

Abstract
Cardiac electrophysiology is regulated by continuous trafficking and internalisation of ion channels occurring over minutes to hours. Kv11.1 (also known as hERG) underlies the rapidly-activating delayed-rectifier K+ current (IKr), which plays a major role in cardiac ventricular repolarization. Experimental characterization of the distinct temporal effects of genetic and acquired modulators on channel trafficking and gating is challenging. Computer models are instrumental to elucidate these effects, but no currently available model incorporates ion-channel trafficking. Here, we present a novel computational model reproducing the experimentally observed production, forward trafficking, internalisation, recycling, and degradation of Kv11.1 channels, as well as their modulation by temperature, pentamidine, dofetilide, and extracellular K+. The acute effects of these modulators on channel gating were also incorporated and integrated with the trafficking model in the O’Hara-Rudy human ventricular cardiomyocyte model. Supraphysiological dofetilide concentrations substantially increased Kv11.1 membrane levels while also producing significant channel block. However, clinically-relevant concentrations did not affect trafficking. Similarly, severe hypokalaemia reduced Kv11.1 membrane levels based on long-term culture data, but had limited effect based on short-term data. By contrast, clinically-relevant elevations in temperature acutely increased IKr due to faster kinetics, while after 24 hours, IKr was decreased due to reduced Kv11.1 membrane levels. The opposite was true for lower temperatures. Taken together, our model reveals a complex temporal regulation of cardiac electrophysiology by temperature, hypokalaemia, and dofetilide through competing effects on channel gating and trafficking, and provides a framework for future studies assessing the role of impaired trafficking in cardiac arrhythmias.
Keywords: cardiac arrhythmia, cardiac cellular electrophysiology, computer model, dynamics, ion channel trafficking, simulation
Graphical Abstract
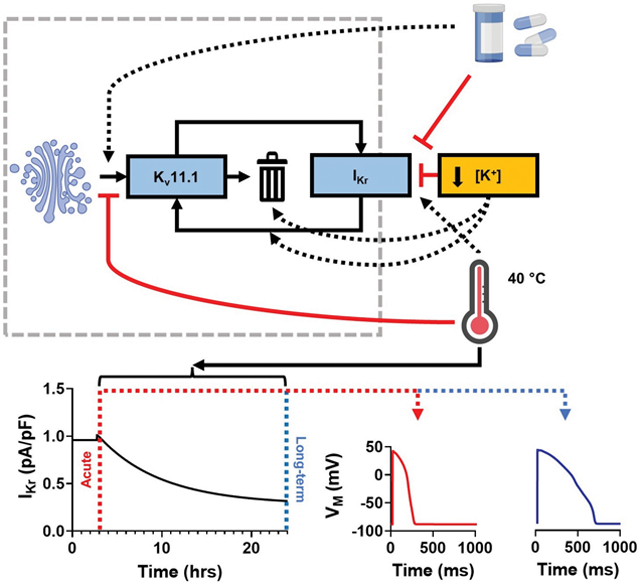
Kv11.1 channels underly the rapidly-activating delayed-rectifier K+ current and are crucial for cardiac ventricular repolarization. Kv11.1 channels are continuously trafficking to and from the plasma membrane, a process which is temperature- and potassium-dependent and modulated by various drugs. Here, we developed a novel computer model of Kv11.1 trafficking that reproduces a wide range of experimental data, including the acute and long-term effects of temperature, extracellular K+ concentration, and drugs. The model reveals complex dynamic regulation of cardiac repolarization through opposing short- and long-term effects of these factors on Kv11.1 channel gating and trafficking, indicating that assessment of their acute effects is insufficient to determine potential proarrhythmic effects. The black dotted arrows represent stimulating effects, while the red solid lines represent inhibitory effects. The Golgi complex, drugs, and thermometer were created with BioRender.com.
Introduction
Regulation of the number and activity of cardiac ion channels occurs over a wide range of timescales and enables dynamic changes in cardiac electrophysiology to adapt to varying circumstances. While posttranslational changes in channel activity can occur within seconds, continuous turnover of ion channels happens over minutes to hours, through trafficking, internalisation, degradation, and recycling processes (Guo et al., 2009; Dennis et al., 2011; Apaja et al., 2013; Ke et al., 2013; Shi et al., 2015; Osterbur Badhey et al., 2017; Ghosh et al., 2018; Kanner et al., 2018; Foo et al., 2019; Li et al., 2022). Proteins are folded and glycosylated in the endoplasmic reticulum (ER) and Golgi complex (GC), after which the channels are transported to- and integrated in the membrane. Once integrated, channels can gate and in their open state allow ions to pass into or out of the cell along the electrochemical gradient, producing an ion current. In the opposite direction, channels are internalised, with some subsequently undergoing lysosomal- or proteasomal degradation, while the others are recycled back to the membrane. This complex dynamic flux of ion channels is a major regulator of cardiac electrophysiology (Balse & Boycott, 2017; van der Heyden et al., 2018; Blandin et al., 2021).
The Kv11.1 or hERG channel underlies the rapidly-activating delayed-rectifier K+ current (IKr). Kv11.1 plays a major role in the electrical recovery (repolarization) of human ventricular cardiomyocytes and its trafficking is highly dynamic (Guo et al., 2009; Dennis et al., 2011; Apaja et al., 2013; Ke et al., 2013; Shi et al., 2015; Osterbur Badhey et al., 2017; Kanner et al., 2018; Foo et al., 2019). Kv11.1 loss-of-function mutations result in prolonged repolarization and are associated with long-QT syndrome type-2 (LQTS2). LQTS2 is the second most prevalent type of LQTS and is associated with life-threatening polymorphic ventricular tachyarrhythmias, notably torsades de pointes (Wallace et al., 2019). Impaired trafficking is the dominant modus operandi of LQTS2-associated Kv11.1 missense mutations (Anderson et al., 2014; Smith et al., 2016). However, the mechanisms and dynamics underlying these trafficking deficiencies can differ markedly between mutations, highlighting the complex, highly dynamic nature of Kv11.1-channel trafficking (Apaja et al., 2013; Ke et al., 2013; Osterbur Badhey et al., 2017; Kanner et al., 2018; Foo et al., 2019). Adding to this complexity are several modulators of Kv11.1 transport like temperature, drugs, and the concentration of extracellular potassium ([K+]).
Febrile temperatures can acutely speed up Kv11.1 gating, resulting in larger currents (Zhou et al., 1998; Vandenberg et al., 2006; Mauerhofer & Bauer, 2016), while long-term exposure to high temperatures reduces the number of Kv11.1 channels in the membrane (Zhao et al., 2016; Foo et al., 2019). The opposite is true for hypothermia, which acutely decreased IKr due to slower Kv11.1 channel gating, but on the long term increased IKr due to increased Kv11.1 trafficking. Moreover, hypokalaemia disrupts Kv11.1 channel gating and trafficking processes by promoting a non-conductive state which is prone to channel internalisation and degradation (Guo et al., 2009; Massaeli et al., 2010). Kv11.1 drug block is an important factor in drug-induced proarrhythmia and therefore plays an important role in cardiac safety pharmacology (Heijman et al., 2014). Interestingly, while several drugs (e.g., E-4031, dofetilide, astemizole) acutely block the Kv11.1 channel and prolong repolarization, these drugs may rescue aberrant channel trafficking when applied for hours (Dennis et al., 2012; de Git et al., 2013; Smith et al., 2013; Varkevisser et al., 2013; Qile et al., 2020). The characterization of these diverse modulating effects is experimentally challenging due to the different timescales involved (milliseconds to hours) and methodologies required (e.g., cell fixation and membrane-specific ion channel staining vs. live-cell patch-clamp recording).
Computer models offer perfect observability and control (Heijman et al., 2021), which may help to characterize the above-mentioned trafficking processes and their modulation by various pathophysiological factors. From the initial computer models of cardiomyocyte electrophysiology developed in the second-half of the 20th century to the more advanced models of the 21st century, much progress has been made with respect to model complexity and scale (Heijman et al., 2016; Heijman et al., 2021). Detailed Markov models (MMs) of ion-channel gating have been developed. For example, the five-state IKr MM by Clancy and Rudy (Clancy & Rudy, 2001) can reproduce a wide range of experimental voltage-clamp data and, when embedded in an action-potential (AP) model like the O’Hara-Rudy (ORd) human ventricular cardiomyocyte model (O’Hara et al., 2011), can simulate the genesis of early afterdepolarizations (EADs) and other arrhythmogenic responses (O’Hara et al., 2011). The ORd model currently serves as a basis for the “comprehensive in-vitro proarrhythmia assay” (CiPA) initiative proposed by the US Food and Drug Administration and pharmaceutical industry for improved screening of the risk of drug-induced ventricular arrhythmias, highlighting the real-world relevance of cellular-electrophysiology models (Dutta et al., 2017; Li et al., 2020). However, none of the currently available AP models incorporate the dynamic trafficking of ion channels, instead they assume that the number of ion channels is fixed, precluding simulation of long-term regulation of cardiac electrophysiology by drugs, fever, hypokalaemia, and other modulators.
Here, we developed a novel Kv11.1-trafficking component that enabled simulation of the regulation of Kv11.1-membrane levels over minutes to hours by temperature, two selected drugs (i.e., the trafficking blocker pentamidine and trafficking rescuer dofetilide), and extracellular [K+], as well as their effects on cardiac cellular electrophysiology.
Methods
Kv11.1 trafficking model
A two-state model with sub-membrane (S) and membrane (M) states and four rates was created to model Kv11.1-channel trafficking (Figure 1A). The state-transitions were modelled by two ordinary differential equations:
| (1) |
| (2) |
where ψ represents the channel production rate, α the forward trafficking rate, β the internalisation rate, and δ the degradation rate. The rates were optimized through stochastic single-channel simulations (Heijman et al., 2013), which generally aimed to minimize the sum of squared errors between the model trafficking behaviour and those observed in literature (Dennis et al., 2011; Apaja et al., 2013). The total amount of Kv11.1 channels in the membrane was calibrated based on estimates from Heijman et al. (2013) which were based on whole-cell and single-channel conductance. The final rate configuration can be found in Table 1.
Figure 1. Model components required to simulate regulation of Kv11.1 trafficking and gating, and its effects on ventricular cardiomyocyte electrophysiology.
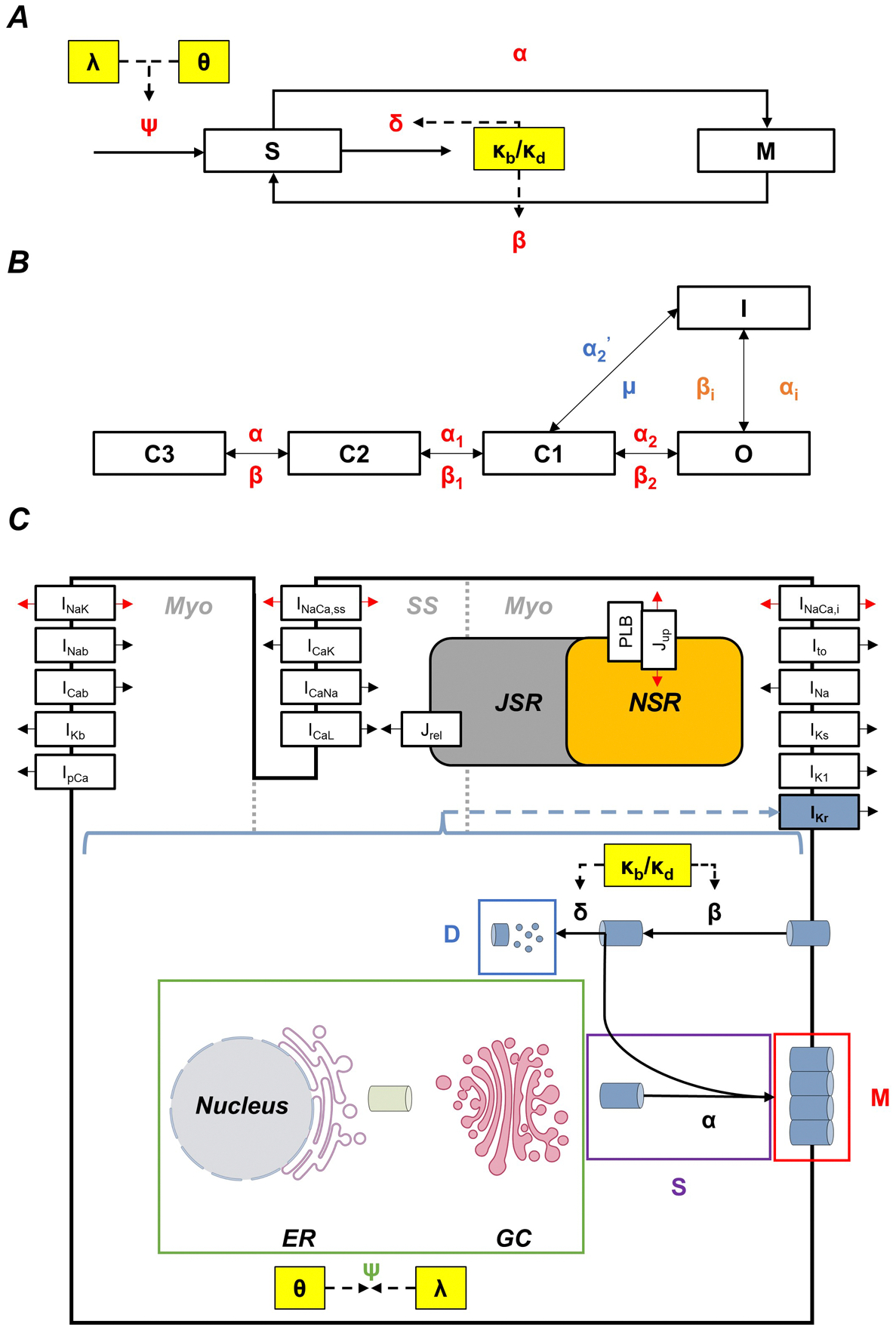
A, The Kv11.1 trafficking model consists of two-states (M: Membrane, S: Sub-membrane) with four rates (ψ: production rate, α: forward trafficking rate, β: internalisation rate, and δ: degradation rate). The temperature (θ), drugs (λ), and extracellular [K+] (κb and κd) parameters are used to scale the ψ, β, and δ rates. B, The Clancy and Rudy (2001) IKr Markov model was used to create a temperature-sensitive model of IKr gating by shifting the voltage dependence of certain rates (see Table 2) and scaling each rate with their respective Q10 values (Clancy & Rudy, 2001). In particular, αn, βn, α2’, and μ rates were scaled with Q10Activation and Q10Deactivation, while αi and βi were scaled with Q10Inactivation and Q10Recovery, respectively. C, The trafficking, temperature, drug, and extracellular [K]+ components controlling IKr were embedded in the O’Hara-Rudy (ORd) human ventricular action potential model (O’Hara et al., 2011). Adapted from O’Hara et al. (2011) and the nucleus, endoplasmic reticulum (ER), and Golgi complex (GC) were created with BioRender.com.
