

Summary
Chimeric antigen receptor (CAR) T cells demonstrate remarkable success in treating hematological malignancies, but their effectiveness in non-hematopoietic cancers remains limited. This study proposes enhancing CAR T cell function and localization in solid tumors by modifying the epigenome governing tissue-residency adaptation and early memory differentiation. We identify that a key factor in human tissue-resident memory CAR T cell (CAR-TRM) formation is activation in the presence of the pleotropic cytokine, transforming growth factor β (TGF-β), which enforces a core program of both “stemness” and sustained tissue residency by mediating chromatin remodeling and concurrent transcriptional changes. This approach leads to a practical and clinically actionable in vitro production method for engineering peripheral blood T cells into a large number of “stem-like” CAR-TRM cells resistant to tumor-associated dysfunction, possessing an enhanced ability to accumulate in situ and rapidly eliminate cancer cells for more effective immunotherapy.
Keywords: chimeric antigen receptor, CAR T-cells3, hematological malignancies, non-hematopoietic cancers, epigenetic modification, tissue residency, memory differentiation, transforming growth factor-β, immunotherapy, solid tumors
Graphical abstract
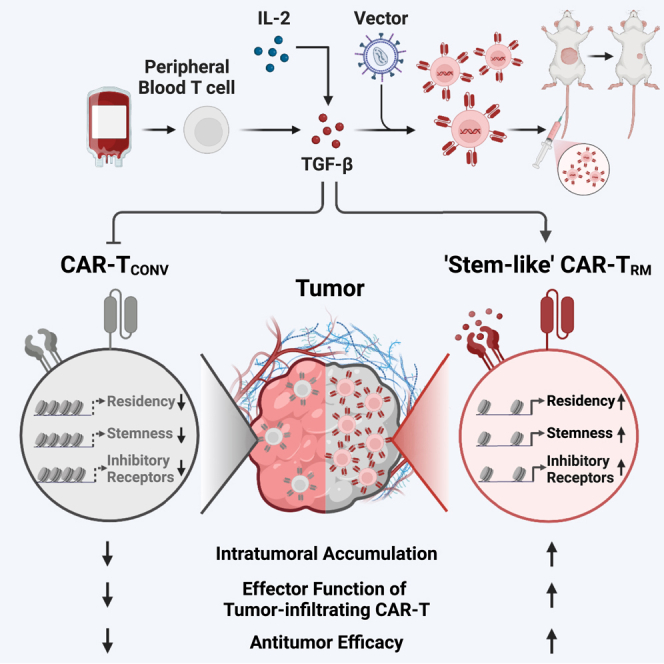
Highlights
-
•
TGF-β induces CAR-TRM cells with enhanced tumor infiltration
-
•
Chromatin changes via TGF-β create a stem-like resident memory fate
-
•
CAR-TRM cells resist tumor-imposed dysfunction
-
•
CAR-TRM cells show potent efficacy in solid and liquid cancers
CAR T cells exhibit promise in cancer immunotherapy. Jung et al. show that combining CAR engineering with TGF-β exposure in ex vivo blood T cells enhances stem-like properties and tissue residency, which is linked to chromatin and transcriptional changes. This results in improved tumor infiltration and enhanced efficacy in various cancer models.
Introduction
The adoptive transfer of chimeric antigen receptor (CAR) T cells has demonstrated significant success in treating hematopoietic malignancies.1,2,3,4,5,6,7,8 Given this advancement in the field of cellular immunotherapy, a growing number of trials now focus on solid tumors, which are responsible for more than three-quarters of cancer-related deaths worldwide.9 However, to date, therapeutic successes in these indications, including results from our own clinical studies, have been modest (reviewed in Nguyen10 and Martinez11). The reasons for poor efficacy are unknown but likely multifactorial. Unlike the situation in hematologic malignancies, CAR T cells must successfully localize from the blood to solid tumor sites despite potential T cell-/tumor-derived chemokine mismatches12,13 and subsequently infiltrate cancer-derived extracellular matrix components to elicit cytotoxicity,14 frequently in the setting of antigen heterogeneity or loss.15 Even with adequate trafficking and infiltration, engineered T cells become rapidly hypofunctional in the tumor microenvironment (TME).16,17 In addition to tumor-mediated immunosuppression, lack of therapeutic levels of in vivo CAR T cell expansion, local persistence at tumor sites, and elicitation of robust effector functions are attributed to T cell-intrinsic mechanisms that hamper intratumoral accumulation, resulting in treatment resistance and disease progression.18,19
Attrition of CAR T cell-intrinsic memory functions has been implicated in poor CAR T cell persistence and lack of durable tumor control.20 Memory CD8+ T cell subsets can be generally categorized into circulating stem cell memory (TSM), central memory (TCM), and effector memory (TEM) T cells,21 in addition to tissue-resident memory (TRM) T cells, which mainly persist in non-lymphoid compartments without recirculation.22,23,24,25 Current CAR T cell production methods incorporating circulating T cells have been heavily influenced by use in hematological malignancies where the ideal CAR T cells would traffic to secondary lymphoid organs and bone marrow. Thus, TSM and TCM with high levels of homing receptor expression and the ability to persist in lymphoid compartments have been favored. However, for solid tumor indications, CAR T cells need to localize within non-lymphoid tissues, persist, and maintain durable antitumor activity. Thus, changing the phenotype of conventional circulating CAR T cells to more efficiently accumulate, persist, and maintain potency in the TME is an important goal.
Large fractions of tumor-infiltrating lymphocytes (TILs) are composed of TRM cells, with the degree of TRM infiltration directly correlating with effective endogenous antitumor responses as well as successful immunotherapy.26,27,28,29 CD8+ TRM cells upregulate multiple adhesion molecules, including CD103 and CD49a, to enable T cell interactions with tumor cells and the surrounding extracellular matrix. In addition, TRM cells decrease expression of migratory pathways governed by Krüppel-like factor 2 (KLF2), which dictate T cell commitment to recirculation.30 These specific molecular features enable TRM cells to accumulate at tumor sites and exert robust effector functions.
Transcription factors such as RUNX3 and cytokines including transforming growth factor beta (TGF-β), interleukin (IL)-7, and IL-15 regulate the differentiation, localization and function of long-lived CD8+ T cells residing in healthy non-lymphoid tissues.31,32,33 In recent studies, TRM cells possessing characteristics of “stemness” were observed in normal peripheral tissues and tumor-draining lymph nodes of syngeneic mice.34,35 These stem-like TRM cells exhibit superior polyfunctionality as well as memory functions relative to their conventional resident T cell counterparts and can give rise to fully differentiated TRM progeny cells in an antigen-dependent manner. Neoantigen-specific TILs possessing these characteristics have been implicated in mediating effective antitumor immunity in the setting of immune checkpoint blockade (ICB) therapies.36 Thus, the differentiation trajectory of CAR T cells can be modulated through targeted chromatin remodeling and subsequent transcriptional changes, a process referred to as epigenetic reprogramming. By inducing these changes, we aim to create populations of CAR T cells that efficiently accumulate in tumors while maintaining robust effector functions and self-renewal capacity, potentially representing a promising treatment modality for certain cancers.
