of the known DNA repair pathways in E. coli and then measured the extent of continuous leading-strand DNA synthesis (82). Nascent leading-strand synthesis proceeded uninterrupted for about 80 kb, with the inactivation of the ribonucleotide excision repair pathway via a mutation of RNase HII (rnhB mutant), providing the largest contribution to the maintenance of continuity. Although 80 kb is roughly 50-fold longer than the length of lagging-strand Okazaki fragments, it nevertheless corresponds to only about 1/30 of the distance between the origin of replication in E. coli (oriC) and the chromosomal termination site (ter). Aside from base and nucleoside excision repair processes, it is well established that other processes that may lead to DNA nicks and gaps include homologous recombination, nonhomologous end joining, and replication fork collapse on damaged and undamaged DNA coupled to replication restart (49). The biochemical basis for the interruption of replication fork progression remains a major area for analysis, but until recently, there was no systematic way to identify the location of ssDNA gaps in E. coli DNA.
There are discontinuities in DNA synthesis and gaps are left behind by the replication fork. Some of these gaps result from lesion skipping when the replisome encounters DNA template lesions. These postreplication gaps are repaired. The concept of repair in postreplication gaps has a 5-decade history in the literature of bacterial DNA metabolism that parallels the investigation of replication mechanisms (60, 70–73). It was first embodied in the concept of postreplication repair, proposed by Howard-Flanders and colleagues (60, 74). These researchers found that low-level UV irradiation of cells defective in nucleotide excision repair did not entirely block replication but instead led to the appearance of shorter nascent DNA strands. The lengths roughly corresponded to the interlesion distance (75, 76). These observations implied that lesions had been left behind in single-strand gaps to be repaired after replication had moved on. With longer incubation times, the shorter DNAs were gradually incorporated into genome-sized DNA molecules. Many predictions of the model were subsequently borne out in vivo (87, 88). As the idea matured, the term daughter strand gap repair became prominent (88, 89), and it became the basis for studies of RecA-mediated gap repair and the specialized functions of proteins such as RecF, RecO, and RecR (71, 90–95). The lesions left behind by lesion skipping would be set up for final repair, with the gap being filled either by TLS (96) or by recombinational DNA repair mediated by the RecFOR system (58, 73, 97). These ideas, and the interplay between the different paths for DNA repair in bacteria, continue to be explored (62, 67, 87, 98–100). However, the role of gaps in DNA repair remains underappreciated amid a continuing focus on TLS polymerase exchange at the replication fork (44, 47, 101) and fork stalling/collapse pathways (39, 100, 102–104). Postreplication gap repair thus requires periodic rediscovery (42, 97, 105).
The importance of DNA gaps in the larger realm of bacterial DNA repair is becoming evident. Whereas the exchange of TLS polymerases into DNA polymerase III replisomes can readily be documented in vitro (47, 101, 106), new work in vivo is telling a different story. Examinations of labeled TLS DNA polymerases IV and V using single-molecule approaches in vivo have revealed that both enzymes spend most of their time at genomic sites well separated from the replication fork (45, 48, 107). The same is true of labeled RecA protein foci that appear after SOS induction (108). Given the substrate preferences for the enzymes being examined, these distal sites may well be postreplication DNA gaps.
Beginning just over a decade ago (109), the repair of postreplication gaps has increasingly been considered in the literature of eukaryotic DNA repair (52, 61), although their provenance is less clear. Postreplication gaps in eukaryotes appear to be repaired by recombination-related template switching (110–112), the more standard recombinational repair (113–115), and/or translesion DNA synthesis by TLS polymerases such as Rev1 or Eta (116–123). The regulation of gap-filling processes involves the ubiquitylation of PCNA (116, 124–126). The gaps themselves are important substrates for DNA damage checkpoint activation (127, 128). A detailed review was published recently (61).
GENERATION OF POSTREPLICATION GAPS
How Much Genomic DNA Is Single Stranded?
The question of how much genomic DNA (gDNA) is single stranded could not be answered until recently. In 2021, a method was developed to map the presence of ssDNA gapped regions throughout the E. coli chromosome to nearly single-nucleotide resolution. A seminal study was carried out under three culture conditions, exponential-phase growth, exponential-phase growth in the presence of high UV fluence, and stationary-phase growth (129). Using a combination of bisulfite treatment, which selectively deaminates cytosines only when they occur in ssDNA, and next-generation DNA sequencing, which allows the deaminated sequence to be read out, Pham et al. provided the first quantitative measurements of the ssDNA content in E. coli cells (129). This approach revealed that log-phase cells grown in relatively rich LB medium at 37°C contain a surprisingly large amount of ssDNA, ~1.3% of the genome at any given moment. It might be expected that the ssDNA would be concentrated on the lagging-strand template of replication due to the lagging-strand gaps generated during replication. However, the lagging-strand gaps generated during replication should encompass <0.1% of the genomic DNA at any given moment. Instead, the ssDNA detected is distributed equally between the Watson-Crick strands and between the leading and lagging strands. In log-phase cells, there is a monotonic reduction in the ssDNA content as a function of the distance from the replication origin, indicating that much of the detected ssDNA is associated with DNA replication despite the lack of lagging-strand predominance. This suggests that much of the detected ssDNA is comprised of postreplication gaps, postreplication mismatch repair gaps, and/or gaps associated with other DNA repair processes (129).
The stable ssDNA gaps exhibited a range of ~50 to 400 nt. Gaps were observed at much higher frequencies in UV-irradiated log-phase cells (encompassing ~6% of the genome) and in stationary-phase cells (11%). The symmetric monotonic reduction in ssDNA occurring between oriC and the replication terminus was absent in stationary-phase cells. High levels of ssDNA were concentrated around oriC, and low levels of ssDNA were located proximal to the replication terminus. Although the average frequencies of gaps were similar on Watson-Crick strands and on leading and lagging strands, their distributions differed markedly for each strand. Hot regions where ssDNA formation was enhanced were concentrated on the lagging strand, while cold regions where ssDNA was relatively scarce were concentrated on the leading strand.
At present, this method has been used to identify a spatially diverse landscape of ssDNA gaps interspersed in the E. coli genome that contain thousands of highly enriched hot ssDNA regions along with fewer cold regions. High-resolution gap mapping has the short-term potential not only to specify which proteins occupy which gaps using chromatin immunoprecipitation sequencing (ChIP-seq) but also to specify which genes are responsible for gap formation, using combinations of deletion mutants.
What Is the Structure of an ssDNA Gap?
Single-stranded DNA gaps are often depicted using simplified diagrams in which an ssDNA links duplex DNA at either end in a colinear arrangement, as done in most of the figures in this review. Although such representations are effective communication tools, they mask the complexity of the structure of ssDNA gaps in the context of the folded three-dimensional nucleoid of the cell and in the context of proteins that assemble on ssDNA. In the same way that chromatin structure is interlinked with genomic metabolism in eukaryotes, nucleoid organization strongly influences genome maintenance in bacterial cells, including the strategies that are required for ssDNA gap repair.
The E. coli chromosome is maintained as a highly condensed nucleoprotein structure called a nucleoid. The condensation of the nucleoid arises from several factors (reviewed in reference 130). These include nucleoid-associated proteins and the structural maintenance of chromosome proteins that bind and condense DNA at both local and global levels (131). Additionally, DNA supercoiling arising from processes such as RNA transcription contribute to nucleoid condensation (132). Finally, the cytoplasm itself contributes to nucleoid condensation. When nongenomic constituents of the cytoplasm are considered the solvent in which the nucleoid resides, experimentation and Monte Carlo simulations indicate that it is a poor chromosomal solvent (133). At physiological DNA concentrations, the nucleoid interacts more strongly with itself than with the surrounding solvent, facilitating condensation (133). Nucleoid condensation in E. coli is neither random nor uniform, but instead, it appears that segments of the genome can assemble into several locally dense chromosome macrodomains and further subdivided into 31 chromosome interaction domains (CIDs) (131, 134–137). Two major macrodomains, each about 1 Mbp in size, encompass the origin (Ori macrodomain) and the terminus region (Ter macrodomain). The impact of nucleoid compaction on ssDNA gap repair has not been investigated. However, given the importance of compaction and nucleoid organization for DNA replication (131), it is likely to be an important feature defining the substrates on which gap repair proteins operate.
Proteins that associate with ssDNA in E. coli also form important cellular features that define the structures of gap DNA substrates, and they present direct challenges (and opportunities) to the gap repair process. Cellular exposure of ssDNA gaps comes with inherent risks since ssDNA is sensitive to chemical and nuclease attacks. Moreover, ssDNA can self-associate to create structures that block gap repair or other genome maintenance reactions. To mediate these risks, bacteria encode oligomeric ssDNA-binding proteins (SSBs) that bind ssDNA with high affinity and cooperativity as it is exposed in cells (138–145).
What might an ssDNA/SSB gap DNA structure look like in cells? Studies of fluorescently labeled SSB at replication sites in E. coli provide a compelling proxy for gap structures. Measurements using fluorescently tagged SSB estimate that its concentration is remarkably high (~500 μM [monomers]) within a 25-nm radius of replication forks in E. coli, a volume that covers the core replisome proteins (146). This is a consequence of SSB binding to the transiently exposed ssDNA lagging-strand template, which can be thousands of bases long in bacteria (147). Due to the flexibility of the lagging strand, SSB foci extend beyond the replisome, with an ~15 μM shell of SSB proteins observed within 100 nm of the fork. In contrast, the overall cytoplasmic SSB concentration is ~2 μM (146). This arrangement creates a striking SSB gradient around ssDNA elements, including lagging-strand and postreplication gap substrates, in E. coli. SSB binding also elicits a significant condensation of the ssDNA (148).
How, then, do gap repair proteins access ssDNA protected in SSB/ssDNA complexes within DNA gaps? In addition to binding ssDNA, SSB acts as a DNA processing hub where ~20 different proteins gain access to genomic substrates by exploiting direct interactions with the C terminus of SSB (SSB-Ct) (143). SSB/ssDNA foci in cells are therefore attractive beacons for binding SSB interaction partners and likely aid in the retention of partner proteins via the high avidity associated with the clustering of many SSB-Ct elements. Notably, several known gap repair proteins interact directly with SSB, including the RecO recombination mediator (149–155), RecQ (151, 156–159), RecG (151, 160, 161), RadD (162, 163), RarA (164), and the RecJ exonuclease (151, 160, 165). In several cases, it appears that partner protein binding to SSB impacts SSB’s binding mode and/or long-range interactions, resulting in the exposure of ssDNA and altering the global condensation of ssDNA/SSB complexes (25, 150, 166). Such interactions could be simultaneously critical for recruiting gap repair proteins to the sites of action in cells and for providing repair enzyme access to ssDNA and ssDNA/double-stranded DNA (dsDNA) junctions that are necessary for proper processing.
How Are Postreplication Gaps Generated?
Lesion skipping to generate a postreplication gap entails replisome stalling at a bulky lesion, disengagement, and reinitiation downstream. The entire process is estimated to take 10 to 20 s (60, 167, 168). As indicated above, the reinitiation process may be different on leading- and lagging-strand templates.
The disengagement process would require either the separation of the replisome from its associated DnaN β-clamp or a conformational change in the β-clamp that breaks the dimeric circle and releases the replisome complex. Disengagement could be a spontaneous event that is inherent to replisome progression, or the process could be catalyzed. These possibilities are not mutually exclusive since some protein or factor could facilitate an intrinsic replisome capacity for replisome disengagement during lesion skipping. On the lagging-strand template, there has been an expectation that encounters with DNA lesions could lead to gap formation via stalling and repriming, utilizing the mechanisms organic to repetitive Okazaki fragment synthesis (105, 169). On the leading strand, lesion skipping has been observed for reconstituted bacterial replisomes in vitro (50, 51).
Replisomes exhibit an intrinsic capacity for leading-strand disengagement during lesion skipping in vitro, along with DnaG-dependent repriming (50, 51). However, uncatalyzed lesion-skipping reactions with UV lesions occur over a period of multiple minutes in vitro. Some forks remain stalled after 6 min (50, 51). This is incompatible with the 10- to 20-s estimate for overall lesion skipping cited above. It is always difficult to relate in vitro conditions to the situation in the cell, where cytoplasm conditions, protein concentrations, genome topology, or the presence of auxiliary factors could increase or decrease the rate of spontaneous lesion skipping by DNA polymerase III. The time between replication initiation events in E. coli cells growing in rich media at 37°C is short (no more than 18 to 20 min [170]). A catalyst to facilitate polymerase disengagement and lesion skipping in vivo seems plausible if not probable. No such catalyst has yet been clearly identified, but we discuss a candidate (RecF) below in this review.
How Often Are Postreplication Gaps Created?
The frequency with which postreplicative gaps are created in bacterial cells is one of the most important unresolved questions in the field of DNA repair. The main reason why this knowledge gap exists is that there are currently no molecular labels available that can specifically mark the positions of postreplication gaps (as opposed to all gaps) in cells. A series of recent studies approached this problem through genetic analyses of gap repair systems, yielding new, and somewhat diverse, estimates for gap creation frequencies in log-phase E. coli cells. These studies have produced estimates ranging from very low (1 gap per every 50 replication cycles) to very high (multiple gaps per replication cycle). Growth conditions may be a key reason for the wide range of estimates. Thus, we highlight the growth conditions when discussing each study. The high estimates predominate.
In recent years, traditional genetics studies have provided new insight into postreplication gap creation. Previous studies had indicated that the inactivation of the recF, recO, and recR genes, each of which encodes a key component of the RecFOR system for loading RecA into SSB-coated ssDNA gaps, has little to no effect on the viability of cells in the absence of exogenous DNA damage (31, 171–175). There are two possible reasons why this would be the case. The first possibility is that gaps are toxic to cells but are extremely rare. In this case, the effects of recF, recO, and recR on cell viability might be difficult to detect. An alternative explanation is that gaps are common but relatively benign. In the absence of the repair protein RecF, RecO, or RecR, gaps might be successfully channeled into alternative repair pathways. The results of five recent genetics studies, by Romero et al. (176, 177), Cooper et al. (178), Jain et al. (179), and Fonville et al. (180), support the second scenario. These studies generally examined log-phase E. coli cells grown at 37°C in rich media.
There are two types of genetic rearrangement reactions—recombination—that are associated with postreplication gaps. The first is RecA-dependent recombinational DNA repair, which represents the primary repair pathway for postreplication gaps and is a major focus of this review. The second is a RecA-independent deletion between tandem repeat sequences that can occur via a template switch process (59, 66, 181–187). The mechanistic origin of template switching within postreplication gaps and its consequences, as described in more detail below in this review, are not yet well understood. As the available evidence suggests that the majority of these deletion reactions take place at postreplication gaps (66), the first of the studies by Romero et al. (176) focused on these genetic rearrangements to both elucidate the activities of two proteins and gain new insight into the frequency of gap generation.
The first study by Romero et al. (176) identified the products of the uup and radD genes as suppressors of the template switch reaction. A wide range of repeat-deletion constructs were assayed, in the context of both plasmids and chromosomal integrants. For all constructs, the deletion of both the uup and radD genes increased the frequency with which RecA-independent template switch deletions occurred at postreplication gaps. For constructs lacking uup and radD function, deletions were increased by almost 100-fold. This phenomenon had become so common that it suggested that postreplication gaps, which are a requisite for the deletions, must be forming very often during log-phase growth. This concept was explored further by comparing the experimental results to simulations. It is important to note that this exercise required strong assumptions to be made and that key uncertainties surrounding the mechanism of the template switch deletion reaction limit the accuracy of the computational approach. The experimental data indicated that the frequency of template switch deletions decreased as a function of the increasing spacing between sequence repeats. The relationship was well fit by an exponential decay function. Working under the assumption that the entirety of both repeats must lie within the same gap (this remains to be tested experimentally), simulations perfectly recapitulated the exponential relationship between repeat spacing and deletion frequency. The simulations suggested that the mean length of postreplication gaps in log-phase E. coli cells is 300 nt. This value lies between previous estimates of 100 to 200 nt (188) and 800 nt (58, 189). Returning to the frequency of postreplication gap creation, the simulated deletion frequencies best matched the experimental data when 300-nt gaps were formed once every 25 thousand nucleotides (knt) if a high deletion probability (50%) was assumed or even more frequently if a low deletion probability was assumed (176). These estimates are subject to considerable uncertainty—they could be strongly overestimated or underestimated—but provide the first indication that postreplication gaps might be formed multiple times per replication cycle.
The remainder of the studies (177–180) focused on the consequences of RecA-dependent recombinational repair of postreplication gaps. When the RecA protein is loaded into such a gap (a step often called presynapsis) and pairs the bound ssDNA with the homologous duplex in the sister chromosome (synapsis), a joint molecule is created behind the replication fork that links the two replicated chromosomes. This joint molecule is resolved in the postsynaptic stages of repair. Failure to resolve this joint molecule can lead to cell death as it can block chromosome segregation at cell division. Thus, the toxic effects observed when the activities of enzymes involved in postsynapsis are ablated can provide additional insight into the frequency of postreplication gap creation. These toxic effects can be traced to the repair of postreplication gaps if they are suppressed by eliminating the activities of the RecF, RecO, or RecR proteins, thus preventing the loading of the RecA protein into the gaps to create potentially problematic joint molecules.
The second study by Romero et al. demonstrates that the loss of two particular postsynaptic gap repair proteins, RecG and RadD, causes a severe growth defect. This defect can be rescued by deleting the recF, recO, or recR gene to prevent RecA loading and joint molecule formation in the gaps (177). The RecG helicase plays a key role in a fork reversal pathway that restarts stalled replication forks (190–196). Under conditions that use protein barriers to promote fork reversal, cells lacking RecG accumulate Holliday junctions (197). Holliday junctions are also intermediates in postreplication gap repair via recombination, and thus, RecG may also play a role in Holliday junction resolution (177, 191). The elimination of the gene encoding Uup suppresses some effects of the recG deletion (177). As Uup binds to Holliday junctions (176), Uup might be a recruitment factor for RecG in the context of Holliday junctions. The RadD helicase is a paralog of RecG (and RecQ) (177, 198). The helicase activity of RadD is RecA dependent, a property that is unique among helicases (198). RadD accelerates RecA-dependent strand exchange reactions (198). The study by Romero et al. demonstrated that cells lacking either the recG or radD gene grew normally, whereas cells lacking both genes exhibited a severe growth defect (177). This indicates that RecG and RadD share a degree of functional redundancy and that one can compensate for the loss of the other. Most importantly, the growth defect observed for the recG radD double mutant was alleviated upon the deletion of recF or recO. This places the RecG/RadD activity that is crucial for cell growth downstream of the presynaptic phase of postreplication gap repair. Moreover, the severity of the growth defect in cells that are able to initiate gap repair (recF+ recO+) but fail to complete gap repair (ΔrecG ΔradD) via pathways utilizing these two proteins strongly suggests that postreplication gap repair is initiated very often, at least once per cell cycle.
Prior to the two studies described above, Cooper et al. (178) showed that a double mutant inactivating radA and recG exhibited synergistic sensitivity to azidothymidine (AZT), UV, and ciprofloxacin. The sensitivity was suppressed by recF inactivation (178) and by the overexpression of the postsynaptic RuvAB proteins, implicating the resolution of recombination intermediates as the role of RadA and RecG in this study. Both the RadA and RadD proteins accelerate RecA-mediated strand exchange (198, 199). As the inactivation of radD with recG leads to a much greater growth defect under normal growth conditions than is the case with the radA recG double mutant, we infer that the RadD enzyme has a greater role in the context of the repair of postreplication gaps. However, radA mutants exhibit synergistic sensitivity to AZT or ciprofloxacin with radD (then called yejH) (178), indicating that the two proteins have complementary cellular functions in at least some situations. The radA gene also exhibits synthetic genetic interactions with respect to AZT and ciprofloxacin with a number of additional helicases and putative helicases, including DinG, Lhr, PriA, Rep, RuvAB, UvrD, and YoaA. The roles of many of these enzymes in the resolution of postreplication gaps remain undefined (178). This work is described in more detail below.
The fourth study (by Jain et al. [179]) explored a situation where all pathways for the resolution or reversal of recombinational joint molecules in postreplication gaps are apparently blocked. This study again indicates that postreplication gaps are formed often, probably multiple times in a replication cycle in rich media (179). The elimination of the ruvB, recG, and rarA genes is synthetically lethal in E. coli. Most of the double mutants produce strong growth defects. As in the previous two studies, these deficiencies are suppressed by the deletion of recF or recO, indicating that the deleterious effects occur downstream of the formation of joint molecules by RecA filaments loaded into gaps by the RecFOR system (179). The SOS response is induced dramatically in triple mutant cells, suggesting the generation of significant amounts of ssDNA (179). Increasingly, these kinds of genetic approaches suggest that postreplication gaps are formed frequently during replication.
The fifth study (by Fonville et al. [180]) explored yet another situation in which recombination mediated by the RecFOR system resulted in cell death when the products were not resolved. This again involved recG inactivation, this time combined with the inactivation of the uvrD helicase gene (180). The recG uvrD combination produced “death by recombination,” which, like the above-described studies, could be suppressed by mutations in recF, recO, or recR. Interestingly, active RecQ helicase was also needed to produce the toxic recombination intermediates in this genetic background (180). The UvrD helicase has several cellular functions, including the removal of RecA protein filaments from the DNA (200–204). It may complement RecG pathways for the resolution of toxic recombination intermediates by reversing and/or preventing their formation.
The studies described above are based on genetics, and the conclusions should be viewed with caution. The latter four of these studies have one thing in common: they all feature mutations in the recG gene, combining these with additional deficiencies. RecG has a clear role in the repair of postreplication gaps. However, RecG also plays important roles in replication termination and chromosome segregation at cell division (205–207). Alternative interpretations are possible. For example, instead of (or in addition to) providing one path to resolving joint molecules behind the replication fork, RecG might have a role in increasing the frequency of gap formation, thus increasing the likelihood that joint molecules might be generated. While this caveat must be considered, the overall evidence suggests that postreplication gaps are generated often under normal growth conditions.
Increasingly, direct observations of proteins associated with postreplication gap repair are adding to our understanding. A recent series of microscopy studies also provided insight into the frequency of postreplication gap formation (208). These studies examined the activities of the gap repair proteins RecF and RecO (173), SSB (146, 209), and the Holliday junction resolvase component RuvC (210).
The RecF and RecO proteins are expressed at quite low levels in cells (approximately 18 and 12 molecules per cell, respectively, in a recent study [173], although estimates of 150 and 85 molecules per cell, respectively, have also been reported [211]). The recent development of live-cell single-molecule fluorescence microscopy techniques made it possible to directly image both proteins at their native expression levels (173). In live-cell single-molecule imaging, the protein of interest is labeled with a fluorophore. This is most commonly done by expressing a fluorescent protein fusion of the target protein. Ideally, the modified gene would replace the endogenous gene on the chromosome where expression at natural abundance is more likely than in plasmid-based systems. Controls to assess functionality are essential.
In single-molecule images, molecules that bind to the bacterial nucleoid present as punctate foci, whereas proteins that are not engaged with the DNA produce only weak, diffuse signals that spread across the entirety of the cell. The RecF and RecO proteins, labeled with C-terminal fusions of green fluorescent protein (GFP) derivatives, are fully functional, while similar fusions of the RecR protein are inactive (173). Cells expressing labeled RecF or RecO produced foci during log-phase growth in relatively rich EZ glucose medium at 37°C (173). Under these rapid-growth conditions, cells were undergoing multifork DNA replication and produced on average 2.5 replisome foci per cell. RecF produced on average 2 foci per cell, while RecO produced 1 focus for every 3 cells. In each case, the images of the RecF and RecO signals captured a tiny portion (<0.5 s) of the cell cycle. The fact that both gap repair proteins were captured as nucleoid-associated foci within these snapshots implies that they bind to the DNA very frequently. This in turn suggests that many postreplication gaps are being processed in each cell cycle.
Microscopy images from two other studies (146, 209) provide further clues that postreplication gaps might form very frequently. In a seminal single-molecule imaging study (146), Reyes-Lamothe et al. labeled, and quantified, almost all of the replisome components in live E. coli cells. These experiments were done using cells grown in M9 glycerol medium at 37°C. For most replisome components, the measured distributions of molecules per replication focus were bimodal, with peaks at values equivalent to one replisome (major peak) or two replisomes (minor peak). The distribution of the SSB protein was a notable exception, indicating much greater heterogeneity. Replication foci contained 5 to 25 SSB tetramers. A second study, by Dubiel et al. (209), also includes images of SSB-labeled E. coli strains grown in 56/2 minimal medium supplemented with glucose at 37°C. The SSB utilized in this study was a novel fusion in which GFP derivatives were fused within a linker region between the oligosaccharide/oligonucleotide binding (OB) fold of SSB and the SSB-Ct. This altered and fluorescent SSB supports E. coli growth in the absence of wild-type SSB. The intensities of these SSB foci are again highly heterogeneous. Postreplication gaps were not the focus of either the study by Reyes-Lamothe et al. (146) or the study by Dubiel et al. (209). However, in light of the new evidence described above (129, 173, 176, 177), it is worth revisiting their data on SSB heterogeneity through the lens of postreplication gaps. Two recent studies indicate that during repair, postreplication gaps are enlarged by the RecJ exonuclease prior to RecA being loaded (179, 212). This step would be expected to create a broad range of ssDNA tracts and, in the process, create additional binding sites for SSB. Such a process would be expected to create particularly bright SSB foci in microscopy images. Given the inherent resolution limits of light microscopes, those SSB molecules bound to postreplication gap repair intermediates would be expected to colocalize with active replisomes so long as repair was instigated soon after gap formation. If the studies by Reyes-Lamothe et al. and Dubiel et al. did inadvertently capture evidence of intermediates that formed during postreplication gap repair in their microscopy snapshots, those repair intermediates must be formed multiple times per cell cycle.
In a study from the Reyes-Lamothe group, the apparent generation of postreplication gaps after UV irradiation showed that replisome encounters with UV lesions result in the formation of new DNA polymerase III foci that do not colocalize with the DnaB helicase (213). This work was again done in cells growing relatively slowly in minimal medium to suppress the complications associated with the multiple replication forks seen in cells grown in rich media. The new foci could represent DNA polymerase III operating in postreplication gaps. Their formation depended on the presence of the χ (chi) replisome subunit (213). DNA polymerase III may have a key role in the gap repair process. The rate of replication decreases after UV irradiation (213), reflecting the required and repeated bypass of unrepaired UV lesions that leads to a temporary replication lag (58).