Table 1.
Calibrated ion channel trafficking parameters.
| Parameter | Value (per hour) |
|---|---|
| α | 6.56850075 |
| β | 2.101663125 ⋅ (κb/κbref) |
| δ | 0.599592 ⋅ (κd/κdref) |
| ψ | 423.26175750000004 ⋅ λ ⋅ θ |
Note, θ, λ, κb, and κd reflect the temperature, drug, and extracellular [K+] modulators from equations 4, 5, 9, and 10, respectively. Moreover, the κbref and κdref are the results from equations 9 and 10 with extracellular [K+] set at 5.4 mmol/L.
Temperature-dependent regulation of IKr gating and trafficking
A temperature-sensitive MM of IKr gating was created by incorporating Q10 values (i.e., change in rate for each 10 °C change in temperature) and voltage shifts in half-maximal activation (V1/2) as scaling factors to the rates in the Clancy and Rudy IKr MM (Figure 1B) (Clancy & Rudy, 2001). In particular, the αn, βn, α2’, and μ rates in the MM were scaled with Q10Activation and Q10Deactivation, while αi and βi were scaled with Q10Inactivation and Q10Recovery, respectively (Figure 1B), whereby, the scaling factor (sf) for each Q10 was calculated as:
| (3) |
The Q10 values and temperature-dependent shift in V1/2 of IKr activation were calibrated based on experimental data (Zhou et al., 1998; Mauerhofer & Bauer, 2016) by replicating the experimental voltage-clamp protocols at different temperatures, calculating the corresponding Q10 values, and optimizing the sum of squared errors between the model values and those found in literature.
Ion-channel trafficking is also affected by temperature, which was modelled using an asymmetrical sigmoid function:
| (4) |
where a is the amplitude, b is the midpoint, c the steepness, s represents the asymmetry around the midpoint (with a value of 1 resulting in a symmetrical sigmoidal function), and d is a constant that prevents the number of channels from going to zero. Again, the temperature effects on trafficking were calibrated based on experimental data (Zhao et al., 2016; Foo et al., 2019). Finally, θ was used to scale the ψ rate in the trafficking model (Figure 1A). The MM equations, parameters, and their temperature-dependent changes can be found in Tables 2 and 3.
Table 2. IKr Markov model transitions and equations adapted from Clancy & Rudy (2001).
The V1/2Activation/Deactivation and V1/2Inactivation/Recovery are abbreviated V1/2AD and V1/2IR, respectively. Note, the temperature (T), voltage (V), and extracellular [K+] are not pre-defined but inputted during simulation. The MM rates are defined in milliseconds−1 (ms).
| Transition equations | |
| C3 → C2 C2 → C3 |
α = sfactivation · p7 · exp(p2 · ((V + V1/2AD · (37 − T)) · FRT)) β = sfdeactivation · p8 · exp(p9 · ((V + V1/2AD · (37 − T)) · FRT)) |
| C2 → C1 C1 → C2 |
α1 = sfactivation · p5 β1 = sfdeactivation · p6 |
| C1 → O O → C1 |
α2 = sfactivation · p1 · exp(p2 · ((V + V1/2AD · (37 − T)) ⋅ FRT)) β2 = sfdeactivation · p3 · exp(p4 · ((V + V1/2AD · (37 − T)) ⋅ FRT)) |
| O → I I → O |
|
| C1 → I I → C1 |
α2′ = p15 · α2
μ = (α2′ · αi · β2)/(α2 · βi) |
| State changes | |
Table 3.
Adapted IKr Markov model parameters from Clancy & Rudy (2001) and calibrated parameters for the effects of temperature on trafficking.
| Baseline MM parameters | Temperature-dependent trafficking regulation | ||
|---|---|---|---|
| Parameter | Value | Parameter | Value |
| p1 | 1.31E-2 | a | 1.2975 |
| p2 | 1.48 | b | 42.2 |
| p3 | 3.3E-3 | c | 1.7 |
| p4 | −5.77E-1 | s | 10 |
| p5 | 2.17 | d | 0.18 |
| p6 | 1.08 | Q 10Activation | 5.64528884 |
| p7 | 3.02E-2 | Q 10Deactivation | 1.19795417 |
| p8 | 2.9E-3 | Q 10Inactivation | 3.41629472 |
| p9 | −9.78E-1 | Q 10Recovery | 2.93346223 |
| p10 | 5.45E-1 | V 1/2Activation/Deactivation | 1.21461546 |
| p11 | −8.17E-1 | V 1/2Inactivation/Recovery | 1.11308848 |
| p12 | 4.5 | ||
| p13 | 8.2E-1 | ||
| p14 | 5.04E-1 | ||
| p15 | 1 | ||
| FRT | 0.037435883507802616 | ||
Modelling drug effects on Kv11.1 gating and trafficking
The effects of pentamidine and dofetilide on acute gating and long-term trafficking were also incorporated in the model. Pentamidine is a Kv11.1 trafficking inhibitor that does not affect channel gating, whereas dofetilide is a potent Kv11.1 channel blocker but long-term exposure (e.g., hours) to dofetilide rescues impaired Kv11.1 trafficking (de Git et al., 2013; Varkevisser et al., 2013; Asahi et al., 2019). The opposing effects of pentamidine and dofetilide were modelled as:
| (5) |
where [P] is the pentamidine concentration in μmol/L, [D] is the dofetilide concentration in μmol/L, h is the Hill factor related to pentamidine, hD is the Hill factor related to dofetilide, a’ is the magnitude of the dofetilide-induced promotion of trafficking, kmD is the affinity of dofetilide, and km is the affinity of pentamidine. We defined km as:
| (6) |
where km’ is the baseline affinity of pentamidine in the absence of dofetilide, [D] is the dofetilide concentration in μmol/L, and R determines the impact of dofetilide on the affinity of pentamidine. Furthermore, we defined a’ as:
| (7) |
with a the magnitude of the dofetilide-induced promotion of trafficking in the presence of pentamidine, [P] is the pentamidine concentration in μmol/L, b is the midpoint of the pentamidine dependence. The parameters from equations 5–7 were calibrated based on the concentration dependence of both pentamidine and dofetilide, as well as the time-dependent effects of dofetilide reported in literature (Varkevisser et al., 2013; Asahi et al., 2019) and λ was used to scale ψ in the trafficking model.
The mean plasma concentration of dofetilide over 24 hours was extracted from literature and converted to μmol/L based on the molecular weight of dofetilide (Allen et al., 2000). This concentration was regarded as a ‘clinical dose’ and used to run simulations to model the effects of dofetilide-induced rescue of channel trafficking. Finally, the acute IKr-blocking effect of dofetilide was modelled based on previously reported IC50 values (Sutanto et al., 2019) as:
| (8) |
where [D] is the dofetilide concentration in μmol/L. The final drug-modelling parameters can be found in Table 4.
Table 4.
Calibrated parameters for the drug effects on trafficking.
| Parameter | Value |
|---|---|
| h | 2.196566 |
| h D | 0.525717 |
| a | 0.730636 |
| km D | 0.329207 |
| km’ | 6.628938 |
| R | 0.126642 |
| b | 6 |
Modelling the effects of extracellular [K+] on Kv11.1 gating and trafficking
The ORd model is sensitive to changes in extracellular [K+], which, besides changing the driving force for all K+ currents, also modulates the gating of IKr and the inward-rectifier K+ current (IK1). However, extracellular [K+] also modulates Kv11.1 trafficking (Guo et al., 2009; Massaeli et al., 2010), which is not part of the original ORd model. Here, we modelled the trafficking effects of hypokalaemia through changes in β and δ, because experimental studies have shown that hypokalaemia primarily affects Kv11.1 channel internalisation and degradation (Guo et al., 2009; Massaeli et al., 2010). In particular, the β rate was scaled by a factor κb (Figure 1A), as follows:
| (9) |
where αk is the magnitude of the extracellular [K+]-induced internalisation, [K+] is the extracellular K+ concentration, km is the affinity for extracellular [K+], and hk is the Hill factor for [K+]. Similarly, the effects of extracellular [K+] on δ were modelled as:
| (10) |
where s is a scalar determining the relative impact of extracellular [K+] on δ vs β. The final parameters related to extracellular [K+] can be found in Table 5.
Table 5.
Calibrated parameters for the effects of extracellular [K+] on trafficking for ‘overnight’ (i.e., 12 hours) and ‘week’ model configurations.
| Parameter | ‘Overnight’ value | ‘Week’ value |
|---|---|---|
| a k | 7.249733 | 7.249733 |
| k m | 0.278542 | 0.871920 |
| h k | 2.895935 | 2.691968 |
| s | 0.226791 | 0.226791 |
Embedding in the O’Hara-Rudy human ventricular action potential model
The trafficking effects of temperature, drugs, and extracellular [K+] were introduced as scaling factors to the appropriate rates in the trafficking model:
| (11) |
where ψbase is the baseline production rate as shown in Table 1, λ represents the opposing effects of pentamidine and dofetilide, and θ represents the temperature-dependent regulation of Kv11.1 channel trafficking. In addition,
| (12) |
where βbase is the baseline internalisation rate (Table 1), κb is the effect of extracellular [K+] on β, and κbref is the reference value of κb at 5.4 mmol/L extracellular [K+]. Similarly,
| (13) |
where δbase is the baseline degradation rate (Table 1), κd is the effect of extracellular [K+] on δ, and κdref is the reference value of κd at 5.4 mmol/L extracellular [K+]. The above-mentioned model components with their respective formulas, parameters, and scaling factors were embedded in the endocardial version of the ORd model (O’Hara et al., 2011) (Figure 1C) to enable evaluation of the effects of Kv11.1-channel trafficking regulation by temperature, extracellular [K+], and drugs on ventricular cellular electrophysiology:
| (14) |
where γ is the acute IKr-blocking effect of dofetilide, M represents the number of Kv11.1 channels in the membrane, Mref represents the number of Kv11.1 membrane channels in steady-state at baseline (i.e., in the absence of drugs, at 37 °C, and with 5.4 mmol/L extracellular [K+]), GKr is the maximum conductance of Kv11.1 channels, O is the open probability of the IKr MM model, VM is the membrane potential, and Erev is the reversal potential of IKr.
Statistics, software and data availability
The experimental data are presented as means and standard deviations. All the simulations were performed through Myokit and Python (version 3.7.6.) (Clerx et al., 2016). The model code, optimization scripts, and data can be found online at: https://github.com/HeijmanLab.
Results
Calibration of the trafficking model
Stochastic simulation of the trafficking model allowed us to track single-channel transitions over time. Figure 2A shows the state transitions over time for two channels, where the top panel shows a channel coming into existence after approximately 20 minutes and then continuously switching between the membrane and sub-membrane states. The lower panel shows another channel that has a much shorter lifespan of approximately 6.5 hours. This single-channel resolution made it possible to replicate experimental data involving tagging of individual channels to quantify internalisation and recycling rates (Dennis et al., 2011; Apaja et al., 2013). After parameter optimization, the model was in good agreement with the experimental behaviour (Figure 2B), with the exception of the first recycling timepoint (Dennis et al., 2011; Apaja et al., 2013). We subsequently evaluated the simulated decay of Kv11.1 channels in the presence of forward trafficking block with these parameters and found substantial agreement with a wide range of experimental data (Figure 2C), providing independent validation of the model fit (Guo et al., 2009; Apaja et al., 2013; Ke et al., 2013; Shi et al., 2015; Osterbur Badhey et al., 2017; Foo et al., 2019).
