

Abstract
Purpose:
Overweight/obese (OW/OB) patients with metastatic melanoma unexpectedly have improved outcomes with immune checkpoint inhibitors (ICIs) and BRAF-targeted therapies. The mechanism(s) underlying this association remain unclear, thus we assessed the integrated molecular, metabolic, and immune profile of tumors, as well as gut microbiome features, for associations with patient BMI.
Experimental Design:
Associations between BMI [normal (NL < 25) or OW/OB (BMI ≥ 25] and tumor or microbiome characteristics were examined in specimens from 782 metastatic melanoma patients across 7 cohorts. DNA associations were evaluated in the TCGA cohort. RNASeq from 4 cohorts (n=357) was batch corrected and gene set enrichment analysis (GSEA) by BMI category was performed. Metabolic profiling was conducted in a subset of patients (x=36) by LC/MS, and in flow-sorted melanoma tumor cells (x=37) and patient-derived melanoma cell lines (x=17) using the Seahorse XF assay. Gut microbiome features were examined in an independent cohort (n=371).
Results:
DNA mutations and copy number variations were not associated with BMI. GSEA demonstrated that tumors from OW/OB patients were metabolically quiescent, with downregulation of oxidative phosphorylation and multiple other metabolic pathways. Direct metabolite analysis and functional metabolic profiling confirmed decreased central carbon metabolism in OW/OB metastatic melanoma tumors and patient-derived cell lines. The overall structure, diversity, and taxonomy of the fecal microbiome did not differ by BMI.
Conclusions:
These findings suggest that the host metabolic phenotype influences melanoma metabolism and provide insight into the improved outcomes observed in OW/OB patients with metastatic melanoma treated with ICIs and targeted therapies.
Keywords: Melanoma, obesity, oxidative phosphorylation, metabolism, microbiome
Introduction
Over the past decade, targeted therapies and immune checkpoint inhibitors (ICI) have dramatically improved survival for patients with metastatic melanoma. BRAF-activating mutations are present in approximately 50% of patients with cutaneous melanoma, and dual inhibition of BRAF and MEK produces a consistent reduction in tumor size and improves overall survival.(1) However, approximately 80% of these patients will progress (median duration of response ~ 1 year). ICI produce much more durable responses, but 40–60% of patients have primary or secondary resistance to ICI, and improved predictors and understanding of resistance remain unmet needs.(2)
There is increasing evidence that tumor-extrinsic factors, such as age, sex, and gut microbiome features, can influence responses to immune and/or targeted therapies.(3–7) In other malignancies, obesity and the associated metabolic phenotype of higher circulating insulin/IGF-1 have been shown to activate tumor PI3K-AKT pathway signaling, which has in turn been implicated in resistance to targeted and immune therapy in melanoma.(8,9) Thus, we were surprised to discover that obesity was associated with significantly improved outcomes in patients with metastatic melanoma who receive ICI or targeted therapy, i.e. an obesity paradox.(10) In our pooled analysis of 2,046 patients with metastatic melanoma, obesity was associated with significantly improved outcomes in patients with metastatic melanoma treated with ICI (n=538) or targeted therapy (n=839). BMI was not associated with outcomes in patients treated with chemotherapy (n=541), suggesting that BMI was predictive, rather than prognostic. (10) The association between high BMI and improved outcomes with these therapies has been validated in multiple independent metastatic melanoma cohorts, as well as in patients with metastatic renal cell carcinoma (RCC) and non-small cell lung cancer (NCSLC) treated with ICIs or targeted therapies.(11–14) To date, the mechanism or mechanisms underlying this obesity paradox, particularly how host-level metabolism influences tumor metabolism, are incompletely understood. This is a critical question given the growing evidence that metabolic pathways in tumor cells can cause resistance to both ICI and BRAF targeted therapy.(15–17)
In order to explore the biological basis for improved outcomes in OW/OB melanoma patients, we assessed and integrated molecular, metabolic, and immune profile of tumors, as well as gut microbiome features, for associations with patient BMI. Our studies reveal that tumors of OW/OB patients with metastatic melanoma exhibit relative downregulation of OXPHOS and other metabolic pathways compared to those from normal BMI patients. As we have previously shown that tumor cell hypermetabolism can create a microenvironment hostile to T cell function, this suggests a potential metabolic driver for the inferior outcomes observed in normal BMI melanoma patients.(15) Conversely, the more metabolically quiescent tumor phenotype observed in OW/OB melanoma patients may render these tumors more vulnerable to targeted and immune therapies.
Materials and Methods
Patient cohorts
As described in Figure 1A, molecular, metabolic, immune, and microbiome profiling were performed on several independent cohorts of metastatic melanoma tumor specimens. These included the TCGA cohort for somatic DNA studies (TCGA, n=202), 4 cohorts for bulk RNA sequencing (TCGA, n=202; Gide, n=68; Hugo, n=26; MDA, n=61), the TCGA cohort for reverse phase protein array (n=202), the University of Pittsburgh cohort for Seahorse Extracellular Flux Assay on patient tumor biopsies (UPMC, n=37), MDA patient-derived cell lines for Seahorse Extracellular Flux Assay (n=17), a TCGA cohort for direct metabolite measurement by LC/MS (n=36), 2 cohorts for IHC testing of immune cells and co-stimulatory/co-inhibitory molecules (Gide, n=83; MDA, n=61), and a distinct MDA cohort for fecal microbiome studies (n=272). For all patients, BMI at time of sample collection was calculated as patient’s weight in kilograms divided by their height in meters squared and categorized into obese (OB, BMI ≥ 30 kg/m2), overweight (OW, 25 kg/m2≤ BMI < 30 kg/m2), and normal weight (NL, BMI < 25 kg/m2). For the TCGA melanoma cohort,(18) we selected a uniform cohort of non-recurrent regionally metastatic melanoma specimens for analysis. We filtered to include patients with biospecimen tissue sites from regional lymph node or regional subcutaneous metastases and excluded patients presenting with stage IV disease. Then, to exclude patients with recurrent stage III disease, we excluded all patients for whom the number of days from the diagnosis of the primary to the accession date was more than 90 days. We downloaded RNA-sequencing (RNA-seq) expression and whole exome sequencing (WES data from the TCGA data portal (www.tcgaportal.org). We obtained BMI from the TCGA clinical database where available (n=121) and obtained BMI from the original contributing center where feasible (n=81), resulting in n=202 samples available for analysis (Supplementary Table 1). We additionally obtained BMI data for two previously published melanoma tumor profiling datasets, the University of California Los Angeles cohort (RNASeq, Hugo, n=26)(19), and the Melanoma Institute of Australia cohort [RNASeq, n=68, and immunohistochemistry (IHC), Gide, n=83](20). We additionally performed RNASeq and IHC for immune markers on resected melanoma lung, liver, or bowel metastases with available BMI from MD Anderson Cancer Center (MDA, n=61). The Institutional Review Board approved the request to analyze tumor tissue from all tumor tissue collection and subsequent analyses. Written informed consent was obtained from patients at time of initial biospecimen collection at the primary institution. The study was conducted in accordance with recognized ethical guidelines (Declaration of Helsinki, CIOMS, The Belmont Report, and the US Common Rule). Baseline characteristics, including sex, age, site of tumor tissue, BMI group, and prior systemic therapy, are described in Supplementary Table 1.
Figure 1:
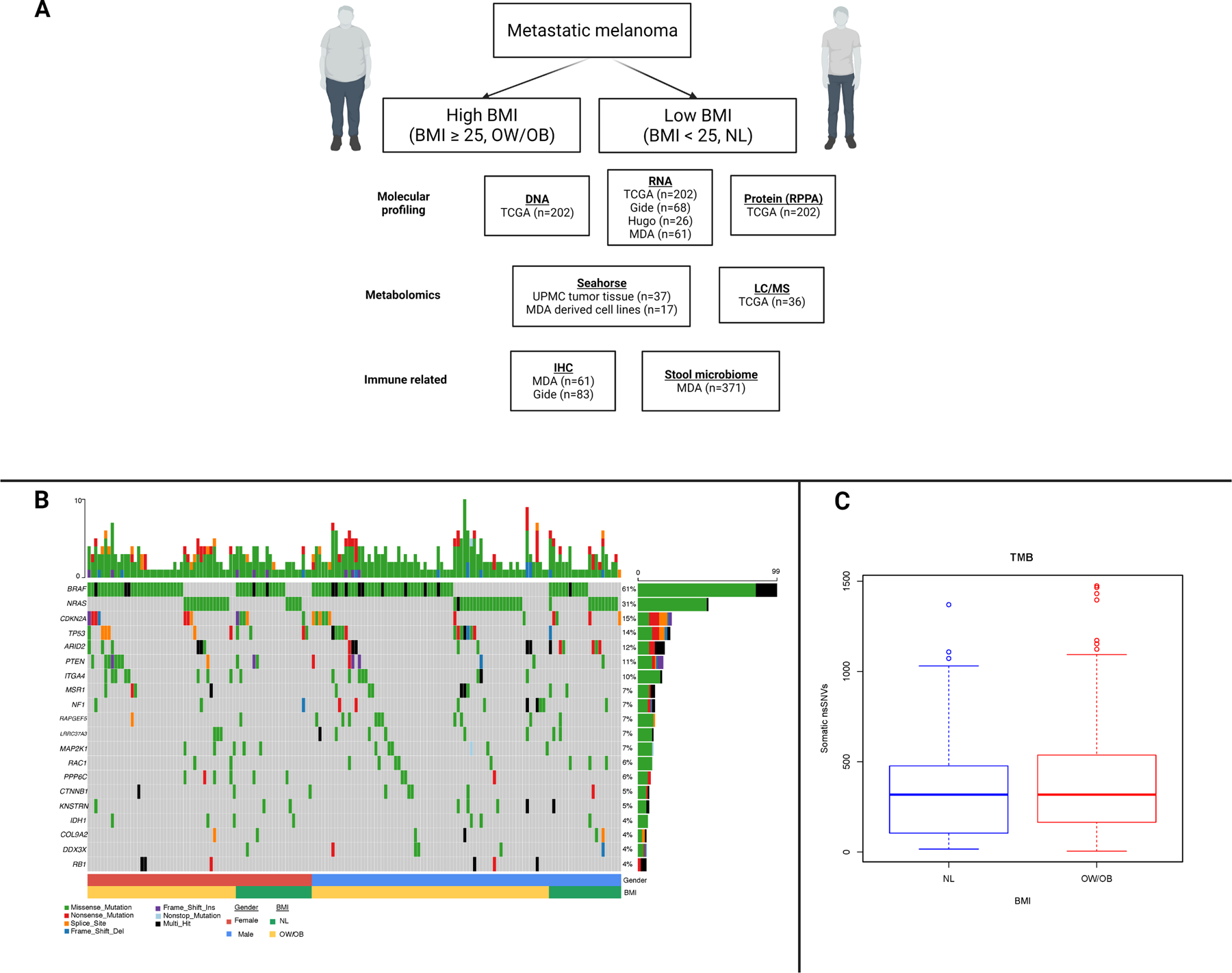
Overview of studies performed by cohort and somatic DNA studies by body mass index.
A. Overview of analyses performed by cohort. BMI = body mass index, OW = overweight, OB = obese, NL = normal, TCGA = The Cancer Genome Atlas, MDA = MD Anderson Cancer Center, RPPA = reverse phase protein array, LC/MS = liquid chromatography/mass spectrometry, IHC = immunohistochemistry.