Not all studies point to the frequent generation of postreplication gaps. Using a cleverly modified version of the RuvC protein to trap, and label, Holliday junctions, Xia et al. found strong evidence that postreplication gaps were being formed and repaired but estimated that this occurred only once every 50 to 100 replication cycles (210). The growth conditions used for those measurements involved supplemented M9 medium at 37°C. The technique developed by Xia et al. represents the most direct method for detecting a likely intermediate in postreplication gap repair, the Holliday junction. However, the low gap creation frequency that it suggests contrasts sharply with the results of the other studies presented in this discussion, including another described above from the Rosenberg group (180). One possibility is that the growth conditions have a strong influence on the rate of postreplication gap formation and that Xia et al. used conditions that yield low frequencies of gaps and Holliday junctions. Alternatively, and perhaps more likely, the rate of Holliday junction detection simply does not adequately correlate with postreplication gap formation and repair. The preponderance of data indicate that postreplication gaps are formed frequently during every replication cycle. However, many may be repaired without the RuvABC Holliday junction resolvase. In addition, many gaps may be resolved without the formation of Holliday junctions that exist long enough for detection. The preferred pathways for intermediate resolution may vary depending on enzyme availability. RecG may resolve at least some of the Holliday junctions, as has been proposed previously (177, 191). The bound RecG could occlude binding by RuvC and thus prevent the detection of many Holliday junction intermediates by the RuvC probe.
In general, there are many indications that postreplication gaps are generated often. In cells growing exponentially in rich media with aeration, multiple postreplication gaps appear to be created in every cell cycle. This is without the addition of DNA-damaging agents. The frequency no doubt decreases if the oxidative damage that accompanies rapid growth is reduced by growing cells in minimal media or under anaerobic conditions.
What Types of DNA Lesions Trigger the Formation of Postreplication Gaps?
The formation of postreplication gaps is caused by replisome encounters with template lesions. Whereas all such lesions will be repaired most often by other repair processes such as nucleotide excision repair and base excision repair, the prevalence of DNA damage, particularly when DNA-damaging agents are added to cause heightened stress, ensures that replisome encounters with unrepaired lesions will occur. What transpires after these encounters no doubt depends on the structure of the lesion that is encountered. So which DNA lesions trigger the formation of postreplication gaps? The answer appears to be lesions that feature bulky adducts on the base or that generate significant structural kinks in the DNA.
The DNA lesion classically associated with the formation of postreplication gaps is the pyrimidine dimer caused by UV irradiation (60, 70–76, 91, 94, 103, 104, 123, 127, 177, 179, 214–219). Another clue to the possible triggers of postreplication gaps resides in the sensitivity of strains lacking one or more components of the RecFOR system to damaging agents. Deletions of any of the recF, recO, and recR genes render cells quite sensitive to nitrofurazone (NFZ) and mitomycin C (173). NFZ introduces N2-deoxyguanosine adducts. Mitomycin C alkylates DNA to create monoadducts as well as intra- and interstrand cross-links. When considered with pyrimidine dimers, bulky lesions and lesions that cause kinks in the DNA are good candidates for lesions that will trigger postreplication gap formation. Mutations in recF and recO confer much more modest sensitivity to agents that cause strand breaks (ciprofloxacin and bleomycin) or that inhibit DNA synthesis (trimethoprim or hydroxyurea) (173). The effects of the latter agents are also more difficult to interpret. Deletions of recO and recR generate modestly increased sensitivity to bleomycin and trimethoprim, but the deletion of recF does not. For ciprofloxacin and hydroxyurea, deletions of recO and recR have little effect on sensitivity, while the deletion of recF actually increases resistance to these agents (173). The limited effects seen for sensitivity to ciprofloxacin, bleomycin, trimethoprim, and hydroxyurea return the overall pattern to the idea that bulky lesions and structural kinks in the DNA trigger postreplication gaps when they are encountered by replisomes. Continuing this pattern, acetylaminofluorene (AAF), which forms bulky lesions on G residues, also reliably triggers the formation of postreplication gaps (212). This pattern has recently been further reinforced with the observation that the ADP-ribosylation of DNA causes gaps that undergo postreplication gap repair (220).
Postreplication gaps are often formed even when cells are not stressed by the addition of DNA-damaging agents. Under normal growth conditions, the lesions that trigger postreplication gap formation are probably some subset of the most prevalent type of damage, oxidative lesions that result in nucleotide base adducts or alterations (221–225).
Artificially Increasing the Generation of Postreplication Gaps with AZT
The use of AZT and other chain-terminating nucleosides as genotoxins provides an opportunity to examine factors that process DNA replication gaps on both leading and lagging strands. Because of the lack of a 3′-hydroxyl group in AZT (226), nascent chains where AZT has been added cannot be extended by normal or translesion synthesis DNA polymerases. In addition, unlike UV photodimers that are scattered throughout the chromosome, including in unreplicating regions, AZT lesions are necessarily present at the 3′-strand terminus at one end of each replication gap created by chain termination. Notably, the gaps thus created do not contain lesions in the adjacent ssDNA (226).
AZT, like UV irradiation, is a strong inducer of the SOS response by the RecFOR pathway and stimulates homologous recombination dependent on RecA and RecFOR (226; J. Grant and S.T. Lovett, unpublished results). Recombination induced by AZT places a high demand on Holliday junction processing by RuvABC: mutants in ruvAB and ruvC are extraordinarily sensitive to AZT (178), much more so than mutants affecting the early stages of homologous recombination, such as recA. Mutations blocking the RecFOR pathway largely suppress this sensitivity of ruvABC (226), indicating that recombination intermediates that accumulate in the absence of RuvABC are toxic and/or interfere with AZT removal or other tolerance pathways. Consistent with this, uvrD mutants, which are hyperrecombinational, are also sensitive to AZT and likewise are suppressed completely by recF and partially by recA (178). The alternative pathways for the resolution of recombination intermediates generated by RecAFOR, utilizing RecG or RuvABC, may rely more on RuvABC after AZT addition. Thus, AZT may provide particularly important insight into the RuvABC resolution path.
The SOS response and cell division arrest assist in the tolerance of replication gaps. Even sublethal levels of AZT induce RecA-dependent cell filamentation (226). Cells reach a length of 15 μm on average, with large unsegregated nucleoids. After the removal of AZT, wild-type cells resume division and return to normal sizes. This filamentation must be important for tolerance since blocking cell division arrest by introducing a sulA mutant causes sensitivity to high levels of AZT (226).
How is AZT removed from DNA? Cells can tolerate a certain amount of AZT by the exonucleolytic or endonucleolytic removal of the chain-terminal azidothymidine monophosphate. Genetic analysis of AZT sensitivity in E. coli implicates exonuclease III, the major abasic site endonuclease and 3′→5′ exonuclease (227), as a factor that removes incorporated azidothymidine monophosphate from the 3′ end of the primer strand. Mutants in xthA (which encodes exonuclease III) are hypersensitive to killing by AZT, while the overproduction of the protein provides additional protection to otherwise wild-type strains (226). Exonuclease III degrades duplex DNA 3′ to 5′ from a nick, gap, or DNA end and can cleave 3′-phosphates or ring-opened sugars such as phosphoglycolates (227, 228). Cell filamentation in xthA mutants persists after the removal of AZT, whereas xthA+ cells resume division, confirming a role for exonuclease III in the direct removal of the AZT moiety from DNA. Mutants in xthA also experience lethal AZT-induced recombination, suppressed by recFOR (226).
Mycobacterial exonuclease III interacts with the β-clamp (229). However, the exonuclease III site required for clamp interaction is not conserved in E. coli, nor has a clamp interaction been established for the E. coli enzyme. Mutants defective in the DNA polymerase III proofreading subunit DnaQ are not sensitive to AZT, suggesting that AZT monophosphate is not efficiently proofread. However, mutants in dnaQ negate the AZT tolerance promoted by the overexpression of the YoaA helicase (230) (see below), indicating that with the assistance of a helicase to unwind DNA, 3′-terminal AZT moieties can be removed by polymerase III proofreading.
REPAIR OF POSTREPLICATION GAPS
The goal of the systems described in this review is not repair but establishing the conditions needed for repair to proceed. A postreplication gap generally includes a lesion in the single-stranded DNA within the gap. To remove this lesion via excision repair or base excision repair, an undamaged cDNA strand must be provided and paired with the lesion-containing single strand. There are multiple pathways by which this cDNA strand can be provided.
Division of Labor
A cDNA strand can be generated by recombination or translesion DNA synthesis (TLS). Multiple studies indicate that recombination, mainly by the RecFOR pathway, is the predominant path for repair (63, 64, 210, 212). This process is a damage avoidance path that is complex but evidently efficient. If successful, it does not produce mutations. Translesion DNA synthesis, by DNA polymerase II, IV, or V, can fill the gap but also can potentially produce mutations. The competition between TLS and recombinational DNA repair occurs largely in the gap-processing phase that occurs before synapsis (212). Mutations that block or constrain recombinational DNA repair result in an increase in TLS and its accompanying mutagenesis (63, 64, 212). The RecFOR pathway is clearly important, but TLS can pick up much of the slack if any of the recF, recO, and recR genes are absent, at least for lesions that are readily bypassed by these enzymes.
Key Proteins
Below, we address the key proteins, most of which are mentioned in Table 1, in more detail.
The single-stranded DNA binding protein, SSB.
Whenever it is exposed in cells, ssDNA is bound by SSB (138–145). Bacterial SSBs function as homooligomers (generally tetramers, such as in E. coli), with functional N- and C-terminal elements bridged by an intrinsically disordered linker (IDL). The N terminus of each monomer within the E. coli SSB tetramer contains one oligosaccharide/oligonucleotide binding (OB) domain that mediates DNA binding and oligomerization. The C-terminal-most region forms an evolutionarily conserved protein interaction motif referred to as the “SSB-Ct,” “tip,” or “C-terminal peptide” (141, 153, 231). In contrast, the SSB IDL is poorly structured, with limited sequence complexity and conservation, and the IDL is disordered in all reported full-length SSB crystal structures (232–241).
E. coli SSB tetramers bind ssDNA with high affinity and with striking positive cooperativity that arises from direct interactions between SSB tetramers (139, 141, 234, 235). This behavior leads to the formation of two different arrangements of SSBs that have been observed in vitro: SSB filaments with unlimited nearest-neighbor (NN) interactions between adjacent SSB tetramers that form under low-salt conditions and less cooperative nucleosome-like SSB beads that form under high-salt conditions (148, 242–245). Both modes condense ssDNA locally. Non-nearest-neighbor (NNN) interactions between SSB tetramers also occur between nonadjacent SSB tetramers, which more globally condense SSB/ssDNA structures (150, 246, 247). NNN interactions require the IDL (246, 247). With its high affinity and highly cooperative binding to ssDNA, SSB creates a protective proteinaceous barrier that protects ssDNA within bacterial cells.
In addition to SSB clustering on ssDNA, SSB proteins have recently been found to form liquid-liquid-phase-separated (LLPS) bodies in which SSBs cluster together through weak multivalent protein-protein interactions (159). The formation of LLPS SSB foci happens in the absence of ssDNA, and it requires all regions of SSB (OB, IDL, and SSB-Ct). LLPS SSB clusters are remarkably protein dense, with SSB diffusing ~5,000 times slower in LLPS bodies than free SSB under physiological protein concentrations and salt conditions. The IDL appears to be particularly important for LLPS formation, and formation is particularly driven by potassium glutamate, the predominant salt in E. coli (159, 248). The presence of ssDNA inhibits LLPS formation. The binding of one SSB interaction partner, RecQ, to LLPS SSB has been examined, and it was found to associate with LLPS SSB (159, 248). Although most microscopy studies of labeled SSB in E. coli have focused on SSB found at DNA replication forks (209, 249–251), one recent study found that SSB foci might also form near cell membranes in E. coli away from sites of replication (252). This may indicate that LLPS SSB foci can form in vivo. Since SSB binding partners can associate with the many SSB C termini within such bodies, it may be that this clustering brings many factors needed for complex reactions, such as gap DNA repair, together to allow their rapid deployment to substrates as SSB transitions from its LLPS form to its ssDNA-bound form. Below, we invoke LLPS as a potential way to constrain the activity of RecO.
The RecF, RecO, and RecR proteins.
Present in widely separated operons, the recF (253), recO (254), and recR (255, 256) genes were discovered over a period of 16 years. The RecF, RecO, and RecR proteins function to target recombinational repair to the appropriate lesion-containing postreplication gaps and to load the RecA protein onto SSB-coated ssDNA within these gaps (202, 257–261). RecF functions as a dimer when ATP is present, and its DNA binding function is highly ATP dependent (262–265). The protein will bind readily to both ssDNA (if it exceeds a minimal length) and dsDNA (95, 263–268). Binding to a wide range of DNA substrates, including those mimicking gap ends, occurs within a narrow low-nanomolar range of dissociation constant (Kd) values (265). RecF exhibits structural similarity to the head domain of eukaryotic Rad50 but lacks a coiled-coil domain (269). The RecO protein (from Deinococcus radiodurans) includes an OB fold, a helical bundle, and a zinc finger (270, 271). The RecO protein interacts with SSB (149–155, 260). The E. coli RecR protein is a dimer in solution (261) but can form a tetramer when interacting with RecF (272). The aggregate RecFOR system has traditionally been associated with gap repair (91–93, 258, 273).
The RecF, RecO, and RecR proteins are part of a defined epistasis group and are often described in aggregate (RecFOR) (202, 258, 274). Gene knockouts of any one of the three genes exhibit very similar phenotypes (275, 276). Collaboration among the three proteins to load RecA onto SSB-coated ssDNA is expected, and in vitro conditions under which that collaboration can be observed have been developed (259, 277). However, a complex that includes all three proteins has never been detected. Instead, there may be a handoff of RecR from RecF to RecO. The RecR protein makes complexes alternatively and in a mutually exclusive manner with RecO (149, 259, 278, 279) or RecF (95, 267, 268, 279). When RecR is limiting, RecF-RecO competition for RecR favors RecF and the formation of RecFR (279). Importantly, the RecOR complex alone is sufficient to load the RecA protein onto SSB-coated ssDNA but requires interactions with both the SSB C terminus and RecR to do so (149, 259, 278). Under many conditions, the addition of RecF to a RecOR reaction will inhibit RecA loading, possibly via the competition between RecF and RecO for the RecR protein (149, 259, 278). The affinity of RecF for RecR may be an important mechanism by which RecO function is constrained so that RecOR does not load the RecA protein onto stretches of SSB-coated ssDNA that do not require recombinational DNA repair.
The RecFR complex, like RecF alone, is an ATPase that binds randomly to single-stranded or duplex DNA (95, 265, 267, 268). The specific binding of RecF or RecFR near a gap end can be engineered in vitro if duplex segments flanking the gaps are very short and the adjacent ssDNA is saturated with SSB such that RecF binding is limited to the short duplex (277). If, instead, the duplex segments are long, the binding of RecFR to the dsDNA becomes random, with no localization near gap junctions (95, 259, 263, 268), consistent with the promiscuous DNA binding observed more recently (265).
Studies demonstrating the RecF-mediated stimulation of RecA loading by RecOR are worth dissecting in detail retrospectively, as they provide potential clues as to how the cell regulates the function of the RecFOR system. The first study, by Sakai and Cox (259), helps establish the importance of the SSB C terminus. When 8 amino acids of the SSB C-terminal end are removed, RecOR loading of RecA is largely blocked (259). However, RecF greatly facilitates the RecOR loading reaction if it is prebound to the ssDNA prior to the binding of the SSBΔC8 protein (259). This indicates that RecF prepositioned on the DNA can recruit RecO, possibly via an activating handoff of RecR from the RecFR complex to RecO, and can trigger RecA loading by RecOR when loading by RecOR alone is blocked due to the inability to interact with SSB. The second study, by Morimatsu et al. (277), uses ssDNA circles with very short duplex regions as a substrate for loading, along with a significant excess of SSB. While not demonstrating the specific binding of RecF to a gap end (RecF effectively has nowhere else to bind), this study does something much more important in again demonstrating that the three proteins can function together, this time with RecF constrained so that it must function near a gap end. Under the conditions of this experiment, the loading of RecA by RecOR alone is not effective. One possible explanation for this is that the available RecO is sequestered by interaction with the excess SSB that is not bound to ssDNA. Positioned on a short duplex, RecFR binding strongly facilitates RecOR-mediated RecA filament formation in the adjacent gaps (277). The DNA-bound RecF is again seen to recruit RecO, possibly via the handoff of RecR. If correct, the functional sequestering of RecO by unbound SSB, coupled with the higher affinity of RecR for RecF, offers a way to prevent independent and inappropriate RecOR function in the cell. As noted above, SSB forms liquid-liquid-phase-separated forms within the cell (159, 248). RecO bound to SSB within the LLPS could simply be unavailable for RecA-loading reactions until recruited by RecFR bound to DNA.
RecF-mediated ATP hydrolysis results in bound RecF complex dissociation (95, 268), but no other function of the ATPase is known. Unlike RecOR, the RecFR complex will not load RecA onto SSB-coated ssDNA on its own. Some bacterial species lack a gene for RecF, but virtually all bacteria appear to have genes encoding RecR and one of two variants of RecO (280, 281). In many genetic backgrounds, the phenotypes of recO and recF deletions have easily measurable differences (90, 173, 174, 282, 283).
If the RecOR complex loads the RecA protein onto SSB-coated DNA, then RecO should be reliably associated with DNA gaps. However, the protein is expressed at quite low levels, and labeled RecO foci are not abundant in single-cell observations (173). The limited availability of RecO may be yet another mechanism by which RecO function is constrained, although attempted RecO overexpression has little measurable effect on cells (282, 284–286).
Considerable mystery still surrounds RecF and the RecFR complex. The recF gene is found in an operon that also includes the dnaA (replication initiator protein), dnaN (the β-clamp), and gyrB (subunit of DNA gyrase) genes (Fig. 4), an arrangement that is quite widely conserved in eubacteria (287–289). RecA filaments, once nucleated, grow primarily in the 5′-to-3′ direction (290, 291). The capacity of RecF to facilitate RecA loading into gaps under some conditions in vitro suggests a role in the nucleation of RecA filament formation, which would place it near the 5′-proximal end of a RecA filament. However, other results suggest a role at the growing end (3′-proximal end) of a RecA filament. If bound near a gap end, RecFR complexes halt RecA filament growth into the duplex DNA adjacent to gaps (95). RecF makes an alternative complex with the RecX protein, interfering with the RecA filament 3′-end-capping function of RecX (292). Given the demonstrated capacity of the RecOR complex to load RecA onto SSB-coated ssDNA on its own, a regulatory and/or targeting role for RecF seems likely to both constrain RecOR (preventing inappropriate RecA loading into gaps not needing repair) and target the RecFOR system specifically to postreplication gaps. A sequence in which RecF and the RecFR complex are involved in system targeting and RecOR is involved in the actual loading of RecA is indicated both by the biochemistry described above and by system genetics (283, 293). As described below, the targeting function of RecF is likely manifested by a recently documented interaction between RecF and the DnaN β-clamp component of the replisome (286).
FIG 4.

The dnaA-dnaN-recF-gyrB operon. The recF locus is shown, along with the neighboring operon genes and putative promoters (small arrows).
RecJ nuclease.
As noted above, RecJ is a 5′→3′ exonuclease with the capacity to expand the size of ssDNA gaps (165, 294–297). This protein was identified in the early 1980s as part of the RecFOR pathway (298). Gap expansion mediated by RecJ appears to be an important early step in postreplication gap repair (212). When combined with the RecQ helicase, the two proteins will unwind and degrade DNA ends in a reaction that can replace the action of RecBCD (299, 300). However, genetic results suggest that the two proteins have distinct roles and functions in distinct phases of recombinational postreplication gap repair (179, 297).
The loss of recJ function is synergistic rather than epistatic with the loss of recF in the context of the recombinational repair of postreplication gaps (212). This may indicate that RecF and RecJ participate in two separate pathways that lead to RecA loading into postreplication gaps. Alternatively, RecF and RecJ may collaborate in a complex fashion in the steps that lead to the RecOR-mediated loading of RecA into the gap. RecJ is thought to act in concert with the RecQ helicase (300), and recQ inactivation had the same effects as recJ inactivation in an assay used by Laureti et al. (212) to detect gap repair events. A recO recJ mutant combination was not included in this study, so it is not clear if the synergistic relationship extends to RecO. As described above, Jain et al. (179) documented the synthetic lethality of deletions in the rarA, ruvB, and recG genes and its suppression by the inactivation of the recF and recO genes. In this work, a deletion of recJ was in all cases as effective as recF or recO deletions in the suppression of the effects of double and triple mutants of the rarA, ruvB, and recG genes. Interestingly, inactivating recQ had the opposite effect, as recQ deletions exhibit synthetic lethality with rarA and ruvB deletions (179). This and other work (301) suggest that the collaboration between RecJ and RecQ is context dependent and that RecJ (but not RecQ) has some direct role that augments the function of RecF in preparing for the RecOR-mediated loading of RecA into gaps.
DnaN β-clamp.
The DnaN β-clamp is the sliding clamp that confers processivity on the replication activities of DNA polymerase III and other E. coli DNA polymerases. The DnaN protein also interacts with and stimulates the function of DNA polymerases II, IV, and V (42, 44, 101, 302–308). The dnaN gene is part of the same operon that encompasses the recF, dnaA, and gyrB genes and is adjacent to recF (309, 310).
RecA recombinase.
The RecA protein is the bacterial recombinase (202, 291, 311–319). It has a central recombination function in virtually all cells, with homologs such as RadA (archaebacteria) and Rad51 (yeast and humans). Studied intensively for several decades, this protein forms right-handed helical nucleoprotein filaments on DNA (320–324). Filament formation occurs in distinct nucleation and extension phases, with nucleation occurring most readily on ssDNA and proceeding primarily in the 5′→3′ direction (290, 291, 313). At the end of a postreplication gap, filament extension will proceed readily into the adjacent dsDNA (290). Bound RecFR will constrain filament extension into the adjacent dsDNA but only if sufficient RecFR is present to saturate the available dsDNA and ensure the placement of at least one RecFR complex near the gap end (95).
Once bound to ssDNA, the nucleoprotein filament will search for homologous sequences in nearby duplex DNA molecules and promote DNA pairing and strand exchange (202, 291, 311–319). A prototypical reaction used in many laboratories is presented in Fig. 5 (325–327). RecA promotes the synapsis phase of postreplication gap repair. In vitro, a RecA-mediated DNA strand exchange reaction requires free DNA ends in the substrates to permit the net interwinding of strands in the duplex product, as is the case in the reaction illustrated in Fig. 5. In the likely DNA substrates utilized during recombinational postreplication gap repair (Fig. 3), no such free ends exist. This reaction requires a topoisomerase to generate a stable and topologically intertwined joint molecule, perhaps supplemented by a nuclease or a helicase. A reaction of this sort, driven by the RecA protein and topoisomerase I, was documented in vitro by the Radding group in 1980 (328). The DNA strand exchange reaction promoted by RecA filaments exhibits a motor-like function in which the progress of the branch is coupled to ATP hydrolysis, allowing the reaction to proceed through heterologous sequence barriers and to continue beyond the single-stranded region of the initiating substrate to encompass a reaction between two duplexes (286, 312, 329–333). This motor may help overcome the substantial topological barrier otherwise barring the formation of a stable and topologically intertwined joint molecule, as shown in Fig. 3.
FIG 5.

DNA strand exchange reactions used to study RecA function in vitro. RecA filaments are formed on the single-stranded or gapped DNA circle. These function to pair the bound DNA with the linear duplex and promote the exchange of DNA strands, as shown.
Following synapsis, the RecA filament must be removed from the DNA so that the postsynaptic processing of the joint molecule intermediates can occur. The UvrD helicase has been implicated in RecA filament removal on the DNA, perhaps during repair, and the removal of RecA filaments on DNA (200–204) that would constitute a barrier to DNA replication (77, 78, 334). UvrD may be especially important in removing RecA from lagging-strand gaps where it has been incorrectly loaded or in aborting recombination after RecA loading in postreplication gaps in favor of TLS or other repair pathways. The RecG and RuvAB proteins, with their important roles in resolving strand exchange intermediates, might also play a role in dispersing RecA filaments (293), although a direct experimental assessment of RecA filament removal by these proteins has not been carried out. The RadD helicase may be especially important in removing RecA during the process of DNA strand exchange (198).
The RecA protein promotes extended strand exchange at a rate of about 360 bp min−1 at 37°C (335), perhaps not fast enough to complete a substantial exchange and process the products in a reaction that must occur in the short 18- to 20-min span between replication forks in cells growing in rich media. Both the RadA and RadD helicases accelerate this process substantially (198, 199). RadD carries out this process in a RecA-dependent manner (198).
The RuvABC proteins.
RuvA is a protein that binds specifically to Holliday junctions (336–343). Two hexameric RuvB helicases are arrayed on DNA arms on either side of RuvA, bound so as to promote DNA translocation reactions that lead to branch migration, movement of the Holliday junction (344). The RuvC protein is an endonuclease that specifically cleaves Holliday junctions (340, 345–347). RuvC binds to the RuvAB complex to bring about this cleavage, potentially leading to joint molecule resolution, as shown in Fig. 3 (210, 340, 347–351). The loss of the RuvA or RuvB protein results in substantially increased sensitivity to an array of DNA-damaging agents (179, 216, 352–354).
The RecG helicase.
The RecG protein is an SF2 multifunctional helicase. Although it can utilize a wide range of branched DNA substrates, RecG exhibits some functional overlap with the RuvAB branch migration complex in that it acts on Holliday junctions. It carries DNA-dependent ATPase activity that facilitates the remodeling of various branched DNA structures. In vitro and in vivo studies of RecG have documented RecG-mediated replication fork reversal (191, 355–358), Holliday junction branch migration activity (194, 195, 359–361), the prevention of overreplication associated with replication termination (205–207), and roles in double-strand break repair (190, 362). The deletion of recG is linked with the increased accumulation of Holliday junctions at a replisome stalling site (180).