Figure 2. Calibration of the trafficking model.
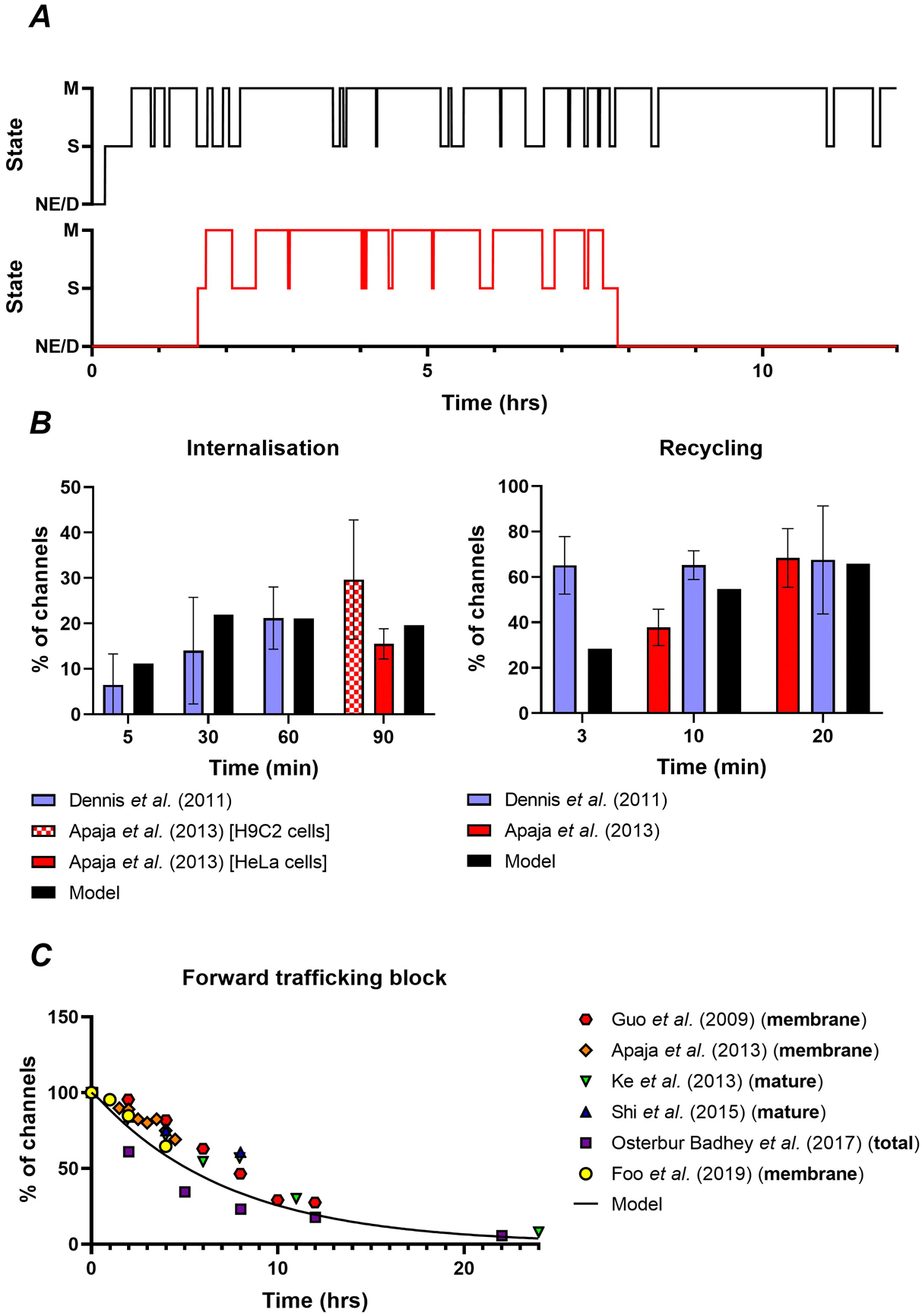
A, Representative examples of single Kv11.1 channel state transitions for channels #3001 and #3600 over 12 hours, showing channel production from non-existing (NE) state, cycling between sub-membrane (S) and membrane states (M), and decay (D). B, Comparison of internalisation and recycling rates derived from single-channel simulations (black bars) to experimental data from biotinylation assays and ELISA methods (Dennis et al., 2011; Apaja et al., 2013). C, Decrease in the relative number of Kv11.1 channels in response to forward trafficking block in the model (black lines, with ψ = 0) and experimental data (symbols) (Guo et al., 2009; Apaja et al., 2013; Ke et al., 2013; Shi et al., 2015; Osterbur Badhey et al., 2017; Foo et al., 2019). Note that some experiments solely focused on membrane channels, whereas others quantified the amount of mature Kv11.1 channels (155 kDa) or total amount of Kv11.1 channels (e.g., mature and immature bands at 155 kDa and 135 kDa, respectively). However, model results based on membrane channels only or membrane and sub-membrane channels combined were superimposable (not shown).
Sensitivity analysis
A sensitivity analysis was performed to obtain a better understanding of the impact of individual rates on Kv11.1 membrane levels and their dynamic interplay. Each parameter was scaled separately (i.e., 4-, 2-, 1-, 0.5-, and 0.25-fold) while the other parameters were kept at their optimized value. As expected, the number of channels in the membrane state increased after augmenting either the production rate or the forward trafficking rate, while augmenting the internalisation or degradation rates decreased the number of membrane channels (Figure 3, left panels). Increasing or decreasing the production and degradation rates (ψ and δ, respectively) had similar effects on the sub-membrane state as the membrane state, with increased production or decreased degradation increasing the total number of channels and vice versa. By contrast, scaling the α and β rates only had a transient effect, with a relatively quick convergence to the original number of channels in the sub-membrane state (Figure 3, right panels). Thus, ψ and δ modulate total Kv11.1 levels, whereas α and β primarily affect the distribution between sub-membrane and membrane states.
Figure 3. Sensitivity analysis of channel numbers in response to changes in parameter values.
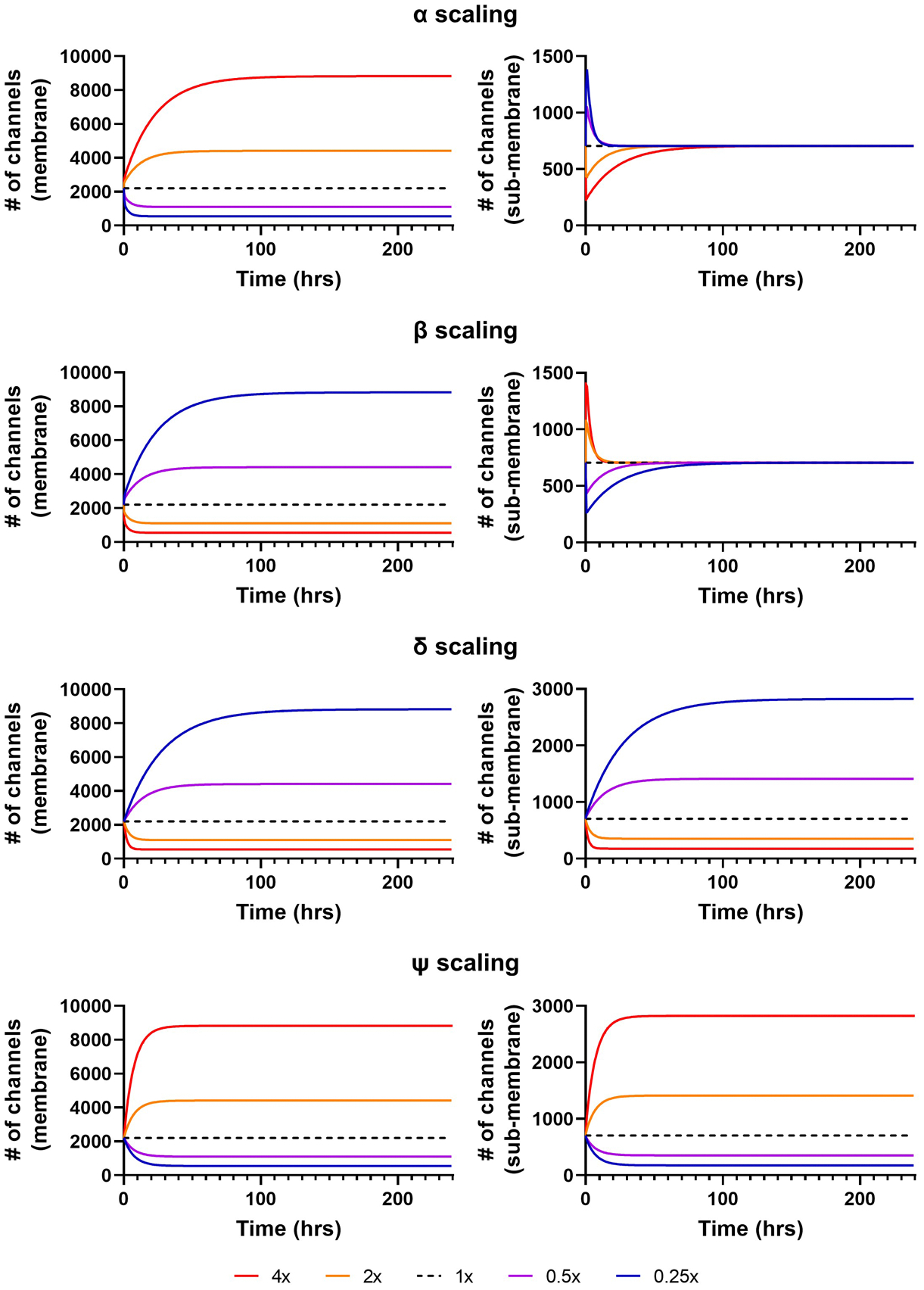
Change in number of channels in the membrane (left panels) and sub-membrane (right panels) states over time in response to a 4-, 2-, 1-, 0.5- or 0.25-fold change in α, β, δ, or ψ rates (top to bottom). The number of channels in the membrane increases for increasing forward rates (α and ψ) or decreasing backward rates (β and δ) and vice versa. Channel levels in the sub-membrane state change parallel to those in the membrane state for changes to ψ and δ, whereas changes to α and β only have a transient effect on sub-membrane state levels.
Modelling trafficking-deficient Kv11.1 mutations
Kv11.1 missense mutations associated with LQTS2 are generally associated with trafficking-deficiencies in the forward trafficking direction, although reduced channel stability, increased retrograde trafficking, and increased degradation have also been reported (Apaja et al., 2013; Ke et al., 2013; Kanner et al., 2018; Foo et al., 2019). We hypothesised that fitting the Kv11.1-trafficking model to experimental data on a Kv11.1 trafficking-deficient mutation would provide information on the primary underlying pathophysiological mechanism, reflected by the most-affected model parameter. The Kv11.1-p.(Ala57Pro) missense mutation (p.(A57P)) characterized by Kanner et al. (2018) was chosen as a representative example to test this hypothesis. Kv11.1-p.(A57P) is a forward-trafficking deficient mutation with normal internalisation (Figure 4A), resulting in an approximately 35% reduction of Kv11.1 membrane levels (Kanner et al., 2018). Because the temporal dynamics of both the wild-type and mutant channel measured by Kanner et al. (2018) were substantially faster (half-time: approximately 12 min) than the majority of the other experimental sources (half-time: approximately 8 hours; Figure 2B), we focused on the relative differences in dynamics between the wild-type and p.(A57P) mutant. Four starting points for parameter optimization were created by scaling each model rate individually to approximate the 35% reduction of Kv11.1 membrane channel levels, while keeping the other rates constant. For example, parameter set 1 was obtained by reducing α by approximately 35% while the other rates were kept at their WT values. Initial values for parameter sets 2–4 were obtained similarly by scaling β, δ, and ψ. Thereafter, all four parameter sets were optimized by updating all the rates from each parameter set. An overview of the optimized parameter sets is presented in Table 6. Despite substantial differences in parameter values, all optimized parameter sets could accurately reproduce the observed difference in membrane stability between wild-type and mutant Kv11.1 (Figure 4A,B). Furthermore, each parameter set resulted in a similar reduction of membrane channels compared to wild-type (~35%; Figure 4C). As expected, the reduction in membrane channels also resulted in AP prolongation (Figure 4D), consistent with the clinical LQTS phenotype of carriers of the Kv11.1-p.(A57P) mutation (Kapplinger et al., 2009; Anderson et al., 2014). A similar IKr reduction in the original ORd model resulted in a comparable APD prolongation of approximately 100 ms (data not shown). Together, the data from Figures 3 and 4 indicate that data on forward trafficking and membrane stability are sufficient to reproduce the changes in Kv11.1 membrane levels and clinical phenotype, but cannot identify the underlying molecular defect.
Figure 4. Simulation of the trafficking-deficient Kv11.1-p.(A57P) mutation associated with long-QT syndrome type 2.
