

Abstract
Interleukin-33 (IL-33) is a crucial nuclear cytokine that induces the type 2 immune response and maintains immune homeostasis. The fine-tuned regulation of IL-33 in tissue cells is critical to control of the type 2 immune response in airway inflammation, but the mechanism is still unclear. Here, we found that healthy individuals had higher phosphate-pyridoxal (PLP, an active form of vitamin B6) concentrations in the serum than asthma patients. Lower serum PLP concentrations in asthma patients were strongly associated with worse lung function and inflammation. In a mouse model of lung inflammation, we revealed that PLP alleviated the type 2 immune response and that this inhibitory effect relied on the activity of IL-33. A mechanistic study showed that in vivo, pyridoxal (PL) needed to be converted into PLP, which inhibited the type 2 response by regulating IL-33 stability. In mice heterozygous for pyridoxal kinase (PDXK), the conversion of PL to PLP was limited, and IL-33 levels were increased in the lungs, aggravating type 2 inflammation. Furthermore, we found that the mouse double minute 2 homolog (MDM2) protein, an E3 ubiquitin-protein ligase, could ubiquitinate the N-terminus of IL-33 and sustain IL-33 stability in epithelial cells. PLP reduced MDM2-mediated IL-33 polyubiquitination and decreased the level of IL-33 through the proteasome pathway. In addition, inhalation of PLP alleviated asthma-related effects in mouse models. In summary, our data indicate that vitamin B6 regulates MDM2-mediated IL-33 stability to constrain the type 2 response, which might help develop a potential preventive and therapeutic agent for allergy-related diseases.
Keywords: Type 2 inflammation, IL-33, Vitamin B6.
Subject terms: Allergy, Interleukins
Introduction
Allergy-related diseases such as asthma, atopic dermatitis, and seasonal rhinitis are already prevalent in the developed world [1]. These diseases often occur when people are persistently or repetitively exposed to allergens. Epithelial cells recognize pathogen-associated molecular patterns (PAMPs) found in microbes and damage-associated molecular patterns (DAMPs) released upon tissue damage by engaging pattern recognition receptors (PRRs) and releasing interleukin-33 (IL-33), IL-25, and thymic stromal lymphopoietin (TSLP) [2]. These cytokines can activate many innate immune cells, including mast cells, eosinophils, and innate lymphoid cells, to enhance the type 2 immune response [3].
IL-33 is a tissue-derived nuclear cytokine identified as a member of the IL-1 family and plays an essential role in regulating and maintaining the immune response and repairing tissue damage [4–6]. Mature IL-33 binds to ST2 on target cells such as mast cells, eosinophils, type 2 innate lymphoid cells (ILC2s), and T helper 2 (TH2) cells [7, 8]. Genome-wide association studies have shown that genetic variation in IL-33 and interleukin 1 receptor-like 1 (IL1RL1/ST2) loci translates into increased asthma susceptibility [9–12]. Therefore, the regulation of IL-33 is vital for IL-33-mediated immune responses in allergic inflammation. Because of its abundant basal expression [13–15], there are multiple mechanisms that limit its activity [16–20]. It has been shown that IL-33 harbors ubiquitination modifications, which are essential for the homeostasis of IL-33 [21–23]. However, evidence for the detailed mechanism by which ubiquitination regulates the stability of IL-33 is limited.
Vitamin B6 is an essential nutrient that humans cannot synthesize de novo. Vitamin B6 exists in six different forms: pyridoxine (PN), pyridoxal (PL), pyridoxamine (PM), and their phosphorylated derivatives [24]. PLP is the active form, transformed by pyridoxal kinase (PDXK) into phosphorylated forms in vivo. Clinical studies have found that adult asthma patients have lower plasma phosphate-pyridoxal (PLP) concentrations and that supplementation with vitamin B6 can decrease the frequency and severity of asthmatic attacks and alleviate proinflammatory responses in rheumatoid arthritis patients [25, 26]. Although increasing amounts of evidence show that vitamin B6 has anti-inflammatory activity, the function of vitamin B6 in the type 2 immune response needs to be further explored.
Mouse double minute 2 homolog (MDM2) is an E3 ligase with a RING finger motif [27]. MDM2, downstream of the p53 signaling pathway, regulates p53 degradation to form negative feedback [28]. MDM2 can interact with and ubiquitinate the RB protein to control cell cycle progression [29]. MDM2 enhances FOXO3a degradation via an MDM2-dependent ubiquitin‒proteasome pathway to inhibit cell proliferation [30]. MDM2 can also target some substrates to mediate nondegradative ubiquitination. Recent studies have shown that MDM2 interacts with Foxp3 and mediates the ubiquitination of Foxp3 to enhance its stability, which positively regulates Treg cell function [31]. MDM2 can mediate the monoubiquitination of HDAC3 to increase its stability [32].
In this study, we demonstrated the effects of vitamin B6 on type 2 lung inflammation via regulation of the homeostasis of IL-33. We found that PLP, phosphorylated by PDXK, controls the stability of IL-33. The active form of PLP reduces MDM2-mediated polyubiquitination of IL-33 and decreases IL-33 levels. This study provides new insights into the potential application value of vitamin B6 in type 2 lung inflammation.
Results
Lower serum PLP levels in asthma patients are strongly correlated with worse lung function and inflammation
Nutrient intake is strongly associated with lung inflammation. Plant foods such as fruits and vegetables permanently reduce the risk of lung inflammation, while high fat intake aggravates lung inflammation [33]. Vitamins have attracted the attention of researchers. Many researchers have reported the protective roles of vitamin A, vitamin D, and vitamin C in asthma development [34–37]. Clinical studies have also found that vitamin B6 deficiency is associated with asthma [25]. However, the mechanism of vitamin B6 in asthma is still unclear. To explore the role of vitamin B6 in type 2 lung inflammation, we detected the PL and PLP levels in the plasma of asthmatic patients and healthy controls by LC‒MS (S-Data 1 and 2). The results showed that the PLP concentration in serum was lower in asthmatic patients than in healthy controls (Fig. 1a). The serum from severe patients had the lowest concentration of PLP, suggesting that the plasma PLP concentration was negatively correlated with asthma severity (Fig. 1b). Moreover, the serum PLP concentration was positively associated with pulmonary ventilation FEV1 (forced expiratory volume) (Fig. 1c) and negatively associated with the percentage and number of eosinophils in asthma patients (Fig. 1d, e). To compare the vitamin B6 levels between asthma and a type 2 inflammation-irrelevant disease, we tracked the serum vitamin B6 concentrations of 311 asthmatic patients and 220 lung cancer patients with the LK3000VI vitamin detector and found that the serum total vitamin B6 concentration in asthmatic patients was lower than that in lung cancer patients (S-Fig. 1a). These results suggest that lower serum PLP levels in asthma patients are strongly correlated with worse lung function and type 2 inflammation.
Fig. 1.

Lower serum PLP levels in asthma patients are strongly correlated with worse lung function and inflammation. a, b LC–MS analysis of serum PL and PLP levels in healthy controls and asthma patients; HC: healthy control (n = 52; male: 26, female: 26); asthma: asthma patients (n = 58; male: 23, female: 35); mild and moderate: mild and moderate asthma patients (n = 29); severe: severe asthma patients (n = 29); **P < 0.01; ****p < 0.0001; not significant (ns); the data were analyzed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. Analysis of the correlations of serum PLP level with the FEV1 percentage (c), percentage of eosinophils in the blood (d) and number of eosinophils in the blood (e) of all asthma patients (n = 58). Each point indicates a sample from one individual. Pearson’s test determined the correlations
Vitamin B6 ameliorates papain-induced acute lung inflammation
Circulating vitamin B6 is usually derived from diet or microbiota metabolites and acts as a cofactor in its active form (PLP). Therefore, we administered PLP daily to mice through the intraperitoneal route three days before papain exposure (Fig. 2a). We found decreased eosinophil infiltration and type 2 cytokine (IL-5 and IL-13) production in bronchoalveolar lavage fluid (BALF) from PLP-treated mice (Fig. 2b and S-Fig. 2a). PLP treatment did not affect the percentage or number of macrophages and neutrophils in BALF from mice after papain exposure (S-Fig. 2b, c). The amelioration of lung inflammation in PLP-treated mice was further proven by PAS and H&E staining of lung tissue, as represented by reduced mucus production and infiltrated cells (Fig. 2c). In addition, the number of ILC2s and the levels of the type 2 cytokines IL-5 and IL-13 from ILC2s were significantly diminished in PLP-treated mice (Fig. 2d, e, and S-Fig. 2d, e). We next used a vitamin B6-deficient diet to produce vitamin B6 deficiency in vivo. The PLP concentration in the serum of mice receiving a vitamin B6-deficient diet was lower than that in the serum of mice receiving a vitamin B6 control diet (Fig. 2f). Eosinophil infiltration in BALF was increased in mice receiving a vitamin B6-deficient diet (Fig. 2g). More severe lung inflammation according to analysis of PAS- and H&E-stained sections (Fig. 2h) and increased numbers of functional ILC2s (IL-5+ ILC2s, IL-13+ ILC2s) (Fig. 2i) were detected in mice that were fed a vitamin B6-deficient diet.
Fig. 2.