The loss of RecG function is accompanied by reduced DNA recombination and repair, reflected in part in sensitivity to DNA-damaging agents (363). As documented by the synergistic toxicity of double and triple mutants, the pathways centered on RecG are complemented by pathways involving RuvB (179, 341, 349, 359), RadD (177), RadA (178), and RarA (179). Many of these effects are suppressed by mutations in the priA gene that eliminate the PriA helicase function but do not compromise the capacity of PriA to load DnaB onto DNA during replication restart (177, 190, 362, 363). A pathway involving RecG and PriA in processing some types of branched DNA substrates is implied by these results (190, 207, 362–364). PriA, normally considered to have a key function in replication restart (25, 26, 365–367), may have an underappreciated role in some aspects of the recombinational repair of postreplication gaps.
The RadA helicase.
Bacterial RadA (also known as “Sms”) is a tripartite protein with an N-terminal C4-type Zn finger, a central RecA-like ATPase, and a C-terminal Lon protein domain (368). The crystal structure of Streptococcus pneumoniae RadA shows a hexamer ring structure of the DnaB helicase family (369). When present in a RecA-mediated strand transfer reaction, RadA greatly speeds up the branch migration phase to yield full-strand assimilation in reactions between 3 or 4 DNA strands (199). Unlike other branch migration helicases, RuvAB or RecG (195), RadA supports directional strand exchange in the presence of the RecA filament; like RuvAB and RecG, RadA can catalyze branch migration on joint molecules in the absence of RecA (195, 199, 343). Pneumococcal RadA is a bona fide 5′-to-3′ helicase on forked or tailed DNA (369).
RadA was isolated as a radiation-sensitive mutant of E. coli (370) and participates in homologous recombination in a wide variety of bacteria (178, 368, 371–376), often in a manner redundant with RuvAB and RecG. Consistent with a role in D-loop extension during homologous recombination, the extent and efficiency of pneumococcal genetic transformation have a strong requirement for RadA (369).
The RadD helicase.
The RadD helicase, encoded by a gene previously called yejH, was discovered as part of a screen for genes required to recover from the effects of ionizing radiation (377). Its name was inspired by further studies implicating its function in the repair of radiation damage, where a radD mutant hypersensitized cells deficient in radA function (378). RadD is structurally related to helicases of the SF2 family, but it exhibits no helicase activity on its own. Instead, like RadA, it accelerates RecA-mediated DNA strand exchange (198). Unlike RadA, the RadD reaction is entirely RecA dependent. RadD also appears to disassemble RecA filaments during the DNA strand exchange process (198). RadD also interacts directly with SSB (163). The RadD ATPase is stimulated by this SSB interaction. The binding pocket on RadD for the SSB C terminus has been defined (162, 379).
The RecQ helicase.
RecQ is a DNA helicase of the SF2 family (380, 381), unwinding DNA by translocating in the 3′→5′ direction along one strand. RecQ can unwind DNA at ends or internally within the DNA (382). The cooperativity in the unwinding process suggests that the unwinding species may have up to three RecQ subunits (382). RecQ and RecJ will collaborate in processing DNA ends for binding by the RecA protein and can substitute to a degree for the absence of the RecBCD enzyme in double-strand break repair (300, 301).
RecQ has a complex involvement in the repair of postreplication gaps. The synthetically lethal combination of deletions in recG, ruvB, and rarA is suppressed by the deletion of recJ but not by the deletion of recQ (179). Instead, a ruvB rarA recQ triple mutant is synthetically lethal, similar to a ruvB rarA recG triple mutant. The former triple mutant is also suppressed by recJ deletion as well as deletions in the recFOR genes (179). This suggests that RecQ is somehow involved in the resolution of recombination intermediates created by RecA, perhaps reversing them (383). Somewhat in contrast, in a slightly different background with deletions of recG and uvrD, Fonville and colleagues have shown that cells endure death by recombination in which branched intermediates behind the replication fork are not resolved and chromosome segregation is blocked (180). The aggregation of interchromosomal exchanges depends on the presence of RecQ along with RecF, RecO, and RecJ (180). This suggests a function early in the repair of postreplication gaps. RecQ may well be involved in gap repair at multiple points and may facilitate either the resolution or the reversal of joint molecules.
HolC: a postreplication recruitment factor?
HolC (also called χ) is a subunit of the DNA polymerase III clamp loader and the product of the holC gene. HolC was identified by the Reyes-Lamothe lab as an enhancing factor in the formation of DNA polymerase III foci that do not colocalize with DnaB after UV irradiation (213). In a genetic screen for factors whose expression improved tolerance to AZT, it was found that HolC and a putative helicase, YoaA, promoted AZT tolerance in a mutually codependent way (230). The loss of the function of either holC or yoaA produced profound AZT sensitivity. By pulldown and yeast two-hybrid analyses, the two proteins were shown to physically interact (230, 384). In DNA polymerase III, HolC interacts with HolD to form an accessory complex to the clamp loader complex (385). The role of this accessory complex has not been entirely clear. HolC/χ is the only protein of the DNA polymerase III holoenzyme that binds to SSB and, through a chain of protein interactions of the clamp loader complex and the core polymerase complex, could tether the DNA polymerase III holoenzyme to an SSB-bound DNA template. The HolC amino acid residues that are required for its interaction with SSB (386) are also required to promote tolerance to AZT with YoaA (230), suggesting that SSB’s recruitment of HolC is required for its function in fork repair.
The residues of HolC required for its interaction with YoaA are the same as those required for the interaction of HolC with HolD (386), suggesting that HolC forms two alternative complexes: one with HolD and the DNA polymerase III holoenzyme and another with YoaA. Indeed, the coexpression of the three proteins HolC, HolD, and YoaA in E. coli yielded distinct HolC-YoaA and HolC-HolD complexes and no ternary complex consisting of all three proteins (386).
Because SSB-bound ssDNA can recruit multiple HolC molecules, we imagine that both HolC complexes, HolC-YoaA and HolC-HolD-DNA polymerase III, may be corecruited. This provides increased access to the 3′ nascent strand, with the helicase aiding in AZT removal and misincorporated nucleotide removal by DnaQ (384; D. J. Glass, J. Grant, and S. T. Lovett, unpublished data).
YoaA is a predicted member of a family of SF2 DNA helicase proteins with 5′-to-3′ polarity and an intrinsic Fe-S cluster. This group includes an E. coli paralog protein, DinG, and a large group of eukaryotic proteins mutated in human DNA repair deficiency syndromes (such as XPD, FANC-J, ChlR1/DDX11, and RTEL [387]). Recent work has confirmed ATP-dependent translocase and helicase activities for the purified Fe-containing YoaA-HolC complex (388). This tight complex, a heterodimer, is likely the active form, with the monomeric YoaA protein being largely insoluble and poorly active. Helicase activity requires a 5′ ssDNA overhang, and activity is more robust on forked substrates with both 3′ and 5′ single-strand overhangs.
Genetic and physical analyses showed that YoaA interacts with HolC/χ via its C terminus, a short 18-amino-acid region not shared with its paralog DinG (384). The deletion of this region yields an AZT-sensitive phenotype as extreme as that of the null yoaA allele, indicating that the interaction with HolC is required for YoaA function in vivo (388). Lesion skipping would produce ssDNA gaps left behind the fork, stably bound by SSB. SSB interactions with both HolC complexes, HolC-HolD-Pol III and HolC-YoaA, recruit factors to process the 3′ end and prime DNA synthesis for gap filling.
As noted above, there is evidence from cytological studies that HolC indeed promotes excursions behind the fork (213). After UV irradiation, fluorescence labeling of the epsilon (ε) (DnaQ) subunit of the core polymerase III replisome and the DnaB fork helicase provided evidence that the replisome relocalizes to chromosomal sites without DnaB present, likely gaps left behind by lesion skipping. These relocalized foci required functional HolC (213). We note that in the repair of lesion-containing postreplication gaps discussed below, a lesion-free template is not available for the intercession of DNA polymerase III until after RecA-mediated DNA strand exchange (Fig. 3). For gaps generated by AZT, recombination may occur, but it is not necessary. In principle, gap resolution could occur by the exonuclease III-mediated removal of the AZT nucleotide followed directly by DNA polymerase III-mediated gap filling. However, the strong effects of ruvABC mutants in a yoaA background suggest that recombination happens in many AZT-generated gaps and that the RuvABC system plays a particularly important role in their resolution.
Recombinational DNA Repair and the RecFOR System
The recombinational repair of postreplication gaps (as outlined in Fig. 3) can be discussed as a succession of phases. First, the RecFOR proteins must be targeted to the newly created postreplication gap. The targeting must be precise, preventing the loading of RecA into lagging-strand gaps or the occasional gaps created during mismatch repair. The gap is presumably bound with SSB the moment it is created. The expansion of the gap by the RecJ exonuclease appears to be a critical part of this process (201, 210, 212), and the effects of deleterious mutant combinations that can be suppressed by the deletion of recF, recO, or recR can also be suppressed by recJ deletions (179). The second phase is presynapsis. RecA is loaded onto the ssDNA gap, a reaction facilitated by RecO and RecR, displacing SSB. Third, the RecA nucleoprotein filament catalyzes synapsis, the pairing of the bound ssDNA with the intact sister duplex. This creates a joint molecule that physically joins the two replicated chromosomes behind the fork. The depiction of this step in Fig. 3 belies its complexity, as the creation of a paired right-handed helical duplex in this pairing reaction requires the collaboration of topoisomerases, nucleases, helicases, or all three. Fourth, and finally, the joint molecule must be resolved. Recent work indicates that there are multiple pathways by which this can occur (176–180). These joint molecules effectively join the two products of replication behind the fork. A failure to resolve or reverse such a joint molecule can prevent daughter chromosome segregation at cell division and lead to cell death.
Targeting RecF to Postreplication Gaps
An often-proposed hypothesis for the targeting of RecFOR to postreplication gaps involves targeting via RecF or RecFR to gap ends followed by an interaction with RecO or RecOR, perhaps a handoff of RecR from RecF to RecO, to facilitate RecA loading onto the adjacent SSB-coated ssDNA (258, 259, 277, 389–391). This general idea is problematic for two reasons. First, as indicated above, there is no component among the RecF, RecO, and RecR proteins that binds to gap ends with the specificity required to find such ends and distinguish them from the bulk of the genomic DNA (95, 259, 263, 265, 268). The caveat is that there may be a component not yet considered, such as RecJ, that might offer the needed specificity and recruit RecF or other proteins. Second, binding to all gap ends, in an environment where most gaps are formed during lagging-strand DNA synthesis and mismatch repair, could direct RecA filament assembly into many places where recombinational DNA repair is not needed and could be deleterious. The formation of unnecessary joint molecules behind the replication fork would need to be resolved. The excessive formation of RecA filaments on the DNA could cause problems in and of itself. If not removed, a bound RecA filament on the DNA represents perhaps the most formidable protein barrier that a replisome might encounter (77). A kind of genomic chaos could result.
For similar reasons, RecOR function must be constrained. RecOR, by itself, can facilitate RecA protein loading into any ssDNA region coated by SSB irrespective of whether that ssDNA occurs in a postreplication gap. No gap end is actually required. Unconstrained, this could also potentially lead to RecA loading into DNA gaps where it is not needed. The sequestering of RecO by interactions with SSB unbound to ssDNA and within LLPS bodies offers one potential way to constrain RecOR within the cell, as noted above. LLPS-mediated sequestration could well be a regulatory mechanism for many other types of SSB-interacting proteins.
The key protein to resolve these issues is most likely RecF. There are increasing cases where the phenotypes of recO and recF mutants do not coincide, suggesting some functional distinctions (173, 392, 393). For example, the overproduction of RecF, but not RecO, is highly toxic to cells (282, 284–286). The functional distinction could come down to the following: RecF is primarily responsible for the targeting of the system to appropriate postreplication gaps, and RecO is primarily responsible for the RecA-loading process. RecF finds the correct gaps and recruits RecO. This general sequence has been established for almost 3 decades (283).
Another factor distinguishing RecO and RecF is the increasing incidence of links between RecF (but not RecO) and the replication fork, both experimental (172–174, 394) and speculative (95, 293). In vivo, RecF often colocalizes with the replication fork and rarely colocalizes with RecO (173). Recent work shows that the toxicity of RecF overexpression can be attributed to replisome destabilization (286). More importantly, the general observations linking RecF to the replisome likely reflect a newly detected direct interaction between RecF and the DnaN β-clamp and also the DnaG primase (286). A RecF-DnaN interaction has been documented by three independent methods (286). McInerney and O’Donnell previously speculated that a RecF-DnaN interaction might exist and be functionally significant (395) with respect to postreplication gaps. Additional speculation about RecF-DnaN interactions was entertained by Kuzminov (58) and Webb et al. (95). This interaction is now acquiring a molecular basis with details remaining to be explored.
The observed interaction of the RecF protein with the replisome via DnaN has the potential to resolve a number of the questions listed above. The polymerase components of the replisome are the ultimate sensors of template strand lesions. In principle, the targeting of RecF uniquely to postreplication gaps could be accomplished by replisome conformational changes triggered by replisome-lesion encounters. Conformational changes triggered by such encounters have been detected (396). The RecF protein, interacting with DnaN, could be left behind on the 3′ end of the resulting gap near the template lesion as the replisome itself disengages from the template. In principle, RecF could also catalyze replisome disengagement by forming a DnaN-RecF complex that effectively breaks the dimeric DnaN β-clamp circle. Leaving RecF behind at the 3′ end of the gap near the template lesion (as suggested previously [95]) would provide prepositioned RecF complexes that would effectively mark the adjacent gap as a lesion-containing postreplication gap in need of repair. Like recF, the recR gene is positioned in an operon otherwise dedicated to replication, near the dnaX gene. If conserved gene proximity can suggest protein-protein interactions, as suggested by Kuzminov (acknowledging that his RecF-DnaN conjecture has proven correct), an interaction between RecR and DnaX would provide proximal access of the deposited RecF protein to RecR. The DNA interaction could be stabilized by the formation of a RecFR complex. RecFR could recruit RecO and activate it for RecA loading by a RecR handoff from RecF to RecO. This general concept is illustrated in Fig. 6. Although RecA loading must occur closer to the 5′ end of the filament away from the location of RecFR in this scheme, the handoff of RecR from RecF to RecO could be facilitated by DNA looping and/or SSB-mediated ssDNA condensation (148). Speculation about a RecF interaction with the replisome appeared in the late 1990s (95, 172, 174, 293), triggered by both suggestive experimental results and consideration of the genomic operon within which the recF gene is expressed (Fig. 4). The proposed sensing role of the replisome and its interaction with RecF have many elements that mirror the quite detailed speculation put forward by Kuzminov more than 25 years ago (293). A variation of the scheme in Fig. 6, proposed by Kuzminov (293), in which the RecF protein is not deposited onto the DNA until the replisome reinitiates DNA synthesis downstream to complete gap formation, would place RecF at the best end of the gap to facilitate RecA filament formation. An interaction between RecF and the DnaG primase has also been detected (286), which might allow such a RecF placement. It is tempting to propose that RecF is deposited at both ends of the gap.
FIG 6.
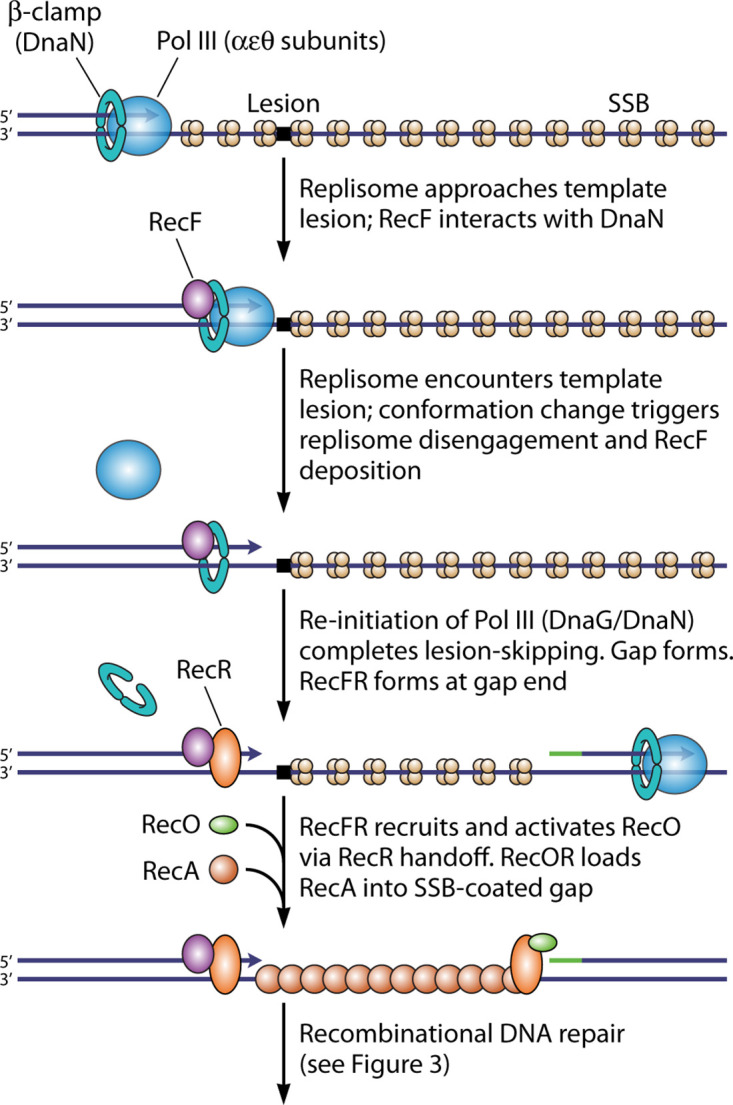
Model for the targeting of the RecA protein specifically to lesion-containing postreplication gaps. In this scenario, the targeting of RecF to the appropriate gaps is mediated by the interaction of the RecF protein with the DnaN β-clamp. An encounter between the replisome and a lesion triggers a conformational change that disengages the replisome for lesion skipping and deposits RecF at the end of the resulting gap where the lesion was encountered. The deposited RecF forms a stabilizing complex with RecR. The RecFR complex recruits RecO, activating RecO by a handoff of RecR from RecF to RecO. RecO loads RecA into the gap to initiate repair. The gap is drawn linearly, but DNA looping could facilitate many of the described interactions. In both cases, the events shown involve sister chromosomes interacting behind the fork.
The new results demonstrating RecF colocalization with replication forks (173) and a direct RecF-DnaN interaction (286) provide a path to convert much of this speculation to molecular substance. Moving from a RecF-DnaN interaction to a deposited RecF protein to RecO activation via RecR handoff to RecA loading is a path with many mechanistic details to work out. Despite the holes remaining to be filled in at present, there is no other plausible mechanism in the literature for the specific targeting of the RecFOR system to the gaps requiring recombinational DNA repair. The demonstrated interactions of RecF with DnaN and DnaG recommend a model of this kind or something similar that involves the replisome as the sensor to facilitate RecFOR system targeting to postreplication gaps.
These studies are obviously at an early stage, and as indicated above, key parts of the scheme outlined above are still speculative. Thus, before closing this discussion, some alternatives for system targeting that have occurred to us are worth mentioning. One alternative for targeting would be a scenario in which RecFOR system components are targeted to the appropriate gaps mainly by a process of elimination. In this case, the RecF, RecO, and RecR proteins would be restricted from lagging-strand replication gaps and mismatch repair gaps by the action of other proteins organic to the replication or repair of such gaps. Many proteins interact with the SSB C terminus and are thus potentially recruited to regions of ssDNA (143). The competition between these proteins for binding sites on the SSB has not yet been explored. Yet another alternative would invoke the existence of some as-yet-unidentified factor that has the necessary specificity to bind to and specifically target lesion-containing postreplication gaps and recruit the RecFOR system to them.
Additional scenarios may be possible. The targeting of the RecFOR system to postreplication gaps remains substantially enigmatic. However, the RecF-DnaN connection provides an attractive and viable path for investigation.
Presynapsis: RecA Loading by RecOR
Once targeting is accomplished, the formation of RecA filaments is perhaps the best-understood phase of the pathway. The RecJ nuclease is needed to enlarge the gaps (212). RecOR overcomes the SSB barrier to RecA nucleation, displacing SSB and facilitating the nucleation of the nucleoprotein RecA filament (259, 278, 283). Once initiated, filament extension occurs spontaneously in vitro, although additional proteins act to constrain it in vivo. RecA filament lengths and lifetimes are highly regulated. The DinI protein can stabilize filaments (397, 398). The RecX protein can limit filament length and stability (292, 397, 399–405). The UvrD helicase will remove RecA filaments from the DNA (200, 201, 203, 204), an activity that may not only control active RecA filaments but also eliminate filaments that appear in gaps that do not require repair.
Synapsis
The RecA protein will promote the DNA-pairing step in synapsis. However, the DNA substrates as presented in Fig. 3 have no free ends, and the major complexity in this phase is topological. In the absence of other proteins, no net interwinding of the two strands being paired can occur. The collaboration of RecA with topoisomerase I, demonstrated by Cunningham et al. (328), may well mimic the reaction needed for recombinational postreplication gap repair. Acting with topoisomerase I, a motor-like function of RecA filaments that allows them to promote DNA strand exchange through heterologous barriers in vitro (and that is required for strand exchange reactions involving two duplexes [329, 330]) may facilitate the formation of an intertwined joint molecule in the difficult topological environment in which this step must take place (312, 332). As noted above, RecQ may also be involved in this phase of the reaction (180). Both the RadA (199) and RadD (198) proteins may accelerate the exchange in this phase. The RadD protein, as it accelerates RecA-mediated DNA strand exchange, appears to dismantle RecA filaments (198). The RdgC protein inhibits RecA-mediated strand exchange in vitro (406). However, this inhibition may simply reflect competitive DNA binding to reaction substrates and could be nongermane to the normal function of the still very enigmatic RdgC protein (407–409).
Postsynapsis
As noted above, the synaptic intermediates created by RecA link the two arms of the replicated DNA and will link the replication products if not resolved. This can lead to cell death (179, 180). Enzymatic complexity is evident in this phase of the process as there appear to be multiple pathways for resolving the intermediates created by RecA. There are at least three pathways, dominated by (i) RuvABC, (ii) RarA, and (iii) either RecG or RecQ (179). RadD and RadA also play roles (176–178). In some cases, resolution may mean a simple reversal of the reaction that created the intermediate by RecQ or UvrD (179, 180). The various pathways doubtless involve a significant number of additional proteins, some of which (SSB, topoisomerases, and helicases) may be involved in multiple pathways.
The RarA protein (also called MgsA) appears to have a role in intermediate processing (179). RarA is closely related to clamp loader proteins (164), although it functions as a tetramer rather than a pentamer. RarA will also bind to the ends of duplex DNA and duplex regions and separate the DNA strands near these ends (410). There is some functional overlap between RarA and RecA based on genetic studies (411).
Replication must occur as part of the gap repair process. The nature of the DNA synthesis that accompanies RecAFOR recombination is not well understood, but we imagine that it involves clamp loading and one or more of the 5 DNA polymerases of E. coli. The most likely template for DNA synthesis is a template lacking a lesion (Fig. 3). This synthesis likely occurs independent of the DnaB fork helicase, and there is then no obligate need for PriA or other fork assembly factors (90). The conserved RarA protein is also at least sometimes required at a late stage in the recombination reaction. The RuvABC and RecG systems provide alternative resolution paths (179). Whether there are factors that constrain DNA synthesis to the gap (rather than initiating a new fork) is not known. There is the potential that RarA could be a clamp loader in particular circumstances, although no demonstration of such a role in vitro has appeared. Once DNA pairing has occurred, branch migration steps will create a replication substrate with an undamaged template that must be filled in. In the absence of the SOS response, this could be accomplished by any E. coli DNA polymerase, but a subcomplex of the DNA polymerase III replisome (412) that loops back to carry out this function is a good candidate for nonmutagenic replication. This step could require the provision of a DnaN β-clamp (perhaps installed by RarA). Even though no role for the DnaB replicative helicase has been demonstrated in this step, the potential topological complexity of DNA synthesis in the context of branched DNA joint molecules could recommend an energetic contribution from DnaB as a way to avoid or resolve congestion. However, the essential role of DnaB in normal replication could preclude obtaining any genetic result that would infer involvement in smaller events outside the replication fork.
Template-Switching Repair of Postreplication Gaps
Having considered gap filling mediated by the RecFOR-RecA-RecJ system, we now turn to RecA-independent template switching.
RecA-independent template switching.
The RecA-independent template-switching pathway is probably the most enigmatic of the gap-filling mechanisms. The properties of this mechanism were deduced from the observation of efficient homology-dependent genetic rearrangements of juxtaposed direct-repeat sequences that occurred concomitantly with crossovers between sister chromosomes (reviewed in reference 66). These crossovers can be resolved to form replicon dimers. Plasmid dimers are viable; chromosomal dimers are resolved by the Dif/XerCD system (69). The intermediates of this reaction are believed to be quite similar to those in RecAFORJ-dependent gap filling, with RuvABC or RecG processing of branched structures invoked in both RecA-dependent and RecA-independent template switching. However, RecA-independent recombination is strongly constrained by proximity (186) and is much more efficient on sister chromosomes (59, 257), possibly because it involves annealing reactions with ssDNA rather than true strand invasion. A model is presented in Fig. 7. The 5′-to-3′ DNA helicases DinG and YoaA may initiate the unwinding of nascent strands, which is a precursor to pairing (384). Because this pairing is behind the fork, this mode of template switching may require a lagging-strand gap (or nick). The RarA protein is required for both RecA-dependent (179) and RecA-independent (413) recombination, possibly as a factor in the common late stages of the reactions, such as the processing of intermediates or the potentiation of DNA synthesis. Mutations in the clamp (DnaN) and the clamp loader (DnaX) reduce the efficiency of RecA-independent template switching (185). RecA-independent template switching is also dependent on the chaperone DnaKJ (185), but the mechanism of this effect is not understood.
FIG 7.

Gap-filling DNA intermediates of homologous recombination versus RecA-independent template-switching. (A) In recombination mediated by the RecFOR system and RecA, the RecA filament extracts and pairs with a strand from a duplex sister chromosome. The 3′ end of the gap can then use the sister chromosome as the template to synthesize the missing information. Pairs of branched DNA junctions are produced and can be resolved by RuvABC or RecG, restoring an intact chromosome. (B) In RecA-independent template switching, the unwinding of the nascent strands of the gapped chromosome and its replicating sister allows them to pair and promote synthesis. Branched molecules are also generated, but the factors that resolve them have not been defined. As shown in the diagram, the resulting chromosomes are noncrossovers, but an alternative resolution in both pathways can produce crossover and sister chromosome exchange.
Template switching in a reversed fork.