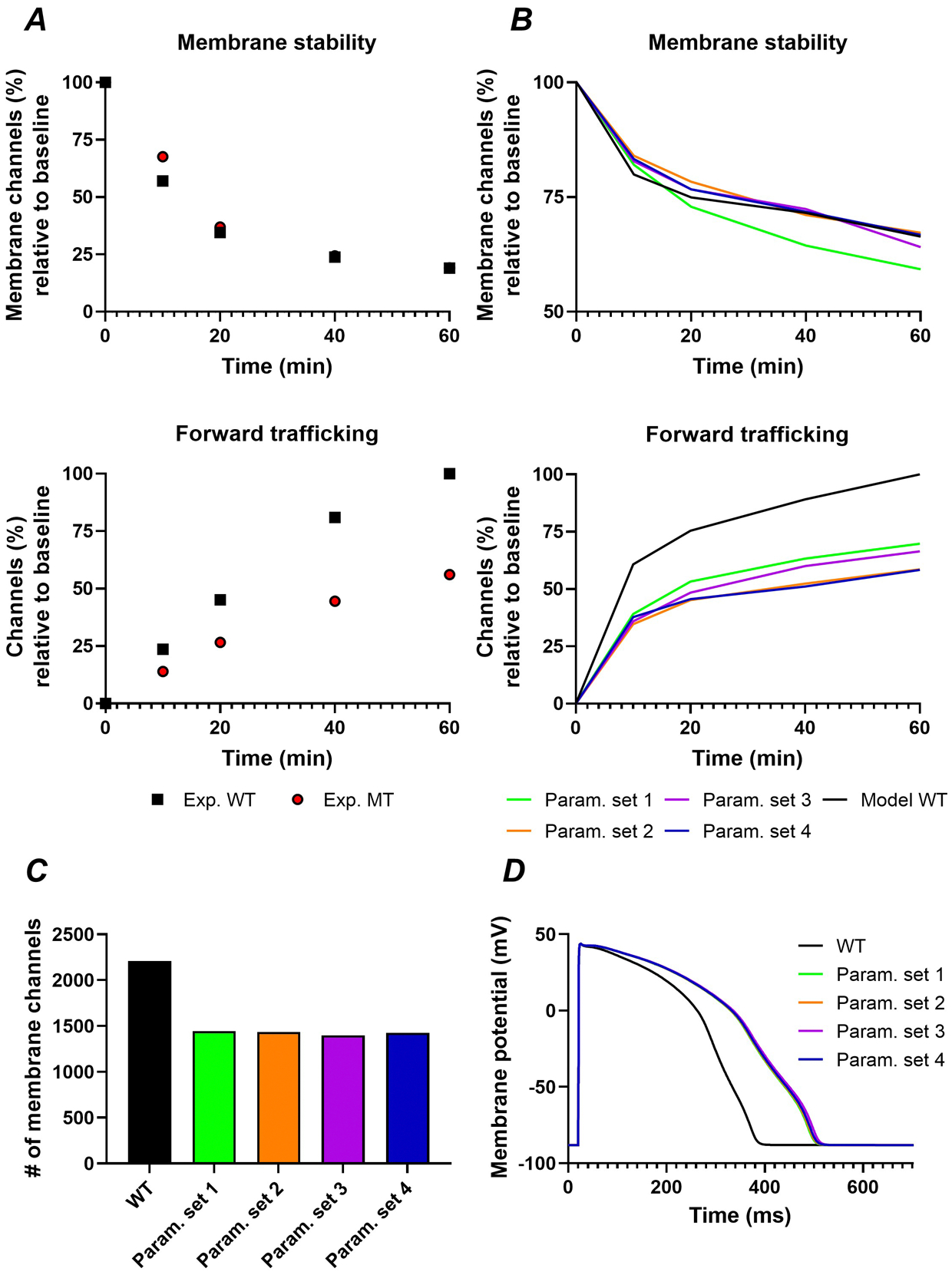
A, Relative changes in membrane stability (top panel) and forward trafficking (bottom panel) over time induced by the Kv11.1-p.(A57P) mutation (red circles, Exp. MT) compared to wild-type Kv11.1 (black squares, Exp. WT) in experimental recordings (Kanner et al., 2018). B, Similar to panel A for simulations with the default parameters representing wild-type Kv11.1 (black line, model WT) and 4 different parameter sets obtained through optimization on the relative differences between wild-type and Kv11.1-p.(A57P) after a perturbation in each of the four rates. C, The number of membrane channels for the wild-type model and each of the 4 parameter sets that was optimized to mimic the Kv11.1-p.(A57P) behaviour. D, Action-potential duration of the human ventricular cardiomyocyte model incorporating the Kv11.1 trafficking component during steady-state pacing at a basic cycle of 60s for the wild-type parameters (black) and each of the 4 parameter sets corresponding to Kv11.1-A57P.
Table 6.
Parameter sets related to Kv11.1 p.(A57P) mutation simulations.
| α | β | δ | ψ | |
|---|---|---|---|---|
| WT parameters | 6.56850075 | 2.101663125 | 0.599592 | 423.2617575 |
| Parameter set 1 | 4.39878871 | 1.93333205 | 0.65697293 | 408.40110484 |
| Parameter set 2 | 7.98396124 | 2.06770286 | 0.81350517 | 298.70774597 |
| Parameter set 3 | 6.71166945 | 2.06613057 | 0.97941628 | 421.38040913 |
| Parameter set 4 | 6.83359678 | 2.05937586 | 0.62798972 | 269.66811756 |
Modelling dofetilide-induced rescue of channel trafficking
After parameter optimization, the model could accurately reproduce the concentration-dependent effect of pentamidine on Kv11.1 membrane levels (Figure 5A), as well as the concentration and time-dependent rescue of mature Kv11.1 levels (155 kDa) by dofetilide in the presence of 10 μmol/L pentamidine (Figures 5B–C). We subsequently employed the model to investigate the combined effect of acute channel inhibition and long-term trafficking promotion by dofetilide in the presence of 5 μmol/L pentamidine (Figure 6, dashed lines) and absence of pentamidine (Figure 6, solid lines). A supraphysiological (1 μmol/L) concentration of dofetilide acutely completely inhibited IKr, while promoting an approximately 50% increase in membrane channels over 24 hours in the presence of pentamidine (Figure 6A, dashed lines). By contrast, in the absence of pentamidine, dofetilide had a negligible effect on Kv11.1 membrane trafficking (Figure 6A, solid lines). The acute inhibition resulted in repolarization failure during dofetilide treatment (25th hour; Figure 6A, bottom panel). However, the rescue of Kv11.1 membrane channels after 24 hours of dofetilide caused a slight AP shortening shortly after dofetilide application was stopped, counteracting the effects of pentamidine (50th hour; Figure 6A, bottom panel). By contrast, a more clinically relevant dofetilide concentration (3.4 nmol/L) produced a modest acute IKr inhibition that prolonged AP duration (APD), while having a minimal rescuing-effect on membrane channel numbers after 24 hours independent of the presence of pentamidine (Figure 6B). As such, there was only a minimal rebound in APD after cessation of simulated dofetilide application. Thus, while dofetilide can rescue Kv11.1 trafficking under pathological conditions (e.g., trafficking blocker pentamidine), this effect appears negligible at clinically relevant concentrations. However, other drugs and trafficking-modulators such as temperature may alter Kv11.1 membrane levels over a physiological range.
Figure 5. Calibration of the effects of pentamidine and dofetilide on Kv11.1 trafficking.
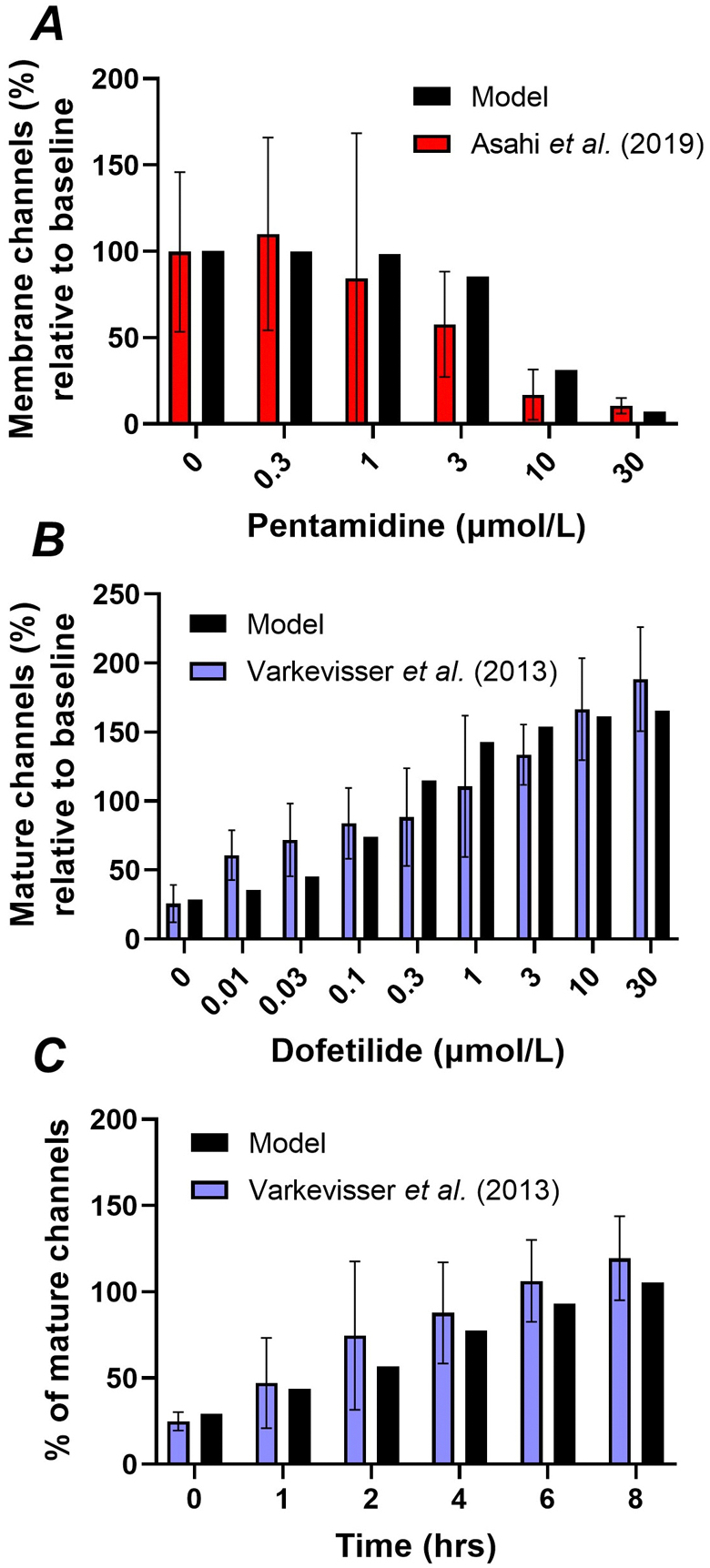
A, The pentamidine concentration dependence of Kv11.1 membrane levels after 24 hours incubation in experimental data from Asahi et al. (2019) (red bars) and model (black bars). B, The concentration dependence of dofetilide-induced rescue of mature Kv11.1 (155 kDa) levels in the presence of pentamidine (10 μmol/L for 48 hours) after 48 hours of incubation in experimental data (Varkevisser et al., 2013) and model. C, The temporal dynamics of dofetilide (1 μmol/L)-induced rescue of mature Kv11.1 levels after 48-hours pre-treatment with 10 μmol/L pentamidine in experimental data (Varkevisser et al., 2013) and model.
Figure 6. Simulated effects of pentamidine and dofetilide on Kv11.1 gating and trafficking.