B. Oncoplot depicting somatic DNA alterations in regionally metastatic melanoma from The Cancer Genome Atlas (TCGA) cohort by BMI and sex. At the top of the figure, the vertical bar from 1–10 shows the number of alterations by individual patient. In the second to bottom row, blue indicates males and red indicates females. In the bottom row, green indicates normal (NL) BMI and yellow indicates overweight/obese (OW/OB) BMI. In the middle rows, the numbers to the left are the frequency of alterations in each gene listed to the right. For these middle rows, green is a missense mutation, red is a nonsense mutation, purple is a frameshift insertion, blue is a frameshift deletion, orange is a splice site alteration, and black is multi-hit.
C. Tumor mutation burden by body mass index (BMI) in regionally metastatic melanoma from TCGA cohort.
Somatic alteration analysis
For somatic mutation analysis of the TCGA cohort, we used the chi-squared test to perform enrichment analysis on mutations of individual genes of OW/OB vs NL. We also performed the differential analysis on somatic copy number alterations (SCNAs) for each gene by t-test. Significance was set at a false discover rate (FDR) of <0.2 which was not met for any genes. An oncoplot was generated based on the top 20 significantly mutated genes (SMGs) in the melanoma TCGA.(18) The differential analysis of tumor mutation burden (TMB) between OW/OB and NL BMI patients was performed by Wilcox test. For mutational signature analysis, we first quantified contribution of the signatures based on the patient mutational profiles. We then performed differential analysis on relative contribution for each signature between OW/OB and NL by t-test. Significance was set at a false discovery rate (FDR) <0.2 under which no significant signature was identified. The mutational signature matrix was downloaded from COSMIC (https://cancer.sanger.ac.uk/cancergenome/assets/signatures_probabilities.txt). The R library we used for the analysis was “MutationalPatterns”.
RNA sequencing
For the MDA cohort, archived paraffin embedded tissue specimens were collected from 61 patients with metastatic melanoma. Sections from paraffin-embedded tissue were reviewed for pathologic diagnosis and dissected, if necessary, to ensure that ≥ 90% of the sample represented tumor. Total cellular RNA was isolated from tissue sections using the High Pure RNA isolation kit according to the manufacturer’s protocol (Roche Diagnostics GmbH, Germany) following de-paraffinization and proteinase K treatment.
Gene set enrichment analyses (GSEA)
For the GSEA, RNA-seq count matrix from different datasets (MDA, TCGA, Gide, Hugo) were merged and batch-corrected by R package sva v3.36.0 ComBat_seq() function. The corrected count matrix were analyzed by R package DESeq2 v1.28.1 for differential gene expression (DGE) analysis among different BMI groups. In addition to DGE analysis on all four batch-corrected datasets (labeled as All), DGE analysis with each of different covariates controlled in DESeq2 modelling (labeled as Ctrl Sex, Ctrl Dataset, and Ctrl Tissue) was performed by using DESeqDataSetFromMatrix() function (e.g., design = ~ Sex + BMI)(Supplementary Table 2). DGE analysis on each of subgroups (Sex: Female, Male; Dataset: MDA, TCGA, Gide, Hugo; and Tissue: lymph node (LN), subcutaneous metastasis (SC), lung) was also performed (Supplementary Table 3). The gene rank lists from DGE analysis were used for GSEA by R package fgsea v1.14.0. The gene lists of hallmark pathways and KEGG pathways were downloaded from MSigDB v7.4 (http://www.gsea-msigdb.org/gsea/msigdb/index.jsp). The results were plotted by R packages ggplot v3.3.3 and ComplexHeatmap v2.4.3. The enriched pathways with adjusted p values (padj) and normalized enrichment score (NES) were visualized.
Reverse phase protein array (RPPA) analysis
For RPPA analysis, the TCGA RPPA dataset was obtained from the TCPA data portal (www.tcpaportal.org), and we calculated the scores of the protein pathways based on the weighted average of the member protein RPPA levels.(21) The differential analysis between OW/OB and NL was performed for each pathway by t-test.
Direct metabolic profiling studies
We performed direct metabolic profiling studies for tricarboxylic acid cycle metabolites on tumor tissue from 36 patients with regionally metastatic melanoma who were part of the TCGA melanoma cohort and had fresh frozen specimens at MDA available for analysis. Metabolites were extracted from tissue samples using the extraction procedure described previously(22,23) and analyzed using a 6495 triple quadrupole mass spectrometer (Agilent Technologies, Santa Clara, CA) coupled to a HPLC system (Agilent Technologies, Santa Clara, CA) via single reaction monitoring. The data were log2-transformed and normalized with internal standards on a per-sample, per-method basis. Statistical analyses were performed with t-Test in R Studio (R Studio Inc., Boston, MA). Differential metabolites were identified by adjusting the p-values for multiple testing at a FDR threshold of < 0.25.
Tumor cells from patient tissue samples metabolic output was measured by Seahorse technology as previously described.(15) We performed assessment of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in 37 fresh frozen melanoma specimens from the University of Pittsburgh Medical Center. Samples were categorized by BMI at the time of resection. Differential analysis of OCR and ECAR by BMI category was performed using the unpaired T test. Samples were also categorized using median values of ECAR and OCR and Fischer’s exact test used to calculate proportion of samples in “low ECAR/low OCR” vs “other.”
Metabolic output was also measured by Seahorse in patient-derived melanoma cell lines (n=17), maintained as low passage cell lines. Patient-derived melanoma cell lines were established according to previously described protocols.(24) All melanoma cell lines were maintained in RPMI 1640 complete medium supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA) and normocin (Invitrogen). All cell lines were verified by short tandem repeat fingerprinting or matching mutational profiles. Cells were routinely monitored for Mycoplasma contamination by using the MycoAlert Kit (Lonza). Cells were counted blindly by two independent investigators and seeded at a density of 2 × 10E4 cells/well in XF96 plates and allowed to stabilize overnight. ECAR and OCR were next measured by the Seahorse XF96 analyzer as described in manufacturer’s instructions for the XF cell mito stress. The results were analyzed using Wave software (Seahorse/Agilent). The OCR and ECAR levels of tumors after adding FCCP were used to determine the mitochondrial metabolism at maximal respiration condition.
Immune cell studies
The levels of key immune markers by BMI was assessed using IHC in formalin-fixed, paraffin-embedded (FFPE) melanoma samples in two cohorts, the Gide and MDA cohorts. The methodology for the Gide cohort was previously described.(20) For the MDA cohort, IHC studies were performed on 4 µm formalin-fixed paraffin-embedded sections using a Leica BOND RXm autostainer. Slides were stained with antibodies targeting human CD3 (Agilent #A045201–2, 1:100), CD4[4B12] (Leica #NCL-L-CD4–368, 1:80), CD8[C8/144B] (Thermo Fisher #MS457S), CD45RO[UCHL1] (Leica #PA0146, RTU), CD68[PG-M1] (Agilent #M087629–2, 1:450), FoxP3[206D] (BioLegend #320102, 1:50), Granzyme B[11F1] (Leica #PA0291, RTU) , LAG3[D2G40] (Cell Signaling #15372, 1:100), PD1[EPR4877(2)] (Abcam #ab137132), and PD-L1[E1L3N] (Cell Signaling #13684, 1:100) using a modified version of either the standard Leica Bond DAB “F” or red “J” IHC protocols. Slides stained for CD4 and PD-L1 were scored by a board-certified pathologist and given an H-score based on percentage and intensity of positivity. All other analyses were performed using Aperio automated analysis as previously described.(25) Differential analysis of immune cell populations was performed using the Mann-Whitney U test.
Fecal microbiome studies
We analyzed the fecal baseline microbial abundance data by 16S rRNA sequencing from the subset of stage III/IV cutaneous patients with metastatic melanoma (n=371) from our previously published MD Anderson melanoma microbiome cohort.(7) We examined in-sample microbial diversity using the alpha-diversity measure and analyzed its association with BMI using an ANOVA analysis after adjusting for sex and age. Beta-diversity was used to measure microbial diversity between samples based on the Bray-Curtis dissimilarity. Each sample is represented by ordination plots based upon the top two principal components extracted from Principal Coordinate Analysis (PCoA) and PERMANOVA analysis with 999 permutations used to examine the impact of BMI on beta-diversity, again controlling for age and sex.
Data and Materials Availability Statement
BMI data corresponding to publicly available genomic profiling (TCGA, Gide, Hugo cohorts) for which BMI was obtained from the contributing investigators to these datasets are provided as Supplementary Table 4 as is the full RNAseq data for the MDACC cohort (Supplementary Table 5) and differential gene expression analyses results for the integrated analyses. The fecal microbiome data and corresponding BMI are publicly available. Direct metabolic and immune profiling data is available upon request from corresponding author.
