

Abstract
Myocardial infarctions (MIs) kickstart an intense inflammatory response resulting in extracellular matrix (ECM) degradation, wall thinning, and chamber dilation that leaves the heart susceptible to rupture. Reperfusion therapy is one of the most effective strategies for limiting adverse effects of MIs, but is a challenge to administer in a timely manner. Late reperfusion therapy (LRT; 3 + hours post-MI) does not limit infarct size, but does reduce incidences of post-MI rupture and improves long-term patient outcomes. Foundational studies employing LRT in the mid-twentieth century revealed beneficial reductions in infarct expansion, aneurysm formation, and left ventricle dysfunction. The mechanism by which LRT acts, however, is undefined. Structural analyses, relying largely on one-dimensional estimates of ECM composition, have found few differences in collagen content between LRT and permanently occluded animal models when using homogeneous samples from infarct cores. Uniaxial testing, on the other hand, revealed slight reductions in stiffness early in inflammation, followed soon after by an enhanced resistance to failure for cases of LRT. The use of one-dimensional estimates of ECM organization and gross mechanical function have resulted in a poor understanding of the infarct’s spatially variable mechanical and structural anisotropy. To resolve these gaps in literature, future work employing full-field mechanical, structural, and cellular analyses is needed to better define the spatiotemporal post-MI alterations occurring during the inflammatory phase of healing and how they are impacted following reperfusion therapy. In turn, these studies may reveal how LRT affects the likelihood of rupture and inspire novel approaches to guide scar formation.
Keywords: Myocardial infarction , Reperfusion therapy, Inflammation, Extracellular matrix, Collagen, Biomechanics
Introduction
Coronary artery occlusion leads to myocardial infarction (MI, or a "heart attack"), irreparable cardiomyocyte damage, and impaired left ventricle (LV) function for nearly a million Americans each year (Tsao et al. 2022). Following coronary artery occlusion, an intense inflammatory response occurs in infarcted myocardium. This post-MI inflammatory response is necessary for removal of necrotic cardiomyocytes, scar formation, and long-term healing, but leaves the infarcted myocardium defenseless against rising LV volumes and pressures during the first week post-MI. This inflammation and subsequent remodeling both contribute to a heightened risk of ventricular rupture during this timeframe (Helpap et al. 2008; Hutchins et al. 2002; W. Roberts et al. 2015; M. Sun et al. 2004).
Reperfusion therapy (RT) is the restoration of blood flow to tissues or organs suffering from a state of ischemia, or insufficient blood flow and oxygenation. Even when administered several hours post-MI, RT is one of the most effective strategies for managing and limiting the adverse effects of MIs, including ventricular rupture (Berger et al. 1999; Gao et al. 2012; Honda et al. 2014; LATE 1993; Lawton et al. 2022; Rentrop and Feit 2015; Van De Werf 2014). While RT has been shown to rescue myocardium and alleviate negative post-MI changes by restoring coronary blood flow, the effectiveness of this strategy depends upon the timeliness of the procedure (Berger et al. 1999; Gao et al. 2012; Jugdutt 1997; Nakatani et al. 2003; Reduto, Smalling, et al. 1981a, b). Administering RT early enough to achieve these benefits can be a challenge for many clinics, especially those serving patients from rural communities (Bhuyan et al. 2013; Cohen et al. 2010; de Villiers and Riley 2020; Gharacholou et al. 2010; Loccoh et al. 2022). Late reperfusion therapy (LRT; ≥ 3 h post-MI) is more common clinically and fails to limit infarct size (Boyle and Weisman 1993; Hale and Kloner 1987, 1988; Hochman and Choo 1987; Jugdutt and Michorowski 1987), but does limit infarct expansion and ventricular rupture between 3 and 5 days post-MI in humans (Becker et al. 1999; Helpap et al. 2008; W. Roberts et al. 2015). Despite the clinical benefits afforded via LRT, the mechanism by which it limits ventricular rupture remains undefined. The objective of this review is to compile and summarize studies discussing the structural and mechanical changes occurring in myocardium during the inflammatory phase of post-MI healing. Our review then identifies areas of research requiring further attention during this phase, particularly the effects of reperfusion therapy on myocardial structure and mechanics, and its potential implications for limiting ventricular rupture.
The healthy myocardial extracellular matrix
In mammals, the healthy myocardial extracellular matrix (ECM) is a three-dimensional network of primarily collagen fibers. It provides the heart with tensile strength and resilience against large deformations at the ultrastructural level, and, at the structural level, it forms supportive architecture for resident cells, enabling the transferal of forces across cellular membranes (Halper and Kjaer 2014; Silva et al. 2021; Whittaker et al. 1991). The myocardial ECM is dynamic, undergoing remodeling to maintain relatively homeostatic stress–strain regimes for resident cells under healthy and pathological circumstances (Frey et al. 2004; Kanekar et al. 1998; Souders et al. 2009). Matricellular proteins present in the extracellular environment can also influence growth and remodeling processes by modulating cell signaling and communication. These proteins are generally found in the interstitium and do not contribute to the mechanical integrity of the ECM (Bornstein 2009; Bornstein and Sage 2002; Frangogiannis 2012).
Fibrillar collagen types I and III form the bulk of the healthy myocardial ECM and are deposited and maintained by local fibroblasts (Silva et al. 2021; Souders et al. 2009). Typically, collagen type I aggregates into thicker fibers, whereas collagen type III forms thinner fibers (Weber 1989). Both collagen I and III fibers run parallel to the cardiomyocytes they surround (Pope et al. 2008) (Fig. 1), occasionally branching off normal to cells to form intermyocyte collagen struts (Jugdutt et al. 1996; Sato et al. 1983; Whittaker et al. 1991). These fibers form the endomysium of the ECM around individual muscle fibers, the perimysium around muscle fiber bundles, and the epimysium around the entire cardiac muscle (Frangogiannis 2012, 2017). Collagens I and III are the best studied and characterized components of the myocardial ECM due to their high abundance, ubiquity, and impressive mechanical behavior. There are also other collagens in the heart, like collagen type IV, a critical structural protein forming the basement membranes of individual cardiomyocytes (Farhadian et al. 1996; Whittaker et al. 1991; H. Yang et al. 2014). Basement membranes are also formed of laminin, a large multi-functional glycoprotein contributing to ECM structure, cellular migration, and differentiation (Halper and Kjaer 2014), as well as fibronectin. Fibronectin is another multi-functional glycoprotein capable of taking on various roles depending on local mechanical and chemical stimuli, but is normally responsible for anchoring cellular integrin receptors to ECM fibers (Farhadian et al. 1996; Valiente-Alandi et al. 2018). This anchoring is essential for transducing extracellular mechanics and deformations to the internal cytoskeletal architecture of individual cells (Farhadian et al. 1996). Other notable structural components of the healthy myocardial ECM include elastin, which is present in minute amounts and contributes to the elastic behavior of the tissue during smaller deformations, as well as glycosaminoglycans (GAGs) and proteoglycans, which endow the tissue with compressive strength, an enhanced ability to retain water, and contribute to the incompressibility of the soft tissue (Christensen et al. 2019; DeLeon et al. 2012; M. Lindsey et al. 2018; Rienks et al. 2014).
Fig. 1.

The orientations of cardiomyocytes and collagen fibers vary transmurally in the healthy rodent LV, but are co-oriented throughout the heart wall. The predominance of collagen fibers and their orientations, imaged here using extended volume confocal microscopy, give rise to nonlinear anisotropic mechanical behavior. Reprinted from the American Journal of Physiology—Heart and Circulatory Physiology, Vol. 295, A. Pope et al., “Three-dimensional transmural organization of perimysial collagen in the heart,” pp. H1243-1252. Copyright (2008), with paid permission from The American Physiological Society
A primary role of the ECM is supporting and structuring the myocardium, which is dominated by cardiomyocytes, the functional and contractile cells of the heart. Cardiomyocytes contract when excited electrically, raising the pressure within the various chambers of the heart and driving blood flow throughout the circulatory system. Although long-lived and robust, these cells have negligible regenerative ability (Bergmann et al. 2015) and contribute minimally to the passive mechanical properties of myocardium (Hiesinger et al. 2012; Zhang et al. 2018); they can, however, undergo hypertrophy in response to a chronic demand for increased contractile force (Frey et al. 2004; Woodcock and Matkovich 2005). In addition to contractile cardiomyocytes, there are a number of other innate cells residing in the heart. Fibroblasts are responsible for remodeling and maintaining the ECM. These flat, spindle-shaped cells deposit structural proteins and also destroy these same proteins by the secretion of matrix metalloproteinases (MMPs) (Kanekar et al. 1998; Shinde and Frangogiannis 2014). Immune cells, primarily macrophages (Epelman et al. 2014; Nahrendorf 2019; Pinto et al. 2012, 2016), serve as vigilant sentinels for the surrounding myocardium, rapidly alerting the peripheral immune system when pathogens or signs of ischemia are detected (Hulsmans et al. 2018; Medzhitov 2001; Meschiari et al. 2018; Nahrendorf et al. 2007). Finally, there are endothelial cells, which line the innermost layer of the heart and coronary vessels to provide a protective, semi-permeable barrier between the blood and myocardium (Gimbrone et al. 2000; Nadaud et al. 1996; Qiu and Tarbell 2000).
The extracellular architecture in healthy myocardium results in passive mechanical behavior (Fig. 2) stereotypical of soft biological tissues: nonlinear force–displacement curves (Chew et al. 1986; Demer and Yin 1983), mechanical anisotropy (Demer and Yin 1983; Gupta et al. 1994; Humphrey et al. 1990; Witzenburg et al. 2012), pronounced hysteresis during ex vivo mechanical testing on a time scale greater than a standard cardiac cycle (Demer and Yin 1983; Holzapfel et al. 2009; Humphrey et al. 1990; Rankin et al. 1977), and regional mechanical heterogeneity (Novak et al. 1994). Throughout the wall of the heart, collagen fibers are roughly co-oriented with cardiomyocytes (Pope et al. 2008): they are oriented longitudinally at the innermost surface of the endocardium (Fig. 1), rotate clockwise towards a circumferential alignment near the mid-wall of the heart, and continue rotating clockwise back to a nearly longitudinal orientation at the outermost surface of the epicardium (Streeter et al. 1969). The predominance of fibrillar collagens in the myocardial ECM, as well as their orientation, gives rise to both nonlinear force–displacement relationships and moderately anisotropic mechanical behavior, best demonstrated by greater equibiaxial stiffnesses and stresses (Fig. 2) in the circumferential direction of LV free wall tissue as opposed to the longitudinal direction (Demer and Yin 1983; Emery et al. 1997; Guccione et al. 1991; Humphrey et al. 1990; Sommer et al. 2015; Witzenburg et al. 2012). Elastin is also present in the myocardial ECM, although its relatively low abundance in the heart and co-location with collagen make its mechanical contribution during large, passive diastolic deformations unclear (Fomovsky et al. 2010). Like many soft tissues, myocardium exhibits both structural and mechanical heterogeneity, resulting in different stiffnesses, elastic constants, or constitutive model parameters for different regions of the heart. As Novak et al. (1994) pointed out, though, the qualitative mechanical behavior does not change drastically throughout the heart: despite regional quantitative differences, nearly all myocardium still exhibits some level of mechanical anisotropy and nonlinearity.
Fig. 2.

The microscopic structure and organization of myocardium’s healthy ECM gives rise to nonlinear force–displacement relationships, hysteresis, and anisotropic mechanical behavior with a preference for the circumferential (main fiber) direction during ex vivo equibiaxial testing. Each of these mechanical attributes can be seen in panel A (nonlinearity, hysteresis, anisotropy) and panel B (anisotropy, impressive passive extensibility), taken from equibiaxial extensions of human myocardium by Sommer et al. (2015). Reprinted from Acta Biomaterialia, Vol. 24, G. Sommer et al., “Biomechanical properties and microstructure of human ventricular myocardium,” pp. 172–192. Copyright (2015), with permission from Elsevier
Structural and mechanical changes following acute myocardial infarction
Acute MI occurs when necessary blood flow to the myocardium is interrupted, creating an imbalance between the supply and demand of oxygen for local cardiomyocytes. Within 30 s of infarction, the ischemic myocardium loses its contractile ability and begins to bulge during systole rather than contracting to drive blood flow (Tennant and Wiggers 1935). In a state of continued ischemia, intracellular levels of adenosine triphosphate are progressively depleted, ion pumps fail as intracellular calcium levels rise, the production of reactive oxygen species is upregulated, and each ischemic cell’s plasma membrane becomes increasingly more susceptible to bursting (Murphy and Steenbergen 2008; Tian et al. 2013). Should these conditions persist, the cardiomyocytes residing in the ischemic region die en masse, establishing a necrotic core surrounded by potentially salvageable myocardium. As the period of occlusion lengthens, the necrotic wave front creeps outward, claiming surrounding viable myocardium and further exacerbating infarct severity (Connelly et al. 1982; Reimer et al. 1977; Reimer and Jennings 1979a, b; Tian et al. 2013).
The infarcted and inflamed myocardial extracellular matrix
During periods of ischemia, cells in the infarct core, both cardiomyocytes and non-cardiomyocytes alike, release danger-associated molecular patterns (DAMPs) (De Haan et al. 2013; Prabhu and Frangogiannis 2016; Rienks and Papageorgiou 2016). Although resident macrophages in the infarct myocardium die as ischemia persists, nearby surviving macrophages, still serving diligently as myocardial sentinels, and circulating monocytes and neutrophils, acting as peripheral reserves capable of detecting DAMPs (Fig. 3), migrate towards the site of infarction, surrounding the infarct border first, then slowly working inwards towards the necrotic core (Bajpai et al. 2019; Gao et al. 2005; O’Rourke et al. 2019; Troidl et al. 2009). As these infiltrating immune cells collide with the encroaching necrotic wave front, they release MMPs that degrade and demolish the collagenous myocardial ECM (DeLeon-Pennell et al. 2017; Etoh et al. 2001; Herzog et al. 1998; Vanhoutte et al. 2006; Webb et al. 2006). Collagenous ECM degradation also results in accentuated immune cell migration that further encourages MMP secretion and activation, resulting in additional degradation (Okada et al. 2017). As the myocardial ECM is dismantled to make way for infiltrating macrophages and neutrophils, biologically active ECM remnants, or matricryptins (G. Davis 2010; de Castro Brás and Frangogiannis 2020; Ricard-Blum and Ballut 2011; Ricard-Blum and Salza 2014), are left behind in the interstitial space of the ischemic region during the first 3 – 4 days post-MI. Matricryptins serve multiple roles in the ensuing inflammatory response, but, most notably, may help attract immune cells and fibroblasts to the infarcted region (Adair-Kirk et al. 2003; Adair-Kirk and Senior 2008; Arslan et al. 2011; Wells et al. 2015). Freshly recruited macrophages, neutrophils, and fibroblasts are all capable of producing and secreting additional MMPs, continuing demolition of the myocardial ECM, both collagenous and non-collagenous components alike (Cavasin et al. 2004; Cleutjens et al. 1995; Danielsen et al. 1998; Etoh et al. 2001; Fang et al. 2007; Forrester et al. 1972; Heymans et al. 1999; Lu et al. 2004; Sahu et al. 2021; Vanhoutte et al. 2006; K. Wang et al. 2021a, b). Immune cells release pro-inflammatory cytokines, such as TNF-α, IL-1, and IL-6, and growth factors, like transforming growth factor-β (TGF-β) and vascular endothelial growth factor (VEGF), that work with extracellular debris to recruit more immune cells, modulate cellular phenotyping, and regulate inflammation (Bujak et al. 2008; Christia et al. 2013; DeLeon-Pennell et al. 2018; Fang et al. 2007; Frangogiannis 2022; Saxena et al. 2013; Silva et al. 2021). Notably, IL-1 and TNF-α also induce MMP production in multiple cell types (Cawston 1996). MMP activity is detectable within 10 min of coronary artery occlusion in pigs, likely reflecting activation of latent MMPs in the ischemic region, and increases steadily for the next several hours as immune cells migrate towards the site of infarction (Etoh et al. 2001; Van Wart and Birkedal-Hansen 1990).
Fig. 3.

Coronary artery occlusion, myocardial infarction, and prolonged ischemia result in an intense post-MI inflammatory response. In the setting of ischemia, cardiomyocytes lose their ability to contract to drive blood flow. Instead, these cells become necrotic, releasing DAMPs and pro-inflammatory cytokines into their interstitial environment. This attracts neutrophils, and macrophages follow, to the site of infarction. Infiltrating immune cells secrete MMPs that degrade the existing ECM to pave a path for immune cells to remove necrotic cardiomyocytes and ECM debris as inflammation persists
MMPs are a family of nearly 30 enzymatic proteins capable of degrading the structural proteins of the ECM. Although a number of MMPs are involved in post-MI remodeling, MMP-2 and MMP-9 are the best-studied facilitators of extracellular destruction during the inflammatory phase of post-MI healing (Cleutjens et al. 1995; DeLeon-Pennell et al. 2017; Etoh et al. 2001; Fang et al. 2007; Herzog et al. 1998; Heymans et al. 1999; Tao et al. 2004). MMP-2 and MMP-9 both exhibit collagenolytic activity (Hojo et al. 2001), and the expression and activation of these proteins have been seen to increase with longer periods of coronary occlusion in the myocardial interstitium. In humans, MMP-9 has been associated with greater LV remodeling, indicated by LV dilation, at 6 weeks post-MI (Agostoni and Banfi 2007). Similarly, plasma MMP-2 (Hojo et al. 2001), MMP-8 (Webb et al. 2006), and MMP-9 (Owolabi et al. 2020; Webb et al. 2006) levels increased post-MI, indicating a likely relationship between MMPs and rates of post-MI ventricular remodeling. In a study of male mice, a high rate of rupture occurred within 3 – 5 days of infarction, corresponding with the temporal expression of MMP-9 (Tao et al. 2004). A temporal relationship between neutrophil infiltration and MMP-9 levels was observed in mice (Tao et al. 2004), as well as in humans (Agostoni and Banfi 2007), implicating a role for neutrophils in MMP secretion and post-MI remodeling. MMPs play a key role in the early degradation of the ECM post-MI (Owolabi et al. 2020), and while these enzymes are present in humans as long as 4 weeks post-MI, their activity is most notable within the first week post-MI (Webb et al. 2006).