Here, we intended to enhance CAR T cell accumulation and function in solid tumors through modulation of TRM fate specificity. In this vein, we explored different methods to endow human CAR T cells with both early memory features and the ability to establish tissue residency. We report the preclinical development of the proposed CAR-TRM cell therapy evaluated in both solid and liquid cancer models, in addition to exploring the molecular signals promoting tissue residency, exhaustion, and early memory T cell differentiation.
Results
TGF-β induces resident memory CAR T cell differentiation
TGF-β has been implicated in TRM cell differentiation in non-lymphoid tissues.37,38,39 Because CAR T cells are generally produced in vitro after activation through CD3 and CD28 ligation, we hypothesized that the presence of TGF-β during CAR T cell production would enhance the formation of engineered TRM cells. To evaluate the impact of TGF-β on the phenotype of CAR T cells, different levels of TGF-β were treated during the manufacture of anti-mesothelin (M5) CAR T cells (Figure S1A). The expression of resident memory markers progressively increased as the concentration of TGF-β was increased and reached saturation at concentrations above 2 ng/mL (Figure S1B). However, the expansion capacity of the CAR T cells was compromised at 10 ng/mL (Figure S1C). To determine the optimal duration of TGF-β treatment, we tested different durations (Figure S1D). The addition of TGF-β after day 3 significantly reduced the development of the resident memory population (Figure S1E), and the CAR T cell expansion was consistent regardless of the duration of TGF-β exposure (Figure S1F). Based on these results, we used a concentration of 2 ng/mL TGF-β starting on day 0 in subsequent experiments. We then characterized the phenotype of M5 CAR T cells produced under these optimal culture conditions. Compared with conventional CAR T cells (CAR-TCONV) cultured in IL-2 alone, CAR T cells that were also exposed to TGF-β (CAR-TRM) upregulate several classical TRM markers, including CD103, CD39, and CD49a (Figures 1A and 1B). In addition to these adhesion molecules, increased expression of inhibitory receptors is a hallmark of TRM cells.37,38,40,41 Consistent with this previous finding, CAR-TRM cells showed elevated expression of PD1, LAG3, and TIM3 inhibitory receptors as well as the exhaustion-related transcription factor, TOX (Figure 1C). Intriguingly, unlike differentiated effector-like resident memory T cells, CAR-TRM products express high levels of proteins that play a central role in maintaining stemness, such as TCF1 and IL7R and the lymph node homing receptor, CCR7, suggesting that TGF-β exposure reprograms T cells toward a stem-like resident memory fate (Figure 1D).31,34,35
Figure 1.

Phenotypic and functional characterization of CAR-TRM cells
CAR T cells were cultured in T cell medium with or without 2 ng/mL TGF-β. (A) Representative flow cytometry plot showing CD39 and CD103 expression on CD8+ CAR T cells after manufacturing.
(B–D) Expression markers associated with (B) residency (CD39, CD103, CD49a), (C) exhaustion (PD1, LAG3, TIM3, TOX), and (D) memory/stemness (TCF1, IL7R, CCR7) in CD8+ CAR T cells.
(E) Anti-mesothelin M5 CAR T cells were co-cultured with AsPC-1 tumor targets at an effector to target (E:T) ratio of 1:2. Cytolytic activity of CAR-TCONV and CAR-TRM cells was measured in real time using the xCELLigence RTCA system. Cytotoxic capacity at 7 h post co-culture is shown.
(F and G) M5 CAR T cells were challenged with AsPC-1 targets at an E:T ratio of 2:1. Supernatant was collected at 18 h after coincubation, and cytokine levels were measured. (F) Granzyme A; (G) effector cytokines (IFN-γ, IL-2, TNFα, IL-6).
(H) CAR T cell expansion levels were measured during ex vivo manufacturing. Data represent results from four independent CAR T cell manufacturing experiments (paired t test). All in vitro experiments were performed using CAR T cells derived from various healthy subjects as biological replicates (n = 4–6). (A–G) Representative results from one donor. ∗p < 0.05; ∗p < 0.01; ∗∗∗p < 0.001; ns, not significant (Mann-Whitney U test).
TGF-β is known to have immunosuppressive effect on T cells.42,43 We co-cultured M5 CAR T cells with the target pancreatic cancer cell, AsPC-1, to assess the antigen-dependent effector function of CAR-TRM cells. Despite a marked increase in the expression of CD103, which is known be associated with enhanced cytolytic activity of resident memory T cells,44 CAR-TRM cell cytotoxic function is diminished (Figures 1E and 1F). However, CAR-TRM cells show enhanced production of multiple effector cytokines (i.e., interferon gamma [IFN-γ], IL-2, tumor necrosis factor alpha [TNFα], IL-6), compared with CAR-TCONV cells and exhibit a comparable level of ex vivo expansion during production, implying that proliferative capacity and certain T cell effector functions are not compromised by TGF-β (Figures 1G and 1H).
The function of CAR-TRM cells was also evaluated during chronic antigen stimulation. Despite having lower cytolytic activity compared with CAR-TCONV cells, CAR-TRM cells showed a high production of IFN-γ, TNFα, and IL-6 and an increased proliferative capacity after repetitive CAR activation (Figures S2A–S2C). To determine the susceptibility of CAR T cells that were exposed to TGF-β during manufacturing to high levels of TGF-β present in the TME, CAR-TCONV, and CAR-TRM cells were co-incubated with target pancreatic cancer cells, with or without exogenous TGF-β. The results showed that both CAR-TCONV and CAR-TRM cells decreased in cytotoxicity and effector cytokine production when exposed to TGF-β (Figures S2D and S2E). However, CAR-TRM cells maintained higher levels of effector cytokine production even after exposure to TGF-β, compared with CAR-TCONV cells. This suggests that adding TGF-β during CAR T cell manufacturing does not make the CAR T cells more susceptible to the immunosuppressive effects of TGF-β.
In recent studies, an increase in the population of regulatory CAR T cells (CAR-TREG) cells has been linked to resistance to CD19 CAR T cell therapy.45,46 To assess whether TGF-β treatment during CAR T cell manufacturing could lead to the development of CAR-TREG cells, we analyzed the frequencies of FOXP3+CD4+ or FOXP3+IL2RA+ CD4+ or CD8+ cells between CAR-TCONV cells and CAR-TRM populations in multiple healthy subjects. Our results showed no significant differences, implying that transient exposure to TGF-β at the 2 ng/mL concentration used to program residency may not be sufficient to induce CAR-TREG cells (Figure S2F), which is concordant with the findings of a previous study.47
CAR-TRM cells exhibit a resident memory transcriptional program
Flow cytometric analysis of CAR-TRM cells reveals acquisition of a resident memory phenotype through TGF-β signaling. To expand this finding, we sought to obtain a comprehensive view of the transcriptional landscape of CAR-TRM cells by performing bulk RNA sequencing (RNA-seq). Principal-component analysis of transcriptome data indicated that TGF-β exposure was the strongest driver of transcriptional variance across multiple donors (Figure 2A). Consistent with our flow cytometry data, CAR-TRM cells are significantly enriched in CD69+CD103+CD8+ resident memory T cell gene signatures and exhibit reduced CD69−CD103−CD8+ non-resident memory T cell transcriptional profiles (Figure 2B). Specifically, CAR-TRM express high levels of resident memory-associated genes encoding cell surface molecules (ITGAE, PDCD1) and transcription factor genes (BHLHE40, EGR2, NR4As, IRF4) while downregulating KLF2, which is essential for T cell recirculation and inhibition of TRM cell differentiation (Figures 2C and 2D).30 This increased resident memory transcriptional program in CAR-TRM cells may be partially attributed to hypoxia pathway upregulation, which is known to facilitate resident memory T cell differentiation, indicating a possible overlap between TGF-β signaling and responses to low oxygen tension (Figures 2E and S3).44,48
Figure 2.