Vitamin B6 ameliorates papain-induced acute lung inflammation. a Schematic diagram of PLP treatment by intraperitoneal (i.p.) delivery in a papain model of lung inflammation in C57BL/6J mice. b Representative flow cytometry analysis and quantification of eosinophils (CD45+SiglecF+CD11c−) in BALF. c Representative images of H&E- and PAS-stained lung sections and semiquantified inflammation of lungs from PBS- or PLP intraperitoneally treated mice (scale bar, 50 µm). d Total number of lung CD45+Lin−CD90.2+ST2+ ILC2s as assessed by flow cytometry. e Quantification of IL-5+ ILC2s and IL-13+ ILC2s by flow cytometry in the lungs of control and PBS- or PLP-treated mice in the papain model. (In b–e, PBS-treated group, n = 3; papain-treated group, n = 5/6) f LC‒MS analysis of serum PLP levels in vitamin B6 control diet- (n = 5) and vitamin B6-deficient diet-fed mice (n = 5). g Number of eosinophils (CD45+ SiglecF + CD11c−) in BALF according to flow analysis from vitamin B6 control diet- and vitamin B6-deficient diet-treated mice in the papain model (n = 6/7). h Representative images of H&E- and PAS-stained lung sections and semiquantified inflammation of lungs from vitamin B6 control diet- and vitamin B6-deficient diet-fed mice in the papain model (n = 6/7) (scale bar, 100 µm). i Numbers of functional ILC2s (IL-5+ ILC2s, IL-13+ ILC2s) in the lungs as determined by flow analysis (n = 6/7). The data are shown as the means ± SDs (error bars) of three independent experiments. The data were analyzed by one-way ANOVA with Dunnett’s multiple comparison test (b‒e) and unpaired Student’s t test (f–i). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Mice were also treated with PLP through the intratracheal route (S-Fig. 2f). Similarly, local administration of PLP reduced eosinophil infiltration (S-Fig. 2g) and the concentrations of IL-5 and IL-13 in BALF (S-Fig. 2h), as well as the post-papain-treatment numbers of lung ILC2s and IL-5/IL-13-producing ILC2s (S-Fig. 2i, j). It has been reported that vitamin B6 can affect CD4 T-cell populations [38]. We then treated Rag1−/− mice lacking mature T and B cells with PLP through the intratracheal route in the papain-induced lung inflammation model. The data showed that PLP treatment also reduced the eosinophil percentage and count in BALF (S-Fig. 2k), inflammatory cell infiltration, and mucus hypersecretion (S-Fig. 2l). These results indicate that circulating and local PLP treatment has an inhibitory effect on lung inflammation.
Vitamin B6 attenuates type 2 inflammation via IL-33
We next explored how vitamin B6 inhibited lung inflammation. The mRNA levels of the type 2 inflammatory cytokines IL-5 and IL-13 were significantly lower in the lungs of the papain model after PLP treatment (Fig. 3a). ILC2s have been implicated as the primary effector cells in papain-induced acute lung inflammation [39]. However, we found no significant difference in IL-5 or IL-13 secretion by ILC2s after 24 h of vitamin B6 treatment in vitro (S-Fig. 3a). These results suggested that vitamin B6 might play a role upstream of ILC2s to reduce lung inflammation rather than having a direct effect on ILC2s. The epithelial-derived cytokine IL-33 is a potent activator of ILC2s [3], and greater expression of IL-33 than of IL-25 or TSLP was induced in the lungs after papain stimulation (Fig. 3b). An IL-33-neutralizing antibody was used for PLP-mediated lung inflammation resolution (S-Fig. 3b). The IL-33 neutralizing antibody attenuated lung inflammation, as demonstrated by reduced numbers of eosinophils (Fig. 3c, d) and levels of type 2 cytokines (Fig. 3e) in BALF, decreased numbers of functional ILC2s (IL-5+ ILC2s, IL-13+ ILC2s) (S-Fig. 3c) in lung tissue, and improved tissue pathology (Fig. 3f). Furthermore, the PLP-mediated reduction in lung inflammation was eliminated by the anti-IL-33 neutralizing antibody (Fig. 3c–f). In summary, these results indicate that the protective effects of PLP in type 2 inflammation occur via IL-33.
Fig. 3.

Vitamin B6 attenuates type 2 inflammation via IL-33. a Q-PCR was used to detect the expression of IL-5 and IL-13 in lungs from PBS-treated mice or PLP-treated papain model mice (n = 6). b Q-PCR was used to detect the expression of IL-25, IL-33, and TSLP in the lungs from the PBS-treated mice or the papain-treated papain model (n = 4/6). c, d Representative flow cytometry analysis and quantification of eosinophils in BALF in an anti-IL-33 antibody-treated papain model. e ELISA was used to determine the amounts of IL-5 and IL-13 in BALF in an anti-IL-33 antibody-treated papain model. f Representative H&E and PAS staining of lung sections (scale bar, 100 µm). Inflammation was determined by semiquantitative scoring. The data are shown as the means ± SDs (error bars) and represent three independent experiments. The data were analyzed by unpaired Student’s t test (a, b) or ordinary one-way ANOVA with Sidak’s test (c–f). Ns not significant. *p < 0.05; **p < 0.01; ****p < 0.0001
Vitamin B6-regulated IL-33 homeostasis is dependent on PDXK
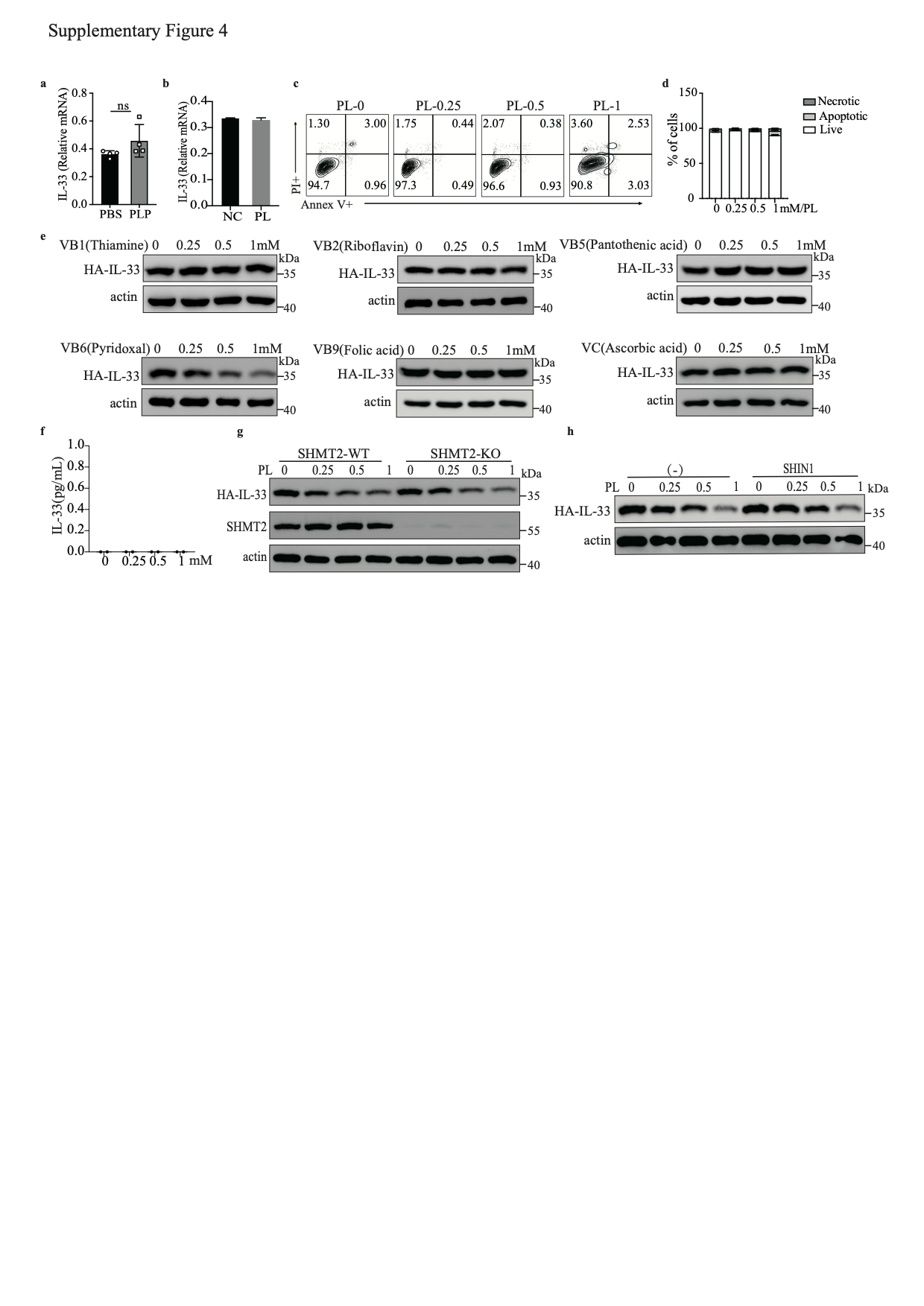
IL-33 production in BALF peaks at 1 h after exposing lung epithelial cells to allergens [40]. We measured the levels of IL-33 in the BALF and lungs of PBS- and PLP-treated mice 1 h after papain exposure (Fig. 4a) and found that the IL-33 concentrations in BALF and lungs were decreased after PLP treatment (Fig. 4b, c). However, the mRNA level of IL-33 was comparable between PBS- and PLP-treated mice (S-Fig. 4a). To better confirm the correlation between PLP and IL-33, we stably expressed full-length IL-33 with an HA tag in a human alveolar basal epithelial cell line, A549 (A549-IL-33). We treated A549-IL-33 cells with PL at different times and found that the intracellular IL-33 level diminished in a time-dependent manner (Fig. 4d, e), but the mRNA level did not change after 24 h of treatment (S-Fig. 4b). More than 90% of A549-IL-33 cells were alive after 1 mM PL treatment for 24 h (S-Fig. 4c, d). We also found that only vitamin B6 could reduce intracellular IL-33 protein levels without promoting IL-33 release, unlike other vitamin B family members and soluble vitamin C (S-Fig. 4e. f). A recent study has shown that PLP, as a cofactor, can shift the SHMT2 oligomeric state to regulate the interaction between BRISC and SHMT2, which mediates the polyubiquitination of type I interferon receptors (IFNAR1 and IFNAR2) [36]. We knocked out SHMT2 in A549-IL-33 cells and found that the PL dose-dependent reduction in IL-33 protein did not change after SHMT2 deletion (S-Fig. 4g). After treatment with SHIN1, a standard inhibitor of SHMT2 and SHMT1, a PL-mediated decrease in IL-33 protein level still occurred (S-Fig. 4h). Thus, vitamin B6-mediated intracellular IL-33 homeostasis is not dependent on SHMT2 or SHMT1.