While the existing extracellular architecture is being dismantled, a provisional matrix enriched with fibrin, fibronectin, and various matricellular proteins is established as scaffolding and support for the burgeoning population of interstitial cells (Dobaczewski et al. 2006; Frangogiannis 2017; Ulrich et al. 1997). This plasma-derived provisional matrix supports infiltrating cells, attracts additional immune cells, and further modulates the inflammatory response (Bashey et al. 1992; Corbett and Schwarzbauer 1998; Dobaczewski et al. 2006; Smiley et al. 2001). In addition to attracting and supporting these cells, the fibrin components of the provisional matrix are capable of binding growth factors and cytokines to their heparin-binding domain, sequestering these molecules until needed (Barker and Engler 2017; Martino et al. 2013; Schultz and Wysocki 2009). While the provisional matrix is essential for modulating healing, it does not offer the same mechanical support as the innate ECM (Connelly et al. 1992; Knowlton et al. 1992). In fact, many of the proteins deposited into interstitial spaces during inflammation do not contribute structurally, but act primarily to modulate the inflammatory response (Bornstein and Sage 2002; Frangogiannis 2017). This is particularly true of matricellular proteins like osteopontin (Murry et al. 1994; Tamaoki et al. 2005; Trueblood et al. 2001), secreted protein acidic and rich in cysteine (SPARC) (Deckx et al. 2019; Harris et al. 2011), thrombospondin-1 (Frangogiannis et al. 2005), and biglycan (Doi et al. 2000; Svensson et al. 1995; Westermann et al. 2008), all of which are upregulated within the first week following MI. Osteopontin and biglycan, two matricellular proteins associated with promoting collagen synthesis and eventual helix assembly (Doi et al. 2000; Murry et al. 1994; Weis et al. 2005; Westermann et al. 2008), are upregulated early, within the first few days post-MI in rodent models, and are necessary for eventual scar formation. Thrombospondin-1 and SPARC are upregulated between 3 and 7 days post-MI in rodents and may influence the organization of the provisional matrix (Frangogiannis et al. 2005; Harris et al. 2011; Schellings et al. 2009). It is worth noting that many of these matricellular proteins are understudied and a full understanding of their contextual actions following MI has not yet been developed (Bornstein 2009; Deckx et al. 2019; Frangogiannis 2017).
Eventually, the provisional plasma-derived matrix is dissolved, making room for a more organized cell-derived provisional matrix enriched with fibronectin and hyaluronan (Dobaczewski et al. 2006; Knowlton et al. 1992; Motley et al. 2016; Ulrich et al. 1997; Welch et al. 1990). Again, this cell-derived provisional matrix contributes less to the passive mechanics of the infarcted myocardium; instead, it attracts additional reparative cells and promotes fibroblast and macrophage proliferation and phenotyping, marking the transition to the proliferative phase of post-MI healing. The proliferative phase of healing and the following phase, scar maturation, are outside the scope of this review. For more information on the proliferative and maturation phases, we recommend excellent discussions of the longer-term dynamic structure and biomechanical behavior of an infarcted heart (Holmes et al. 2005; Witzenburg and Holmes 2017), depictions of the extracellular environment and various interstitial cells contributing to post-infarction healing (Frangogiannis 2017; Shinde and Frangogiannis 2014; Takemura and Fujiwara 2004), and the process and implications of scar maturation (Richardson et al. 2015; Y. Sun and Weber 2000; Talman and Ruskoaho 2016).
Myocardial extracellular mechanics and rupture
After weeks of healing, infarcted myocardium will eventually be replaced with a stiff, dense collagenous scar, but, prior to this scar formation, the initial inflammatory response demolishes the existing collagenous ECM and leaves behind a mechanically compromised infarct (Cannon et al. 1983; Connelly et al. 1985, 1992; Fang et al. 2007). This is best demonstrated by an increased risk of ventricular rupture, which occurs most frequently in humans within the first 3 – 5 days following infarction (Becker et al. 1999; Helpap et al. 2008; W. Roberts et al. 2015). Multiple studies have reported an early decrease in post-MI myocardial passive stiffness (Becker et al. 1999; Gao et al. 2005; Helpap et al. 2008; M. Sun et al. 2004), though not all (Diamond and Forrester 1972; Laird and Vellekoop 1977; Przyklenk et al. 1987). The altered extracellular architecture has not been shown to affect the nonlinear elasticity of the tissue (Akaishi et al. 1986; Gupta et al. 1994; Sirry et al. 2016; Voorhees et al. 2015). Anisotropy is also preserved (Brazile et al. 2021; Gupta et al. 1994; Holmes et al. 1997; Sirry et al. 2016; Whittaker et al. 1989), but reports suggest this may depend on the animal model, as well as infarct topography and location (Fomovsky et al. 2012; Fomovsky and Holmes 2010). Studies indicate large animals, like pigs and canines, develop anisotropic infarcts (Brazile et al. 2021; Gupta et al. 1994; Holmes et al. 1997; Sirry et al. 2016; Whittaker et al. 1989). In rats, however, both isotropic (Fomovsky and Holmes 2010) and anisotropic infarcts (Sirry et al. 2016) have been reported. This latter study (Sirry et al. 2016), as well as work by Gupta et al. (1994) on ovine infarcts, reported gradual post-MI increases in longitudinal stiffnesses, but still greater circumferential stiffnesses, leading these groups to conclude some degree of the infarcted myocardium’s innate mechanical anisotropy was preserved.
As aforementioned, not every study has reported decreased stiffness in the first few days following MI. In fact, some have reported moderate increases in stiffness, expressed in uniaxial tension (Laird and Vellekoop 1977), pressure-segment length curves (Pirzada et al. 1976), pressure–volume curves (Diamond and Forrester 1972), and non-invasive shear wave imaging (Pernot et al. 2016). These reports can seem confounding, as it is well established that MMPs are destroying the existing collagenous ECM as infiltrating immune cells lead a relentless inflammatory effort. It is possible that early increases in edema and water content (Amirhamzeh et al. 1997; Fishbein et al. 1978b, 1978a; Ghugre et al. 2017; Pfeffer et al. 1979; Waldenström et al. 1991), led by an upregulation of matricellular proteins and proteoglycans (Drobnik et al. 2013; Huebener and Frangogiannis 2006; X. Wang et al. 2019), may contribute to apparent increases in stiffness and changes in geometry and wall thickness of the LV (Holmes et al. 2005; Tyberg et al. 1974). Since both decreases and increases in stiffness have been reported during the timeframe that ventricular rupture, a local phenomenon, also commonly occurs, researchers have considered regional alterations to ECM mechanics and structure as another possible factor contributing to rupture (Cannon et al. 1983; Tao et al. 2004; Troidl et al. 2009; Whittaker et al. 1991).
Perhaps the most interesting change following infarction is the sudden creation of mechanical and structural heterogeneity around the infarcted zone. The presence of an infarct borderzone has been debated since the late 1960s when Cox et al. (1968), Lushnikov et al. (1963), and Vihert et al. (1971) observed regions of moderately damaged myocardium associated with decreased enzyme activity surrounding the central core of infarcts that developed within 2 – 7 days of infarction. These results were subsequently questioned by Factor et al. (1978) and Marcus et al. (1975), both of whom found minimal evidence of a clearly defined borderzone in canine infarcts, as well as Barlow and Chance (1976), who claimed any region separating infarcted and remote myocardium must be “quite small.” Histological studies from Fishbein et al. (1980) and Gottlieb et al. (1981) in the 1980s added to the pool of conflicting findings by again demonstrating notable borderzones in rats and canines, respectively, within the first 3 days post-MI; these studies followed similar reports from Vokonas et al. (1978) just a few years earlier. In 1981, Hearse and Yellon (1981a) published a review of pertinent borderzone literature, eventually deciding that an identifiable borderzone in the lateral plane was improbable as it was likely less than 2 mm wide and potentially “as little as the dimensions of one cell,” but the existence of a borderzone in the transmural plane of the infarct was less certain. A few years later, Sakai et al. (1985) and Gallagher et al. (1986) reported early depressed mechanical function in the regions surrounding infarcts in pigs and canines, respectively, and in 2006, Berry et al. (2006) utilized atomic force microscopy to estimate the spatial distribution of the elastic modulus within infarcts. Although these measurements were obtained at 2 weeks post-infarction in rats, they depicted a clear transitional region, approximately 5 mm in width, from the stiff infarct core to the more compliant remote myocardium (Berry et al. 2006). More recently, spatiotemporal transcriptome analyses were used to study protein and gene expression in murine infarcts (Calcagno et al. 2022; Yamada et al. 2022), revealing the presence of a narrow borderzone within the first week post-MI characterized by abundant mechano-sensing gene expression that may help regulate LV remodeling.
While the existence of an infarct borderzone is still debated (Hayat and Kramann 2022), one thing is certain: MI results in drastic, malignant changes to the affected cardiomyocytes and their supporting connective tissue within minutes of ischemia’s onset (Etoh et al. 2001; Tennant and Wiggers 1935). As inflammation sets in and the innate ECM is progressively demolished, the risk of ventricular rupture rises. Rupture most commonly occurs between 3 and 5 days post-MI (Becker et al. 1999; Helpap et al. 2008; M. Sun et al. 2004), when the full effects of post-MI immune cell infiltration, MMP activation, and early adverse remodeling have occurred. Rupture carries a high mortality rate and requires prompt medical attention and surgical intervention for an effective recovery (Matteucci et al. 2019). Severe hypotension is the cardinal manifestation of rupture, but many patients also suffer sudden death (Koklu et al. 2017; Varghese and Ohlow 2019). Although advances in post-MI treatment have decreased rupture’s prevalence, it remains a devastating, fatal, and poorly understood post-MI complication accounting for a third of post-MI in-hospital deaths (Ma et al. 2022; Nakamura et al. 1992).
What is reperfusion therapy?
Since the late 1970s, coronary artery occlusion has been widely accepted as the cause of MI (Clark et al. 1936; Herrick 1912; Rentrop and Feit 2015). There was debate, however, in the 1950s, 1960s, and 1970s about whether occlusion was the primary cause of acute MI or merely a consequence (Baroldi 1965; Friedberg and Horn 1939; Miller et al. 1951; Oliva and Breckinridge 1977; W. C. Roberts 1971; Sherry 1989). This confusion resulted in nebulous guidelines for managing MIs, best demonstrated by an ambivalence towards practices aiming to re-open occluded coronary arteries, deemed reperfusion therapies, for several decades (Rentrop and Feit 2015; Van De Werf 2014). Despite early controversies, reperfusion therapy (RT) is one of the most effective MI treatments. Studies have shown it reduces mortality rates within the first month post-MI (Berger et al. 1999; Fibrinolytic Therapy Trialists’ (FTT) (1994); LATE 1993; Yusuf et al. 1985), preserves left ventricular function and geometry (Harrison et al. 1993; Jugdutt 1997; Kereiakes et al. 1991; Reduto, Freund, et al., 1981; Reduto, Smalling, et al. 1981a, b; Ward et al. 1997), and limits instances of ventricular rupture (Bates 2014; Gao et al. 2012; Nakatani et al. 2003).
Manual reperfusion of the affected myocardium can be achieved pharmacologically or mechanically. Fibrinolytic therapies have traditionally relied on the administration of streptokinase (Chazov et al. 1976; Fletcher et al. 1958, 1959; Ganz et al. 1981; Jinatongthai et al. 2017; Van De Werf 2014), but other drugs provide comparable effects (Gruppo Italiano per lo Studio 1986; Simari et al. 1994; Van De Werf 2014; Van De Werf et al. 1984). Currently, percutaneous coronary intervention (PCI), a mechanical approach involving the forced opening of occluded vessels, is the recommended revascularization technique when clinically feasible (Ibanez et al. 2018; Lawton et al. 2022). Fibrinolytic therapies may also be administered en route to a facility capable of PCI, coined “facilitated PCI,” but benefits of this tag-team approach have not yet been defined (Van De Werf 2014). Although it is a fairly straightforward management strategy, RT has some risks and limitations. Pharmacological approaches may lead to internal hemorrhaging or subpar coronary vessel patency rates. PCI avoids these concerns, but requires a trained physician and catheterization facility. The effectiveness of RT also depends on the amount of elapsed time between the initial onset of ischemia and re-opening of the occluded coronary artery (Maroko et al. 1971), prompting some to describe PCI with the aphorism, “time is muscle.” Various groups have observed differences in early and late reperfusion, typically defined as 3 + h post-MI, and have reached similar conclusions: although LRT does not reduce infarct size, infarct transmurality, or offer the same cardioprotective effects as early reperfusion therapy (ERT), it provides some long-term benefits to the patient (Althaus et al. 1977; Boyle and Weisman 1993; Christia et al. 2013; Hale and Kloner 1987; Hochman and Choo 1987; Nakagawa et al. 2008; Takemura et al. 2009) and may reduce the incidence of ventricular rupture. In general, post-MI changes in immune cell mobilization, MMP activation, and ECM destruction have been well-studied for cases of permanent coronary artery occlusion (PO). The effects of RT are less clear. For those interested, the historical context of RT, the developments and discoveries that made it possible, and current recommendations for its employment are thoroughly summarized and discussed by Van de Werf (2014), Rentrop and Feit (2015), and Lawton et al. (2022).
Infarct size and shape
Usually expressed as a ratio between the area, length, or mass of the infarcted tissue relative to the whole LV (Hale and Kloner 1988; Morita et al. 1993; Schuster and Bulkley 1979), infarct size is a simple measure of severity, where larger infarcts have been associated with congestive heart failure, reduced cardiac output, and elevated filling pressures (Pfeffer et al. 1979). Many studies (Hale and Kloner 1987, 1988; Hochman and Choo 1987; Morita et al. 1993) observed beneficial effects of ERT on limiting infarct size and expansion, a measure of LV wall thinning and chamber dilation (Hale and Kloner 1988; Leong et al. 2021). Conversely, Hochman and Choo (1987), Hale and Kloner (1988), Boyle and Weisman (1993), and Jugdutt (1997) found that LRT (defined individually by these groups as anywhere from 1.5 to 8 h post-MI) did not limit infarct size or its transmural extent. LRT did tend to limit infarct expansion, though, especially during 4 – 7 days post-MI (Boyle and Weisman 1993).
In a seminal study, Schuster and Bulkley posited a connection between infarct expansion and ventricular rupture in humans (Schuster and Bulkley 1979). This theory was corroborated by Hochman and Choo (1987) in the late 1980s, who noted greater rates of aneurysm formation in rats, supporting a link between rupture risk and infarct expansion. Hale and Kloner (1988), Boyle and Weisman (1993), and Jugdutt (1997) also determined that, like ERT, LRT limited the degree of LV cavity dilatation, scar thinning, and hypertrophy of nearby surviving myocardium. These benefits of LRT, namely a reduction of infarct expansion and preserved LV function, may limit wall stresses and adverse remodeling, instances of ventricular rupture, and mortality rates (GUSTO 1993; Lambert et al. 2010; LATE 1993; Nepper-Christensen et al. 2021). As many of the classic studies concerned with infarct borderzones typically relied on PO, however, the effects of LRT on borderzone topography are still unknown (Berry et al. 2006; Calcagno et al. 2022; Cox et al. 1968; Factor et al. 1978; Fishbein et al. 1980; Gallagher et al. 1986; Gottlieb et al. 1981; Marcus et al. 1975; Sakai et al. 1985; Vokonas et al. 1978; Yamada et al. 2022).
Structural changes to the reperfused myocardium
Post-MI tissue swelling, attributed to hemorrhage or edema, is a common concern following RT. Early work from Pirzada et al. (1978) observed an apparent stiffening of the canine LV following LRT (6 h post-MI), which the authors contributed to likely increases in edema or myofibrillar contracture, although no comparisons to ERT were made and no structural mechanisms were explicitly identified. Several years later, Roberts et al. (1983) noted significantly more hemorrhaging in cases of LRT (4 h post-MI) when compared to PO in canines, particularly during the first day following MI. They also observed the presence of a rim of necrotic, but non-hemorrhagic tissue, surrounding the hemorrhagic region. Increases in proteoglycan and hyaluronan content have been proposed as possible drivers of swelling, but this has not been confirmed.
Hydroxyproline is a unique component of fibrillar collagen promoting fiber stability (Ramachandran et al. 1973; Xu et al. 2019), and hydroxyproline assays provide a convenient estimate of collagen content in a sample. Roberts et al. (1983) measured similar hydroxyproline concentrations in PO and LRT models at 2 weeks post-MI and concluded they had comparable collagen content (Fig. 4). Building on studies concerning ECM architecture during periods of ischemia (Factor et al. 1987; Sato et al. 1983; Whittaker et al. 1989, 1991; Zhao et al. 1987), Wiggers et al. (1997) conducted hydroxyproline assays roughly 3 h following LRT (6 h post-MI) in pigs. There were no differences in hydroxyproline content from various regions of the heart in comparison to controls. There was also minimal evidence of collagen degradation products (PIIINP and ICTP), prompting them to conclude that extensive ECM degradation occurs later on during inflammation or that their methods were insufficient to detect damage (Wiggers et al. 1997). Hydroxyproline assays produce a one-dimensional measure of collagen content and typically do not distinguish between different fibrillar collagen types, do not offer information about collagen crosslinking, or do not quantify the integrity of the collagenous ECM. These limitations were highlighted by Connelly et al. (1985) in their study of PO, ERT (1 h post-MI), and LRT (3 h post-MI) in rabbits. At 3 weeks post-MI, PO, ERT, and LRT samples all had similar levels of collagen content, as quantified through hydroxyproline assays. However, ERT contributed to the formation of a thicker and more muscular scar, whereas PO and LRT resulted in thin, stiff collagenous scars containing few cardiomyocytes. Uniaxial tensile testing at this time point revealed apparent disparities between the structural and mechanical changes occurring in reperfused myocardium. The ERT and LRT samples exhibited comparable stiffnesses, but were more compliant than PO samples, allowing Connelly et al. (1985) to conclude collagen content alone cannot fully explain disparities in mechanical performance. Aldol, an intramolecular crosslink found in collagen, was present in significantly greater amounts in PO samples, indicating collagen crosslinking may be more useful for explaining differences in post-MI mechanics. It is worth noting that Connelly et al. (1985) made measurements at 3 weeks post-MI, long after inflammation has resolved. Additionally, samples were oriented longitudinally and taken from the infarct core, effectively neglecting any effects of structural and mechanical anisotropy or heterogeneity.