CAR-TRM cells possess a core resident memory transcriptomic signature
Bulk RNA-seq experiments were conducted using CD8+ CAR-TCONV and CAR-TRM cells.
(A) Principal-component analysis was carried out with RNA expression data.
(B) Gene set enrichment analysis (GSEA) using gene sets associated with resident memory T cells.
(C) Heatmap showing differentially expressed genes from CD69+CD103+ and CD69−CD103−CD8+ T cell signatures.
(D) Heatmap displaying expression of transcription factor genes known to promote and suppress resident memory T cell differentiation.
(E) GSEA (hypoxia pathways: MSigDB C2).
(F) Pathway analysis incorporating genes upregulated in CAR-TRM-cells using Enrichr.
(G) GSEA analysis (migration pathways: MSigDB C2). GSEA with gene signatures of tumor-infiltrating T cells associated with response to ICB. Bulk RNA-seq experiments were conducted using CAR T cells derived from n = 3 distinct healthy donors, serving as biological replicates.
One of the major barriers that limits the antitumor potency of cell therapies derived from circulating lymphocytes is poor trafficking and infiltration to tumors surrounded by an extracellular matrix (ECM). CAR-TRM cells upregulate genes encoding integrins, laminin, type I heparan sulfate proteoglycan, and matrix metalloproteinases (MMPs), which degrade various ECM proteins and enriched pathways related to ECM regulation and cell migration (Figures 2F and 2G; Table S1). Accordingly, tumor-infiltrating T cells (TILs) that clonally expand in response to ICB therapy exhibit a tissue-resident memory transcriptional program in responding oral cancer patients.49 We thus sought to examined whether TGF-β-generated CAR-TRM cells share transcriptional features of these favorable resident memory subsets observed in ICB responders. Gene set enrichment analysis indicates that CAR-TRM cells significantly upregulate signatures enriched in pre- and post-treatment TILs that expand upon response to anti-programmed cell death protein 1 (PD-1)/cytotoxic T lymphocyte–associated protein 4 (CTLA-4) treatment, while downregulating gene sets found in tumor-infiltrating T cells that fail to respond to ICB (Figure 2H). Altogether, our bulk RNA-seq analyses suggest that CAR-TRM cells acquire a favorable and clinically relevant transcriptional program of tissue residency.
CAR-TRM cells show transcriptional features of stem-like resident memory T cells
Our bulk analyses indicate that CAR-TRM cells elevate markers of both tissue residency and T cell stemness (Figures 1B and 1D). We further explored how these residency- and stemness-related transcriptional programs are regulated in CAR-TRM cells using single-cell RNA-seq (scRNA-seq). CAR-TCONV and CAR-TRM were harvested after 9 days of expansion and processed for scRNA-seq. After removal of low-quality cells, 12,230 cells were obtained. Uniform Manifold Approximation and Projection (UMAP) clustering of CD8+ T cells reveal three clusters with discrete transcriptional features (Figure 3A). Notably, a TCF7+CD8+ CAR-TRM subset expresses high levels of stemness-related genes such as TCF7 and IL7R (Figures 3B, S4A, and S4B). Consistent with this gene expression profile, TCF7+CD8+ CAR-TRM cells are enriched in a core transcriptional program of stem-like CD8+ T cells possessing self-renewal potential (Figure S4C). In addition, MKI67+CD8+ CAR-TRM cells upregulate G2M and S phase pathways as well as other cell cycle-related genes (MKI67, CDCA8, CDCA6, CDKN3, CDK1, CDC25B) while downregulating memory-related genes and signatures associated with successive linear T cell differentiation (Figures 3A, 3B, and S4A–S4C). These findings are consistent with the proposed model and support the idea that MKI67+CD8+ CAR T cells predominantly represent the replicating fraction. CD27+CD8+ CAR-TRM cells exhibit intermittent transcriptional features between those of TCF7+CD8+ and MKI67+CD8+ populations, as manifested by elevated expression of central memory T cell genes (CD27, SELL), along with T cell activation and cytotoxic effector genes (HLA-B, GZMA, HLA-B, HLA-A, CD69) (Figure 3B). In accordance with a previous report of TGF-β preserving TCF1+ precursor cells in chronic infection,50 CAR-TRM cells are enriched in stem-like TCF7+CD8+ T cells with concordant gene signatures and composed of reduced frequencies of more differentiated subsets (e.g., CD27+CD8+ and MKI67+CD8+) compared with the CAR-TCONV counterpart (Figures 3A and S4C).
Figure 3.

TGF-β-generated CAR-TRM cells transcriptionally resemble stem-like TRM cells
(A) UMAP imbedding of CD8+CAR T cell (top, CD8 subclusters; middle, CAR T cell samples; bottom, cell cycle). Frequencies of three CD8+ clusters in CAR-TCONV and CAR-TRM cells are shown.
(B) Bubble plot displaying expression levels of marker genes in each CD8+ subcluster.
(C) Violin plots showing expression of KLF2 and ITGAE.
(D) CD69−CD103− and CD69+CD103+CD8+ T cell signature scores calculated for CAR-TCONV and CAR-TRM cells.
(E) A UMAP plot showing CAR T cell sample origin in TCF7+CD8+T cells.
(F) Expression of resident memory (ITGAE, BHLHE40, CCL5) and early memory (IL7R) markers in TCF7+CD8+ T cells.
(G) Module score comparison for CD69−CD103− and CD69+CD103+ CD8+ T cell signatures between CAR-TCONV and CAR-TRMTCF7+CD8+ T cells.
