

Summary
Dysregulated host-microbial interactions play critical roles in initiation and perpetuation of gut inflammation in Crohn’s disease (CD). However, the spatial distribution and interaction network across the intestine and its accessory tissues are still elusive. Here, we profile the host proteins and tissue microbes in 540 samples from the intestinal mucosa, submucosa-muscularis-serosa, mesenteric adipose tissues, mesentery, and mesenteric lymph nodes of 30 CD patients and spatially decipher the host-microbial interactions. We observe aberrant antimicrobial immunity and metabolic processes across multi-tissues during CD and determine bacterial transmission along with altered microbial communities and ecological patterns. Moreover, we identify several candidate interaction pairs between host proteins and microbes associated with perpetuation of gut inflammation and bacterial transmigration across multi-tissues in CD. Signature alterations in host proteins (e.g., SAA2 and GOLM1) and microbes (e.g., Alistipes and Streptococcus) are further imprinted in serum and fecal samples as potential diagnostic biomarkers, thus providing a rationale for precision diagnosis.
Keywords: Crohn disease, proteome, microbiome, interaction pattern, spatial characteristics
Graphical abstract
Highlights
-
•
Spatial omics reveals host-microbe interactions across multiple tissues in CD
-
•
Dysregulated antimicrobial immunity and metabolism occur in multiple tissues during CD
-
•
Microbial transmission is identified in the intestine and its accessory tissues
-
•
A panel of host proteins and microbes highlights the non-invasive diagnostic potential
Gao et al. delineate the spatial characteristics of host proteins and gut microbiota and their interactions via integrative multi-omics analysis of the intestine and its accessory tissues from patients with Crohn’s disease. This study provides an in-depth understanding of the pathogenesis and diagnostic potential of signature proteins and microbes.
Introduction
Crohn disease (CD), a type of inflammatory bowel disease (IBD), is a chronic intestinal disorder involving any part of the gastrointestinal tract and is characterized by transmural inflammation,1 globally afflicting more than 3.5 million individuals.2 Multiple factors, including genetic susceptibility, immune dysregulation, and microbiota disturbance, have been considered to be drivers of CD.3,4 However, the etiology and pathophysiology of CD are still not fully understood.
It is generally considered that inflammatory lesions are initially induced in the intestinal mucosa (MUC) and that transmural inflammation enables to involve in the submucosa-muscularis-serosa (SMS) of CD patients. Accumulated evidence reveals that intestinal accessory tissues, such as mesenteric adipose tissue (MAT), mesentery (MES), and mesenteric lymph nodes (MLNs), are closely associated with the clinical course and pathophysiology of CD.5,6 Under steady-state conditions, MES comprises various cells, including adipocytes, fibrocytes, immune cells, and a vast network of blood vessels and lymphatic vessels.7 However, CD MES is characterized by mesenteric hypervascularity and edema, increased fat density, fibrofatty proliferation, mesenteric lymphadenopathy, connective tissue thickening, and densely infiltrating inflammatory cells and hyperplastic adipocytes. Chronic inflammation in the adjacent intestine significantly correlates with fat wrapping.7,8 Inflamed MAT (iMAT) wrapping the involved intestinal segments and encroaching into the adjacent intestine is also called “creeping fat” (CrF), a macroscopical phenomenon of CD and an important source of proinflammatory cytokines.8,9 Consistently, postoperative recurrence and reoperation incidence is significantly lower in CD patients who undergo partial MES resection concurrently.7 However, a recent study has illustrated that the specific translocation of the bacterial consortium in MAT promotes formation of CrF, which prevents systemic bacterial translocation and limits the perpetuation of gut inflammation.10 Collectively, the dichotomous roles of intestinal accessory tissues appear in the development of CD beneficially11 or harmfully.8 Therefore, it is crucial to elucidate the exact roles of the intestine and its accessory tissues in treating and managing CD.
The gut microbiome is classically an appreciated factor involved in the pathogenesis of CD.10,12 Hazardous conditions, such as intestinal inflammation, transmural inflammation,13 and an impaired gut vascular barrier,14,15 facilitate bacterial transmission into the intestine and extraintestinal tissues and further promote the initiation and perpetuation of intestinal inflammation, but the underlying mechanisms regulating bacterial transmission as well as intestinal inflammation remain elusive. Recently, large-scale genome-wide association studies (GWASs) have identified more than 240 IBD susceptibility alleles implicated in bacterial sensing and innate immunity, especially for CD-specific susceptibility genes.16,17 Multi-omics analysis, including single-cell RNA sequencing and metagenomics, also reveals important roles of the gut microbiota in the pathogenesis of CD.18,19 However, the transcriptome cannot fully dissect the functional interactions between the host and gut microbiota during CD.20 Hence, attention has shifted mainly toward proteomics profiling, and several studies have provided protein-level insights into the pathogenesis of CD21,22 and identified a variety of protein biomarkers in serum, stool, and mucosal biopsies from IBD patients.23,24 However, it is still largely unknown how the host proteome interacts with the gut microbiome across the intestine and extraintestinal tissues; shedding light on this would allow us to better understand the underlying mechanisms involved in bacterial transmission during CD.25,26
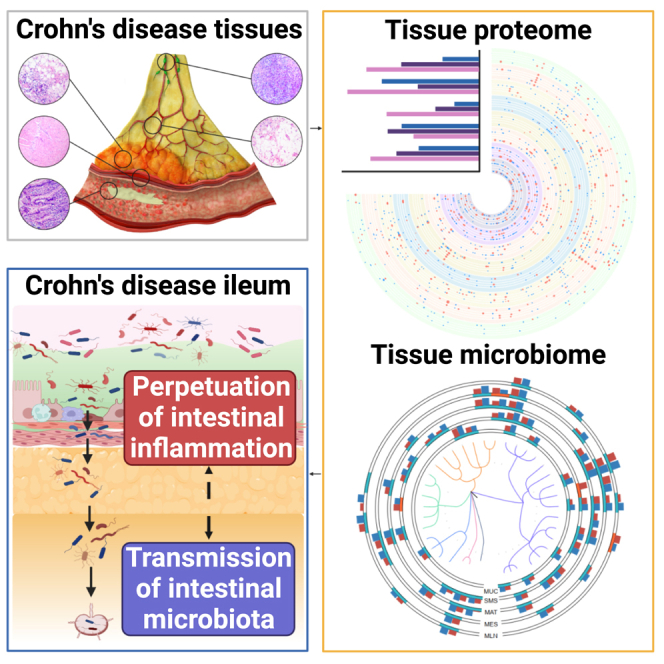
Here, we collected 540 tissue samples from 30 CD patients and 50 tissue samples from 5 non-CD controls (NCs) for paired tissue proteome and microbiome, including MUC, SMS, MAT, MES, and MLN specimens. We mapped the spatial distribution of proteins and gut microbiota and deciphered the interactions between the host proteome and gut microbiome across different tissues involved in the pathogenesis of the disease. Additionally, we also observed that a distinct alteration in the host proteome (e.g., SAA2, GOLM1, IGFBP2, GBP1, and DPP4) and gut microbiome (e.g., Alistipes, Dialister, and Streptococcus) could be imprinted in serum and fecal samples from another cohort of 30 patients with active CD, thus providing a rationale of omics datasets for precision diagnosis and an in-depth understanding of the pathogenesis.
Results
To investigate the characteristics of the host proteins and gut microbes in multi-tissues from CD patients, we collected 540 surgical resection samples from 30 CD patients for paired proteome and microbiome, respectively, including inflamed terminal ileal MUC (iMUC) and adjacent uninflamed terminal ileal MUC (uMUC), inflamed ileal SMS (iSMS) and adjacent uninflamed ileal SMS (uSMS), iMAT and adjacent uninflamed MAT (uMAT), inflamed MES (iMES) and adjacent uninflamed MES (uMES), as well as MLN (Figure 1A; STAR Methods). Additionally, 50 normal tissue samples analogous to the corresponding sites (including MUC, SMS, MAT, MES, and MLN) were collected from five NCs for paired proteome and microbiome, respectively, as described above. Details regarding tissue sample collection and information of patients, including age, gender, clinical characteristics, and medications, are shown in STAR Methods and Table S1. Paired tissue proteomes and microbiomes were performed on multi-regional tissue samples from 30 CD patients and 5 NCs (Figure 1A). A total of 10,135 unique proteins were quantified with a false discovery rate (FDR) of less than 1% (Figure S1A; Table S2), and the microbiome compositions were mapped across our cohort by 16S rRNA gene amplicon sequencing, yielding 32,330 ± 17,916 reads (Figure 1A).
Figure 1.

The strategy for collecting multi-tissues and the general design of this study
(A) An overview of the experimental design and sample collection of the proteome and microbiome. The schematic presents the sample collection strategy of inflamed and adjacent uninflamed tissues from CD patients. iMUC, inflamed mucosa (H&E, ×100); uMUC, adjacent uninflamed MUC (H&E, ×100); iSMS, inflamed submucosa-muscularis-serosa (H&E, ×100); uSMS, adjacent uninflamed SMS (H&E, ×100); iMAT, inflamed mesenteric adipose tissue (i.e., CrF) (H&E, ×200); uMAT, adjacent uninflamed MAT (H&E, ×200); iMES, inflamed mesentery (H&E, ×200); uMES, adjacent uninflamed MES (H&E, ×200); MLN, mesenteric lymph node (H&E, ×100).
(B) Quantified and dysregulated proteins across multiple tissues. The outermost labels represent the different types of tissues (i.e., MUC, SMS, MAT, MES, and MLN). The number of samples from specific tissue is indicated in the outmost or first sector (gray). The second sector (green) refers to undetected proteins in a specific tissue. The third sector (light red) refers to upregulated proteins in a specific tissue. The fourth sector (light blue) refers to downregulated proteins in a specific tissue (B-H-adjusted p < 0.05; |log2[FC of CD versus non-CD]| > log2[1.2]), and the fifth or innermost sector (dark blue) refers to unregulated proteins in a specific tissue.
To investigate whether the distinct signature alterations in the proteome and microbiome obtained from multi-tissues of CD patients could be imprinted in serum and feces from CD patients, we collected paired serum and fecal samples from an independent cohort of 30 active CD patients and 30 healthy donors for TMTpro-based untargeted proteomics and 16S rRNA sequencing analysis, respectively (Figure 1A; STAR Methods).
The landscape of spatial protein characteristics across different tissues of CD patients
To obtain a global view of spatial profiles of host proteins during CD, we leveraged proteomics analysis on multi-tissues from CD patients, including inflamed and adjacent uninflamed ileal MUC, SMS, MAT, MES, and MLN. T-distributed stochastic neighbor embedding (t-SNE) analysis found no apparent technical bias from different batches or mass spectrometry equipment (Figure S1B), showing the high quality of the proteomic data. t-SNE analysis also revealed similar proteomes between inflamed and adjacent uninflamed tissues of CD patients (Figure S1B). In addition, the differential analysis also observed no significant differences in proteomes between inflamed and corresponding uninflamed MUC, SMS, MAT, and MES of CD patients, in which proteins with log2(fold change [FC]) beyond 0.25 or below −0.25 as well as an adjusted p value of 0.05 or less were considered to be significantly differentially expressed (Figure S1C). Hence, the comparisons were assessed between inflamed tissues from CD patients and normal tissues from NCs after that.
A total of 2,817 dysregulated proteins were identified from five types of tissues (i.e., MUC, SMS, MAT, MES, and MLN) between CD patients and NCs (Figure 1B and Table S2). Of them, only seven proteins were observed to be altered in all five types of inflamed tissues with different expression patterns, including serum amyloid A2 (SAA2), arginase 2 (ARG2), D-aminoacyl-tRNA deacylase 1 (DTD1), Fas apoptotic inhibitory molecule 1 (FAIM), heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1), keratin 15 (KRT15), and POTE ankyrin domain family member K, pseudogene (POTEKP) (Figure S2B).
According to the known pathophysiology of CD,1 we mapped all differentially expressed proteins (Figure S2A) into eight functional clusters, including IBD susceptibility gene-encoded proteins, antimicrobial immunity-associated proteins, innate immunity-associated proteins, adaptive immunity-associated proteins, immune exhaustion-associated proteins, cytokines, cellular metabolism-associated proteins, and fibrosis-associated proteins (Figure 2A). More details of eight cluster-related proteins are described in Table S3.