Fig. 4.

Structural constituent expression varies temporally for cases of PO (solid line) and LRT (dotted lines). For PO models, Collagen I and III content decreases at the start of inflammation (), then increases drastically at the end of inflammation (). Collagen I and III both appear earlier in LRT models, but by the end of inflammation, the overall content is similar to PO (Connelly et al. 1985, 1992; Fang et al. 2007; Knowlton et al. 1992; Lerman et al. 1983). Collagen IV, an important component of the basement membrane, increases for both PO and LRT models, but this increase typically occurs sooner in LRT (Knowlton et al. 1992; Morishita et al. 1996). Fibronectin expression increases for both PO and LRT models. In LRT models, however, expression is shifted forward temporally (Carlyle et al. 1997; Knowlton et al. 1992). For PO, elastin is degraded while hyaluronan content increases (Petz et al. 2019; Rienks et al. 2014; Skjøt-Arkil et al. 2013; Waldenström et al. 1991; Yu et al. 2018). These constituents have not been well-studied and defined for cases of LRT. All axes rely on arbitrary units, and curves are qualitative representations of the studies summarized within this review. The start of inflammation () is defined as immediately following MI and before the application of LRT. The end of inflammation () is defined by mass migration and proliferation of fibroblasts in the infarcted region
Following MI, the application of RT may alter MMP expression and collagen degradation. Some studies actually show MMP activity is unchanged or, in some cases, accentuated following RT. In humans, MMP-1 levels increased 5 days post-MI despite ERT (Hirohata et al. 1997). In porcine hearts subjected to ischemia and ERT, a membrane-type MMP found in the myocardium, called MT1-MMP, increased in a time-dependent manner (Deschamps et al. 2005). In another study using a porcine model of ischemia and LRT (6 h of occlusion and 3 h of reperfusion), gelatinolytic and collagenolytic activities increased due to rising levels of MMP-9 and MMP-1 following reperfusion (Danielsen et al. 1998). Additionally, MMP-9 increased by approximately 200% after 1 h of ischemia and 5 h of reperfusion in canines (M. Lindsey et al. 2001), and MMP-9 activity has also been shown to be an avid marker of infarct size and a risk factor for heart failure in post-ERT patients (Wagner et al. 2006). Other research groups have found more beneficial effects of RT on MMP activity. Namely, MMP activity has been reported to decrease with ERT in both porcine and rodent models (M. L. Lindsey and Zamilpa 2012; Lu et al. 2000). In rats, MMP-1 (measured at day 7 post-MI), MMP-2 (measured at day 3 and day 7 post-MI), and MMP-9 (measured at day 1 post-MI) activity were all reduced in ERT hearts vs. PO hearts (M. L. Lindsey and Zamilpa 2012). In addition, LRT (150 min post-MI) has been reported to reduce MMP levels when compared to PO. Carlyle et al. (1997) reported MMP-1 and MMP-2 were reduced by 50% and 60%, respectively, at 7 days post-MI, and MMP-9 by 55% and 84% at 24 and 48 h, respectively (Fig. 5). Of these, MMP-9 is of particular interest because this decrease occurred during the inflammatory phase of post-MI healing, whereas MMP-1 and MMP-2 decreased during the proliferative phase. Since rupture typically occurs during the inflammatory phase, when the ECM is being broken down (Tao et al. 2004), MMP activity measurements taken during this period of time may be more relevant for ventricular rupture.
Fig. 5.

In rodent infarcts, LRT (2.5 h post-MI), reduced MMP-1, -2, and -9 activity. MMP activity occurring in non-reperfused (closed circles) and LRT (open circles) infarcts are compared at days 1, 2, 3, and 7 post-MI for MMP-1 (a) and MMP-2 (both unreduced form—62 kDa (b), and reduced form—65 kDa (c)) using zymography. No MMP-9 activity was detected in the control rodent myocardium, so MMP-9 activity is shown as a ratio of reperfused compared to non-reperfused infarct zones. Additionally, the absolute increase in MMP-9 activity for non-reperfused (closed circles) and reperfused (open circles) infarct zones is in the inset (d). Reprinted from J. Mol. Cell. Cardiol., Vol. 29, no. 9, W. C. Carlyle et al., “Delayed reperfusion alters matrix metalloproteinase activity and fibronectin mRNA expression in the infarct zone of the ligated rat heart,” pp. 2451–2463. Copyright (1997), with permission from Elsevier
The aforementioned studies almost exclusively focus on the content and structure of the collagenous architecture in the ECM following MI. There are only a few studies concerning changes in the content of other constituents (Fig. 4) following MI and LRT. Elastin content steadily declines following PO (Yu et al. 2018), whereas hyaluronan, a GAG encouraging water retention that plays an important role in inflammation resolution during wound healing, is upregulated (Fig. 4) (Huebener et al. 2008; Petz et al. 2019; Rienks et al. 2014; Taylor et al. 2004). Knowlton et al. (Knowlton et al. 1992) focused on fibronectin, a key component of the provisional matrix connecting cells to the ECM and promoting cellular migration and phenotyping. They detected fibronectin earlier (within 3 vs. 4 days post-MI) and to a greater degree in LRT samples than in PO samples (Fig. 4). For instances of ERT, Echtermeyer et al. (Echtermeyer et al. 2011) identified the importance of syndecan-4, a transmembrane heparan sulfate proteoglycan, for proper removal of granulation tissue and collagen deposition in mice. Lumican, a small leucine-rich proteoglycan (SLRP) implicated in collagen fiber formation, was upregulated 3 days post-MI in mice subjected to ERT (Baba et al. 2001). Other members of the SLRP family regulating collagen matrix formation, like biglycan (Westermann et al. 2008) and decorin (Doi et al. 2000; Weis et al. 2005), were also upregulated within the first week following MI, but their expressions following LRT are not well-defined. Thrombospondin-1, an inhibitor of angiogenesis and an activator of TGF-β, was upregulated within the first day of MI and ERT, and was localized to the ECM of the borderzone, potentially forming a protective barrier limiting expansion of granulation tissue (Christia et al. 2013; Frangogiannis et al. 2005). Finally, osteopontin, a diverse matricellular protein often associated with collagen fiber synthesis and deposition, was upregulated 10- to 20-fold in the first three days post-MI for mice subjected to PO and ERT (Christia et al. 2013; Singh et al. 2010; Trueblood et al. 2001). As many of these matricellular proteins, GAGs, and proteoglycans govern processes concerning the formation and structure of the infarct scar, a better understanding of their contextual actions may reveal mechanisms by which LRT guards against ventricular rupture and adverse remodeling, as well as potential therapeutic targets for guiding infarct scar formation.
Du and co. (Fang et al. 2007; Gao et al. 2005, 2012) extensively studied rupture and its connection to ECM structure following PO in mice, an animal model frequently experiencing spontaneous rupture. Rupture typically occurred within the first week (Fang et al. 2007; Gao et al. 2005), was located at the center or border of an infarct (Gao et al. 2005), and was associated with pronounced hemorrhaging and immune cell infiltration in the borderzone (Fang et al. 2007; Gao et al. 2005). In their study, Fang et al. (2007) conducted an assay to distinguish between soluble (non-crosslinked) and insoluble (crosslinked) collagen. They observed a decrease in crosslinked collagen and an increase in MMP activity within the first 4 days post-MI, the period when rupture was most common. Decreased collagen crosslinking has been proposed as a risk factor for rupture, although aged mice exhibit increased crosslinking and higher rupture rates (Y. Yang et al. 2008). No form of RT was employed in these studies; however, Gao et al. (2012) did report drastic decreases in ventricular rupture (~ 30% vs. 0%) following ERT in mice in an excellent review article (Gao et al. 2012). The nuanced nature of post-MI damage necessitates more advanced tools capable of better quantifying the properties of the ECM to determine the effects of RT on LV function and rupture post-MI (Carlyle et al. 1997; Connelly et al. 1985; C. Roberts et al. 1983; Wiggers et al. 1997).
Mechanical changes to the reperfused myocardium
The initial inflammatory response to MI leaves myocardium susceptible to rupture as LV walls thin (Connelly et al. 1992; Fishbein et al. 1978b, 1980), LV chamber volume increases (Capasso et al. 1992), and the ECM is dismantled (Etoh et al. 2001; McCurdy et al. 2011). For cases of PO, the stiffness of the LV initially declines, rises during the proliferative phase of healing (Fang et al. 2007; Theroux et al. 1977; Vokonas et al. 1976), and climbs the next several weeks (Arunachalam et al. 2018; Connelly et al. 1985; Laird and Vellekoop 1977; Walker et al. 2005). Pernot et al. (2016) observed an increase in diastolic stiffness following MI, which was further increased with ERT. Their shear wave imaging measurements of samples from the infarct core (Pernot et al. 2016) corresponded to the circumferential direction, supplementing results from Pislaru et al. (2014) taken in the longitudinal direction several years prior. Although Pernot et al. (2016) used sheep and Pislaru et al. (2014) used pigs in their respective studies, the efforts of these two groups revealed ERT resulted in statistically significant stiffening in the circumferential and longitudinal directions, respectively, about 6 h post-MI. At 1 week post-MI, after the inflammatory phase, Connelly et al. (1985) observed that PO and ERT longitudinally oriented samples from the infarct core exhibited increased uniaxial tensile strength (~ 500 kPa) compared to control samples (~ 150 kPa). By 3 weeks post-MI, ERT samples continued to exhibit increased uniaxial tensile strength (~ 800 kPa) and resistance to failure (~ 500 g) relative to control levels (~ 300 kPa and ~ 130 g, respectively), but reduced values in comparison to PO (~ 1.6 MPa and ~ 640 g, respectively).
To our knowledge, there are only two studies assessing post-MI mechanics following the application of LRT. Following LRT at 6 h post-MI, infarcted myocardium stiffens more drastically, likely due to edema and hemorrhage (Pirzada et al. 1978; C. Roberts et al. 1983). These estimates of stiffness were derived from catheter and pressure-segment length measurements from mercury-in-Silastic gauges placed within the infarcted region; unfortunately, Pirzada et al. (1978) did not mention strain gauge orientation. One day post-MI, the uniaxial tensile strength of longitudinally oriented rabbit LRT samples was initially depressed compared to control and PO samples (Connelly et al. 1992). In this study, Connelly et al. (1992) expanded their past experimental techniques to include inflation-to-rupture and tear-testing, loading modalities more representative of physiological failure and ventricular rupture. At 1 day post-MI, results from these tests were similar between PO, LRT, and control samples. At 3 days post-MI, however, LRT samples displayed an enhanced resistance to failure when compared to both control and PO samples despite still having uniaxial tensile strengths (~ 200 kPa) less than those of PO samples (~ 300 kPa). In addition, the location of failure in inflation-to-rupture tests of LRT hearts was now exclusively in the remote myocardium (Fig. 6). Finally, after a full week post-MI, PO and LRT samples had comparable tensile strengths (~ 500 kPa), much larger than those of the control samples (~ 200 kPa), and exhibited greater resistance to tearing than control samples. Based on catheter pressure measurements, chamber radii, and LV wall thicknesses, Connelly et al. (1992) then calculated wall stresses at the location of each rupture site following inflation-to-rupture testing. They reported elevated stresses in the borderzone of infarcts at 1 day post-MI for both PO and LRT samples; however, this observation did not hold for day 3 post-MI hearts, when no LRT hearts ruptured at the infarct core or borderzone (Fig. 6). Connelly et al. (1992) also pointed out that all of the measured pressures and applied stresses during their ex vivo testing were much larger than any pressures or stresses experienced by these samples in vivo. This observation, also made elsewhere (Arunachalam et al. 2018; Connelly et al. 1985; Lerman et al. 1983), suggests infarcted myocardium should never rupture, which is not the case, and highlights the need for advanced, physiologically relevant techniques to quantify infarct rupture mechanics.
Fig. 6.
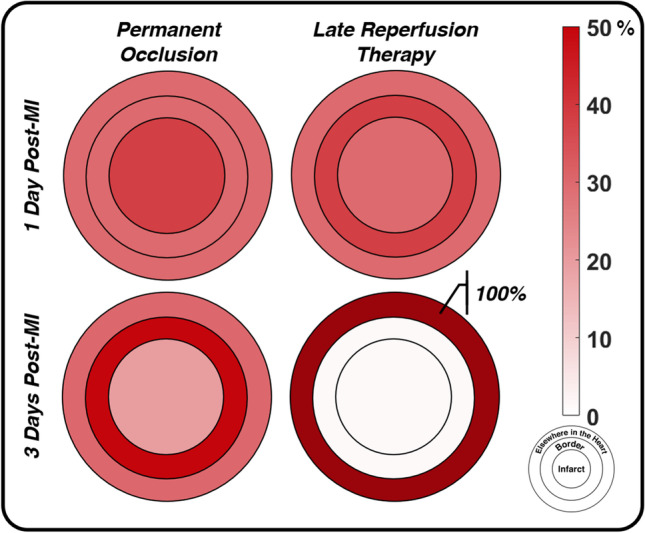
Soon after MI (day 1 post-MI), PO and LRT rabbit hearts tend to fail during ex vivo inflation testing at similar rates in similar locations, primarily the infarct core and the borderzone. By day 3 post-MI, LRT hearts have gained an improved resistance to failure and failure did not occur in the infarct or borderzone regions. Conversely, PO samples exhibited the largest rate of rupture in the understudied borderzone. Circles corresponding to the infarct, border, and elsewhere in the heart are not drawn to scale. Data replotted from Connelly et al. (1992)
Late reperfusion therapy and rupture
Despite offering minimal benefits for reducing infarct size (Boyle and Weisman 1993; Hale and Kloner 1987, 1988; Hochman and Choo 1987; Jugdutt and Michorowski 1987), infarct transmurality (Hale and Kloner 1988; Hochman and Choo 1987; Jugdutt 1997), and cellular necrosis (Connelly et al. 1982), LRT still improves post-MI outcomes and limits rupture in animal models (Gao et al. 2012; Michael et al. 1999) and humans (Honda et al. 2014; Ikeda et al. 2004; Late 1993; Nakamura et al. 1992). The mechanism by which this clinical tool reduces rupture, however, is unclear (Barlow and Chance 1976; Cox et al. 1968; Gallagher et al. 1986; Hearse and Yellon, 1981b; Lushnikov 1963; Sakai et al. 1985). LRT reduces MMP activity during the inflammatory phase (Fig. 7) (Carlyle et al. 1997). Assessments of collagen content and collagen crosslinking, though, have produced mixed results (Connelly et al. 1985, 1992; Knowlton et al. 1992; Lerman et al. 1983), suggesting that content and/or crosslinking follows a similar course as PO or is not depleted for as long (Fig. 7). In direct contrast to these structural observations, uniaxial mechanical studies (Connelly et al. 1985, 1992) indicate tissue stiffness is initially reduced by LRT (Fig. 7). Past work has relied heavily on assays to examine ECM structure and uniaxial testing to quantify mechanics, offering one-dimensional estimates of mechanical and structural function (Connelly et al. 1985, 1992; Fang et al. 2008; C. Roberts et al. 1983). Only one study, Connelly et al. (1992), utilized physiologically motivated mechanical testing techniques focused on failure. While their failure stresses were supraphysiological, they did report LRT increased resistance to rupture (Fig. 7). Additionally, there were clear spatial trends in rupture incidence (Fig. 6), suggesting future studies should include techniques designed to capture spatial variations in mechanical behavior and microscopic structure. Better quantification of full-field properties will also offer insight into the behavior of the neglected borderzone myocardium (Batts et al. 1990; Fang et al. 2007; Gao et al. 2005; Lerman et al. 1983). Despite having observed immune cell infiltration around infarct borders (Bajpai et al. 2019; Fang et al. 2007; Gao et al. 2005; O’Rourke et al. 2019; Troidl et al. 2009), depressed function (Cox et al. 1968; Lushnikov 1963), notable thinning (Clarke et al. 2016; Jackson et al. 2005; Leong et al. 2021; Mazhari et al. 2000), accentuated mechano-sensing gene expression (Calcagno et al. 2022; Yamada et al. 2022), and the common occurrence of rupture in this region (Batts et al. 1990; Fang et al. 2007; Gao et al. 2005; Lerman et al. 1983), past studies have largely focused on the homogeneous infarct core.
Fig. 7.

LRT does not reduce infarct size (Boyle and Weisman 1993; Hale and Kloner 1988; Hochman and Choo 1987; Jugdutt 1997), significantly alter collagen content (Connelly et al. 1992; Fang et al. 2007; Knowlton et al. 1992; Lerman et al. 1983), or increase uniaxial metrics of stiffness when compared to PO models (Connelly et al. 1985, 1992; Fang et al. 2007), but may reduce MMP activity (Carlyle et al. 1997; Fang et al. 2007) and improve the myocardium’s resistance to rupture (Connelly et al. 1985, 1992) during post-MI inflammation. All axes rely on arbitrary units, and all curves are qualitative representations of the studies summarized within this review. The start of inflammation () is defined as immediately following MI and before the application of LRT. The end of inflammation () is defined by mass migration and proliferation of fibroblasts in the infarcted region
Conclusions and future directions
Characterizations of reperfused myocardium are complicated by the temporal changes in its unique mechanical and structural properties. In particular, the tissue’s nonlinear elasticity, mechanical and structural anisotropy, and spatially variable composition demand advanced loading and imaging modalities for proper full-field characterizations. For structural studies, quantitative histological analyses can be used to visualize and quantify the organization and abundance of various ECM components (Gratz et al. 2020; Hanna et al. 2020; Whittaker et al. 1994). Optical coherence tomography (Goergen et al. 2016; Hendon et al. 2019; Pinkert et al. 2018) and second harmonic generation (SHG) imaging can also provide high-resolution images of collagenous architecture (Pinkert et al. 2018; Quinn et al. 2016; Sahu et al. 2021; Sommer et al. 2015). SHG, in particular, enables quantitative comparisons of collagen fibers from various regions of the heart. When paired with open-source software like CurveAlign or CT-Fire (Bredfeldt et al. 2014; Liu et al. 2017), fiber length, width, straightness, and direction may be quantified. SHG can also be used to image elastin (Fig. 4), an important extracellular structural protein that has been understudied compared to its collagenous counterparts (Thimm et al. 2015; Tilbury et al. 2014).