Fork reversal (or fork regression) involves the re-pairing of the template strands at the fork, which extrudes the paired nascent strands. If the extruded “nub” escapes degradation by the RecBCD nuclease and the lagging nascent strand is longer than the leading strand, the leading strand can prime synthesis using the lagging nascent strand as the template, as presented in Fig. 8. Such a situation might be encountered after a stalled leading strand where the fork and the lagging strand proceed ahead for some short distance. After the template switch, branch migration can restore the fork, with the leading strand block being bypassed. Although it is clear that fork reversal does occur in vivo (reviewed in references 100 and 103), it is still unclear how often it is associated with this form of template switching and what role this might play in repair.
FIG 8.

Template switching after fork reversal. (A) A lesion on the leading strand (triangle) blocks DNA synthesis. (B) Branch migration at the fork produces a reversed fork in which both nascent strands anneal to form a nub. From this point, there are three alternative outcomes. (C) Leading nascent strand extended by DNA synthesis from the lagging-nascent-strand template. (D) Branch migration to restore the fork with lesion bypass. (C′) Alternatively, the exonucleolytic degradation of the nub by RecBCD resets the fork, backward from the original position. (D′) Because the lesion is now in dsDNA, it can be removed by excision repair. (C″) In a third scenario, the 4-strand Holliday junction (HJ) is cleaved, leaving a single-ended broken chromosome. This broken chromosome can be repaired by RecA during RecBCD-mediated double-strand break repair with the intact chromosome (not shown).
Further consideration of the division of labor in gap repair pathways.
As noted above, there is competition among RecA-independent template switching, TLS, and RecA-dependent recombination mediated by the RecFOR system, with the last process taking precedence (212). Defects in the early steps of recombination (RecAFORJ) lead to an increase in TLS at an engineered lesion. In contrast, defects in the late steps (RuvAB and RecG) do not, indicating that after synapsis and strand transfer, the capacity for TLS is lost. Blocks to both RecA-dependent and RecA-independent template switch/recombination pathways are synthetically lethal (recA dnaK), indicating functional overlap (185). The Uup and RadD proteins appear to channel events away from the RecA-independent template-switching pathway: in uup radD mutants, the frequency of RecA-independent template switching is increased many orders of magnitude, and these events are more likely to be associated with crossovers (176).
Translesion DNA Synthesis
When recombinational repair falters or is unavailable, translesion DNA synthesis takes up at least some of the slack (63, 64, 212). The increase in mutagenesis is tolerated in cellular emergencies. DNA polymerase II (Pol II) and Pol IV are available in cells, although there are only limited data available to indicate where and how each polymerase acts in this process.
DNA polymerase V would presumably have a limited role, reflecting its limited availability during normal growth in the absence of an SOS response. As much of what the TLS polymerases do takes place in gaps, their potential contributions are included here.
DNA polymerases II and IV are present constitutively at moderate levels in the cell, increasing to significantly higher levels (~10-fold) within about 1 min following the induction of the SOS response (67). In contrast, Pol V is barely detectable in the absence of external stressors (107). Tight transcriptional and posttranslational regulation is responsible for the absence of Pol V until 30 to 45 min after the SOS response is induced (414, 415), to provide the cell with ample opportunity to repair a damaged chromosome.
Seemingly, E. coli goes to great lengths to avoid the presence of Pol V in rapidly dividing cells, except perhaps as a last resort to allow the copying of replication-blocking lesions that have failed to undergo repair. A striking illustration of why Pol V is kept tightly under wraps can be seen in a RecA730(E38K) mutant that is constitutively induced for SOS. Here, Pol V-induced mutations are increased by 100-fold in log-phase cells in the absence of DNA damage and by an additional 10-fold in cells irradiated with UV light (416). It seems possible, and perhaps even likely, that sufficiently high levels of Pol V can undermine polymerase III by catalyzing error-prone chromosomal replication. Genetic data from Maliszewska-Tkaczyk and colleagues (417) suggest that Pol V mutagenesis in RecA730 cells occurs predominantly on the lagging strand, which could easily be envisioned to happen when the polymerase III holoenzyme dissociates from the β-sliding clamp during the time interval between the completion of one Okazaki fragment and the initiation of a new Okazaki fragment.
In SOS-induced cells, in strains lacking Pol V (umuDC-null mutants), chromosomal mutations do not appear to exceed spontaneous background levels (418–420), suggesting the absence of a prominent role for either Pol IV or Pol II in copying past UV-induced or other lesions found in postreplication gaps. Both polymerases have been shown to play central roles in the occurrence of adaptive mutations in stationary-phase cells (421–425), and Pol II has been shown to play a role in replication restart in UV-irradiated cells (426). Biochemical data suggest that Pol IV and Pol II can act in tandem during RecA-mediated D-loop extension underlying error-prone recombination such that the exonuclease proofreading activity of Pol II competes with Pol IV within D-loops to reduce errors (427, 428). Indeed, determining the location of RecA, generally throughout the cell and specifically within gapped regions throughout the chromosome, is central to developing a fine-grained picture of all processes in DNA metabolism.
Pol V has especially intimate connections to both the nucleoprotein filament RecA and, even more so, individual RecA monomers. Pol V’s connection to RecA is defined initially via the induction of an SOS response whereby RecA, acting as a coprotease, cleaves and thereby inactivates the LexA repressor (429). RecA nucleoprotein filaments further act as a coprotease to cleave UmuD→UmuD′ (430–432) to enable the formation of Pol V=UmuD′2C (433). RecA is further required to transfer a RecA monomer from its 3′ tip to Pol V, which, along with the binding of ATP, results in the assembly of Pol V into its active mutasomal form Pol V Mut=UmuD′2C-RecA-ATP (434). Where each of these events takes place, be they on or off the chromosome, is anyone’s guess.
A recent highly perceptive review article by Fujii and Fuchs (435) paints a broad yet detailed picture of the types of molecular models that could support TLS. The models include a variety of error-prone and high-fidelity TLS mechanisms such as direct lesion copying, perhaps involving DNA polymerase exchange; lesion skipping; replication restart; and replication fork regression. The models invoke the presence of a variety of recombination proteins (e.g., RecA and RecBCD); each of the TLS polymerases V, II, and IV; polymerase III; the β-clamp; and SSB. Clearly, the devil is in the details. Using high-resolution gap mapping (129) in conjunction with ChIP-seq, it is now feasible to identify each of these proteins bound within the ssDNA gapped regions of gDNA. A noteworthy subtle application of gap mapping with ChIP-seq would be to compare the location of Pol V Mut in a recA1730(S117F) mutant (418) with that of Pol V Mut in a wild-type recA background. All of the RecA functions are retained (albeit somewhat reduced) in recA1730 cells, except that SOS mutations are completely suppressed to spontaneous background levels (418). Thus, the recA1730 phenotype is the same as that for ΔumuDC cells (418–420).
The biochemical data reflect the genetic data; Pol V Mut with RecA1730 is catalytically dead (436). Assuming that Pol V Mutwt can be mapped to a subset of ssDNA repair gaps, the question is, Where is Pol V Mut1730 in relation to Pol V Mutwt? And where might both polymerases map in the ssDNA gaps in relation to RecA?
TLS takes place most often in rapidly dividing cells; the SOS polymerases, acting either alone or in combination, have been shown to strongly enhance cell fitness in nondividing E. coli cells in deep stationary phase in the absence of UV- or chemical-induced DNA damage (437, 438). Gap mapping can be used to address the formation and resolution of ssDNA gaps in nutrient-deprived cells that are not dividing. Perhaps one of the most important challenges to address is related to the less advertised of the current pandemics, namely, the generation of multidrug-resistant pathogenic bacteria. Might exposure to antibiotics that interact with DNA cause ssDNA gaps to form? Those that alter the DNA to cause kinks or bulky base lesions may well do this. Might the TLS polymerases contribute to a mutational landscape that leads first to the acquisition of drug tolerance and then to drug resistance (439)? The processes leading to antibiotic resistance are complex (424), and work on deciphering them is just beginning. Might it be relevant that pathogenic strains of bacteria often encode homologs of Pol V (440)? Clinical isolates of Acinetobacter baumannii were found to have as many as four umuC homologs (440). Recently, it was shown that Rum polymerase, which is a highly mutagenic homolog of Pol V (441, 442) encoded on an integrative conjugative R plasmid, is active in vitro and in vivo only when present as a Rum mutasome (RumA′2B-RecA-ATP) (443). Rum Mut was found to be activated by individual RecA homologs donated by each recipient bacterium (443). The use of gap mapping to investigate the location of the TLS polymerases on gDNA provides an important new tool to alleviate a gulf in our understanding of where and how these SOS polymerases act.
GENOMICALLY PROGRAMMED POSTREPLICATION GAPS
There may be cases where postreplication gap formation is programmed into the genome at a particular location to facilitate some aspect of genome function. Two examples that illustrate this phenomenon are now apparent in the literature. Both examples feature sequence elements embedded in the genome that can form elaborate and very stable secondary structures that are difficult to replicate when present in the template. Both trigger the formation of postreplication gaps on the lagging strand.
The first example was detected in the course of a study documenting the patterns of RecA and SSB deposition on ssDNA in the E. coli genome, using a new method called ssGAP-seq (444). Two sharp peaks of enhanced RecA deposition onto ssDNA were detected, each spanning about 2 kbp, or the length of a long Okazaki fragment (Fig. 9A). The peaks of RecA deposition corresponded closely with a pair of genomic-repeat sequences, 222 bp in length, that have been largely overlooked to date (Fig. 9B). The repeats are highly GC rich and are capable of forming elaborate secondary structures (Fig. 9C). These repeats are symmetrically arrayed around the dif sequence at the replication terminus, and each is about 650 kbp from dif (Fig. 9D). They encompass the entirety of the genomic Ter macrodomain (131, 134–137) (Fig. 9D). When cloned into plasmids, they cannot be sequenced using standard commercial Sanger sequencing protocols unless special procedures involving added heat are applied.
FIG 9.

The RRS genomic element. (A) Discovery of the RRS (Replication Risk Sequence) element. Shown are genome-wide binding patterns of RecA protein to ssDNA, generated by ssGAP-Seq (444). Sharp peaks of RecA binding to ssDNA are indicated by green arrows. The RRS elements were discovered near the end of each of these regions. The RRS are positioned near the dusC and lysO genes. Both RRS are intergenic. The letters A, B, C, and D denote ter sites that are bound by the protein Tus. (B) The sequence of the RRS element. The two RRS elements in the E. coli genome differ at only 3 positions out of 222 (shown). (C) One of several predicted folding patterns that can be taken up by the RRS sequence. (D) Positioning of the RRS in the E. coli genome is shown in the center panel. RIGHT, LEFT, TER, and ORI refer to defined chromosomal macrodomains. NSL and NSR refer to left and right non-structural regions, respectively. W-strand and C-strand refers to Watson and Crick strands, respectively. Each RRS is equidistant (about 650,000 bp) from dif and symmetrically arranged around the ter macrodomain. To the right and left are shown close-ups of the RecA binding patterns in the peak adjacent to the RRS. In each case, the highest levels of RecA binding are seen near the RRS, then trailing off over a range of about 2 kbp. Enhanced RecA binding to ssDNA is seen only on the lagging strand in each case.
The positioning suggests that after they are unwound by DnaB and the lagging-strand replisome encounters them, lagging-strand DNA synthesis is blocked, and the replisome disengages, leaving a programmed gap into which RecA is deposited. In this scenario, the length of the gaps created should vary, with a maximum length equal to a long Okazaki fragment (~2 kbp). Accordingly, RecA deposition is greatest near the repeat, trailing off over a range of 2 kbp (Fig. 9D).
The RecA deposition, along with the difficulties encountered in Sanger sequencing, recommended the name replication risk sequence (RRS) (444). One RRS or the other can be deleted without a detectable effect on cell growth or viability. However, the construction of a strain lacking both RRSs has so far proven impossible, suggesting that the possession of at least one RRS is essential. The RRS elements are highly conserved in both sequence and positioning in enterobacterial species, although some species have more than two RRSs (Fig. 10).
FIG 10.

RRS conservation in enterobacteria. RRS elements in several other enterobacterial species are shown. They are highly conserved with respect to both sequence and positioning. Some species have more than two RRSs, but they always have one pair flanking the Ter region that encompasses the matS sites that are critical for Ter macrodomain function (450, 451).
The RRS elements thus appear to be important functional features of the E. coli genome. Given the extraordinary topological issues that must be overcome to facilitate replication termination and successful chromosome segregation in bacteria (69, 445), we speculate that the RRS elements act as a kind of topological relief valve. The region near the RRS is rendered single stranded when replisomes pass. In this state, every phosphodiester bond can act as a swivel. The elaborate secondary structure may help protect the sequence from nucleases. RecA loading may be incidental. However, the sequences must somehow be replicated once each replication cycle, and RecA may somehow facilitate that process.
The second possible example of an element that may trigger the programmed formation of a postreplication gap is found in Neisseria gonorrhoeae. This pathogen undergoes pilin-based antigenic variation that allows it to evade the human immune system (446). Whatever pilin subunit that is encoded by the pilE locus is expressed, while several variant sequences encoded at other chromosomal locations are silent. At intervals, sequence information from the silent loci (pilS) is transferred to pilE via recombination (447–449). A sequence near pilE forms a G quadruplex that is required for this recombination event. The G quadruplex can trigger gap formation during replication. A small RNA transcribed locally can form an RNA-DNA hybrid duplex, helping to stabilize the formation of the single strand where the G quadruplex is formed. Recombination itself has requirements (including RecA, RecO, RecR, and RecJ) that mirror the requirements for postreplication gap repair. It is tempting to speculate that the mechanisms are very similar and that gap formation triggered by the G quadruplex is what gets the process started.
Single-stranded gaps formed at particular chromosomal sites could be destabilizing genomic features. We thus assume that the evolution of such elements is rare, occurring only when some major biological purpose is addressed. However, the detection of such elements is nontrivial. The RRSs have been overlooked for decades despite ready access to genomic sequences. The G quadruplex in Neisseria was detected only with focused attention on a particular cellular system. Attendant biological purposes employing such elements might be more numerous than we anticipate. Analogous genomic features might be plentiful in eukaryotic genomes, where chromosomes are larger and more elaborately structured. Perhaps the elucidation of these examples will trigger a wider search for more such elements.
CONCLUSION
Since their discovery in the 1960s, the DNA metabolism of postreplication gaps has received substantial attention. However, the status of our understanding of the molecular mechanisms in this area remains unsatisfying. The mechanics of lesion skipping by the replisome, the facile identification and specific targeting of a lesion-containing postreplication gap, the prevention of RecOR function in gaps that do not require recombinational repair, and the complex resolution reactions that resolve or reverse recombination intermediates are all processes replete with mechanistic questions that still need answers. There are a number of enzymes that have been implicated in these processes by genetics (RarA, Uup, YoaA, RadA, and RadD, etc.) but which are still in need of much additional functional understanding. Genomic elements are being discovered that trigger the formation of programmed postreplication gaps. Are these elements rare, or are they widespread genomic features? And what is the range of functions that they serve? In general, the field is in need of much additional work. That work is not limited to bacterial DNA replication. Similar processes are increasingly being recognized as important features of DNA replication in eukaryotes (61).
ACKNOWLEDGMENTS
We thank Camille Henry for work on Fig. 4 and Phuong Pham for work on Fig. 9 and 10. We also recognize the substantial contributions of our three reviewers (including Andrei Kuzminov, who revealed his identity), whose comments greatly improved the final form of this article.
Biographies

Michael M. Cox is the Evelyn M. Mercer Professor of Biochemistry at the University of Wisconsin—Madison, where he has worked since 1983. He holds a B.A. in Biology from the University of Delaware. His graduate studies were carried out with William P. Jencks at Brandeis University, completed with a Ph.D. in Biochemistry. Postdoctoral studies were carried out with I. Robert Lehman at Stanford University. For the past 40 years, he has headed a laboratory focused on recombinational DNA repair in bacteria. He is a Fellow of the American Association for the Advancement of Science and of the American Society of Biochemistry and Molecular Biology. He is also Editor in Chief of Critical Reviews in Biochemistry and Molecular Biology and a coauthor of 8 editions of Lehninger Principles in Biochemistry.

Myron F. Goodman was raised in Brooklyn and Queens, NY; attended Queens College and Columbia University Engineering School; and obtained a Ph.D. in Electrical Engineering from Johns Hopkins University in 1968. Following a postdoc with Maurice J. Bessman in Biology at Johns Hopkins University, he was appointed as Assistant Professor in Biological Sciences at the University of Southern California in 1973 and Professor in 1980 and received a joint appointment in Chemistry in 1998. He served as Head of Molecular and Computational Biology from 1998 to 2018. His research centers on the biochemical basis of “hypermutation,” SOS mutagenesis in E. coli involving DNA polymerase V identified in his lab in 1999, and somatic hypermutation of immunoglobulin genes involving activation-induced deoxycytidine deaminase (AID), whose activity on single-stranded DNA was identified in his lab in 2003.

James L. Keck trained at the University of Massachusetts—Amherst, the University of California—Berkeley, and Harvard University and has been a professor of Biomolecular Chemistry at the University of Wisconsin—Madison since 2001. His lab studies the physical mechanisms that support genome maintenance in bacteria, with an emphasis on structure determination and structure-guided biochemical and genetic studies. His lab is currently focused on single-stranded DNA binding proteins, gap repair proteins, DNA replication restart factors, and G-quadruplex processing enzymes.

Antoine van Oijen obtained his undergraduate degree and Ph.D. in the Netherlands, where he was trained as a physicist. He later moved into biology and chemistry, using his multidisciplinary background to develop biophysical tools to visualize and study genomic-maintenance pathways at the single-molecule level. He has run research labs at Harvard Medical School in the United States, Groningen University in the Netherlands, and the University of Wollongong in Australia.

Susan T. Lovett is the Abraham S. and Gertrude Burg Professor of Biology at Brandeis University in Waltham, MA, where she has worked since 1989. She holds a B.A. in Chemistry from Cornell University and a Ph.D. in Molecular Biology from the University of California—Berkeley, studying with A. John Clark. She was a postdoctoral fellow with Bob Mortimer at Lawrence Berkeley Laboratory and with Richard Kolodner at Harvard Medical School. Her current research concerns the mechanisms of homologous recombination, DNA repair, and genomic instability. She is a Fellow of the American Academy of Microbiology, the American Association for the Advancement of Science, the American Academy of Arts and Sciences, and the National Academy of Sciences. Outside Brandeis, she serves as Editor in Chief of the journal EcoSal Plus published by the American Society for Microbiology and is on the editorial board of the Proceedings of the National Academy of Sciences of the United States of America.

Andrew Robinson received his Ph.D. from Macquarie University, Australia, in 2008. He completed postdoctoral appointments with Prof. Nicholas Dixon at the University of Wollongong, Australia, and Prof. Antoine van Oijen at the University of Groningen, the Netherlands, before becoming a Group Leader at the Molecular Horizons Institute, University of Wollongong, Australia. He is now an Honorary Research Fellow at the University of Wollongong. He is an expert in single-molecule fluorescence imaging in live bacterial cells and has used this technique to gain new insight into the mechanisms of DNA replication and repair in bacteria.