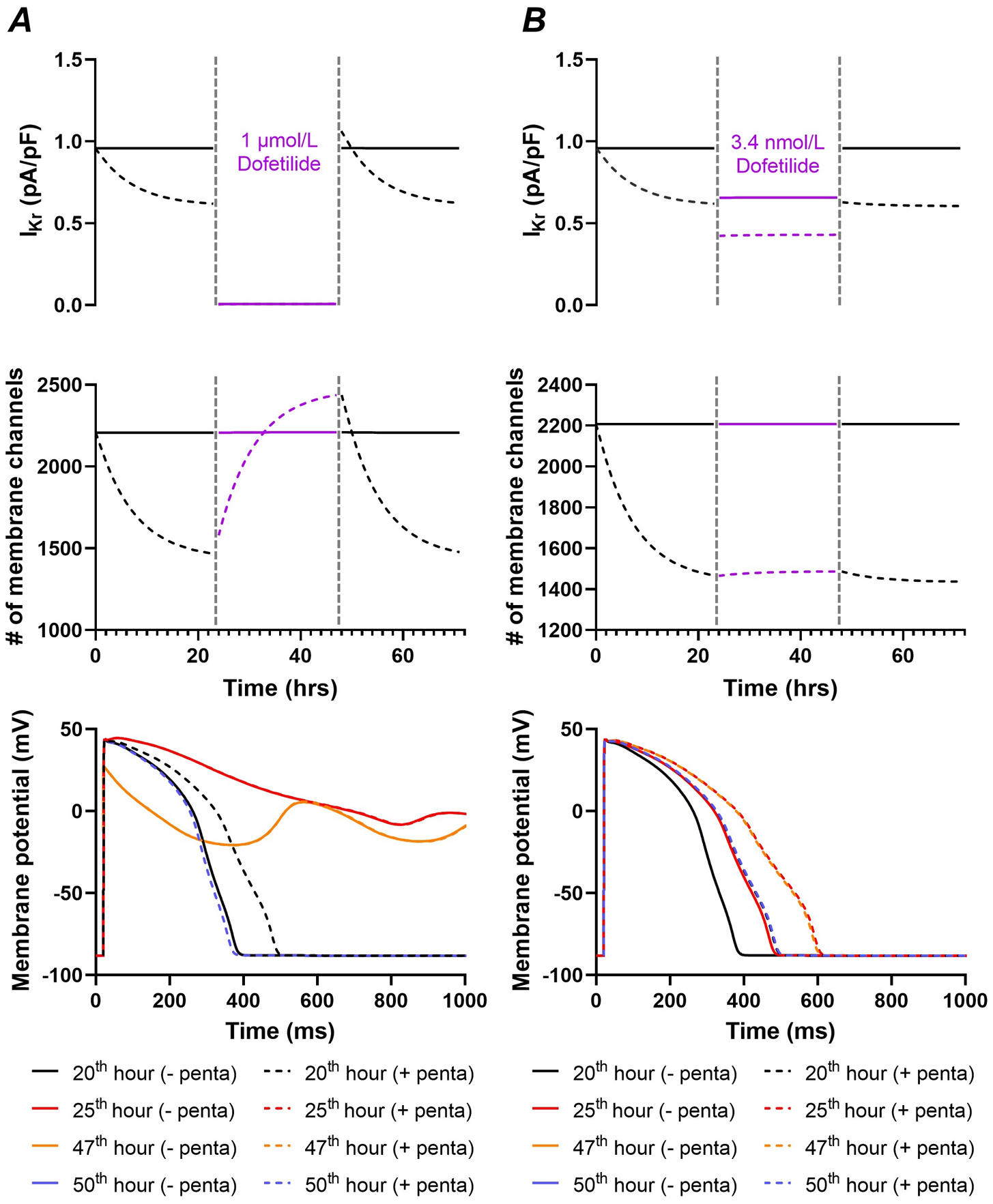
A, Time course of IKr (top panel) and Kv11.1 membrane levels (middle panel) during 24 hours at baseline (solid lines) or with 5 μmol/L pentamidine (dashed lines), followed by 24 hours with simulated dofetilide application (1 μmol/L) and 24 hours at baseline, revealing acute inhibition and long-term rescue of Kv11.1 membrane levels only during the presence of pentamidine. The dashed vertical lines (grey) indicate the start and end time of dofetilide application. The bottom panel shows the action-potential morphology at various points in the time course shown in the other panels for the simulations with 1 μmol/L dofetilide with pentamidine (solid lines) and without pentamidine (dashed lines), showing acute prolongation of repolarization duration for both conditions, but only a slight shortening of repolarization after cessation of simulated dofetilide application due to the complete rescue and minor increase in Kv11.1 membrane levels. B, Similar to panel A, for 3.4 nmol/L dofetilide. The bottom panel shows action potential prolongation when pentamidine is present due to a reduction of the number of membrane channels. The subsequent simulated dofetilide administration (3.4 nmol/L) had minimal effects on Kv11.1 trafficking.
Modelling temperature-dependent modulation of Kv11.1 channel trafficking
Similar to dofetilide, temperature acutely affects Kv11.1 gating, while modulating channel trafficking over hours. Figure 7A shows the calibration of the acute effects of temperature on gating through Q10 values and shifts in V1/2 (Zhou et al., 1998; Mauerhofer & Bauer, 2016). Combined, these effects resulted in faster gating and larger IKr at elevated temperatures (Figure 7B), consistent with experimental data (Amin et al., 2008) (Figure 7C). To model the long-term effects of temperature on trafficking, the parameters of equation 4 were calibrated to experimental data with temperatures ranging from 30 °C to 41 °C (Figure 8A) (Zhao et al., 2016; Foo et al., 2019). Overall, higher temperatures resulted in fewer channels in the membrane, while lower temperatures increased the number of membrane channels. The combined acute and long-term effects of altered temperatures are shown in Figure 8B. Higher temperatures (fever) produced a slight acute increase in IKr due to faster channel gating. However, after 24 hours, IKr decreased substantially due to reduced membrane expression of Kv11.1 channels. This was also reflected in a slight acute shortening of APD and extreme prolongation of APD after 24 hours (Figure 8C). The opposite was true for lower temperatures, albeit less pronounced.
Figure 7. Temperature-dependent regulation of Kv11.1 gating.
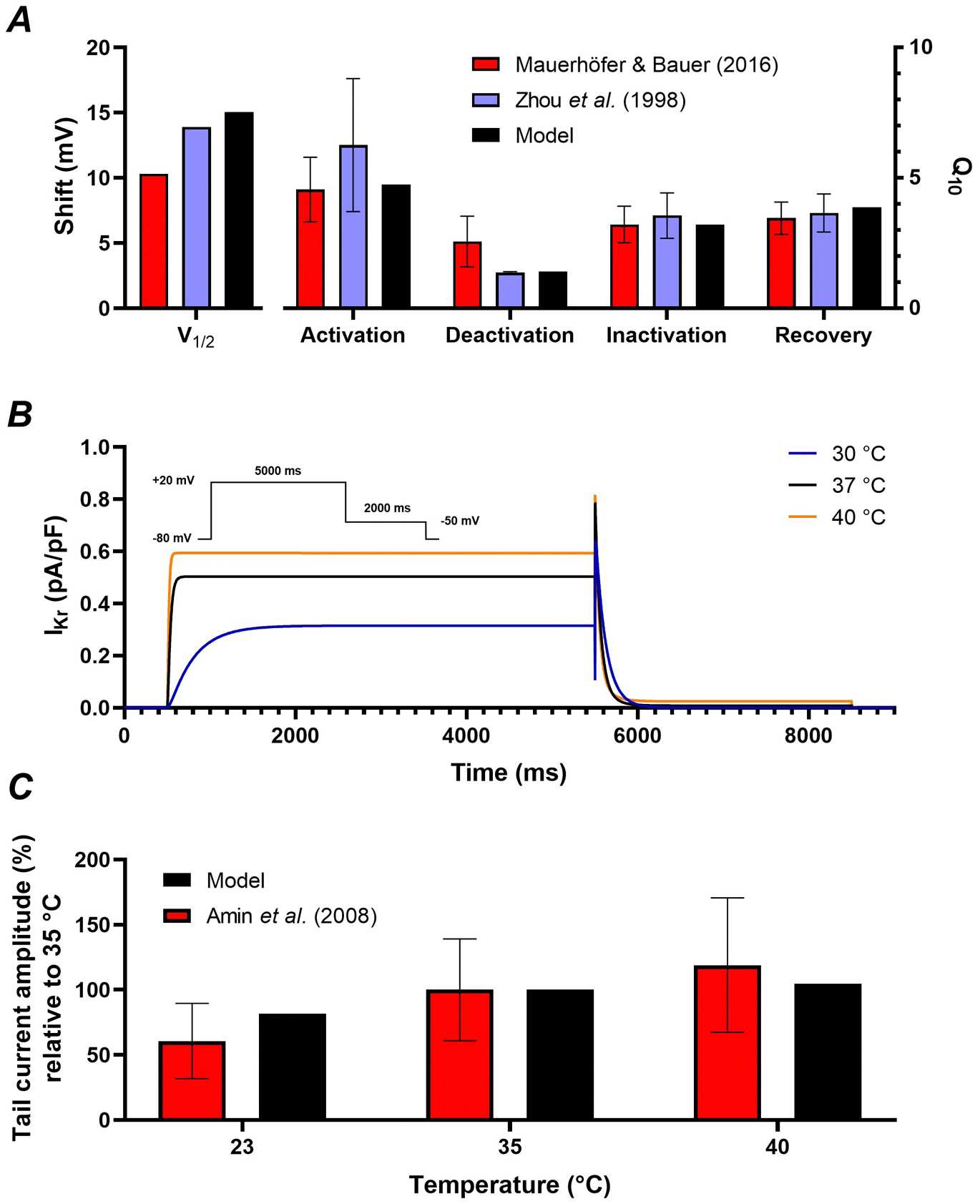
A, Calibration of the shift in midpoint of voltage dependence and Q10 values for activation, deactivation, inactivation and recovery of IKr in experimental recordings (Zhou et al., 1998; Mauerhofer & Bauer, 2016) and model. B, Combined effects of temperature-dependent changes in midpoint and Q10 on IKr at 30, 37, and 40 °C. Inset shows voltage-clamp protocol for steady-state and tail IKr. C, Relative tail current amplitudes at 23, 35 and 40 °C normalised to 35 °C obtained with the voltage-clamp protocol from panel B in experimental recordings (Amin et al., 2008) and model.
Figure 8. Temperature-dependent regulation of Kv11.1 gating and trafficking and its effect on ventricular cardiomyocyte repolarization.
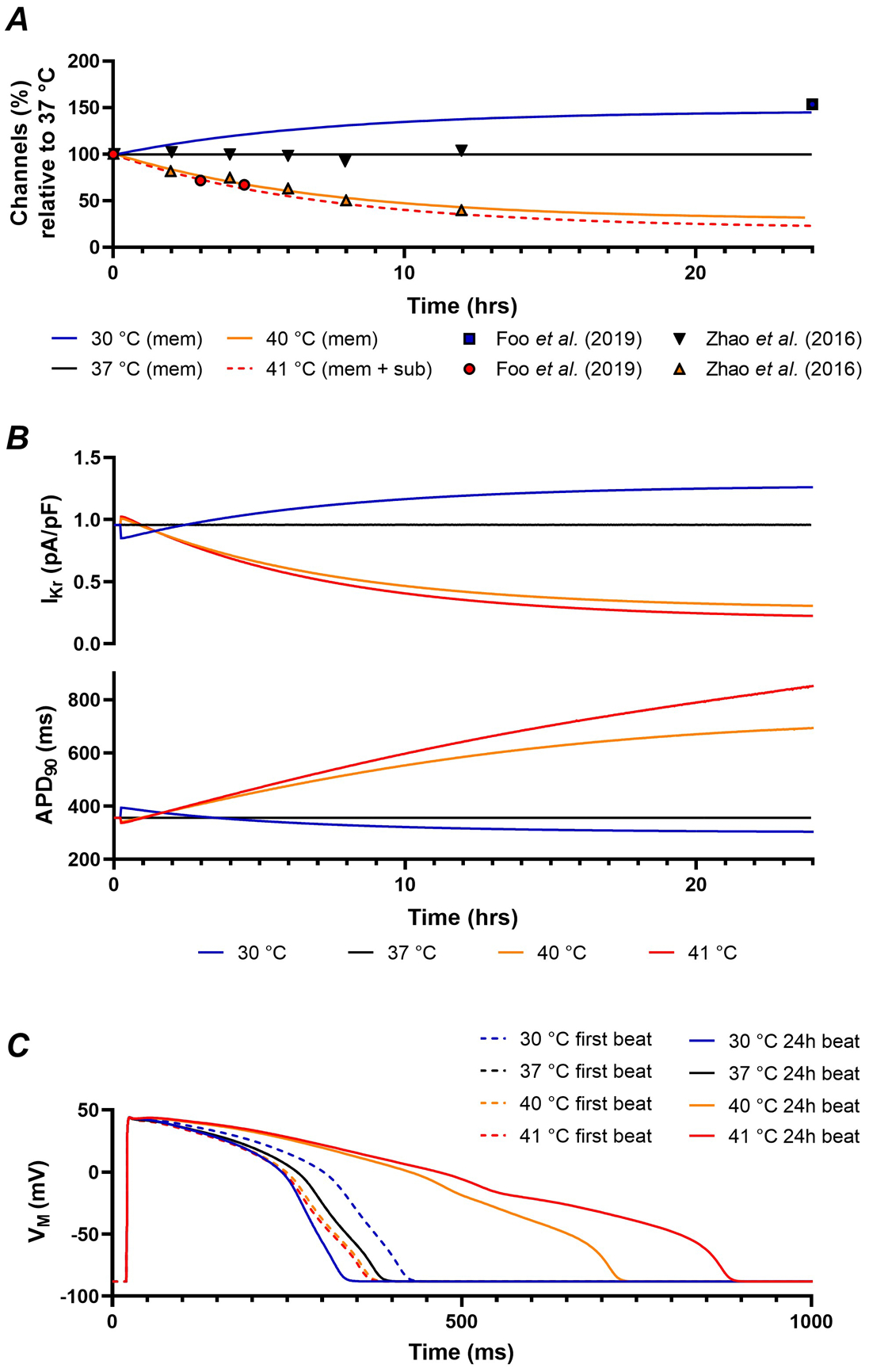
A, Time course of changes in Kv11.1 membrane or membrane and sub-membrane levels in response to temperature changes in experiments (symbols) (Zhao et al., 2016; Foo et al., 2019) and model (lines). B, Combined effect of temperature-dependent changes on Kv11.1 gating and trafficking. Higher temperatures acutely increase IKr due to faster gating, but after 24 hours IKr is decreased due to reduced membrane expression of Kv11.1 channels, producing significant prolongation of action potential (AP) duration. C, AP morphology for the first AP after a change in temperature (dashed lines) or after 24 hours (solid lines) at 30, 37, 40, and 41 °C, showing the opposing acute and long-term effects of temperature-dependent regulation of Kv11.1.