Mechanically, while uniaxial testing has been used to quantify stiffness (Connelly et al. 1985, 1992; Y. Yang et al. 2008), it is limited in its ability to characterize anisotropy (Gupta et al. 1994). Furthermore, uniaxial and indentation testing produce a single metric of stiffness, failing to describe spatially heterogeneous mechanical behavior. Biaxial and inflation testing offer more physiologically relevant loading mechanisms for myocardium. When used in tandem with digital image correlation, biaxial testing can produce full-field displacements and boundary forces to describe fiber and cross-fiber contributions to gross mechanical function (Lanir and Fung 1974; Pearce et al. 2022; Sacks 2000). This information can be used to perform inverse characterizations (F. Davis et al. 2015; Genovese et al. 2014; Witzenburg et al. 2012) of samples that provide full-field estimates of stiffness, mechanical anisotropy, and the spatial variability of these properties. Triaxial testing is another comprehensive testing modality that can be used to capture a sample’s three-dimensional mechanical response to shear (Avazmohammadi et al. 2018; Sommer et al. 2015). Advanced full-field laser micrometry (Pearce et al. 2022) can be used in combination with any of these approaches to provide full-field descriptions of sample thickness, allowing groups to better estimate stresses within tissues, to discriminate between areas of geometrical and mechanical heterogeneity, and to better visualize regions where stress concentrations may form in vivo, giving rise to an increased risk of rupture.
Studies exploring spatial distributions of cellular mobilization can also provide valuable insights, as variations in rates of cellular infiltration and MMP activation likely affect ECM destruction and reparation. A promising post-MI therapeutic approach is MMP inhibition, which is speculated to improve post-MI outcomes and prevent rupture by limiting ECM degradation and LV chamber dilation (Rohde et al. 1999). By specifically targeting MMP activity post-MI, LV enlargement, and obtrusive ventricular remodeling were diminished in rodents, rabbits, and pigs (Ikonomidis et al. 2005; Krishnamurthy et al. 2009; M. Lindsey et al. 2002; Mukherjee et al. 2003; Rohde et al. 1999; Wu et al. 2018; Yarbrough et al. 2003a, b; Zavadzkas et al. 2014). In other studies, however, MMP inhibition inhibited angiogenesis and impaired scar formation in mice (Heymans et al. 1999) and rats (Tessone et al. 2005). In one study, MMP-12 inhibition exacerbated LV dysfunction and led to prolonged inflammation (Iyer et al. 2015). Pig models of MMP inhibition did not preserve LV end-diastolic volume, ejection fraction, regional wall stresses, or peak pressures compared to control levels; however, the MMP inhibitor did lead to decreased end-diastolic volumes and regional wall stresses when compared to the MI-only group (Yarbrough et al. 2003a, b). Given the mixed results from animal studies, questions remain about the clinical relevance of MMP inhibition. Additionally, translating these results to humans should be done cautiously. No improvements in LV remodeling or long-term outcomes were observed in a clinical trial using PG-116800, an MMP inhibitor, on a group of 203 MI patients (Hudson et al. 2006).
The inflammatory phase of post-MI healing is historically understudied, but presents important opportunities and implications for long-term timeframes corresponding to scar formation and maturation. Studies addressing spatiotemporal differences in structure, mechanics, and cellular infiltration during this phase could reveal therapeutic targets to improve post-MI outcomes and address key gaps in LRT and rupture literature. Thorough mechanical and structural characterizations of reperfused myocardium will also aid the development of informed constitutive models for this tissue, improving our ability to simulate ventricular deformation, failure, and growth during post-MI inflammation and beyond. Additionally, while ERT is common in research studies, LRT is more common and clinically feasible, especially for patients from rural and less developed communities (Bhuyan et al. 2013; Cohen et al. 2010; de Villiers and Riley 2020; Gharacholou et al. 2010; Loccoh et al. 2022). A better understanding of its protective actions against adverse remodeling and LV rupture may further emphasize the importance of employing LRT following MI and other occlusive pathologies, like strokes (Imran et al. 2021). Finally, revelation of the mechanism by which LRT limits ventricular rupture may lead to advances in post-MI therapies complementing and potentially supplementing LRT. Should borderzone topography and composition play such a crucial role in post-MI ventricular rupture, therapeutic approaches utilizing implantable cardiac patches or stem cell injections may be delivered in a more spatially conscious and intentional way to promote optimal borderzone topography and healing (Botleroo et al. 2021; Cui et al. 2020; Mei and Cheng 2020; L. Wang et al. 2021a, b).
Acknowledgements
The authors would also like to thank Michael Chiariello, Elizabeth Gunderson, and Shreya Sreedhar for their assistance with various projects and concepts contributing to this effort.
Author contribution
All authors contributed to the conceptualization and organization of this work. DPP and MTN conducted the initial literature searches, crafted the first drafts of the document, generated figures, and requested permissions for reprinted figures. DPP, MTN, and CMW then reviewed, revised, and rewrote the document together.
Funding
This work was funded by a grant from the National Science Foundation Division of Civil, Mechanical and Manufacturing Innovation (ID, 2030173) to CMW.
Declarations
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol. 2008;40(6–7):1101–1110. doi: 10.1016/j.biocel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair-Kirk TL, Atkinson JJ, Broekelmann TJ, Doi M, Tryggvason K, Miner JH, Mecham RP, Senior RM. A site on laminin α5, AQARSAASKVKVSMKF, induces inflammatory cell production of matrix metalloproteinase-9 and chemotaxis. J Immunol. 2003;171(1):398–406. doi: 10.4049/jimmunol.171.1.398. [DOI] [PubMed] [Google Scholar]
- Agostoni P, Banfi C. Matrix metalloproteinase and heart failure: is it time to move from research to clinical laboratories? Eur Heart J. 2007;28(6):659–660. doi: 10.1093/eurheartj/ehl574. [DOI] [PubMed] [Google Scholar]
- Akaishi M, Weintraub WS, Schneider RM, Klein LW, Agarwal JB, Helfant RH. Analysis of systolic bulging. Mechanical characteristics of acutely ischemic myocardium in the conscious dog. Circ Res. 1986;58(2):209–217. doi: 10.1161/01.RES.58.2.209. [DOI] [PubMed] [Google Scholar]
- Althaus U, Gurtner HP, Baur H, Hamburger S, Roos B. Consequences of myocardial reperfusion following temporary coronary occlusion in pigs: effects on morphologic, biochemical and haemodynamic findings. Eur J Clin Invest. 1977;7(5):437–443. doi: 10.1111/j.1365-2362.1977.tb01631.x. [DOI] [PubMed] [Google Scholar]
- Amirhamzeh MMR, Hsu DT, Cabreriza SE, Jia CX, Spotnitz HM. Myocardial edema: comparison of effects on filling volume and stiffness of the left ventricle in rats and pigs. Ann Thorac Surg. 1997;63(5):1293–1297. doi: 10.1016/S0003-4975(97)00080-5. [DOI] [PubMed] [Google Scholar]
- Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, Peters JH, Hoefer IE, Doevendans PA, Pasterkamp G, De Kleijn DP. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ Res. 2011;108(5):582–592. doi: 10.1161/CIRCRESAHA.110.224428. [DOI] [PubMed] [Google Scholar]
- Arunachalam SP, Arani A, Baffour F, Rysavy JA, Rossman PJ, Glaser KJ, Lake DS, Trzasko JD, Manduca A, McGee KP, Ehman RL, Araoz PA. Regional assessment of in vivo myocardial stiffness using 3D magnetic resonance elastography in a porcine model of myocardial infarction. Magn Reson Med. 2018;79(1):361–369. doi: 10.1002/MRM.26695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avazmohammadi R, Li DS, Leahy T, Shih E, Soares JS, Gorman JH, Gorman RC, Sacks MS. An integrated inverse model-experimental approach to determine soft tissue three-dimensional constitutive parameters: application to post-infarcted myocardium. Biomech Model Mechanobiol. 2018;17(1):31–53. doi: 10.1007/s10237-017-0943-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba H, Ishiwata T, Takashi E, Xu G, Asano G. Expression and localization of lumican in the ischemic and reperfused rat heart. Jpn Circ J. 2001;65(5):445–450. doi: 10.1253/JCJ.65.445. [DOI] [PubMed] [Google Scholar]
- Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, Kovacs A, Epelman S, Artyomov M, Kreisel D, Lavine KJ. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. 2019;124(2):263–278. doi: 10.1161/CIRCRESAHA.118.314028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker TH, Engler AJ. The provisional matrix: setting the stage for tissue repair outcomes. Matrix Biol. 2017;60–61:1–4. doi: 10.1016/j.matbio.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow CH, Chance B. Ischemic areas in perfused rat hearts: measurement by NADH fluorescence photography. Science. 1976;193(4256):909–910. doi: 10.1126/science.181843. [DOI] [PubMed] [Google Scholar]
- Baroldi G. Acute coronary occlusion as a cause of myocardial infarct and sudden coronary heart death. Am J Cardiol. 1965;16(6):859–880. doi: 10.1016/0002-9149(65)90704-6. [DOI] [PubMed] [Google Scholar]
- Bashey RI, Martinez-Hernandez A, Jimenez SA. Isolation, characterization, and localization of cardiac collagen type VI. Associations with other extracellular matrix components. Circ Res. 1992;70(5):1006–1017. doi: 10.1161/01.RES.70.5.1006. [DOI] [PubMed] [Google Scholar]
- Bates ER (2014) Reperfusion therapy reduces the risk of myocardial rupture complicating ST-elevation myocardial infarction. J Am Heart Assoc, 3(5). 10.1161/JAHA.114.001368 [DOI] [PMC free article] [PubMed]
- Batts KP, Ackermann DM, Edwards WD. Postinfarction rupture of the left ventricular free wall: clinicopathologic correlates in 100 consecutive autopsy cases. Hum Pathol. 1990;21(5):530–535. doi: 10.1016/0046-8177(90)90010-3. [DOI] [PubMed] [Google Scholar]
- Becker RC, Hochman JS, Cannon CP, Spencer FA, Ball SP, Rizzo MJ, Antman EM. Fatal cardiac rupture among patients treated with thrombolytic agents and adjunctive thrombin antagonists observations from the thrombolysis and thrombin inhibition in myocardial infarction 9 study. J Am Coll Cardiol. 1999;33(2):479–487. doi: 10.1016/S0735-1097(98)00582-8. [DOI] [PubMed] [Google Scholar]
- Berger PB, Ellis SG, Holmes DR, Granger CB, Criger DA, Betriu A, Topol EJ, Califf RM. Relationship between delay in performing direct coronary angioplasty and early clinical outcome in patients with acute myocardial infarction. Circulation. 1999;100(1):14–20. doi: 10.1161/01.CIR.100.1.14. [DOI] [PubMed] [Google Scholar]
- Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, Malm T, Andrä M, Jashari R, Nyengaard JR, Possnert G, Jovinge S, Druid H, Frisén J. Dynamics of cell generation and turnover in the human heart. Cell. 2015;161(7):1566–1575. doi: 10.1016/J.CELL.2015.05.026. [DOI] [PubMed] [Google Scholar]
- Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V, Morine KJ, Gardner TJ, Discher DE, & Sweeney HL (2006) Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol-Heart Circ Physiol, 290(6). 10.1152/ajpheart.01017.2005 [DOI] [PubMed]
- Bhuyan SS, Wang Y, Opoku S, Lin G. Rural–urban differences in acute myocardial infarction mortality: evidence from Nebraska. J Cardiovasc Dis Res. 2013;4(4):209. doi: 10.1016/J.JCDR.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P. Matricellular proteins: an overview. J Cell Commun Signal. 2009;3(3–4):163. doi: 10.1007/S12079-009-0069-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002;14(5):608–616. doi: 10.1016/S0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- Botleroo RA, Bhandari R, Ahmed R, Kareem R, Gyawali M, Venkatesan N, Ogeyingbo OD, & Elshaikh AO (2021) Stem cell therapy for the treatment of myocardial infarction: how far are we now? Cureus, 13(8). 10.7759/CUREUS.17022 [DOI] [PMC free article] [PubMed]
- Boyle MP, Weisman HF. Limitation of infarct expansion and ventricular remodeling by late reperfusion: study of time course and mechanism in a rat model. Circulation. 1993;88(6):2872–2883. doi: 10.1161/01.CIR.88.6.2872. [DOI] [PubMed] [Google Scholar]
- Brazile BL, Butler JR, Patnaik SS, Claude A, Prabhu R, Williams LN, Perez KL, Nguyen KT, Zhang G, Bajona P, Peltz M, Yang Y, Hong Y, Liao J. Biomechanical properties of acellular scar ECM during the acute to chronic stages of myocardial infarction. J Mech Behav Biomed Mater. 2021;116:104342. doi: 10.1016/J.JMBBM.2021.104342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredfeldt JS, Liu Y, Pehlke CA, Conklin MW, Szulczewski JM, Inman DR, Keely PJ, Nowak RD, Mackie TR, Eliceiri KW. Computational segmentation of collagen fibers from second-harmonic generation images of breast cancer. J Biomed Opt. 2014;19(1):016007. doi: 10.1117/1.JBO.19.1.016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173(1):57–67. doi: 10.2353/AJPATH.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagno DM, Taghdiri N, Ninh VK, Mesfin JM, Toomu A, Sehgal R, Lee J, Liang Y, Duran JM, Adler E, Christman KL, Zhang K, Sheikh F, Fu Z, King KR. Single-cell and spatial transcriptomics of the infarcted heart define the dynamic onset of the border zone in response to mechanical destabilization. Nat Cardiovasc Res. 2022;1(11):1039–1055. doi: 10.1038/s44161-022-00160-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon RO, Butany JW, McManus BM, Speir E, Kravitz AB, Bolli R, Ferrans VJ. Early degradation of collagen after acute myocardial infarction in the rat. Am J Cardiol. 1983;52(3):390–395. doi: 10.1016/0002-9149(83)90145-5. [DOI] [PubMed] [Google Scholar]
- Capasso JM, Li P, Zhang X, Anversa P. Heterogeneity of ventricular remodeling after acute myocardial infarction in rats. Am J Physiol-Heart Circ Physiol. 1992;262(2):31–2. doi: 10.1152/ajpheart.1992.262.2.H486. [DOI] [PubMed] [Google Scholar]
- Carlyle WC, Jacobson AW, Judd DL, Tian B, Chu C, Hauer KM, Hartman MM, McDonald KM. Delayed reperfusion alters matrix metalloproteinase activity and fibronectin mRNA expression in the infarct zone of the ligated rat heart. J Mol Cell Cardiol. 1997;29(9):2451–2463. doi: 10.1006/JMCC.1997.0482. [DOI] [PubMed] [Google Scholar]
- Cavasin MA, Tao Z, Menon S, Yang XP. Gender differences in cardiac function during early remodeling after acute myocardial infarction in mice. Life Sci. 2004;75:2181–2192. doi: 10.1016/j.lfs.2004.04.024. [DOI] [PubMed] [Google Scholar]
- Cawston TE. Metalloproteinase inhibitors and the prevention of connective tissue breakdown. Pharmacol Ther. 1996;70(3):163–182. doi: 10.1016/0163-7258(96)00015-0. [DOI] [PubMed] [Google Scholar]
- Chazov EI, Matveeva LS, Mazaev AV. Intracoronary administration of fibrinolysin in acute myocardial infarction (Russian) Ter Arkh. 1976;48(4):8–19. [PubMed] [Google Scholar]
- Chew PH, Yin FCP, Zeger SL. Biaxial stress-strain properties of canine pericardium. J Mol Cell Cardiol. 1986;18(6):567–578. doi: 10.1016/S0022-2828(86)80965-8. [DOI] [PubMed] [Google Scholar]
- Christensen G, Herum KM, Lunde IG. Sweet, yet underappreciated: Proteoglycans and extracellular matrix remodeling in heart disease. Matrix Biol. 2019;75–76:286–299. doi: 10.1016/J.MATBIO.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Christia P, Bujak M, Gonzalez-Quesada C, Chen W, Dobaczewski M, Reddy A, Frangogiannis NG. Systematic characterization of myocardial inflammation, repair, and remodeling in a mouse model of reperfused myocardial infarction. J Histochem Cytochem. 2013;61(8):555–570. doi: 10.1369/0022155413493912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E, Graef I, Chasis H (1936) Thrombosis of the aorta and coronary arteries, with special reference to the fibrinoid lesions. Athlerosclerosis 22(2):183–212
- Clarke SA, Richardson WJ, & Holmes JW (2016) Modifying the mechanics of healing infarcts: is better the enemy of good? J Mol Cell Cardiol 93, 115–124. Academic Press. 10.1016/j.yjmcc.2015.11.028 [DOI] [PMC free article] [PubMed]
- Cleutjens JPM, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27(6):1281–1292. doi: 10.1016/S0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- Cohen M, Boiangiu C, Abidi M. Therapy for ST-segment elevation myocardial infarction patients who present late or are ineligible for reperfusion therapy. J Am Coll Cardiol. 2010;55(18):1895–1906. doi: 10.1016/J.JACC.2009.11.087. [DOI] [PubMed] [Google Scholar]