As observed in bulk RNA-seq analysis, CAR-TRM cells increase expression of resident memory T cell genes and related signatures and reduce KLF2 and CD69−CD103−CD8+ gene set expression (Figures 3C and 3D; Table S2). A series of recent studies demonstrated that stem-like progenitor and resident memory cells that persist in tumors give rise to the proliferative burst of neoantigen-specific T cells and drive antitumor activity in response to ICB blockade.27,36,49,51,52 These reports suggest that stemness and resident memory programs could be optimal transcriptional features for cellular immunotherapies. Thus, we asked whether the resident memory transcriptional program is preserved in the stem-like CAR-TRM cells by conducting gene expression and pathway analysis in the TCF7+CD8+ cluster. Similar to what we observed in bulk CD8+ populations, CAR-TRM increased resident memory-associated genes (e.g., ITGAE, BHLHE40, CCL5) and TRM transcriptional signatures as well as early memory genes (IL7R, STAT3), whereas it downregulated genes associated with circulation (e.g., KLF2) and non-resident memory signatures (Figures 3E–3G; Table S2). These data suggest that TGF-β-produced CAR-TRM cells harbor a pool of stem-like cells, which could potentially persist and give rise to large numbers of differentiated resident memory progeny cells in tumors.34,35
Epigenetic reprogramming of stem-like CAR-TRM cells sustains a persistent resident memory phenotype during chronic antigen stimulation
Epigenetic modification leads to durable changes in gene expression, sets thresholds for future gene transcription, and plays an important role stabilizing T cell differentiation fate.53,54,55,56 In this regard, we investigated whether transient exposure to TGF-β during CAR T cell production can induce epigenetic rewiring of CAR-TRM cells to sustain the acquired stem-like resident memory phenotype. We thus conducted assay for transposase-accessible chromatin with sequencing (ATAC-seq) to assess the global chromatin accessibility of CAR-TCONV and CAR-TRM cells. As seen in our transcriptional studies, CAR-TRM differentiation mediates the largest epigenetic variance across donors and induces global chromatin remodeling (Figures 4A and 4B). Gene set enrichment analysis with differentially accessible regions suggests that CAR-TRM cells are enriched in a CD69+CD103+CD8+ signature while downregulating a CD69−CD103−CD8+ chromatin landscape profile (Figures 4C and 4D). Consistent with these observations, chromatin accessibility is significantly increased at resident memory gene loci (e.g., ITGAE, ITGAV, ENTPD1, IRF4) and decreased at non-resident memory genes (e.g., KLF2) in CAR-TRM cells (Figures 4E and 4F). Additionally, peaks were enriched in stemness-related gene regions such as TCF7 and IL7R in CAR-TRM cells (Figure 4G). Based on these findings, we then asked whether this epigenetic rewiring of stem-like CAR-TRM cells can sustain a resident memory phenotype during chronic antigen stimulation. We therefore repetitively challenged mesothelin-directed CAR-TRM cells with irradiated AsPC-1 tumor targets without additional supplementation of exogenous TGF-β and assessed their resident memory phenotype over time. Notably, TGF-β-generated CAR-TRM cells maintain both tissue-resident (CD103+CD39+CD8+ and CD103+CD49a+CD8+) and stem-like resident memory (CD103+IL7R+CD8+) phenotypes following repetitive tumor challenge, suggesting that epigenetic reprogramming may contribute to the stability of these differentiation fates (Figures 4H and S5).
Figure 4.

Chronically stimulated stem-like CAR-TRM cells sustain a resident memory phenotype in association with epigenetic reprogramming
(A) Principal-component analysis of ATAC-seq data.
(B) Volcano plot demonstrating differentially accessible peaks between CAR-TRM and CAR-TCONV CD8+ cells.
(C–G) GSEA showing enrichment of (C) CD69+CD103+CD8+ and (D) CD69−CD103−CD8+ T cell signatures. ATAC-seq tracks of genes associated with (E) resident memory T cells (ITGAE, ITGAV, ENTPD1, IRF4), (F) circulating T cells (KLF2), and (G) stemness (TCF7, IL7R). Differentially accessible regions are highlighted in blue.
(H) M5 CAR T cells were repetitively challenged with AsPC1 cells, and frequencies of resident memory (CD103+CD39+CD8+) and stem-like resident memory (CD103+IL7R+CD8+) T cells are shown over time. Bulk ATAC-seq experiments were performed using manufactured CD8+ CAR T cells from n = 2 healthy donors as biological replicates. Restimulation assays were performed with CAR T cells generated from n = 3 distinct healthy subjects, also serving as biological replicates. Results from one representative donor are shown.
CAR-TRM cells exhibit transcriptional and epigenetic features of exhaustion but show enhanced effector function
One of the major barriers to efficacious CAR T cell therapy is T cell exhaustion, which is characterized by sustained expression of multiple inhibitory checkpoint receptors, reduced proliferative capacity, and diminished antitumor effector functions.57,58,59 TRM cells are known to exhibit certain transcriptional features of T cell exhaustion, such as upregulation of inhibitory receptor encoding genes.44,60 Given that CAR-TRM cells show increased expression of PD1, LAG3, TIM3, as well as the exhaustion-related transcription factor, TOX (Figure 1C), we sought to further investigate the transcriptional and epigenetic properties of exhaustion in CAR-TRM cells. We first conducted motif analysis to identify transcription factors involved in CAR-TRM cell differentiation and found that basic leucine zipper (bZIP) and interferon regulatory factor (IRF) binding sites were the most significantly enriched in CAR-TRM cells, which is an epigenetic hallmark of exhausted CAR T cells (Figure 5A).61 Consistent with these findings, transcriptional analysis reveals that several bZIP and IRF transcription factor genes (ATF3, IRF4, ATF4, BATF3, ATF2, ATF6) were highly elevated in CAR-TRM cells, while canonical AP-1 transcription factors (JUN, FOS), which are required for optimal T cell expansion and effector function, were downregulated (Figure 5B). Transcriptional signatures associated with human T cell exhaustion were also enriched in CAR-TRM cells (Figure 5C). We investigated whether the enrichment of exhaustion signatures is specific to a particular subpopulation of CD8+ CAR-TRM cells. We compared the exhaustion signature scores of CAR-TCONV and CAR-TRM cells within each CD8+ CAR T cell subset identified in scRNA-seq. Our results showed that, in all three CD8 subsets, CAR-TRM cells displayed an upregulation of exhaustion pathways compared with CAR-TCONV cells. This suggests that the programming of TRM cells regulates exhaustion independently of the cell state (Figures S6A and S6B). Altogether, these results indicate that despite sustained antigen-specific proliferation and enhancement of effector cytokine production (Figures 1G and 1H), CAR-TRM cells exhibit several epigenetic and transcriptional signs of exhaustion (Figures 5B and 5C). The enhanced effector cytokine production capability of CAR-TRM cells can be partly attributed to the distinctive transcriptional and epigenetic characteristics that set CAR-TRM cells apart from terminally exhausted T cells. Notably, CAR-TRM cells exhibit decreased expression of PRDM1 (logFC = −1.26, p < 0.001), ID2 (logFC = −0.34, p = 0.007), and TOX2 (logFC = −0.73, p = 0.002), and increased transcript levels of ID3 (logFC = 0.61, p = 0.007) (Table S1). PRDM1, ID2, and TOX2 are known to promote terminal differentiation, T cell exhaustion, CAR T cell dysfunction, and loss of stemness and polyfunctionality,19,62,63,64,65,66 while ID3 has been shown to preserve stemness, enhance IL-2 and TNFα production, and prevent terminal exhaustion.34,67 Correspondingly, CAR-TRM cells demonstrate reduced chromatin accessibility at PRDM1, ID2, and TOX2 gene regions (Figure S6C).
Figure 5.

Highly functional CAR-TRM cells appear to be transcriptionally and epigenetically exhausted
(A) Top transcription factor motifs enriched in CAR-TRM cells.
(B) Heatmap showing expression of bZIP and IRF transcription factor genes (left) and genes associated with CAR T cell exhaustion in CD8+CAR T cells (right).
(C) GSEA showing enrichment of exhaustion signatures.
(D) The frequency of exhausted CAR T cells (PD1+TOX+ CD8+) is shown (two-way ANOVA, n = 3 biological replicates).
(E and F) M5 CAR T cells were challenged with AsPC-1 tumor cells at an E:T ratio of 2:1. Supernatants were collected at 18 h after coincubation and cytokine levels measured. (E) IL-2 production levels (two-way ANOVA, n = 4 biological replicates). (F) Heatmap showing effector cytokine and cytotoxic molecule production levels.