Modulation of Kv11.1 gating and trafficking by hypokalaemia
Extracellular [K+] is a prominent regulator of ventricular electrophysiology, with both hyper- and hypokalaemia being associated with increased risk of cardiac arrhythmias. Previously, hypokalaemia has been shown to negatively regulate Kv11.1 channel gating and membrane stability in a concentration-dependent manner through increased internalisation and degradation (Guo et al., 2009; Massaeli et al., 2010). The model’s extracellular [K+] dependence was calibrated to experimental data from Guo et al. (2009), which revealed a distinct half-maximal concentration after overnight (i.e., 12 hours) incubation compared to incubation for a week (Figure 9A, left vs. right panel). The rate of decrease in Kv11.1 membrane levels in the presence of low (0.1 mmol/L) extracellular [K+] and the rate of recovery of Kv11.1 membrane expression after switching back to 5.0 mmol/L extracellular [K+] following overnight incubation at 0.1 mmol/L were also calibrated based on experimental data (Figure 9B). The corresponding ‘overnight’ and ‘week’ parameter sets can be found in Table 5. Subsequently, we performed similar simulations to those in Figure 6 to evaluate the combined acute and long-term (trafficking) effects of hypokalaemia. After 24 hours, the [K+] was reduced from 5.4 mmol/L to 2.5 mmol/L, reflecting a clinically-relevant hypokalaemia. The ‘overnight’ parameter set resulted in an approximately 20% reduction in IKr, however, the amount of Kv11.1 membrane channels remained stable, reflecting the acute effects of hypokalaemia on channel gating over time (Figure 9C). For the ‘week’ parameter set, the reduction in IKr was much more pronounced (e.g., approximately 45%) due to an additional 25% reduction in Kv11.1 membrane channels (Figure 9C). This is also reflected in differences between APD prolongation immediately after extracellular [K+] was reduced to 2.5 mmol/L (25th hour; Figure 9D) and towards the end of the hypokalaemic period (47th hour). With the ‘overnight’ parameters, APD remained mostly stable after the first hour of hypokalaemia (Figure 9D, left panel). By contrast, the APD related to the ‘week’ parameters substantially increased during hypokalaemia (Figure 9D, right panel).
Figure 9. Modelling the effects of hypokalaemia.
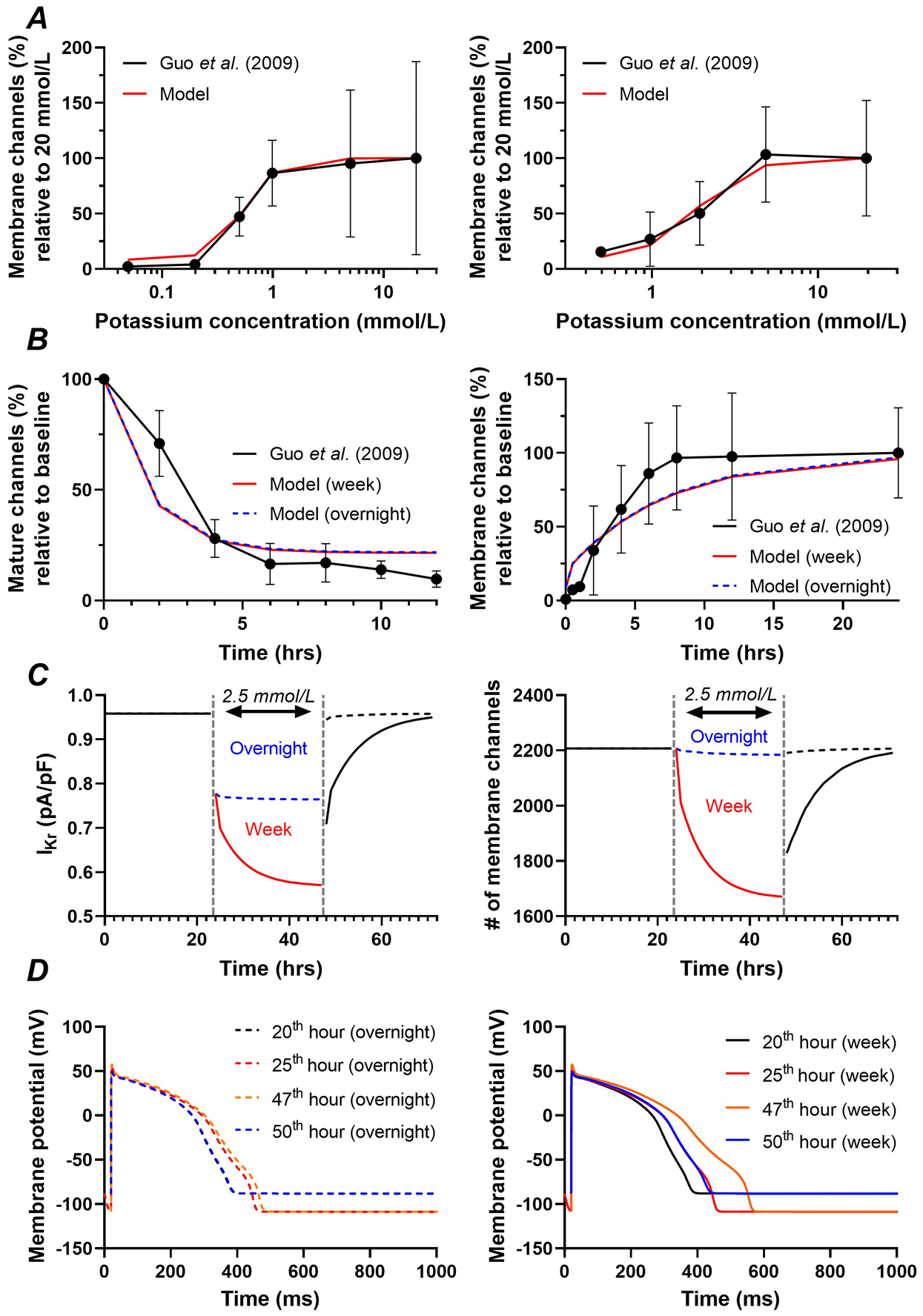
A, Concentration-dependence of extracellular [K+] on Kv11.1 membrane levels based on overnight incubation (i.e., 12 hours; left) or incubation for one week (right) in experimental data (Guo et al., 2009) (black line/symbols) and corresponding model versions (red lines). Experimental data were based on IKr recordings in 5 mmol/L [K+] after incubation at the indicated concentration for 12 hours or one week, which were used as a proxy for Kv11.1 membrane levels. B, Time course of reduction in Kv11.1 membrane levels in response to incubation in low (0 mmol/L in experiments, 0.1 mmol/L in model) extracellular [K+] (left) or recovery after 12 hours at low extracellular [K+] following re-exposure to 5 mmol/L extracellular [K+] (right) in experimental data (Guo et al., 2009) as well as ‘overnight’ and ‘week’ model configurations. C, Simulated time course of IKr (left) and Kv11.1 membrane levels (right) during 24 hours at baseline (i.e., 5.4 mmol/L [K+]), followed by 24 hours with hypokalaemia (2.5 mmol/L), and 24 hours at baseline, revealing acute inhibition for both the ‘overnight’ and ‘week’ model configurations, and long-term decrease of Kv11.1 membrane levels for the ‘week’ parameters. The dashed vertical lines (grey) indicate the start and end of hypokalaemia. D, Action-potential morphology at various time points from the simulations in panel C for the ‘overnight’ parameters (dashed lines) and ‘week’ parameters (solid lines), showing the acute prolongation of repolarization duration for both parameter sets and subsequent additional APD prolongation for the ‘week’ parameters, which remains present after cessation of hypokalaemia due to the decrease in Kv11.1 membrane levels (compare blue vs. black curves in right panel).
Discussion
Here, we developed a novel computational model of ion-channel trafficking that can reproduce a wide range of experimental data, including the temperature-, extracellular [K+]-, and/or drug-dependent regulation of Kv11.1 gating and trafficking. Our sensitivity analyses revealed that multiple mechanisms, all producing a similar steady-state decrease in Kv11.1 membrane levels, can underlie the phenotypic effects of LQTS2-associated missense mutations. Moreover, the model indicates that simulated application of dofetilide (in the presence of pentamidine), changes in temperature or hypokalaemia produce complex dynamic changes in IKr, and consequently APD, due to interactions between acute gating and long-term trafficking effects. These results underscore the importance of evaluating the time course of dynamic regulation of cardiac electrophysiology, rather than only studying acute or steady-state effects.
Computational modelling of ion-channel trafficking
The trafficking model was designed for Kv11.1 channels because the data on the temporal dynamics of ion-channel trafficking are most abundant for this channel. However, the molecular basis of ion-channel trafficking is highly complex, with numerous regulators that are crucial for normal trafficking (Blandin et al., 2021). These include, but are not limited to, the motifs involved in the ER-associated degradation system, the Ras-associated binding proteins, and the anchoring-, tethering-, and scaffolding proteins (Basheer & Shaw, 2016; Blandin et al., 2021). Moreover, native Kv11.1 channels are composed of two α-subunits (Kv11.1a and Kv11.1b) and channel trafficking depends on the exact subunit composition (Phartiyal et al., 2008). In particular Kv11.1b subunits are retained in the ER, unless they are co-assembled with Kv11.1a (Phartiyal et al., 2008).
Our model presents a simplified representation of this complex trafficking paradigm, where all processes taking place at the nucleus, ER, and GC are lumped together in a single rate (ψ) and no other regulating proteins/processes of forward trafficking besides temperature, pentamidine, and dofetilide were considered. In this model structure, ψ reflects both channel production and the first part of forward trafficking (e.g., microtubule-mediated trafficking between ER and GC), so we cannot distinguish between the effects of modulators on these two components. Finally, we also ignored dynamics of subunit composition and channel assembly. Nevertheless, this simplified and partially phenomenological representation of the trafficking paradigm is able to reproduce the vast majority of the experimentally observed characteristics of Kv11.1 trafficking in a computationally efficient manner. In the future, our model could be extended by compartmentalizing the ψ rate into an ER and GC state, but this would require experimental data on the trafficking dynamics between these compartments, which, to the best of our knowledge, are currently not available for Kv11.1.
Of note, the underlying structure of this trafficking model can be applied more broadly, given that most channels rely on a similar trafficking paradigm. For example, Ghosh et al. (2018) and Li et al. (2022) showed that L-type Ca2+ channels and Kir2.1 channels also dynamically circulate between the cytoplasm and plasma membrane. Both studies also highlighted the importance of the cytoskeleton in trafficking, where disruption of actin and tubulin was associated with impaired trafficking of both Kir2.1 and L-type Ca2+ channels, while inhibition of dynamin motor-proteins resulted in reduced Kir2.1 channel internalisation. Similarly, small-conductance Ca2+-activated K+-channels display highly dynamic trafficking behaviour (Heijman & Dobrev, 2017), which is partially regulated by atrial rate (Ozgen et al., 2007) and dependent on cytoskeletal proteins (Rafizadeh et al., 2014; Zhang et al., 2017), adding another layer of complexity to the trafficking paradigm that could be incorporated in future versions of the model when quantitative data on the impact on channel trafficking are available. The integration of trafficking models for multiple ion channels would also make it possible to model reciprocities due to α-α subunit interactions between channels. For example, Kv11.1 channel trafficking and functioning can be modulated by Kv7.1 (KCNQ1), the α-subunit of the slow-activating delayed-rectifier K+ current (IKs) (Ehrlich et al., 2004; Guo et al., 2011), whereas Nav1.5 expression reduces Kir2.1 internalisation in rodents (Milstein et al., 2012).
Potential limitations
Experimental data on ion channel trafficking are relatively scarce and the data that are available were obtained with different methodologies, under varying conditions, and often in distinct heterologous expression systems (e.g., HEK-293, HeLa, and H9C2 cells). Despite these sources of heterogeneity, the model was consistent with the vast majority of data, suggesting that the temporal dynamics are in the same order of magnitude regardless of cell type and detection method. The only notable exception for Kv11.1 was the high-throughput flow cytometry data from Kanner et al. (2018), where the half-time for both forward trafficking and internalisation were in the order of minutes rather than hours. The field of ion-channel trafficking is evolving rapidly and novel methodologies with higher spatial and temporal resolution may cause reconsideration of the current conceptual framework (Ghosh et al., 2018; Kanner et al., 2018; Li et al., 2022), warranting updates to the model. Moreover, the distinct acute and long-term effects identified in the present study would likely still apply even if future experiments would show faster time courses, just for different time points (e.g., 1 vs. 12 hours could be 10 vs. 60 minutes). The ‘long-term’ effects of trafficking modulators would then be observable after a couple of hours instead of 24–48 hours. However, at present our model provides a parsimonious representation of the current understanding of Kv11.1 trafficking.