Results
An overview of the analyses performed on the multiple cohorts included in this study is provided for reference (Figure 1A). We began by evaluating somatic DNA alterations by BMI in patients with regionally metastatic melanoma included in The Cancer Genome Atlas (TCGA) melanoma cohort (n=202)(18) (Supplementary Table 6). BMI was not associated with the prevalence of DNA mutations or copy number variations of any gene, including known driver oncogenes (e.g. BRAF and NRAS) and tumor suppressors (Figure 1B), nor with mutational signatures. There was also no difference in estimated tumor mutational burden (TMB), which has been associated with response to ICI, between OW/OB (BMI ≥ 25) and normal (NL, BMI < 25) BMI patients (Figure 1C).(26)
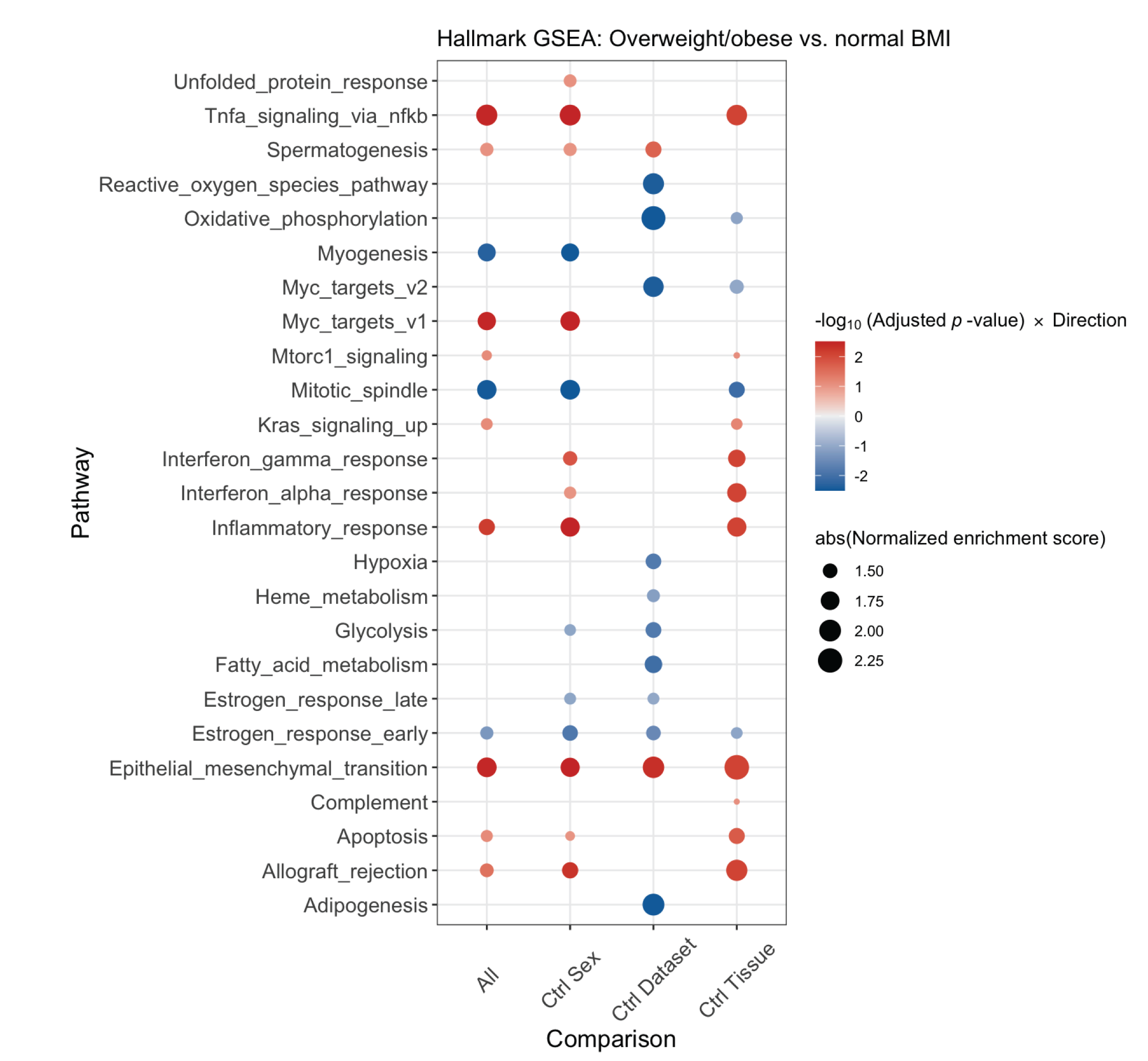
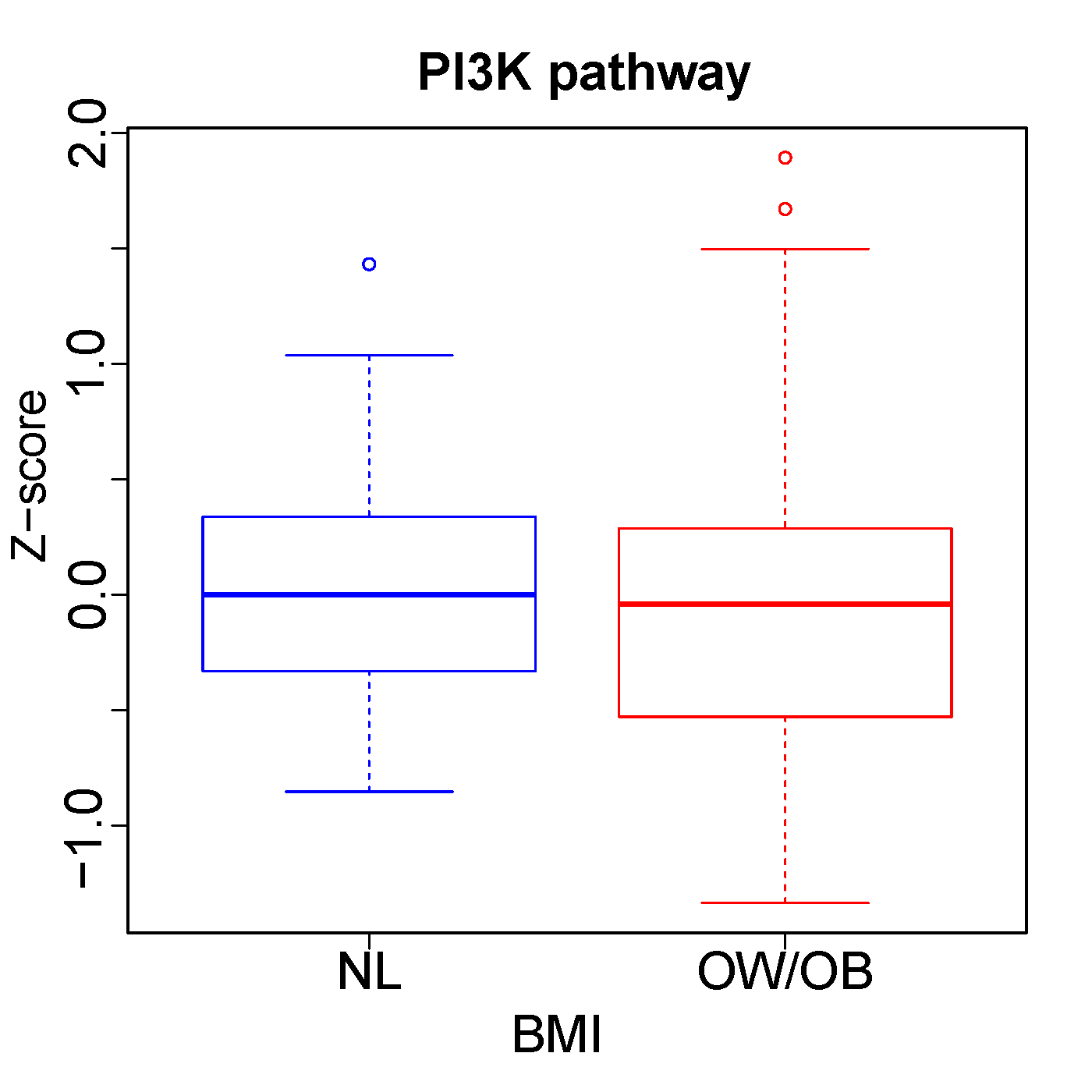
To assess for associations of BMI with gene expression, we performed a gene set enrichment analysis (GSEA) of an expanded cohort of melanoma metastases analyzed by RNAseq, including the TCGA regionally metastatic melanoma cohort (n=202)(18), as well as cohorts reported by Hugo (n=26)(19) and Gide (n=68)(20), and a new MD Anderson Cancer Center cohort (MDA, n=61, Supplementary Table 1). We performed an integrated GSEA of these 4 cohorts, correcting for batch effects, to compare mRNA expression of 50 hallmark pathways between OW/OB and NL BMI patients (Figure 2). Across the integrated cohort, we observed downregulation of metabolic pathways in OW/OB tumors, including oxidative phosphorylation [OXPHOS, padj < 0.01, normalized enrichment score (NES) −2.11] and glycolysis (padj < 0.1, NES −1.45). Myogenesis (padj < 0.01, NES −1.91), adipogenesis (padj < 0.01, NES – 1.78), reactive oxygen species (padj < 0.1, NES −1.58), and hypoxia pathways (padj < 0.1, NES −1.40) were also downregulated in OW/OB tumors. These associations were attenuated when S-phase correction was performed, supporting a link between metabolic activity and proliferation, as would be expected (Supplementary Figure 1). Interestingly, although prior studies have shown that lipid transfer from adipocytes to melanocytes in the tumor microenvironment (TME) can fuel beta oxidation and OXPHOS, fatty acid metabolism was not significantly different in metastatic OW/OB vs NL BMI tumors (Supplementary Table 7 and 8). Obesity is associated with increased production of insulin and growth factors, which may activate the PI3K pathway; however, PI3K pathway gene expression was similar between OW/OB and NL BMI patients, as was PI3K pathway protein expression in the TCGA cohort (Supplementary Figure 2).(27,28) Due to our previous finding that the influence of BMI on the efficacy of ICI and BRAF targeted therapy may differ by sex, we also performed GSEA controlling for sex as well as cohort and tumor tissue site (Figure 2).(10) The association of BMI with OXPHOS, myogenesis, adipogenesis, glycolysis, reactive oxygen species, and hypoxia pathways was observed in each analysis, and there was no evidence that the association between BMI and metabolic pathways differed by sex. (Supplementary Figure 3).(10)
Figure 2:
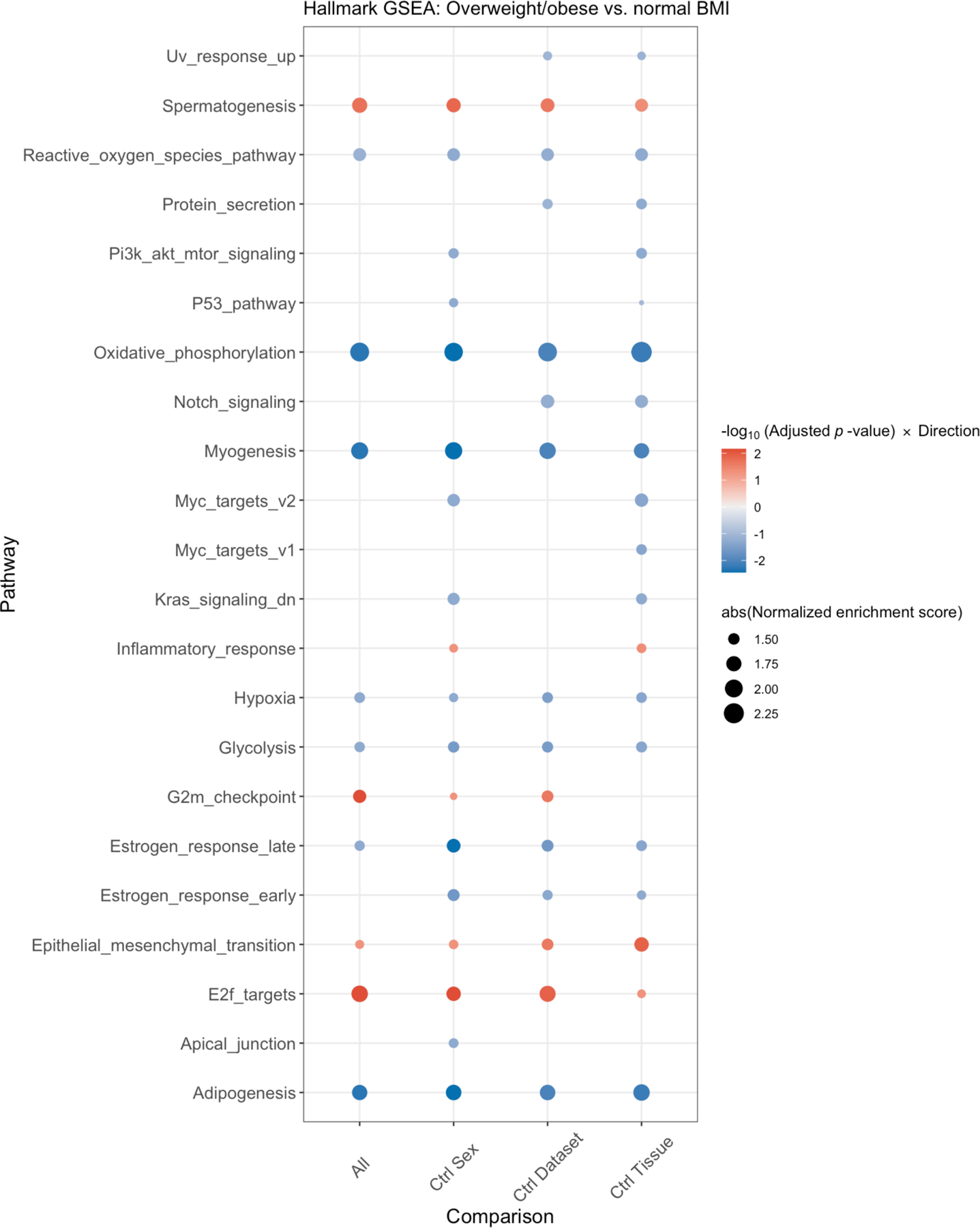
Integrated gene set enrichment analysis by body mass index (BMI). This figure presents a dotplot of genes differentially up- or downregulated in OW/OB patients versus NL BMI patients. Red indicates upregulation in OW/OB verse NL. The far left column is all patients, and then, an analysis controlling for sex, cohort, and tissue site is shown in the 3 columns to the right.