(G) Cytolytic activity of M5 CAR T cells was determined by coculturing the cells with Capan-2 cells at an effector to target ratio of 1:6 after three antigen stimulations. The normalized cell index at 31 h post co-culture was measured using the xCELLigence RTCA system. Results are shown as mean ± SEM of replicates (two-way ANOVA).
(H) Frequencies of resident memory CAR T cells (CD103+ or CD49a+ CD8+) are shown (two-way ANOVA, n = 3 biological replicates). ∗p < 0.05; ∗p < 0.01; ∗∗∗p < 0.001; ns., not significant.
Resident memory and exhaustion-related transcriptomic programs share several transcription factors, such as IRF4, NR4A family genes, and EGR2 (Figure 5B).38,68,69,70 To gain a deeper understanding of the role of these transcription factors on the regulation of T cell residency and exhaustion programs, we deleted these transcription factor genes in CAR-TCONV and CAR-TRM cells and assessed the immunophenotype of these knockout variants. Interestingly, IRF4 deletion significantly decreased the number of PD1+TOX+CD8+ T cells (Figure 5D). Furthermore, IRF4 knockout resulted in higher production of effector cytokines (IL-2, IFN-γ) and cytotoxic molecules (perforin, granzyme A) in CAR-TRM cells, enhancing their cytolytic capability, although IRF4-deficient CAR T cells showed a decreased capacity to proliferate (Figures 5E–5G and S6D). Moreover, IRF4 knockout CAR-TRM cells maintained a resident memory phenotype, showing a comparable level of CD103 expression and increased expression of CD49a compared with the AAVS1-deleted CAR-TRM control (Figure 5H). Collectively, these studies demonstrate that IRF4 promotes T cell exhaustion and is dispensable for induction of the resident memory phenotype in CAR-TRM cells.
Stem-like CAR-TRM cells demonstrate superior antitumor potency against solid and liquid tumors
We next examined the in vivo antitumor activity of CAR-TRM cells in both solid and blood cancer models. M5 anti-mesothelin CAR-TRM cells exhibit remarkable tumor control compared with CAR-TCONV cells, as demonstrated in both a mesothelin-expressing xenogeneic AsPC-1 pancreatic tumor model (Figure 6A) and in the suppression of mesothelin-expressing Capan-2 pancreatic ductal adenocarcinoma (Figure 6B). To understand the phenotype of intratumoral CAR T cells, we harvested tumors on day 13 post CAR T cell injection, when the tumor size was similar among experimental groups (∼600 mm3). We found that the improved antitumor activity was due to a significant increase in the number of CAR-TRM cells within the tumors (Figure 6C). This increase was partially due to improved early localization of the T cells in the tumor, as a higher number of CAR-TRM cells were observed as early as 2 days after CAR T cell infusion (Figure S7A). Both CAR-TCONV and CAR-TRM cells are highly enriched with CD103+CD39+CD8+ TILs, suggesting that CAR T cells with a resident memory phenotype preferentially accumulate at tumor sites (Figure 6D). Notably, mice treated with CAR-TRM cells show a significant increase in the absolute number of tumor-infiltrating CD103+CD39+CD8+ cells (Figure 6D), which indicates that the TGF-β-induced stem-like resident memory population can give rise to a large number of resident memory CAR-TILs. Consistent with our in vitro results, CAR-TRM cells in the tumor show upregulated expression of IL7R and other early memory markers (Figures 6E and S7B). Additionally, CAR-TRM cells increased frequencies of T cells co-expressing multiple inhibitory receptors (Figure 6F). Despite this exhaustion-like phenotype, CAR-TRM cells harvested from pancreatic tumors demonstrate enhanced TNFα, perforin, and granzyme B production compared with CAR-TCONV products, further underscoring that the exhaustion-like phenotype and CAR T cell antitumor effector function are decoupled (Figures 6G and S7C). Given the high expression of memory markers in CAR-TRM cells, we investigated their ability to provide durable antitumor activity against a secondary tumor challenge. Mice that had previously rejected Capan-2 tumors and received CAR-TRM cell treatment were re-challenged with Capan-2 cells, while a control group of CAR-TRM-naive mice was also given Capan-2 cells (Figure 6H, left). Notably, the CAR-TRM cells successfully prevented tumor growth, while the control group did not (Figure 6H, right). These results suggest that CAR-TRM cells confer long-lasting protection against tumors. Next, to investigate the contribution of the resident memory cell population to the degree of antitumor activity, we depleted the CD103+ population in CAR-TRM cell products and assessed their potency in vivo (Figure 6I). The removal of CD103+ T cells partially impairs tumor control mediated by CAR-TRM cells, suggesting that this subset is required for optimal efficacy (Figure 6J). We also tested CAR-TRM cells in an aggressive B cell leukemia xenograft model. Anti-CD19 CAR-TRM cells prevent tumor growth and markedly improve mouse survival (Figures 6K and 6L). Analysis of peripheral blood revealed that CAR-TRM cells increase in vivo CAR T cell expansion capacity due to enhanced early memory differentiation (Figures 6M and 6N).
Figure 6.

CAR-TRM cells with stem-like features mediated superior antitumor efficacy
(A) NOD scid gamma (NSG) mice were subcutaneously engrafted with 4 × 106 AsPC-1 pancreatic cancer cells. On day 32 post tumor implantation, M5 CAR-TCONV, CAR-TRM, or irrelevant (control) CD19-targeting CAR T cells (19BBz) were infused (n = 7 biological replicates for CAR-TCONV and CAR-TRM, n = 2 biological replicates for 19BBz). Tumor size was measured by taking caliper measurements over time.
(B–G) 5 × 106 Capan-2 pancreatic adenocarcinoma cells were subcutaneously injected into NSG mice. M5 CAR-TCONV and CAR-TRM were intravenously administered on day 29 following tumor engraftment (n = 6 biological replicates).
(B) Longitudinal tumor growth was monitored. Tumors were collected to characterize the immunophenotype of tumor-infiltrating CAR T cells (Mann-Whitney U test, n = 7 biological replicates).
(C) Absolute number of human T cells in the tumor.
(D) Frequency (left) and absolute number (right) of CD103+CD39+CD8+ T cells in harvested tumors.
(E and F) Frequencies of hCD45+CD8+ cells expressing (E) IL7R and (F) PD1, LAG3, and TIM3.
(G) Tumor-infiltrating CAR T cells were reactivated ionomycin and phorbol 12-myristate 13-acetate and ionomycin, followed by measurement of effector molecule (TNFα, perforin, granzyme B) production.
(H) 4 × 106 Capan-2 cells were subcutaneously inoculated into NSG mice that had cleared an initial Capan-2 tumor and had survived until day 138 (left) or into CAR T-naive mice as a control group. Longitudinal tumor growth after the second tumor challenge was monitored (right).
(I) Flow cytometry plots showing CD103 expression on CAR-TRM cells with or without magnetic depletion of CD103+ cells.
(J) CAR-TCONV, CAR-TRM, and CD103+ cell-depleted CAR-TRM cells were intravenously administered to NSG mice harboring subcutaneous Capan-2 tumors. Tumor growth was monitored over time (Mann-Whitney U test, n = 7 biological replicates).