Figure 2.

Functional clusters of dysregulated proteins related to the pathogenesis of CD across multi-tissues
(A) The landscape of dysregulated proteins in multiple tissues of CD patients. Counts of dysregulated proteins in 8 clusters of protein molecules are shown in a bar chart, including IBD and other autoimmune disease-associated susceptibility gene-encoded proteins, antimicrobial immunity-associated proteins, innate immunity-associated proteins, adaptive immunity-associated proteins, immune exhaustion-associated proteins, cytokines, metabolism-associated proteins, and fibrosis-associated proteins. The horizontal columns represent the number of proteins in different functional clusters. The dysregulated proteins in 8 clusters are indicated as circles (red spots, upregulated proteins; blue spots, downregulated proteins). The size of the spots indicates absolute log2(FC).
(B) The chord diagram shows dysregulated proteins encoded by IBD and autoimmune disease-associated susceptibility genes in different tissues between CD patients and NCs. The length of the outermost brick representing each protein corresponds to the sum of |log2(FC)| in different types of tissues, and the length of the outermost brick representing each type of tissue corresponds to the sum of |log2(FC)| in one or more proteins.
(C) Protein expression of ATG16L1 and GSDMB across different tissues. The y axis indicates protein abundance. A pairwise comparison of each protein between inflamed tissues of CD patients and normal tissues of NCs was performed using a two-sided unpaired Welch’s t test. B-H-adjusted p values: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Because genetic susceptibility is indispensable for development of CD, we first identified 40 proteins associated with IBD susceptibility and 62 proteins related to other autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis (Figure 2B). Among them, eight IBD susceptibility gene-encoded proteins were differentially expressed in at least two types of inflamed tissues from CD patients (Figure 2B). ATG16L1, a canonical autophagy-regulating protein, was downregulated in iMUC, iMAT, iMES, and MLN of CD patients (Figure 2C), suggesting an impairment of autophagy-mediated microbial clearance.27 The pore-forming cytolysin gasdermin B (GSDMB), an executioner of inflammatory cell death programs that restricts intracellular bacterial growth mediated by natural killer (NK) cells,28 was upregulated in iSMS but downregulated in MLN of CD patients (Figure 2C). These results suggest that dysregulated host defense exists across the intestine and its accessory tissues during CD and is related to genetic susceptibility.
To gain an in-depth understanding of the functional changes in the host proteome, we took advantage of ingenuity pathway analysis (IPA) to illustrate dysregulated proteins in each tissue site. We found that the urea cycle pathway was enriched in all five types of inflamed tissues (Figure S2C) and that several pathways associated with tryptophan metabolism were enriched in different inflamed tissues,29 such as xenobiotic metabolism signaling and aryl hydrocarbon receptor (AHR) signaling (Figure S2C). Moreover, we also observed an enrichment of immunity-associated pathways in different inflamed tissues with distinct functional alterations. For example, B cell receptor signaling was suppressed in iMUC but significantly activated in iSMS of CD patients (Figure S2C).
Compromised antimicrobial immunity across different tissues of CD patients
Given the fundamental importance of antimicrobial immunity in the maintenance of intestinal homeostasis, we identified 100 signature dysregulated proteins related to antimicrobial immunity based on the ImmPort database,30 25 of which were shared in at least two types of inflamed tissues from CD patients (Figure S3A). Multiple immune response processes related to antimicrobial immunity were particularly compromised during CD, including pathogen invasion, immune cell migration, antigen recognition and presentation, and pathogen-killing function (Figure 3A). A large number of proteins related to host antimicrobial immunity were found to be downregulated in different types of tissues during CD (Figure 3A). For example, C-X-C motif chemokine ligand 12 (CXCL12), an essential chemokine related to leukocyte trafficking,31,32 was downregulated in iMUC, iSMS, and iMES of CD patients (Figures 3A and 3C). High-mobility group box 1 (HMGB1), synergistic with CXCL12 to induce lymphocyte activation,33 was also reduced in iMUC of CD patients (Figures 3A and S3B). Arrestin beta 1 (ARRB1), which is involved in the autoimmune pathology of experimental autoimmune encephalomyelitis (EAE),34 was downregulated in iMUC and iMES of CD patients (Figure 3A), whereas growth differentiation factor 15 (GDF15), a secreted ligand that interacts with ARRB1,35 was significantly upregulated in iSMS and MLN of CD patients (Figure 3A). Moreover, we also identified several well-known antimicrobial peptides, as exemplified by upregulated lysozyme (LYZ), s100 calcium-binding proteins A8 and A9 (S100A8 and S100A9, respectively), and defensins alpha 5 and 6 (DEFA5 and DEFA6, respectively), in iMUC of CD patients (Figure 3A), suggesting a greatly enhanced bactericidal capacity of myeloid cells and Paneth cells.
Figure 3.

Antimicrobial immunity- and metabolism-associated proteins are dysregulated in different types of tissues
(A) The diagram summarizes protein signatures and different pathways involved in host antimicrobial immunity during CD. Pathways are displayed in pink boxes. Dysregulated proteins are displayed in yellow boxes, and alterations of these proteins are labeled in the boxes below the protein-labeled boxes, which represent the proteins derived from 5 different types of tissues (MUC, SMS, MAT, MES, and MLN, respectively) (red, upregulation; blue, downregulation).
(B) The diagram summarizes the crucial characteristics and pathways involved in metabolism during CD. Pathways are displayed in light green boxes. Dysregulated proteins are displayed in yellow boxes, and alterations of these proteins are labeled in the neighboring boxes, which represent the proteins derived from 5 different types of tissues (MUC, SMS, MAT, MES, and MLN, respectively) (red, upregulation; blue, downregulation). Crucial metabolites in the different metabolic processes are labeled in gray boxes.
(C) Proteins associated with antimicrobial immunity and metabolism across multi-tissues. The y axis indicates protein abundance. A pairwise comparison of each protein between the inflamed tissues of CD patients and normal tissues of NCs was performed using a two-sided unpaired Welch’s t test. B-H-adjusted p values: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Dysregulated metabolic processes across different tissues in CD
Because aberrant metabolic pathways have been reported to be involved in development of CD,36,37 we did observe that multifarious metabolic processes, including amino acid and glucose metabolism as well as nucleotide and fatty acid synthesis, were disturbed in different types of tissues from CD patients (Figure S3C). Of note, a total of 164 dysregulated proteins involved in metabolic processes were identified across different inflamed tissues, 25 of which were differentially expressed in at least two types of inflamed tissues from CD patients (Figure S3C).
In recent years, accumulating evidence has shown that aberrant amino acid metabolism exacerbates intestinal mucosal inflammation, particularly in IBD.38,39 We sought to investigate the alterations in protein metabolism during CD and observed 39 dysregulated proteins associated with metabolic pathways of amino acids in different types of tissues, including methionine, tyrosine, tryptophan, and arginine (Figures 3B and S3C). A series of enzymes in the urea cycle is involved in the metabolism of various amino acids.40 ARG2, which facilitates oxidative phosphorylation of inflammatory macrophages,41 was generally upregulated in all five types of inflamed tissues (Figure 3B), and other urea cycle-related proteins, including ASS1, OAT, and OTC, were dysregulated in multi-tissues during CD (Figure 3B). Carbamoyl-phosphate synthase 1 (CPS1), an initial enzyme of glutamine metabolism,42 was upregulated in iMUC and iMES of CD patients (Figures 3B and 3C). In addition, we identified that a number of proteins modulating glucose metabolism were dysregulated across different tissues during CD (Figure 3B). For example, superoxide dismutase 2 (SOD2), a mitochondrion-specific antioxidant enzyme,43 was significantly upregulated in iSMS of CD patients (Figure 3B). Pyruvate dehydrogenase kinase 4 (PDK4), a crucial regulator of glycolysis and oxidative phosphorylation,44 was downregulated in iMUC and iSMS of CD patients (Figure 3B), which parallels our recent report.45 Enolase 3 (ENO3), a key upstream regulator of glycolysis,46 was significantly upregulated in iMES of CD patients and correlated with lactate dehydrogenase A (LDHA) (Figures 3B and S3D), an enzyme that was significantly decreased in iMES and is responsible for conversion of pyruvate into lactate in glycolysis (Figure 3B). Moreover, we also observed that dysregulated proteins predominantly participated in fatty acid synthesis in iMAT and iMES of CD patients (Figure S3C). Enoyl-coenzyme A hydratase and 3-hydroxyacyl CoA dehydrogenase (EHHADH), a key enzyme of the fatty acid β-oxidation pathway,47 was downregulated in iMES of CD patients (Figures 3B and S3D). Acetyl-CoA catalyzed by acetyl-CoA carboxylase alpha (ACACA), a rate-limiting enzyme of de novo fatty acid synthesis,48 was downregulated and correlated with EHHADH in iMES of CD patients (Figures 3B and S3D). NAD(P)H:quinone oxidoreductase 1 (NQO1) was also decreased in iMAT and iMES of CD patients (Figures 3B and 3C), suggesting that cellular protection against oxidative damage is compromised in the intestinal accessory tissues of CD patients.49 Collectively, these findings suggest that metabolic reprogramming occurs during CD.
Global views of microbial characteristics across different tissues in CD
We then characterized the spatial landscape of the microbiota across different tissues from CD patients (Figure 4A). No significant difference in microbial composition was seen among the intestine and extraintestinal tissues (Figures S4A and S4B), in line with a previous study demonstrating a comparable alteration in the microbiome between MUC, SMS, and MAT.10,12 In addition, similar microbial compositions were also observed between the inflamed and adjacent uninflamed tissues regardless of regional sites in alpha diversity (Figure S4C) and beta diversity (Figure S4D), consistent with the scenarios in the proteome as described above (Figure S1C).
Figure 4.

The distinctive patterns of spatial microbial compositions and functional genes across different tissues of CD patients
(A) A phylogenetic tree shows the union of the differential genera (38 in total), mainly grouped by the phyla Firmicutes, Bacteroidota, Proteobacteria, and Actinobacteria. Significantly different genera (p < 0.05, two-tailed Wilcoxon test) in each tissue site are labeled in the outer circles. Each outer circle track represents one type of tissue site (i.e., MUC, SMS, MAT, MES, and MLN). The outer bar plots on the tracks represent the normalized relative abundance (log2) of each differential genus between the inflamed tissues of CD patients (red) and normal tissues of NCs (blue).
(B) The bar plots show the number of KO genes with differential relative abundance (p < 0.05 identified by two-tailed Wilcoxon test) in each tissue site. The numbers of increased and decreased KO genes in inflamed tissues of CD patients are labeled in orange and green, respectively.
(C) Venn diagrams of the differential KO genes across five tissue sites. The number in each circle represents the amount of KO genes with differential relative abundance between the inflamed tissues of CD patients and normal tissues of NCs. The overlapping numbers indicate the mutual differential KO genes between different types of tissues, and nonoverlapping numbers specify the KO genes unique to each tissue site.
See also Figures S4 and S5 and Table S5.
To clarify the characteristics of the spatial microbial communities in CD patients, we thoroughly investigated the microbiome across different tissues from multiple dimensions. We found that microbial alpha diversity was decreased in inflamed tissues (i.e., MUC, SMS, MAT, MES, and MLN) of CD patients compared with that in normal tissues of NCs (Figures S5A and S5B). At the phylum level, the microbiota was mainly composed of Bacteroidota, Firmicutes, Actinobacteria, and Proteobacteria (Figure S5C). More alterations were observed in the iMUC of CD patients, including an increase in the relative abundance of the dominant Bacteroidota but a decrease in the relative abundance of Firmicutes and Proteobacteria (Figure S5C). We then profiled the spatial characteristics of the microbiota at the genus level in different types of tissues from CD patients (Figure 4A), and surprisingly, a reduction in the relative abundance of short chain fatty acid (SCFA) producers was observed in all five types of inflamed tissues from CD patients (Figure 4A). Notably, the SCFA-producing Granulicatella and Eubacterium were significantly decreased in all inflamed tissues from CD patients, while distinctive SCFA-producing genera were also observed in different tissues, such as Coprococcus in iMUC and iMES; Blautia, Clostridium, Dorea, and Faecalibacterium in iSMS; Faecalibacterium in iMAT; and Bulleidia in MLN (Figure 4A). Moreover, Streptococcus, one of the dominant microbes involved in intestinal inflammation,50 was remarkably reduced in all inflamed tissues of CD patients except iSMS (Figure 4A). Conversely, a significant increase in Lactobacillus, which enables metabolism of tryptophan into AHR ligands,51 was present in iSMS (Figure 4A). Intriguingly, the proteome data displayed increased expression of IDO1, a regulator of AHR responses, in the iMUC of CD patients (Figure 3B), suggesting a potential association between microbiome and host proteome during CD.