Fig. 4.

Vitamin B6-regulated IL-33 homeostasis is dependent on PDXK. a Schematic diagram of PBS or PLP treatment by the intratracheal route for 3 days. BALF and lung homogenate were collected 1 h after challenge with papain at Day 4. b The amount of IL-33 in BALF was determined by ELISA (n = 4). The results of one of three experiments are shown. Unpaired Student’s t test was used to analyze the data. *p < 0.05. c The lung tissue of mice was weighted and addded 2.5 mL/g PBS to homogenize, and then the amounts of IL-33 in the supernatant of lung homogenates were determined by ELISA (n = 4). The results of one of three experiments is are. Unpaired Student’s t test was used to analyze the data. *p < 0.05. d, e Western blot detection of the protein level of IL-33 in A549-IL-33 cells treated with 0.5 mM PL for the duration indicated. e The ratio of HA-IL-33/actin is shown (n = 3). The results for one of three experiments are shown. Ordinary one-way ANOVA with Dunnett’s multiple comparisons test was used to analyze the data. ****p < 0.0001. f A549-IL-33 cells or PDXK-knockout A549-IL-33 cells were treated with the indicated concentrations of PL for 24 h, and western blotting was used to analyze intracellular IL-33 protein levels. The data are representative of three independent experiments. g The ratio of HA-IL-33/actin in (a) is shown (n = 3). Ordinary one-way ANOVA with Dunnett’s multiple comparisons test was used to analyze the data. ***p < 0.001; ns not significant. h HEK293FT cells were transfected with HA-IL-33 or His-PDXK for 24 h, treated with or without 0.5 mM PL for 24 h, and analyzed by western blotting. The results of one of three experiments are shown. i The HA-IL-33/actin ratio in (c) is shown (n = 3). Ordinary one-way ANOVA with Tukey’s multiple comparisons test was used to analyze the data. *p < 0.05; **p < 0.01. j PLP levels in the serum of PDXK+/+ (n = 6) and PDXK+/− (n = 4) mice according to LC‒MS. Unpaired Student’s t test was used to analyze the data. *p < 0.05. The data are representative of three independent experiments. k ELISA was used to detect the IL-33 levels in the supernatant of lung homogenates of PDXK+/+ (n = 7) and PDXK+/− (n = 6) mice. Unpaired Student’s test was used to analyze the data. **p < 0.01. The data are representative of two independent experiments
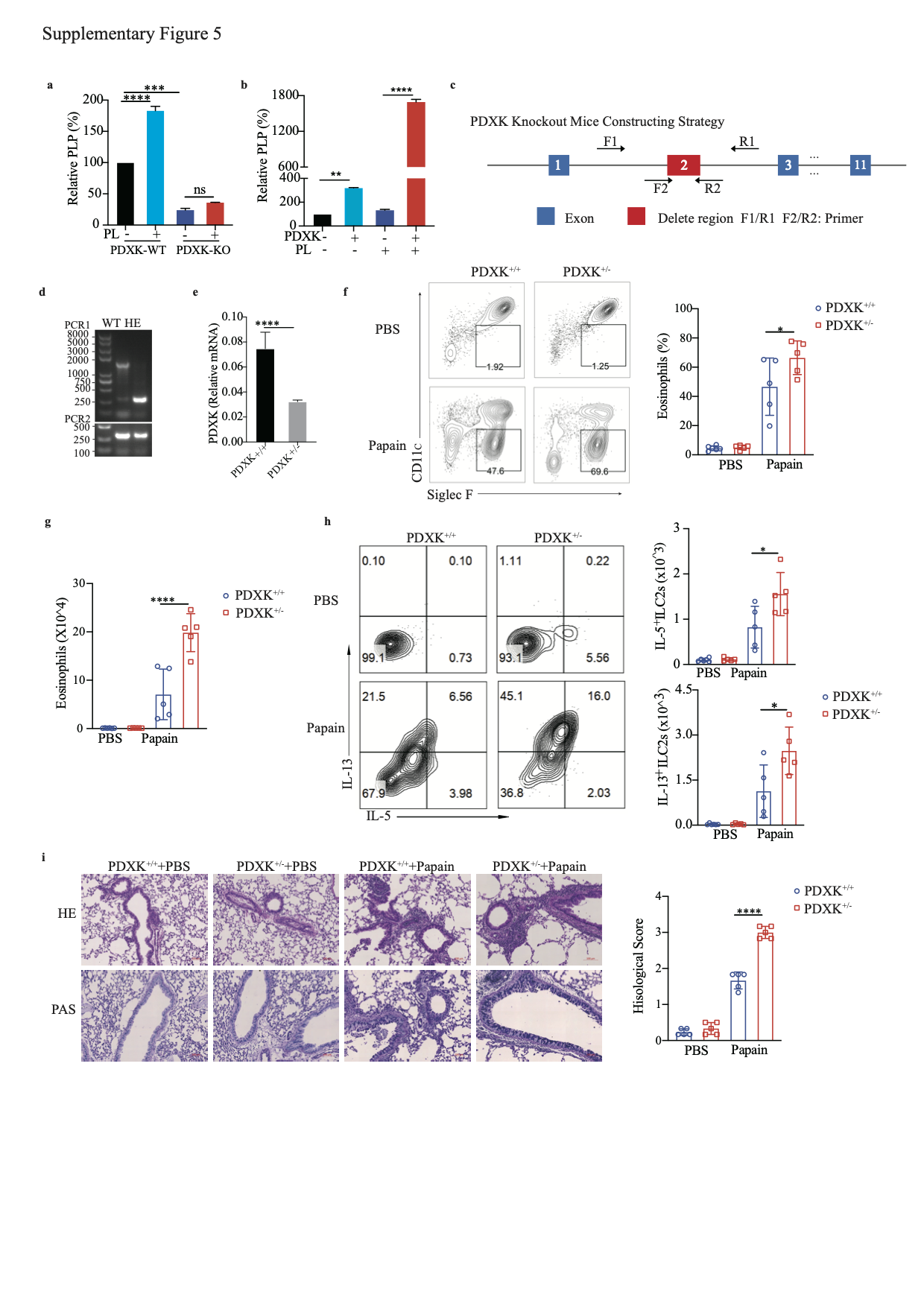
PDXK is a crucial enzyme responsible for producing the active form of vitamin B6, PLP [40, 41]. We knocked out PDXK in A549-IL-33 cells and found that once the PDXK gene was knocked out, the generation of intracellular PLP was almost blocked, and PL could not restore the intracellular PLP level (S-Fig. 5a). In addition, the PL-mediated decrease in IL-33 protein levels was nearly abolished (Fig. 4f, g). Overexpression of PDXK increased intracellular PLP levels and promoted the reduction in the IL-33 protein level in HEK293FT cells, especially in the PL-treated group (S-Fig. 5b and Fig. 4h, i). Because homozygous global PDXK gene knockout in mice is lethal [42], we used PDXK-haploinsufficient mice (PDXK+/−) to validate the function of PLP in IL-33 homeostasis and IL-33-mediated type 2 inflammation in vivo (S-Fig. 5c, d). PDXK gene expression was lower in the lung tissues of PDXK+/− mice than in those of wild-type (WT) mice (S-Fig. 5e). The serum PLP concentration was also decreased in PDXK+/− mice (Fig. 4j). Accordingly, the IL-33 concentration in lung tissue was increased in PDXK+/− mice compared with WT mice (Fig. 4k). Increased eosinophil infiltration in BALF and more functional ILC2s (IL-5+ ILC2s, IL-13+ ILC2s) were observed in the lungs of papain-challenged PDXK+/− mice than in those of WT mice (S-Fig. 5f–h). Moreover, histological staining of lung tissue revealed more severe lung inflammation in the lungs of PDXK+/− mice (S-Fig. 5i). These data suggest that the PDXK-mediated active form of vitamin B6 (PLP) is critical in IL-33 homeostasis.
Vitamin B6 inhibits IL-33 stability through ubiquitin modification
We studied how vitamin B6 regulates IL-33 homeostasis and found that PL still accelerated intracellular IL-33 degradation in the presence of cycloheximide (CHX), which inhibits protein synthesis (Fig. 5a). PL treatment led to a dose-dependent reduction in intracellular IL-33 protein level that could be prevented by the proteasome inhibitor MG132 but not by the lysosome inhibitor NH4Cl (Fig. 5b, c), which indicated the proteasome-dependent degradation of IL-33. IL-33 could be ubiquitinated, and PL reduced the ubiquitination of IL-33 (Fig. 5d). We also expressed HA-tagged full-length IL-33 and mature IL-33 (112–270 aa) in HEK293FT cells treated with different concentrations of PL and found that PL treatment did not affect mature IL-33 (112–270 aa) (Fig. 5e, f). In contrast, the ubiquitylation of mature IL-33 (112–270 aa) was significantly decreased compared with that of full-length IL-33 (Fig. 5g) [43]. Together, these results indicate that vitamin B6 inhibits IL-33 stability through ubiquitin modification of the N-terminal domain of IL-33 (1–111 aa).
Fig. 5.