Despite the model’s simplicity, our sensitivity analyses (Figures 3 and 4) revealed that similar phenotypic behaviour of LQTS2 mutations can be obtained through markedly different parameter combinations, even for a mutation (p.(A57P)) for which experimental data indicate that only forward trafficking is impaired. Mutations with more complex phenotypes (Kanner et al., 2018) are even less likely to provide a unique parameter set. It cannot be excluded that these different parameter sets show distinct behaviour under other (patho)physiological conditions, or a distinct response to interventions. This aspect could be assessed in more detail through a population-of-models approach, which would make it possible to assess the likelihood of outcomes such as the formation of afterdepolarizations, rather than considering only a binary (yes/no) result (Sobie, 2009; Ni et al., 2018; Heijman et al., 2021).
In general, the model was able to closely mimic experimental data, with only minor deviations from the experimentally observed range in Figures 2B, 5B, and 9B. In particular, the experimental data from Dennis et al. (2011) in Figure 2B showed approximately 60% Kv11.1 recycling within 3 minutes after 30 minutes of experimental channel internalisation. Thereafter, the amount of channel recycling remains stable. Our model shows a more sigmoidal increase in channel recycling, with the model recycling rate falling within the experimental standard deviation after 10 minutes. This difference might lead to an underestimate of the short-term (occurring within 5 minutes) effects of modulators of channel recycling and should be taken into consideration when interpreting our findings. The model also slightly underestimated the effect of dofetilide on Kv11.1 trafficking at low doses (Figure 5B), since we emphasised the 1 μmol/L concentration, which is what is primarily used experimentally. As such, the effect of clinically relevant concentrations may have been slightly underestimated. In Figure 9B, the reduction in membrane Kv11.1 channels during hypokalaemia was slightly faster and less pronounced in the model compared to experiments, whereas, the recovery after hypokalaemia was a bit too slow, although it still was within the standard deviation of the experimental data. More complex models might be able to approximate these data even more closely, but the general agreement with numerous data sets is noteworthy given the relative simplicity of the model.
The effects of temperature, pentamidine, dofetilide, and hypokalaemia were implemented in a phenomenological way by scaling the production-, internalisation-, and degradation rates. There are many other known pharmacological modulators of Kv11.1 trafficking, including cardiac glycosides, tricyclic anti-depressants, and E-4031 (van der Heyden et al., 2008; Dennis et al., 2011; Apaja et al., 2013; de Git et al., 2013). Similarly, other (patho)physiological factors such as hyperglycaemia are known to modulate ion channel trafficking, which may contribute to their proarrhythmic risk (Shi et al., 2015).
Implications for cardiac arrhythmogenesis
Here, we focused on dofetilide as a modulator of Kv11.1 trafficking because of its clinical relevance in the treatment of cardiac arrhythmias, in particular atrial fibrillation. Previous experimental work has shown that high concentrations of dofetilide can rescue Kv11.1-trafficking deficiencies induced by pentamidine (Varkevisser et al., 2013). Similarly, our model only rescues channels when pentamidine is present, however, our results also suggest that the impact of dofetilide on trafficking is likely limited for clinically relevant concentrations, even in the presence of pentamidine. In general, drugs that rescue ion-channel trafficking primarily seem to have an effect during aberrant conditions (e.g., trafficking deficient mutations, trafficking blockers), but may not be able to increase Kv11.1-trafficking under physiological conditions (Wible et al., 2005; Anderson et al., 2006; Varkevisser et al., 2013). However, a recent study has shown an approximately 60% increase in WT Kv11.1 levels after 24 hours of incubation with 5 μmol/L E-4031 (Al-Moubarak et al., 2020) in the absence of pentamidine or other compounds impairing Kv11.1 trafficking. More research on the effects of trafficking rescuers under physiological conditions is therefore warranted. Moreover, distinct short- and long-term effects of dofetilide have been reported and were attributed to modulation of phosphoinositide 3-kinase signalling, promoting an increase in late Na+ current over hours (Yang et al., 2014). For other drugs, effects on Kv11.1 trafficking could manifest at physiological concentrations and may similarly lead to differences between acute and long-term behaviour. Alternatively, combined application of dofetilide and LUF7244, an allosteric Kv11.1 activator that counteracts the direct inhibitory effects of high concentrations of dofetilide, has been proposed as an approach to rescue impaired Kv11.1 trafficking without drug-induced proarrhythmia due to excessive IKr inhibition (Qile et al., 2020).
It is known that fever can be an important trigger of arrhythmias when a vulnerable substrate is present, e.g., in Brugada Syndrome (Adler et al., 2013; Roterberg et al., 2020) or for certain LQT2 mutations (Amin et al., 2008). Our model predicted extreme APD prolongation after 24 hours of simulated febrile temperatures (Figure 8C), which would be expected to promote arrhythmogenesis. By contrast, a study comparing ECG parameters in patients presenting with fever to the emergency department with a comparison ECG obtained within 30 days without fever suggested a shortening of QTc interval duration in the presence of fever (Drew et al., 2017). Conversely, long-term hypothermia decreased APD in our model, whereas clinical studies in patients with therapeutic hypothermia after cardiac arrest instead suggest QTc prolongation (Khan et al., 2010; Nishiyama et al., 2012). This inconsistency is likely due to the fact we only implemented the temperature-dependent regulation of Kv11.1 in the ORd model, while other channels also undergo dynamic temperature-dependent modulation of gating and trafficking. When sufficient quantitative data become available, these effects on other channels should also be implemented in the ORd to gain more realistic insights into the temperature-dependent effects on cardiac repolarization and arrhythmogenesis. In addition, fever is often associated with inflammation which itself has complex direct and indirect electrophysiological effects. For example, inflammatory mediators such as interleukin-1, interleukin-6, and tumour necrosis factor α have been shown to induce oxidative stress and proarrhythmic calcium-handling abnormalities, as well as affecting K+- and Ca2+-channel functioning (Lazzerini et al., 2015; Heijman et al., 2020; Dobrev et al., 2022). Similarly, therapeutic hypothermia is performed in severely ill patients and has numerous systemic effects, including the induction of hypokalaemia (Khan et al., 2010), which, as shown in Figure 9, may also dynamically modulate repolarization and arrhythmogenic risk. Thus, the presence of several other confounding factors during changes in body temperature preclude a direct comparison of our results with clinical data. As such, the goal of Figure 8 was not to show the macroscopic effects of fever, but rather the temporal effects of temperature on gating and trafficking, and their combined effect on APD.
Hypokalaemia affects several key repolarizing K+ channels and is a known risk factor for cardiac arrhythmogenesis (Pezhouman et al., 2015). Our simulations show an acute prolongation of APD and hyperpolarization of the resting membrane potential in response to hypokalaemia (Figure 9), in line with experimental data (Pezhouman et al., 2015). In addition, severe hypokalaemia may induce additional APD prolongation over time due to a decrease in Kv11.1 membrane levels. Whether this effect occurs at clinically relevant concentrations depends on the affinity of Kv11.1 trafficking for extracellular [K+]. Guo et al. (2009) identified a half-maximal effect on Kv11.1 internalisation of 0.5 mmol/L for 12-hour incubation and 2.1 mmol/L for 1-week incubation. Thus, while short periods of clinically relevant hypokalaemia are unlikely to affect Kv11,1 membrane levels, longer periods may reduce Kv11.1 levels, potentially contributing to excessive APD prolongation.
Our results highlight the complexity of the many factors affecting cardiac electrophysiology over timescales from milliseconds to hours, with several factors having opposing acute and long-term effects. As such, the acute evaluation of drug effects alone may not be sufficient for safety screening of compounds that will be administered over longer periods of time in clinical practice, as repolarization abnormalities may only show up after hours.
Conclusion
In conclusion, we presented a simple and computationally efficient mathematical framework of Kv11.1-channel trafficking that makes it possible to study medium- to long-term regulation of cardiac electrophysiology. This framework highlights the distinct effects of acute modulation of channel gating and long-term regulation of channel trafficking induced by temperature changes, pharmacological interventions, and hypokalaemia. It provides a foundation to integrate other modulators of ion-channel trafficking (e.g., hyperglycaemia and other drugs) and study trafficking-deficient mutations and/or interventions that rescue ion-channel trafficking, which may facilitate a better understanding of arrhythmogenic disorders. Finally, our model can be used to optimize experimental protocols which normally are challenging to perform over longer timescales (e.g., hours to days) and to generate new hypotheses about arrhythmia mechanisms.
Supplementary Material
Key points.
Kv11.1 channels underlying the rapidly-activating delayed-rectifier K+ current are important for ventricular repolarization and are continuously shuttled from the cytoplasm to the plasma membrane and back over minutes to hours.
Kv11.1 gating and trafficking are modulated by temperature, drugs, and extracellular K+ concentration but experimental characterization of their combined effects is challenging. Computer models may facilitate these analyses, but no currently available model incorporates ion-channel trafficking.
We introduce a new two state ion-channel trafficking model able to reproduce a wide range of experimental data, along with the effects of modulators of Kv11.1 channel functioning and trafficking.
The model reveals complex dynamic regulation of ventricular repolarization by temperature, extracellular K+ concentration, and dofetilide through opposing acute (millisecond) effects on Kv11.1 gating and long-term (hours) modulation of Kv11.1 trafficking.
This in-silico trafficking framework provides a tool to investigate the roles of acute and long-term processes on arrhythmia promotion and maintenance.
Funding
The authors’ work is supported by the Netherlands Organization for Scientific Research (NWO/ZonMW Vidi 09150171910029 to J.H.), the National Institutes of Health (R01HL136389, R01HL131517, R01HL089598, and R01HL163277 to D.D.), and the European Union (large-scale integrative project MEASTRIA, No. 965286 to D.D.), and by The Netherlands CardioVascular Research Initiative (CVON 2018-30 PREDICT2 to P.G.A.V.).
Biography

Stefan Meier obtained his Master’s degree in ‘Systems Biology’ at Maastricht University in 2021 and is currently pursuing a PhD in computational electro-cardiology at CARIM (Maastricht University) under the supervision of Dr. Jordi Heijman and Prof. Dr. Paul G.A. Volders. Previously, he primarily focused on heart failure with preserved ejection fraction, but he transitioned to cardiac electrophysiology and arrhythmias for his PhD. He is especially interested in the temporal dynamics of cardiac ion-channel trafficking and how to incorporate this in computer/mathematical models.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Data availability
The model code, optimization scripts, and data can be found online at: https://github.com/HeijmanLab.