Consistent with microbial taxonomical compositions, we also found that the predicted microbial functions at the Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) gene level were altered in inflamed tissues of CD patients and that distinct microbial KO genes with differential relative abundance were identified between CD patients and NCs across multi-tissues. Particularly, significant microbial dysbiosis was observed in the iMUC of CD patients, including 359 genes with increased abundance and 737 genes with decreased abundance compared with normal MUC from NCs (Figure 4B), while fewer differential microbial genes were identified in iSMS, iMAT, iMES, and MLN of CD patients (Figure 4B) compared with controls. Moreover, only a few genes were altered across all five types of inflamed tissues from CD patients (Figure 4C). For instance, murM, encoding a protein essential for high-level penicillin resistance to Streptococcus,52 decreased across all five types of inflamed tissues from CD patients (Figure S5D). The phenylacetate degradation-related gene paaD decreased in all inflamed tissues but increased in iMAT (Figure S5D). Moreover, an enrichment analysis revealed that the two-component system and peptidoglycan biosynthesis were the dominantly enriched microbial pathways across all five types of inflamed tissues (Table S5), suggesting essential roles in microbial growth and development during CD.
We further observed that bacterial co-abundance associations appeared to be positive among the majority of these microbial communities, showing tight competition among taxa. Of note, the microbial community in iMES of CD patients included 56 genera and 84 correlations, which was much denser than that in other tissues (Figure 5A). However, the co-abundance network in iSMS of CD patients was the smallest and sparsest, with several isolated subnetworks, indicating a less stable microbial community (Figure 5A). Besides, relatively complex and connected communities were observed in iMUC, iMAT, and MLN of CD patients (Figure 5A). We also found that SCFA producers were common keystone species in each tissue site, with different participants, such as Enterobacteriaceae in iMUC, Oscillospira, Parabacteroides and Eubacterium in iSMS and iMAT, Aggregatibacter in iMES, and Ruminococcus in MLN (Figure 5A). In addition, tissue-specific keystone species were identified, such as Blautia and Alistipes in iSMS; Sutterella, Bilophila, and Ruminococcus in iMAT; Parvimonas and Mogibacterium in iMES; and Deinococcus in MLN (Figure 5A). Taken together, these results demonstrate the spatial distinctiveness of the microbial ecosystem at multiple levels and identify the important roles of SCFA producers in multi-tissues of CD patients.
Figure 5.

The spatial heterogeneity of microbial ecosystems and distributions of potential microbial transmission sources across different tissues
(A) The co-abundance networks involve prevalent genera in different inflamed tissues (MUC, SMS, MAT, and MES) and MLNs of CD patients and their keystone distributions. The colors of nodes indicate the phylum to which each node belongs. Only significant absolute correlations above 0.4 are shown (FDR <0.01, 1,000 permutations). The orange lines indicate positive species interactions, and the gray lines indicate negative species interactions. Genera identified as keystones by hyperlink-induced topic search (HIST) algorithms are labeled with their genus name.
(B) Potential microbial transmission sources in each tissue site of CD patients via FEAST analysis. The percentage represents the contribution from the indicated tissue site and unknown sources.
The trajectory of microbial community across different tissues in CD
The similarity of microbiota has indicated no novel microbial communities in the intestinal accessory tissues (i.e., MAT, MES, and MLN) but rather a translocation from the gut,10 which prompted us to further explore the source of residential microorganisms in different types of tissues from CD patients via fast expectation maximization for microbial source tracking (FEAST) at the community level. Considering the shared ecological niches in inflamed and adjacent uninflamed tissues, we also considered adjacent uninflamed tissues as potential sources. We found that 60%–70% of the microbiota of each inflamed tissue site could be tracked by its source from other tissue sites independent of inflammatory status (Figure 5B). For example, the microbiota in iMUC was mainly derived from uMUC (12.60% on average), iMAT (8.16%), uMAT (8.87%), and iMES (13.36%), and around 38.67% was derived from an unknown source environment (Figure 5B). As expected, a mutual contribution pattern of source compositions was observed between adjacent tissue sites. In detail, the microbiota of MAT contributed over 20% of sources (11.72% from iMAT and 10.69% from uMAT) to the microbiota of SMS, and nearly 30% of the microbiota of iMAT was sourced from SMS (17.06% from iSMS and 13.03% from uSMS) (Figure 5B). Unexpectedly, relatively fewer bidirectional contributions were shown between MUC and SMS, where the microbiota of MUC traced only 9% of sources from SMS and, vice versa, only 7% of the microbiota of SMS from MUC (Figure 5B). Moreover, frequent microbial transmission was also observed among non-adjacent tissue sites, such as MUC and MES. Near 20% of the MUC microbiota was tracked from MES, and nearly 30% of the MES microbiota was tracked from MUC (Figure 5B), suggesting that specific microbes have potent movement ability to continuously transmit across different tissues and display a colonization preference because of gut vascular barrier impairment.14,15 We also observed that nearly 30% of the microbiota was tracked from unknown sources, probably tissue-resident microorganisms (Figure 5B). Our data reveal that the microbiota spatially transmits across different tissues during CD.
The host-microbiota interactions across different tissues in CD
Given the essential role of host-microbiota interactions in the pathogenesis of CD, we sought to explore the relationships between microbes and host proteins in different tissues of CD patients. We first utilized Fuzzy C-means (FCM) clustering to characterize the spatial dynamics of the significantly altered proteins in different tissues from CD patients and identified seven clusters (protein clusters [P.Cs]) (Figures 6A and S6A) that displayed dominant expression patterns with diverse functions (Table S6). The differential microbial genes were then grouped into five clusters with distinct expression patterns (microbial clusters (M.Cs]). The specific species in different tissues dominated different clusters (Figure 6A and S6A). The microbial genes in M.Cs were commonly enriched in the two-component system (Table S6), suggesting adaption to the microenvironment among different types of tissues.53
Figure 6.

The interaction networks between host proteome and gut microbiome in CD patients
(A) mfuzz clustering of differential host proteins and gut microbial genes across different tissue sites of CD patients. Membership scores indicate the degree to which the microbial gene or host protein belongs in each cluster. M.C, microbial cluster; P.C, protein cluster.
(B) A concept map of the host-microbiota interaction patterns involved in the pathogenesis of CD. During CD, the intestinal inflammation initially occurs in the mucosal area and gradually involves extraintestinal tissues, including the SMS, MAT, MES, and MLN, leading to impaired host defense and dysregulation of immune response in these areas, which contribute to transmission of the gut microbiota from the intestinal lumen into the intestinal accessory tissues.
To investigate the potential host-gut microbiota interactions, we quantified the associations between P.Cs and M.Cs and revealed a significantly positive association between P.C1 and M.C1 (Figure S6B). The functional annotations of M.C1 were enriched in several categories associated with bacterial nutrients and energy metabolism as well as basic biosynthesis (i.e., propanoate), suggesting a significant association with intestinal homeostasis and immunology (Table S6). Propionate is one of the most abundant SCFAs produced by the gut microbiota.54,55 Consistent with this, we also found alterations in SCFA producers in microbial abundance (Figure 4A) and critical roles of SCFA producers in microbial communities (Figure 5A). Moreover, KEGG pathway enrichment analysis revealed that manifold metabolism- and immune-related pathways associated with CD pathogenesis were enriched in P.C1, including nitrogen metabolism and extracellular matrix (ECM)-receptor interaction (Table S6).
Additionally, we identified several key candidate proteins and microbial genes likely associated (through Spearman correlation) with perpetuation and deterioration of gut inflammation and bacterial transmigration across different types of tissues during CD, which were accompanied by impaired barrier function and reduced bactericidal capacity (Figures 6B and S6D). For example, dysregulated expression of cell adhesion molecules, mucus-associated proteins (e.g., CEACAM1 and MUC5B), and antimicrobial peptides (e.g., LYZ and REG4) contributed to restricted microbial growth and reduced defense and healing functions (Figure 6B), which allows unrestrained flux of bacteria into the adjacent SMS layer. Moreover, looking through the components in M.C1 and P.C1, we identified several interactions between microbial genes and host proteins across the intestine and its accessory tissues (Figure 6B). A few bacterial core genes were observed to be involved in bacterial migration, including liaS and phoB, belonging to the two-component system of bacteria (Figure S6D). The migration of bacteria into the SMS provoked an adaptive immune response magnified by SAA2, a significant contributor to Th17 cell-mediated immunopathology.56 In addition, metabolic reprogramming and certain types of programmed cell death pathways, such as pyroptosis, as evidenced by aberrant expression of GSDMB and CPB1 (Figure S6D), appeared to enable bacteria to preferentially cross the SMS into the MAT in synergy with associated bacterial genes (e.g., ompC and zapD). Bacterial translocation into the MAT might direct an influx of immune cells that gather in MAT and elicit ECM destruction. For example, MMP7, a protease to cleave ECM proteins,57 and ECE1 were strongly correlated with bacterial energy metabolism-associated genes, including thrH and SPP. The inefficiency of confining bacterial growth and motility in MAT allowed bacteria to translocate into MES, where bacterial encroachment drove immune responses and destroyed host cell morphology and ECM organization (Figure 6B). Correspondingly, the altered genes of bacterial virulence and metabolism were closely correlated with several essential host proteins, such as ZG16, which limits bacterial translocation,58 and ITGA8, which is likely to attenuate fibrosis formation.59 In MLN, several proteins involved in immune responses, such as GSDMB and SAA2, were highly correlated with bacterial genes belonging to the two-component system and energy metabolism, characterized by Propionibacterium, Enterobacteriaceae, Acinetobacter, and Bacteroides. Altogether, our data suggest that dysfunction of host physiological processes, especially barrier function and host immune response in the intestine and its accessory tissues, is associated with bacterial transmission and that host-microbiota interactions contribute to initiation and perpetuation of intestinal inflammation during CD.
Signature proteins and microbes in serum and feces of active CD patients as potential diagnostic biomarkers
To investigate whether the unique host-microbiota interaction pairs scattered across various tissues could converge in the serum and feces of CD patients and could be used for potential diagnostic biomarkers (namely, liquid biopsies), we performed paired TMTpro-based untargeted proteomics of serum samples and 16S rRNA sequencing of fecal samples from an independent cohort of 30 patients with active CD and 30 healthy donors. We identified 2,477 proteins in serum samples (Figure 1A and Table S2), and 247 of them were differentially expressed (adjusted p < 0.05; Table S2). Pathway enrichment analysis of differentially expressed proteins in the serum of these active CD patients appeared to have significant similarities to those present in tissue samples, as described above (Figure S7A). Importantly, 60 differentially expressed proteins in serum between active CD patients and healthy donors overlapped with those derived from different types of tissues (Table S7). Further comparisons of candidate proteins in P.C1 were performed, and 5 common signature proteins (i.e., IGFBP2, DPP4, SAA2, GOLM1, and GBP1) were screened out (Figures 7A and 7B).
Figure 7.

The signature host proteins and microbes in serum and fecal samples of CD patients and diagnostic potential
(A) The top plot shows the number of differentially expressed proteins shared in tissue samples and serum of CD patients through integrative analysis. The number on top of each column represents the size of differentially expressed proteins. The bar plot at the bottom right represents the number of differentially expressed proteins (set size) in each tissue site, and the connected dot plot at the bottom left represents the common dysregulated proteins across connected tissue sites.
(B) The intensity (log2) of representative differential proteins in sera. All panels represent the 25th–75th percentiles of the distribution, and the median is shown as a thick line in the middle of the box. The whiskers indicate the range of 1.5-fold interquartile range (IQR). The p values were calculated by a two-tailed Wilcoxon test.
(C) The top plot shows the number of differential microbial genera shared in tissue samples and feces of CD patients through integrative analysis. The number on top of each column represents the size of the differential genera. The bar plot at the bottom right represents the number of differential proteins (set size) in each tissue site, and the connected dot plot at the bottom left represents the common differential genera across the connected tissue sites.