- Connelly C, Vogel WM, Wiegner AW. Effects of reperfusion after coronary artery occlusion on post-infarction scar tissue. Circ Res. 1985;57(4):562–577. doi: 10.1161/01.RES.57.4.562. [DOI] [PubMed] [Google Scholar]
- Connelly C, Ngoy S, Schoen FJ, Apstein CS. Biomechanical properties of reperfused transmural myocardial infarcts in rabbits during the first week after infarction: implications for left ventricular rupture. Circ Res. 1992;71(2):401–413. doi: 10.1161/01.RES.71.2.401. [DOI] [PubMed] [Google Scholar]
- Connelly C, Vogel WM, Hernandez YM, & Apstein CS (1982) Movement of necrotic wavefront after coronary artery occlusion in rabbit. Am J Physiol-Heart Circ Physiol, 12(5). 10.1152/ajpheart.1982.243.5.h682 [DOI] [PubMed]
- Corbett SA, Schwarzbauer JE. Fibronectin-fibrin cross-linking: a regulator of cell behavior. Trends Cardiovasc Med. 1998;8(8):357–362. doi: 10.1016/S1050-1738(98)00028-0. [DOI] [PubMed] [Google Scholar]
- Cox JL, McLaughlin VW, Flowers NC, Horan LG. The ischemic zone surrounding acute myocardial infarction. Its morphology as detected by dehydrogenase staining. Am Heart J. 1968;76(5):650–659. doi: 10.1016/0002-8703(68)90164-6. [DOI] [PubMed] [Google Scholar]
- Cui H, Liu C, Esworthy T, Huang Y, Yu ZX, Zhou X, San H, Lee SJ, Hann SY, Boehm M, Mohiuddin M, Fisher JP, & Zhang LG (2020) 4D physiologically adaptable cardiac patch: a 4-month in vivo study for the treatment of myocardial infarction. Sci Adv 6(26). 10.1126/SCIADV.ABB5067/SUPPL_FILE/ABB5067_SM.PDF [DOI] [PMC free article] [PubMed]
- Danielsen CC, Wiggers H, Andersen HR. Increased amounts of collagenase and gelatinase in porcine myocardium following ischemia and reperfusion. J Mol Cell Cardiol. 1998;30(7):1431–1442. doi: 10.1006/JMCC.1998.0711. [DOI] [PubMed] [Google Scholar]
- Davis G. Matricryptic sites control tissue injury responses in the cardiovascular system: relationships to pattern recognition receptor regulated events. J Mol Cell Cardiol. 2010;48(3):454–460. doi: 10.1016/j.yjmcc.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis F, Luo Y, Avril S, Duprey A, & Lu J (2015) Pointwise characterization of the elastic properties of planar soft tissues: application to ascending thoracic aneurysms. Biomech Model Mechanobiol 14, 967–978. https://hal.archives-ouvertes.fr/hal-01215247. Accessed 14 Jan 2022 [DOI] [PubMed]
- de Castro Brás LE, Frangogiannis NG. Extracellular matrix-derived peptides in tissue remodeling and fibrosis. Matrix Biol. 2020;91–92:176–187. doi: 10.1016/j.matbio.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villiers C, Riley PR (2020) Mouse models of myocardial infarction: comparing permanent ligation and ischaemia-reperfusion. The Company of Biologists 13(11). https://journals.biologists.com/dmm/article/13/11/dmm046565/225770/Mouse-models-of-myocardial-infarction-comparing. Accessed 21 Jul 2021 [DOI] [PMC free article] [PubMed]
- Deckx S, Johnson DM, Rienks M, Carai P, Van Deel E, Van der Velden J, Sipido KR, Heymans S, Papageorgiou AP. Extracellular SPARC increases cardiomyocyte contraction during health and disease. Plos One. 2019;14(4):e0209534. doi: 10.1371/JOURNAL.PONE.0209534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeon KY, De Castro Brás LE, Lange RA, & Lindsey ML (2012) Extracellular matrix proteomics in cardiac ischemia/reperfusion: the search is on. Circulation, 125(6). 10.1161/CIRCULATIONAHA.111.086835 [DOI] [PMC free article] [PubMed]
- DeLeon-Pennell KY, Meschiari CA, Jung M, Lindsey ML. Matrix metalloproteinases in myocardial infarction and heart failure. Prog Mol Biol Transl Sci. 2017;147:75–100. doi: 10.1016/BS.PMBTS.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeon-Pennell KY, Mouton AJ, Ero OK, Ma Y, Padmanabhan Iyer R, Flynn ER, Espinoza I, Musani SK, Vasan RS, Hall ME, Fox ER, Lindsey ML. LXR/RXR signaling and neutrophil phenotype following myocardial infarction classify sex differences in remodeling. Basic Res Cardiol. 2018;113(5):40. doi: 10.1007/S00395-018-0699-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demer LL, Yin FC. Passive biaxial mechanical properties of isolated canine myocardium. J Physiol. 1983;339(1):615–630. doi: 10.1113/jphysiol.1983.sp014738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps AM, Yarbrough WM, Squires CE, Allen RA, McClister DM, Dowdy KB, McLean JE, Mingoia JT, Sample JA, Mukherjee R, Spinale FG. Trafficking of the membrane type-1 matrix metalloproteinase in ischemia and reperfusion: relation to interstitial membrane type-1 matrix metalloproteinase activity. Circulation. 2005;111(9):1166–1174. doi: 10.1161/01.CIR.0000157149.71297.3A. [DOI] [PubMed] [Google Scholar]
- Diamond G, Forrester JS. Effect of coronary artery disease and acute myocardial infarction on left ventricular compliance in man. Circulation. 1972;45(1):11–19. doi: 10.1161/01.CIR.45.1.11. [DOI] [PubMed] [Google Scholar]
- Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324(3):475–488. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- Doi M, Kusachi S, Murakami T, Ninomiya Y, Murakami M, Nakahama M, Takeda K, Komatsubara I, Naito I, Tsuji T. Time-dependent changes of decorin in the infarct zone after experimentally induced myocardial infarction in rats: comparison with biglycan. Pathol Res Pract. 2000;196(1):23–33. doi: 10.1016/S0344-0338(00)80018-7. [DOI] [PubMed] [Google Scholar]
- Drobnik J, Tosik D, Piera L, Szczepanowska A, Olczak S, Zielinska A, Liberski PP, Ciosek J. Melatonin-induced glycosaminoglycans augmentation in myocardium remote to infarction. J Physiol Pharmacol. 2013;64(6):737–744. [PubMed] [Google Scholar]
- Echtermeyer F, Harendza T, Hubrich S, Lorenz A, Herzog C, Mueller M, Schmitz M, Grund A, Larmann J, Stypmann J, Schieffer B, Lichtinghagen R, Hilfiker-Kleiner D, Wollert KC, Heineke J, Theilmeier G. Syndecan-4 signalling inhibits apoptosis and controls NFAT activity during myocardial damage and remodelling. Cardiovasc Res. 2011;92(1):123–131. doi: 10.1093/CVR/CVR149. [DOI] [PubMed] [Google Scholar]
- Emery JL, Omens JH, McCulloch AD. Biaxial mechanics of the passively overstretched left ventricle. Am J Physiol-Heart Circ Physiol. 1997;272(5):41–5. doi: 10.1152/ajpheart.1997.272.5.h2299. [DOI] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40(1):91–104. doi: 10.1016/J.IMMUNI.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etoh T, Joffs C, Deschamps AM, Davis J, Dowdy K, Hendrick J, Baicu S, Mukherjee R, Manhaini M, Spinale FG (2001) Myocardial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs. Am J Physiol. Heart Circ Physiol, 281(3). 10.1152/AJPHEART.2001.281.3.H987 [DOI] [PubMed]
- Factor SM, Sonnenblick EH, Kirk ES. The histologic border zone of acute myocardial infarction. Islands or peninsulas? Am J Pathol. 1978;92(1):111–124. [PMC free article] [PubMed] [Google Scholar]
- Factor SM, Robinson TF, Dominitz R, Cho SH (1987) Alterations of the myocardial skeletal framework in acute myocardial infarction with and without ventricular rupture. A preliminary report. Am J Cardiovasc 1(1), 91–97. https://europepmc.org/article/med/2458117. Accessed 14 Jan 2022 [PubMed]
- Fang L, Gao X, Moore XL, Kiriazis H, Su Y, Ming Z, Lim YL, Dart AM, Du XJ. Differences in inflammation, MMP activation and collagen damage account for gender difference in murine cardiac rupture following myocardial infarction. J Mol Cell Cardiol. 2007;43(5):535–544. doi: 10.1016/j.yjmcc.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Fang L, Gao X, Samuel CS, Su Y, Lim YL, Dart AM, Du XJ. Higher levels of collagen and facilitated healing protect against ventricular rupture following myocardial infarction. Clin Sci. 2008;115(3–4):99–106. doi: 10.1042/CS20070365. [DOI] [PubMed] [Google Scholar]
- Farhadian F, Contard F, Sabri A, Samuel JL, Rappaport L. Fibronectin and basement membrane in cardiovascular organogenesis and disease pathogenesis. Cardiovasc Res. 1996;32(3):433–442. doi: 10.1016/0008-6363(96)00119-8. [DOI] [PubMed] [Google Scholar]
- Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients Fibrinolytic Therapy Trialists’ (FTT) Collaborative. Lancet. 1994;343(8893):311–322. doi: 10.1016/S0140-6736(94)91161-4. [DOI] [PubMed] [Google Scholar]
- Fishbein M, Maclean D, Maroko PR. Experimental myocardial infarction in the rat. Qualitative and quantitative changes during pathologic evolution. Am J Pathol. 1978;90(1):57–70. [PMC free article] [PubMed] [Google Scholar]
- Fishbein M, Maclean D, Maroko PR. The histopathologic evolution of myocardial infarction. Chest. 1978;73(6):843–849. doi: 10.1378/chest.73.6.843. [DOI] [PubMed] [Google Scholar]
- Fishbein M, Hare CA, Gissen SA, Spadaro J, Maclean D, Maroko PR (1980) Identification and quantification of histochemical border zones during the evolution of myocardial infarction in the rat. In Cardiovascular Research (Vol. 14, Issue 1, pp. 41–49). Oxford University Press. 10.1093/cvr/14.1.41 [DOI] [PubMed]
- Fletcher AP, Sherry S, Alkjaersig N, Jick S. The maintenance of a sustained thrombolytic state in man II Clinical observations on patients with myocardial infarction and other thromboembolic disorders. J Clin Investig. 1959;38(7):1111–1119. doi: 10.1172/JCI103887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher AP, Alkjaersig N, Smyrniotis FE, Sherry S (1958) The treatment of patients suffering from early myocardial infarction with massive and prolonged streptokinase therapy. Transactions of the Association of American Physicians, 71, 287–296. http://www.ncbi.nlm.nih.gov/pubmed/13603526. Accessed 14 Jan 2022 [PubMed]
- Fomovsky GM, Thomopoulos S, Holmes JW. Contribution of extracellular matrix to the mechanical properties of the heart. J Mol Cell Cardiol. 2010;48(3):490–496. doi: 10.1016/j.yjmcc.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomovsky GM, Rouillard AD, Holmes JW. Regional mechanics determine collagen fiber structure in healing myocardial infarcts. J Mol Cell Cardiol. 2012;52(5):1083–1090. doi: 10.1016/j.yjmcc.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomovsky GM, Holmes JW (2010) Evolution of scar structure, mechanics, and ventricular function after myocardial infarction in the rat. Am J Physiol Heart Circ Physiol 298(1). 10.1152/ajpheart.00495.2009 [DOI] [PMC free article] [PubMed]
- Forrester JS, Diamond G, Parmley WW, Swan HJ. Early increase in left ventricular compliance after myocardial infarction. J Clin Investig. 1972;51(3):598–603. doi: 10.1172/JCI106849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92(2):635. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Investig. 2017;127(5):1600–1612. doi: 10.1172/JCI87491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. Transforming growth factor-β in myocardial disease. Nature Reviews: Cardiology; 2022. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111(22):2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109(13):1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- Friedberg CK, Horn H. Acute myocardial infarction not due to coronary artery occlusion. J Am Med Assoc. 1939;112(17):1675–1679. doi: 10.1001/JAMA.1939.02800170021007. [DOI] [Google Scholar]
- Gallagher KP, Gerren RA, Stirling MC, Choy M, Dysko RC, McManimon SP, Dunham WR. The distribution of functional impairment across the lateral border of acutely ischemic myocardium. Circ Res. 1986;58(4):570–583. doi: 10.1161/01.RES.58.4.570. [DOI] [PubMed] [Google Scholar]
- Ganz W, Buchbinder N, Marcus H, Mondkar A, Maddahi J, Charuzi Y, O’Connor L, Shell W, Fishbein M, Kass R, Miyamoto A, Swan HJC. Intracoronary thrombolysis in evolving myocardial infarction. Am Heart J. 1981;101(1):4–13. doi: 10.1016/0002-8703(81)90376-8. [DOI] [PubMed] [Google Scholar]
- Gao X, Xu Q, Kiriazis H, Dart AM, Du XJ. Mouse model of post-infarct ventricular rupture: time course, strain- and gender-dependency, tensile strength, and histopathology. Cardiovasc Res. 2005;65(2):469–477. doi: 10.1016/j.cardiores.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Gao X, White DA, Dart AM, Du X-J. Post-infarct cardiac rupture: recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther. 2012;134(2):156–179. doi: 10.1016/j.pharmthera.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Genovese K, Casaletto L, Humphrey JD, Lu J (2014) Digital image correlation-based point-wise inverse characterization of heterogeneous material properties of gallbladder in vitro. Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences, 470(2167). 10.1098/rspa.2014.0152
- Gharacholou SM, Alexander KP, Chen AY, Wang TY, Melloni C, Gibler WB, Pollack CV, Ohman EM, Peterson ED, Roe MT. Implications and reasons for the lack of use of reperfusion therapy in patients with ST-segment elevation myocardial infarction: findings from the CRUSADE initiative. Am Heart J. 2010;159(5):757–763. doi: 10.1016/J.AHJ.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Ghugre NR, Pop M, Thomas R, Newbigging S, Qi X, Barry J, Strauss BH, Wright GA. Hemorrhage promotes inflammation and myocardial damage following acute myocardial infarction: insights from a novel preclinical model and cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2017;19(1):1–13. doi: 10.1186/s12968-017-0361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci. 2000;902:230–240. doi: 10.1111/J.1749-6632.2000.TB06318.X. [DOI] [PubMed] [Google Scholar]
- Goergen CJ, Chen HH, Sakadžić S, Srinivasan VJ, Sosnovik DE (2016) Microstructural characterization of myocardial infarction with optical coherence tractography and two‐photon microscopy. Physiol Rep 4(18). 10.14814/PHY2.12894 [DOI] [PMC free article] [PubMed]
- Gottlieb GJ, Kubo SH, Alonso DR (1981) Ultrastructural characterization of the border zone surrounding early experimental myocardial infarcts in dogs. Am J Physiol. [PMC free article] [PubMed]
- Gratz D, Winkle AJ, Dalic A, Unudurthi SD, Hund TJ. Computational tools for automated histological image analysis and quantification in cardiac tissue. MethodsX. 2020;7:100755. doi: 10.1016/J.MEX.2019.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruppo Italiano per lo Studio Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Lancet. 1986;327(8478):397–402. doi: 10.1016/S0140-6736(86)92368-8. [DOI] [PubMed] [Google Scholar]
- Guccione JM, McCulloch AD, Waldman LK. Passive material properties of intact ventricular myocardium determined from a cylindrical model. J Biomech Eng. 1991;113(1):42–55. doi: 10.1115/1.2894084. [DOI] [PubMed] [Google Scholar]
- Gupta KB, Ratcliffe MB, Fallert MA, Edmunds LH, Bogen DK. Changes in passive mechanical stiffness of myocardial tissue with aneurysm formation. Circulation. 1994;89(5):2315–2326. doi: 10.1161/01.CIR.89.5.2315. [DOI] [PubMed] [Google Scholar]
- GUSTO An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med. 1993;329(10):673–682. doi: 10.1056/nejm199309023291001. [DOI] [PubMed] [Google Scholar]
- De Haan JJ, Smeets MB, Pasterkamp G, Arslan F (2013) Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediators of Inflammation, 2013. 10.1155/2013/206039 [DOI] [PMC free article] [PubMed]
- Hale SL, Kloner RA. Effect of early coronary artery reperfusion on infarct development in a model of low collateral flow. Cardiovasc Res. 1987;21(9):668–673. doi: 10.1093/CVR/21.9.668. [DOI] [PubMed] [Google Scholar]
- Hale SL, Kloner RA. Left ventricular topographic alterations in the completely healed rat infarct caused by early and late coronary artery reperfusion. Am Heart J. 1988;116(6):1508–1513. doi: 10.1016/0002-8703(88)90736-3. [DOI] [PubMed] [Google Scholar]
- Halper J, Kjaer M. Basic components of connective tissues and extracellular matrix: elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv Exp Med Biol. 2014;802:31–47. doi: 10.1007/978-94-007-7893-1_3. [DOI] [PubMed] [Google Scholar]
- Hanna A, Shinde AV, Frangogiannis NG. Validation of diagnostic criteria and histopathological characterization of cardiac rupture in the mouse model of nonreperfused myocardial infarction. Am J Physiol - Heart Circ Physiol. 2020;319(5):H948–H964. doi: 10.1152/AJPHEART.00318.2020/ASSET/IMAGES/LARGE/ZH40102032060008.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris BS, Zhang Y, Card L, Rivera LB, Brekken RA, Bradshaw AD. SPARC regulates collagen interaction with cardiac fibroblast cell surfaces. Am J Physiol. 2011;301(3):H841. doi: 10.1152/AJPHEART.01247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JK, Califf RM, Woodlief LH, Kereiakes D, George BS, Stack RS, Ellis SG, Lee KL, O’Neill W, Topol EJ. Systolic left ventricular function after reperfusion therapy for acute myocardial infarction. Analysis of determinants of improvement. TAMI Study Group. Circ. 1993;87(5):1531–1541. doi: 10.1161/01.CIR.87.5.1531. [DOI] [PubMed] [Google Scholar]