Vitamin B6 inhibits IL-33 stability through ubiquitin modification. a A549-IL-33 cells were cultured with or without 0.5 mM PL treatment for 24 h and then treated with CHX (20 µg/ml) for the indicated duration. The HA-IL-33/actin ratio is shown (n = 3). b, c A549-IL-33 cells were treated with PL at the indicated concentrations for 20 h and then with or without MG132 (10 μM) (a) or NH4Cl (20 mM) (b). The intracellular IL-33 level was analyzed by western blotting. The HA-IL-33/actin ratio is shown (n = 3). d Determination of the effect of PL treatment on IL-33 ubiquitination. HEK293FT cells were transfected with the indicated plasmids and then treated with 0.5 mM PL (PL) for 24 h before being subjected to His pulldown. The data are representative of three independent experiments. e, f HEK293FT cells were transfected with full-length HA-IL-33 or mature IL-33 (112–270 aa) for 24 h and then treated with PL at the indicated concentrations. The western blot shows IL-33 expression. f Ratio of HA-IL-33 to actin expression (n = 2). g HEK293FT cells were transfected with full-length IL-33, mature IL-33 (112–270 aa) and Myc-Ub for 48 h and immunoprecipitated with an anti-HA antibody. The data are representative of three independent experiments. The data were analyzed by two-way ANOVA with Sidak’s multiple comparisons test (a–c) or one-way ANOVA with Dunnett’s multiple comparisons test (f). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant
MDM2 interacts with IL-33 to facilitate IL-33 stability
We performed RNA sequencing (RNA-seq) analysis on A549-IL-33 cells with or without PL treatment and found enrichment of the p53 signaling pathway after PL treatment (Fig. 6a). Vitamin B6 has also been reported to be associated with p53 signaling [44, 45]. A comprehensive database for proteome-wide known and predicted ubiquitin ligase/deubiquitinase-substrate interactions shows that mouse double minute 2 homolog (MDM2) is one of the possible E3 candidates that interacts with IL-33 (S-Fig. 6a) (http://ubibrowser.ncpsb.org). MDM2 is an E3 ligase with a RING finger motif [27] that is involved in p53 signaling. Recent studies have shown that MDM2 enhances the stability of Foxp3 [31], HDAC3 [32], and STAT5 [46] through ubiquitination. IL-33 colocalized with MDM2 in the nucleus (Fig. 6b). In addition, endogenous (Fig. 6c) and exogenous MDM2 interacted with IL-33, but PL treatment reduced the interaction between MDM2 and IL-33 in HEK293FT cells (Fig. 6d, e). MDM2 knockout decreased the IL-33 protein level in A549-IL-33 cells, and a PL-mediated decrease in the IL-33 protein level was not detected (Fig. 6f). Consistently, knockout of MDM2 caused reduced IL-33 expression in HEK293FT cells (S-Fig. 6b). MDM2 expression increased full-length IL-33 protein levels in a dose-dependent manner, but not mature IL-33 (112–270 aa) levels (Fig. 6g, h). Coimmunoprecipitation assays confirmed that MDM2 interacted with full-length IL-33 and the N-terminal domain of IL-33 (1–111 aa) but not mature IL-33 (112–270 aa) (Fig. 6i). We then found that the 102–288 aa domain of MDM2, which contained the acidic domain, was essential for the interaction between MDM2 and IL-33 (Fig. 6j, k). Collectively, these results indicate that MDM2 interacts with IL-33 to facilitate IL-33 stability.
Fig. 6.

MDM2 interacts with IL-33 to facilitate IL-33 stability. a RNA-seq analysis of A549-IL-33 cells treated with or without 0.5 mM PL for 24 h. KEGG pathway analysis showed enriched signaling pathways. b A confocal assay was performed in A549 cells stably expressing full-length IL-33 with GFP. DAPI (blue) and an anti-MDM2 antibody (red) were used (scale bar = 5 μm). c Coimmunoprecipitation of endogenous MDM2 and IL-33 in A549-IL-33 cells. d, e HEK293FT cells were transfected with the indicated plasmids for 24 h and then treated with or without 0.5 mM PL for 24 h before being subjected to immunoprecipitation with an HA antibody. f A549-IL-33 or MDM2-knockout A549-IL-33 cells treated with different concentrations of PL for 24 h. Western blot analysis of intracellular IL-33 levels. The HA-IL-33/actin ratio is shown (n = 3). Ordinary one-way ANOVA with Dunnett’s multiple comparisons test was used to analyze the data. ****p < 0.0001; ns not significant. g, h HEK293FT cells were transfected with full-length HA-IL-33 or mature IL-33 (112–270 aa) and increasing concentrations of His-MDM2. g Western blot showing IL-33 and MDM2 expression. h Ratio of HA-IL-33 to actin expression (n = 3). Ordinary one-way ANOVA with Dunnett’s multiple comparisons test was used to analyze the data. ****p < 0.0001; ns not significant. i Coimmunoprecipitation of HEK293FT cells transfected with IL-33 deletion mutants and V5-MDM2. j Schematic representation of MDM2 and its truncation mutant. k Immunoprecipitation and western blot analysis of HEK293FT cells transfected with HA-IL-33 and truncated MDM2. The data in b–i and k are representative of three independent experiments
Vitamin B6 regulates IL-33 homeostasis by attenuating MDM2-mediated IL-33 ubiquitination
Cotransfection of MDM2 and IL-33 into HEK293FT cells resulted in substantial ubiquitination of IL-33. PL treatment reduced the MDM2-mediated ubiquitination level of IL-33 (Fig. 7a). Knockout of MDM2 reduced IL-33 ubiquitination in HEK293FT cells (S-Fig. 7a). An in vitro ubiquitination assay also showed that MDM2 mediated the ubiquitination of IL-33 (Fig. 7b). Protein ubiquitylation has several ubiquitin-molecule linkage types, namely, Lys6-, Lys11-, Lys27-, Lys29-, Lys33-, Ly48-, Lys63- and Met1-linked chains [47, 48]. Distinct ubiquitin modifications have different functions and outcomes [49–51]. We found that the K63R mutation in Ub significantly reduced MDM2-mediated IL-33 ubiquitination (Fig. 7c). Moreover, MDM2-mediated ubiquitination of IL-33 was observed with K63-only ubiquitin, not with K48-only ubiquitin (Fig. 7d). PL treatment abolished MDM2-mediated K63-linked ubiquitination of IL-33 (Fig. 7e). MDM2 and PLP also mediated K27- and K29-linked ubiquitination of IL-33 (S-Fig. 7b). Because the RING domain is indispensable for the ubiquitin ligase activity of MDM2 [27], truncation of the MDM2 RING domain led to the loss of IL-33 ubiquitination mediated by MDM2 (Fig. 7f and S-Fig. 7c). Moreover, the absence of the RING domain caused MDM2 to fail to promote the stability of the IL-33 protein (Fig. 7g and S-Fig. 7d). The truncated 102–288 aa domain of MDM2, which was essential for the interaction between MDM2 and IL-33 (Fig. 6j, k), could also not sustain IL-33 ubiquitination and IL-33 stability (Fig. 7h, i and S-Fig. 7e). These data suggest that MDM2 catalyzes various types of ubiquitination (K27, K29, K63) of IL-33 to sustain IL-33 homeostasis, which is downregulated by vitamin B6.
Fig. 7.

Vitamin B6 regulates IL-33 homeostasis by attenuating MDM2-mediated IL-33 ubiquitination. a HEK293FT cells were transfected with the indicated plasmids for 24 h, treated with or without 0.5 mM PL for 24 h, and immunoprecipitated with an HA antibody. Western blot analysis of IL-33 ubiquitination. b Western blot analysis of IL-33 ubiquitination in vitro. c, d Role of MDM2 in IL-33 ubiquitination. HEK293FT cells were transfected with the indicated plasmids and lysed before being immunoprecipitated with an anti-HA antibody. e Role of PL treatment in MDM2-mediated IL-33 ubiquitination. HEK293FT cells were transfected with the indicated plasmids for 24 h, treated with or without 0.5 mM PL for 24 h, lysed, and immunoprecipitated with an HA antibody. The V5-Ub/HA-IL-33 ratios in the immunoprecipitated groups are shown (n = 3). Two-way ANOVA with Sidak’s multiple comparisons test was used to analyze the data. ****p < 0.0001; ***p < 0.001; ns not significant. f Immunoblot analysis of lysates from HEK293FT cells transfected with the indicated plasmids and His-tagged MDM2 or the MDM2 RING domain truncation mutant and then treated as in (c). g HEK293FT cells were transfected with HA-IL-33 and increasing concentrations of full-length His-MDM2 and the truncated RING domain of His-MDM2 and analyzed by western blotting. The HA-IL-33/actin ratio is shown (n = 3). Ordinary one-way ANOVA with the Brown-Forsythe test was used to analyze the data. ****p < 0.0001; ns not significant. h Effect of the truncated 102–288 aa domain of MDM2 on IL-33 ubiquitination. HEL293FT cells were transfected with full-length MDM2 or truncated MDM2 and the indicated plasmids and immunoprecipitated with an HA antibody. i HEK293FT cells were transfected with HA-IL-33, and increasing concentrations of full-length His-MDM2 and the truncated 102–288 aa domain mutant of His-MDM2 were analyzed by western blotting. The HA-IL-33/actin ratio is shown (n = 3). One-way ANOVA with the Brown-Forsythe test was used to analyze the data. ****p < 0.0001; ns not significant. The results of one of three experiments are shown
Vitamin B6 relieves experimental type 2 lung inflammation
To further evaluate the therapeutic effect of vitamin B6 on type 2 inflammation, we used a vitamin B6-supplemented diet in a clinically relevant model of house dust mite (HDM)-induced chronic allergic lung inflammation. We found that the number of eosinophils in BALF did not differ between mice receiving a vitamin B6-supplemented diet and those receiving a control diet (S-Fig. 8a), nor did the functional CD4 T-cell and ILC2 numbers in the lungs (S-Fig. 8b, c). The LC‒MS data showed that a vitamin B6-supplemented diet only increased the serum PLP concentration in wild-type mice approximately 1.5-fold, from 200 to 300 nM (S-Fig. 8d), which might not have guaranteed high concentrations of PLP in the lungs. These data indicated that oral vitamin B6 might have low efficiency in type 2 inflammation suppression because of relatively low delivery efficiency to the lungs.