REFERENCES
- 1.De Bont R, van Larebeke N. 2004. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T. 1993. Instability and decay of the primary structure of DNA. Nature 362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 3.Sancar A. 1996. DNA excision repair. Annu Rev Biochem 65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 4.Reardon JT, Sancar A. 2005. Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol 79:183–235. doi: 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- 5.Wozniak KJ, Simmons LA. 2022. Bacterial DNA excision repair pathways. Nat Rev Microbiol 20:465–477. doi: 10.1038/s41579-022-00694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stingele J, Bellelli R, Boulton SJ. 2017. Mechanisms of DNA-protein crosslink repair. Nat Rev Mol Cell Biol 18:563–573. doi: 10.1038/nrm.2017.56. [DOI] [PubMed] [Google Scholar]
- 7.Rasouly A, Pani B, Nudler E. 2017. A magic spot in genome maintenance. Trends Genet 33:58–67. doi: 10.1016/j.tig.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 9.Cline SD, Hanawalt PC. 2003. Who’s on first in the cellular response to DNA damage? Nat Rev Mol Cell Biol 4:361–372. doi: 10.1038/nrm1101. [DOI] [PubMed] [Google Scholar]
- 10.Norbury CJ, Hickson ID. 2001. Cellular responses to DNA damage. Annu Rev Pharmacol Toxicol 41:367–401. doi: 10.1146/annurev.pharmtox.41.1.367. [DOI] [PubMed] [Google Scholar]
- 11.Kunkel TA, Erie DA. 2005. DNA mismatch repair. Annu Rev Biochem 74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 12.Kolodner RD, Marsischky GT. 1999. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev 9:89–96. doi: 10.1016/s0959-437x(99)80013-6. [DOI] [PubMed] [Google Scholar]
- 13.Kunkel TA, Erie DA. 2015. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet 49:291–313. doi: 10.1146/annurev-genet-112414-054722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolodner RD. 1995. Mismatch repair—mechanisms and relationship to cancer susceptibility. Trends Biochem Sci 20:397–401. doi: 10.1016/s0968-0004(00)89087-8. [DOI] [PubMed] [Google Scholar]
- 15.Modrich P, Lahue R. 1996. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 16.Jiricny J. 2006. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 17.Kuzminov A. 2001. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci USA 98:8241–8246. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avemann K, Knippers R, Koller T, Sogo JM. 1988. Camptothecin, a specific inhibitor of type I DNA topoisomerase, induces DNA breakage at replication forks. Mol Cell Biol 8:3026–3034. doi: 10.1128/MCB.8.8.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gourlie BB, Pigiet VP. 1983. Poluoma virus minichromosomes: characterization of the products of in vitro DNA synthesis. J Virol 45:585–593. doi: 10.1128/JVI.45.2.585-593.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kowalczykowski SC. 2000. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem Sci 25:156–165. doi: 10.1016/s0968-0004(00)01569-3. [DOI] [PubMed] [Google Scholar]
- 21.Kuzminov A. 1995. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol 16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 22.Kuzminov A. 2001. DNA replication meets genetic exchange: chromosomal damage and its repair by homologous recombination. Proc Natl Acad Sci USA 98:8461–8468. doi: 10.1073/pnas.151260698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson DG, Churchill JJ, Kowalczykowski SC. 1999. A single mutation, RecB(D1080A), eliminates RecA protein loading but not Chi recognition by RecBCD enzyme. J Biol Chem 274:27139–27144. doi: 10.1074/jbc.274.38.27139. [DOI] [PubMed] [Google Scholar]
- 24.Anderson DG, Kowalczykowski SC. 1997. The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a chi-regulated manner. Cell 90:77–86. doi: 10.1016/s0092-8674(00)80315-3. [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharyya B, George NP, Thurmes TM, Zhou R, Jani N, Wessel SR, Sandler SJ, Ha T, Keck JL. 2014. Structural mechanisms of PriA-mediated DNA replication restart. Proc Natl Acad Sci USA 111:1373–1378. doi: 10.1073/pnas.1318001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marians KJ. 2000. PriA-directed replication fork restart in Escherichia coli. Trends Biochem Sci 25:185–189. doi: 10.1016/s0968-0004(00)01565-6. [DOI] [PubMed] [Google Scholar]
- 27.Sandler SJ, Marians KJ. 2000. Role of PriA in replication fork reactivation in Escherichia coli. J Bacteriol 182:9–13. doi: 10.1128/JB.182.1.9-13.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sivaramakrishnan P, Sepulveda LA, Halliday JA, Liu J, Nunez MAB, Golding I, Rosenberg SM, Herman C. 2017. The transcription fidelity factor GreA impedes DNA break repair. Nature 550:214–218. doi: 10.1038/nature23907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shee C, Cox BD, Gu F, Luengas EM, Joshi MC, Chiu L-Y, Magnan D, Halliday JA, Frisch RL, Gibson JL, Nehring RB, Do HG, Hernandez M, Li L, Herman C, Hastings PJ, Bates D, Harris RS, Miller KM, Rosenberg SM. 2013. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. Elife 2:e01222. doi: 10.7554/eLife.01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capaldo FN, Ramsey G, Barbour SD. 1974. Analysis of the growth of recombination-deficient strains of Escherichia coli K-12. J Bacteriol 118:242–249. doi: 10.1128/jb.118.1.242-249.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miranda A, Kuzminov A. 2003. Chromosomal lesion suppression and removal in Escherichia coli via linear DNA degradation. Genetics 163:1255–1271. doi: 10.1093/genetics/163.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha AK, Durand A, Desfontaines JM, Iurchenko I, Auger H, Leach DRF, Barre FX, Michel B. 2017. Division-induced DNA double strand breaks in the chromosome terminus region of Escherichia coli lacking RecBCD DNA repair enzyme. PLoS Genet 13:e1006895. doi: 10.1371/journal.pgen.1006895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michel B, Sinha AK, Leach DRF. 2018. Replication fork breakage and restart in Escherichia coli. Microbiol Mol Biol Rev 82:e00013-18. doi: 10.1128/MMBR.00013-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iliakis G. 1991. The role of DNA double strand breaks in ionizing radiation-induced killing of eukaryotic cells. Bioessays 13:641–648. doi: 10.1002/bies.950131204. [DOI] [PubMed] [Google Scholar]
- 35.Bonura T, Smith KC. 1975. Quantitative evidence for enzymatically induced DNA double-strand breaks as lethal lesions in UV irradiated Pol+ and PolA1 strains of E. coli K-12. Photochem Photobiol 22:243–248. doi: 10.1111/j.1751-1097.1975.tb06743.x. [DOI] [PubMed] [Google Scholar]
- 36.Sakofsky CJ, Saini N, Klimczak LJ, Chan K, Malc EP, Mieczkowski PA, Burkholder AB, Fargo D, Gordenin DA. 2019. Repair of multiple simultaneous double-strand breaks causes bursts of genome-wide clustered hypermutation. PLoS Biol 17:e3000464. doi: 10.1371/journal.pbio.3000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Syeda AH, Hawkins M, McGlynn P. 2014. Recombination and replication. Cold Spring Harb Perspect Biol 6:a016550. doi: 10.1101/cshperspect.a016550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeiranian HA, Schalow BJ, Courcelle CT, Courcelle J. 2013. Fate of the replisome following arrest by UV-induced DNA damage in Escherichia coli. Proc Natl Acad Sci USA 110:11421–11426. doi: 10.1073/pnas.1300624110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeeles JTP, Poli J, Marians KJ, Pasero P. 2013. Rescuing stalled or damaged replication forks. Cold Spring Harb Perspect Biol 5:a012815. doi: 10.1101/cshperspect.a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox MM. 2002. The nonmutagenic repair of broken replication forks via recombination. Mutat Res 510:107–120. doi: 10.1016/s0027-5107(02)00256-7. [DOI] [PubMed] [Google Scholar]
- 41.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. 2000. The importance of repairing stalled replication forks. Nature 404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 42.Gabbai CB, Yeeles JTP, Marians KJ. 2014. Replisome-mediated translesion synthesis and leading strand template lesion skipping are competing bypass mechanisms. J Biol Chem 289:32811–32823. doi: 10.1074/jbc.M114.613257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henrikus SS, Wood EA, McDonald JP, Cox MM, Woodgate R, Goodman MF, van Oije AM, Robinson A. 2018. DNA polymerase IV primarily operates outside of DNA replication forks in Escherichia coli. PLoS Genet 14:e1007161. doi: 10.1371/journal.pgen.1007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikeda M, Furukohri A, Philippin G, Loechler E, Akiyama MT, Katayama T, Fuchs RP, Maki H. 2014. DNA polymerase IV mediates efficient and quick recovery of replication forks stalled at N2-dG adducts. Nucleic Acids Res 42:8461–8472. doi: 10.1093/nar/gku547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henrikus SS, van Oijen AM, Robinson A. 2018. Specialised DNA polymerases in Escherichia coli: roles within multiple pathways. Curr Genet 64:1189–1196. doi: 10.1007/s00294-018-0840-x. [DOI] [PubMed] [Google Scholar]
- 46.Fuchs RP, Fujii S, Wagner J. 2004. Properties and functions of Escherichia coli: Pol IV and Pol V. Adv Protein Chem 69:229–264. doi: 10.1016/S0065-3233(04)69008-5. [DOI] [PubMed] [Google Scholar]
- 47.Kath JE, Chang S, Scotland MK, Wilbertz JH, Jergic S, Dixon NE, Sutton MD, Loparo JJ. 2016. Exchange between Escherichia coli polymerases II and III on a processivity clamp. Nucleic Acids Res 44:1681–1690. doi: 10.1093/nar/gkv1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thrall ES, Kath JE, Chang S, Loparo JJ. 2017. Single-molecule imaging reveals multiple pathways for the recruitment of translesion polymerases after DNA damage. Nat Commun 8:2170. doi: 10.1038/s41467-017-02333-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marians KJ. 2018. Lesion bypass and the reactivation of stalled replication forks. Annu Rev Biochem 87:217–238. doi: 10.1146/annurev-biochem-062917-011921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yeeles JTP, Marians KJ. 2011. The Escherichia coli replisome is inherently DNA damage tolerant. Science 334:235–238. doi: 10.1126/science.1209111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeeles JTP, Marians KJ. 2013. Dynamics of leading-strand lesion skipping by the replisome. Mol Cell 52:855–865. doi: 10.1016/j.molcel.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bainbridge LJ, Teague R, Doherty AJ. 2021. Repriming DNA synthesis: an intrinsic restart pathway that maintains efficient genome replication. Nucleic Acids Res 49:4831–4847. doi: 10.1093/nar/gkab176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heller RC, Marians KJ. 2006. Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439:557–562. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 54.Heller RC, Marians KJ. 2006. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol 7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 55.Conley EC, Muller BM, West SC. 1990. Molecular mechanism of post-replication repair: formation and resolution of recombination intermediates in vitro. Prog Clin Biol Res 340A:121–133. [PubMed] [Google Scholar]
- 56.West SC, Cassuto E, Howard-Flanders P. 1981. Mechanism of E. coli RecA protein directed strand exchanges in post-replication repair of DNA. Nature 294:659–662. doi: 10.1038/294659a0. [DOI] [PubMed] [Google Scholar]
- 57.Yang W, Seidman MM, Rupp WD, Gao Y. 2019. Replisome structure suggests mechanism for continuous fork progression and post-replication repair. DNA Repair (Amst) 81:102658. doi: 10.1016/j.dnarep.2019.102658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63:751–813. doi: 10.1128/MMBR.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lovett ST, Drapkin PT, Sutera VA, Jr, Gluckman-Peskind TJ. 1993. A sister-strand exchange mechanism for RecA-independent deletion of repeated DNA sequences in Escherichia coli. Genetics 135:631–642. doi: 10.1093/genetics/135.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rupp WD, Howard-Flanders P. 1968. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J Mol Biol 31:291–304. doi: 10.1016/0022-2836(68)90445-2. [DOI] [PubMed] [Google Scholar]
- 61.Wong RP, Petriukov K, Ulrich HD. 2021. Daughter-strand gaps in DNA replication—substrates of lesion processing and initiators of distress signalling. DNA Repair (Amst) 105:103163. doi: 10.1016/j.dnarep.2021.103163. [DOI] [PubMed] [Google Scholar]
- 62.Fuchs RP. 2016. Tolerance of lesions in E. coli: chronological competition between translesion synthesis and damage avoidance. DNA Repair (Amst) 44:51–58. doi: 10.1016/j.dnarep.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 63.Laureti L, Demol J, Fuchs RP, Pages V. 2015. Bacterial proliferation: keep dividing and don’t mind the gap. PLoS Genet 11:e1005757. doi: 10.1371/journal.pgen.1005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Naiman K, Pages V, Fuchs RP. 2016. A defect in homologous recombination leads to increased translesion synthesis in E. coli. Nucleic Acids Res 44:7691–7699. doi: 10.1093/nar/gkw488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dutra BE, Sutera VA, Jr, Lovett ST. 2007. RecA-independent recombination is efficient but limited by exonucleases. Proc Natl Acad Sci USA 104:216–221. doi: 10.1073/pnas.0608293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lovett ST. 2017. Template-switching during replication fork repair in bacteria. DNA Repair (Amst) 56:118–128. doi: 10.1016/j.dnarep.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goodman MF, Woodgate R. 2013. Translesion DNA polymerases. Cold Spring Harb Perspect Biol 5:a010363. doi: 10.1101/cshperspect.a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jaszczur M, Bertram JG, Robinson A, van Oijen AM, Woodgate R, Cox MM, Goodman MF. 2016. Mutations for worse or better: low-fidelity DNA synthesis by SOS DNA polymerase V is a tightly regulated double-edged sword. Biochemistry 55:2309–2318. doi: 10.1021/acs.biochem.6b00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barre FX, Soballe B, Michel B, Aroyo M, Robertson M, Sherratt D. 2001. Circles: the replication-recombination-chromosome segregation connection. Proc Natl Acad Sci USA 98:8189–8195. doi: 10.1073/pnas.111008998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Howard-Flanders P. 1975. Repair by genetic recombination in bacteria: overview. Basic Life Sci 5A:265–274. doi: 10.1007/978-1-4684-2895-7_35. [DOI] [PubMed] [Google Scholar]
- 71.Rothman RH, Clark AJ. 1977. The dependence of postreplication repair on uvrB in a recF mutant of Escherichia coli K-12. Mol Gen Genet 155:279–286. doi: 10.1007/BF00272806. [DOI] [PubMed] [Google Scholar]
- 72.Rothman RH, Clark AJ. 1977. Defective excision and postreplication repair of UV-damaged DNA in a recL mutant strain of E. coli K-12. Mol Gen Genet 155:267–277. doi: 10.1007/BF00272805. [DOI] [PubMed] [Google Scholar]
- 73.Rupp WD, Wilde CE, III, Reno DL, Howard-Flanders P. 1971. Exchanges between DNA strands in ultraviolet-irradiated Escherichia coli. J Mol Biol 61:25–44. doi: 10.1016/0022-2836(71)90204-x. [DOI] [PubMed] [Google Scholar]
- 74.Howard-Flanders P, Rupp WD, Wilkins BM, Cole RS. 1968. DNA replication and recombination after UV irradiation. Cold Spring Harb Symp Quant Biol 33:195–207. doi: 10.1101/sqb.1968.033.01.023. [DOI] [PubMed] [Google Scholar]
- 75.Sedgwick SG. 1975. Genetic and kinetic evidence for different types of postreplication repair in Escherichia coli B. J Bacteriol 123:154–161. doi: 10.1128/jb.123.1.154-161.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Youngs DA, Smith KC. 1976. Genetic control of multiple pathways of post-replicational repair in uvrB strains of Escherichia coli K-12. J Bacteriol 125:102–110. doi: 10.1128/jb.125.1.102-110.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim T, Chitteni-Pattu S, Cox BL, Wood EA, Sandler SJ, Cox MM. 2015. Directed evolution of RecA variants with enhanced capacity for conjugational recombination. PLoS Genet 11:e1005278. doi: 10.1371/journal.pgen.1005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cox JM, Li H, Wood EA, Chitteni-Pattu S, Inman RB, Cox MM. 2008. Defective dissociation of a “slow” RecA mutant protein imparts an Escherichia coli growth defect. J Biol Chem 283:24909–24921. doi: 10.1074/jbc.M803934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Okazaki R, Okazaki T, Sakabe K, Sugimoto K, Kainuma R, Sugino A, Iwatsuki N. 1968. In vivo mechanism of DNA chain growth. Cold Spring Harb Symp Quant Biol 33:129–143. doi: 10.1101/SQB.1968.033.01.017. [DOI] [Google Scholar]
- 80.Okazaki T, Okazaki R. 1969. Mechanism of DNA chain growth. 4. Direction of synthesis of T4 short DNA chains as revealed by exonucleolytic degradation. Proc Natl Acad Sci USA 64:1242–1248. doi: 10.1073/pnas.64.4.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kurosawa Y, Okazaki R. 1975. Mechanism of DNA chain growth. 13. Evidence for discontinuous replication of both strands of P2 phage DNA. J Mol Biol 94:229–241. doi: 10.1016/0022-2836(75)90080-7. [DOI] [PubMed] [Google Scholar]
- 82.Cronan GE, Kouzminova EA, Kuzminov A. 2019. Near-continuously synthesized leading strands in Escherichia coli are broken by ribonucleotide excision. Proc Natl Acad Sci USA 116:1251–1260. doi: 10.1073/pnas.1814512116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Konrad EB, Lehman IR. 1975. Novel mutants of Escherichia coli that accumulate very small DNA replicative intermediates. Proc Natl Acad Sci USA 72:2150–2154. doi: 10.1073/pnas.72.6.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tye BK, Chien J, Lehman IR, Duncan BK, Warner HR. 1978. Uracil incorporation: a source of pulse-labeled DNA fragments in the replication of the Escherichia coli chromosome. Proc Natl Acad Sci USA 75:233–237. doi: 10.1073/pnas.75.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tye BK, Lehman IR. 1977. Excision repair of uracil incorporated in DNA as a result of a defect in dUTPase. J Mol Biol 117:293–306. doi: 10.1016/0022-2836(77)90128-0. [DOI] [PubMed] [Google Scholar]
- 86.Tye BK, Nyman PO, Lehman IR, Hochhauser S, Weiss B. 1977. Transient accumulation of Okazaki fragments as a result of uracil incorporation into nascent DNA. Proc Natl Acad Sci USA 74:154–157. doi: 10.1073/pnas.74.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Courcelle J, Hanawalt PC. 2003. RecA-dependent recovery of arrested DNA replication forks. Annu Rev Genet 37:611–646. doi: 10.1146/annurev.genet.37.110801.142616. [DOI] [PubMed] [Google Scholar]
- 88.Hanawalt PC, Cooper PK, Ganesan AK, Smith CA. 1979. DNA repair in bacteria and mammalian cells. Annu Rev Biochem 48:783–836. doi: 10.1146/annurev.bi.48.070179.004031. [DOI] [PubMed] [Google Scholar]
- 89.Bridges BA, Sedgwick SG. 1974. Effect of photoreactivation on the filling of gaps in deoxyribonucleic acid synthesized after exposure of Escherichia coli to ultraviolet light. J Bacteriol 117:1077–1081. doi: 10.1128/jb.117.3.1077-1081.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grompone G, Sanchez N, Ehrlich SD, Michel B. 2004. Requirement for RecFOR-mediated recombination in priA mutant. Mol Microbiol 52:551–562. doi: 10.1111/j.1365-2958.2004.03997.x. [DOI] [PubMed] [Google Scholar]
- 91.Tseng Y-C, Hung J-L, Wang T-CV. 1994. Involvement of RecF pathway recombination genes in postreplication repair in UV-irradiated Escherichia coli cells. Mutat Res 315:1–9. doi: 10.1016/0921-8777(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 92.Wang T-CV, Smith KC. 1983. Mechanisms for recF-dependent and recB-dependent pathways of postreplication repair in UV-irradiated Escherichia coli uvrB. J Bacteriol 156:1093–1098. doi: 10.1128/jb.156.3.1093-1098.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang T-CV, Smith KC. 1984. recF-dependent and recF recB-independent DNA gap-filling repair processes transfer dimer-containing parental strands to daughter strands in Escherichia coli K-12 uvrB. J Bacteriol 158:727–729. doi: 10.1128/jb.158.2.727-729.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang T-CV, Smith KC. 1986. recA (Srf) suppression of recF deficiency in the postreplication repair of UV-irradiated Escherichia coli K-12. J Bacteriol 168:940–946. doi: 10.1128/jb.168.2.940-946.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Webb BL, Cox MM, Inman RB. 1997. Recombinational DNA repair—the RecF and RecR proteins limit the extension of RecA filaments beyond single-strand DNA gaps. Cell 91:347–356. doi: 10.1016/s0092-8674(00)80418-3. [DOI] [PubMed] [Google Scholar]
- 96.Pages V, Fuchs RP. 2003. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science 300:1300–1303. doi: 10.1126/science.1083964. [DOI] [PubMed] [Google Scholar]
- 97.Lehmann AR, Fuchs RP. 2006. Gaps and forks in DNA replication: rediscovering old models. DNA Repair (Amst) 5:1495–1498. doi: 10.1016/j.dnarep.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 98.Courcelle J, Belle JJ, Courcelle CT. 2004. When replication travels on damaged templates: bumps and blocks in the road. Res Microbiol 155:231–237. doi: 10.1016/j.resmic.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 99.Kuzminov A. 2014. The precarious prokaryotic chromosome. J Bacteriol 196:1793–1806. doi: 10.1128/JB.00022-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Michel B, Grompone G, Flores MJ, Bidnenko V. 2004. Multiple pathways process stalled replication forks. Proc Natl Acad Sci USA 101:12783–12788. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kath JE, Jergic S, Heltzel JM, Jacob DT, Dixon NE, Sutton MD, Walker GC, Loparo JJ. 2014. Polymerase exchange on single DNA molecules reveals processivity clamp control of translesion synthesis. Proc Natl Acad Sci USA 111:7647–7652. doi: 10.1073/pnas.1321076111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Septenville AL, Duigou S, Boubakri H, Michel B. 2012. Replication fork reversal after replication-transcription collision. PLoS Genet 8:e1002622. doi: 10.1371/journal.pgen.1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Michel B, Boubakri H, Baharoglu Z, LeMasson M, Lestini R. 2007. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst) 6:967–980. doi: 10.1016/j.dnarep.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 104.Khan SR, Kuzminov A. 2012. Replication forks stalled at ultraviolet lesions are rescued via RecA and RuvABC protein-catalyzed disintegration in Escherichia coli. J Biol Chem 287:6250–6265. doi: 10.1074/jbc.M111.322990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Indiani C, O’Donnell M. 2013. A proposal: source of single strand DNA that elicits the SOS response. Front Biosci (Landmark Ed) 18:312–323. doi: 10.2741/4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao G, Gleave ES, Lamers MH. 2017. Single-molecule studies contrast ordered DNA replication with stochastic translesion synthesis. ELife 6:e32177. doi: 10.7554/eLife.32177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Robinson A, McDonald JP, Caldas VEA, Patel M, Wood EA, Punter CM, Ghodke H, Cox MM, Woodgate R, Goodman MF, van Oijen AM. 2015. Regulation of mutagenic DNA polymerase V activation in space and time. PLoS Genet 11:e1005482. doi: 10.1371/journal.pgen.1005482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ghodke H, Paudel BP, Lewis JS, Jergic S, Gopal K, Romero ZJ, Wood EA, Woodgate R, Cox MM, van Oijen AM. 2019. Spatial and temporal organization of RecA in the Escherichia coli DNA-damage response. Elife 8:e42761. doi: 10.7554/eLife.42761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lopes M, Foiani M, Sogo JM. 2006. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 110.Branzei D, Szakal B. 2017. Building up and breaking down: mechanisms controlling recombination during replication. Crit Rev Biochem Mol Biol 52:381–394. doi: 10.1080/10409238.2017.1304355. [DOI] [PubMed] [Google Scholar]
- 111.Giannattasio M, Zwicky K, Follonier C, Foiani M, Lopes M, Branzei D. 2014. Visualization of recombination-mediated damage bypass by template switching. Nat Struct Mol Biol 21:884–892. doi: 10.1038/nsmb.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D. 2010. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet 6:e1001205. doi: 10.1371/journal.pgen.1001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Adar S, Izhar L, Hendel A, Geacintov N, Livneh Z. 2009. Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res 37:5737–5748. doi: 10.1093/nar/gkp632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boiteux S, Jinks-Robertson S. 2013. DNA repair mechanisms and the bypass of DNA damage in Saccharomyces cerevisiae. Genetics 193:1025–1064. doi: 10.1534/genetics.112.145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Heyer WD, Ehmsen KT, Liu J. 2010. Regulation of homologous recombination in eukaryotes. Annu Rev Genet 44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Edmunds CE, Simpson LJ, Sale JE. 2008. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 117.Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Verspuy J, Gali H, Haracska L, de Wind N. 2009. Mammalian polymerase zeta is essential for post-replication repair of UV-induced DNA lesions. DNA Repair (Amst) 8:1444–1451. doi: 10.1016/j.dnarep.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 118.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. 2009. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev 73:134–154. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Villani G, Hubscher U, Gironis N, Parkkinen S, Pospiech H, Shevelev I, di Cicco G, Markkanen E, Syväoja JE, Tanguy Le Gac N. 2011. In vitro gap-directed translesion DNA synthesis of an abasic site involving human DNA polymerases epsilon, lambda, and beta. J Biol Chem 286:32094–32104. doi: 10.1074/jbc.M111.246611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Diamant N, Hendel A, Vered I, Carell T, Reissner T, de Wind N, Geacinov N, Livneh Z. 2012. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res 40:170–180. doi: 10.1093/nar/gkr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Livneh Z, Ziv O, Shachar S. 2010. Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle 9:729–735. doi: 10.4161/cc.9.4.10727. [DOI] [PubMed] [Google Scholar]