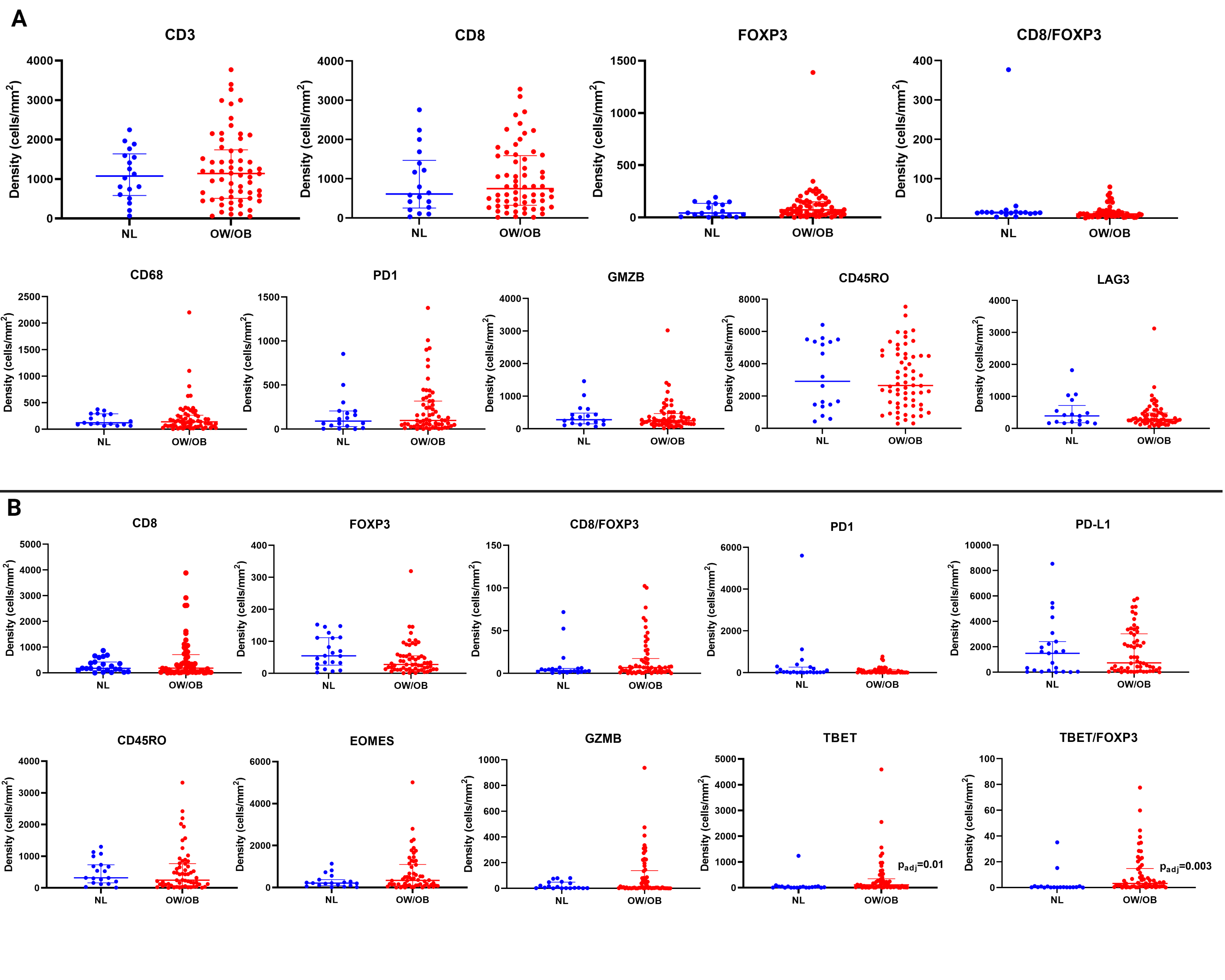
The presence of immune infiltrates and signatures of activated immune cells are associated with improved responses to both ICI and targeted therapy. However, we did not observe differences in immune pathways by BMI in Hallmark GSEA analysis (Figure 2). We further interrogated the presence of immune cells and co-stimulatory/co-inhibitory molecules by immunohistochemistry (IHC) in the MDA and Gide cohorts. We did not observe any differences in immune infiltrates (CD3, CD8, CD68, CD45RO, or FOXP3+ cells) by BMI (Supplementary Figure 3A–B). With the exception of higher TBET expression observed in the OW/OB patients (padj=0.01) and higher ratio of TBET+/FOXP3+ cells (padj=0.003), we did not observe any differences in co-stimulatory/co-inhibitory molecules in either of the full cohorts (Supplementary Figure 4A–B), nor in sex-stratified analyses (Supplementary Figures 5A–B).
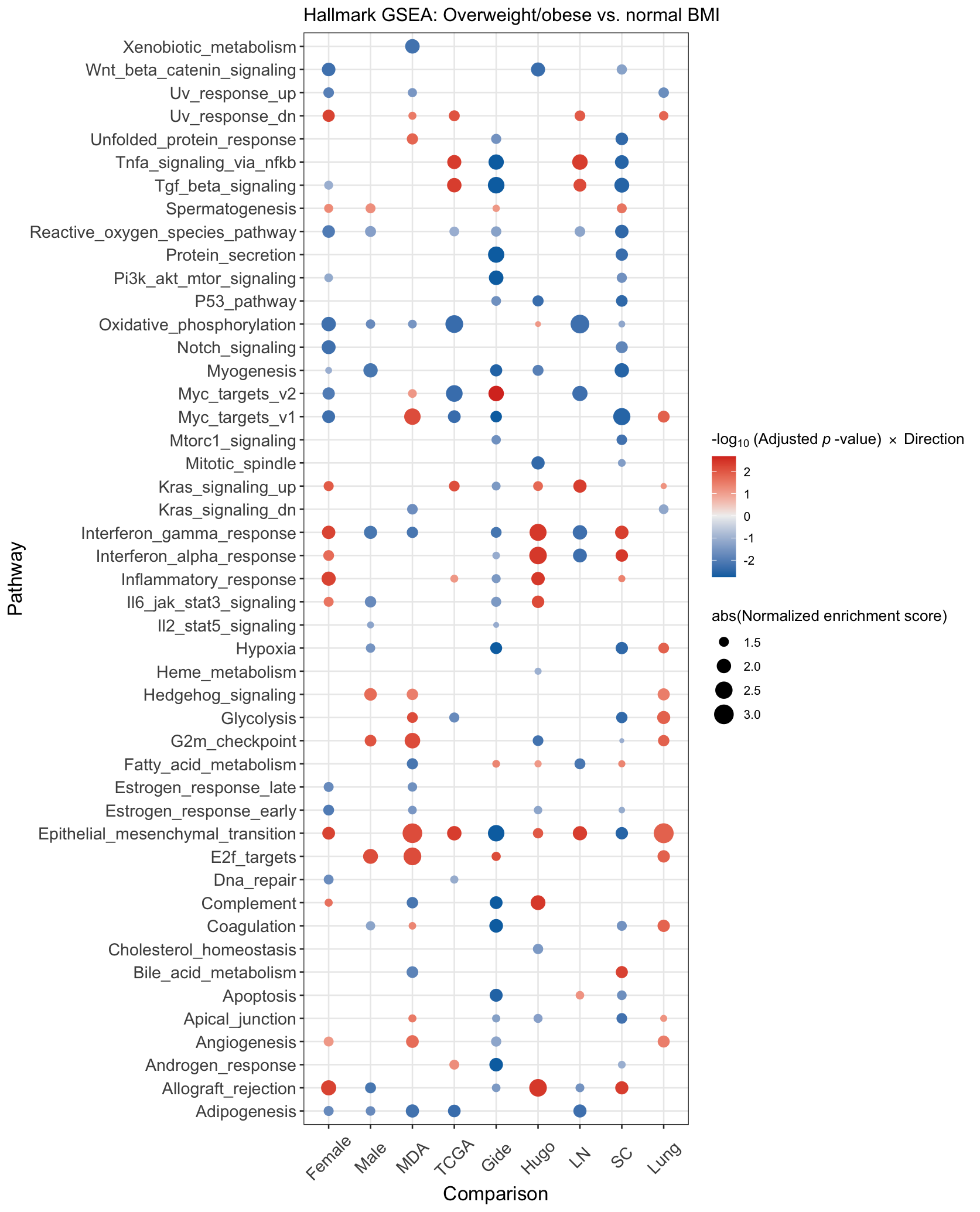
Given the intriguing finding from our hallmark GSEA that OW/OB tumors had downregulation of metabolic pathways, e.g. OXPHOS and glycolysis, we further interrogated metabolic pathways by performing GSEA for 57 KEGG pathways involved in biosynthesis and metabolism (Figure 3). OW/OB tumors were metabolically quiescent globally, with downregulation of multiple metabolic pathways without compensatory upregulation of alternative metabolic pathways except terpenoid and fatty acid biosynthesis. Liquid chromatography/mass spectrometry (LC/MS) analysis of tricarboxylic acid (TCA) metabolites in available frozen TCGA melanoma specimens (n=36) identified significantly lower concentrations of citrate (logfold difference −1.31, p=0.012) and succinate (logfold difference −0.91, p=0.048) in OW/OB tumors (Figure 3, Supplementary Figure 6), suggesting a suppression of the tricarboxylic acid cycle.