(K–N) 106 NALM6 B cell leukemia cells were intravenously injected into NSG mice, and anti-CD19 CAR T cells were infused on day seven.
(K) Tumor growth measured by bioluminescent imaging is shown.
(L) Kaplan-Meier curves depicting survival of mice (log rank test). On day 12 after CAR T cell injection, peripheral blood samples were collected for T cell immunophenotyping.
(M) Peripheral blood human T cell count is shown.
(N) Expression of early memory markers (TCF1, CD62L). All in vivo efficacy data are representative of two independent experiments. ∗p < 0.05; ∗p < 0.01; ∗∗∗p < 0.001; ns., not significant.
Building on the findings from previously examined solid and blood cancer models, we evaluated the in vivo antitumor activity of CAR-TRM cells in an intraosseous prostate tumor model, using PC3 prostate tumor cells engineered to express PSMA (PC3-PSMA). A single infusion of PSMA CAR-TRM cells led to a significant reduction in bioluminescent tumor burden compared with CAR-TCONV cells in mice bearing intraosseous tumors generated by PC3-PSMA cell engraftment (Figure S7D). Moreover, CAR-TRM cells not only effectively controlled tumor growth but also resulted in prolonged survival for the mice compared with CAR-TCONV cells (Figure S7E), further highlighting the potential therapeutic advantages of CAR-TRM cells.
RUNX3 is a key transcription factor essential for the establishment of tissue-resident T cells, and ectopic overexpression of this transcription factor enhances CD103+CD8+ T cell differentiation in murine models.31 We therefore compared forced expression of RUNX3 with TGF β treatment in the context of generating optimally potent CAR-TRM cells. To compare these engineering strategies, we employed an in vivo CAR T cell stress test based on the use of a tonically signaling anti-mesothelin CAR construct (SS1)71 and our previously characterized, highly treatment-resistant mesothelioma model incorporating a human mesothelioma cell line transduced to express mesothelin (EMMESO cells).72 Clinically, this CAR construct demonstrates limited clinical efficacy against malignant pleural mesothelioma, pancreatic ductal adenocarcinoma, and ovarian cancer due to poor persistence and a low level of tumor infiltration.73 Human RUNX3 was successfully overexpressed in SS1 CAR T cells through lentiviral transduction (Figure 7A). Notably, unlike TGF-β-generated CAR-TRM products enriched with resident memory CD8+ T cells, RUNX3 overexpression was unable to induce expression of tissue-resident markers such as CD103, CD39, and CD49a despite an increase in T cell activation as indicated by CD25 expression (Figure 7B). We next assessed the in vivo antitumor potency of these differentially engineered CAR T cells in mice engrafted with EMMESO cells. While TGF-β-reprogrammed CAR-TRM cells successfully enhance tumor control, CAR T cells overexpressing RUNX3 fail to suppress tumor growth (Figure 7C).
Figure 7.

Comparison of CAR-TRM cell differentiation methods
(A) RUNX3 expression measured in various anti-mesothelin SS1 CAR T cell products: standard T cell medium culture (control CAR T), standard T cell medium supplemented with TGF-β (TGF-β CAR T) and engineered to overexpress RUNX3 (RUNX3 OE CAR T) (MFI, geometric mean fluorescence intensity).
(B) TRM (CD103, CD39, CD49a) and activation (CD25) markers were assessed after CAR T cell manufacturing.
(C) NSG mice (n = 7 biological replicates per group) were injected with the EMMESO cell line. Once tumors reached a size of ∼120 mm3, mice received a single dose of 1 × 107 SS1 CAR T cells. Activated non-transduced T cells served as a negative control. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, not significant (one-way ANOVA).
Discussion
In this study, we demonstrate that epigenetic rewiring of human peripheral blood T cells can produce stem-like TRM CAR T cells with improved ability to accumulate in solid tumors and resist dysfunction. We explored different methods to endow CAR T cells with both early memory features and the ability to establish tissue residency. Although TGF-β is traditionally thought of as an inhibitor of T cell function,74 we found that human T cell exposure to TGF-β during the ex vivo engineering process alters the epigenetic landscape of CAR T cells to a stem-like fate, which enables CAR-TRM products to sustain a resident memory phenotype during experimental stress tests involving chronic antigen challenge. Furthermore, we found that although CAR-TRM cells exhibit epigenetic and transcriptional characteristics commonly associated with exhaustion, these T cells were highly resistant to tumor-imposed dysfunction. Through evaluation of CAR-TRM cells in both solid and liquid cancer models, we show that these products exhibit superior antitumor potency over conventional CAR T cells, as manifested by enhanced accumulation at tumor sites and elicitation of robust effector functions.
Current CAR T cell production methods rely on peripheral-blood-derived lymphocytes as the starting point for manufacturing. In the treatment of hematological malignancies, protocols that lead to enrichment of TSM and TCM cells with high levels of CCR7 and CD62L expression for persistence within secondary lymphoid organs and bone marrow are desired. In contrast, for solid tumors, CAR T cells need to acquire the capacity to localize and expand within non-lymphoid tissues to exert sustained antitumor activity. TRM cells that naturally reside in healthy non-hematopoietic tissues and tumors may represent a promising CAR-engineered cell population for adoptive transfer to treat these latter indications. However, TRM-based cell therapy approaches have been limited to date due to the technical challenges associated with the isolation and expansion of TRM cells.
We sought to reprogram readily abundant circulating T cells to CAR-TRM cells using a simple and actionable manufacturing process. Accordingly, we show that the presence of TGF-β during CAR T cell production promotes the robust expansion of a TRM-like population. This strategy for generating CAR-TRM cells induces CD103 upregulation and promotes CD103+ CD8 TRM cell differentiation, which is important for the long-term retention of T cells in tissues.37,38,75 The observed partial attenuation of tumor control following depletion of CD103+ T cells from adoptively transferred products supports the conclusion that acquisition of a residency phenotype is a critical element of increased CAR-TRM cell potency.
We found that CAR-TRM cells are enriched in a core TRM transcriptomic signature composed of a transcription factor gene expression profile, including BHLHE40, NR4A family genes, AHR, IRF4, and EGR2,44,76,77,78 with a concomitant downregulation of EOMES79 and KLF2,30 which normally function to counter T cell residency programs and promote egress from tissues/recirculation. Additional molecules whose upregulation is associated with tissue retention, such as CD39 and CD49a, are also highly expressed by TGF-β-generated CAR-TRM cells, independently of other environmental cues, such as low oxygen tension.48 These findings suggest that TGF-β alone is sufficient to induce transcriptional and functional attributes of tissue residency in the setting of CAR-engineered T cells.
Although it was previously reported that RUNX3 programs murine CD8+ TRM cell differentiation,31 forced expression of RUNX3 in human CAR-TRM cells was not sufficient to promote formation or enhance in vivo antitumor efficacy. Recently, it was reported that RUNX3 promotes a CD8+ T cell tissue-residency program in a TGF-β-signaling-dependent manner.80 Thus, additional work is needed to determine if the combination of RUNX3 overexpression and TGF-β signaling increases the potency of CAR-TRM cells. There are appreciable similarities and critical differences between the phenotypes and functional attributes of mouse and human TRM cells,41,81,82,83 which highlights the need to investigate human TRM cell differentiation further to understand the molecular underpinnings of fate programs and enable effective translational application. Thus, these findings contribute to the advancement of the use of tissue-resident T cells in cancer therapy.