- 122.Livneh Z, Cohen IS, Paz-Elizur T, Davidovsky D, Carmi D, Swain U, Mirlas-Neisberg N. 2016. High-resolution genomic assays provide insight into the division of labor between TLS and HDR in mammalian replication of damaged DNA. DNA Repair (Amst) 44:59–67. doi: 10.1016/j.dnarep.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 123.Quinet A, Vessoni AT, Rocha CRR, Gottifredi V, Biard D, Sarasin A, Menck CFM, Stary A. 2014. Gap-filling and bypass at the replication fork are both active mechanisms for tolerance of low-dose ultraviolet-induced DNA damage in the human genome. DNA Repair (Amst) 14:27–38. doi: 10.1016/j.dnarep.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 124.Becker JR, Pons C, Nguyen HD, Costanzo M, Boone C, Myers CL, Bielinsky A-K. 2015. Genetic interactions implicating postreplicative repair in Okazaki fragment processing. PLoS Genet 11:e1005659. doi: 10.1371/journal.pgen.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Daigaku Y, Etheridge TJ, Nakazawa Y, Nakayama M, Watson AT, Miyabe I, Ogi T, Osborne MA, Carr AM. 2017. PCNA ubiquitylation ensures timely completion of unperturbed DNA replication in fission yeast. PLoS Genet 13:e1006789. doi: 10.1371/journal.pgen.1006789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang K, Weinacht CP, Zhuang Z. 2013. Regulatory role of ubiquitin in eukaryotic DNA translesion synthesis. Biochemistry 52:3217–3228. doi: 10.1021/bi400194r. [DOI] [PubMed] [Google Scholar]
- 127.Callegari AJ, Clark E, Pneuman A, Kelly TJ. 2010. Postreplication gaps at UV lesions are signals for checkpoint activation. Proc Natl Acad Sci USA 107:8219–8224. doi: 10.1073/pnas.1003449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Novarina D, Amara F, Lazzaro F, Plevani P, Muzi-Falconi M. 2011. Mind the gap: keeping UV lesions in check. DNA Repair (Amst) 10:751–759. doi: 10.1016/j.dnarep.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pham P, Shao J, Cox MM, Goodman MF. 2022. Genomic landscape of single-stranded DNA gapped intermediates in Escherichia coli. Nucleic Acids Res 50:937–951. doi: 10.1093/nar/gkab1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Surovtsev IV, Jacobs-Wagner C. 2018. Subcellular organization: a critical feature of bacterial cell replication. Cell 172:1271–1293. doi: 10.1016/j.cell.2018.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dame RT, Rashid FZM, Grainger DC. 2020. Chromosome organization in bacteria: mechanistic insights into genome structure and function. Nat Rev Genet 21:227–242. doi: 10.1038/s41576-019-0185-4. [DOI] [PubMed] [Google Scholar]
- 132.Dorman CJ. 2019. DNA supercoiling and transcription in bacteria: a two-way street. BMC Mol Cell Biol 20:26. doi: 10.1186/s12860-019-0211-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xiang Y, Surovtsev IV, Chang Y, Govers SK, Parry BR, Liu J, Jacobs-Wagner C. 2021. Interconnecting solvent quality, transcription, and chromosome folding in Escherichia coli. Cell 184:3626–3642.e14. doi: 10.1016/j.cell.2021.05.037. [DOI] [PubMed] [Google Scholar]
- 134.Boccard F, Esnault E, Valens M. 2005. Spatial arrangement and macrodomain organization of bacterial chromosomes. Mol Microbiol 57:9–16. doi: 10.1111/j.1365-2958.2005.04651.x. [DOI] [PubMed] [Google Scholar]
- 135.Cagliero C, Grand RS, Jones MB, Jin DJ, O’Sullivan JM. 2013. Genome conformation capture reveals that the Escherichia coli chromosome is organized by replication and transcription. Nucleic Acids Res 41:6058–6071. doi: 10.1093/nar/gkt325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Valens M, Penaud S, Rossignol M, Cornet F, Boccard F. 2004. Macrodomain organization of the Escherichia coli chromosome. EMBO J 23:4330–4341. doi: 10.1038/sj.emboj.7600434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Verma SC, Qian Z, Adhya SL. 2019. Architecture of the Escherichia coli nucleoid. PLoS Genet 15:e1008456. doi: 10.1371/journal.pgen.1008456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. 1994. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev 58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lohman TM, Ferrari ME. 1994. Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and cooperativities. Annu Rev Biochem 63:527–570. doi: 10.1146/annurev.bi.63.070194.002523. [DOI] [PubMed] [Google Scholar]
- 140.Meyer RR, Glassberg J, Kornberg A. 1979. An Escherichia coli mutant defective in single-strand binding protein is defective in DNA replication. Proc Natl Acad Sci USA 76:1702–1705. doi: 10.1073/pnas.76.4.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Meyer RR, Laine PS. 1990. The single-stranded DNA-binding protein of Escherichia coli. Microbiol Rev 54:342–380. doi: 10.1128/mr.54.4.342-380.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Molineux IJ, Gefter ML. 1975. Properties of the Escherichia coli DNA-binding (unwinding) protein interaction with nucleolytic enzymes and DNA. J Mol Biol 98:811–825. doi: 10.1016/s0022-2836(75)80012-x. [DOI] [PubMed] [Google Scholar]
- 143.Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL. 2008. SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol 43:289–318. doi: 10.1080/10409230802341296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mackay V, Linn S. 1976. Selective inhibition of the DNase activity of the recBC enzyme by the DNA binding protein from Escherichia coli. J Biol Chem 251:3716–3719. doi: 10.1016/S0021-9258(17)33402-6. [DOI] [PubMed] [Google Scholar]
- 145.Meyer RR, Glassberg J, Scott JV, Kornberg A. 1980. A temperature-sensitive single-stranded DNA-binding protein from Escherichia coli. J Biol Chem 255:2897–2901. doi: 10.1016/S0021-9258(19)85824-6. [DOI] [PubMed] [Google Scholar]
- 146.Reyes-Lamothe R, Sherratt DJ, Leake MC. 2010. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science 328:498–501. doi: 10.1126/science.1185757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alberts B, Morris CF, Mace D, Sinha N, Bittner M, Moran L. 1975. Reconstruction of the T4 bacteriophage DNA replication apparatus from purified components, p 241–269. In Goulian M, Hanawalt PC, Fox CF (ed), DNA synthesis and its regulation. WA Benjamin, Menlo Park, CA. [Google Scholar]
- 148.Chrysogelos S, Griffith J. 1982. Escherichia coli single-strand binding protein organizes single-stranded DNA in nucleosome-like units. Proc Natl Acad Sci USA 79:5803–5807. doi: 10.1073/pnas.79.19.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hobbs MD, Sakai A, Cox MM. 2007. SSB protein limits RecOR binding onto single-stranded DNA. J Biol Chem 282:11058–11067. doi: 10.1074/jbc.M611007200. [DOI] [PubMed] [Google Scholar]
- 150.Bell JC, Liu B, Kowalczykowski SC. 2015. Imaging and energetics of single SSB-ssDNA molecules reveal intramolecular condensation and insight into RecOR function. Elife 4:e08646. doi: 10.7554/eLife.08646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Costes A, Lecointe F, McGovern S, Quevillon-Cheruel S, Polard P. 2010. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet 6:e1001238. doi: 10.1371/journal.pgen.1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ryzhikov M, Koroleva O, Postnov D, Tran A, Korolev S. 2011. Mechanism of RecO recruitment to DNA by single-stranded DNA binding protein. Nucleic Acids Res 39:6305–6314. doi: 10.1093/nar/gkr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shinn MK, Kozlov AG, Nguyen B, Bujalowski WM, Lohman TM. 2019. Are the intrinsically disordered linkers involved in SSB binding to accessory proteins? Nucleic Acids Res 47:8581–8594. doi: 10.1093/nar/gkz606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hwang J, Kim J-Y, Kim C, Park S, Joo S, Kim SK, Lee NK. 2020. Single-molecule observation of ATP-independent SSB displacement by RecO in Deinococcus radiodurans. Elife 9:e50945. doi: 10.7554/eLife.50945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Inoue J, Nagae T, Mishima M, Ito Y, Shibata T, Mikawa T. 2011. A mechanism for single-stranded DNA-binding protein (SSB) displacement from single-stranded DNA upon SSB-RecO interaction. J Biol Chem 286:6720–6732. doi: 10.1074/jbc.M110.164210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shereda RD, Bernstein DA, Keck JL. 2007. A central role for SSB in Escherichia coli RecQ DNA helicase function. J Biol Chem 282:19247–19258. doi: 10.1074/jbc.M608011200. [DOI] [PubMed] [Google Scholar]
- 157.Shereda RD, Reiter NJ, Butcher SE, Keck JL. 2009. Identification of the SSB binding site on E. coli RecQ reveals a conserved surface for binding SSB’s C-terminus. J Mol Biol 386:612–625. doi: 10.1016/j.jmb.2008.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Suski C, Marians KJ. 2008. Resolution of converging replication forks by RecQ and topoisomerase III. Mol Cell 30:779–789. doi: 10.1016/j.molcel.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Harami GM, Kovacs ZJ, Pancsa R, Palinkas J, Barath V, Tarnok K, Malnasi-Csizmadia A, Kovacs M. 2020. Phase separation by ssDNA binding protein controlled via protein-protein and protein-DNA interactions. Proc Natl Acad Sci USA 117:26206–26217. doi: 10.1073/pnas.2000761117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Butland G, Peregrin-Alvarez MJ, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, Davey M, Parkinson J, Greenblatt J, Emili A. 2005. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 161.Buss JA, Kimura Y, Bianco PR. 2008. RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res 36:7029–7042. doi: 10.1093/nar/gkn795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Osorio Garcia MA, Wood EA, Keck JL, Cox MM. 2023. Interaction with single-stranded DNA-binding protein (SSB) modulates Escherichia coli RadD DNA repair activities. J Biol Chem 2023:104773. doi: 10.1016/j.jbc.2023.104773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen SH, Byrne-Nash RT, Cox MM. 2016. Escherichia coli RadD protein functionally interacts with the single-stranded DNA-binding protein. J Biol Chem 291:20779–20786. doi: 10.1074/jbc.M116.736223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Page AN, George NP, Marceau AH, Cox MM, Keck JL. 2011. Structure and biochemical activities of Escherichia coli MgsA. J Biol Chem 286:12075–12085. doi: 10.1074/jbc.M110.210187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Han ES, Cooper DL, Persky NS, Sutera VA, Jr, Whitaker RD, Montello ML, Lovett ST. 2006. RecJ exonuclease: substrates, products and interaction with SSB. Nucleic Acids Res 34:1084–1091. doi: 10.1093/nar/gkj503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wessel SR, Marceau AH, Massoni SC, Zhou R, Ha T, Sandler SJ, Keck JL. 2013. PriC-mediated DNA replication restart requires PriC complex formation with the single-stranded DNA-binding protein. J Biol Chem 288:17569–17578. doi: 10.1074/jbc.M113.478156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fujii S, Isogawa A, Fuchs RP. 2018. Chronological switch from translesion synthesis to homology-dependent gap repair in vivo. Toxicol Res 34:297–302. doi: 10.5487/TR.2018.34.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rudolph CJ, Upton AL, Lloyd RG. 2007. Replication fork stalling and cell cycle arrest in UV-irradiated Escherichia coli. Genes Dev 21:668–681. doi: 10.1101/gad.417607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Langston LD, O’Donnell M. 2006. DNA replication: keep moving and don’t mind the gap. Mol Cell 23:155–160. doi: 10.1016/j.molcel.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 170.Campbell JL, Kleckner N. 1990. Escherichia coli OriC and the dnaA gene promoter are sequestered from Dam methyltransferase following the passage of the chromosomal replication fork. Cell 62:967–979. doi: 10.1016/0092-8674(90)90271-f. [DOI] [PubMed] [Google Scholar]
- 171.Chow K-H, Courcelle J. 2004. RecO acts with RecF and RecR to protect and maintain replication forks blocked by UV-induced DNA damage in Escherichia coli. J Biol Chem 279:3492–3496. doi: 10.1074/jbc.M311012200. [DOI] [PubMed] [Google Scholar]
- 172.Courcelle J, Carswell-Crumpton C, Hanawalt P. 1997. recF and recR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc Natl Acad Sci USA 94:3714–3719. doi: 10.1073/pnas.94.8.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Henrikus SS, Henry C, Ghodke H, Wood EA, Mbele N, Saxena R, Upasana B, van Oijen AM, Cox MM, Robinson A. 2019. RecFOR epistasis group: RecF and RecO have distinct localizations and functions in Escherichia coli. Nucleic Acids Res 47:2946–2965. doi: 10.1093/nar/gkz003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sandler SJ. 1996. Overlapping functions for recF and priA in cell viability and UV-inducible SOS expression are distinguished by dnaC809 in Escherichia coli K-12. Mol Microbiol 19:871–880. doi: 10.1046/j.1365-2958.1996.429959.x. [DOI] [PubMed] [Google Scholar]
- 175.Lloyd RG, Buckman C. 1995. Conjugational recombination in Escherichia coli: genetic analysis of recombinant formation in Hfr × F− crosses. Genetics 139:1123–1148. doi: 10.1093/genetics/139.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Romero ZJ, Armstrong TJ, Henrikus SS, Chen SH, Glass DJ, Ferrazzoli AE, Wood EA, Chitteni-Pattu S, van Oijen AM, Lovett ST, Robinson A, Cox MM. 2020. Frequent template switching in postreplication gaps: suppression of deleterious consequences by the Escherichia coli Uup and RadD proteins. Nucleic Acids Res 48:212–230. doi: 10.1093/nar/gkz960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Romero ZJ, Chen SH, Armstrong T, Wood EA, van Oijen A, Robinson A, Cox MM. 2020. Resolving toxic DNA repair intermediates in every E. coli replication cycle: critical roles for RecG, Uup and RadD. Nucleic Acids Res 48:8445–8460. doi: 10.1093/nar/gkaa579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cooper DL, Boyle DC, Lovett ST. 2015. Genetic analysis of Escherichia coli RadA: functional motifs and genetic interactions. Mol Microbiol 95:769–779. doi: 10.1111/mmi.12899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jain K, Wood EA, Cox MM. 2021. The rarA gene as part of an expanded RecFOR recombination pathway: negative epistasis and synthetic lethality with ruvB, recG, and recQ. PLoS Genet 17:e1009972. doi: 10.1371/journal.pgen.1009972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Fonville NC, Blankschien MD, Magner DB, Rosenberg SM. 2010. RecQ-dependent death-by-recombination in cells lacking RecG and UvrD. DNA Repair (Amst) 9:403–413. doi: 10.1016/j.dnarep.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mazin AV, Kuzminov AV, Dianov GL, Salganik RI. 1991. Mechanisms of deletion formation in Escherichia coli plasmids. 2. Deletions mediated by short direct repeats. Mol Gen Genet 228:209–214. doi: 10.1007/BF00282467. [DOI] [PubMed] [Google Scholar]
- 182.Bi X, Liu LF. 1994. RecA-independent and RecA-dependent intramolecular plasmid recombination. Differential homology requirement and distance effect. J Mol Biol 235:414–423. doi: 10.1006/jmbi.1994.1002. [DOI] [PubMed] [Google Scholar]
- 183.Bi X, Liu LF. 1996. A replicational model for DNA recombination between direct repeats. J Mol Biol 256:849–858. doi: 10.1006/jmbi.1996.0131. [DOI] [PubMed] [Google Scholar]
- 184.Dianov GL, Kuzminov AV, Mazin AV, Salganik RI. 1991. Molecular mechanisms of deletion formation in Escherichia coli plasmids. 1. Deletion formation mediated by long direct repeats. Mol Gen Genet 228:153–159. doi: 10.1007/BF00282460. [DOI] [PubMed] [Google Scholar]
- 185.Goldfless SJ, Morag AS, Belisle KA, Sutera VA, Jr, Lovett ST. 2006. DNA repeat rearrangements mediated by DnaK-dependent replication fork repair. Mol Cell 21:595–604. doi: 10.1016/j.molcel.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 186.Lovett ST, Gluckman TJ, Simon PJ, Sutera VA, Jr, Drapkin PT. 1994. Recombination between repeats in Escherichia coli by a RecA-independent, proximity-sensitive mechanism. Mol Gen Genet 245:294–300. doi: 10.1007/BF00290109. [DOI] [PubMed] [Google Scholar]
- 187.Morag AS, Saveson CJ, Lovett ST. 1999. Expansion of DNA repeats in Escherichia coli: effects of recombination and replication functions. J Mol Biol 289:21–27. doi: 10.1006/jmbi.1999.2763. [DOI] [PubMed] [Google Scholar]
- 188.Wang T-CV, Chen S-H. 1992. Similar sized daughter-strand gaps are produced in the leading and lagging strands of DNA in UV-irradiated Escherichia coli uvrA cells. Biochem Biophys Res Commun 184:1496–1503. doi: 10.1016/s0006-291x(05)80052-x. [DOI] [PubMed] [Google Scholar]
- 189.Iyer VN, Rupp WD. 1971. Usefulness of benzoylated naphthoylated DEAE-cellulose to distinguish and fractionate double-stranded DNA bearing different extents of single-stranded regions. Biochim Biophys Acta 228:117–126. doi: 10.1016/0005-2787(71)90551-x. [DOI] [PubMed] [Google Scholar]
- 190.Gregg AV, McGlynn P, Jaktaji RP, Lloyd RG. 2002. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol Cell 9:241–251. doi: 10.1016/s1097-2765(02)00455-0. [DOI] [PubMed] [Google Scholar]
- 191.Lloyd RG, Rudolph CJ. 2016. 25 years on and no end in sight: a perspective on the role of RecG protein. Curr Genet 62:827–840. doi: 10.1007/s00294-016-0589-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.McGlynn P, Lloyd RG. 2001. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci USA 98:8227–8234. doi: 10.1073/pnas.111008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Singleton MR, Scaife S, Wigley DB. 2001. Structural analysis of DNA replication fork reversal by RecG. Cell 107:79–89. doi: 10.1016/s0092-8674(01)00501-3. [DOI] [PubMed] [Google Scholar]
- 194.Whitby MC, Lloyd RG. 1998. Targeting Holliday junctions by the RecG branch migration protein of Escherichia coli. J Biol Chem 273:19729–19739. doi: 10.1074/jbc.273.31.19729. [DOI] [PubMed] [Google Scholar]
- 195.Whitby MC, Ryder L, Lloyd RG. 1993. Reverse branch migration of Holliday junctions by RecG protein: a new mechanism for resolution of intermediates in recombination and DNA repair. Cell 75:341–350. doi: 10.1016/0092-8674(93)80075-p. [DOI] [PubMed] [Google Scholar]
- 196.Warren GM, Stein RA, McHaourab HS, Eichman BF. 2018. Movement of the RecG motor domain upon DNA binding is required for efficient fork reversal. Int J Mol Sci 19:3049. doi: 10.3390/ijms19103049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Weaver GM, Mettrick KA, Corocher T-A, Graham A, Grainge I. 2019. Replication fork collapse at a protein-DNA roadblock leads to fork reversal, promoted by the RecQ helicase. Mol Microbiol 111:455–472. doi: 10.1111/mmi.14166. [DOI] [PubMed] [Google Scholar]
- 198.Bonde NJ, Romero ZJ, Chitteni-Pattu S, Cox MM. 2022. RadD is a RecA-dependent accessory protein that accelerates DNA strand exchange. Nucleic Acids Res 50:2201–2210. doi: 10.1093/nar/gkac041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cooper DL, Lovett ST. 2016. Recombinational branch migration by the RadA/Sms paralog of RecA in Escherichia coli. Elife 5:e10807. doi: 10.7554/eLife.10807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Centore RC, Leeson MC, Sandler SJ. 2009. UvrD303, a hyperhelicase mutant that antagonizes RecA-dependent SOS expression by a mechanism that depends on its C terminus. J Bacteriol 191:1429–1438. doi: 10.1128/JB.01415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Centore RC, Sandler SJ. 2007. UvrD limits the number and intensities of RecA-green fluorescent protein structures in Escherichia coli K-12. J Bacteriol 189:2915–2920. doi: 10.1128/JB.01777-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cox MM. 2007. Regulation of bacterial RecA function. Crit Rev Biochem Mol Biol 42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- 203.Petrova V, Chen SH, Molzberger ET, Tomko EJ, Chitteni-Pattu S, Jia H, Ordabayev Y, Lohman TM, Cox MM. 2015. Active displacement of RecA filaments by UvrD translocase activity. Nucleic Acids Res 43:4133–4149. doi: 10.1093/nar/gkv186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Veaute X, Delmas P, Selva M, Jeusset J, Le Cam E, Matic I, Fabre F, Petit MA. 2005. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J 24:180–189. doi: 10.1038/sj.emboj.7600485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Rudolph CJ, Upton AL, Lloyd RG. 2009. Replication fork collisions cause pathological chromosomal amplification in cells lacking RecG DNA translocase. Mol Microbiol 74:940–955. doi: 10.1111/j.1365-2958.2009.06909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Rudolph CJ, Upton AL, Harris L, Lloyd RG. 2009. Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol 73:352–366. doi: 10.1111/j.1365-2958.2009.06773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rudolph CJ, Mahdi AA, Upton AL, Lloyd RG. 2010. RecG protein and single-strand DNA exonucleases avoid cell lethality associated with PriA helicase activity in Escherichia coli. Genetics 186:473–492. doi: 10.1534/genetics.110.120691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Henry C, Henrikus SS. 2021. Elucidating recombination mediator function using biophysical tools. Biology (Basel) 10:288. doi: 10.3390/biology10040288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dubiel K, Henry C, Spenkelink LM, Kozlov AG, Wood EA, Jergic S, Dixon NE, van Oijen AM, Cox MM, Lohman TM, Sandler SJ, Keck JL. 2020. Development of a single-stranded DNA-binding protein fluorescent fusion toolbox. Nucleic Acids Res 48:6053–6067. doi: 10.1093/nar/gkaa320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Xia J, Chen L-T, Mei Q, Ma C-H, Halliday JA, Lin H-Y, Magnan D, Pribis JP, Fitzgerald DM, Hamilton HM, Richters M, Nehring RB, Shen X, Li L, Bates D, Hastings PJ, Herman C, Jayaram M, Rosenberg SM. 2016. Holliday junction trap shows how cells use recombination and a junction-guardian role of RecQ helicase. Sci Adv 2:e1601605. doi: 10.1126/sciadv.1601605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li G-W, Burkhardt D, Gross C, Weissman JS. 2014. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Laureti L, Lee L, Philippin G, Kahi M, Pages V. 2022. Single strand gap repair: the presynaptic phase plays a pivotal role in modulating lesion tolerance pathways. PLoS Genet 18:e1010238. doi: 10.1371/journal.pgen.1010238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Soubry N, Wang A, Reyes-Lamothe R. 2019. Replisome activity slowdown after exposure to ultraviolet light in Escherichia coli. Proc Natl Acad Sci USA 116:11747–11753. doi: 10.1073/pnas.1819297116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Altshuler M. 1993. Recovery of DNA replication in UV-damaged Escherichia coli. Mutat Res 294:91–100. doi: 10.1016/0921-8777(93)90017-b. [DOI] [PubMed] [Google Scholar]
- 215.Bichara M, Meier M, Wagner J, Cordonnier A, Lambert IB. 2011. Postreplication repair mechanisms in the presence of DNA adducts in Escherichia coli. Mutat Res 727:104–122. doi: 10.1016/j.mrrev.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 216.Kuzminov A. 1993. RuvA, RuvB and RuvC proteins: cleaning-up after recombinational repairs in E. coli. Bioessays 15:355–358. doi: 10.1002/bies.950150511. [DOI] [PubMed] [Google Scholar]
- 217.Smith KC. 2004. Recombinational DNA repair: the ignored repair systems. Bioessays 26:1322–1326. doi: 10.1002/bies.20109. [DOI] [PubMed] [Google Scholar]
- 218.Smith KC, Wang T-CV, Sharma RC. 1987. recA-dependent DNA repair in UV-irradiated Escherichia coli. J Photochem Photobiol B 1:1–11. doi: 10.1016/1011-1344(87)80002-7. [DOI] [PubMed] [Google Scholar]
- 219.Tomer G, Cohen-Fix O, O’Donnell M, Goodman M, Livneh Z. 1996. Reconstitution of repair-gap UV mutagenesis with purified proteins from Escherichia coli: a role for DNA polymerases III and II. Proc Natl Acad Sci USA 93:1376–1380. doi: 10.1073/pnas.93.4.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lawaree E, Jankevicius G, Cooper C, Ahel I, Uphoff S, Tang CM. 2020. DNA ADP-ribosylation stalls replication and is reversed by RecF-mediated homologous recombination and nucleotide excision repair. Cell Rep 30:1373–1384.e4. doi: 10.1016/j.celrep.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wilson DM, Sofinowski TM, McNeill DR. 2003. Repair mechanisms for oxidative DNA damage. Front Biosci 8:D963–D981. doi: 10.2741/1109. [DOI] [PubMed] [Google Scholar]
- 222.Henry C, Loiseau L, Vergnes A, Vertommen D, Merida-Floriano A, Chitteni-Pattu S, Wood EA, Casadesus J, Cox MM, Barras F, Ezraty B. 2021. Redox controls RecA protein activity via reversible oxidation of its methionine residues. Elife 10:e63747. doi: 10.7554/eLife.63747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Demple B. 1990. Oxidative DNA damage: repair and inducible cellular responses. Prog Clin Biol Res 340A:155–167. [PubMed] [Google Scholar]
- 224.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Slupphaug G, Kavli B, Krokan HE. 2003. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res 531:231–251. doi: 10.1016/j.mrfmmm.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 226.Cooper DL, Lovett ST. 2011. Toxicity and tolerance mechanisms for azidothymidine, a replication gap-promoting agent, in Escherichia coli. DNA Repair (Amst) 10:260–270. doi: 10.1016/j.dnarep.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Shida T, Noda M, Sekiguchi J. 1996. Cleavage of single- and double-stranded DNAs containing an abasic residue by Escherichia coli exonuclease III (AP endonuclease VI). Nucleic Acids Res 24:4572–4576. doi: 10.1093/nar/24.22.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Demple B, Johnson A, Fung D. 1986. Exonuclease III and endonuclease IV remove 3′ blocks from DNA synthesis primers in H2O2-damaged Escherichia coli. Proc Natl Acad Sci USA 83:7731–7735. doi: 10.1073/pnas.83.20.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Khanam T, Rai N, Ramachandran R. 2015. Mycobacterium tuberculosis class II apurinic/apyrimidinic-endonuclease/3′-5′ exonuclease III exhibits DNA regulated modes of interaction with the sliding DNA beta-clamp. Mol Microbiol 98:46–68. doi: 10.1111/mmi.13102. [DOI] [PubMed] [Google Scholar]
- 230.Brown LT, Sutera VA, Jr, Zhou S, Weitzel CS, Cheng Y, Lovett ST. 2015. Connecting replication and repair: YoaA, a helicase-related protein, promotes azidothymidine tolerance through association with Chi, an accessory clamp loader protein. PLoS Genet 11:e1005651. doi: 10.1371/journal.pgen.1005651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Curth U, Genschel J, Urbanke C, Greipel J. 1996. In vitro and in vivo function of the C-terminus of Escherichia coli single-stranded DNA binding protein. Nucleic Acids Res 24:2706–2711. doi: 10.1093/nar/24.14.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Bernstein DA, Eggington JM, Killoran MP, Misic AM, Cox MM, Keck JL. 2004. Crystal structure of the D. radiodurans single-stranded DNA binding protein suggests a novel mechanism for coping with DNA damage. Proc Natl Acad Sci USA 101:8575–8580. doi: 10.1073/pnas.0401331101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Matsumoto T, Morimoto Y, Shibata N, Kinebuchi T, Shimamoto N, Tsukihara T, Yasuoka N. 2000. Roles of functional loops and the C-terminal segment of a single-stranded DNA binding protein elucidated by X-ray structure analysis. J Biochem 127:329–335. doi: 10.1093/oxfordjournals.jbchem.a022611. [DOI] [PubMed] [Google Scholar]
- 234.Raghunathan S, Kozlov AG, Lohman TM, Waksman G. 2000. Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat Struct Biol 7:648–652. doi: 10.1038/77943. [DOI] [PubMed] [Google Scholar]