Figure 3:
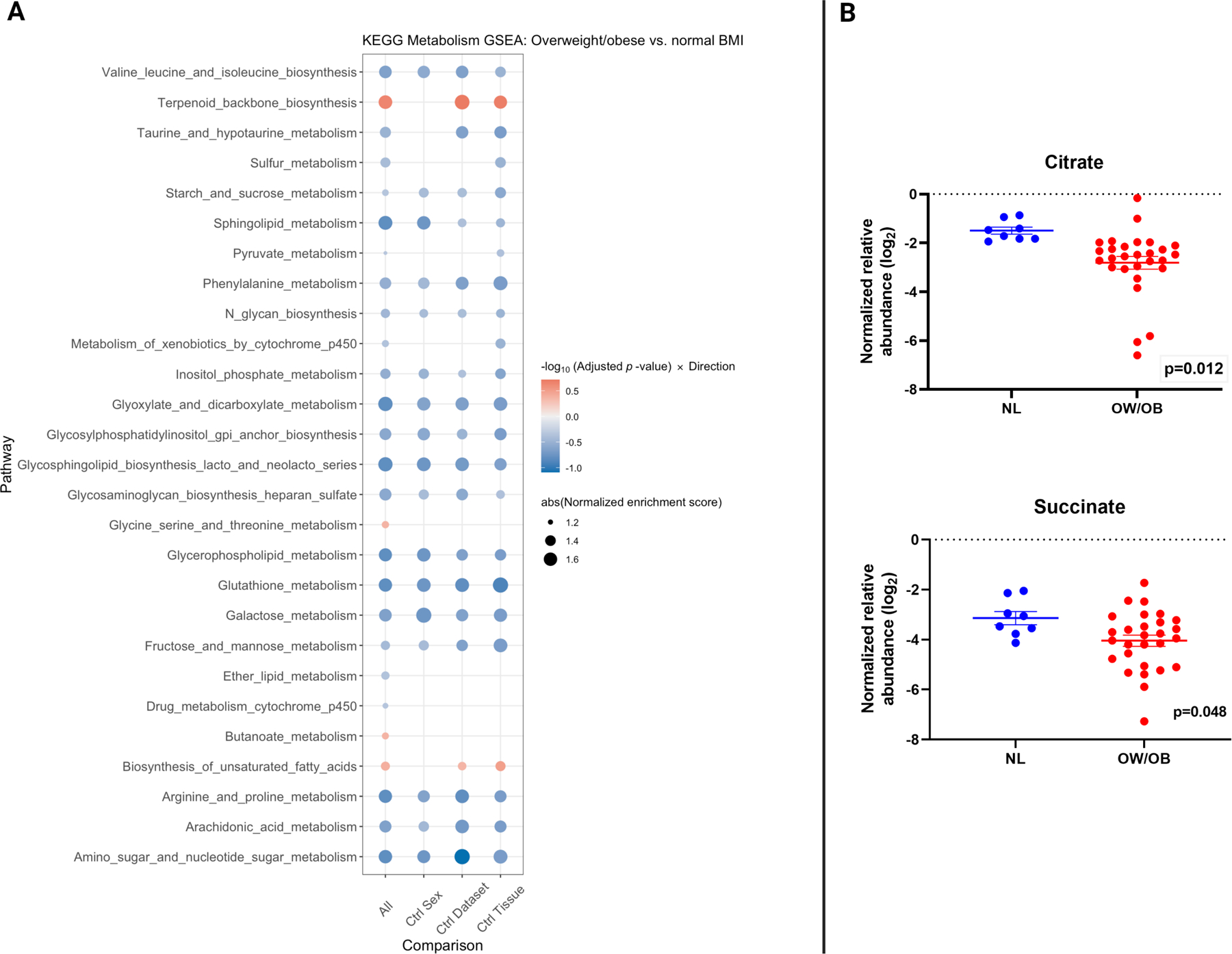
Gene expression of KEGG metabolism pathways and direct metabolic profiling of tumor samples by BMI.
A. Integrated gene set enrichment analysis comparing KEGG biosynthesis and metabolism pathways by body mass index (BMI) in patients with metastatic melanoma. Blue indicates downregulation in OW/OB verse NL BMI patients. The far left column is all patients, and then, an analysis controlling for sex, cohort, and tissue site is shown in the 3 columns to the right.
B. Comparison of the tricarboxylic acid cycle metabolites citrate and succinate as measured by liquid chromatography/mass spectrometry between metastatic melanoma tumors from overweight/obese (OW/OB) patients by body mass index (BMI) verse normal (NL) BMI from The Cancer Genome Atlas (TCGA). Lines represent mean +/− SEM; each dot represents a single tumor.
To functionally assess OXPHOS and glycolysis, we performed Seahorse Extracellular Flux Assay on melanoma cells isolated from fresh patient tumor biopsies (n=45) (Figure 4A). Consistent with gene expression and LC/MS results, the oxygen consumption rate (OCR; surrogate for OXPHOS) was significantly lower in tumors from OW/OB versus NL (p=0.018, Figure 4B). Similar to the gene expression data, melanoma tumors from OW/OB tended to be more metabolically quiescent overall (47.8% below median for both OCR and ECAR among OW/OB tumors vs. 21.4%% of NL BMI, p=0.166) (Figure 4B). We also performed Seahorse analysis on 17 patient-derived melanoma cell lines, which had been kept at low passage, and observed lower basal ECAR (p=0.047) and OCR (p=0.035) in OW/OB verse NL BMI patient-derived melanoma cell lines [55.6% low ECAR & OCR in OW/OB vs. 12.5% among normal BMI, p=0.131 (Figure 4C)]. The fact that the tumor metabolic phenotypes associated with host BMI were conserved in patient-derived cell lines indicates cancer cell autonomy of the phenotype, and further suggests potential reprogramming at the epigenetic level.
Figure 4:
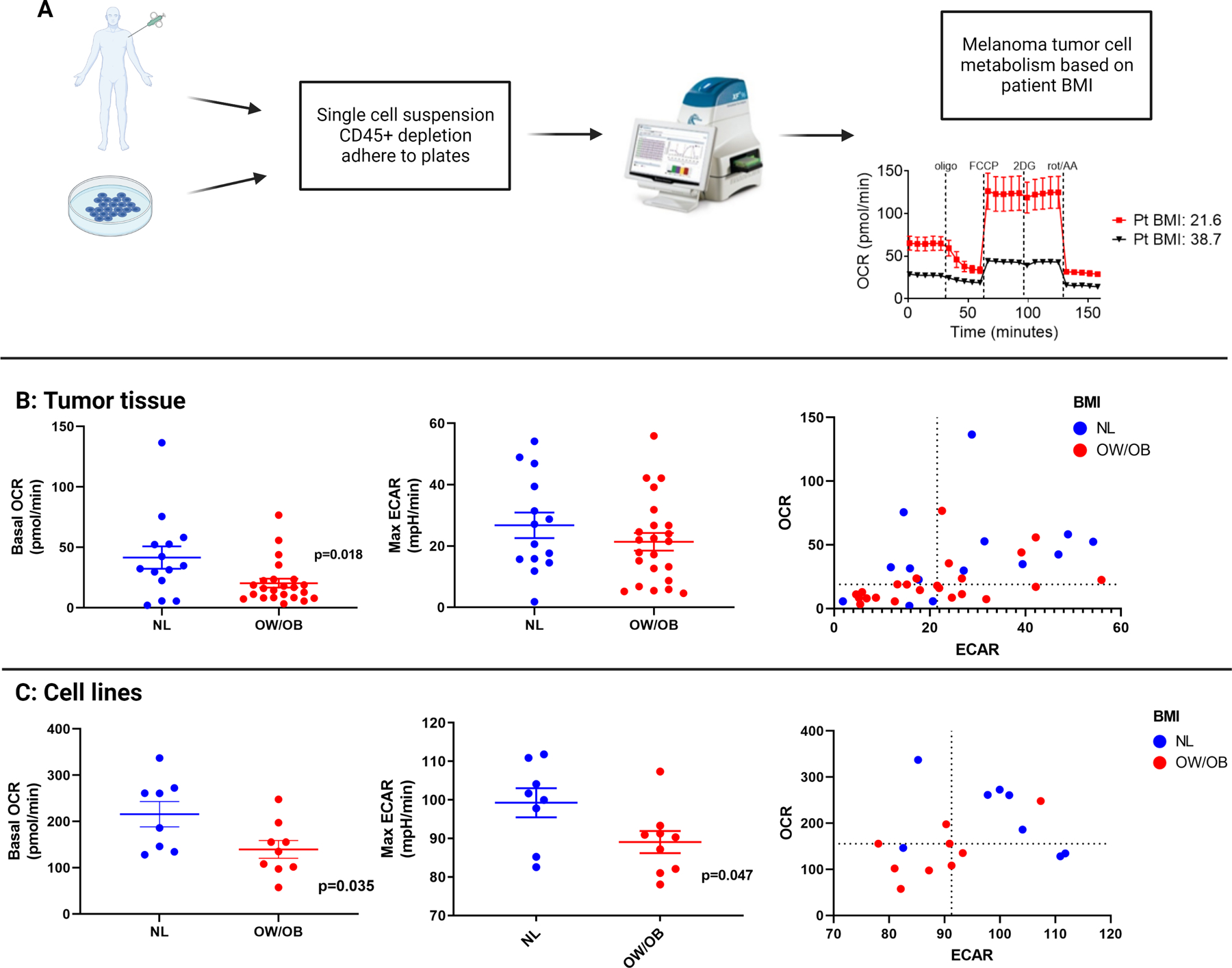
Direct measurement of mitochondrial metabolism in tumor tissue specimens and patient-derived melanoma cell lines by BMI.
A. Overview of the process to measure mitochondrial metabolism by Seahorse Extracellular Flux Assay from either tumor tissue biopsy or patient-derived melanoma cell lines. OCR = oxygen consumption rate, ECAR = extracellular acidification rate.
B. The first figure shows OCR measurements and the second figure shows ECAR measurements by BMI in tumor tissue of patients with metastatic melanoma. The third figure in Panel B shows one dot per patient reflecting ECAR measurements on the X axis and OCR measurements on the Y axis. Lines represent mean +/− SEM; each dot represents a single tumor. Dashed lines represent the median value in the third figure.
C. The first figure in Panel C shows OCR measurements and the second figure shows ECAR measurements by BMI in patient-derived melanoma cell lines. The third figure in Panel shows one dot per unique cell line representing ECAR measurements on the X axis and OCR measurements on the Y axis. Lines represent mean +/− SEM; each dot represents a single tumor. Dashed lines represent the median value in the third figure
Several studies have demonstrated that the gut microbiome influences outcomes with ICI.(5–7,29,30) As the microbiome has a bidirectional relationship with host metabolism and energy balance, we examined the associations between BMI and fecal microbiome features in a cohort of 371 patients with metastatic melanoma.(31) The overall structure (beta diversity, padj=0.10), diversity (padj=0.15), and taxonomy of the fecal microbiome was similar between OW/OB and NL BMI patients (Figure 5A–C) in the overall cohort as well as in sex-stratified analyses (Supplementary Figure 7–8). There were also no differences by BMI in the abundance of taxa we have previously found to be associated with ICI response (Figure 5D).(5,7) These findings suggest that differences in the gut microbiome are unlikely to explain the improved outcomes seen with ICI treatment in OW/OB patients with metastatic melanoma.