(D) The relative abundance (percent) of representative differential genera in feces. All panels represent the 25th–75th percentiles of the distribution, and the median is shown as a thick line in the middle of the box. The whiskers indicate the range of 1.5-fold IQR. The p values were calculated by a two-tailed Wilcoxon test.
(E) The AUC plot shows the diagnostic efficacy of identified signature proteins and genera in predicting CD patients. The shadow denotes a 95% confidence interval for AUC values. The colors represent combinations of biomarkers, including circulating proteins (protein), fecal genera (microbiome), and the combination of proteins and genera (P+M). AUC: area under the curve.
As for the microbiome, the alpha diversity of the fecal samples from these active CD patients was significantly decreased (Figure S7B), and microbial compositions were also significantly altered between active CD patients and healthy donors (p < 0.001; Figure S7C). We identified 19 differential genera, including Alistipes, Dialister, and Fusobacterium, and SCFA-producing bacteria (e.g., Butyricimonas and Dorea), commonly present in different types of tissue (Figures 7C and 7D; Table S7). In addition, we observed 28 decreased microbial genes and 967 increased microbial genes in the fecal samples of these active CD patients (Figure S7D). Enrichment of these elevated genes in feces was consistent with the microbial functions identified in the tissue samples, including enrichment of the two-component system and biosynthesis of amino acids (Figure S7E).
Intriguingly, the circulating proteins correlated with microbial functional genes and microbial genera (Table S7) in these CD patients. Particularly, the circulating protein SAA2 was positively associated with the microbial transporters ugpE and ugpC but negatively associated with microbial gerKC and flaG (Table S7). SAA2 was also positively associated with the abundance of SCFA-producing genera, such as Dorea and Butyricicoccus (Table S7). Furthermore, five overlapped signature proteins (AUC = 0.91) and 19 key genera of microbiota (AUC = 0.91) displayed powerful diagnostic capability in distinguishing active CD patients from healthy controls, and the combination of these protein and bacterial biomarkers significantly improved the classification accuracy (AUC = 0.96), which was higher than that of C-reactive protein (CRP) (Figure 7E). Taken together, these findings unequivocally confirm the tight interactions between host proteome and microbiome across different types of tissues during CD and, more importantly, provide a rationale for omics datasets for precision diagnosis.
Discussion
Accumulating evidence demonstrates that intestinal inflammation initiates in the mucosal area and transmits to intestinal accessory tissues, including MAT, MES, and MLN, in the progression of CD, with ensuing translocation of the gut microbiota. However, little is known about the substantial spatial changes in host proteins and microbiota among these tissues and the potential mechanisms underlying the host-gut microbiota cross-talk. Here, for the first time, we utilized high-throughput omics of different types of tissues from CD patients, including MUC, SMS, MAT, MES, and MLN, and systematically identified the spatial characteristics of microbiota and host proteins spanning multiple tissues. Importantly, we unraveled the complex interactions between the host proteins and microbial genes in different tissues and clarified the underlying mechanisms involved in impaired barrier function and dysregulated immune response that cause progressive intrusion of bacteria into different tissue layers in CD. Unexpectedly, our study revealed that there was no significant difference in proteome between inflamed and adjacent uninflamed tissue sites from CD patients, suggesting that the translational signatures of CD might precede intestinal inflammation. Adjacent uninflamed tissues of CD patients are probably in a vulnerable state and become preferentially inflamed when exposed to harmful external factors such as opportunistic pathogens or dietary antigens. Moreover, we also found that the microbial community was similar between the inflamed and adjacent uninflamed regions but differed markedly from the microbiomes of NCs at the phylum level, which was consistent with a previous study.12 Currently, many studies have demonstrated the landscape of transcriptional profiles of tissues from IBD patients and have shown similar single-cell profiles and cell subtypes between inflamed and adjacent uninflamed mucosa samples of IBD patients.60,61 Our data were consistent with previous studies at translational levels. Collectively, these results provide spatial insight into the pathophysiology of CD and, more importantly, help us to implement precision medicine in disease management.
Through detailed proteome analysis on multi-tissues of CD patients, we unraveled the translational levels of IBD susceptibility genes in different types of tissues and identified 40 dysregulated IBD susceptibility gene-encoded proteins, suggesting preferential expression of susceptibility gene-encoded proteins in different tissues during CD. Particularly ATG16L1 and GSDMB, the two key proteins associated with IBD susceptibility genes, were dysregulated in different types of tissues, suggesting that aberrant antimicrobial immunity exists across multi-tissues during disease and is related to IBD genetic susceptibility. In addition, we also observed that a large number of proteins, especially those related to antimicrobial immunity and metabolic processes, were differentially expressed in at least two types of tissue layers, suggesting that the expression patterns and interplay networks of the same protein molecule appear differently in different microenvironments across multi-tissues.
Another highlight of our study is the multi-site sampling, which allows us to gain access to microbial profiles in different types of tissues from CD patients. Although a previous study has shown that bacteria could translocate from the MUC into the MAT,10 we observed the existence of microbes across multi-tissues, displaying a comprehensive profile of bacterial transmission across different tissue layers in the presence of CD. Through FEAST, we observed that the majority of microbiota could transmit across different tissue sites, with cross-sectional exchanges between the inflamed and adjacent uninflamed sites and longitudinal translocations among different tissues. However, we could not further identify specific taxa of microbiota translocating across different tissues because of the limitation of FEAST. We observed frequent microbial transmission among non-adjacent tissue sites, such as MUC and MES. We presume that some bacteria preferentially colonize among different tissue sites and then enter the bloodstream because of gut vascular barrier impairment, migrating into non-adjacent tissues, as reported by previous studies.14,15 Regardless of the distinctive characteristics of microbial composition, SCFA-producing bacteria, such as Granulicatella and Eubacterium, decreased in all five types of tissues from CD patients, indicating dysbiosis with microbiota-associated inflammation. Taken together, our data reveal that SCFA-producing bacteria play crucial roles in the microbial community across intestinal and extraintestinal tissues and that bacterial transmission exists across different tissues during CD.
Because interactions between the bacterial community and host proteins are integral for maintaining and regulating intestinal homeostasis, we investigated correlative expression patterns of host proteins and bacteria within different tissue sites in the context of CD. Interestingly, some signature proteins were closely associated with specific bacterial genes and functions within distinct tissues. For example, olfactomedin 4 (OLFM4), a protein molecule expressed in intestinal epithelia and neutrophils that is associated with bacterial clearance,62 was correlated with the bacterial genes liaS, thrH, and citXG, which belong to the two-component system and are mainly attributed to Enterococcus, Clostridium, and Enterobacteriaceae. Another notable example is that SAA2 was differentially expressed across all five types of tissues. It is ubiquitously correlated with diverse bacterial genes ranging from MUC to MLN, suggesting its extensive interaction with various bacteria and effects on the pathogenesis of CD. SAA2 is expressed in different immune cells, such as macrophages and T cells, and plays a crucial role in the development of inflammation.63,64 Our findings demonstrated and underscored the diversity of the interactions between host SAA2 and a variety of bacteria across multiple tissues, suggesting SAA2 as a comprehensive marker mirroring inflammatory injury in the intestine. Therefore, our study revealed that the interaction pairs between host proteins and microbial genes are responsible for barrier function and immune responses, preventing transmission of microbes across the intestine and its accessory tissues.
Increasing sensitivity and accuracy prompted us to take advantage of clinical applications of liquid biopsy based on reliable markers in blood and stool. Thus, we considered whether unique interaction pairs detected in tissues by multi-omics could converge in blood and stool in light of their collective and circulating properties. A new cohort of serum and fecal samples from active CD patients and healthy donors was set up, and, more importantly, we identified 60 differentially expressed proteins and 19 differential genera in serum and feces of active CD patients, respectively, which overlapped with those detected in the intestine and its accessory tissues. Moreover, the circulating proteins were found to be correlated with microbial genera in feces, and a panel of key circulating proteins (e.g., SAA2, GOLM1, DPP4, IGFBP2, and GBP1) and fecal bacteria (e.g., Alistipes, Dialister, Fusobacterium, and Streptococcus) illustrated powerful diagnostic efficiency, suggesting that dysregulation of molecular and bacterial signatures in multi-tissues accumulates in serum and feces, highlighting the non-invasive diagnostic potential in CD.
Limitations of the study
First we performed a microbial analysis of the bacterial genome by 16S rRNA gene sequencing but not in combination with metagenomes within different tissues, especially in SMS, MAT, MES, and MLN. However, considering the relatively low biomass of the microbiota and a high proportion of host DNA in tissue samples, especially in intestinal accessory tissues, 16S rRNA gene sequencing is more suitable for primary exploration of tissue microbiotas. Second, host-microbiota interactions were only preliminarily explored by multi-omics, and the specific functions and underlying mechanisms of key interaction pairs in the pathophysiology of CD are still elusive. Further study is warranted to define the functions and underlying mechanisms of critical interaction pairs from different tissues of the intestine and its accessory tissues in CD by shotgun metagenomics and advanced technologies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Surgical tissue samples from 30 CD patients and 5 non-CD controls | Shanghai 10th People’s Hospital | This paper (Table S1) |
| Serum and fecal samples from 30 active CD patients and 30 healthy donors | Shanghai 10th People’s Hospital | This paper (Table S1) |
| Chemicals, peptides, and recombinant proteins | ||
| Triethylammonium bicarbonate buffer (TEAB) | Sigma-Aldrich | Cat #T7408 |
| Urea | Sigma-Aldrich | Cat #U1250 |
| Thiourea | Sigma-Aldrich | Cat #T8656 |
| Tris (2-carboxyethyl) phosphine (TCEP) | Adamas-beta | Cat # 61820E |
| Iodoacetamide (IAA) | Sigma-Aldrich | Cat #I6125 |
| Trypsin | Hualishi Tech | Cat # HLS TRY001C |
| Trifluoroacetic acid (TFA) | Thermo Fisher Scientific | Cat # 85183 |
| Water | Thermo Fisher Scientific | Cat #W6-4 |
| Acetonitrile | Thermo Fisher Scientific | Cat # A955-4 |
| Formic acid (FA) | Thermo Fisher Scientific | Cat # A117-50 |
| Ammonium hydroxide solution | Sigma-Aldrich | Cat # 221228 |
| Methanol | Sigma-Aldrich | Cat # 3486 |
| E.Z.N.A.® stool kit | Omega Bio-tek | Cat #D4015-02 |
| TransStart FastPfu DNA Polymerase | TransGen | Cat # AP221-03 |
| Miseq Reagent Kit V3 | Illumina | Cat # MS-102-3003 |
| Critical commercial assays | ||
| Ultracel-3 regenerated cellulose membrane | Millipore | Cat # UFC500324 |
| XBridge Peptide BEH C18 column | Waters | Cat # 186003570 |
| High SelectTM Top-14 Abundant Protein Depletion Mini Spin Columns | Thermo Fisher Scientific | Cat # A36370 |
| 96-well SOLAμ™ SPE plate | Thermo Fisher Scientific | Cat # 60209-001 |
| μ-Precolumn™ Catridge | Thermo Fisher Scientific | Cat # DX163593 |
| Pierce™ C18 Spin Column | Thermo Fisher Scientific | Cat # 89870 |
| TMTpro 16plex reagents | Thermo Fisher Scientific | Cat # A44520 |
| Emitter column, 15 cm × 75 μm | New Objective | N/A |
| C18 resin, 1.9 μm | Fresh Biosciences | N/A |
| Deposited data | ||
| Mass spectrometry data | This paper | https://www.iprox.org |
| 16S amplicon sequencing (tissue) | NODE: OEP003464 | https://www.biosino.org/node/ |
| 16S amplicon sequencing (feces) | NODE: OEP003463 | https://www.biosino.org/node/ |
| Software and algorithms | ||
| QIIME2 v2019.07 | Bolyen et al., 2019a65 | https://qiime2.org |
| PICRUSt v2.0 | Douglas et al., 2020b66 | https://github.com/picrust/picrust2 |
| Sparcc | Friedman and Alm, 2012a67 | https://bitbucket.org/yonatanf/sparcc |
| FEAST | Shenhav et al., 201968 | https://github.com/cozygene/FEAST |
| R package ClusterProfile v4.0 | Wu et al., 2021a69 | https://bioconductor.org/packages/clusterProfiler/ |
| R package decontam | Davis et al., 201870 | https://github.com/benjjneb/decontam |
| R package phyloseq v1.22.3 | McMurdie and Holmes, 201371 | https://github.com/joey711/phyloseq |
| R package mfuzz v2.50.0 | Futschik and Carlisle, 200572 | http://mfuzz.sysbiolab.eu |
| R package vegan v2.5-7 | Dixon, 200373 | https://github.com/vegandevs/vegan |
| Proteome Discoverer (Version 2.4.0.305) | Thermo Fisher Scientific | N/A |
| Spectronaut (Version 14.6.201001.47784) | Biognosys | N/A |
| R v4.0.2 | R project | https://www.r-project.org/ |
| String | Szklarczyk et al., 202174 | https://string-db.org/ |
| Cytoscape (version 3.6.1) | Shannon et al., 200375 | NA |
| Ingenuine pathway analysis (version 51963813) | Kramer et al., 201476 | https://www.qiagen.com/cn/ |
| R v3.6.3 | R project | https://www.r-project.org/ |
| Python package scikit-learn v0.21 | Pedregosa et al., 201177 | https://scikit-learn.org/0.21/ |
| Other | ||
| timsTOF Pro mass spectrometer | Bruker Daltonics | N/A |
| nanoElute liquid chromatographer | Bruker Daltonics | N/A |
| DIONEX UltiMate 3000 RSLCnano liquid chromatographer | Thermo Fisher Scientific | N/A |
| Q Exactive HF-Orbitrap | Thermo Fisher Scientific | N/A |
| NanoDrop Eight | Thermo Fisher Scientific | N/A |
| QuantiFluor™-ST | Promega | N/A |
| MiSeq System | Illumina | N/A |
Resource availability
Lead contact
Further information should be directed to and will be fulfilled by the lead contact, Zhanju Liu (zhanjuliu@tongji.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Patient collection and sampling strategy
This study was approved by the Medical Ethics Committee of the Shanghai 10th Hospital affiliated to Tongji University (No. 20KT863) and Chinese Clinical Trial Registry (ChiCTR2000036115).