References
- Adler A, Topaz G, Heller K, Zeltser D, Ohayon T, Rozovski U, Halkin A, Rosso R, Ben-Shachar S, Antzelevitch C & Viskin S. (2013). Fever-induced Brugada pattern: how common is it and what does it mean? Heart Rhythm 10, 1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Moubarak E, Zhang Y, Dempsey CE, Zhang H, Harmer SC & Hancox JC. (2020). Serine mutation of a conserved threonine in the hERG K(+) channel S6-pore region leads to loss-of-function through trafficking impairment. Biochem Biophys Res Commun 526, 1085–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MJ, Nichols DJ & Oliver SD. (2000). The pharmacokinetics and pharmacodynamics of oral dofetilide after twice daily and three times daily dosing. Br J Clin Pharmacol 50, 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin AS, Herfst LJ, Delisle BP, Klemens CA, Rook MB, Bezzina CR, Underkofler HA, Holzem KM, Ruijter JM, Tan HL, January CT & Wilde AA. (2008). Fever-induced QTc prolongation and ventricular arrhythmias in individuals with type 2 congenital long QT syndrome. J Clin Invest 118, 2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ & January CT. (2006). Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 113, 365–373. [DOI] [PubMed] [Google Scholar]
- Anderson CL, Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP & January CT. (2014). Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat Commun 5, 5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apaja PM, Foo B, Okiyoneda T, Valinsky WC, Barriere H, Atanasiu R, Ficker E, Lukacs GL & Shrier A. (2013). Ubiquitination-dependent quality control of hERG K+ channel with acquired and inherited conformational defect at the plasma membrane. Mol Biol Cell 24, 3787–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi Y, Nomura F, Abe Y, Doi M, Sakakura T, Takasuna K & Yasuda K. (2019). Electrophysiological evaluation of pentamidine and 17-AAG in human stem cell-derived cardiomyocytes for safety assessment. Eur J Pharmacol 842, 221–230. [DOI] [PubMed] [Google Scholar]
- Balse E & Boycott HE. (2017). Ion channel trafficking: control of ion channel density as a target for arrhythmias? Front Physiol 8, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basheer WA & Shaw RM. (2016). Connexin 43 and CaV1.2 ion channel trafficking in healthy and diseased myocardium. Circ Arrhythm Electrophysiol 9, e001357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandin CE, Gravez BJ, Hatem SN & Balse E. (2021). Remodeling of ion channel trafficking and cardiac arrhythmias. Cells 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE & Rudy Y. (2001). Cellular consequences of HERG mutations in the long QT syndrome: precursors to sudden cardiac death. Cardiovasc Res 50, 301–313. [DOI] [PubMed] [Google Scholar]
- Clerx M, Collins P, de Lange E & Volders PG. (2016). Myokit: a simple interface to cardiac cellular electrophysiology. Prog Biophys Mol Biol 120, 100–114. [DOI] [PubMed] [Google Scholar]
- de Git KC, de Boer TP, Vos MA & van der Heyden MA. (2013). Cardiac ion channel trafficking defects and drugs. Pharmacol Ther 139, 24–31. [DOI] [PubMed] [Google Scholar]
- Dennis AT, Nassal D, Deschenes I, Thomas D & Ficker E. (2011). Antidepressant-induced ubiquitination and degradation of the cardiac potassium channel hERG. J Biol Chem 286, 34413–34425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis AT, Wang L, Wan H, Nassal D, Deschenes I & Ficker E. (2012). Molecular determinants of pentamidine-induced hERG trafficking inhibition. Mol Pharmacol 81, 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D, Heijman J, Hiram R, Li N & Nattel S. (2022). Inflammatory signalling in atrial cardiomyocytes: a novel unifying principle in atrial fibrillation pathophysiology. Nat Rev Cardiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew D, Baranchuk A, Hopman W & Brison RJ. (2017). The impact of fever on corrected QT interval. J Electrocardiol 50, 570–575. [DOI] [PubMed] [Google Scholar]
- Dutta S, Chang KC, Beattie KA, Sheng J, Tran PN, Wu WW, Wu M, Strauss DG, Colatsky T & Li Z. (2017). Optimization of an in silico cardiac cell model for proarrhythmia risk assessment. Front Physiol 8, 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich JR, Pourrier M, Weerapura M, Ethier N, Marmabachi AM, Hebert TE & Nattel S. (2004). KvLQT1 modulates the distribution and biophysical properties of HERG. A novel alpha-subunit interaction between delayed rectifier currents. J Biol Chem 279, 1233–1241. [DOI] [PubMed] [Google Scholar]
- Foo B, Barbier C, Guo K, Vasantharuban J, Lukacs GL & Shrier A. (2019). Mutation-specific peripheral and ER quality control of hERG channel cell-surface expression. Scientific Reports 9, 6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Nieves-Cintron M, Tajada S, Brust-Mascher I, Horne MC, Hell JW, Dixon RE, Santana LF & Navedo MF. (2018). Dynamic L-type CaV1.2 channel trafficking facilitates CaV1.2 clustering and cooperative gating. Biochim Biophys Acta Mol Cell Res 1865, 1341–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N & Zhang S. (2009). Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest 119, 2745–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Wang T, Yang T, Xu J, Li W, Fridman MD, Fisher JT & Zhang S. (2011). Interaction between the cardiac rapidly (IKr) and slowly (IKs) activating delayed rectifier potassium channels revealed by low K+-induced hERG endocytic degradation. J Biol Chem 286, 34664–34674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J & Dobrev D. (2017). Inhibition of small-conductance Ca2+-activated K+ channels: the long-awaited breakthrough for antiarrhythmic drug therapy of atrial fibrillation? Circ Arrhythm Electrophysiol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J, Erfanian Abdoust P, Voigt N, Nattel S & Dobrev D. (2016). Computational models of atrial cellular electrophysiology and calcium handling, and their role in atrial fibrillation. J Physiol 594, 537–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, Wang Q, Abu-Taha IH, Gorka M, Kunzel S, El-Armouche A, Reichenspurner H, Kamler M, Nikolaev V, Ravens U, Li N, Nattel S, Wehrens XHT & Dobrev D. (2020). Atrial myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation. Circ Res 127, 1036–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J, Sutanto H, Crijns H, Nattel S & Trayanova NA. (2021). Computational models of atrial fibrillation: achievements, challenges, and perspectives for improving clinical care. Cardiovasc Res 117, 1682–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J, Voigt N, Carlsson LG & Dobrev D. (2014). Cardiac safety assays. Curr Opin Pharmacol 15, 16–21. [DOI] [PubMed] [Google Scholar]
- Heijman J, Zaza A, Johnson DM, Rudy Y, Peeters RL, Volders PG & Westra RL. (2013). Determinants of beat-to-beat variability of repolarization duration in the canine ventricular myocyte: a computational analysis. PLoS Comput Biol 9, e1003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner SA, Jain A & Colecraft HM. (2018). Development of a high-throughput flow cytometry assay to monitor defective trafficking and rescue of long QT2 mutant hERG channels. Front Physiol 9, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AA & Ackerman MJ. (2009). Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 6, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Ng CA, Hunter MJ, Mann SA, Heide J, Hill AP & Vandenberg JI. (2013). Trafficking defects in PAS domain mutant Kv11.1 channels: roles of reduced domain stability and altered domain-domain interactions. Biochem J 454, 69–77. [DOI] [PubMed] [Google Scholar]
- Khan JN, Prasad N & Glancy JM. (2010). QTc prolongation during therapeutic hypothermia: are we giving it the attention it deserves? Europace 12, 266–270. [DOI] [PubMed] [Google Scholar]
- Lazzerini PE, Capecchi PL & Laghi-Pasini F. (2015). Long QT syndrome: an emerging role for inflammation and immunity. Front Cardiovasc Med 2, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Loen V, van Ham WB, Kool W, van der Heyden MAG & Takanari H. (2022). Quantitative analysis of the cytoskeleton's role in inward rectifier KIR2.1 forward and backward trafficking. Front Physiol 12, 812572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Mirams GR, Yoshinaga T, Ridder BJ, Han X, Chen JE, Stockbridge NL, Wisialowski TA, Damiano B, Severi S, Morissette P, Kowey PR, Holbrook M, Smith G, Rasmusson RL, Liu M, Song Z, Qu Z, Leishman DJ, Steidl-Nichols J, Rodriguez B, Bueno-Orovio A, Zhou X, Passini E, Edwards AG, Morotti S, Ni H, Grandi E, Clancy CE, Vandenberg J, Hill A, Nakamura M, Singer T, Polonchuk L, Greiter-Wilke A, Wang K, Nave S, Fullerton A, Sobie EA, Paci M, Musuamba Tshinanu F & Strauss DG. (2020). General principles for the validation of proarrhythmia risk prediction models: an extension of the CiPA in silico strategy. Clin Pharmacol Ther 107, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaeli H, Guo J, Xu J & Zhang S. (2010). Extracellular K+ is a prerequisite for the function and plasma membrane stability of HERG channels. Circ Res 106, 1072–1082. [DOI] [PubMed] [Google Scholar]
- Mauerhofer M & Bauer CK. (2016). Effects of temperature on heteromeric Kv11.1a/1b and Kv11.3 channels. Biophys J 111, 504–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR & Jalife J. (2012). Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 109, E2134–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H, Morotti S & Grandi E. (2018). A heart for diversity: simulating variability in cardiac arrhythmia research. Front Physiol 9, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama N, Sato T, Aizawa Y, Nakagawa S & Kanki H. (2012). Extreme QT prolongation during therapeutic hypothermia after cardiac arrest due to long QT syndrome. Am J Emerg Med 30, 638 e635–638. [DOI] [PubMed] [Google Scholar]
- O'Hara T, Virag L, Varro A & Rudy Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7, e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterbur Badhey ML, Bertalovitz AC & McDonald TV. (2017). Express with caution: epitope tags and cDNA variants effects on hERG channel trafficking, half-life and function. J Cardiovasc Electrophysiol 28, 1070–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgen N, Dun W, Sosunov EA, Anyukhovsky EP, Hirose M, Duffy HS, Boyden PA & Rosen MR. (2007). Early electrical remodeling in rabbit pulmonary vein results from trafficking of intracellular SK2 channels to membrane sites. Cardiovasc Res 75, 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezhouman A, Singh N, Song Z, Nivala M, Eskandari A, Cao H, Bapat A, Ko CY, Nguyen T, Qu Z, Karagueuzian HS & Weiss JN. (2015). Molecular basis of hypokalemia-induced ventricular fibrillation. Circulation 132, 1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phartiyal P, Sale H, Jones EM & Robertson GA. (2008). Endoplasmic reticulum retention and rescue by heteromeric assembly regulate human ERG 1a/1b surface channel composition. J Biol Chem 283, 3702–3707. [DOI] [PubMed] [Google Scholar]
- Qile M, Ji Y, Golden TD, Houtman MJC, Romunde F, Fransen D, van Ham WB, AP IJ, January CT, Heitman LH, Stary-Weinzinger A, Delisle BP & van der Heyden MAG. (2020). LUF7244 plus dofetilide rescues aberrant Kv11.1 trafficking and produces functional IKv11.1. Mol Pharmacol 97, 355–364. [DOI] [PubMed] [Google Scholar]
- Rafizadeh S, Zhang Z, Woltz RL, Kim HJ, Myers RE, Lu L, Tuteja D, Singapuri A, Bigdeli AA, Harchache SB, Knowlton AA, Yarov-Yarovoy V, Yamoah EN & Chiamvimonvat N. (2014). Functional interaction with filamin A and intracellular Ca2+ enhance the surface membrane expression of a small-conductance Ca2+-activated K+ (SK2) channel. Proc Natl Acad Sci U S A 111, 9989–9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roterberg G, El-Battrawy I, Veith M, Liebe V, Ansari U, Lang S, Zhou X, Akin I & Borggrefe M. (2020). Arrhythmic events in Brugada syndrome patients induced by fever. Ann Noninvasive Electrocardiol 25, e12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YQ, Yan M, Liu LR, Zhang X, Wang X, Geng HZ, Zhao X & Li BX. (2015). High glucose represses hERG K+ channel expression through trafficking inhibition. Cell Physiol Biochem 37, 284–296. [DOI] [PubMed] [Google Scholar]
- Smith JL, Anderson CL, Burgess DE, Elayi CS, January CT & Delisle BP. (2016). Molecular pathogenesis of long QT syndrome type 2. J Arrhythm 32, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JL, Reloj AR, Nataraj PS, Bartos DC, Schroder EA, Moss AJ, Ohno S, Horie M, Anderson CL, January CT & Delisle BP. (2013). Pharmacological correction of long QT-linked mutations in KCNH2 (hERG) increases the trafficking of Kv11.1 channels stored in the transitional endoplasmic reticulum. Am J Physiol Cell Physiol 305, C919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobie EA. (2009). Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys J 96, 1264–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutanto H, Laudy L, Clerx M, Dobrev D, Crijns H & Heijman J. (2019). Maastricht antiarrhythmic drug evaluator (MANTA): a computational tool for better understanding of antiarrhythmic drugs. Pharmacol Res 148, 104444. [DOI] [PubMed] [Google Scholar]
- van der Heyden MA, Smits ME & Vos MA. (2008). Drugs and trafficking of ion channels: a new pro-arrhythmic threat on the horizon? Br J Pharmacol 153, 406–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heyden MAG, Delisle BP & Abriel H. (2018). Editorial: Ion channel trafficking and cardiac arrhythmias. Front Physiol 9, 1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg JI, Varghese A, Lu Y, Bursill JA, Mahaut-Smith MP & Huang CL. (2006). Temperature dependence of human ether-a-go-go-related gene K+ currents. Am J Physiol Cell Physiol 291, C165–175. [DOI] [PubMed] [Google Scholar]
- Varkevisser R, Houtman MJ, Linder T, de Git KC, Beekman HD, Tidwell RR, Ijzerman AP, Stary-Weinzinger A, Vos MA & van der Heyden MA. (2013). Structure-activity relationships of pentamidine-affected ion channel trafficking and dofetilide mediated rescue. Br J Pharmacol 169, 1322–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace E, Howard L, Liu M, O'Brien T, Ward D, Shen S & Prendiville T. (2019). Long QT syndrome: genetics and future perspective. Pediatr Cardiol 40, 1419–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wible BA, Hawryluk P, Ficker E, Kuryshev YA, Kirsch G & Brown AM. (2005). HERG-Lite: a novel comprehensive high-throughput screen for drug-induced hERG risk. J Pharmacol Toxicol Methods 52, 136–145. [DOI] [PubMed] [Google Scholar]
- Yang T, Chun YW, Stroud DM, Mosley JD, Knollmann BC, Hong C & Roden DM. (2014). Screening for acute IKr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation 130, 224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ledford HA, Park S, Wang W, Rafizadeh S, Kim HJ, Xu W, Lu L, Lau VC, Knowlton AA, Zhang XD, Yamoah EN & Chiamvimonvat N. (2017). Distinct subcellular mechanisms for the enhancement of the surface membrane expression of SK2 channel by its interacting proteins, alpha-actinin2 and filamin A. J Physiol 595, 2271–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang T, Guo J, Yang T, Li W, Koichopolos J, Lamothe SM, Kang Y, Ma A & Zhang S. (2016). Febrile temperature facilitates hERG/IKr degradation through an altered K(+) dependence. Heart Rhythm 13, 2004–2011. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA & January CT. (1998). Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J 74, 230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The model code, optimization scripts, and data can be found online at: https://github.com/HeijmanLab.