Figure 5:
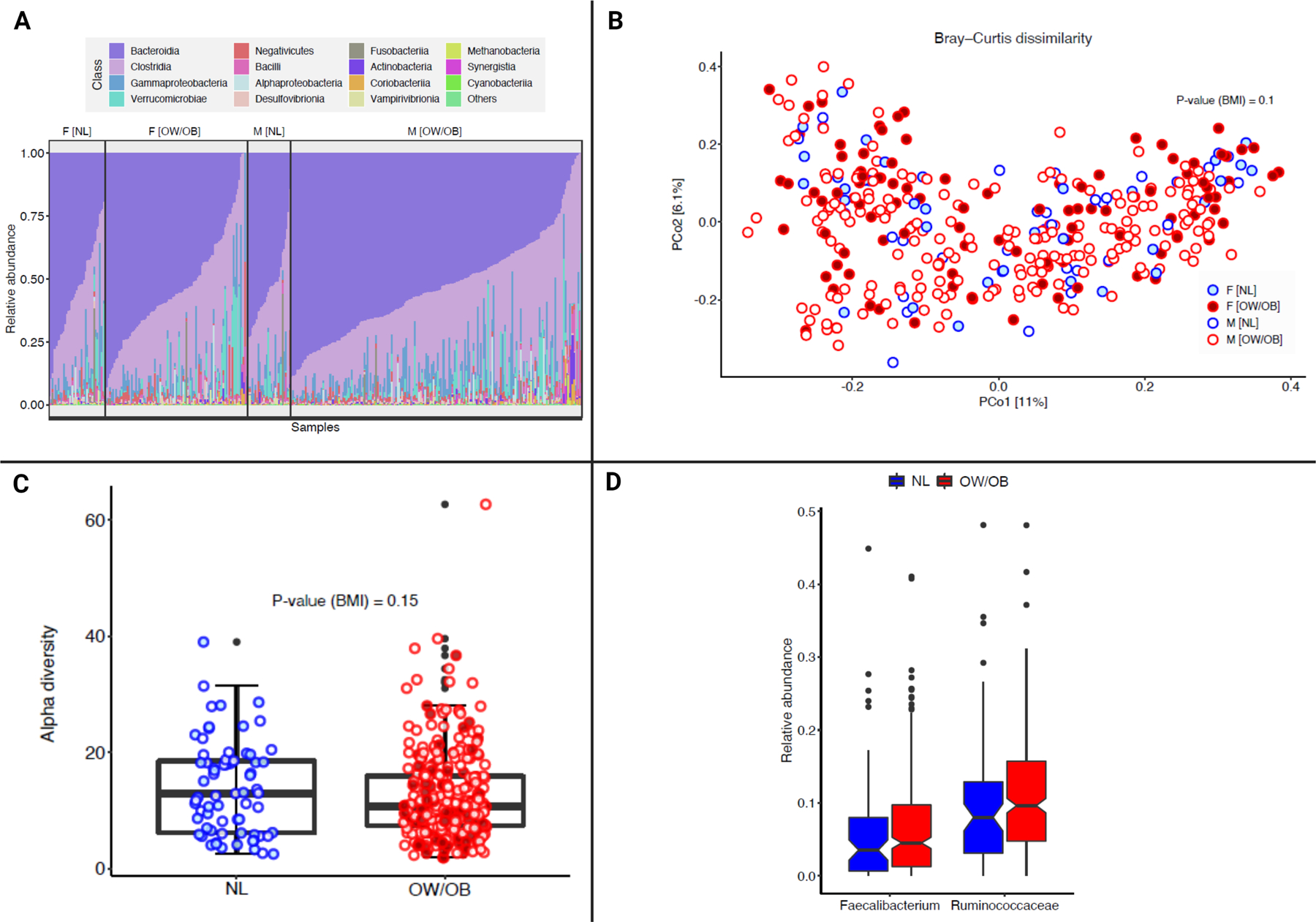
Fecal microbiome diversity and composition by body mass index (BMI).
A. The composition plot depicts the microbiome samples at the class level in the taxonomy, where samples are ordered by BMI and sex in the MD Anderson melanoma microbiome cohort (n=371).
B. Beta-diversity analysis based on Bray-Curtis dissimilarity represents microbiome samples with metastatic melanoma in terms of the two top principal components (explaining around 17% of variance) obtained from the principal coordinate analysis. Red dots are from overweight/obese (OW/OB) BMI patients, and blue dots are from normal (NL) BMI patients. Shading inside the dots indicates the female gender. PERMANOVA analysis analyzes the significant (p-value reported) impact of BMI on beta-diversity after controlling for the batch effect of age and sex.
C. Inverse Simpson alpha diversity scores of the fecal microbiome by BMI in patients with metastatic melanoma. Box plot represents the median bar with the box bounding the IQR and whiskers the most extreme points within 1.5 x IQR. The red dots represent OW/OB patients and the blue dots represent NL patients by BMI.
D. Relative abundance of Faecalibacterium and Ruminococcaceae by BMI. The blue columns are NL BMI patients, and the red columns are OW/OB BMI patients.
Discussion
Though multiple studies have found that a high BMI is associated with improved outcomes with immune and targeted therapies, the biology underlying this association remains unclear. The relationship between host and tumor metabolism has not been well studied in this context, despite mounting evidence that tumor and immunocyte metabolism can play a critical role in the response to immune and targeted therapies.(15,16) Intriguingly, our molecular, metabolic, and immune characterization of multiple cohorts of metastatic melanoma biospecimens demonstrated that OW/OB metastatic melanoma tumors had significant downregulation of OXPHOS and other metabolic pathways. Importantly, evidence of the relative metabolic quiescence of OW/OB tumors was supported by multiple lines of investigation, including gene expression from multiple independent cohorts and direct metabolic profiling by both LC/MS and Seahorse bioenergetics analyses.
High tumor OXPHOS, as well as a hypermetabolic phenotype characterized by upregulation of both OXPHOS and glycolysis, can drive resistance to ICI by creating a hypoxic microenvironment hostile to T cell function.(15,17) OXPHOS can similarly drive resistance to MAPK pathway directed targeted therapy in melanoma.(16) However, the determinants of tumor metabolic phenotype are poorly understood. Here, we found that host metabolic phenotype was associated with tumor metabolic phenotype, but in a direction that was unexpected and perhaps counterintuitive. The obese phenotype, which is characterized by systemic overabundance of nutrients and growth factors, seems to engender a more metabolically quiescent tumor. The maintenance of this phenotype on flux assays of tumors and established patient-derived cell lines supports the durability of the phenotype. Our analysis of DNA and RNA sequencing does not provide a clear mechanism for the association between host and tumor metabolic phenotypes, but it provides a rationale to interrogate other potential drivers, particularly environmentally-responsive epigenetic factors.
Interestingly, prior work has demonstrated that stromal adipocytes can directly transfer lipids to melanocytes in the TME, driving increased OXPHOS via fatty acid oxidation and melanoma progression (32). However, we did not observe any differences in genes or pathways involved in fatty acid uptake or oxidation by BMI. While these results may seem contradictory, this may result from a distinction in what is observed when stromal adipocytes are in direct contact with melanoma cells (e.g. in melanomas invasive into subcutaneous tissues) vs the systemic effects of host global adiposity on metastatic melanoma tumors where adipocytes are not typically prominent features of the microenvironment, a hypothesis that could be directly interrogated.
Our immunologic findings contrast to a prior study that reported that obesity was associated with higher expression of PD-1-on T-cells, which conferred increased sensitivity to anti-PD1 in preclinical models.(33) However, analysis of human tumor tissue was limited in that study. The authors found that colorectal tumors from obese patients had lower CD3+ infiltrates, and in the TCGA melanoma cohort, they found increased expression of markers of immune activation/exhaustion (e.g. PD-1, LAG-3) in elderly (age>60) OB individuals compared to NL BMI counterparts. However, when we examined immune signatures across all ages in TCGA and with additional BMI data gathered from TCGA contributing investigators, we did not observe differences by BMI. We also failed to observe such differences in an integrated analysis of RNAseq data from other cohorts of melanoma patients (n=155 in total). Further, we did not observe significant differences in immune cell populations and T cell activation markers by IHC when stratified by BMI and/or sex in two large melanoma cohorts other than higher TBET expression and a higher ratio of TBET+/FOXP3+ cells in OW/OB tumors. Obesity, via leptin, is known to skew helper T cells towards a T helper 1 phenotype (for which TBET is a marker), and FOXP3 is a marker for immunosuppressive, regulatory T cells.(34,35) Thus, this could indicate a skewing of CD4 cells towards phenotypes conducive to anti-tumor immunity, though this needs further study.
Our gene expression findings in patient tumor material are limited by the use of bulk RNASeq, which does not allow us to differentiate between metabolic changes in the tumor cells versus other cells in the microenvironment. However, our flux analysis of patient tumors and patient-derived cell lines was specifically performed on isolated melanoma cells, which showed both lower OXPHOS and glycolysis in OW/OB patient derived melanoma cells. As tumor metabolism can influence immune metabolism and functionality, and the influence of host metabolism on immunometabolism is currently unknown, interrogation of the metabolic phenotype of immunocytes within the tumor microenvironment of obese patients is an important future direction.(15) While we did not observe quantitative differences in most immune cell populations or co-stimulatory molecules by BMI by either IHC or bulk gene expression analysis for immune pathways, direct assessment of tumor microenvironment interactions by single-cell and/or spatial transcriptomics should be used to further evaluate this in future studies.
In conclusion, our findings suggest that host energy balance influences tumor metabolism in metastatic melanoma, with downregulation of OXPHOS and other metabolic pathways in tumors of OW/OB patients. These findings were consistent across multiple independent patient cohorts profiled by complementary techniques. As OXPHOS has previously been associated with resistance to targeted and immune therapies, this suggests a potential mechanism whereby obesity is associated with improved outcomes with these therapies. Further studies are needed to better understand the mechanism by which host metabolism influences melanoma metabolism and how this change impacts the metabolism and function of tumor infiltrating immunocytes. However, these unexpected and provocative findings that melanoma tumors from obese individuals are more metabolically quiescent have significant implications for understanding how host-level metabolism may shape tumor metabolism.
Supplementary Material
Supplementary Figure 1 Legend: Integrated gene set enrichment analysis by body mass index (BMI) with correction for S phase. This figure presents a dotplot of genes differentially up- or downregulated in OW/OB patients versus NL BMI patients. Red indicates upregulation in OW/OB verse NL. The far left column is all patients, and then, an analysis controlling for sex, cohort, and tissue site is shown in the 3 columns to the right.
{kind=link}
Supplementary Figure 2 Legend: Reverse phase protein array of the PI3K pathway expression by BMI. Box plot represents the median bar with the box bounding IQR and whiskers the minimum and maximum values.
{kind=link}
Supplementary Figure 3 Legend: Integrated gene set enrichment analysis of subgroups by body mass index BMI. This figure compares gene expression between overweight/obese (OW/OB) and normal (NL) BMI patients with metastatic melanoma across subgroups by sex, cohort, and tissue site. Supplementary Figure 2 presents a dotplot of genes differentially up- or downregulated in OW/OB patients versus NL BMI patients by subgroups. Red indicates upregulation in OW/OB verse NL.
{kind=link}
Supplementary Figure 4 Legend: Immune cell analysis by BMI.
A. Immunohistochemistry (IHC) analysis for CD8-, CD45RO-, FOXP3-, CD68-, GZMB-, PD-1-, LAG-3-, and CD3-positive cells in overweight/obese (OW/OB) verse normal (NL) tumors as defined by body mass index from the MD Anderson Cancer Center (MDA) cohort. Line represents median +/− interquartile range; each dot represents a single tumor.
B. IHC analysis for PD-L1-, CD45RO-, FOXP3-, EOMES-, GZMB-, PD-1-, TBET-, and TBET:FOXP3-positive cells in OW/OB verse NL tumors as defined by body mass index from the Gide cohort. Line represents median +/− interquartile range; each dot represents a single tumor.
{kind=link}
Supplementary Figure 5 Legend: Immune cell analysis by BMI and sex.
A. Immunohistochemistry (IHC) analysis of the MDA cohort stratified by BMI and sex. Line represents median +/− interquartile range; each dot represents a single tumor.
B. IHC analysis of the Gide cohort stratified by BMI and sex. Line represents median +/− interquartile range; each dot represents a single tumor.
{kind=link}
Supplementary Figure 6 Legend: Direct metabolite measurements from a subset of The Cancer Genome Atlas (TCGA) cohort.
Comparison of additional tricarboxylic acid cycle metabolites measured by liquid chromatography/mass spectrometry between metastatic melanoma tumors from overweight/obese (OW/OB) patients by body mass index (BMI) verse normal (NL) BMI from The Cancer Genome Atlas (TCGA). Lines represent mean +/− SEM; each dot represents a single tumor.