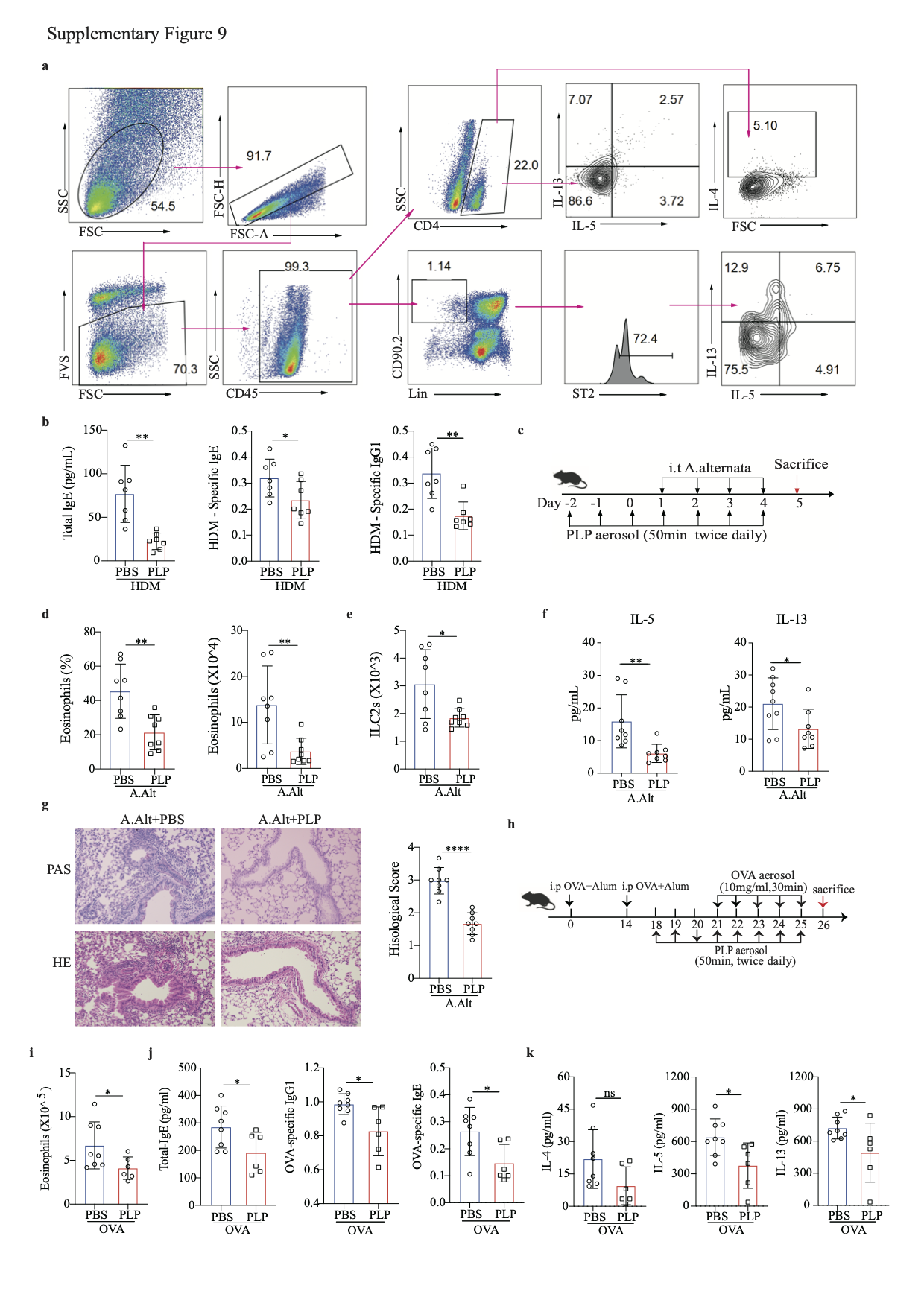
We next used the inhalation method to treat HDM-induced chronic allergic lung inflammation (Fig. 8a). PLP-treated mice had a reduced frequency of eosinophils in BALF and decreased lung inflammation (Fig. 8b, c). Flow cytometry analysis further revealed that the numbers of type 2 cytokine-producing CD4 T cells and ILC2s in the lungs were reduced after PLP treatment (Fig. 8d, e and S-Fig. 9a). The production of serum IgE, HDM-specific IgE and IgG1 was also reduced in PLP-treated mice (S-Fig. 9b). In the Alternaria alternata mouse model (S-Fig. 9c), the frequency and total number of eosinophils in the BALF of mice receiving PLP were significantly lower than those in the BALF of mice receiving PBS (S-Fig. 9d), as were the total number of lung ILC2s (S-Fig. 9e) and the level of type 2 lung inflammation (S-Fig. 9f, g). In the OVA mouse model (S-Fig. 9h), PLP treatment reduced the number of eosinophils in BALF and the production of total serum IgE, OVA-specific IgE, and IgG1 (S-Fig. 9i, j). The levels of IL-5 and IL-13 were significantly reduced in the supernatant of lung monocytes from PLP-treated mice cultured with OVA for 72 h (S-Fig. 9k). These data indicate a promising strategy for allergic inflammation prevention and treatment.
Fig. 8.

Vitamin B6 relieves HDM-induced type 2 inflammation. a Schematic diagram of PLP aerosol in the HDM model of lung inflammation in C57BL/6J mice (n = 7). b Representative flow analysis and quantification of the percentage of eosinophils in BALF. c Representative images of H&E- and PAS-stained lung sections and semiquantification of inflammation in the lungs (scale bar, 50 µm). d Flow cytometry quantification of IL-4+ T cells, IL-5+ T cells, and IL-13+ T cells in the lungs. e Flow cytometry quantification of IL-5+IL-13+ ILC2s in the lungs. The data are shown as the means ± SDs (error bars) from three independent experiments. Unpaired Student’s t test was used to analyze the data. *p < 0.05; **p < 0.01; ****p < 0.0001
Discussion
Our study demonstrates the importance of the PLP/MDM2/IL-33 axis in type 2 lung inflammation (S-Fig. 10). Our findings reveal that PLP attenuates the MDM2-mediated ubiquitination of IL-33 and subsequently regulates IL-33 stability. This study provides a new perspective for preventing and treating allergy-related diseases such as asthma.
Our results showed that PLP levels were lower in the plasma of asthma patients than in that of healthy controls, which is consistent with the results of a previous study [25]. We also found that the concentration of PLP in plasma decreased with the severity of asthma. As an active form of vitamin B6, PLP is also the main form existing in plasma [52]. In our lung inflammation model, local and systemic administration of PLP alleviated allergen-induced type 2 inflammation. In contrast, a vitamin B6-deficient diet led to a lower serum PLP concentration and severe type 2 lung inflammation. ILC2 culture treatment with vitamin B6 in vitro did not reveal a direct effect of vitamin B6 on ILC2s. IL-33 expression was mainly induced in the lungs after papain stimulation, and the suppressive effect of PLP was not observed in anti-IL-33 antibody-treated papain-induced lung inflammation. These data indicate the effect of PLP on IL-33 in epithelial cells. These findings are supported by the data that IL-33 protein levels were reduced after vitamin B6 treatment in vitro and in vivo. Furthermore, we showed, for the first time, that vitamin B6 regulates IL-33 homeostasis. An unreported mechanistic study demonstrated that MDM2 can interact with IL-33 and mediate various types of ubiquitination to facilitate IL-33 stability. Lys27-linkages have been related to mitochondrial biology [48]. Lys29-linkages are associated with proteasomal degradation [47]. Lys63-linkages can regulate signal transduction and protect proteins from degradation [47, 50]. While mixed and branched chains on proteins endow ubiquitin with a new function [53], more methods and techniques are needed to explore the roles of multiple linkages of ubiquitin. In general, as an E3 ubiquitin ligase, MDM2 mediates p53, Rb, and FOXO3a degradation in the ubiquitin‒proteasome pathway [28–30]. Recent studies have shown that MDM2 can increase FOXP3, HDAC3, and STAT5 stability via nondegradative ubiquitination [31, 32, 46]. Here, we showed that MDM2 mediated the ubiquitination of IL-33 by the RING domain to facilitate IL-33 stability. Moreover, PL treatment attenuated MDM2-mediated ubiquitination of IL-33. Therefore, we revealed that MDM2 sustains IL-33 stability via ubiquitination modification and that PLP negatively regulates MDM2-mediated IL-33 stability, reducing type 2 inflammation.
Asthma and allergic inflammation have become prevalent in developed countries [1]. The primary treatments for asthma are inhaled corticosteroids and long-acting beta antagonists, but the disease often develops steroid resistance, resulting in severe uncontrolled disease. IL-33 is a crucial cytokine for initiating the type 2 immune response and is becoming a promising therapeutic target [54]. In a murine experimental model of asthma, IL-33 blockade has been found to inhibit allergic airway inflammation [55]. IL-33 inhibition in atopic dermatitis (AD) patients shows a rapid and persistent clinical benefit. A phase 2a clinical trial for COPD with itepekimab, an IL-33-targeting antibody, has confirmed the efficacy and safety of itepekimab [56]. The inhalation route for drug delivery locally to the lungs is commonly used to treat lung diseases. We found that PLP inhalation constrained type 2 lung inflammation in clinically related asthma models, indicating possible clinical application. In addition to treating it, vitamin B6 can also prevent allergic disease in susceptible patients. Because of the safety and low economic cost of vitamin B6, it is a valuable tool for the development of PLP inhalation agents for allergic inflammation.