Surgical specimens were collected from the terminal ileum of CD patients and adjacent normal terminal ileum of non-CD controls who underwent ileocolectomy. All CD patients were diagnosed according to the ECCO-ESGAR Guideline for Diagnostic Assessment in IBD78,79 and accepted surgical therapy due to severe complications (e.g., stenosis, perforation, and massive bleeding) or failure of medical treatment. Exclusion criteria included patients under 18 years old, inability or unwillingness to provide informed consent, antibiotics or antifungal use within one month prior to surgery, or individuals with other diseases affecting the intestines. Non-CD controls were diagnosed with early-stage colon cancer (involved in the cecum and/or ascending colon) according to their clinical symptoms, serological tests, and imaging examinations. Patients provided informed consent before the operation. To prevent contamination of parenteral tissues with the luminal contents, the specimens were obtained under sterile conditions in the operating room by professional surgeons within 20 min post-operation. Intestinal accessory tissues, including mesenteric adipose tissues, mesentery, and mesenteric lymph nodes, were also obtained during the operation. We gently washed specimens with sterile PBS until all surface blood and luminal contents were removed. The specimens were transported immediately in liquid nitrogen and stored at −80°C freezers for later use. We collected tissue samples from the inflamed areas (including inflamed mucosa, submucosa-muscularis-serosa, mesenteric adipose tissue, and mesentery; 4 tissue regions) and the adjacent uninflamed areas (including adjacent uninflamed mucosa, submucosa-muscularis-serosa, mesenteric adipose tissue, and mesentery; 4 tissue regions) of each CD patient. In addition, mesenteric lymph nodes were also collected, and 9 tissue samples were collected from each CD patient. We recruited 30 CD patients in our study and collected a total of 270 tissue samples, including the inflamed mucosa (iMUC) (n = 30; n represents the number of samples), adjacent uninflamed mucosa (uMUC) (n = 30), inflamed submucosa-muscularis-serosa (iSMS) (n = 30), adjacent uninflamed submucosa-muscularis-serosa (uSMS) (n = 30), inflamed mesenteric adipose tissues (iMAT) (n = 30), adjacent uninflamed mesenteric adipose tissues (uMAT) (n = 30), inflamed mesentery (iMES) (n = 30), uninflamed mesentery (uMES) (n = 30), and mesenteric lymph nodes (MLN) (n = 30). We then divided each tissue sample into two parts for paired host proteome and tissue microbiome, respectively, under sterile conditions. In addition, 5 adjacent normal specimens (including MUC, SMS, MAT, MES, and MLN; 5 tissue samples) were collected from each NC. We recruited 5 NCs in our study and collected 25 tissue samples, including mucosa (n = 5), submucosa-muscularis-serosa (n = 5), mesenteric adipose tissue (n = 5), mesentery (n = 5), and mesenteric lymph nodes (n = 5). We then divided each tissue sample into two parts for paired host proteome and tissue microbiome, respectively, under sterile conditions. All NCs' specimens were histologically stained, and at least two professional pathologists verified them as relatively normal tissues without tumor invasion. More details of the clinical characteristics of these patients are detailed in Table S1.
60 pairs of serum samples and fecal samples were collected from 30 active CD patients and 30 healthy donors from the Shanghai 10th People’s Hospital. In the pre-sample interview, informed consent was obtained from CD patients. Disease severity was evaluated based on their clinical CDAI scores, SES-CD scores, serological results, and radiological findings. Individuals with other intestinal diseases or those under 18 years old were excluded, and those who refused to provide informed consent or had taken antibiotics within one month of sampling were also excluded. Additionally, 30 age- and gender-matched healthy volunteers were recruited in our study. More details of the clinical characteristics of these patients are detailed in Table S1.
Method details
Proteomic analyses of tissue specimens
Sample preparation and data acquisition
295 tissue specimens and 23 mouse liver (ML) QC samples were collected. 1 mg of tissue block from each sample was taken for peptide extraction using an accelerated pressure cycling technology (PCT) method as described previously.80 The derived peptides were desalted using 96-well SOLAμ SPE plates (Cat. No. 60209-001, Thermo Fisher Scientific; Waltham, MA, USA) and quantitated using NanoDrop Eight (Cat. No. 912A1100, Thermo Fisher Scientific) according to the manufacturer’s protocols. 300 ng of the peptides from 20 randomly selected specimens covering all types of samples were taken and mixed to generate a pooled sample.
23 batches were designed, including 295 tissue samples, and 19 samples were randomly selected as technical replicates. A pooled sample and an ML sample were added to each batch as technical and biological replicates, respectively.
For the LC-MS/MS data acquisition, the tryptic peptides were analyzed by a nanoElute (Bruker Daltonics Inc.; Blerika, MA, USA) liquid chromatography coupled to a timsTOF Pro (Bruker Daltonics Inc.) mass spectrometer. HPLC separation was carried out by gradient elution using a two-column system, wherein the trap column adopts μ-Precolumn Cartridge (Thermo Fisher Scientific), and the analytical column adopts an in-house emitter column (15 cm × 75 μm, New Objective; Littleton, MA, USA) home-packed with C18 resin (1.9 μm, Fresh Biosciences; Shanghai, China). Mobile phase A was 0.1% formic acid in the water, and mobile phase B was 0.1% formic acid in acetonitrile. LC separation was achieved at a flow of 300 nL/min using a 70 min gradient (5%–27% of phase B at 0−50 min, 27%–40% of phase B at 50−60 min, 40%–80% of phase B at 60−61 min, 80% of phase B from 61 to 70 min). Mass-spectrometric measurements for pool samples were carried out using a data-independent acquisition parallel accumulation serial fragmentation (diaPASEF) acquisition method.81 The electrospray ionization (ESI) source settings were as follows: 4500 V capillary voltage, 500 V endplate offset, and 3.0 L/min of dry gas at the temperature of 180°C. The measurements were carried out over the m/z range from 100 to 1700 Th. The range of ion mobilities included values from 0.70 to 1.30 V s/cm2 (1/k0). The number of PASEF MS/MS scans was set to 14 m/z. Mass-spectrometric measurements for clinical specimens were carried out using a pulseDIA scheme82 based on the diaPASEF acquisition method (pulse-diaPASEF), which uses gas-phase fractionation (GPF) to segregate the m/z and ion mobility acquisition range into two injections. The other parameters in the pulse-diaPASEF method were the same as those in the diaPASEF method. Mass-spectrometric measurements for clinical specimens were carried out using a data-dependent acquisition PASEF (ddaPASEF) method.83 The range of ion mobilities included values from 0.60 to 1.60 V s/cm2 (1/k0), and the m/z inclusion windows were unspecified. Other parameters were the same as those in the diaPASEF method.
Spectral library generation
Two spectral libraries were generated for this study, namely libraries A and B respectively. For library A, 10 μg of peptides was extracted from 30 randomly selected samples, including 10 MUC samples, 10 SMS samples, and 10 MLN samples, and combined to generate peptide pools. For library B, 10 μg of peptides was extracted from 30 randomly selected samples, including 15 MAT samples and 15 MES samples, and combined to generate peptide pools. The peptide pools for each library were fractionated using a nanoflow DIONEX UltiMate 3000 RSLCnano System (Thermo Fisher Scientific) with an XBridge Peptide BEH C18 column (300 Å, 5 μm × 4.6 mm × 250 mm) (Waters; Milford, MA, USA). The samples were separated using a gradient from 5% to 35% acetonitrile (ACN) in 10 mM ammonia (pH = 10.0) at a flow rate of 1 mL/min. Peptides were separated into 20 fractions, which were subsequently dried and re-dissolved in 2% ACN/0.1% formic acid (FA). The re-dissolved peptides were analyzed in a ddaPASEF mode with the same LC-MS/MS settings as in the ML samples.
We acquired 40 data-dependent acquisition (DDA)-MS files to generate a spectral library using Spectronaut (version: 14.6.201001.47784). The FDR cutoff was set to 1% at the precursor and protein levels. The SwissProt Human database (downloaded on 15 July 2020, containing 20368 reviewed protein sequences) was used for protein inference. The Decoy generation method, as set as inverse and Cross Run Normalization, was not performed for quantification. Other parameter settings were default. The result generated a library containing 149865 precursors, 104067 peptides, and 10135 proteins.
Data analysis into the protein matrix
The acquired data for the tissue specimens were analyzed by Spectronaut using a library-based workflow. The FDR cutoff was set to 1% at the precursor and protein levels, and the other parameter settings were default. 130582 unique precursors, 109727 modified peptides, 96865 stripped peptides, and 8948 protein groups were quantified from the 295 tissue samples together with the 19 technical replicates. 3388 unique proteins of 3452 protein groups were quantified from the 23 pooled samples by independent Spectronaut analysis. The peptide matrixes were processed into protein matrixes for clinical specimens and pools (Table S2) using ProteomeExpert.84
Quality control
The quality of proteomic data was assessed at multiple levels. For LC-MS/MS acquisition, blank samples (buffer A) were injected after every four LC-MS/MS injections to avoid carryovers. For data processing, pooled samples were used to evaluate quantitative accuracy.
After filtering 8948 proteins with protein groups in all samples, 8860 unique proteins of 314 samples (including 295 tissue samples and 19 technical replicates) with a missing ratio rate of 40.47% were used for QC. We assessed the quality of proteomics data of proteins in pooled samples and technical replicates by Pearson correlation analysis. The Pearson correlation was computed by the cor function in R (version: 4.0.2). "pairwise.complete.obs" was used to compute covariances in the presence of missing values. Besides, t-SNE unsupervised clustering was performed in all samples for different MS machines, diseases, spatial locations, and batches. 8807 proteins were obtained after excluding contaminants in the data matrix for further bioinformatics analysis.
Pathway and network analysis
IPA software was used to perform the pathway enrichment. All the enriched pathways were demonstrated to be significant (p value <0.05) by IPA analysis.