- 235.Raghunathan S, Ricard CS, Lohman TM, Waksman G. 1997. Crystal structure of the homo-tetrameric DNA binding domain of Escherichia coli single-stranded DNA-binding protein determined by multiwavelength X-ray diffraction on the selenomethionyl protein at 2.9-Å resolution. Proc Natl Acad Sci USA 94:6652–6657. doi: 10.1073/pnas.94.13.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Savvides SN, Raghunathan S, Futterer K, Kozlov AG, Lohman TM, Waksman G. 2004. The C-terminal domain of full-length E. coli SSB is disordered even when bound to DNA. Protein Sci 13:1942–1947. doi: 10.1110/ps.04661904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Chan K-W, Lee Y-J, Wang C-H, Huang H, Sun Y-J. 2009. Single-stranded DNA-binding protein complex from Helicobacter pylori suggests an ssDNA-binding surface. J Mol Biol 388:508–519. doi: 10.1016/j.jmb.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 238.Huang Y-H, Guan H-H, Chen C-J, Huang C-Y. 2017. Staphylococcus aureus single-stranded DNA-binding protein SsbA can bind but cannot stimulate PriA helicase. PLoS One 12:e0182060. doi: 10.1371/journal.pone.0182060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Jedrzejczak R, Dauter M, Dauter Z, Olszewski M, Długołecka A, Kur J. 2006. Structure of the single-stranded DNA-binding protein SSB from Thermus aquaticus. Acta Crystallogr D Biol Crystallogr 62:1407–1412. doi: 10.1107/S0907444906036031. [DOI] [PubMed] [Google Scholar]
- 240.Kaushal PS, Singh P, Sharma A, Muniyappa K, Vijayan M. 2010. X-ray and molecular-dynamics studies on Mycobacterium leprae single-stranded DNA-binding protein and comparison with other eubacterial SSB structures. Acta Crystallogr D Biol Crystallogr 66:1048–1058. doi: 10.1107/S0907444910032208. [DOI] [PubMed] [Google Scholar]
- 241.Saikrishnan K, Jeyakanthan J, Venkatesh J, Acharya N, Sekar K, Varshney U, Vijayan M. 2003. Structure of Mycobacterium tuberculosis single-stranded DNA-binding protein. Variability in quaternary structure and its implications. J Mol Biol 331:385–393. doi: 10.1016/s0022-2836(03)00729-0. [DOI] [PubMed] [Google Scholar]
- 242.Lohman TM, Overman LB, Datta S. 1986. Salt-dependent changes in the DNA binding co-operativity of Escherichia coli single strand binding protein. J Mol Biol 187:603–615. doi: 10.1016/0022-2836(86)90338-4. [DOI] [PubMed] [Google Scholar]
- 243.Bujalowski W, Lohman TM. 1987. Limited co-operativity in protein-nucleic acid interactions. A thermodynamic model for the interactions of Escherichia coli single strand binding protein with single-stranded nucleic acids in the “beaded,” (SSB)65 mode. J Mol Biol 195:897–907. doi: 10.1016/0022-2836(87)90493-1. [DOI] [PubMed] [Google Scholar]
- 244.Bujalowski W, Lohman TM. 1989. Negative co-operativity in Escherichia coli single strand binding protein-oligonucleotide interactions. II. Salt, temperature and oligonucleotide length effects. J Mol Biol 207:269–288. doi: 10.1016/0022-2836(89)90455-5. [DOI] [PubMed] [Google Scholar]
- 245.Bujalowski W, Lohman TM. 1989. Negative co-operativity in Escherichia coli single strand binding protein-oligonucleotide interactions. I. Evidence and a quantitative model. J Mol Biol 207:249–268. doi: 10.1016/0022-2836(89)90454-3. [DOI] [PubMed] [Google Scholar]
- 246.Kozlov AG, Shinn MK, Weiland EA, Lohman TM. 2017. Glutamate promotes SSB protein-protein interactions via intrinsically disordered regions. J Mol Biol 429:2790–2801. doi: 10.1016/j.jmb.2017.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Kozlov AG, Weiland E, Mittal A, Waldman V, Antony E, Fazio N, Pappu RV, Lohman TM. 2015. Intrinsically disordered C-terminal tails of E. coli single-stranded DNA binding protein regulate cooperative binding to single-stranded DNA. J Mol Biol 427:763–774. doi: 10.1016/j.jmb.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kozlov AG, Cheng X, Zhang H, Shinn MK, Weiland E, Nguyen B, Shkel IA, Zytkiewicz E, Finkelstein IJ, Record MT, Jr, Lohman TM. 2022. How glutamate promotes liquid-liquid phase separation and DNA binding cooperativity of E. coli SSB protein. J Mol Biol 434:167562. doi: 10.1016/j.jmb.2022.167562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Reyes-Lamothe R, Nicolas E, Sherratt DJ. 2012. Chromosome replication and segregation in bacteria. Annu Rev Genet 46:121–143. doi: 10.1146/annurev-genet-110711-155421. [DOI] [PubMed] [Google Scholar]
- 250.Reyes-Lamothe R, Possoz C, Danilova O, Sherratt DJ. 2008. Independent positioning and action of Escherichia coli replisomes in live cells. Cell 133:90–102. doi: 10.1016/j.cell.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Spenkelink LM, Lewis JS, Jergic S, Xu Z-Q, Robinson A, Dixon NE, van Oijen AM. 2019. Recycling of single-stranded DNA-binding protein by the bacterial replisome. Nucleic Acids Res 47:4111–4123. doi: 10.1093/nar/gkz090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Zhao T, Liu Y, Wang Z, He R, Zhang JX, Xu F, Lei M, Deci MB, Nguyen J, Bianco PR. 2019. Super-resolution imaging reveals changes in Escherichia coli SSB localization in response to DNA damage. Genes Cells 24:814–826. doi: 10.1111/gtc.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Horii Z, Clark AJ. 1973. Genetic analysis of the RecF pathway to genetic recombination in Escherichia coli K12: isolation and characterization of mutants. J Mol Biol 80:327–344. doi: 10.1016/0022-2836(73)90176-9. [DOI] [PubMed] [Google Scholar]
- 254.Kolodner R, Fishel RA, Howard M. 1985. Genetic recombination of bacterial plasmid DNA: effect of RecF pathway mutations on plasmid recombination in Escherichia coli. J Bacteriol 163:1060–1066. doi: 10.1128/jb.163.3.1060-1066.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Mahdi AA, Lloyd RG. 1989. The recR locus of Escherichia coli K-12: molecular cloning, DNA sequencing and identification of the gene product. Nucleic Acids Res 17:6781–6794. doi: 10.1093/nar/17.17.6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Mahdi AA, Lloyd RG. 1989. Identification of the recR locus of Escherichia coli K-12 and analysis of its role in recombination and DNA repair. Mol Gen Genet 216:503–510. doi: 10.1007/BF00334397. [DOI] [PubMed] [Google Scholar]
- 257.Lovett ST, Hurley RL, Sutera VA, Jr, Aubuchon RH, Lebedeva MA. 2002. Crossing over between regions of limited homology in Escherichia coli: RecA-dependent and RecA-independent pathways. Genetics 160:851–859. doi: 10.1093/genetics/160.3.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Morimatsu K, Kowalczykowski SC. 2003. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol Cell 11:1337–1347. doi: 10.1016/s1097-2765(03)00188-6. [DOI] [PubMed] [Google Scholar]
- 259.Sakai A, Cox MM. 2009. RecFOR and RecOR as distinct RecA loading pathways. J Biol Chem 284:3264–3272. doi: 10.1074/jbc.M807220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Umezu K, Chi NW, Kolodner RD. 1993. Biochemical interaction of the Escherichia coli RecF, RecO, and RecR proteins with RecA protein and single-stranded DNA binding protein. Proc Natl Acad Sci USA 90:3875–3879. doi: 10.1073/pnas.90.9.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Umezu K, Kolodner RD. 1994. Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J Biol Chem 269:30005–30013. doi: 10.1016/S0021-9258(18)43981-6. [DOI] [PubMed] [Google Scholar]
- 262.Madiraju MV, Clark AJ. 1992. Evidence for ATP binding and double-stranded DNA binding by Escherichia coli RecF protein. J Bacteriol 174:7705–7710. doi: 10.1128/jb.174.23.7705-7710.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Makharashvili N, Mi T, Koroleva O, Korolev S. 2009. RecR-mediated modulation of RecF dimer specificity for single- and double-stranded DNA. J Biol Chem 284:1425–1434. doi: 10.1074/jbc.M806378200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Tang Q, Liu Y-P, Shan H-H, Tian L-F, Zhang J-Z, Yan X-X. 2018. ATP-dependent conformational change in ABC-ATPase RecF serves as a switch in DNA repair. Sci Rep 8:2127. doi: 10.1038/s41598-018-20557-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Henry C, Mbele N, Cox MM. 1 May 2023. RecF protein targeting to postreplication (daughter strand) gaps I: DNA binding by RecF and RecFR. Nucleic Acids Res . doi: 10.1093/nar/gkad311. [DOI] [PMC free article] [PubMed]
- 266.Griffin TJ, IV, Kolodner RD. 1990. Purification and preliminary characterization of the Escherichia coli K-12 RecF protein. J Bacteriol 172:6291–6299. doi: 10.1128/jb.172.11.6291-6299.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Webb BL, Cox MM, Inman RB. 1995. An interaction between the Escherichia coli RecF and RecR proteins dependent on ATP and double-stranded DNA. J Biol Chem 270:31397–31404. doi: 10.1074/jbc.270.52.31397. [DOI] [PubMed] [Google Scholar]
- 268.Webb BL, Cox MM, Inman RB. 1999. ATP hydrolysis and DNA binding by the Escherichia coli RecF protein. J Biol Chem 274:15367–15374. doi: 10.1074/jbc.274.22.15367. [DOI] [PubMed] [Google Scholar]
- 269.Koroleva O, Makharashvili N, Courcelle CT, Courcelle J, Korolev S. 2007. Structural conservation of RecF and Rad50: implications for DNA recognition and RecF function. EMBO J 26:867–877. doi: 10.1038/sj.emboj.7601537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Leiros I, Timmins J, Hall DR, McSweeney S. 2005. Crystal structure and DNA-binding analysis of RecO from Deinococcus radiodurans. EMBO J 24:906–918. doi: 10.1038/sj.emboj.7600582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Makharashvili N, Koroleva O, Bera S, Grandgenett DP, Korolev S. 2004. A novel structure of DNA repair protein RecO from Deinococcus radiodurans. Structure 12:1881–1889. doi: 10.1016/j.str.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 272.Honda M, Fujisawa T, Shibata T, Mikawa T. 2008. RecR forms a ring-like tetramer that encircles dsDNA by forming a complex with RecF. Nucleic Acids Res 36:5013–5020. doi: 10.1093/nar/gkn471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Keller KL, Overbeck-Carrick TL, Beck DJ. 2001. Survival and induction of SOS in Escherichia coli treated with cisplatin, UV-irradiation, or mitomycin C are dependent on the function of the RecBC and RecFOR pathways of homologous recombination. Mutat Res 486:21–29. doi: 10.1016/s0921-8777(01)00077-5. [DOI] [PubMed] [Google Scholar]
- 274.Sandler SJ. 19 April 2001. RecFOR protein. In eLS. John Wiley & Sons Ltd, Chichester, United Kingdom. doi: 10.1038/npg.els.0000588. [DOI] [Google Scholar]
- 275.Liu Y-H, Cheng A-J, Wang T-CV. 1998. Involvement of recF, recO, and recR genes in UV-radiation mutagenesis of Escherichia coli. J Bacteriol 180:1766–1770. doi: 10.1128/JB.180.7.1766-1770.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wang T-CV, Chang H-Y, Hung J-L. 1993. Cosuppression of recF, recR and recO mutations by mutant recA alleles in Escherichia coli cells. Mutat Res 294:157–166. doi: 10.1016/0921-8777(93)90024-b. [DOI] [PubMed] [Google Scholar]
- 277.Morimatsu K, Wu Y, Kowalczykowski SC. 2012. RecFOR proteins target RecA protein to a DNA gap with either DNA or RNA at the 5′ terminus: implication for repair of stalled replication forks. J Biol Chem 287:35621–35630. doi: 10.1074/jbc.M112.397034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Bork JM, Cox MM, Inman RB. 2001. The RecOR proteins modulate RecA protein function at 5′ ends of single-stranded DNA. EMBO J 20:7313–7322. doi: 10.1093/emboj/20.24.7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Honda M, Inoue J, Yoshimasu M, Ito Y, Shibata T, Mikawa T. 2006. Identification of the RecR toprim domain as the binding site for both RecF and RecO. A role of RecR in RecFOR assembly at double-stranded DNA-single-stranded DNA junctions. J Biol Chem 281:18549–18559. doi: 10.1074/jbc.M512658200. [DOI] [PubMed] [Google Scholar]
- 280.Marsin S, Mathieu A, Kortulewski T, Guérois R, Radicella JP. 2008. Unveiling the other RecO family of homologous recombination proteins. PLoS Genet 4:e1000146. doi: 10.1371/journal.pgen.1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Rocha EPC, Cornet E, Michel B. 2005. Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet 1:e15. doi: 10.1371/journal.pgen.0010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Sandler SJ. 1994. Studies on the mechanism of reduction of UV-inducible sulAp expression by recF overexpression in Escherichia coli K-12. Mol Gen Genet 245:741–749. doi: 10.1007/BF00297281. [DOI] [PubMed] [Google Scholar]
- 283.Sandler SJ, Clark AJ. 1994. RecOR suppression of recF mutant phenotypes in Escherichia coli K-12. J Bacteriol 176:3661–3672. doi: 10.1128/jb.176.12.3661-3672.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Sandler SJ, Clark AJ. 1993. Use of high and low level overexpression plasmids to test mutant alleles of the recF gene of Escherichia coli K-12 for partial activity. Genetics 135:643–654. doi: 10.1093/genetics/135.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Xia J, Chiu L-Y, Nehring RB, Nunez MAB, Mei Q, Perez M, Zhai Y, Fitzgerald DM, Pribis JP, Wang Y, Hu CW, Powell RT, LaBonte SA, Jalali A, Guzman MLM, Lentzsch AM, Szafran AT, Joshi MC, Richters M, Gibson JL, Frisch RL, Hastings PJ, Bates D, Queitsch C, Hilsenbeck SG, Coarfa C, Hu JC, Siegele DA, Scott KL, Liang H, Mancini MA, Herman C, Miller KM, Rosenberg SM. 2019. Bacteria-to-human protein networks reveal origins of endogenous DNA damage. Cell 176:127–143.e24. doi: 10.1016/j.cell.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Henry C, Kaur G, Cherry ME, Henrikus SS, Bonde NJ, Sharma N, Beyer HA, Wood EA, Chitteni-Pattu S, van Oijen AM, Robinson A, Cox MM. 1 May 2023. RecF protein targeting to post-replication (daughter strand) gaps II: RecF interaction with replisomes. Nucleic Acids Res . doi: 10.1093/nar/gkad310. [DOI] [PMC free article] [PubMed]
- 287.Fujita MQ, Yoshikawa H, Ogasawara N. 1990. Structure of the dnaA region of Micrococcus luteus: conservation and variations among eubacteria. Gene 93:73–78. doi: 10.1016/0378-1119(90)90138-h. [DOI] [PubMed] [Google Scholar]
- 288.Fujita MQ, Yoshikawa H, Ogasawara N. 1992. Structure of the dnaA and dnaA-box region in the Mycoplasma capricolum chromosome: conservation and variations in the course of evolution. Gene 110:17–23. doi: 10.1016/0378-1119(92)90439-v. [DOI] [PubMed] [Google Scholar]
- 289.Margerrison EEC, Hopewell R, Fisher LM. 1992. Nucleotide sequence of the Staphylococcus aureus gyrB-gyrA locus encoding the DNA gyrase A and B proteins. J Bacteriol 174:1596–1603. doi: 10.1128/jb.174.5.1596-1603.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Shan Q, Bork JM, Webb BL, Inman RB, Cox MM. 1997. RecA protein filaments: end-dependent dissociation from ssDNA and stabilization by RecO and RecR proteins. J Mol Biol 265:519–540. doi: 10.1006/jmbi.1996.0748. [DOI] [PubMed] [Google Scholar]
- 291.Bell JC, Plank JL, Dombrowski CC, Kowalczykowski SC. 2012. Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature 491:274–278. doi: 10.1038/nature11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Lusetti SL, Hobbs MD, Stohl EA, Chitteni-Pattu S, Inman RB, Seifert HS, Cox MM. 2006. The RecF protein antagonizes RecX function via direct interaction. Mol Cell 21:41–50. doi: 10.1016/j.molcel.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Kuzminov A, Stahl FW. 1999. Double-strand end repair via the RecBC pathway in Escherichia coli primes DNA replication. Genes Dev 13:345–356. doi: 10.1101/gad.13.3.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Cheng K, Xu H, Chen X, Wang L, Tian B, Zhao Y, Hua Y. 2016. Structural basis for DNA 5′-end resection by RecJ. Elife 5:e14294. doi: 10.7554/eLife.14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Lovett ST, Clark AJ. 1985. Cloning of the Escherichia coli recJ chromosomal region and identification of its encoded proteins. J Bacteriol 162:280–285. doi: 10.1128/jb.162.1.280-285.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Lovett ST, Kolodner RD. 1991. Nucleotide sequence of the Escherichia coli recJ chromosomal region and construction of recJ-overexpression plasmids. J Bacteriol 173:353–364. doi: 10.1128/jb.173.1.353-364.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Lovett ST, Sutera VA Jr. 1995. Suppression of recJ exonuclease mutants of Escherichia coli by alterations in DNA helicases II (UvrD) and IV (HelD). Genetics 140:27–45. doi: 10.1093/genetics/140.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Lovett ST, Clark AJ. 1984. Genetic analysis of the recJ gene of Escherichia coli K-12. J Bacteriol 157:190–196. doi: 10.1128/jb.157.1.190-196.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Courcelle J, Hanawalt PC. 1999. RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol Gen Genet 262:543–551. doi: 10.1007/s004380051116. [DOI] [PubMed] [Google Scholar]
- 300.Morimatsu K, Kowalczykowski SC. 2014. RecQ helicase and RecJ nuclease provide complementary functions to resect DNA for homologous recombination. Proc Natl Acad Sci USA 111:E5133–E5142. doi: 10.1073/pnas.1420009111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Ivancic-Bace I, Salaj-Smic E, Brcic-Kostic K. 2005. Effects of recJ, recQ, and recFOR mutations on recombination in nuclease-deficient recB recD double mutants of Escherichia coli. J Bacteriol 187:1350–1356. doi: 10.1128/JB.187.4.1350-1356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Sutton MD, Farrow MF, Burton BM, Walker GC. 2001. Genetic interactions between the Escherichia coli umuDC gene products and the β processivity clamp of the replicative DNA polymerase. J Bacteriol 183:2897–2909. doi: 10.1128/JB.183.9.2897-2909.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Beuning PJ, Sawicka D, Barsky D, Walker GC. 2006. Two processivity clamp interactions differentially alter the dual activities of UmuC. Mol Microbiol 59:460–474. doi: 10.1111/j.1365-2958.2005.04959.x. [DOI] [PubMed] [Google Scholar]
- 304.Klemperer N, Zhang D, Skangalis M, O’Donnell M. 2000. Cross-utilization of the beta sliding clamp by replicative polymerases of evolutionary divergent organisms. J Biol Chem 275:26136–26143. doi: 10.1074/jbc.M002566200. [DOI] [PubMed] [Google Scholar]
- 305.Stukenberg PT, O’Donnell M. 1995. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. V. Four different polymerase-clamp complexes on DNA. J Biol Chem 270:13384–13391. doi: 10.1074/jbc.270.22.13384. [DOI] [PubMed] [Google Scholar]
- 306.Stukenberg PT, Turner J, O’Donnell M. 1994. An explanation for lagging strand replication: polymerase hopping among DNA sliding clamps. Cell 78:877–887. doi: 10.1016/s0092-8674(94)90662-9. [DOI] [PubMed] [Google Scholar]
- 307.Turner J, Hingorani MM, Kelman Z, O’Donnell M. 1999. The internal workings of a DNA polymerase clamp-loading machine. EMBO J 18:771–783. doi: 10.1093/emboj/18.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Sikand A, Jaszczur M, Bloom LB, Woodgate R, Cox MM, Goodman MF. 2021. The SOS error-prone DNA polymerase V mutasome and β-sliding clamp acting in concert on undamaged DNA and during translesion synthesis. Cells 10:1083. doi: 10.3390/cells10051083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Blanar MA, Sandler SJ, Armengod ME, Ream LW, Clark AJ. 1984. Molecular analysis of the recF gene of Escherichia coli. Proc Natl Acad Sci USA 81:4622–4626. doi: 10.1073/pnas.81.15.4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Perez-Roge RL, Garcia-Sogo M, Navarro-Avino J, Lopez-Acedo C, Macian F, Armengod ME. 1991. Positive and negative regulatory elements in the dnaA-dnaN-recF operon of Escherichia coli. Biochimie 73:329–334. doi: 10.1016/0300-9084(91)90220-u. [DOI] [PubMed] [Google Scholar]
- 311.Cox MM. 1999. Recombinational DNA repair in bacteria and the RecA protein. Prog Nucleic Acid Res Mol Biol 63:311–366. doi: 10.1016/s0079-6603(08)60726-6. [DOI] [PubMed] [Google Scholar]
- 312.Cox MM. 2003. The bacterial RecA protein as a motor protein. Annu Rev Microbiol 57:551–577. doi: 10.1146/annurev.micro.57.030502.090953. [DOI] [PubMed] [Google Scholar]
- 313.Cox MM. 2007. The bacterial RecA protein: structure, function, and regulation, p 53–94. In Rothstein R, Aguilera A (ed), Molecular genetics of recombination. Topics in current genetics, vol 17. Springer-Verlag, Heidelberg, Germany. [Google Scholar]
- 314.Kowalczykowski SC. 1991. Biochemical and biological function of Escherichia coli RecA protein: behavior of mutant RecA proteins. Biochimie 73:289–304. doi: 10.1016/0300-9084(91)90216-n. [DOI] [PubMed] [Google Scholar]
- 315.Kowalczykowski SC, Roman LJ. 1990. Reconstitution of homologous pairing activity dependent upon the combined activities of purified E. coli RecA, RecBCD, and SSB proteins, p 357–373. In Richardson CC, Lehman IR (ed), Molecular mechanisms in DNA replication and recombination, vol 127. Alan R Liss, New York, NY. [Google Scholar]
- 316.Lehman IR. 1979. On the role of the RecA protein of Escherichia coli in general recombination. UCLA Forum Med Sci 1979:23–37. [DOI] [PubMed] [Google Scholar]
- 317.Lusetti SL, Cox MM. 2002. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem 71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- 318.Radding CM. 1988. Homologous pairing and strand exchange promoted by Escherichia coli RecA protein, p 193–229. In Kucherlapati R, Smith GR (ed), Genetic recombination. American Society for Microbiology, Washington, DC. [Google Scholar]
- 319.West SC, Cassuto E, Howard-Flanders P. 1982. Role of SSB protein in RecA promoted branch migration reactions. Mol Gen Genet 186:333–338. doi: 10.1007/BF00729451. [DOI] [PubMed] [Google Scholar]
- 320.Chen Z, Yang H, Pavletich NP. 2008. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 453:489–494. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- 321.Egelman EH, Stasiak A. 1986. Structure of helical RecA-DNA complexes. Complexes formed in the presence of ATPγS or ATP. J Mol Biol 191:677–697. doi: 10.1016/0022-2836(86)90453-5. [DOI] [PubMed] [Google Scholar]
- 322.Griffith JD, Harris LD, Register J, III.. 1984. Visualization of SSB-ssDNA complexes active in the assembly of stable RecA-DNA filaments. Cold Spring Harb Symp Quant Biol 49:553–559. doi: 10.1101/sqb.1984.049.01.062. [DOI] [PubMed] [Google Scholar]
- 323.Stasiak A, Egelman EH, Howard-Flanders P. 1988. Structure of helical RecA-DNA complexes. III. The structural polarity of RecA filaments and functional polarity in the RecA-mediated strand exchange reaction. J Mol Biol 202:659–662. doi: 10.1016/0022-2836(88)90293-8. [DOI] [PubMed] [Google Scholar]
- 324.Xing X, Bell CE. 2004. Crystal structures of Escherichia coli RecA in a compressed helical filament. J Mol Biol 342:1471–1485. doi: 10.1016/j.jmb.2004.07.091. [DOI] [PubMed] [Google Scholar]
- 325.Das Gupta C, Shibata T, Cunningham RP, Radding CM. 1980. The topology of homologous pairing promoted by RecA protein. Cell 22:437–446. doi: 10.1016/0092-8674(80)90354-2. [DOI] [PubMed] [Google Scholar]
- 326.Cox MM, Lehman IR. 1981. Directionality and polarity in RecA protein-promoted branch migration. Proc Natl Acad Sci USA 78:6018–6022. doi: 10.1073/pnas.78.10.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Cox MM, Lehman IR. 1981. RecA protein of Escherichia coli promotes branch migration, a kinetically distinct phase of DNA strand exchange. Proc Natl Acad Sci USA 78:3433–3437. doi: 10.1073/pnas.78.6.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Cunningham RP, Wu AM, Shibata T, Das Gupta C, Radding CM. 1981. Homologous pairing and topological linkage of DNA molecules by combined action of E. coli RecA protein and topoisomerase I. Cell 24:213–223. doi: 10.1016/0092-8674(81)90517-1. [DOI] [PubMed] [Google Scholar]
- 329.MacFarland KJ, Shan Q, Inman RB, Cox MM. 1997. RecA as a motor protein. Testing models for the role of ATP hydrolysis in DNA strand exchange. J Biol Chem 272:17675–17685. doi: 10.1074/jbc.272.28.17675. [DOI] [PubMed] [Google Scholar]
- 330.Shan Q, Cox MM. 1998. On the mechanism of RecA-mediated repair of double-strand breaks: no role for four-strand DNA pairing intermediates. Mol Cell 1:309–317. doi: 10.1016/s1097-2765(00)80031-3. [DOI] [PubMed] [Google Scholar]
- 331.Cox MM. 1994. Why does RecA protein hydrolyze ATP? Trends Biochem Sci 19:217–222. doi: 10.1016/0968-0004(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 332.Cox MM. 2007. Motoring along with the bacterial RecA protein. Nat Rev Mol Cell Biol 8:127–138. doi: 10.1038/nrm2099. [DOI] [PubMed] [Google Scholar]
- 333.Arenson TA, Tsodikov OV, Cox MM. 1999. Quantitative analysis of the kinetics of end-dependent disassembly of RecA filaments from ssDNA. J Mol Biol 288:391–401. doi: 10.1006/jmbi.1999.2705. [DOI] [PubMed] [Google Scholar]
- 334.Bakhlanova IV, Dudkina AV, Wood EA, Lanzov VA, Cox MM, Baitin DM. 2016. DNA metabolism in balance: rapid loss of a RecA-based hyperrec phenotype. PLoS One 11:e0154137. doi: 10.1371/journal.pone.0154137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Cox JM, Tsodikov OV, Cox MM. 2005. Organized unidirectional waves of ATP hydrolysis within a RecA filament. PLoS Biol 3:e52. doi: 10.1371/journal.pbio.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Iwasaki H, Takahagi M, Nakata A, Shinagawa H. 1992. Escherichia coli RuvA and RuvB proteins specifically interact with Holliday junctions and promote branch migration. Genes Dev 6:2214–2220. doi: 10.1101/gad.6.11.2214. [DOI] [PubMed] [Google Scholar]
- 337.Parsons CA, Tsaneva I, Lloyd RG, West SC. 1992. Interaction of Escherichia coli RuvA and RuvB proteins with synthetic Holliday junctions. Proc Natl Acad Sci USA 89:5452–5456. doi: 10.1073/pnas.89.12.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.van Gool AJ, Hajibagheri NM, Stasiak A, West SC. 1999. Assembly of the Escherichia coli RuvABC resolvasome directs the orientation of Holliday junction resolution. Genes Dev 13:1861–1870. doi: 10.1101/gad.13.14.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Yamada K, Kunishima N, Mayanagi K, Ohnishi T, Nishino T, Iwasaki H, Shinagawa H, Morikawa K. 2001. Crystal structure of the Holliday junction migration motor protein RuvB from Thermus thermophilus HB8. Proc Natl Acad Sci USA 98:1442–1447. doi: 10.1073/pnas.98.4.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Zerbib D, Mezard C, George H, West SC. 1998. Coordinated actions of RuvABC in Holliday junction processing. J Mol Biol 281:621–630. doi: 10.1006/jmbi.1998.1959. [DOI] [PubMed] [Google Scholar]
- 341.McGlynn P, Lloyd RG. 2001. Action of RuvAB at replication fork structures. J Biol Chem 276:41938–41944. doi: 10.1074/jbc.M107945200. [DOI] [PubMed] [Google Scholar]
- 342.Rafferty JB, Sedelnikova SE, Hargreaves D, Artymiuk PJ, Baker PJ, Sharples GJ, Mahdi AA, Lloyd RG, Rice DW. 1996. Crystal structure of DNA recombination protein RuvA and a model for its binding to the Holliday junction. Science 274:415–421. doi: 10.1126/science.274.5286.415. [DOI] [PubMed] [Google Scholar]
- 343.Tsaneva IR, Muller B, West SC. 1992. ATP-dependent branch migration of Holliday junctions promoted by the RuvA and RuvB proteins of E. coli. Cell 69:1171–1180. doi: 10.1016/0092-8674(92)90638-s. [DOI] [PubMed] [Google Scholar]