- Hayat S, Kramann R. Mapping the border zone in myocardial infarction. Nat Cardiovasc Res. 2022;1(11):978–979. doi: 10.1038/s44161-022-00161-2. [DOI] [PubMed] [Google Scholar]
- Hearse DJ, Yellon DM. The “border zone” in evolving myocardial infarction: controversy or confusion? Am J Cardiol. 1981;47(6):1321–1334. doi: 10.1016/0002-9149(81)90266-6. [DOI] [PubMed] [Google Scholar]
- Helpap B, Féaux de Lacroix W, Langewitz W. Die Herzruptur: Histologische Untersuchungen am Myokard rupturierter und nicht rupturierter Herzinfarkte. DMW - Deutsche Medizinische Wochenschrift. 2008;105(15):515–519. doi: 10.1055/s-2008-1070698. [DOI] [PubMed] [Google Scholar]
- Hendon CP, Lye TH, Yao X, Gan Y, Marboe CC. Optical coherence tomography imaging of cardiac substrates. Quant Imaging Med Surg. 2019;9(5):882–904. doi: 10.21037/qims.2019.05.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick JB. Clinical features of sudden obstruction of the coronary arteries. J Am Med Assoc. 1912;23:2015–2022. doi: 10.1001/jama.1912.04270120001001. [DOI] [PubMed] [Google Scholar]
- Herzog E, Gu A, Kohmoto T, Burkhoff D, Hochman JS. Early Activation of metalloproteinases after experimental myocardial infarction occurs in infarct and non-infarct zones. Cardiovasc Pathol. 1998;7(6):307–312. doi: 10.1016/S1054-8807(98)00008-8. [DOI] [PubMed] [Google Scholar]
- Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JPM, Shipley M, Angellilo A, Levi M, Nübe O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JFM, Shapiro SD, Baes M, … Carmeliet P (1999) Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nature Medicine, 5(10), 1135–1142. 10.1038/13459 [DOI] [PubMed]
- Hiesinger W, Brukman MJ, McCormick RC, Fitzpatrick JR, Frederick JR, Yang EC, Muenzer JR, Marotta NA, Berry MF, Atluri P, Woo YJ. Myocardial tissue elastic properties determined by atomic force microscopy after stromal cell-derived factor 1a Angiogenic therapy for acute myocardial infarction in a murine model. J Thorac Cardiovasc Surg. 2012;143(4):962–966. doi: 10.1016/j.jtcvs.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirohata S, Kusachi S, Murakami M, Murakami T, Sano I, Watanabe T, Komatsubara I, Kondo J, Tsuji T, Hirohata S, Kusachi S, Murakami M, Murakami T, Sano I, Watanabe T, Komatsubara I, Kondo J, Tsuji T. Time dependent alterations of serum matrix metalloproteinase-1 and metalloproteinase-1 tissue inhibitor after successful reperfusion of acute myocardial infarction. Heart. 1997;78(3):278–284. doi: 10.1136/HRT.78.3.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochman JS, Choo H. Limitation of myocardial infarct expansion by reperfusion independent of myocardial salvage. Circulation. 1987;75(1):299–306. doi: 10.1161/01.CIR.75.1.299. [DOI] [PubMed] [Google Scholar]
- Hojo Y, Ikeda U, Ueno S, Arakawa H, Shimada K. Expression of matrix metalloproteinases in patients with acute myocardial infarction. Jpn Circ J. 2001;65(2):71–75. doi: 10.1253/JCJ.65.71. [DOI] [PubMed] [Google Scholar]
- Holmes JW, Nuñez JA, Covell JW. Functional Implications of Myocardial Scar Structure. 1997 doi: 10.1152/Ajpheart.1997.272.5.H2123,272(541-5). [DOI] [PubMed] [Google Scholar]
- Holmes JW, Borg TK, Covell JW. Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng. 2005;7(1):223–253. doi: 10.1146/annurev.bioeng.7.060804.100453. [DOI] [PubMed] [Google Scholar]
- Holzapfel GA, Ogden RW. Constitutive modelling of passive myocardium: a structurally based framework for material characterization. Philos Trans R Soc. 2009;367(1902):3445–3475. doi: 10.1098/rsta.2009.0091. [DOI] [PubMed] [Google Scholar]
- Honda S, Asaumi Y, Yamane T, Nagai T, Miyagi T, Noguchi T, Anzai T, Goto Y, Ishihara M, Nishimura K, Ogawa H, Ishibashi-Ueda H, Yasuda S (2014) Trends in the clinical and pathological characteristics of cardiac rupture in patients with acute myocardial infarction over 35 years. J Am Heart Assoc 3(5). 10.1161/JAHA.114.000984 [DOI] [PMC free article] [PubMed]
- Hudson MP, Armstrong PW, Ruzyllo W, Brum J, Cusmano L, Krzeski P, Lyon R, Quinones M, Theroux P, Sydlowski D, Kim HE, Garcia MJ, Jaber WA, Weaver WD. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction. Results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J Am Coll Cardiol. 2006;48(1):15–20. doi: 10.1016/j.jacc.2006.02.055. [DOI] [PubMed] [Google Scholar]
- Huebener P, Frangogiannis N. Matricellular proteins in myocardial infarction. Curr Cardiol Rev. 2006;2(3):163–171. doi: 10.2174/157340306778019432. [DOI] [Google Scholar]
- Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, Frangogiannis NG. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J Immunol. 2008;180(4):2625–2633. doi: 10.4049/JIMMUNOL.180.4.2625. [DOI] [PubMed] [Google Scholar]
- Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, … Nahrendorf M (2018) Cardiac macrophages promote diastolic dysfunction. J Exp Med 215(2), 423–440. 10.1084/jem.20171274 [DOI] [PMC free article] [PubMed]
- Humphrey JD, Strumpf RK, Yin FCP. Biaxial mechanical behavior of excised ventricular epicardium. Am J Physiol - Heart Circ Physiol. 1990;259(1):28–1. doi: 10.1152/AJPHEART.1990.259.1.H101. [DOI] [PubMed] [Google Scholar]
- Hutchins KD, Skurnick J, Lavenhar M, Natarajan GA. Cardiac rupture in acute myocardial infarction. Am J Forensic Med Pathol. 2002;23(1):78–82. doi: 10.1097/00000433-200203000-00017. [DOI] [PubMed] [Google Scholar]
- Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, Caforio ALP, Crea F, Goudevenos JA, Halvorsen S, Hindricks G, Kastrati A, Lenzen MJ, Prescott E, Roffi M, Valgimigli M, Varenhorst C, Vranckx P, Widimský P, … Gale CP (2018) 2017 ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J, 39(2), 119–177. 10.1093/EURHEARTJ/EHX393
- Ikeda N, Yasu T, Kubo N, Hirahara T, Sugawara Y, Kobayashi N, Hashimoto S, Tsuruya Y, Fujii M, Saito M. Effect of reperfusion therapy on cardiac rupture after myocardial infarction in Japanese. Circ J. 2004;68(5):422–426. doi: 10.1253/CIRCJ.68.422. [DOI] [PubMed] [Google Scholar]
- Ikonomidis JS, Hendrick JW, Parkhurst AM, Herron AR, Escobar PG, Dowdy KB, Stroud RE, Hapke E, Zile MR, Spinale FG (2005) Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: effects of exogenous MMP inhibition. Am J Physiol Heart Circ Physiol 288(1). 10.1152/AJPHEART.00370.2004 [DOI] [PubMed]
- Imran R, Mohamed GA, Nahab F (2021) Acute reperfusion therapies for acute ischemic stroke. J Clin Med 10(16). 10.3390/jcm10163677 [DOI] [PMC free article] [PubMed]
- Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, De Castro Brás LE, Lindsey ML. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Cardiol. 2015;185:198. doi: 10.1016/J.IJCARD.2015.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson BM, Parish LM, Gorman JH, III, Enomoto Y, Sakamoto H, Plappert T, Sutton MG, Salgo I, Gorman RC. Borderzone geometry after acute myocardial infarction: a three-dimensional contrast enhanced echocardiographic study. Ann Thorac Surg. 2005;80(6):2250–2255. doi: 10.1016/J.ATHORACSUR.2005.05.103. [DOI] [PubMed] [Google Scholar]
- Jinatongthai P, Kongwatcharapong J, Foo CY, Phrommintikul A, Nathisuwan S, Thakkinstian A, Reid CM, Chaiyakunapruk N. Comparative efficacy and safety of reperfusion therapy with fibrinolytic agents in patients with ST-segment elevation myocardial infarction: a systematic review and network meta-analysis. Lancet. 2017;390(10096):747–759. doi: 10.1016/S0140-6736(17)31441-1. [DOI] [PubMed] [Google Scholar]
- Jugdutt B, Michorowski B. Role of infarct expansion in rupture of the ventricular septum after acute myocardial infarction: a two-dimensional echocardiographic study. Clin Cardiol. 1987;10(11):641–652. doi: 10.1002/CLC.4960101109. [DOI] [PubMed] [Google Scholar]
- Jugdutt B, Joljart MJ, Khan MI. Rate of collagen deposition during healing and ventricular remodeling after myocardial infarction in rat and dog models. Circulation. 1996;94(1):94–101. doi: 10.1161/01.CIR.94.1.94. [DOI] [PubMed] [Google Scholar]
- Jugdutt B (1997). Effect of reperfusion on ventricular mass, topography, and function during healing of anterior infarction. Am J Physiol - Heart Circ Physiol 41(3). 10.1152/ajpheart.1997.272.3.h1205 [DOI] [PubMed]
- Kanekar S, Hirozanne T, Terracio L, Borg TK. Cardiac fibroblasts. Cardiovasc Pathol. 1998;7(3):127–133. doi: 10.1016/S1054-8807(97)00119-1. [DOI] [PubMed] [Google Scholar]
- Kereiakes DJ, Califf RM, George BS, Ellis S, Samaha J, Stack R, Martin LH, Young S, Topol EJ. Coronary bypass surgery improves global and regional left ventricular function following thrombolytic therapy for acute myocardial infarction. Am Heart J. 1991;122(2):390–399. doi: 10.1016/0002-8703(91)90991-P. [DOI] [PubMed] [Google Scholar]
- Knowlton A, Connelly C, Romo GM, Mamuya W, Apstein CS, Brecher P, Ngoy S. Rapid expression of fibronectin in the rabbit heart after myocardial infarction with and without reperfusion. J Clin Investig. 1992;89(4):1060–1068. doi: 10.1172/JCI115685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koklu E, Arslan S, Yuksel IO, Bayar N, Yilmaz GM, Kucukseymen S. Management of left ventricular free wall rupture associated with acute myocardial infarction. Jacme. 2017;7(1):31. doi: 10.6705/J.JACME.2017.0701.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy P, Peterson JT, Subramanian V, Singh M, Singh K. Inhibition of matrix metalloproteinases improves left ventricular function in mice lacking osteopontin after myocardial infarction. Mol Cell Biochem. 2009;322(1–2):53–62. doi: 10.1007/S11010-008-9939-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird JD, Vellekoop HP. Time course of passive elasticity of myocardial tissue following experimental infarction in rabbits and its relation to mechanical dysfunction. Circ Res. 1977;41(5):715–721. doi: 10.1161/01.RES.41.5.715. [DOI] [PubMed] [Google Scholar]
- Lambert L, Brown K, Segal E, Brophy J, Rodes-Cabau J, Bogaty P. Association between timeliness of reperfusion therapy and clinical outcomes in ST-elevation myocardial infarction. JAMA. 2010;303(21):2148–2155. doi: 10.1001/JAMA.2010.712. [DOI] [PubMed] [Google Scholar]
- Lanir Y, Fung YC. Two-dimensional mechanical properties of rabbit skin—I. Experimental system. J Biomech. 1974;7(1):29–34. doi: 10.1016/0021-9290(74)90067-0. [DOI] [PubMed] [Google Scholar]
- LATE Late Assessment of Thrombolytic Efficacy (LATE) study with alteplase 6–24 hours after onset of acute myocardial infarction. The Lancet. 1993;342(8874):759–766. doi: 10.1016/0140-6736(93)91538-W. [DOI] [PubMed] [Google Scholar]
- Lawton JS, Tamis-Holland JE, Bangalore S, Bates ER, Beckie T. M, Bischoff JM, Bittl JA, Cohen MG, Dimaio JM, Don CW, Fremes SE, Gaudino MF, Goldberger ZD, Grant MC, Jaswal JB, Kurlansky PA, Mehran R, Metkus TS, Nnacheta LC, … Zwischenberger BA (2022) 2021 ACC/AHA/SCAI guideline for coronary artery revascularization: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation, 145(3), E18–E114. 10.1161/CIR.0000000000001038/FORMAT/EPUB [DOI] [PubMed]
- Leong CO, Leong CN, Liew YM, Al Abed A, Aziz YFA, Chee KH, Sridhar GS, Dokos S, Lim E. The role of regional myocardial topography post-myocardial infarction on infarct extension. Intl J Numer Methods Biomed Eng. 2021;37(8):e3501. doi: 10.1002/CNM.3501. [DOI] [PubMed] [Google Scholar]
- Lerman RH, Apstein CS, Kagan HM, Osmers EL, Chichester CO, Vogel WM, Connelly CM, Steffee WP. Myocardial healing and repair after experimental infarction in the rabbit. Circ Res. 1983;53(3):378–388. doi: 10.1161/01.RES.53.3.378. [DOI] [PubMed] [Google Scholar]
- Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther. 2012;30(1):31–41. doi: 10.1111/j.1755-5922.2010.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey M, Wedin K, Brown MD, Keller C, Evans AJ, Smolen J, Burns AR, Rossen RD, Michael L, Entman M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation. 2001;103(17):2181–2187. doi: 10.1161/01.CIR.103.17.2181. [DOI] [PubMed] [Google Scholar]
- Lindsey M, Gannon J, Aikawa M, Schoen FJ, Rabkin E, Lopresti-Morrow L, Crawford J, Black S, Libby P, Mitchell PG, Lee RT. Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation. 2002;105(6):753–758. doi: 10.1161/HC0602.103674. [DOI] [PubMed] [Google Scholar]
- Lindsey M, Jung M, Hall ME, DeLeon-Pennell KY (2018) Proteomic analysis of the cardiac extracellular matrix: clinical research applications. In Expert Review of Proteomics 15(2):105–112. Expert Rev Proteomics. 10.1080/14789450.2018.1421947 [DOI] [PMC free article] [PubMed]
- Liu Y, Keikhosravi A, Mehta GS, Drifka CR, Eliceiri KW. Methods for quantifying fibrillar collagen alignment. Methods Mol Biol. 2017;1627:429–451. doi: 10.1007/978-1-4939-7113-8_28/FIGURES/8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loccoh EC, Joynt Maddox KE, Wang Y, Kazi DS, Yeh RW, Wadhera RK. Rural-urban disparities in outcomes of myocardial infarction, heart failure, and stroke in the United States. J Am Coll Cardiol. 2022;79(3):267–279. doi: 10.1016/J.JACC.2021.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Gunja-Smith Z, Frederick Woessner J, Ursell PC, Nissen T, Galardy RE, Xu Y, Zhu P, Schwartz GG. Matrix metalloproteinases and collagen ultrastructure in moderate myocardial ischemia and reperfusion in vivo. Am J Physiol - Heart Circ Physiol. 2000;279(2):48–2. doi: 10.1152/AJPHEART.2000.279.2.H601/ASSET/IMAGES/LARGE/H40800075001.JPEG. [DOI] [PubMed] [Google Scholar]
- Lu L, Zhang JQ, Ramires FJ, Sun Y. Molecular and cellular events at the site of myocardial infarction: from the perspective of rebuilding myocardial tissue. Biochem Biophys Res Commun. 2004;320(3):907–913. doi: 10.1016/J.BBRC.2004.06.034. [DOI] [PubMed] [Google Scholar]
- Lushnikov EF. Histochemical study of experimentally produced myocardial infarction. Fed Procl. 1963;4(1):55. [PubMed] [Google Scholar]
- Ma S, Bai L, Liu P, She G, Deng XL, Song AQ, Du XJ, Lu Q (2022) Pathogenetic link of cardiac rupture and left ventricular thrombus following acute myocardial infarction: a joint preclinical and clinical study. Front Cardiovasc Med 9. 10.3389/FCVM.2022.858720 [DOI] [PMC free article] [PubMed]
- Marcus ML, Kerber RE, Ehrhardt J, Abboud FM. Three dimensional geometry of acutely ischemic myocardium. Circulation. 1975;52(2):254–263. doi: 10.1161/01.CIR.52.2.254. [DOI] [PubMed] [Google Scholar]
- Maroko PR, Kjekshus JK, Sobel BE, Watanabe T, Covell JW, Ross J, Braunwald E. Factors influencing infarct size following experimental coronary artery occlusions. Circulation. 1971;43(1):67–82. doi: 10.1161/01.CIR.43.1.67. [DOI] [PubMed] [Google Scholar]
- Martino MM, Briquez PS, Ranga A, Lutolf MP, Hubbell JA. Heparin-binding domain of fibrin(ogen) binds growth factors and promotes tissue repair when incorporated within a synthetic matrix. Proc Natl Acad Sci USA. 2013;110(12):4563–4568. doi: 10.1073/PNAS.1221602110/SUPPL_FILE/PNAS.201221602SI.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteucci M, Fina D, Jiritano F, Meani P, Blankesteijn WM, Raffa GM, Kowaleski M, Heuts S, Beghi C, Maessen J, Lorusso R. Treatment strategies for post-infarction left ventricular free-wall rupture. Eur Heart J Acute Cardiovasc Care. 2019;8(4):379–387. doi: 10.1177/2048872619840876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhari R, Omens JH, Covell JW, McCulloch AD. Structural basis of regional dysfunction in acutely ischemic myocardium. Cardiovasc Res. 2000;47(2):284–293. doi: 10.1016/S0008-6363(00)00089-4. [DOI] [PubMed] [Google Scholar]
- McCurdy SM, Dai Q, Zhang J, Zamilpa R, Ramirez TA, Dayah T, Nguyen N, Jin YF, Bradshaw AD, Lindsey ML. SPARC mediates early extracellular matrix remodeling following myocardial infarction. Am J Physiol - Heart Circ Physiol. 2011;301(2):497–505. doi: 10.1152/AJPHEART.01070.2010/ASSET/IMAGES/LARGE/ZH40081199640005.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Mei X, Cheng K. Recent development in therapeutic cardiac patches. Front Cardiovasc Med. 2020;7:294. doi: 10.3389/FCVM.2020.610364/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meschiari CA, Jung M, Iyer RP, Yabluchanskiy A, Toba H, Garrett MR, Lindsey ML. Macrophage overexpression of matrix metalloproteinase-9 in aged mice improves diastolic physiology and cardiac wound healing after myocardial infarction. Am J Physiol Heart Circ Physiol. 2018;314(2):224–235. doi: 10.1152/AJPHEART.00453.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael LH, Ballantyne CM, Zachariah JP, Gould KE, Pocius JS, Taffet GE, Hartley CJ, Pham TT, Daniel SL, Funk E, Entman ML. Myocardial infarction and remodeling in mice: effect of reperfusion. Am J Physiol - Heart Circ Physiol. 1999;277(2):46–2. doi: 10.1152/ajpheart.1999.277.2.H660. [DOI] [PubMed] [Google Scholar]