Proteomic analyses of serum samples
Sample preparation
10 μL of serum from each sample was processed using a High Select Top-14 Abundant Protein Depletion Mini Spin Column (Thermo Fisher Scientific) equipped with 175 μL of packing material and 300 μL of phosphate buffer saline, then incubated on a shaker for 20 min. After removing the caps and tips, the columns were placed into 2 mL EP tubes and centrifuged at 2000 rpm for 2 min at room temperature to derive the samples. 500 μL of 1× PBS was added to a 3 kDa Millipore super filtration membrane column and centrifuged at 12000 g for 10 min to remove glycerol until less than 100 μL of PBS was left. To enrich the proteins, we added the samples into the Millipore super filtration membrane columns and centrifuged at 12000 g for 30 min at room temperature. 50 μL of the eluate was transferred to a 1.5 mL EP tube and denatured in 50 μL lysis buffer (8 M urea in 100 mM triethylammonium bicarbonate, TEAB) at 600 rpm for 30 min at 31.5°C. The proteins were reduced with 5 μL of 200 mM tris (2-carboxyethyl) phosphine (TCEP) for 30 min at 32°C, 600 rpm, then alkylated for 30 min with 5 μL of 800 mM iodoacetamide in darkness at 32°C, 600 rpm. The protein extracts were diluted with 200 μL 100 mM TEAB and digested with double-step trypsinization. The reaction was quenched by adding 31.5 μL 10% trifluoroacetic acid (TFA) and confirmed that the pH of the solution was between 2 and 3. Digested peptides were cleaned-up with SOLAμ (Thermo Fisher Scientific) and labeled with TMTpro 16plex label reagents (Thermo Fisher Scientific) according to the manufacturer’s instructions. The labeled peptides in each batch were combined and fractionated using a DIONEX U3000 System (Thermo Fisher Scientific) with an XBridge Peptide BEH C18 column (300 Å, 5 μm × 4.6 mm × 250 mm) (Waters). Fractionation was performed with a gradient from 5% to 35% buffer (98% acetonitrile in water containing 10 mM ammonia, pH = 10.0) at a flow rate of 0.5 mL/min in 60 min 60 fractions were collected and consolidated into 30 fractions. After dryness, the fractions were re-dissolved with 2% acetonitrile (ACN)/0.1% formic acid (FA) of MS grade in preparation for LC-MS/MS analysis.
Data acquisition
The peptides were analyzed with a nanoflow DIONEX UltiMate 3000 RSLCnano system coupled to a Q Exactive HF-Orbitrap (Thermo Fisher Scientific) in data-dependent acquisition (DDA) mode. For LC-MS/MS acquisition, peptides were loaded onto a pre-column (3 μm, 100 Å, 20 mm∗75 μm i.d.) at a flow rate of 6 μL/min and then analyzed using an analytical column (1.9 mm, 120 Å, 150 mm∗75 mm i.d.) with a 60 min LC gradient at a flow rate of 300 nL/min. Buffer A was 2% ACN and 98% H2O containing 0.1% FA, and buffer B was 98% ACN and 2% H2O containing 0.1% FA. All reagents were MS grade. The m/z range of MS1 was 350-1,800 m/z with the resolution at 60000 (at 200 m/z). The top 20 precursors were selected for MS/MS acquisition. The resolution of MS2 was set as 30000. AGC target was set at 3e6 for MS1 and 2e5 for MS2, respectively. The maximum injection time (max IT) was set as 50 ms for MS1 and 100 ms for MS2, respectively. The isolation window of the selected precursor was 0.7 m/z.
Data analyses into the protein matrix
The raw proteomic data for the 60 serum samples from 4 batches were analyzed using Proteome Discoverer software (Version 2.4.0.305, Thermo Fisher Scientific). The fasta file was downloaded from the Uniprot database (SwissProt Human database was downloaded from UniProtKB on 15 July 2020) containing sequences from 20368 reviewed proteins of Homo Sapiens and 286 contaminants. Sequest HT was set as the search engine, and the enzyme was set to trypsin with two missed cleavage tolerance. Static modifications were set to carbamidomethylation (+57.021464) of cysteine, TMTpro (+304.207145) of lysine residues, and peptides’ N-terminus. Variable modifications were set to oxidation (+15.994915) of methionine and acetylation (+42.010565) of peptides’ N- terminus. Precursor ion mass tolerance was set to 10 ppm, and product ion mass tolerance was set to 0.02 Da. The peptide-spectrum-match was evaluated at a 1% target false discovery rate (FDR) (strict) and a 5% target FDR (relaxed). Other parameters were set as default. 2074 proteins were identified in total (Table S2).
Microbiome analyses of tissue and fecal specimens
DNA extraction for tissue and fecal 16S rRNA amplicon sequencing
Frozen tissue samples and fecal samples were processed for DNA extraction using an E.Z.N.A. stool kit (Omega Bio-tek; Norcross, GA, USA) according to the manufacturer’s instructions. The hypervariable V3-V4 region (338F_806R) of the 16S rRNA gene was amplified by PCR with the following primer: forward 338F (5′-ACTCCTACGGGAGGCAGCAG-3′ and reverse 806R (5′-GGACTACHVGGGTWTCTAAT-3′). PCR products were purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences; Union City, CA, USA) and quantified by QuantiFluor-ST (Promega; Madison, WI, USA). The constructed DNA libraries were sequenced on Illumina MiSeq 3000 (Illumina, Inc.; San Diego, CA, USA) platform with 2 × 300 bp paired-end reads.
Bioinformatic analysis of 16S rRNA gene sequencing data
The procession of 16S rRNA sequencing data was performed in the Quantitative Insights Into Microbial Ecology 2 (QIIME2 V.2019.07) platform.85 Briefly, barcode sequences were first removed by Cutadapt.86 Then paired reads were joined, and low-quality reads (Q < 20) were filtered out. Deblur algorithm plugin wrapped in QIIME2 was used to obtain sub-operational-taxonomic-unit (sOTU), which obtained a single-nucleotide resolution from sequencing data by inferring the putative true sequences. The average number of tissue and fecal 16S rRNA reads per sample was 32330 and 18880, respectively. Samples with less than 2000 reads were removed from further analyses. Representative sequences of each sOTU were aligned using Fast Fourier Trans-form in Multiple Alignment (MAFFT)87 in q2-phylogeny plug-in, and the phylogenetic tree was constructed by the Fast-Tree plugin.88 Finally, sOTUs were assigned taxonomy via the Naive Bayes classifier89 trained on 99% clustered sequences in Greengenes 13.8 99% full-length reference database.90
Decontamination and microbial taxonomic analysis
For tissue samples, sOTU counts of each sample were stringently decontaminated using the “decontam” package in R. Putative contaminants were identified using the negative blank samples (“method = prevalence” in decontam; p = 0.5), where sOTUs were labeled as putative contaminants for being more abundant in negative blanks than in biological samples. Collectively, 184 sOTUs were removed as putative contaminants. Then, negative blanks and 6 biological samples that had few remaining microbial reads were removed. Furthermore, contaminating DNA reads from extraction kits and other reagents were removed to ensure a robust analysis in low microbial biomass tissue samples.91
Microbial taxonomic and functional analyses
To avoid the bias of unbalanced sequencing depth, alpha and beta diversities were calculated based on the rarefied count table at a depth of 2075. Bray-Curtis distance was used to estimate microbiota beta diversity, and microbial compositions of different tissue sites were measured by Anosim. The sOTU count table was then organized into different levels and converted into the relative abundance to perform downstream analysis. The differential genus was identified with a non-parameter Wilcoxon test, and p values less than 0.05 were identified as the significantly differential genus.
The functional composition of the gut microbiome was inferred from 16S rRNA sequences with Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) as described previously.92 Microbial functions were annotated according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database at KEGG Orthology (KO) gene and pathway levels. Differential analyses were performed based on a microbial functional profile via a non-parameter Wilcoxon test. To evaluate the contribution of each genus to overall differential KO genes, it was defined as a ratio of the sum of the functional abundance of sOTUs assigned as a specific genus to the total functional abundance of all sOTUs in a given KO gene using a “per_sequence_contrib” mode.93 We thus determined the major contribution genera of key KO genes selected in the host protein-microbial function associations analyses.
To study the structure of the microbial community in each tissue site, we performed a co-abundance analysis at the genus level. Only genera with prevalence in more than 10 samples were included in this analysis. Associations between genera were calculated using the SparCC algorithm, which is known for its robustness for compositional data characterized by the members' diversity and sparsity.67 Correlation coefficients were estimated at the average of 50 inference iterations with the default strength threshold. p values were calculated from 1,000 bootstrap correlations. Correlation coefficients with p values <0.01 (defined as significant), as well as a magnitude above 0.4, were selected for further visualization in Gephi (V 0.9.2). The importance of genus in the community was evaluated using Hyperlink-Induced Topic Search (HITS) algorithms in the Python package ‘networkx’, which takes the information of both the seed node and its linked nodes into consideration. The importance of the node was indicated in terms of node size.
Microbial source tracking analysis
FEAST is a highly efficient expectation-maximization-based method68 that uses community structure to measure the similarity between sink samples and potential source environments. Besides, FEAST is agnostic to the sequencing data type and performs well across all levels of sequencing depth. Therefore, we adopted FEAST to investigate the potential microbiota source of the inflamed tissue sites from CD patients based on 16S rRNA microbial data. The statistical model used by FEAST assumes each sink is a convex combination of known and unknown sources. In detail, the microbial composition profile of each inflamed sample site was treated as a sink, and microbial profiles of other tissue sites were treated as potential sources. To this end, the relative contributions from the different source environments (i.e., uMUC, iSMS, uSMS) to the sink environment (i.e., iMUC) were assessed using FEAST with default parameters.
Host proteome and gut microbiome interaction analyses
Changes in the abundance of all differential microbial functional KO genes across the intestinal and its accessory tissue regions were standardized and clustered using Fuzzy C-Means clustering implemented in the “mfuzz” R package. The final number of clusters was determined based on an inspection of the coherence of the profiles contained in each cluster. The clustering assigned a membership between 0 and 1 to each genus for each cluster. A similar analysis was done against the host proteome, and seven protein clusters were identified. Pathway enrichment analyses were performed via clusterProfile 4.0 against the KEGG database.69
The associations between microbial genes and host proteins in the corresponding clusters were calculated via spearman correlation analysis based on the relative abundance of microbial genes and the expression amount of host proteins. Permutation tests were used to calculate the exact p value of each association via perm.cor.test (10000 permutations), and associations with permutation p values <0.05 (defined as significant) were selected for further visualization with a heatmap and labeled with a star.
Construction of classification model
To assess the potential diagnostic efficiency of serum circulating proteins and fecal genera, we constructed a classification model for diagnosing patients with active CD using the five overlapped differential proteins and 19 overlapped differential genera detected in both different tissue as serum and fecal samples. We first tuned hyperparameters (e.g., the number of estimated trees, the maximum depth of the trees, and the number of features per tree of the RF classifier) via a Bayesian optimization method. To avoid overfitting, we built the classification models with stratified 5-fold cross-validation against the selected optimal hyperparameters. The general performance of the model was reported with cross-validation AUC values. All analyses were performed in the scikit-learn (v 0.21) package.77
Quantification and statistical analysis
Missing values were imputed with the 0.8-fold minimum value in the proteomics dataset. Fold-change (FC) was calculated at a ratio of the mean value of protein intensity to each pair of comparing groups. A two-sided unpaired Welch’s t-test was performed for each pair of comparing groups. Adjusted p values were calculated by the method of Benjamini & Hochberg correction. Differentially expressed proteins were selected by the stated cutoff: adjusted p value <0.05 and |log2(FC)| > 1. Differential analyses of the microbiome were performed as described above. All statistical analysis was based on R (version: 4.0.2). Most microbiome plots were performed using the vegan package.73 Other plots were generated using the ggplot2 package.94
Asterisks denote statistical significance based on unpaired two-sided Welch’s t-test: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Acknowledgments
We thank Dr. Qing Wei for the professional pathological diagnosis. We appreciate all physicians, nurses, and PhD students from the Center for IBD Research at Shanghai Tenth People’s Hospital of Tongji University for assistance with the sampling work and comments on this study, the Guomics team for assistance with proteome data quality control and analysis, and the Zhu Lab team for assistance with microbiome data quality control and analysis. This work was supported by the National Natural Science Foundation of China (91942312, 81630017, 82170542, and 82000536). The omics study for validation of unique host-microbiota interaction pairs across the intestine and its accessory tissues as potential diagnostic biomarkers for CD patients was registered with the Chinese Clinical Trial Registry (registration number ChiCTR2000036115).