An interesting aspect of this study is that human CAR T cells activated and expanded in the presence of TGF-β are epigenetically reprogramed toward a stem-like memory state, manifested at least in part by increased expression of the stemness transcription factor TCF1 (encoded by TCF7) in association with chromatin accessibility at the TCF7 locus. TGF-β-generated stem-like CAR-TRM cells display an epigenetic program composed of tissue-residency signatures. Although this epigenetically rewired stem-like program allowed CAR-TRM cells to maintain a TRM phenotype during chronic antigen stimulation, additional investigations are needed to better determine whether these imprints are fixed or amenable to epigenetic remodeling following CAR-TRM cell trafficking to specific tumor tissues.
Exhaustion-related transcriptional programs are often shared between TRM cells and dysfunctional T cells.44 We demonstrate that human CAR-TRM cells not only upregulate surface inhibitory receptors but also share high correlation among their chromatin profiles with those of dysfunctional CAR T cells, including increased accessibility of bZIP and IRF motifs and overexpression exhaustion-related transcription factor genes. In reference to our findings, as shown in Figure 5, the bZIP and IRF transcription factor genes (ATF3, IRF4, ATF4, BATF3, ATF2, ATF6) were found to be highly elevated in CAR-TRM cells, while the canonical AP-1 transcription factor genes (JUN, FOS) were downregulated. However, as a recent study reported, the enrichment of AP-1, bZIP, and IRF motifs and upregulation of BATF, JUNB, and IRF8 in CAR T cells was associated with enhanced tumor control, despite exposure to tonic CAR signaling.84 This suggests that the presence or absence of specific transcription factors alone may not determine the function of CAR T cells, but rather a complex interplay of multiple factors is involved.
These findings suggest that the epigenetic and transcriptional programs of exhausted CAR T cells and CAR-TRM cells are intertwined; however, despite these hallmarks of exhaustion, CAR-TRM cells display enhanced in vitro cytokine production compared with conventional CAR T cells and exhibit resistance to cancer-imposed dysfunction, which leads to superior in vivo antitumor efficacy. This indicates that certain epigenetic and transcriptional features of classical T cell exhaustion are decoupled from CAR-TRM cell functionality. Furthermore, we identified IRF4 as a transcription factor implicated in both exhaustion and residency programming, specifically regulating exhaustion without promoting a resident memory phenotype. Our study of transcription factor KOs thus provides valuable insight into the co-regulation of tissue-residency and exhaustion pathways and indicates that exhaustion and tissue-residency features may be uncoupled for further improvement of CAR-TRM cells.
In conclusion, our study demonstrates the potential of CAR-TRM cells to overcome the limitations of current CAR T cell therapies by providing a source of immune cells with enhanced tumor accumulation and sustained resistance to dysfunction. Our study also provides a straightforward and practical method for generating these human stem-like CAR-TRM cells, which stands apart from previous attempts to create resident memory cells in murine T cells (e.g., RUNX3 transcription factor overexpression31 or Von Hippel Lindau [VHL] knockout44 methods). Our findings also provide valuable insights into the regulation of tissue-residency and exhaustion pathways in human TRM cells and how these two features can be uncoupled for further improvement of CAR-TRM-based therapies. Thus, this work collectively supports the use of TGF-β-generated CAR-TRM cells in early-phase clinical trials currently underway at our center, with the potential to revolutionize the treatment of various diseases, including infectious diseases, autoimmunity, and cardiac disease.
Limitations of the study
Potential limitations of our study include the use of xenograft models for human T cells, lacking the presence of host-derived cells such as regulatory T cells and myeloid-derived suppressor cells, which may affect the results. To better reflect human pathophysiology, further testing of CAR-TRM cells in immunocompetent models is required. However, transitioning to an immunocompetent model would necessitate the use of mouse T cells, which exhibit numerous dissimilarities from their human counterparts. Despite these limitations, xenogeneic mouse models still provide good methods for assessment of potential CAR T cell-augmenting strategies, with subsequent direct testing in humans being critical. The pleiotropic roles of TGF-β in CAR T cell therapy should also be further examined in the future, as previous studies have shown that TGF-β inhibition and ICB produce the best responses and T cell re-invigoration.85,86 In addition, our proposed strategy must be evaluated in light of previous research that demonstrates the enhancement of antitumor function through blocking TGF-β signaling in CAR T cells.42,87,88,89 Notably, our findings indicate that exposure to TGF-β during the manufacturing process of CAR T cells, optimized for concentration and exposure time, leads to an epigenetic reprogramming that enforces a durable T cell tissue-residency state without compromising antitumor immunity. Thus, while our study has some limitations, it provides a foundation for further exploring the potential of CAR-TRM cells as a promising therapy for various diseases.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Brilliant Violet (BV) 421 anti-human CD279 (PD-1) | BioLegend | Cat# 329920; RRID: AB_10960742 |
| BV 711 anti-human CD39 | BioLegend | Cat# 328228; RRID: AB_2632894 |
| BV 570 anti-human CD45RO | BioLegend | Cat# 304225; RRID: AB_11126987 |
| BV 650 anti-human CD8a | BioLegend | Cat# 301041; RRID: AB_11125174 |
| BV 785 anti-human CD4 | BioLegend | Cat# 317442; RRID: AB_2563242 |
| BV 570 anti-human CD127 (IL-7Ralpha) | BioLegend | Cat# 351308; RRID: AB_2832685 |
| BV 570 anti-human IFN-gamma | BioLegend | Cat# 502534; RRID: AB_2563880 |
| BV 421 anti-human Perforin | BioLegend | Cat# 353307; RRID: AB_11149688 |
| Allophycocyanin (APC) anti-human Perforin | BioLegend | Cat# 308112; RRID: AB_2252843 |
| Alexa Fluor (AF) 700 anti-human TNF-alpha | BioLegend | Cat# 502928; RRID: AB_2561315 |