- Miller RD, Burchell HB, Edwards JE. Myocardial infarction with and without acute coronary occlusion: a pathologic study. A.M.A Arch Int Med. 1951;88(5):597–604. doi: 10.1001/ARCHINTE.1951.03810110049005. [DOI] [PubMed] [Google Scholar]
- Morishita N, Kusachi S, Yamasaki S, Kondo J, Tsuji T. Sequential changes in laminin and type IV collagen in the infarct zone—immunohistochemical study in rat myocardial infarction. Circ J. 1996;60(2):108–114. doi: 10.1253/jcj.60.108. [DOI] [PubMed] [Google Scholar]
- Morita M, Kawashima S, Ueno M, Kubota A, Iwasaki T. Effects of late reperfusion on infarct expansion and infarct healing in conscious rats. Am J Pathol. 1993;143(2):419–430. [PMC free article] [PubMed] [Google Scholar]
- Motley MP, Madsen DH, Jürgensen HJ, Spencer DE, Szabo R, Holmbeck K, Flick MJ, Lawrence DA, Castellino FJ, Weigert R, Bugge TH. A CCR2 macrophage endocytic pathway mediates extravascular fibrin clearance in vivo. Blood. 2016;127(9):1085–1096. doi: 10.1182/blood-2015-05-644260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Baskin JM, Deschamps AM, Lowry AS, Escobar GP, Lucas DG, Yarbrough WM, Zile MR, Spinale FG. Myocardial infarct expansion and matrix metalloproteinase inhibition. Circulation. 2003;107(4):618–625. doi: 10.1161/01.CIR.0000046449.36178.00. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88(2):581–609. doi: 10.1152/PHYSREV.00024.2007/ASSET/IMAGES/LARGE/Z9J0030824730011.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Giachelli CM, Schwartz SM, Vracko R. Macrophages express osteopontin during repair of myocardial necrosis. Am J Pathol. 1994;145(6):1450. [PMC free article] [PubMed] [Google Scholar]
- Nadaud S, Philippe M, Arnal JF, Michel JB, Soubrier F. Sustained increase in aortic endothelial nitric oxide synthase expression in vivo in a model of chronic high blood flow. Circ Res. 1996;79(4):857–863. doi: 10.1161/01.RES.79.4.857. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M. Myeloid cells in cardiovascular organs. J Intern Med. 2019;285(5):491. doi: 10.1111/JOIM.12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Sosnovik DE, Waterman P, Swirski FK, Pande AN, Aikawa E, Figueiredo JL, Pittet MJ, Weissleder R. Dual channel optical tomographic imaging of leukocyte recruitment and protease activity in the healing myocardial infarct. Circ Res. 2007;100(8):1218–1225. doi: 10.1161/01.RES.0000265064.46075.31. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Takemura G, Kanamori H, Goto K, Maruyama R, Tsujimoto A, Ohno T, Okada H, Ogino A, Esaki M, Miyata S, Li L, Ushikoshi H, Aoyama T, Kawasaki M, Nagashima K, Fujiwara T, Minatoguchi S, Fujiwara H. Mechanisms by which late coronary reperfusion mitigates postinfarction cardiac remodeling. Circ Res. 2008;103(1):98–106. doi: 10.1161/CIRCRESAHA.108.177568. [DOI] [PubMed] [Google Scholar]
- Nakamura F, Nagano M, Higaki J, Ogihara T, Minamino T, Higashino Y, Ito H, Fujii K, Fujita T. Cardiac free wall rupture in acute myocardial infarction: ameliorative effect of coronary reperfusion. Clin Cardiol. 1992;15(4):244–250. doi: 10.1002/clc.4960150405. [DOI] [PubMed] [Google Scholar]
- Nakatani D, Sato H, Kinjo K, Mizuno H, Hishida E, Hirayama A, Mishima M, Ito H, Matsumura Y, Hori M. Effect of successful late reperfusion by primary coronary angioplasty on mechanical complications of acute myocardial infarction. Am J Cardiol. 2003;92(7):785–788. doi: 10.1016/S0002-9149(03)00883-X. [DOI] [PubMed] [Google Scholar]
- Nepper-Christensen L, Lønborg J, Høfsten DE, Sadjadieh G, Schoos MM, Pedersen F, Jørgensen E, Kelbaek H, Haahr-Pedersen S, Lassen JF, Køber L, Holmvang L, Engstrøm T. Clinical outcome following late reperfusion with percutaneous coronary intervention in patients with ST-segment elevation myocardial infarction. Eur Heart J Acute Cardiovasc Care. 2021;10(5):523–531. doi: 10.1177/2048872619886312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak VP, Yin FCP, Humphrey JD. Regional mechanical properties of passive myocardium. J Biomech. 1994;27(2):403–412. doi: 10.1016/0021-9290(94)90016-7. [DOI] [PubMed] [Google Scholar]
- O’Rourke SA, Dunne A, Monaghan MG. The role of macrophages in the infarcted myocardium: orchestrators of ECM remodeling. Front Cardiovas Med. 2019;6:101. doi: 10.3389/FCVM.2019.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Murata N, Yamawaki H. Canstatin stimulates migration of rat cardiac fibroblasts via secretion of matrix metalloproteinase-2. Am J Phys Cell Physiol. 2017;312(3):C199–C208. doi: 10.1152/AJPCELL.00329.2015/ASSET/IMAGES/LARGE/ZH00021780900007.JPEG. [DOI] [PubMed] [Google Scholar]
- Oliva PB, Breckinridge JC. Arteriographic evidence of coronary arterial spasm in acute myocardial infarction. Circulation. 1977;56(3):366–374. doi: 10.1161/01.CIR.56.3.366. [DOI] [PubMed] [Google Scholar]
- Owolabi US, Amraotkar AR, Coulter AR, Singam NSV, Aladili BN, Singh A, Trainor PJ, Mitra R, DeFilippis AP. Change in matrix metalloproteinase 2, 3, and 9 levels at the time of and after acute atherothrombotic myocardial infarction. J Thromb Thrombolysis. 2020;49(2):235–244. doi: 10.1007/S11239-019-02004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce D, Nemcek M, Witzenburg C. Combining unique planar biaxial testing with full-field thickness and displacement measurement for spatial characterization of soft tissues. Curr Protoc. 2022;2(7):e493. doi: 10.1002/CPZ1.493. [DOI] [PubMed] [Google Scholar]
- Pernot M, Lee WN, Bel A, Mateo P, Couade M, Tanter M, Crozatier B, Messas E. Shear wave imaging of passive diastolic myocardial stiffness stunned versus infarcted myocardium. JACC: Cardiovasc Imaging. 2016;9(9):1023–1030. doi: 10.1016/j.jcmg.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petz A, Grandoch M, Gorski DJ, Abrams M, Piroth M, Schneckmann R, Homann S, Müller J, Hartwig S, Lehr S, Yamaguchi Y, Wight TN, Gorressen S, Ding Z, Kötter S, Krüger M, Heinen A, Kelm M, Gödecke A, … Fischer JW (2019) Cardiac hyaluronan synthesis is critically involved in the cardiac macrophage response and promotes healing after ischemia reperfusion injury. Circ Res 124(10), 1433–1447. 10.1161/CIRCRESAHA.118.313285 [DOI] [PubMed]
- Pfeffer MA, Pfeffer JM, Fishbein M, Fletcher PJ, Spadaro J, Kloner RA, Braunwald E. Myocardial infarct size and ventricular function in rats. Circ Res. 1979;44(4):503–512. doi: 10.1161/01.RES.44.4.503. [DOI] [PubMed] [Google Scholar]
- Pinkert MA, Hortensius RA, Ogle BM, Eliceiri KW. Imaging the cardiac extracellular matrix. Adv Exp Med Biol. 2018;1098:21. doi: 10.1007/978-3-319-97421-7_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD. Revisiting cardiac cellular composition. Circ Res. 2016;118(3):400–409. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, Godwin JW, Rosenthal NA (2012) An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PloS One 7(5). 10.1371/JOURNAL.PONE.0036814 [DOI] [PMC free article] [PubMed]
- Pirzada F, Ekong EA, Vokonas PS, Apstein CS, Hood WB. Experimental myocardial infarction. XIII. Sequential changes in left ventricular pressure-length relationships in the acute phase. Circulation. 1976;53(6):970–975. doi: 10.1161/01.CIR.53.6.970. [DOI] [PubMed] [Google Scholar]
- Pirzada F, Weiner JM, Hood WB. Experimental myocardial infarction. XIV. Accelerated myocardial stiffening related to coronary reperfusion following ischemia. Chest. 1978;74(2):190–195. doi: 10.1378/chest.74.2.190. [DOI] [PubMed] [Google Scholar]
- Pislaru C, Urban MW, Pislaru SV, Kinnick RR, Greenleaf JF. Viscoelastic properties of normal and infarcted myocardium measured by a multifrequency shear wave method: comparison with pressure-segment length method. Ultrasound Med Biol. 2014;40(8):1785–1795. doi: 10.1016/j.ultrasmedbio.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope AJ, Sands GB, Smaill BH, LeGrice IJ. Three-dimensional transmural organization of perimysial collagen in the heart. Am J Physiol - Heart Circ Physiol. 2008;295(3):H1243. doi: 10.1152/AJPHEART.00484.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119(1):91. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przyklenk K, Connelly C, McLaughlin RJ, Kloner RA, Apstein CS. Effect of myocyte necrosis on strength, strain, and stiffness of isolated myocardial strips. Am Heart J. 1987;114(6):1349–1359. doi: 10.1016/0002-8703(87)90536-9. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Tarbell JM. Interaction between wall shear stress and circumferential strain affects endothelial cell biochemical production. J Vasc Res. 2000;37(3):147–157. doi: 10.1159/000025726. [DOI] [PubMed] [Google Scholar]
- Quinn KP, Sullivan KE, Liu Z, Ballard Z, Siokatas C, Georgakoudi I, Black LD. Optical metrics of the extracellular matrix predict compositional and mechanical changes after myocardial infarction. Sci Rep. 2016;6(1):35823. doi: 10.1038/srep35823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran GN, Bansal M, Bhatnagar RS. A hypothesis on the role of hydroxyproline in stabilizing collagen structure Biochimica et Biophysica Acta Protein. Struct Biochim et Biophys Acta (BBA) Protein Struct. 1973;322(1):166–171. doi: 10.1016/0005-2795(73)90187-6. [DOI] [PubMed] [Google Scholar]
- Rankin JS, Arentzen CE, McHale PA, Ling D, Anderson RW. Viscoelastic properties of the diastolic left ventricle in the conscious dog. Circ Res. 1977;41(1):37–45. doi: 10.1161/01.RES.41.1.37. [DOI] [PubMed] [Google Scholar]
- Reduto LA, Freund GC, Gaeta JM, Smalling RW, Lewis B, Gould KL. Coronary artery reperfusion in acute myocardial infarction: beneficial effects of intracoronary streptokinase on left ventricular salvage and performance. Am Heart J. 1981;102(6):1168–1177. doi: 10.1016/0002-8703(81)90648-7. [DOI] [PubMed] [Google Scholar]
- Reduto LA, Smalling RW, Freund GC, Gould KL. Intracoronary infusion of streptokinase in patients with acute myocardial infarction: effects of reperfusion on left ventricular performance. Am J Cardiol. 1981;48(3):403–409. doi: 10.1016/0002-9149(81)90066-7. [DOI] [PubMed] [Google Scholar]
- Reimer KA, Jennings RB. The changing anatomic reference base of evolving myocardial infarction. Underestimation of myocardial collateral blood flow and overestimation of experimental anatomic infarct size due to tissue edema, hemorrhage and acute inflammation. Circulation. 1979;60(4):866–876. doi: 10.1161/01.CIR.60.4.866. [DOI] [PubMed] [Google Scholar]
- Reimer KA, Jennings RB. The “wavefront phenomenon” of myocardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and collateral flow. Lab Invest. 1979;40(6):633–644. [PubMed] [Google Scholar]
- Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation. 1977;56(5):786–794. doi: 10.1161/01.CIR.56.5.786. [DOI] [PubMed] [Google Scholar]
- Rentrop KP, Feit F. Reperfusion therapy for acute myocardial infarction: concepts and controversies from inception to acceptance. Am Heart J. 2015;170(5):971–980. doi: 10.1016/J.AHJ.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Ricard-Blum S, Ballut L. Matricryptins derived from collagens and proteoglycans. Front Biosci - Landmark. 2011;16:674–697. doi: 10.2741/3712. [DOI] [PubMed] [Google Scholar]
- Ricard-Blum S, Salza R. Matricryptins and matrikines: biologically active fragments of the extracellular matrix. Exp Dermatol. 2014;23(7):457–463. doi: 10.1111/EXD.12435. [DOI] [PubMed] [Google Scholar]
- Richardson WJ, Clarke SA, Quinn TA, Holmes JW, Alexander Quinn T, Holmes JW. Physiological implications of myocardial scar structure. Compr Physiol. 2015;5(4):1877–1909. doi: 10.1002/cphy.c140067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rienks M, Papageorgiou AP. Novel regulators of cardiac inflammation: matricellular proteins expand their repertoire. J Mol Cell Cardiol. 2016;91:172–178. doi: 10.1016/J.YJMCC.2016.01.008. [DOI] [PubMed] [Google Scholar]
- Rienks M, Papageorgiou A-P, Frangogiannis NG, Heymans S. Myocardial extracellular matrix. Circ Res. 2014;114(5):872–888. doi: 10.1161/CIRCRESAHA.114.302533. [DOI] [PubMed] [Google Scholar]
- Roberts WC. The pathology of acute myocardial infarction. Hosp Pract. 1971;6(12):89–104. doi: 10.1080/21548331.1971.11706704. [DOI] [Google Scholar]
- Roberts W, Burks KH, Ko JM, Filardo G, Guileyardo JM. Commonalities of cardiac rupture (left ventricular free wall or ventricular septum or papillary muscle) during acute myocardial infarction secondary to atherosclerotic coronary artery disease. Am J Cardiol. 2015;115(1):125–140. doi: 10.1016/j.amjcard.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Roberts C, Schoen FJ, Kloner RA (1983) Effect of coronary reperfusion on myocardial hemorrhage and infarct healing. Am J Cardiol 52(5):610–614. 10.1016/0002-9149(83)90036-x [DOI] [PubMed]
- Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez-Anaya A, McClure KF, Mitchell PG, Libby P, Lee RT. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999;99(23):3063–3070. doi: 10.1161/01.CIR.99.23.3063. [DOI] [PubMed] [Google Scholar]
- Sacks MS. Biaxial mechanical evaluation of planar biological materials. J Elast Phys Sci Solids. 2000;61(1):199–246. doi: 10.1023/A:1010917028671. [DOI] [Google Scholar]
- Sahu SP, Liu Q, Prasad A, Hasan SMA, Liu Q, Rodriguez MXB, Mukhopadhyay O, Burk D, Francis J, Mukhopadhyay S, Fu X, Fu X, Gartia MR, Gartia MR. Characterization of fibrillar collagen isoforms in infarcted mouse hearts using second harmonic generation imaging. Biomed Opt Express. 2021;12(1):604–618. doi: 10.1364/BOE.410347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Watanabe K, Millard RW. Defining the mechanical border zone: a study in the pig heart. Am J Physiol-Heart Circ Physiol. 1985;249(1):H88–H94. doi: 10.1152/ajpheart.1985.249.1.H88. [DOI] [PubMed] [Google Scholar]
- Sato S, Ashraf M, Millard RW, Fujiwara H, Schwartz A. Connective tissue changes in early ischemia of porcine myocardium: an ultrastructural study. J Mol Cell Cardiol. 1983;15(4):261–275. doi: 10.1016/0022-2828(83)90281-X. [DOI] [PubMed] [Google Scholar]
- Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, Frangogiannis NG. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. 2013;191(9):4838–4848. doi: 10.4049/JIMMUNOL.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellings MWM, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, Van Leeuwen REW, D’Hooge J, De Van Werf F, Carmeliet P, Pinto YM, Sage EH, Heymans S. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med. 2009;206(1):113–123. doi: 10.1084/JEM.20081244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz GS, Wysocki A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009;17(2):153–162. doi: 10.1111/J.1524-475X.2009.00466.X. [DOI] [PubMed] [Google Scholar]
- Schuster EH, Bulkley BH. Expansion of transmural myocardial infarction: a pathophysiologic factor in cardiac rupture. Circulation. 1979;60(7):1532–1538. doi: 10.1161/01.CIR.60.7.1532. [DOI] [PubMed] [Google Scholar]
- Sherry S. The origin of thrombolytic therapy. J Am Coll Cardiol. 1989;14(4):1085–1092. doi: 10.1016/0735-1097(89)90493-2. [DOI] [PubMed] [Google Scholar]
- Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. 2014;70:74–82. doi: 10.1016/J.YJMCC.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AC, Pereira C, Fonseca ACRG, Pinto-do-ÓP, Nascimento DS (2021) Bearing my heart: the role of extracellular matrix on cardiac development, homeostasis, and injury response. Front Cell Dev Biol 0, 1705. 10.3389/FCELL.2020.621644 [DOI] [PMC free article] [PubMed]
- Simari RD, Berger PB, Bell MR, Gibbons RJ, Holmes DR. Coronary angioplasty in acute myocardial infarction: primary, immediate adjunctive, rescue, or deferred adjunctive approach? Mayo Clin Proc. 1994;69(4):346–358. doi: 10.1016/s0025-6196(12)62220-4. [DOI] [PubMed] [Google Scholar]
- Singh M, Foster CR, Dalal S, Singh K. Osteopontin: role in extracellular matrix deposition and myocardial remodeling post-MI. J Mol Cell Cardiol. 2010;48(3):538–543. doi: 10.1016/J.YJMCC.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirry MS, Butler JR, Patnaik SS, Brazile B, Bertucci R, Claude A, McLaughlin R, Davies NH, Liao J, Franz T. Characterisation of the mechanical properties of infarcted myocardium in the rat under biaxial tension and uniaxial compression. J Mech Behav Biomed Mater. 2016;63:252–264. doi: 10.1016/J.JMBBM.2016.06.029. [DOI] [PubMed] [Google Scholar]