Author contributions
Z.L., T.G., and R.Z. designed and supervised the project. X.G., G.L., C.C., M.Y., and X.S. collected tissue samples in cohort 1 and serum and fecal samples in cohort 2. X.G., X.L., and R.S. conducted proteomics analyses. X.G., N.J., and R.S. conducted the microbiome analyses. X.G., N.J., and R.S. conducted integrative analyses of proteome and microbiome data. X.G., L.C., W.W., and H.G. organized the clinical data. X.G., R.S., N.J., X.L., and Z.L. wrote the manuscript with input from co-authors. X.G., R.S., T.G., R.Z., and Z.L. interpreted the data with input from all co-authors and revised the manuscript. Y.C. improved the language and logic of the manuscript. All authors reviewed and approved the manuscript before submission.
Declaration of interests
T.G. is a shareholder of Westlake Omics Inc.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 11, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101050.
Contributor Information
Ruixin Zhu, Email: rxzhu@tongji.edu.cn.
Tiannan Guo, Email: guotiannan@westlake.edu.cn.
Zhanju Liu, Email: zhanjuliu@tongji.edu.cn.
Supplemental information
Data and code availability
All data are available in this paper. The proteomics data reported in this paper have been deposited in ProteomeXchange Consortium with accession iProX: IPX0004848000. The 16S rRNA sequencing data reported in this paper have been deposited in The National Omics Data Encyclopedia (NODE) with accession ID OEP003464 (intestine-associated tissue samples) and OEP003463 (fecal samples), respectively. Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
References
- 1.Roda G., Chien Ng S., Kotze P.G., Argollo M., Panaccione R., Spinelli A., Kaser A., Peyrin-Biroulet L., Danese S. Crohn's disease. Nat. Rev. Dis. Primers. 2020;6:22. doi: 10.1038/s41572-020-0156-2. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan G.G. The global burden of IBD: from 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015;12:720–727. doi: 10.1038/nrgastro.2015.150. [DOI] [PubMed] [Google Scholar]
- 3.Graham D.B., Xavier R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature. 2020;578:527–539. doi: 10.1038/s41586-020-2025-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd-Price J., Arze C., Ananthakrishnan A.N., Schirmer M., Avila-Pacheco J., Poon T.W., Andrews E., Ajami N.J., Bonham K.S., Brislawn C.J., et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coffey J.C., O'Leary D.P. The mesentery: structure, function, and role in disease. Lancet. Gastroenterol. Hepatol. 2016;1:238–247. doi: 10.1016/S2468-1253(16)30026-7. [DOI] [PubMed] [Google Scholar]
- 6.Coffey J.C., Sehgal R., Walsh D. CRC Press; 2017. Mesenteric Principles of Gastrointestinal Surgery: Basic and Applied Science. [Google Scholar]
- 7.Rivera E.D., Coffey J.C., Walsh D., Ehrenpreis E.D. The mesentery, systemic inflammation, and crohn's disease. Inflamm. Bowel Dis. 2019;25:226–234. doi: 10.1093/ibd/izy201. [DOI] [PubMed] [Google Scholar]
- 8.Peyrin-Biroulet L., Chamaillard M., Gonzalez F., Beclin E., Decourcelle C., Antunes L., Gay J., Neut C., Colombel J.F., Desreumaux P. Mesenteric fat in Crohn's disease: a pathogenetic hallmark or an innocent bystander? Gut. 2007;56:577–583. doi: 10.1136/gut.2005.082925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto K., Kiyohara T., Murayama Y., Kihara S., Okamoto Y., Funahashi T., Ito T., Nezu R., Tsutsui S., Miyagawa J.I., et al. Production of adiponectin, an anti-inflammatory protein, in mesenteric adipose tissue in Crohn's disease. Gut. 2005;54:789–796. doi: 10.1136/gut.2004.046516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ha C.W.Y., Martin A., Sepich-Poore G.D., Shi B., Wang Y., Gouin K., Humphrey G., Sanders K., Ratnayake Y., Chan K.S.L., et al. Translocation of viable gut microbiota to mesenteric adipose drives formation of creeping fat in humans. Cell. 2020;183:666–683.e17. doi: 10.1016/j.cell.2020.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batra A., Heimesaat M.M., Bereswill S., Fischer A., Glauben R., Kunkel D., Scheffold A., Erben U., Kühl A., Loddenkemper C., et al. Mesenteric fat - control site for bacterial translocation in colitis? Mucosal Immunol. 2012;5:580–591. doi: 10.1038/mi.2012.33. [DOI] [PubMed] [Google Scholar]
- 12.Pedamallu C.S., Bhatt A.S., Bullman S., Fowler S., Freeman S.S., Durand J., Jung J., Duke F., Manzo V., Cai D., et al. Metagenomic characterization of microbial communities in situ within the deeper layers of the ileum in Crohn’s disease. Cell. Mol. Gastroenterol. Hepatol. 2016;2:563–566.e5. doi: 10.1016/j.jcmgh.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suau R., Pardina E., Domènech E., Lorén V., Manyé J. The complex relationship between microbiota, immune response and creeping fat in Crohn’s disease. J. Crohns Colitis. 2022;16:472–489. doi: 10.1093/ecco-jcc/jjab159. [DOI] [PubMed] [Google Scholar]
- 14.Spadoni I., Zagato E., Bertocchi A., Paolinelli R., Hot E., Di Sabatino A., Caprioli F., Bottiglieri L., Oldani A., Viale G., et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science. 2015;350:830–834. doi: 10.1126/science.aad0135. [DOI] [PubMed] [Google Scholar]
- 15.Bertocchi A., Carloni S., Ravenda P.S., Bertalot G., Spadoni I., Lo Cascio A., Gandini S., Lizier M., Braga D., Asnicar F., et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell. 2021;39:708–724.e11. doi: 10.1016/j.ccell.2021.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L.P., Sharma Y., Anderson C.A., et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T., et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee M., Chang E.B. Inflammatory bowel diseases (IBD) and the microbiome-searching the crime scene for clues. Gastroenterology. 2021;160:524–537. doi: 10.1053/j.gastro.2020.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong L., Pokatayev V., Lefkovith A., Carter G.T., Creasey E.A., Krishna C., Subramanian S., Kochar B., Ashenberg O., Lau H., et al. The landscape of immune dysregulation in Crohn's disease revealed through single-cell transcriptomic profiling in the ileum and colon. Immunity. 2023;56:444–458.e5. doi: 10.1016/j.immuni.2023.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bludau I., Aebersold R. Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat. Rev. Mol. Cell Biol. 2020;21:327–340. doi: 10.1038/s41580-020-0231-2. [DOI] [PubMed] [Google Scholar]
- 21.Amatullah H., Fraschilla I., Digumarthi S., Huang J., Adiliaghdam F., Bonilla G., Wong L.P., Rivard M.E., Beauchamp C., Mercier V., et al. Epigenetic reader SP140 loss of function drives Crohn's disease due to uncontrolled macrophage topoisomerases. Cell. 2022;185:3232–3247.e18. doi: 10.1016/j.cell.2022.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pierre N., Salée C., Massot C., Blétard N., Mazzucchelli G., Smargiasso N., Morsa D., Baiwir D., De Pauw E., Reenaers C., et al. Proteomics highlights common and distinct pathophysiological processes associated with ileal and colonic ulcers in crohn's disease. J. Crohns Colitis. 2020;14:205–215. doi: 10.1093/ecco-jcc/jjz130. [DOI] [PubMed] [Google Scholar]
- 23.Torres J., Petralia F., Sato T., Wang P., Telesco S.E., Choung R.S., Strauss R., Li X.J., Laird R.M., Gutierrez R.L., et al. Serum biomarkers identify patients who will develop inflammatory bowel diseases up to 5 Years before diagnosis. Gastroenterology. 2020;159:96–104. doi: 10.1053/j.gastro.2020.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Starr A.E., Deeke S.A., Ning Z., Chiang C.K., Zhang X., Mottawea W., Singleton R., Benchimol E.I., Wen M., Mack D.R., et al. Proteomic analysis of ascending colon biopsies from a paediatric inflammatory bowel disease inception cohort identifies protein biomarkers that differentiate Crohn's disease from UC. Gut. 2017;66:1573–1583. doi: 10.1136/gutjnl-2015-310705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y., Bhosle A., Bae S., McIver L.J., Pishchany G., Accorsi E.K., Thompson K.N., Arze C., Wang Y., Subramanian A., et al. Discovery of bioactive microbial gene products in inflammatory bowel disease. Nature. 2022;606:754–760. doi: 10.1038/s41586-022-04648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen R.Y., Kung V.L., Das S., Hossain M.S., Hibberd M.C., Guruge J., Mahfuz M., Begum S.M.K.N., Rahman M.M., Fahim S.M., et al. Duodenal microbiota in stunted undernourished children with enteropathy. N. Engl. J. Med. 2020;383:321–333. doi: 10.1056/NEJMoa1916004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murthy A., Li Y., Peng I., Reichelt M., Katakam A.K., Noubade R., Roose-Girma M., DeVoss J., Diehl L., Graham R.R., van Lookeren Campagne M. A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506:456–462. doi: 10.1038/nature13044. [DOI] [PubMed] [Google Scholar]
- 28.Hansen J.M., de Jong M.F., Wu Q., Zhang L.-S., Heisler D.B., Alto L.T., Alto N.M. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell. 2021;184:3178–3191.e18. doi: 10.1016/j.cell.2021.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cervenka I., Agudelo L.Z., Ruas J.L. Kynurenines: tryptophan's metabolites in exercise, inflammation, and mental health. Science. 2017;357 doi: 10.1126/science.aaf9794. [DOI] [PubMed] [Google Scholar]
- 30.Bhattacharya S., Dunn P., Thomas C.G., Smith B., Schaefer H., Chen J., Hu Z., Zalocusky K.A., Shankar R.D., Shen-Orr S.S., et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci. Data. 2018;5 doi: 10.1038/sdata.2018.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan L.K., Gerstenlauer M., Konukiewitz B., Steiger K., Weichert W., Wirth T., Maier H.J. Epithelial NEMO/IKKgamma limits fibrosis and promotes regeneration during pancreatitis. Gut. 2017;66:1995–2007. doi: 10.1136/gutjnl-2015-311028. [DOI] [PubMed] [Google Scholar]
- 32.Rueda P., Richart A., Récalde A., Gasse P., Vilar J., Guérin C., Lortat-Jacob H., Vieira P., Baleux F., Chretien F., et al. Homeostatic and tissue reparation defaults in mice carrying selective genetic invalidation of CXCL12/proteoglycan interactions. Circulation. 2012;126:1882–1895. doi: 10.1161/CIRCULATIONAHA.112.113290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schiraldi M., Raucci A., Muñoz L.M., Livoti E., Celona B., Venereau E., Apuzzo T., De Marchis F., Pedotti M., Bachi A., et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012;209:551–563. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Y., Feng Y., Kang J., Liu C., Li Z., Li D., Cao W., Qiu J., Guo Z., Bi E., et al. Critical regulation of CD4+ T cell survival and autoimmunity by beta-arrestin 1. Nat. Immunol. 2007;8:817–824. doi: 10.1038/ni1489. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Z., Xu X., Tian W., Jiang R., Lu Y., Sun Q., Fu R., He Q., Wang J., Liu Y., et al. ARRB1 inhibits non-alcoholic steatohepatitis progression by promoting GDF15 maturation. J. Hepatol. 2020;72:976–989. doi: 10.1016/j.jhep.2019.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Bushman F.D., Conrad M., Ren Y., Zhao C., Gu C., Petucci C., Kim M.-S., Abbas A., Downes K.J., Devas N., et al. Multi-omic analysis of the interaction between Clostridioides difficile infection and pediatric inflammatory bowel disease. Cell Host Microbe. 2020;28:422–433.e7. doi: 10.1016/j.chom.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adolph T.E., Meyer M., Schwärzler J., Mayr L., Grabherr F., Tilg H. The metabolic nature of inflammatory bowel diseases. Nat. Rev. Gastroenterol. Hepatol. 2022;19:753–767. doi: 10.1038/s41575-022-00658-y. [DOI] [PubMed] [Google Scholar]
- 38.Scott S.A., Fu J., Chang P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA. 2020;117:19376–19387. doi: 10.1073/pnas.2000047117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu X., Deng J., Yu T., Chen S., Ge Y., Zhou Z., Guo Y., Ying H., Zhai Q., Chen Y., et al. ATF4 deficiency promotes intestinal inflammation in mice by reducing uptake of glutamine and expression of antimicrobial peptides. Gastroenterology. 2019;156:1098–1111. doi: 10.1053/j.gastro.2018.11.033. [DOI] [PubMed] [Google Scholar]
- 40.Morris S.M., Jr. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002;22:87–105. doi: 10.1146/annurev.nutr.22.110801.140547. [DOI] [PubMed] [Google Scholar]