Our results indicate that PLP reduces the interaction of MDM2 and IL-33 to attenuate MDM2-mediated IL-33 ubiquitination. However, how PLP decreases the interaction is unclear. It has been reported that the MDM2 RING domain can bind ATP and that the 2 and 3 hydroxyls of the ribose are responsible for binding. Additionally, phosphate groups of ATP can contribute to binding [57, 58]. PLP has phosphate and hydroxy groups. Therefore, we speculate that PLP might bind to MDM2 to change the conformation of MDM2, decreasing the interaction of MDM2 and IL-33. However, the mechanism and whether it applies to other MDM2-mediated proteins still need to be confirmed.
In our current work, PL-treated cells showed p53 signaling pathway activation, consistent with other research reports [45]. MDM2, downstream of the p53 signaling pathway, regulates p53 stability to form a negative feedback loop [28]. Because we found that PLP/MDM2 could regulate IL-33 stability, PLP might function as an MDM2 antagonist, inhibiting the degradation of the MDM2-mediated tumor suppressor p53 and promoting the p53 signaling pathway in tumor therapy. However, there is still a lack of evidence for this hypothesis in our current work. We will focus our attention on this in the future.
Overall, our study underlines the importance of vitamin B6 in asthma patients. We revealed that MDM2 can interact with IL-33 to sustain IL-33 stability in epithelial cells and that vitamin B6 attenuates the IL-33 ubiquitination mediated by the E3 ligase MDM2 to promote IL-33 degradation and regulate type 2 lung inflammation, which depends on PDXK-mediated PLP production. Our results might aid in the use of vitamin B6 as a potential preventative and therapeutic agent in allergic inflammation.
Materials and methods
Mice
C57BL/6 mice were purchased from Shanghai SLAC Laboratory Animal Limited Liability Company. Rag1−/− mice were maintained at the Chinese Academy of Science (Shanghai, China). PDXK+/− mice were purchased from GemPharmatech Co., Ltd and generated by knocking out exon 2 of the mouse PDXK gene using the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9). PCR confirmed the PDXK-knockout effect. All animals were kept in a specific-pathogen-free room. Age- (6–10 weeks old) and sex-matched experimental mice were used. All mouse experiments were performed in compliance with the guidelines of the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (accession number: SIBCB-S303–2005–006).
Human samples
Fifty-eight asthmatic subjects (n = 58; male: 23, female: 35; aged 21–74 y) and 52 healthy subjects (n = 52; male: 26, female: 26; aged 20~70 y) were enrolled to measure PL/PLP concentrations in serum with LC‒MS. A total of 311 asthmatic patients (n = 311) and 220 lung cancer patients (n = 220) were tracked to analyze serum vitamin B6 detected by an LK3000VI vitamin detector. All human samples were collected by the Department of Respiratory and Critical Care Medicine, Shanghai Pulmonary Hospital, Tongji University. Ethical approval was provided by the hospital Ethics Committee (accession number: 2021LY0413). All of the patients provided informed consent before use during the study.
The inclusion criteria for asthma patients were as follows: (1) has physician-diagnosed asthma based on the “Global Strategy for Asthma Management and Prevention” [59], (2) aged between 18 and 75, (3) demonstrates good compliance and cooperation to complete lung function tests, (4) has no other diagnosed pulmonary disease or allergic disease (such as allergic rhinitis, food allergy, or atopic dermatitis), and (5) approved of participation in the study and gave written informed consent after learning about the research purposes and methods. Asthma patients were classified as having mild to moderate or severe asthma based on the guidelines mentioned previously.
The inclusion criteria for healthy controls were as follows: (1) has no diagnosed illness, (2) aged between 18 and 75, (3) has a normal thoracic image, (4) has a normal eosinophil count, and (5) gave written informed consent.
Antibodies and reagents
Anti-hemagglutinin (Sigma-Aldrich, H6908), horseradish peroxidase (HRP)-conjugated 6X His (Proteintech, HRP-66005), anti-V5 tag (Cell Signaling Technology, 13202S), anti-actin (Sigma-Aldrich, A2066), anti-Ub (Santa Cruz, sc-8017), anti-PDXK (Sigma-Aldrich, HPA030196), anti-SHMT2 (Sigma-Aldrich, HPA020549), anti-MDM2 (Abcam, ab259265), HRP-conjugated goat anti-mouse IgG (R&D, HAF007), and HRP-conjugated goat anti-rabbit IgG (R&D, HAF007) antibodies were used. MG132 (Selleck, S2619), a protein synthesis inhibitor; cycloheximide (CHX) (Selleck, S7418), NH4Cl (Sangon Biotech), and SHIN1 (MCE, HY112066) were purchased and used, and papain (Sigma Aldrich, P3125), A. alternata (Greer Labs, XPM1D3A25), HDM (Greer Labs, XPB91D3A25), recombinant IL-33 (BioLegend, 580506), and an anti-IL-33 antibody (Shanghai Immune Biotech, S730-8) were purchased and used.
Various enzyme-linked immunosorbent assay (ELISA) kits were used in this study, including mouse IL-33 (R&D, DY3626), mouse IL-5 (Thermo Fisher Scientific, 88–5054–76), mouse IL-13 (Thermo Fisher, 88–7137–76) kits and human IL-33 (R&D, D3300B) kits.
Similarly, the following antibodies were used in flow cytometry: anti-CD11b (Bioscience, clone M1/70), anti-NK1.1 (eBioscience, PK136), anti-CD11c (eBioscience, clone N418), anti-SiglecF (BD Biosciences, clone E50-2440), anti-Ly-6G (eBioscience, clone RB6–8C5), anti-CD4 (eBioscience, clone GK1.5), anti-CD3 (eBioscience, clone 2C11), anti-Ter119 (eBioscience, clone Ter119), anti-FcεR1 (eBioscience, clone MAR-1), anti-CD45R (BD Biosciences, clone RA3–6B2), anti-TCR γ/δ (BioLegend, clone GL3), anti-CD45.2 (BioLegend, clone 104), anti-CD90.2 (BD Biosciences, clone 53–2.1), anti-IL-33R (T1/ST2, MD Biosciences, clone DJ8), and anti-CD127 (eBioscience, clone A7R34). Eosinophils were identified as CD45+SiglecF+CD11c−. Lung ILC2s were identified as Lin- (TCRγ/δ, CD45R, NK1.1, CD3, CD11b, CD11c, Ter119, Ly6G, FcεR1, Gr1) CD45+CD90.2+ST2+. For intracellular cytokine staining, lung leukocytes were stimulated with PMA (50 ng/ml, Merck Millipore), ionomycin (10 µg/mL, Merck) and brefeldin (10 µg/mL, BD Bioscience) for 4 h at 37 °C and then stained to determine intracellular IL-5 and IL-13 levels.
Lung inflammation model
Mice were anesthetized by inhalation of 4% isoflurane, intratracheally administered papain (4 µg in 40 µl of PBS) or saline (as a control) daily for five days and sacrificed at Day 6 for papain-induced lung inflammation. For IL-33-induced lung inflammation, recombinant mouse IL-33 (10 ng/mouse) was intratracheally administered to mice for three consecutive days, and the mice were sacrificed 24 h after the final treatment. House dust mites (HDMs) (25 µg) were administered intratracheally to mice for three consecutive days for HDM-induced allergic inflammation. The mice were challenged with HDM (5 µg) at Day 14 for four continuous days. The mice were sacrificed 24 h after the last challenge. For ovalbumin (OVA)-induced lung inflammation, mice were intraperitoneally treated with 20 µg OVA complexed with 1 mg aluminum (alum) in a 200 µl volume on Days 1 and 14. On Days 21–25, mice were challenged with an aerosol of 10 mg/ml OVA inhalation daily for 30 min. On Day 26, the mice were sacrificed.
In vivo PLP treatment
For systemic administration, mice were given 20 mg/kg PLP (Selleck) or saline (as a control) intraperitoneally three days before being exposed to papain or IL-33 and throughout the study. For local administration, intratracheal administration of 4 mg/kg PLP or saline was performed daily three days before exposure to papain and throughout the study. For PLP aerosol, mice were treated with 20 mM aerosol PLP for 50 min twice daily for 3 days before being exposed to A. alternata or HDM and throughout the study.
Vitamin B6 diet treatment
The mice were divided into three groups that differed in PN concentration: (1) a vitamin B6 control group (7 mg/kg), which was fed a standard diet; (2) a vitamin B6 deficiency group (0 mg/kg), which was subjected to complete vitamin B6 deficiency; and (3) a vitamin B6-supplemented group (70 mg/kg) that was subjected to excessive vitamin B6 supplementation. The diet concentration was selected as shown in S-Data 3. The mice were reared on these diets for two weeks and then challenged with papain or HDM.
Lung cell isolation and in vitro culture
Mouse lung cells were isolated as described previously [60]. Mouse lung tissue was cut into pieces and digested in Roswell Park Memorial Institute (RPMI) medium containing 10% serum and 5% collagenase I at 37 °C for 30 min. After centrifugation, the red blood cells were removed with red blood cell (RBC) lysis buffer. The remaining cells were separated by a 40%/80% Percoll density gradient, and the cells were collected from a layer between the 40% and 80% fractions for fluorescence-activated cell sorting (FACS) analysis.