- 344.Parsons CA, Stasiak A, Bennett RJ, West SC. 1995. Structure of a multisubunit complex that promotes DNA branch migration. Nature 374:375–378. doi: 10.1038/374375a0. [DOI] [PubMed] [Google Scholar]
- 345.Bennett RJ, West SC. 1995. RuvC protein resolves Holliday junctions via cleavage of the continuous (noncrossover) strands. Proc Natl Acad Sci USA 92:5635–5639. doi: 10.1073/pnas.92.12.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Connolly B, Parsons CA, Benson FE, Dunderdale HJ, Sharples GJ, Lloyd RG, West SC. 1991. Resolution of Holliday junctions in vitro requires the Escherichia coli ruvC gene product. Proc Natl Acad Sci USA 88:6063–6067. doi: 10.1073/pnas.88.14.6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.West SC. 1997. Processing of recombination intermediates by the RuvABC proteins. Annu Rev Genet 31:213–244. doi: 10.1146/annurev.genet.31.1.213. [DOI] [PubMed] [Google Scholar]
- 348.Davies AA, West SC. 1998. Formation of RuvABC-Holliday junction complexes in vitro. Curr Biol 8:725–727. doi: 10.1016/s0960-9822(98)70282-9. [DOI] [PubMed] [Google Scholar]
- 349.Müller B, West SC. 1994. Processing of Holliday junctions by the Escherichia coli RuvA, RuvB, RuvC and RecG proteins. Experientia 50:216–222. doi: 10.1007/BF01924004. [DOI] [PubMed] [Google Scholar]
- 350.West SC. 1996. The RuvABC proteins and Holliday junction processing in Escherichia coli. J Bacteriol 178:1237–1241. doi: 10.1128/jb.178.5.1237-1241.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Michel B, Recchia GD, Penel-Colin M, Ehrlich SD, Sherratt DJ. 2000. Resolution of Holliday junctions by RuvABC prevents dimer formation in rep mutants and UV-irradiated cells. Mol Microbiol 37:180–191. doi: 10.1046/j.1365-2958.2000.01989.x. [DOI] [PubMed] [Google Scholar]
- 352.Iwasaki H, Shiba T, Nakata A, Shinagawa H. 1989. Involvement in DNA repair of the ruvA gene of Escherichia coli. Mol Gen Genet 219:328–331. doi: 10.1007/BF00261196. [DOI] [PubMed] [Google Scholar]
- 353.Sargentini NJ, Smith KC. 1989. Role of ruvAB genes in UV- and gamma-radiation and chemical mutagenesis in Escherichia coli. Mutat Res 215:115–129. doi: 10.1016/0027-5107(89)90224-8. [DOI] [PubMed] [Google Scholar]
- 354.Sharples GJ, Benson FE, Illing GT, Lloyd RG. 1990. Molecular and functional analysis of the ruv region of Escherichia coli K-12 reveals three genes involved in DNA repair and recombination. Mol Gen Genet 221:219–226. doi: 10.1007/BF00261724. [DOI] [PubMed] [Google Scholar]
- 355.Dillingham MS, Kowalczykowski SC. 2001. A step backward in advancing DNA replication: rescue of stalled replication forks by RecG. Mol Cell 8:734–736. doi: 10.1016/s1097-2765(01)00358-6. [DOI] [PubMed] [Google Scholar]
- 356.Gupta S, Yeeles JTP, Marians KJ. 2014. Regression of replication forks stalled by leading-strand template damage. I. Both RecG and RuvAB catalyze regression, but RuvC cleaves the Holliday junctions formed by RecG preferentially. J Biol Chem 289:28376–28387. doi: 10.1074/jbc.M114.587881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.McGlynn P, Lloyd RG, Marians KJ. 2001. Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc Natl Acad Sci USA 98:8235–8240. doi: 10.1073/pnas.121007798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.McGlynn P, Lloyd RG. 2002. Genome stability and the processing of damaged replication forks by RecG. Trends Genet 18:413–419. doi: 10.1016/s0168-9525(02)02720-8. [DOI] [PubMed] [Google Scholar]
- 359.Lloyd RG, Sharples GJ. 1993. Processing of recombination intermediates by the RecG and RuvAB proteins of Escherichia coli. Nucleic Acids Res 21:1719–1725. doi: 10.1093/nar/21.8.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Sharples GJ, Ingleston SM, Lloyd RG. 1999. Holliday junction processing in bacteria: insights from the evolutionary conservation of RuvABC, RecG, and RusA. J Bacteriol 181:5543–5550. doi: 10.1128/JB.181.18.5543-5550.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Whitby MC, Vincent SD, Lloyd RG. 1994. Branch migration of Holliday junctions: identification of RecG protein as a junction specific DNA helicase. EMBO J 13:5220–5228. doi: 10.1002/j.1460-2075.1994.tb06853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Azeroglu B, Mawer JSP, Cockram CA, White MA, Hasan AMM, Filatenkova M, Leach DRF. 2016. RecG directs DNA synthesis during double-strand break repair. PLoS Genet 12:e1005799. doi: 10.1371/journal.pgen.1005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Al Deib AA, Mahdi AA, Lloyd RG. 1996. Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K-12. J Bacteriol 178:6782–6789. doi: 10.1128/jb.178.23.6782-6789.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.McGlynn P, Al Deib AA, Liu J, Marians KJ, Lloyd RG. 1997. The DNA replication protein PriA and the recombination protein RecG bind D-loops. J Mol Biol 270:212–221. doi: 10.1006/jmbi.1997.1120. [DOI] [PubMed] [Google Scholar]
- 365.Jaktaji RP, Lloyd RG. 2003. PriA supports two distinct pathways for replication restart in UV-irradiated Escherichia coli cells. Mol Microbiol 47:1091–1100. doi: 10.1046/j.1365-2958.2003.03357.x. [DOI] [PubMed] [Google Scholar]
- 366.Manhart CM, McHenry CS. 2013. The PriA replication restart protein blocks replicase access prior to helicase assembly and directs template specificity through its ATPase activity. J Biol Chem 288:3989–3999. doi: 10.1074/jbc.M112.435966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Sandler SJ, McCool JD, Do TT, Johansen RU. 2001. PriA mutations that affect PriA-PriC function during replication restart. Mol Microbiol 41:697–704. doi: 10.1046/j.1365-2958.2001.02547.x. [DOI] [PubMed] [Google Scholar]
- 368.Beam CE, Saveson CJ, Lovett ST. 2002. Role for radA/sms in recombination intermediate processing in Escherichia coli. J Bacteriol 184:6836–6844. doi: 10.1128/JB.184.24.6836-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Marie L, Rapisarda C, Morales V, Berge M, Perry T, Soulet AL, Gruget C, Remaut H, Fronzes R, Polard P. 2017. Bacterial RadA is a DnaB-type helicase interacting with RecA to promote bidirectional D-loop extension. Nat Commun 8:15638. doi: 10.1038/ncomms15638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Diver WP, Sargentini NJ, Smith KC. 1982. A mutation (radA100) in Escherichia coli that selectively sensitizes cells grown in rich medium to X- or U.V.-radiation, or methyl methanesulphonate. Int J Radiat Biol Relat Stud Phys Chem Med 42:339–346. doi: 10.1080/09553008214551251. [DOI] [PubMed] [Google Scholar]
- 371.Burghout P, Bootsma HJ, Kloosterman TG, Bijlsma JJE, de Jongh CE, Kuipers OP, Hermans PWM. 2007. Search for genes essential for pneumococcal transformation: the RadA DNA repair protein plays a role in genomic recombination of donor DNA. J Bacteriol 189:6540–6550. doi: 10.1128/JB.00573-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Carrasco B, Cozar MC, Lurz R, Alonso JC, Ayora S. 2004. Genetic recombination in Bacillus subtilis 168: contribution of Holliday junction processing functions in chromosome segregation. J Bacteriol 186:5557–5566. doi: 10.1128/JB.186.17.5557-5566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Castellanos M, Romero D. 2009. The extent of migration of the Holliday junction is a crucial factor for gene conversion in Rhizobium etli. J Bacteriol 191:4987–4995. doi: 10.1128/JB.00111-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Kruger E, Msadek T, Ohlmeier S, Hecker M. 1997. The Bacillus subtilis clpC operon encodes DNA repair and competence proteins. Microbiology (Reading) 143:1309–1316. doi: 10.1099/00221287-143-4-1309. [DOI] [PubMed] [Google Scholar]
- 375.Lovett ST. 2006. Microbiology: resurrecting a broken genome. Nature 443:517–519. doi: 10.1038/443517b. [DOI] [PubMed] [Google Scholar]
- 376.Slade D, Lindner AB, Paul G, Radman M. 2009. Recombination and replication in DNA repair of heavily irradiated Deinococcus radiodurans. Cell 136:1044–1055. doi: 10.1016/j.cell.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 377.Byrne RT, Chen SH, Wood EA, Cabot EL, Cox MM. 2014. Surviving extreme exposure to ionizing radiation: Escherichia coli genes and pathways. J Bacteriol 196:3534–3545. doi: 10.1128/JB.01589-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Chen SH, Byrne RT, Wood EA, Cox MM. 2015. Escherichia coli radD (yejH) gene: a novel function involved in radiation resistance and double-strand break repair. Mol Microbiol 95:754–768. doi: 10.1111/mmi.12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Osorio Garcia MA, Satyshur KA, Cox MM, Keck JL. 2022. X-ray crystal structure of the Escherichia coli RadD DNA repair protein bound to ADP reveals a novel zinc ribbon domain. PLoS One 17:e0266031. doi: 10.1371/journal.pone.0266031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Umezu K, Nakayama H. 1993. RecQ DNA helicase of Escherichia coli. Characterization of the helix-unwinding activity with emphasis on the effect of single-stranded DNA-binding protein. J Mol Biol 230:1145–1150. doi: 10.1006/jmbi.1993.1231. [DOI] [PubMed] [Google Scholar]
- 381.Umezu K, Nakayama K, Nakayama H. 1990. Escherichia coli RecQ protein is a DNA helicase. Proc Natl Acad Sci USA 87:5363–5367. doi: 10.1073/pnas.87.14.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Harmon FG, Kowalczykowski SC. 2001. Biochemical characterization of the DNA helicase activity of the Escherichia coli RecQ helicase. J Biol Chem 276:232–243. doi: 10.1074/jbc.M006555200. [DOI] [PubMed] [Google Scholar]
- 383.Harmon FG, Kowalczykowski SC. 1998. RecQ helicase, in concert with RecA and SSB proteins, initiates and disrupts DNA recombination. Genes Dev 12:1134–1144. doi: 10.1101/gad.12.8.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Sutera VA, Sass TH, Leonard SE, Wu L, Glass DJ, Giordano GG, Zur Y, Lovett ST. 2021. Genetic analysis of DinG family helicase YoaA and its interaction with replication clamp loader protein HolC in Escherichia coli. J Bacteriol 203:e00228-21. doi: 10.1128/JB.00228-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Gulbis JM, Kazmirski SL, Finkelstein J, Kelman Z, O’Donnell M, Kuriyan J. 2004. Crystal structure of the chi:psi sub-assembly of the Escherichia coli DNA polymerase clamp-loader complex. Eur J Biochem 271:439–449. doi: 10.1046/j.1432-1033.2003.03944.x. [DOI] [PubMed] [Google Scholar]
- 386.Sutera VA, Weeks SJ, Dudenhausen EE, Baggett HBR, Shaw MC, Brand KA, Glass DJ, Bloom LB, Lovett ST. 2021. Alternative complexes formed by the Escherichia coli clamp loader accessory protein HolC (x) with replication protein HolD (ψ) and repair protein YoaA. DNA Repair (Amst) 100:103006. doi: 10.1016/j.dnarep.2020.103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.White MF. 2009. Structure, function and evolution of the XPD family of iron-sulfur-containing 5′→3′ DNA helicases. Biochem Soc Trans 37:547–551. doi: 10.1042/BST0370547. [DOI] [PubMed] [Google Scholar]
- 388.Weeks-Pollenz SJ, Ali Y, Morris L, Sutera VA, Dudenhausen EE, Hibnick M, Lovett ST, Bloom LB. 2023. Characterization of the Escherichia coli XPD/Rad3 iron-sulfur helicase, YoaA, in complex with the DNA polymerase III clamp loader subunit, chi (χ). J Biol Chem 299:102786. doi: 10.1016/j.jbc.2022.102786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Gupta R, Shuman S, Glickman MS. 2015. RecF and RecR play critical roles in the homologous recombination and single-strand annealing pathways of mycobacteria. J Bacteriol 197:3121–3132. doi: 10.1128/JB.00290-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Myka KK, Marians KJ. 2020. Two components of DNA replication-dependent LexA cleavage. J Biol Chem 295:10368–10379. doi: 10.1074/jbc.RA120.014224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Park MS, O’Donnell M. 2009. The clamp loader assembles the beta clamp onto either a 3′ or 5′ primer terminus: the underlying basis favoring 3′ loading. J Biol Chem 284:31473–31483. doi: 10.1074/jbc.M109.050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Lenhart JS, Brandes ER, Schroeder JW, Sorenson RJ, Showalter HD, Simmons LA. 2014. RecO and RecR are necessary for RecA loading in response to DNA damage and replication fork stress. J Bacteriol 196:2851–2860. doi: 10.1128/JB.01494-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Odsbu I, Skarstad K. 2014. DNA compaction in the early part of the SOS response is dependent on RecN and RecA. Microbiology (Reading) 160:872–882. doi: 10.1099/mic.0.075051-0. [DOI] [PubMed] [Google Scholar]
- 394.Kogoma T. 1997. Is RecF a DNA replication protein? Proc Natl Acad Sci USA 94:3483–3484. doi: 10.1073/pnas.94.8.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.McInerney P, O’Donnell M. 2007. Replisome fate upon encountering a leading strand block and clearance from DNA by recombination proteins. J Biol Chem 282:25903–25916. doi: 10.1074/jbc.M703777200. [DOI] [PubMed] [Google Scholar]
- 396.Chang S, Naiman K, Thrall ES, Kath JE, Jergic S, Dixon NE, Fuchs RP, Loparo JJ. 2019. A gatekeeping function of the replicative polymerase controls pathway choice in the resolution of lesion-stalled replisomes. Proc Natl Acad Sci USA 116:25591–25601. doi: 10.1073/pnas.1914485116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Lusetti SL, Drees JC, Stohl EA, Seifert HS, Cox MM. 2004. The DinI and RecX proteins are competing modulators of RecA function. J Biol Chem 279:55073–55079. doi: 10.1074/jbc.M410371200. [DOI] [PubMed] [Google Scholar]
- 398.Lusetti SL, Voloshin ON, Inman RB, Camerini-Otero RD, Cox MM. 2004. The DinI protein stabilizes RecA protein filaments. J Biol Chem 279:30037–30046. doi: 10.1074/jbc.M403064200. [DOI] [PubMed] [Google Scholar]
- 399.Baitin DM, Gruenig MC, Cox MM. 2008. SSB antagonizes RecX-RecA interaction. J Biol Chem 283:14198–14204. doi: 10.1074/jbc.M801511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Drees JC, Lusetti SL, Chitteni-Pattu S, Inman RB, Cox MM. 2004. A RecA filament capping mechanism for RecX protein. Mol Cell 15:789–798. doi: 10.1016/j.molcel.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 401.Drees JC, Lusetti SL, Cox MM. 2004. Inhibition of RecA protein by the Escherichia coli RecX protein: modulation by the RecA C terminus and filament functional state. J Biol Chem 279:52991–52997. doi: 10.1074/jbc.M409050200. [DOI] [PubMed] [Google Scholar]
- 402.Gruenig MC, Stohl EA, Chitteni-Pattu S, Seifert HS, Cox MM. 2010. Less is more: Neisseria gonorrhoeae RecX protein stimulates recombination by inhibiting RecA. J Biol Chem 285:37188–37197. doi: 10.1074/jbc.M110.171967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Pages V, Koffel-Schwartz N, Fuchs RP. 2003. recX, a new SOS gene that is co-transcribed with the recA gene in Escherichia coli. DNA Repair (Amst) 2:273–284. doi: 10.1016/s1568-7864(02)00217-3. [DOI] [PubMed] [Google Scholar]
- 404.Stohl EA, Brockman JP, Burkle KL, Morimatsu K, Kowalczykowski SC, Seifert HS. 2003. Escherichia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. J Biol Chem 278:2278–2285. doi: 10.1074/jbc.M210496200. [DOI] [PubMed] [Google Scholar]
- 405.VanLoock MS, Yu X, Yang S, Galkin VE, Huang H, Rajan SS, Anderson WF, Stohl EA, Seifert HS, Egelman EH. 2003. Complexes of RecA with LexA and RecX differentiate between active and inactive RecA nucleoprotein filaments. J Mol Biol 333:345–354. doi: 10.1016/j.jmb.2003.08.053. [DOI] [PubMed] [Google Scholar]
- 406.Drees JC, Chitteni-Pattu S, McCaslin DR, Inman RB, Cox MM. 2006. Inhibition of RecA protein function by the RdgC protein from Escherichia coli. J Biol Chem 281:4708–4717. doi: 10.1074/jbc.M513592200. [DOI] [PubMed] [Google Scholar]
- 407.Briggs GS, McEwan PA, Yu J, Moore T, Emsley J, Lloyd RG. 2007. Ring structure of the Escherichia coli DNA-binding protein RdgC associated with recombination and replication fork repair. J Biol Chem 282:12353–12357. doi: 10.1074/jbc.C700023200. [DOI] [PubMed] [Google Scholar]
- 408.Moore T, McGlynn P, Ngo H-P, Sharples GJ, Lloyd RG. 2003. The RdgC protein of Escherichia coli binds DNA and counters a toxic effect of RecFOR in strains lacking the replication restart protein PriA. EMBO J 22:735–745. doi: 10.1093/emboj/cdg048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Tessmer I, Moore T, Lloyd RG, Wilson A, Erie DA, Allen S, Tendler SJ. 2005. AFM studies on the role of the protein RdgC in bacterial DNA recombination. J Mol Biol 350:254–262. doi: 10.1016/j.jmb.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 410.Stanage TH, Page AN, Cox MM. 2017. DNA flap creation by the RarA/MgsA protein of Escherichia coli. Nucleic Acids Res 45:2724–2735. doi: 10.1093/nar/gkw1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Shibata T, Hishida T, Kubota Y, Han Y-W, Iwasaki H, Shinagawa H. 2005. Functional overlap between RecA and MgsA (RarA) in the rescue of stalled replication forks in Escherichia coli. Genes Cells 10:181–191. doi: 10.1111/j.1365-2443.2005.00831.x. [DOI] [PubMed] [Google Scholar]
- 412.Georgescu RE, Kurth I, O’Donnell ME. 2012. Single-molecule studies reveal the function of a third polymerase in the replisome. Nat Struct Mol Biol 19:113–116. doi: 10.1038/nsmb.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Jain K, Wood EA, Romero ZJ, Cox MM. 2021. RecA-independent recombination: dependence on the Escherichia coli RarA protein. Mol Microbiol 115:1122–1137. doi: 10.1111/mmi.14655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Goodman MF, McDonald JP, Jaszczur MM, Woodgate R. 2016. Insights into the complex levels of regulation imposed on Escherichia coli DNA polymerase V. DNA Repair (Amst) 44:42–50. doi: 10.1016/j.dnarep.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Woodgate R, Ennis DG. 1991. Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol Gen Genet 229:10–16. doi: 10.1007/BF00264207. [DOI] [PubMed] [Google Scholar]
- 416.Sweasy JB, Witkin EM, Sinha N, Roegner-Maniscalco V. 1990. RecA protein of Escherichia coli has a third essential role in SOS mutator activity. J Bacteriol 172:3030–3036. doi: 10.1128/jb.172.6.3030-3036.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Maliszewska-Tkaczyk M, Jonczyk P, Bialoskorska M, Schaaper RM, Fijalkowska IJ. 2000. SOS mutator activity: unequal mutagenesis on leading and lagging strands. Proc Natl Acad Sci USA 97:12678–12683. doi: 10.1073/pnas.220424697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Dutreix M, Moreau PL, Bailone A, Galibert F, Battista JR, Walker GC, Devoret R. 1989. New recA mutations that dissociate the various RecA protein activities in Escherichia coli provide evidence for an additional role for RecA protein in UV mutagenesis. J Bacteriol 171:2415–2423. doi: 10.1128/jb.171.5.2415-2423.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Kato T, Shinoura Y. 1977. Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol Gen Genet 156:121–131. doi: 10.1007/BF00283484. [DOI] [PubMed] [Google Scholar]
- 420.Woodgate R, Sedgwick SG. 1992. Mutagenesis induced by bacterial UmuDC proteins and their plasmid homologs. Mol Microbiol 6:2213–2218. doi: 10.1111/j.1365-2958.1992.tb01397.x. [DOI] [PubMed] [Google Scholar]
- 421.McKenzie GJ, Harris RS, Lee PL, Rosenberg SM. 2000. The SOS response regulates adaptive mutation. Proc Natl Acad Sci USA 97:6646–6651. doi: 10.1073/pnas.120161797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.McKenzie GJ, Lee PL, Lombardo MJ, Hastings PJ, Rosenberg SM. 2001. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Mol Cell 7:571–579. doi: 10.1016/s1097-2765(01)00204-0. [DOI] [PubMed] [Google Scholar]
- 423.Petrosino JF, Galhardo RS, Morales LD, Rosenberg SM. 2009. Stress-induced β-lactam antibiotic resistance mutation and sequences of stationary-phase mutations in the Escherichia coli chromosome. J Bacteriol 191:5881–5889. doi: 10.1128/JB.00732-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Pribis JP, Garcia-Villada L, Zhai Y, Lewin-Epstein O, Wang AZ, Liu J, Xia J, Mei Q, Fitzgerald DM, Bos J, Austin RH, Herman C, Bates D, Hadany L, Hastings PJ, Rosenberg SM. 2019. Gamblers: an antibiotic-induced evolvable cell subpopulation differentiated by reactive-oxygen-induced general stress response. Mol Cell 74:785–800.e7. doi: 10.1016/j.molcel.2019.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Shee C, Gibson JL, Darrow MC, Gonzalez C, Rosenberg SM. 2011. Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc Natl Acad Sci USA 108:13659–13664. doi: 10.1073/pnas.1104681108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Rangarajan S, Woodgate R, Goodman MF. 1999. A phenotype for enigmatic DNA polymerase II: a pivotal role for pol II in replication restart in UV-irradiated Escherichia coli. Proc Natl Acad Sci USA 96:9224–9229. doi: 10.1073/pnas.96.16.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Pomerantz RT, Goodman MF, O’Donnell ME. 2013. DNA polymerases are error-prone at RecA-mediated recombination intermediates. Cell Cycle 12:2558–2563. doi: 10.4161/cc.25691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Pomerantz RT, Kurth I, Goodman MF, O’Donnell ME. 2013. Preferential D-loop extension by a translesion DNA polymerase underlies error-prone recombination. Nat Struct Mol Biol 20:748–755. doi: 10.1038/nsmb.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Little JW. 1984. Autodigestion of LexA and phage lambda repressors. Proc Natl Acad Sci USA 81:1375–1379. doi: 10.1073/pnas.81.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Burckhardt SE, Woodgate R, Scheuermann RH, Echols H. 1988. UmuD mutagenesis protein of Escherichia coli: overproduction, purification, and cleavage by RecA. Proc Natl Acad Sci USA 85:1811–1815. doi: 10.1073/pnas.85.6.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Nohmi T, Battista JR, Dodson LA, Walker GC. 1988. RecA-mediated cleavage activates UmuD for mutagenesis: mechanistic relationship between transcriptional derepression and posttranslational activation. Proc Natl Acad Sci USA 85:1816–1820. doi: 10.1073/pnas.85.6.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Shinagawa H, Iwasaki H, Kato T, Nakata A. 1988. RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc Natl Acad Sci USA 85:1806–1810. doi: 10.1073/pnas.85.6.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Woodgate R, Rajagopalan M, Lu C, Echols H. 1989. UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD′. Proc Natl Acad Sci USA 86:7301–7305. doi: 10.1073/pnas.86.19.7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Jiang Q, Karata K, Woodgate R, Cox MM, Goodman MF. 2009. The active form of DNA polymerase V is UmuD′2C·RecA′·ATP. Nature 460:359–363. doi: 10.1038/nature08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Fujii S, Fuchs RP. 2020. A comprehensive view of translesion synthesis in Escherichia coli. Microbiol Mol Biol Rev 84:e00002-20. doi: 10.1128/MMBR.00002-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Gruber AJ, Erdem AL, Sabat G, Karata K, Jaszczur MM, Vo DD, Olsen TM, Woodgate R, Goodman MF, Cox MM. 2015. A RecA protein surface required for activation of DNA polymerase V. PLoS Genet 11:e1005066. doi: 10.1371/journal.pgen.1005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Corzett CH, Goodman MF, Finkel SE. 2013. Competitive fitness during feast and famine: how SOS DNA polymerases influence physiology and evolution in Escherichia coli. Genetics 194:409–420. doi: 10.1534/genetics.113.151837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Yeiser B, Pepper ED, Goodman MF, Finkel SE. 2002. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc Natl Acad Sci USA 99:8737–8741. doi: 10.1073/pnas.092269199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ. 2017. Antibiotic tolerance facilitates the evolution of resistance. Science 355:826–830. doi: 10.1126/science.aaj2191. [DOI] [PubMed] [Google Scholar]
- 440.Norton MD, Spilkia AJ, Godoy VG. 2013. Antibiotic resistance acquired through a DNA damage-inducible response in Acinetobacter baumannii. J Bacteriol 195:1335–1345. doi: 10.1128/JB.02176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Ho C, Kulaeva OI, Levine AS, Woodgate R. 1993. A rapid method for cloning mutagenic DNA repair genes: isolation of umu-complementing genes from multidrug resistant plasmids R391, R446B, and R471A. J Bacteriol 175:5411–5419. doi: 10.1128/jb.175.17.5411-5419.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Mead S, Vaisman A, Valjavec-Gratian M, Karata K, Vandewiele D, Woodgate R. 2007. Characterization of polV(R391): a Y-family polymerase encoded by rumA′B from the IncJ conjugative transposon, R391. Mol Microbiol 63:797–810. doi: 10.1111/j.1365-2958.2006.05561.x. [DOI] [PubMed] [Google Scholar]
- 443.Ojha D, Jaszczur MM, Sikand A, McDonald JP, Robinson A, van Oijen AM, Mak CH, Pinaud F, Cox MM, Woodgate R, Goodman MF. 2022. Host cell RecA activates a mobile element-encoded mutagenic DNA polymerase. Nucleic Acids Res 50:6854–6869. doi: 10.1093/nar/gkac515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Pham P, Wood EA, Cox MM, Goodman MF. 18 April 2023. RecA and SSB genome-wide distribution in ssDNA gaps and ends in Escherichia coli. Nucleic Acids Res . doi: 10.1093/nar/gkad263. [DOI] [PMC free article] [PubMed]
- 445.Midgley-Smith SL, Dimude JU, Taylor T, Forrester NM, Upton AL, Lloyd RG, Rudolph CJ. 2018. Chromosomal over-replication in Escherichia coli recG cells is triggered by replication fork fusion and amplified if replichore symmetry is disturbed. Nucleic Acids Res 46:7701–7715. doi: 10.1093/nar/gky566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Vink C, Rudenko G, Seifert HS. 2012. Microbial antigenic variation mediated by homologous DNA recombination. FEMS Microbiol Rev 36:917–948. doi: 10.1111/j.1574-6976.2011.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Palmer GH, Bankhead T, Seifert HS. 2016. Antigenic variation in bacterial pathogens. Microbiol Spectr 4:VMBF-0005-2015. doi: 10.1128/microbiolspec.VMBF-0005-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Prister LL, Xu J, Seifert HS. 2019. A double-strand break does not promote Neisseria gonorrhoeae pilin antigenic variation. J Bacteriol 201:e00256-19. doi: 10.1128/JB.00256-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Xu J, Seifert HS. 2018. Analysis of pilin antigenic variation in Neisseria meningitidis by next-generation sequencing. J Bacteriol 200:e00465-18. doi: 10.1128/JB.00465-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Mercier R, Petit M-A, Schbath S, Robin S, El Karoui M, Boccard F, Espeli O. 2008. The MatP/matS site-specific system organizes the terminus region of the E. coli chromosome into a macrodomain. Cell 135:475–485. doi: 10.1016/j.cell.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 451.Nolivos S, Upton AL, Badrinarayanan A, Muller J, Zawadzka K, Wiktor J, Gill A, Arciszewska L, Nicolas E, Sherratt D. 2016. MatP regulates the coordinated action of topoisomerase IV and MukBEF in chromosome segregation. Nat Commun 7:10466. doi: 10.1038/ncomms10466. [DOI] [PMC free article] [PubMed] [Google Scholar]