- 41.Dowling J.K., Afzal R., Gearing L.J., Cervantes-Silva M.P., Annett S., Davis G.M., De Santi C., Assmann N., Dettmer K., Gough D.J., et al. Mitochondrial arginase-2 is essential for IL-10 metabolic reprogramming of inflammatory macrophages. Nat. Commun. 2021;12:1460. doi: 10.1038/s41467-021-21617-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakagawa T., Lomb D.J., Haigis M.C., Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma S., Bhattarai S., Ara H., Sun G., St Clair D.K., Bhuiyan M.S., Kevil C., Watts M.N., Dominic P., Shimizu T., et al. SOD2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox Biol. 2020;37 doi: 10.1016/j.redox.2020.101740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Z., Peng Y., Li J., Chen Z., Chen F., Tu J., Lin S., Wang H. N6-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat. Commun. 2020;11:2578. doi: 10.1038/s41467-020-16306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu H., Lin J., Xu C., Sun M., Zuo K., Zhang X., Li M., Huang H., Li Z., Wu W., et al. Cyclosporine modulates neutrophil functions via the SIRT6-HIF-1alpha-glycolysis axis to alleviate severe ulcerative colitis. Clin. Transl. Med. 2021;11:e334. doi: 10.1002/ctm2.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mzoughi S., Fong J.Y., Papadopoli D., Koh C.M., Hulea L., Pigini P., Di Tullio F., Andreacchio G., Hoppe M.M., Wollmann H., et al. PRDM15 is a key regulator of metabolism critical to sustain B-cell lymphomagenesis. Nat. Commun. 2020;11:3520. doi: 10.1038/s41467-020-17064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Houten S.M., Denis S., Argmann C.A., Jia Y., Ferdinandusse S., Reddy J.K., Wanders R.J.A. Peroxisomal L-bifunctional enzyme (Ehhadh) is essential for the production of medium-chain dicarboxylic acids. J. Lipid Res. 2012;53:1296–1303. doi: 10.1194/jlr.M024463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lally J.S.V., Ghoshal S., DePeralta D.K., Moaven O., Wei L., Masia R., Erstad D.J., Fujiwara N., Leong V., Houde V.P., et al. Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. 2019;29:174–182.e5. doi: 10.1016/j.cmet.2018.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimura A., Kitajima M., Nishida K., Serada S., Fujimoto M., Naka T., Fujii-Kuriyama Y., Sakamato S., Ito T., Handa H., et al. NQO1 inhibits the TLR-dependent production of selective cytokines by promoting IkappaB-zeta degradation. J. Exp. Med. 2018;215:2197–2209. doi: 10.1084/jem.20172024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van den Bogert B., Meijerink M., Zoetendal E.G., Wells J.M., Kleerebezem M. Immunomodulatory properties of Streptococcus and Veillonella isolates from the human small intestine microbiota. PLoS One. 2014;9 doi: 10.1371/journal.pone.0114277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexander M., Turnbaugh P.J. Deconstructing mechanisms of diet-microbiome-immune interactions. Immunity. 2020;53:264–276. doi: 10.1016/j.immuni.2020.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.York A., Lloyd A.J., Del Genio C.I., Shearer J., Hinxman K.J., Fritz K., Fulop V., Dowson C.G., Khalid S., Roper D.I. Structure-based modeling and dynamics of MurM, a Streptococcus pneumoniae penicillin resistance determinant present at the cytoplasmic membrane. Structure. 2021;29:731–742.e6. doi: 10.1016/j.str.2021.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tierney A.R., Rather P.N. Roles of two-component regulatory systems in antibiotic resistance. Future Microbiol. 2019;14:533–552. doi: 10.2217/fmb-2019-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Furusawa Y., Obata Y., Fukuda S., Endo T.A., Nakato G., Takahashi D., Nakanishi Y., Uetake C., Kato K., Kato T., et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 55.Kim M.H., Kang S.G., Park J.H., Yanagisawa M., Kim C.H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. 2013;145:396–406.e1-10. doi: 10.1053/j.gastro.2013.04.056. [DOI] [PubMed] [Google Scholar]
- 56.Lee J.Y., Hall J.A., Kroehling L., Wu L., Najar T., Nguyen H.H., Lin W.Y., Yeung S.T., Silva H.M., Li D., et al. Serum amyloid A proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell. 2020;183:2036–2039. doi: 10.1016/j.cell.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puthenedam M., Wu F., Shetye A., Michaels A., Rhee K.J., Kwon J.H. Matrilysin-1 (MMP7) cleaves galectin-3 and inhibits wound healing in intestinal epithelial cells. Inflamm. Bowel Dis. 2011;17:260–267. doi: 10.1002/ibd.21443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bergström J.H., Birchenough G.M.H., Katona G., Schroeder B.O., Schütte A., Ermund A., Johansson M.E.V., Hansson G.C. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc. Natl. Acad. Sci. USA. 2016;113:13833–13838. doi: 10.1073/pnas.1611400113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marek I., Lichtneger T., Cordasic N., Hilgers K.F., Volkert G., Fahlbusch F., Rascher W., Hartner A., Menendez-Castro C. Alpha8 integrin (Itga8) signalling attenuates chronic renal interstitial fibrosis by reducing fibroblast activation, not by interfering with regulation of cell turnover. PLoS One. 2016;11 doi: 10.1371/journal.pone.0150471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smillie C.S., Biton M., Ordovas-Montanes J., Sullivan K.M., Burgin G., Graham D.B., Herbst R.H., Rogel N., Slyper M., Waldman J., et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell. 2019;178:714–730.e22. doi: 10.1016/j.cell.2019.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Erickson A., He M., Berglund E., Marklund M., Mirzazadeh R., Schultz N., Kvastad L., Andersson A., Bergenstråhle L., Bergenstråhle J., et al. Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature. 2022;608:360–367. doi: 10.1038/s41586-022-05023-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alder M.N., Opoka A.M., Lahni P., Hildeman D.A., Wong H.R. Olfactomedin 4 is a candidate marker for a pathogenic neutrophil subset in septic shock. Crit. Care Med. 2017;45:e426–e432. doi: 10.1097/CCM.0000000000002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Buck M., Gouwy M., Wang J.M., Van Snick J., Opdenakker G., Struyf S., Van Damme J. Structure and expression of different serum amyloid A (SAA) variants and their concentration-dependent functions during host insults. Curr. Med. Chem. 2016;23:1725–1755. doi: 10.2174/0929867323666160418114600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye R.D., Sun L. Emerging functions of serum amyloid A in inflammation. J. Leukoc. Biol. 2015;98:923–929. doi: 10.1189/jlb.3VMR0315-080R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F., et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Douglas G.M., Maffei V.J., Zaneveld J.R., Yurgel S.N., Brown J.R., Taylor C.M., Huttenhower C., Langille M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020;38:685–688. doi: 10.1038/s41587-020-0548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Friedman J., Alm E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012;8 doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shenhav L., Thompson M., Joseph T.A., Briscoe L., Furman O., Bogumil D., Mizrahi I., Pe'er I., Halperin E. FEAST: fast expectation-maximization for microbial source tracking. Nat. Methods. 2019;16:627–632. doi: 10.1038/s41592-019-0431-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davis N.M., Proctor D.M., Holmes S.P., Relman D.A., Callahan B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McMurdie P.J., Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8 doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Futschik M.E., Carlisle B. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 2005;3:965–988. doi: 10.1142/s0219720005001375. [DOI] [PubMed] [Google Scholar]
- 73.Dixon P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003;14:927–930. [Google Scholar]
- 74.Szklarczyk D., Gable A.L., Nastou K.C., Lyon D., Kirsch R., Pyysalo S., Doncheva N.T., Legeay M., Fang T., Bork P., et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49:D605–D612. doi: 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krämer A., Green J., Pollard J., Jr., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- 78.Sturm A., Maaser C., Calabrese E., Annese V., Fiorino G., Kucharzik T., Vavricka S.R., Verstockt B., van Rheenen P., Tolan D., et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 2: IBD scores and general principles and technical aspects. J. Crohns Colitis. 2019;13:273–284. doi: 10.1093/ecco-jcc/jjy114. [DOI] [PubMed] [Google Scholar]
- 79.Maaser C., Sturm A., Vavricka S.R., Kucharzik T., Fiorino G., Annese V., Calabrese E., Baumgart D.C., Bettenworth D., Borralho Nunes P., et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: initial diagnosis, monitoring of known IBD, detection of complications. J. Crohns Colitis. 2019;13:144–164. doi: 10.1093/ecco-jcc/jjy113. [DOI] [PubMed] [Google Scholar]
- 80.Gao H., Zhang F., Liang S., Zhang Q., Lyu M., Qian L., Liu W., Ge W., Chen C., Yi X., et al. Accelerated lysis and proteolytic digestion of biopsy-level fresh-frozen and FFPE tissue samples using pressure cycling technology. J. Proteome Res. 2020;19:1982–1990. doi: 10.1021/acs.jproteome.9b00790. [DOI] [PubMed] [Google Scholar]
- 81.Meier F., Brunner A.D., Frank M., Ha A., Bludau I., Voytik E., Kaspar-Schoenefeld S., Lubeck M., Raether O., Bache N., et al. diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods. 2020;17:1229–1236. doi: 10.1038/s41592-020-00998-0. [DOI] [PubMed] [Google Scholar]
- 82.Cai X., Ge W., Yi X., Sun R., Zhu J., Lu C., Sun P., Zhu T., Ruan G., Yuan C., et al. PulseDIA: data-independent acquisition mass spectrometry using multi-injection pulsed gas-phase fractionation. J. Proteome Res. 2021;20:279–288. doi: 10.1021/acs.jproteome.0c00381. [DOI] [PubMed] [Google Scholar]
- 83.Meier F., Brunner A.D., Koch S., Koch H., Lubeck M., Krause M., Goedecke N., Decker J., Kosinski T., Park M.A., et al. Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteomics. 2018;17:2534–2545. doi: 10.1074/mcp.TIR118.000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu T., Chen H., Yan X., Wu Z., Zhou X., Xiao Q., Ge W., Zhang Q., Xu C., Xu L., et al. ProteomeExpert: a docker image based web-server for exploring, modeling, visualizing, and mining quantitative proteomic data sets. Bioinformatics. 2021;37:273–275. doi: 10.1093/bioinformatics/btaa1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F., et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:3. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 87.Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Price M.N., Dehal P.S., Arkin A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bokulich N.A., Kaehler B.D., Rideout J.R., Dillon M., Bolyen E., Knight R., Huttley G.A., Gregory Caporaso J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2's q2-feature-classifier plugin. Microbiome. 2018;6:90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Salter S.J., Cox M.J., Turek E.M., Calus S.T., Cookson W.O., Moffatt M.F., Turner P., Parkhill J., Loman N.J., Walker A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12 doi: 10.1186/s12915-014-0087-z. 87–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Douglas G.M., Maffei V.J., Zaneveld J., Yurgel S.N., Brown J.R., Taylor C.M., Huttenhower C., Langille M.G.I. PICRUSt2: an improved and extensible approach for metagenome inference. bioRxiv. 2020 doi: 10.1101/672295. Preprint at. [DOI] [Google Scholar]
- 93.Wu Y., Jiao N., Zhu R., Zhang Y., Wu D., Wang A.J., Fang S., Tao L., Li Y., Cheng S., et al. Identification of microbial markers across populations in early detection of colorectal cancer. Nat. Commun. 2021;12:3063. doi: 10.1038/s41467-021-23265-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ginestet C. ggplot2: elegant graphics for data analysis. J. R. Stat. Soc. 2011;174:245–246. doi: 10.1111/j.1467-985X.2010.00676_9.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in this paper. The proteomics data reported in this paper have been deposited in ProteomeXchange Consortium with accession iProX: IPX0004848000. The 16S rRNA sequencing data reported in this paper have been deposited in The National Omics Data Encyclopedia (NODE) with accession ID OEP003464 (intestine-associated tissue samples) and OEP003463 (fecal samples), respectively. Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.