- Skjøt-Arkil H, Clausen RE, Rasmussen LM, Wang W, Wang Y, Zheng Q, Mickley H, Saaby L, Diederichsen ACP, Lambrechtsen J, Martinez FJ, Hogaboam CM, Han ML, Larsen MR, Nawrocki A, Vainer B, Krustrup D, Bjørling-Poulsen M, Karsdal MA, Leeming DJ. Acute myocardial infarction and pulmonary diseases result in two different degradation profiles of elastin as quantified by two novel ELISAs. PLOS ONE. 2013;8(6):e60936. doi: 10.1371/JOURNAL.PONE.0060936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167(5):2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- Sommer G, Schriefl AJ, Andrä M, Sacherer M, Viertler C, Wolinski H, Holzapfel GA. Biomechanical properties and microstructure of human ventricular myocardium. Acta Biomater. 2015;24:172–192. doi: 10.1016/j.actbio.2015.06.031. [DOI] [PubMed] [Google Scholar]
- Souders CA, Bowers SLK, Baudino TA (2009) Cardiac fibroblast: the renaissance cell. Circ Res 105(12). 10.1161/CIRCRESAHA.109.209809 [DOI] [PMC free article] [PubMed]
- Streeter DD, Spotnitz HM, Patel DP, Ross J, Sonnenblick EH. Fiber orientation in the canine left ventricle during diastole and systole. Circ Res. 1969;24(3):339–347. doi: 10.1161/01.RES.24.3.339. [DOI] [PubMed] [Google Scholar]
- Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46(2):250–256. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- Sun M, Dawood F, Wen WH, Chen M, Dixon I, Kirshenbaum LA, Liu PP. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation. 2004;110(20):3221–3228. doi: 10.1161/01.CIR.0000147233.10318.23. [DOI] [PubMed] [Google Scholar]
- Svensson L, Heinegard D, Oldberg A. Decorin-binding sites for collagen type I are mainly located in leucine-rich repeats 4–5. J Biol Chem. 1995;270(35):20712–20716. doi: 10.1074/JBC.270.35.20712. [DOI] [PubMed] [Google Scholar]
- Takemura G, Nakagawa M, Kanamori H, Minatoguchi S, Fujiwara H (2009) Benefits of reperfusion beyond infarct size limitation. Cardiovasc Res 83(2), 269–276. https://academic.oup.com/cardiovascres/article/83/2/269/320731. Accessed 14 Jan 2022 [DOI] [PubMed]
- Takemura G, Fujiwara H. Role of apoptosis in remodeling after myocardial infarction. Pharmacol Ther. 2004;104(1):1–16. doi: 10.1016/j.pharmthera.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. 2016;365(3):563–581. doi: 10.1007/s00441-016-2431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167(1):71. doi: 10.1016/S0002-9440(10)62954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao ZY, Cavasin MA, Yang F, Liu YH, Yang XP. Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci. 2004;74(12):1561–1572. doi: 10.1016/j.lfs.2003.09.042. [DOI] [PubMed] [Google Scholar]
- Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279(17):17079–17084. doi: 10.1074/JBC.M310859200. [DOI] [PubMed] [Google Scholar]
- Tennant R, Wiggers CJ. The effect of coronary occlusion on myocardial contraction. Am J Physiol-Legacy Content. 1935;112(2):351–361. doi: 10.1152/ajplegacy.1935.112.2.351. [DOI] [Google Scholar]
- Tessone A, Feinberg MS, Barbash IM, Reich R, Holbova R, Richmann M, Mardor Y, Leor J. Effect of matrix metalloproteinase inhibition by doxycycline on myocardial healing and remodeling after myocardial infarction. Cardiovasc Drugs Ther. 2005;19(6):383–390. doi: 10.1007/s10557-005-5201-6. [DOI] [PubMed] [Google Scholar]
- Theroux P, Ross J, Franklin D, Covell JW, Bloor CM, Sasayama S. Regional myocardial function and dimensions early and late after myocardial infarction in the unanesthetized dog. Circ Res. 1977;40(2):158–165. doi: 10.1161/01.RES.40.2.158. [DOI] [PubMed] [Google Scholar]
- Thimm TN, Squirrell JM, Liu Y, Eliceiri KW, Ogle BM. Endogenous optical signals reveal changes of elastin and collagen organization during differentiation of mouse embryonic stem cells. Tissue Eng. Part C, Methods. 2015;21(10):995. doi: 10.1089/ten.tec.2014.0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X-F, Cui M-X, Yang S-W, Zhou Y-J, Hu D-Y. Cell death, dysglycemia and myocardial infarction. Biomed Rep. 2013;1(3):341–346. doi: 10.3892/br.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilbury K, Hocker J, Wen BL, Sandbo N, Singh V, Campagnola PJ. Second harmonic generation microscopy analysis of extracellular matrix changes in human idiopathic pulmonary fibrosis. J Biomed Optics. 2014;19(8):086014. doi: 10.1117/1.JBO.19.8.086014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troidl C, Möllmann H, Nef H, Masseli F, Voss S, Szardien S, Willmer M, Rolf A, Rixe J, Troidl K, Kostin S, Hamm C, Elsässer A. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J Cell Mol Med. 2009;13(9b):3485–3496. doi: 10.1111/J.1582-4934.2009.00707.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueblood NA, Xie Z, Communal C, Sam F, Ngoy S, Liaw L, Jenkins AW, Wang J, Sawyer DB, Bing OHL, Apstein CS, Colucci WS, Singh K. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res. 2001;88(10):1080–1087. doi: 10.1161/HH1001.090842. [DOI] [PubMed] [Google Scholar]
- Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, Elkind M SV, Evenson KR, Eze-Nliam C, Ferguson JF, Generoso G, Ho JE, Kalani R, Khan SS, Kissela BM, … Martin SS (2022) Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circulation 145(8), e153–e639. 10.1161/CIR.0000000000001052 [DOI] [PubMed]
- Tyberg JV, Forrester JS, Wyatt HL, Goldner SJ, Parmley WW, Swan HJ. An analysis of segmental ischemic dysfunction utilizing the pressure-length loop. Circulation. 1974;49(4):748–754. doi: 10.1161/01.CIR.49.4.748. [DOI] [PubMed] [Google Scholar]
- Ulrich MMW, Janssen AMH, Daemen MJAP, Rappaport L, Samuel J-L, Contard F, Smits JFM, Cleutjens JPM. Increased expression of fibronectin isoforms after myocardial infarction in rats. J Mol Cell Cardiol. 1997;29(9):2533–2543. doi: 10.1006/jmcc.1997.0486. [DOI] [PubMed] [Google Scholar]
- Valiente-Alandi I, Potter SJ, Salvador AM, Schafer AE, Schips T, Carrillo-Salinas F, Gibson AM, Nieman ML, Perkins C, Sargent MA, Huo J, Lorenz JN, DeFalco T, Molkentin JD, Alcaide P, Blaxall BC. Inhibiting fibronectin attenuates fibrosis and improves cardiac function in a model of heart failure. Circulation. 2018;138(12):1236. doi: 10.1161/CIRCULATIONAHA.118.034609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Werf F. The history of coronary reperfusion. Eur Heart J. 2014;35(37):2510–2515. doi: 10.1093/eurheartj/ehu268. [DOI] [PubMed] [Google Scholar]
- Van De Werf F, Ludbrook PA, Bergmann SR, Tiefenbrunn AJ, Fox KAA, de Geest H, Verstraete M, Collen D, Sobel BE. Coronary thrombolysis with tissue-type plasminogen activator in patients with evolving myocardial infarction. N Engl J Med. 1984;310(10):609–613. doi: 10.1056/NEJM198403083101001. [DOI] [PubMed] [Google Scholar]
- Van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci USA. 1990;87(14):5578. doi: 10.1073/PNAS.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte D, Schellings M, Pinto Y, Heymans S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc Res. 2006;69(3):604–613. doi: 10.1016/J.CARDIORES.2005.10.002/2/M_69-3-604-UFIG2.GIF. [DOI] [PubMed] [Google Scholar]
- Varghese S, Ohlow M-A. Left ventricular free wall rupture in myocardial infarction: a retrospective analysis from a single tertiary center. JRSM Cardiovasc Dis. 2019;8:204800401989669. doi: 10.1177/2048004019896692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihert A, Cherpachenko N. Some aspects of myocardial metabolism outside the zone of experimental myocardial infarction. Virchows Arch Abt A Path Anat. 1971;354:293–304. doi: 10.1007/BF00545722. [DOI] [PubMed] [Google Scholar]
- Vokonas PS, Pirzada F, Hood WB. Experimental myocardial infarction: XII. Dynamic changes in segmental mechanical behavior of infarcted and non-infarcted myocardium. Am J Cardiol. 1976;37(6):853–859. doi: 10.1016/0002-9149(76)90109-0. [DOI] [PubMed] [Google Scholar]
- Vokonas PS, Malsky PM, Paul SJ, Robbins SL, Hood WB. Radioautographic studies in experimental myocardial infarction: profiles of ischemic blood flow and quantification of infarct size in relation to magnitude of ischemic zone. Am J Cardiol. 1978;42(1):67–75. doi: 10.1016/0002-9149(78)90987-6. [DOI] [PubMed] [Google Scholar]
- Voorhees AP, DeLeon-Pennell KY, Ma Y, Halade GV Yabluchanskiy A, Padmanabhan R, Chao H (2015) Building a better infarct: modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction 43. 10.1016/j.yjmcc.2015.06.006 [DOI] [PMC free article] [PubMed]
- Wagner DR, Delagardelle C, Ernens I, Rouy D, Vaillant M, Beissel J. Matrix metalloproteinase-9 is a marker of heart failure after acute myocardial infarction. J Cardiac Fail. 2006;12(1):66–72. doi: 10.1016/j.cardfail.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Waldenström A, Martinussen HJ, Gerdin B, Hällgren R. Accumulation of hyaluronan and tissue edema in experimental myocardial infarction. J Clin Investig. 1991;88(5):1622–1628. doi: 10.1172/JCI115475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JC, Ratcliffe MB, Zhang P, Wallace AW, Fata B, Hsu EW, Saloner D, Guccione JM. MRI-based finite-element analysis of left ventricular aneurysm. Am J Physiol - Heart Circ Physiol. 2005;289(2):692–700. doi: 10.1152/AJPHEART.01226.2004. [DOI] [PubMed] [Google Scholar]
- Wang X, Lu Y, Xie Y, Shen J, Xiang M (2019) Emerging roles of proteoglycans in cardiac remodeling. Int J Cardiol 278, 192–198. https://pubmed.ncbi.nlm.nih.gov/30528626/. Accessed 14 Jan 2022 [DOI] [PubMed]
- Wang K, Meng X, Guo Z (2021a) Elastin structure, synthesis, regulatory mechanism and relationship with cardiovascular diseases. Front Cell Dev Biol, 9. 10.3389/FCELL.2021.596702 [DOI] [PMC free article] [PubMed]
- Wang L, Serpooshan V, Zhang J (2021b) Engineering human cardiac muscle patch constructs for prevention of post-infarction LV remodeling. Front Cardiovasc Med, 8. 10.3389/FCVM.2021.621781/FULL [DOI] [PMC free article] [PubMed]
- Ward SR, Sutton JM, Pieper KS, Schwaiger M, Califf RM, Topol EJ. Effects of thrombolytic regimen, early catheterization, and predischarge angiographic variables on six-week left ventricular function. Am J Cardiol. 1997;79(5):539–544. doi: 10.1016/S0002-9149(96)00812-0. [DOI] [PubMed] [Google Scholar]
- Webb CS, Bonnema DD, Ahmed SH, Leonardi AH, McClure CD, Clark LL, Stroud RE, Corn WC, Finklea L, Zile MR, Spinale FG. Specific temporal profile of matrix metalloproteinase release occurs in patients after myocardial infarction: relation to left ventricular remodeling. Circulation. 2006;114(10):1020–1027. doi: 10.1161/CIRCULATIONAHA.105.600353. [DOI] [PubMed] [Google Scholar]
- Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13(7):1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- Weis SM, Zimmerman SD, Shah M, Covell JW, Omens JH, Ross J, Dalton N, Jones Y, Reed CC, Iozzo RV, McCulloch AD. A role for decorin in the remodeling of myocardial infarction. Matrix Biol. 2005;24(4):313–324. doi: 10.1016/J.MATBIO.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Welch MP, Odland GF, Clark RAF. Fibronectin receptor expression to wound contraction. Cell. 1990;110:133–145. doi: 10.1083/jcb.110.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JM, Gaggar A, Blalock JE. MMP generated matrikines. Matrix Biol. 2015;44–46(5):122–129. doi: 10.1016/j.matbio.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann D, Mersmann J, Melchior A, Freudenberger T, Petrik C, Schaefer L, Lüllmann-Rauch R, Lettau O, Jacoby C, Schrader J, Brand-Herrman SM, Young MF, Schultheiss HP, Levkau B, Baba HA, Unger T, Zacharowski K, Tschöpe C, Fischer JW. Biglycan is required for adaptive remodeling after myocardial infarction. Circulation. 2008;117(10):1269–1276. doi: 10.1161/CIRCULATIONAHA.107.714147. [DOI] [PubMed] [Google Scholar]
- Whittaker P, Boughner DR, Kloner RA. Analysis of healing after myocardial infarction using polarized light microscopy. Am J Pathol. 1989;134(4):879–893. [PMC free article] [PubMed] [Google Scholar]
- Whittaker P, Boughner DR, Kloner RA. Role of collagen in acute myocardial infarct expansion. Circulation. 1991;84(5):2123–2134. doi: 10.1161/01.CIR.84.5.2123. [DOI] [PubMed] [Google Scholar]
- Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Res Cardiol. 1994;89(5):397–410. doi: 10.1007/BF00788278. [DOI] [PubMed] [Google Scholar]
- Wiggers H, Klebe T, Heickendorff L, Høst NB, Danielsen CC, Baandrup U, Andersen HR. Ischemia and reperfusion of the porcine myocardium: effect on collagen. J Mol Cell Cardiol. 1997;29(1):289–299. doi: 10.1006/jmcc.1996.0274. [DOI] [PubMed] [Google Scholar]
- Witzenburg C, Raghupathy R, Kren SM, Taylor DA, Barocas VH. Mechanical changes in the rat right ventricle with decellularization. J Biomech. 2012;45(5):842–849. doi: 10.1016/j.jbiomech.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzenburg C, Holmes JW (2017). Biomechanics of myocardial ischemia and infarction. In Studies in Mechanobiology, Tissue Engineering and Biomaterials 20:233–269. Springer. 10.1007/978-3-319-41475-1_6
- Woodcock EA, Matkovich SJ. Cardiomyocytes structure, function and associated pathologies. Int J Biochem Cell Biol. 2005;37(9):1746–1751. doi: 10.1016/J.BIOCEL.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Wu X, Chen Z, Yang Y, Dong Y, Liu H, Kuang S, Luo K. Impact of proteasome inhibitor MG-132 on expression of NF-κB, IL-1β and histological remodeling after myocardial infarction. Exp Ther Med. 2018;16(2):1365. doi: 10.3892/ETM.2018.6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Gu M, Wu K, Li G. Unraveling the role of hydroxyproline in maintaining the thermal stability of the collagen triple helix structure using simulation. J Phys Chem B. 2019;123(36):7754–7763. doi: 10.1021/ACS.JPCB.9B05006/ASSET/IMAGES/LARGE/JP9B05006_0008.JPEG. [DOI] [PubMed] [Google Scholar]
- Yamada S. Ko T, Hatsuse S, Nomura S, Zhang B, Dai Z, Inoue S, Kubota M, Sawami K, Yamada T, Sassa T, Katagiri M, Fujita K, Katoh M, Ito M, Harada M, Toko H, Takeda N, Morita H, … Komuro I (2022) Spatiotemporal transcriptome analysis reveals critical roles for mechano-sensing genes at the border zone in remodeling after myocardial infarction. Nature Cardiovascular Research 2022 1:11, 1(11), 1072–1083. 10.1038/s44161-022-00140-7 [DOI] [PMC free article] [PubMed]
- Yang Y, Ma Y, Han W, Li J, Xiang Y, Liu F, Ma X, Zhang JF, Fu Z, Su YD, Du XJ, Gao XM. Age-related differences in postinfarct left ventricular rupture and remodeling. Am J Physiol - Heart Circ Physiol. 2008;294(4):1815–1822. doi: 10.1152/ajpheart.00831.2007. [DOI] [PubMed] [Google Scholar]
- Yang H, Borg TK, Wang Z, Ma Z, Gao BZ. Role of the basement membrane in regulation of cardiac electrical properties. Ann Biomed Eng. 2014;42(6):1148–1157. doi: 10.1007/s10439-014-0992-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarbrough WM, Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Escobar GP, Joffs C, Lucas DG, Crawford FA, Spinale FG, Damiano RJ, Yacoub MH, Sellke FW. Matrix metalloproteinase inhibition modifies left ventricular remodeling after myocardial infarction in pigs. J Thorac Cardiovasc Surg. 2003;125(3):602–610. doi: 10.1067/mtc.2003.197. [DOI] [PubMed] [Google Scholar]
- Yarbrough WM, Mukherjee R, Escobar GP, Mingoia JT, Sample JA, Hendrick JW, Dowdy KB, McLean JE, Lowry AS, O’Neill TP, Spinale FG. Selective targeting and timing of matrix metalloproteinase inhibition in post-myocardial infarction remodeling. Circulation. 2003;108(14):1753–1759. doi: 10.1161/01.CIR.0000091087.78630.79. [DOI] [PubMed] [Google Scholar]
- Yu Y, Yin G, Bao S, Guo Z. Kinetic alterations of collagen and elastic fibres and their association with cardiac function in acute myocardial infarction. Mol Med Rep. 2018;17(3):3519–3526. doi: 10.3892/MMR.2017.8347/HTML. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf S, Collins R, Peto R, Furberg C, Stampfer MJ, Goldhaber SZ, Hennekens CH. Intravenous and intracoronary fibrinolytic therapy in acute myocardial infarction: overview of results on mortality, reinfarction and side-effects from 33 randomized controlled trials. Eur Heart J. 1985;6(7):556–585. doi: 10.1093/oxfordjournals.eurheartj.a061905. [DOI] [PubMed] [Google Scholar]
- Zavadzkas JA, Stroud RE, Bouges S, Mukherjee R, Jones JR, Patel RK, McDermott PJ, Spinale FG. Targeted overexpression of tissue inhibitor of matrix metalloproteinase-4 modifies post-myocardial infarction remodeling in mice. Circ Res. 2014;114(9):1435–1445. doi: 10.1161/CIRCRESAHA.114.303634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wang W, He W, Xi N, Wang Y, Liu L. Dynamic model for characterizing contractile behaviors and mechanical properties of a cardiomyocyte. Biophys J . 2018;114(1):188–200. doi: 10.1016/j.bpj.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Zhang H, Robinson TF, Factor SM, Sonnenblick EH, Eng C. Profound structural alterations of the extracellular collagen matrix in postischemic dysfunctional (“stunned”) but viable myocardium. J Am Coll Cardiol. 1987;10(6):1322–1334. doi: 10.1016/S0735-1097(87)80137-7. [DOI] [PubMed] [Google Scholar]