For ILC2 culture in vitro, mice were treated with recombinant IL-33 (50 ng/mouse) for three consecutive days and then allowed to rest for three days. Mice were sacrificed on Day 7 to isolate lung monocytes. Then, sorted ILC2s were identified as Lin-CD90.2+ST2+ and cultured with IL-2 (BioLegend, 10 ng/ml) and IL-7 (PeproTech, 20 ng/ml) with or without IL-33 (BioLegend, 1 ng/ml) for 24 h in the presence or absence of PL. Then, the levels of IL-5 and IL-13 in the culture supernatant were measured by ELISA.
Lung histology
Lung tissue was fixed in 4% paraformaldehyde (PFA) for 24 h and then dehydrated and embedded in paraffin. The tissue sections were stained with periodic acid-Schiff (PAS) and hematoxylin and eosin (H&E) stain. The histopathological score for each section was determined by semiquantitative scoring as described previously ([61, 62]). In brief, inflammation was quantified by a treatment-blind observer and graded on the following scale: 0, none; 1, mild; 2, moderate; 3, marked; and 4, severe. An increment of 0.5 was used when the inflammation was between two levels. The histopathological score of each section was the average of three selected random fields.
Plasmid constructs
Human IL-33, MDM2 and PDXK were amplified via PCR using PBMC or A549 cell cDNA libraries and then cloned into pcDNA3 vectors with HA or a 6X His-tag. MDM2 was also cloned into a PHAGE vector with V5 tags. V5-Ub and its mutant were inserted into the PHAGE vector. The IL-33 gene was inserted into a pLVX vector with puromycin resistance for screening. SgRNAs for SHMT2, PDXK and MDM2 knockout were inserted into a lenti-CRISPR v.2 vector with puromycin resistance for screening. The lentivirus-packaging plasmids psPAX2 and PMD2.G were kindly provided by Hai Jiang as a gift (Shanghai Institute of Biochemistry and Cell Biology, Shanghai, China). All plasmids were confirmed by sequencing.
Generation of genetically modified cell lines
For knock-in and knockout experiments, HEK293FT cells were transfected with packing plasmids (psPAX2 and PMD2.G) and the pLVX vector containing IL-33-HA (knock-in) or a lenti-CRISPR v.2-based vector (knockout) with HighGene transfection reagent (ABclonal). Virus-containing supernatants were collected 72 h after transfection. Then, A549 cells were cultured with the virus supernatant for 24 h and screened with 10 µg/ml puromycin. The remaining cells were selected as single-cell clones according to the dilution method. Knock-in and knockout clones were identified by sequencing and immunoblotting. The sgRNA sequences used for knocking out are listed in S-Data 4.
qRT–PCR
RNA from lung tissue and A549-IL-33 cells was extracted using TRIzol reagent (Invitrogen). The purified RNA was quantitated and reverse-transcribed with a cDNA synthesis kit (Vazyme, R323-01). Gene expression was analyzed via qRT–PCR, and the primers used are listed in S-Data 5.
Cell culture and treatment
HEK293FT and A549 cells were cultured in Dulbecco’s modified Eagle’s medium. All media were supplemented with 10% FBS, 100 U/ml penicillin, and 100 U/ml streptomycin, and the cells were cultured at 37 °C with 5% CO2.
A549-IL-33 cells were treated with 0.5 mM PL for 0, 3, 6, 12, and 24 h or with 0, 0.25, 0.5, and 1 mM PL (Selleck) for 24 h. Then, the cells were collected for immunoblotting to measure intracellular IL-33 levels.
A549-IL-33 cells were stimulated with PL for 18 h and then incubated together with a proteasome inhibitor (MG132, 10 μM) for 8 h or a lysosome inhibitor (NH4Cl, 20 mM) for 10 h to measure intracellular IL-33 levels after proteasome or lysosome activity.
Immunoprecipitation and immunoblot analysis
HEK293FT cells were transfected with plasmids. Forty-eight hours after transfection, the cells were lysed in RIPA buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 1 mM EDTA, 0.25% sodium deoxycholate, 0.1% SDS) supplemented with 1% proteinase inhibitor cocktail (Roche). The cell lysates were subsequently centrifuged, and the supernatant was collected. The indicated Abs were added together with protein A/G Plus-agarose immunoprecipitation reagent (Sigma-Aldrich, A2095) at 4 °C overnight. The beads were washed with the above RIPA buffer five times, and the immunoprecipitate was boiled in SDS loading buffer for 10 min and then analyzed by immunoblotting.
In vitro ubiquitination assays
IL-33 and MDM2 proteins were expressed with a TNT Quick Coupled Transcription/Translation System Kit (Promega). An in vitro ubiquitination assay was performed with a ubiquitination kit (Enzo Life Science) according to the manufacturer’s instructions. The ubiquitination system was immunoprecipitated with HA antibodies and analyzed by western blotting.
Sample processing for LC‒MS
Plasma was deproteinized via 1% perchloric acid, vortexed, and centrifuged at 20,000 × g and 4 °C for 15 min. Methanol (75%, v/v) containing 0.2% formic acid (v/v) was added to the supernatant. The mixture was vortexed, after which the sample was incubated for 1 h at −20 °C. The supernatant after centrifugation was analyzed via LC‒MS.
To detect intracellular PLP levels, cells were digested with trypsin and then washed twice with phosphate-buffered saline (PBS). Cells were lysed with methanol (75%, v/v) containing 0.2% formic acid (v/v), and the lysates were then vortexed. The supernatant was deproteinized with 1% perchloric acid, vortexed and centrifuged at 20,000 × g and 4 °C for 15 min. Then, the supernatant was collected for analysis via LC‒MS.
LC‒MS
LC‒MS analysis was performed with a QTRAP 6500+ LC‒MS/MS system (AB SCIEX). A QTRAP 6500+ LC‒MS/MS system equipped with an electrospray ionization (ESI) source and an IonDriveTM high-energy detector was used. Separation was performed with an Agilent Poroshell 120 column (100 × 2.1 mm, 2.7 µm). The mobile phase consisted of solution A (0.2% formic acid and 5 mM ammonium formate in water) and solution B (methanol). Samples were injected every 9 min with gradient separation: 2–2.2 min (95% A and 5% B), 4.0 min (0% A and 100% B), and 4.1–9.0 min (95% A and 5% B). All of the gradients were linear. The flow rate was 0.3 ml/min. The sample compartment of the autosampler was maintained at 4 °C, and the column temperature was 35 °C. Quantitation of PLP or PL in the samples was performed using a calibration standard curve as previously described [63].
Detection of serum vitamins
The serum of asthma patients (S-Data 6) and lung cancer patients (S-Data 7) was collected and analyzed using the electrochemical stripping voltammetry method with the LK3000VI vitamin detector (Tianjin Lanbiao Electronic Technology Development Co., Ltd, China) according to the manufacturer’s instructions.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism. The data are presented as the means ± SDs. Significant differences were determined by unpaired Student’s test or one-way analysis of variance (ANOVA) (to compare three groups or more) or two-way ANOVA (to compare curves). Detailed information on the statistical analyses is included in the figure legends. A value of P < 0.05 was considered to indicate significance.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We are grateful to Guomei Lin for animal breeding and management. We thank Prof. Ronggui Hu and Prof. Bin Li for their suggestions to organize the manuscript. We also acknowledge the individuals who provided technical support at the Core Facility for Cell Biology and the Animal Core Facility.
Author contributions
SZhu, SZhong, KC, and LZ performed the experiments and analyzed the data. JB and ZC collected the clinical samples. WC, SC, LM, YH, and MZ provided protocols and suggestions. SW, ZL, WG, XS, and CY prepared the cell lines and reagents. SZhu, YZ, BS, JX, and SL designed the study and wrote the manuscript. YZ, BS, JX, and SL supervised the project and revised the manuscript.
Funding
This work was supported by the Ministry of Science and Technology of China (2018YFA0507402), the National Natural Science Foundation of China (32000667, 81925001, and 82170051), the Shanghai Science and Technology Innovation Action (21ZR1470600, 21JC1405800 and 20S11901800), the Key Scientific Innovation project of the Shanghai Municipal Education Commission (No. 202101070007-E00097), and the Youth Innovation Promotion Association of the Chinese Academy of Sciences (2022264).
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Songling Zhu, Shufen Zhong, Kebin Cheng, Li-Sha Zhang.
Contributor Information
Shuo Liang, Email: liangshuo79@163.com.
Jin-Fu Xu, Email: jfxu@tongji.edu.cn.
Bing Sun, Email: bsun@sibs.ac.cn.
Yaguang Zhang, Email: zhangyaguang@sibcb.ac.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-023-01029-6.