{kind=link}
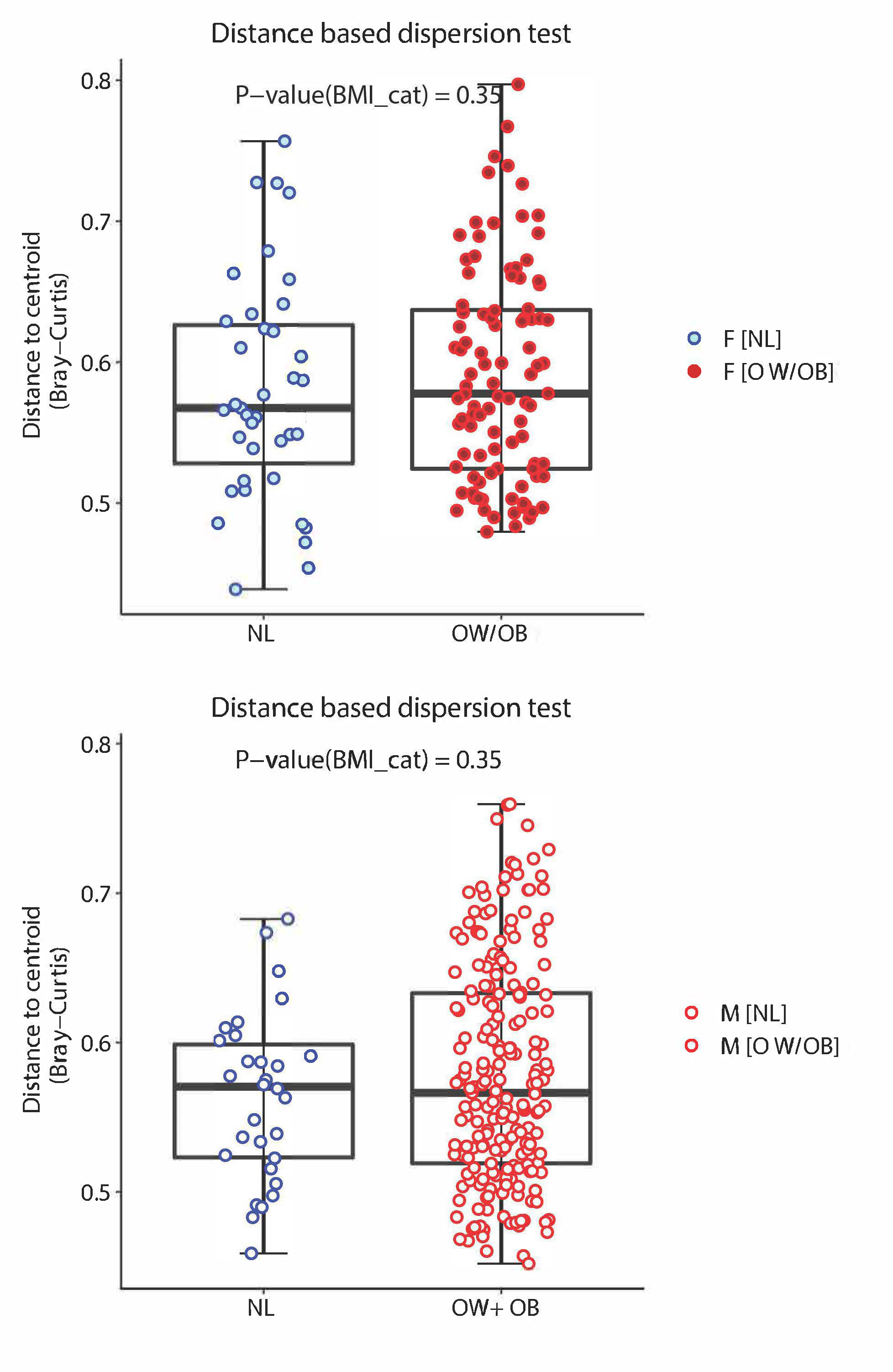
Supplementary Figure 7 Legend: Beta-diversity analysis stratified by sex based on Bray-Curtis dissimilarity represents microbiome samples with metastatic melanoma in terms of the two top principal components (explaining around 17% of variance) obtained from the principal coordinate analysis. Red dots are from overweight/obese (OW/OB) BMI patients, and blue dots are from normal (NL) BMI patients. Shading inside the dots indicates the female gender. This is a subgroup analysis of Figure 5B.
{kind=link}
Supplementary Figure 8 Legend: Inverse Simpson alpha diversity scores of the fecal microbiome by BMI in patients with metastatic melanoma stratified by sex. Box plot represents the median bar with the box bounding the IQR and whiskers the most extreme points within 1.5 x IQR. The red dots represent OW/OB patients and the blue dots represent NL patients by BMI. This is a subgroup analysis of Figure 5C.
{kind=link}
Translational relevance.
Overweight/obese (OW/OB) patients with metastatic melanoma unexpectedly have improved outcomes with immune checkpoint inhibitors (ICIs) and BRAF-targeted therapies. The biologic basis for the “obesity paradox” in metastatic melanoma is unknown. In our multi-omic analysis, gene expression from multiple independent cohorts and direct metabolic profiling by both LC/MS and Seahorse bioenergetics analyses found that host energy balance influences tumor metabolism, with downregulation of oxidative phosphorylation (OXPHOS) and other metabolic pathways in tumors from overweight/obese (OW/OB) patients. As OXPHOS has previously been associated with resistance to targeted and immune therapies, this suggests a potential mechanism whereby obesity is associated with improved outcomes with these therapies. Further work is needed to elucidate the mechanism whereby host metabolism influences melanoma metabolism.
Financial support:
This work was supported by American Society of Clinical Oncology and Conquer Cancer Foundation Young Investigator Award and Career Development Award to JLM. JLM also acknowledges the Transdisciplinary Research in Energetics and Cancer Research Training Workshop R25CA203650 and the MD Anderson Cancer Center (MDA) Center for Energy Balance in Cancer Prevention and Survivorship and is supported by the Melanoma Research Alliance, the Elkins Foundation, Seerave Foundation, Rising Tide Foundation, the Mark Foundation for Cancer Research, MDA Melanoma SPORE Developmental Research Program Award, MDA Physician Scientist Program and MDA Moonshot Program.
AWH is supported by an American Society of Clinical Oncology and Conquer Cancer Foundation Young Investigator Award and the Rob Heyvaert and Paul Heynen Prostate Cancer Foundation Young Investigator Award.
This work was supported by CPRIT Proteomics and Metabolomics Core Facility (N.P.; RP210227), NIH (P30 CA125123), and Dan L. Duncan Cancer Center.
RJD is supported by HHMI, NIH/NCI (R35CA22044901), and CPRIT (RP180778).
GMD was supported by DP2AI136598, a Mark Foundation Emerging Leader Award, and a CRI Lloyd Old STAR Award. GMD and YGN are supported by DoD CA170483 and the Melanoma Research Alliance.
AF was supported by the Welch Foundation, G-0040–20010628.
MAD is supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the AIM at Melanoma Foundation, the NIH/NCI (1 P50 CA221703–02), Cancer Fighters of Houston, the Anne and John Mendelsohn Chair for Cancer Research, and philanthropic contributions to the Melanoma Moon Shots Program of MD Anderson
JEL is supported by the Irving and Nadine Mansfield and Robert David Levitt Cancer Research Chair, the NIH/NCI (1 P50 CA221703–02), and philanthropic contributions to the MD Anderson Marit Peterson Fund for Melanoma Research.
JEG is supported by the John M. Skibber Professorship, the Booker Family Foundation, the NIH/NCI (1 P50 CA221703–02), and philanthropic contributions to the Melanoma Moon Shots Program of MD Anderson.
C.R.D. is supported by the American Cancer Society (RSG-17–049-01-NEC), the Melanoma Research Alliance, Andrew Sabin Family Fellows Program, and the National Cancer Institute (CCSG 5P30 CA016672–37 to MD Anderson) and acknowledges the MDACC Bionurtrition Core and Center for Energy Balance in Cancer Prevention and Survivorship.
TSN is supported by the NIH grant K08 CA241088, the Tower Cancer Research Foundation Career Development Award, and the Hyundai Hope on Wheels Young Investigator Award.
AJL is supported by the MDA Melanoma SPORE and the MDA Melanoma Moonshot Program.
TNG and CQ are supported by Cancer Institute NSW Early Career Fellowships.
JAW is supported by the NIH (1 R01 CA219896–01A1), U.S- Israel Binational Science Foundation (201332), the Melanoma Research Alliance (4022024), American Association for Cancer Research Stand Up To Cancer (SU2C-AACR-IRG-19–17), Department of Defense (W81XWH-16–1-0121), MD Anderson Cancer Center Multidisciplinary Research Program Grant, Andrew Sabin Family Fellows Program, and MD Anderson Cancer Center’s Melanoma Moon Shots Program. JAW is a member of the Parker Institute for Cancer Immunotherapy at MD Anderson Cancer Center.
RAS and JFT are supported an Australian National Health and Medical Research Council (NHMRC) Program Grant (APP1093017)
RAS is supported by an NHMRC Practitioner Fellowship (APP1141295). Support from Deborah McMurtrie and John McMurtrie is also gratefully acknowledged.
Footnotes
Conflicts of interest statement:
TSN has received honoraria from consulting with Allogene Therapeutics, PACT Pharma, and Adaptive Biotechnologies.
PH is a scientific advisory board member for Dragonfly and Immatics
JFT has received honoraria for advisory board participation from Bristol-Myers Squibb Australia, MSD Australia, GlaxoSmithKline, and Provectus Inc, and travel and conference support from GlaxoSmithKline, Provectus Inc., and Novartis.
JEG has been a consultant to and/or advisory board member of Merck, Bristol-Myers Squibb, Novartis, and Regeneron.
RAS has received fees for professional services from F. Hoffmann-La Roche Ltd, Evaxion, Provectus Biopharmaceuticals Australia, Qbiotics, Novartis, Merck Sharp & Dohme, NeraCare, AMGEN Inc., Bristol-Myers Squibb, Myriad Genetics, and GlaxoSmithKline.
AL has been a consultant for AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Genentech, Merck, and Novartis.
DS is a consultant/scientific advisor to Array, Pfizer, Novartis, Bristol-Myers Squibb, MSD, Sanofi-Aventis, Neracare, Immunocore, Replimune, Daiichi-Sanyo, Labcorp, InFlarX, AstraZeneca, OncoSec, Merck-EMD, Nektar, Philogen, Pierre-Fabre, and Sun Pharma. DS has received institutional funding for research grants from Merck, Amgen, Novartis, MSD, Array, and Roche.
JAW reports compensation for speaker’s bureau and honoraria from Imedex, Dava Oncology, Omniprex, Illumina, Gilead, PeerView, Physician Education Resource, MedImmune, and Bristol-Myers Squibb. JAW serves as a consultant and/or advisory board member for Roche/Genentech, Novartis, AstraZeneca, GlaxoSmithKline, Bristol-Myers Squibb, Merck, Biothera Pharmaceuticals, Microbiome DX, and Micronoma. J.A.W. also receives research support from GlaxoSmithKline, Roche/Genentech, Bristol-Myers Squibb, and Novartis.
JAW and VG are inventors on patent WO2020106983A1 submitted by the Board of Regents, The University of Texas System, and Institut Gustav Roussy that covers methods and compositions for treating cancer and for predicting a subject’s response to combination checkpoint inhibitor therapy. JAW and VG are inventors and CNS is a collaborator on a US patent application (PCT/US17/53.717) submitted by the University of Texas MD Anderson Cancer Center that covers methods to Enhance immune checkpoint blockade responses by modulating the microbiome. JAW is an inventor on patent WO2020150429A1 submitted by the Board of Regents, The University of Texas System, that covers methods and compositions for treating immune checkpoint inhibitor–associated colitis in a subject through the administration of fecal matter from a healthy donor to the subject.
VG reports honoraria from ExpertConnect and Kansas Society of Clinical Oncology.
RJD is a founder and advisor for Atavistik Bioscience and an advisor for Agios Pharmaceuticals, Vida Ventures and Nirogy Therapeutics.
HL is a scientific advisor for Precision Scientific.
AF is on the scientific advisory board of Scorpion.
MAD has been a consultant to Roche/Genentech, Array, Pfizer, Novartis, BMS, GSK, Sanofi-Aventis, Vaccinex, Apexigen, Eisai, and ABM Therapeutics, and he has been the PI of research grants to MD Anderson Cancer Center by Roche/Genentech, GSK, Sanofi-Aventis, Merck, Myriad, and Oncothyreon.
GMD declares conflicts of interests and has submitted patents covering the use of metabolic reprogramming and immunotherapy that are licensed or pending and is entitled to a share in net income generated from licensing of these patent rights for commercial development. GMD consults for and/or is on the scientific advisory board of BlueSphere Bio, Century Therapeutics, Novasenta, Pieris Pharmaceuticals, and Western Oncolytics/Kalivir; has grants from Bluebird Bio, Novasenta, Pfizer, Pieris Pharmaceuticals, TCR2, and Western Oncolytics/Kalivir; GMD owns stock in Novasenta and BlueSphere Bio.
YGN is a consultant for Novartis, Merck and Array Biopharma, and has (institutional) research funding from Merck, Pfizer and Bristol-Myers Squibb.
JLM is a consultant for Bristol-Myers Squibb, Roche, and Merck.
All remaining authors have no conflicts of interest to report.
References:
- 1.Long GV, Eroglu Z, Infante J, Patel S, Daud A, Johnson DB, et al. Long-Term Outcomes in Patients With BRAF V600-Mutant Metastatic Melanoma Who Received Dabrafenib Combined With Trametinib. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018;36(7):667–73 doi 10.1200/jco.2017.74.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2019;381(16):1535–46 doi 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 3.Kugel CH, 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin Cancer Res 2018;24(21):5347–56 doi 10.1158/1078-0432.Ccr-18-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conforti F, Pala L, Bagnardi V, De Pas T, Martinetti M, Viale G, et al. Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. The Lancet Oncology 2018;19(6):737–46 doi 10.1016/s1470-2045(18)30261-4. [DOI] [PubMed] [Google Scholar]
- 5.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 2018;359(6371):97–103 doi 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrews MC, Duong CPM, Gopalakrishnan V, Iebba V, Chen WS, Derosa L, and PD-1 blockade. Nat Med 2021;27(8):1432–41 doi 10.1038/s41591-021-01406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 2021;374(6575):1632–40 doi 10.1126/science.aaz7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klil-Drori AJ, Azoulay L, Pollak MN. Cancer, obesity, diabetes, and antidiabetic drugs: is the fog clearing? Nat Rev Clin Oncol 2017;14(2):85–99 doi 10.1038/nrclinonc.2016.120. [DOI] [PubMed] [Google Scholar]
- 9.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 2016;6(2):202–16 doi 10.1158/2159-8290.Cd-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McQuade JL, Daniel CR, Hess KR, Mak C, Wang DY, Rai RR, et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. The Lancet Oncology 2018;19(3):310–22 doi 10.1016/s1470-2045(18)30078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.An Y, Wu Z, Wang N, Yang Z, Li Y, Xu B, et al. Association between body mass index and survival outcomes for cancer patients treated with immune checkpoint inhibitors: a systematic review and meta-analysis. Journal of translational medicine 2020;18(1):235 doi 10.1186/s12967-020-02404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lalani AA, Bakouny Z, Farah S, Donskov F, Dudani S, Heng DYC, et al. Assessment of Immune Checkpoint Inhibitors and Genomic Alterations by Body Mass Index in Advanced Renal Cell Carcinoma. JAMA oncology 2021. doi 10.1001/jamaoncol.2021.0019. [DOI] [PMC free article] [PubMed]
- 13.Kichenadasse G, Miners JO, Mangoni AA, Rowland A, Hopkins AM, Sorich MJ. Association Between Body Mass Index and Overall Survival With Immune Checkpoint Inhibitor Therapy for Advanced Non-Small Cell Lung Cancer. JAMA oncology 2020;6(4):512–8 doi 10.1001/jamaoncol.2019.5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cortellini A, Ricciuti B, Tiseo M, Bria E, Banna GL, Aerts JG, et al. Baseline BMI and BMI variation during first line pembrolizumab in NSCLC patients with a PD-L1 expression ≥ 50%: a multicenter study with external validation. J Immunother Cancer 2020;8(2) doi 10.1136/jitc-2020-001403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Najjar YG, Menk AV, Sander C, Rao U, Karunamurthy A, Bhatia R, et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI insight 2019;4(5) doi 10.1172/jci.insight.124989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gopal YN, Rizos H, Chen G, Deng W, Frederick DT, Cooper ZA, et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1α and oxidative phosphorylation in melanoma. Cancer research 2014;74(23):7037–47 doi 10.1158/0008-5472.Can-14-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaiswal AR, Liu AJ, Pudakalakatti S, Dutta P, Jayaprakash P, Bartkowiak T, et al. Melanoma Evolves Complete Immunotherapy Resistance through the Acquisition of a Hypermetabolic Phenotype. Cancer immunology research 2020;8(11):1365–80 doi 10.1158/2326-6066.Cir-19-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akbani R, Akdemir KC, Arman Aksoy B, Albert M, Ally A, Amin SB. Genomic Classification of Cutaneous Melanoma. Cell 2015;161(7):1681–96 doi 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016;165(1):35–44 doi 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer cell 2019;35(2):238–55.e6 doi 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun 2014;5:3887 doi 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, et al. KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer cell 2020;37(4):599–617.e7 doi 10.1016/j.ccell.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Putluri N, Shojaie A, Vasu VT, Vareed SK, Nalluri S, Putluri V, et al. Metabolomic profiling reveals potential markers and bioprocesses altered in bladder cancer progression. Cancer research 2011;71(24):7376–86 doi 10.1158/0008-5472.Can-11-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab 2018;27(5):977–87.e4 doi 10.1016/j.cmet.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer GM, Jalali A, Kircher DA, Lee WC, McQuade JL, Haydu LE, et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov 2019;9(5):628–45 doi 10.1158/2159-8290.Cd-18-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marabelle A, Fakih MG, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with select advanced solid tumours treated with pembrolizumab in KEYNOTE-158. Annals of Oncology 2019;30:v477–v8 doi 10.1093/annonc/mdz253.018. [DOI] [PubMed] [Google Scholar]
- 27.Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018;560(7719):499–503 doi 10.1038/s41586-018-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Insulin Pollak M. and insulin-like growth factor signalling in neoplasia. Nature reviews Cancer 2008;8(12):915–28 doi 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 29.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, et al. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018;359(6371):104–8 doi 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018;359(6371):91–7 doi 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 31.Xiao H, Kang S. The Role of the Gut Microbiome in Energy Balance With a Focus on the Gut-Adipose Tissue Axis. Frontiers in genetics 2020;11:297 doi 10.3389/fgene.2020.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov 2018;8(8):1006–25 doi 10.1158/2159-8290.Cd-17-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med 2019;25(1):141–51 doi 10.1038/s41591-018-0221-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998;394(6696):897–901 doi 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 35.Werneck MB, Lugo-Villarino G, Hwang ES, Cantor H, Glimcher LH. T-bet plays a key role in NK-mediated control of melanoma metastatic disease. Journal of immunology (Baltimore, Md : 1950) 2008;180(12):8004–10 doi 10.4049/jimmunol.180.12.8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Legend: Integrated gene set enrichment analysis by body mass index (BMI) with correction for S phase. This figure presents a dotplot of genes differentially up- or downregulated in OW/OB patients versus NL BMI patients. Red indicates upregulation in OW/OB verse NL. The far left column is all patients, and then, an analysis controlling for sex, cohort, and tissue site is shown in the 3 columns to the right.
Supplementary Figure 2 Legend: Reverse phase protein array of the PI3K pathway expression by BMI. Box plot represents the median bar with the box bounding IQR and whiskers the minimum and maximum values.
Supplementary Figure 3 Legend: Integrated gene set enrichment analysis of subgroups by body mass index BMI. This figure compares gene expression between overweight/obese (OW/OB) and normal (NL) BMI patients with metastatic melanoma across subgroups by sex, cohort, and tissue site. Supplementary Figure 2 presents a dotplot of genes differentially up- or downregulated in OW/OB patients versus NL BMI patients by subgroups. Red indicates upregulation in OW/OB verse NL.
Supplementary Figure 4 Legend: Immune cell analysis by BMI.
A. Immunohistochemistry (IHC) analysis for CD8-, CD45RO-, FOXP3-, CD68-, GZMB-, PD-1-, LAG-3-, and CD3-positive cells in overweight/obese (OW/OB) verse normal (NL) tumors as defined by body mass index from the MD Anderson Cancer Center (MDA) cohort. Line represents median +/− interquartile range; each dot represents a single tumor.
B. IHC analysis for PD-L1-, CD45RO-, FOXP3-, EOMES-, GZMB-, PD-1-, TBET-, and TBET:FOXP3-positive cells in OW/OB verse NL tumors as defined by body mass index from the Gide cohort. Line represents median +/− interquartile range; each dot represents a single tumor.
Supplementary Figure 5 Legend: Immune cell analysis by BMI and sex.
A. Immunohistochemistry (IHC) analysis of the MDA cohort stratified by BMI and sex. Line represents median +/− interquartile range; each dot represents a single tumor.
B. IHC analysis of the Gide cohort stratified by BMI and sex. Line represents median +/− interquartile range; each dot represents a single tumor.
Supplementary Figure 6 Legend: Direct metabolite measurements from a subset of The Cancer Genome Atlas (TCGA) cohort.
Comparison of additional tricarboxylic acid cycle metabolites measured by liquid chromatography/mass spectrometry between metastatic melanoma tumors from overweight/obese (OW/OB) patients by body mass index (BMI) verse normal (NL) BMI from The Cancer Genome Atlas (TCGA). Lines represent mean +/− SEM; each dot represents a single tumor.
Supplementary Figure 7 Legend: Beta-diversity analysis stratified by sex based on Bray-Curtis dissimilarity represents microbiome samples with metastatic melanoma in terms of the two top principal components (explaining around 17% of variance) obtained from the principal coordinate analysis. Red dots are from overweight/obese (OW/OB) BMI patients, and blue dots are from normal (NL) BMI patients. Shading inside the dots indicates the female gender. This is a subgroup analysis of Figure 5B.
Supplementary Figure 8 Legend: Inverse Simpson alpha diversity scores of the fecal microbiome by BMI in patients with metastatic melanoma stratified by sex. Box plot represents the median bar with the box bounding the IQR and whiskers the most extreme points within 1.5 x IQR. The red dots represent OW/OB patients and the blue dots represent NL patients by BMI. This is a subgroup analysis of Figure 5C.