References
- 1.Locksley RM. Asthma and allergic inflammation. Cell. 2010;140:777–783. doi: 10.1016/j.cell.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. 2012;18:684–692. doi: 10.1038/nm.2737. [DOI] [PubMed] [Google Scholar]
- 3.Roan F, Obata-Ninomiya K, Ziegler SF. Epithelial cell-derived cytokines: more than just signaling the alarm. J Clin Investig. 2019;129:1441–1451. doi: 10.1172/JCI124606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16:676–689. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 5.Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity. 2015;42:1005–1019. doi: 10.1016/j.immuni.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drake LY, Kita H. IL-33: biological properties, functions, and roles in airway disease. Immunol Rev. 2017;278:173–184. doi: 10.1111/imr.12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De la Fuente M, MacDonald TT, Hermoso MA. The IL-33/ST2 axis: role in health and disease. Cytokine Growth Factor Rev. 2015;26:615–623. doi: 10.1016/j.cytogfr.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 8.Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol. 2016;17:122–131. doi: 10.1038/ni.3370. [DOI] [PubMed] [Google Scholar]
- 9.Grotenboer NS, et al. Decoding asthma: translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J Allergy Clin Immunol. 2013;131:856–865. doi: 10.1016/j.jaci.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 10.Moffatt MF, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savenije OE, et al. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J Allergy Clin Immunol. 2014;134:170–177. doi: 10.1016/j.jaci.2013.12.1080. [DOI] [PubMed] [Google Scholar]
- 12.Backman JD, et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature. 2021;599:628–634. doi: 10.1038/s41586-021-04103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tjota MY, et al. IL-33-dependent induction of allergic lung inflammation by FcgammaRIII signaling. J Clin Invest. 2013;123:2287–2297. doi: 10.1172/JCI63802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardman CS, Panova V, McKenzie AN. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur J Immunol. 2013;43:488–498. doi: 10.1002/eji.201242863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS ONE. 2010;5:e11944. doi: 10.1371/journal.pone.0011944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bessa J, et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun. 2014;55:33–41. doi: 10.1016/j.jaut.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 17.Luthi AU, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA. 2009;106:9021–9026. doi: 10.1073/pnas.0812690106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen ES, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. 2015;6:8327. doi: 10.1038/ncomms9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayakawa H, et al. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. 2007;282:26369–26380. doi: 10.1074/jbc.M704916200. [DOI] [PubMed] [Google Scholar]
- 21.Tao LQ, et al. Deubiquitination and stabilization of IL-33 by USP21. Int J Clin Exp Pathol. 2014;7:4930–4937. [PMC free article] [PubMed] [Google Scholar]
- 22.Ni Y, et al. The deubiquitinase USP17 regulates the stability and nuclear function of IL-33. Int J Mol Sci. 2015;16:27956–27966. doi: 10.3390/ijms161126063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, et al. MG149 inhibits histone acetyltransferase KAT8-mediated IL-33 acetylation to alleviate allergic airway inflammation and airway hyperresponsiveness. Signal Transduct Target Ther. 2021;6:321. doi: 10.1038/s41392-021-00667-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ueland PM, et al. Inflammation, vitamin B6 and related pathways. Mol Asp Med. 2017;53:10–27. doi: 10.1016/j.mam.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 25.Reynolds RD, Natta CL. Depressed plasma pyridoxal phosphate concentrations in adult asthmatics. Am J Clin Nutr. 1985;41:684-8. [DOI] [PubMed]
- 26.Huang SC, et al. Vitamin B(6) supplementation improves pro-inflammatory responses in patients with rheumatoid arthritis. Eur J Clin Nutr. 2010;64:1007–1013. doi: 10.1038/ejcn.2010.107. [DOI] [PubMed] [Google Scholar]
- 27.Poyurovsky MV, et al. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 2007;26:90–101. doi: 10.1038/sj.emboj.7601465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nag S, et al. The MDM2-p53 pathway revisited. J Biomed Res. 2013;27:254–271. doi: 10.7555/JBR.27.20130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernandez-Monge J, et al. MDM2 regulates RB levels during genotoxic stress. EMBO Rep. 2021;22:e50615. doi: 10.15252/embr.202050615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang JY, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang A, et al. Mouse double minute 2 homolog-mediated ubiquitination facilitates forkhead box P3 stability and positively modulates human regulatory T cell function. Front Immunol. 2020;11:1087. doi: 10.3389/fimmu.2020.01087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi YM, et al. Mdm2 is required for HDAC3 monoubiquitination and stability. Biochem Biophys Res Commun. 2019;517:353–358. doi: 10.1016/j.bbrc.2019.07.052. [DOI] [PubMed] [Google Scholar]
- 33.Alwarith J, et al. The role of nutrition in asthma prevention and treatment. Nutr Rev. 2020;78:928–938. doi: 10.1093/nutrit/nuaa005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talaei M, Hughes DA, Mahmoud O, et al. Dietary intake of vitamin A, lung function and incident asthma in childhood. Eur Respir J. 2021;58:2004407. [DOI] [PMC free article] [PubMed]
- 35.Majak P, et al. Vitamin D supplementation in children may prevent asthma exacerbation triggered by acute respiratory infection. J Allergy Clin Immunol. 2011;127:1294–1296. doi: 10.1016/j.jaci.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 36.Jolliffe DA, et al. Vitamin D metabolism is dysregulated in asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2020;202:371–382. doi: 10.1164/rccm.201909-1867OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Milan SJ, Hart A, Wilkinson M. Vitamin C for asthma and exercise-induced bronchoconstriction. Cochrane Database Syst Rev. 2013;2013:CD010391. [DOI] [PMC free article] [PubMed]
- 38.Qian B, et al. Effects of vitamin B6 deficiency on the composition and functional potential of T cell populations. J Immunol Res. 2017;2017:2197975. doi: 10.1155/2017/2197975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agoro R, et al. IL-1R1-MyD88 axis elicits papain-induced lung inflammation. Eur J Immunol. 2016;46:2531–2541. doi: 10.1002/eji.201646366. [DOI] [PubMed] [Google Scholar]
- 40.Uchida M, et al. Oxidative stress serves as a key checkpoint for IL-33 release by airway epithelium. Allergy. 2017;72:1521–1531. doi: 10.1111/all.13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parra M, Stahl S, Hellmann H. Vitamin B(6) and its role in cell metabolism and physiology. Cells. 2018;7:84. [DOI] [PMC free article] [PubMed]
- 42.Kumar V, et al. Pyridoxal kinase: A vitamin B6 salvage pathway enzyme from Leishmania donovani. Int J Biol Macromol. 2018;119:320–334. doi: 10.1016/j.ijbiomac.2018.07.095. [DOI] [PubMed] [Google Scholar]
- 43.Chelban V, et al. PDXK mutations cause polyneuropathy responsive to pyridoxal 5’-phosphate supplementation. Ann Neurol. 2019;86:225–240. doi: 10.1002/ana.25524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schernhammer ES, Ogino S, Fuchs CS. Folate and vitamin B6 intake and risk of colon cancer in relation to p53 expression. Gastroenterology. 2008;135:770–780. doi: 10.1053/j.gastro.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang P, et al. Vitamin B(6) activates p53 and elevates p21 gene expression in cancer cells and the mouse colon. Oncol Rep. 2014;31:2371–2376. doi: 10.3892/or.2014.3073. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J, et al. The ubiquitin ligase MDM2 sustains STAT5 stability to control T cell-mediated antitumor immunity. Nat Immunol. 2021;22:460–470. doi: 10.1038/s41590-021-00888-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26:399–422. doi: 10.1038/cr.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- 49.Grice GL, Nathan JA. The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci. 2016;73:3497–3506. doi: 10.1007/s00018-016-2255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shmuel-Galia L, et al. Dysbiosis exacerbates colitis by promoting ubiquitination and accumulation of the innate immune adaptor STING in myeloid cells. Immunity. 2021;54:1137–1153.e8. doi: 10.1016/j.immuni.2021.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang S, et al. The E3 ubiquitin ligase Pellino3 protects against obesity-induced inflammation and insulin resistance. Immunity. 2014;41:973–987. doi: 10.1016/j.immuni.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 52.Spinneker A, et al. Vitamin B6 status, deficiency and its consequences-an overview. Nutr Hosp. 2007;22:7–24. [PubMed] [Google Scholar]
- 53.Tracz M, Bialek W. Beyond K48 and K63: non-canonical protein ubiquitination. Cell Mol Biol Lett. 2021;26:1. doi: 10.1186/s11658-020-00245-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Braun H, et al. Dichotomous function of IL-33 in health and disease: from biology to clinical implications. Biochem Pharmacol. 2018;148:238–252. doi: 10.1016/j.bcp.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 55.Liu X, et al. Anti-IL-33 antibody treatment inhibits airway inflammation in a murine model of allergic asthma. Biochem Biophys Res Commun. 2009;386:181–185. doi: 10.1016/j.bbrc.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 56.Rabe KF, et al. Safety and efficacy of itepekimab in patients with moderate-to-severe COPD: a genetic association study and randomised, double-blind, phase 2a trial. Lancet Respir Med. 2021;9:1288–1298. doi: 10.1016/S2213-2600(21)00167-3. [DOI] [PubMed] [Google Scholar]
- 57.Priest C, Prives C, Poyurovsky MV. Deconstructing nucleotide binding activity of the Mdm2 RING domain. Nucleic Acids Res. 2010;38:7587–7598. doi: 10.1093/nar/gkq669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poyurovsky MV, Jacq X, Ma C, Karni-Schmidt O, Parker PJ, Chalfie M, et al. Nucleotide binding by the MDM2 RING domain facilitates Arf-Independent MDM2 nucleolar localization. Mol Cell. 2003;12:875–887. doi: 10.1016/S1097-2765(03)00400-3. [DOI] [PubMed] [Google Scholar]
- 59.Reddel HK, et al. Global initiative for asthma strategy 2021: executive summary and rationale for key changes. Am J Respir Crit Care Med. 2022;205:17–35. doi: 10.1164/rccm.202109-2205PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moro K, et al. Isolation and analysis of group 2 innate lymphoid cells in mice. Nat Protoc. 2015;10:792–806. doi: 10.1038/nprot.2015.047. [DOI] [PubMed] [Google Scholar]
- 61.Dubin PJ, Kolls JK. IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol. 2007;292:L519–L528. doi: 10.1152/ajplung.00312.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McKay A, et al. A novel anti-inflammatory role of simvastatin in a murine model of allergic asthma. J Immunol. 2004;172:2903–2908. doi: 10.4049/jimmunol.172.5.2903. [DOI] [PubMed] [Google Scholar]
- 63.Kushnir MM, et al. Development and clinical evaluation of a high-throughput LC-MS/MS assay for vitamin B6 in human plasma and serum. J Appl Lab Med. 2021;6:702–714. doi: 10.1093/jalm/jfaa166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.