| APC/Fire 750 anti-human CD49a | BioLegend | Cat# 328318; RRID: AB_2810495 |
| Phycoerythrin (PE) anti-human CD366 (Tim-3) | BioLegend | Cat# 345006; RRID: AB_2116576 |
| PE/Cyanine 5 anti-human CD62L | BioLegend | Cat# 304808; RRID: AB_314468 |
| Peridinin-Chlorophyll-Protein (PerCP)/Cyanine 5.5 anti-mouse CD45 | BioLegend | Cat# 103132; RRID: AB_893340 |
| Fluorescein (FITC) anti-human CD103 (Integrin alphaE) | BioLegend | Cat# 350204; RRID: AB_10639865 |
| Biotin anti-human CD103 (Integrin alphaE) | BioLegend | Cat# 350220; RRID: AB_2629646 |
| APC-H7 anti-human CD8 | BD Biosciences | Cat# 641409; RRID: AB_1645737 |
| PE-CF594 anti-human CD197 (CCR7) | BD Biosciences | Cat# 562381; RRID: AB_11153301 |
| PE-CF594 anti-human IL-2 | BD Biosciences | Cat# 562384; RRID: AB_11154601 |
| APC-H7 anti-human CD45 | BD Biosciences | Cat# 560274; RRID: AB_1645480 |
| PE anti-human RUNX3 | BD Biosciences | Cat# 564814; RRID: AB_2738969 |
| APC anti-human CD25 | ThermoFisher Scientific | Cat# 17-0259-42; RRID: AB_1582219 |
| PE/Cyanine 5.5 anti-human Granzyme B | ThermoFisher Scientific | Cat# GRB18; RRID: AB_2536541 |
| APC anti-human TOX | Miltenyi Biotec | Cat# 130-118-335; RRID: AB_2751485 |
| PE/Cyanine 7 anti-human CD223 (LAG-3) | eBioscience | Cat# 25-2239-42; RRID: AB_2573430 |
| APC anti-FMC63 CAR19 | ACROBiosytems | Cat# FM3-AY54P1 |
| Biological samples | ||
| Human Healthy Donor PBMC | Human Immunology Core, University of Pennsylvania | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| PE-conjugated Mesothelin Protein | AcroBiosystems | Cat# MSN-HP2H5 |
| PSMA Protein, Human, Recombinant (His Tag), Biotinylated | Sino Biological | Cat# 15877-H07H-B |
| Recombinant Human TGF-beta 1 Protein | R&D Systems | Cat# 240-B-002 |
| Recombinant Human Interleukin-2 (IL-2) Protein | R&D Systems | Cat# 202-IL-050 |
| Human TGF-β1 Recombinant Protein | Cell Signaling Technology | Cat# 75362S |
| D-Luciferin | PerkinElmer | Cat# 122799 |
| TRIzol | ThermoFisher Scientific | Cat# 15596026 |
| Truecut™ Cas9 Protein v2 | ThermoFisher Scientific | Cat# A36498 |
| Matrigel Basement Membrane Matrix | Corning | Cat# 356234 |
| Alt-R® CRISPR-Cas9 tracrRNA | Integrated DNA Technologies | Cat# 1072534 |
| Collagenase Type IV | STEMCELL Technologies | Cat# 07909 |
| DNase I Solution | STEMCELL Technologies | Cat# 07900 |
| 123count eBeads™ | ThermoFisher Scientific | Cat# 01-1234-42 |
| AMPure Beads | Beckman Coulter | Cat# E6116 |
| Dynabeads human T-activator CD3/CD28 | ThermoFisher Scientific | Cat# 11131D |
| Critical commercial assays | ||
| 96-well RTCA E-plates | Agilent | Cat# 300601010 |
| MycoAlert® Mycoplasma Detection Kit | Lonza | Cat# LT07-318 |
| Pan T cell Isolation Kit, human | Miltenyi Biotec | Cat# 130-096-535 |
| RNA Clean & Concentrator™ Kits | Zymo Research | Cat# R1015 |
| P3 Primary Cell 4D-nucleofector Kit | Lonza | Cat# V4XP-3032 |
| 13-plex LEGENDplex™ Human CD8 Panel | BioLegend | Cat# 740267 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | ThermoFisher Scientific | Cat# L34957 |
| Foxp3 Transcription Factor Staining Buffer Set | ThermoFisher Scientific | Cat# 00-5523-00 |
| Chromium Next GEM Single Cell 5′ Kit v2 | 10X Genomics | Cat# PN-1000263 |
| Dead Cell Removal Kit | Miltenyi Biotec | Cat# 130-090-101 |
| Deposited data | ||
| RNA- and ATAC-seq data | NCBI Gene expression omnibus (GEO) | GSE224866 |
| Experimental models: Cell lines | ||
| NALM6, clone G5 | ATCC | CRL-3273 |
| Capan-2 | ATCC | HTB-80 |
| AsPC-1 | ATCC | CRL-1682 |
| PC3 | ATCC | CRL-1435 |
| HEK 293T | ATCC | CRL-3216 |
| EMMESO | Steven M. Albelda Lab | N/A |
| Experimental models: Organisms/strains | ||
| NOD/SCID/IL-2Rγ-null (NSG) mice | University of Pennsylvania Stem Cell and Xenograft Core | N/A |
| Oligonucleotides | ||
| Please see Table S3. | This study | N/A |
| Recombinant DNA | ||
| pTRPE-RUNX3 | Steven M. Albelda Lab | N/A |
| pTRPE-M5 (anti-mesothelin) CAR (4-1BB/CD3ζ) | Carl H. June Lab | N/A |
| pTRPE-SS1 (anti-mesothelin) CAR (4-1BB/CD3ζ) | Carl H. June Lab | N/A |
| pTRPE-PSMA CAR (4-1BB/CD3ζ) | Carl H. June Lab | N/A |
| pTRPE-CD19 CAR (4-1BB/CD3ζ) | Carl H. June Lab | N/A |
| Software and algorithms | ||
| FlowJo v10 | FlowJo, LLC | https://www.flowjo.com; RRID:SCR_008520 |
| GraphPad Prism v9.1.0 | GraphPad | https://www.graphpad.com; RRID:SCR_002798 |
| RTCA 2.0 | Agilent | https://www.agilent.com |
| Inference of CRISPR Edits (ICE) Tool | Synthego | https://ice.synthego.com |
| Cell Ranger v6.1.2 | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/tutorial_ct |
| Seurat v4.1.1 | Satija Lab | https://github.com/satijalab/seurat |
| SCTransform (part of Seurat) | Christoph Hafemeister, Rahul Satija | https://github.com/ChristophH/sctransform |
| Functions within Seurat (SelectIntegrationFeatures, PrepSCTIntegration, FindIntegrationAnchors, IntegrateData, RunPCA, FindNeighbors, FindClusters, runUMAP, FindMarkers, FindAllMarkers, AddModuleScore) | Satija Lab | https://satijalab.org/seurat/reference/index.html |
| Cutadapt | Marcel Martin | https://github.com/marcelm/cutadapt |
| Bowtie2 | Ben Langmead, Cole Trapnell, Mihai Pop, Steven Salzberg | https://github.com/BenLangmead/bowtie2 |
| removeChrom | John M. Gaspar | https://github.com/jsh58/harvard/blob/master/removeChrom.py |
| Samtools | Heng Li, Bob Handsaker, and colleagues | https://github.com/samtools/samtools |
| The Integrative Genome Viewer (IGV) | Broad Institute | https://github.com/igvteam/igv |
| MACS2 | Tao Liu and colleagues | https://github.com/taoliu/MACS |
| ENCODE blacklisted regions | Anshul Kundaje Lab | https://sites.google.com/site/anshulkundaje/projects/blacklists |
| findMotifsGenome script from HOMER | Chris Benner and colleagues | http://homer.ucsd.edu/homer/ngs/peakMotifs.html |
| edgeR v3.34.0 | Gordon Smyth, Yunshun Chen, Matthew Ritchie, and colleagues | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| enrichr | Ma’ayan Lab | https://maayanlab.cloud/Enrichr |
| GSEABase | Martin Morgan, Dan Tenenbaum, and colleagues | https://github.com/Bioconductor/GSEABase |
| kallisto v0.46.0 | Páll Melsted, Lior Pachter, and colleagues | https://github.com/pachterlab/kallisto |
| tximport 1.24.0 | Michael Love, Charlotte Soneson, Mark Robinson, and colleagues | https://github.com/mikelove/tximport |
Resource availability
Lead contact
Additional information and inquiries regarding resources and reagents should be directed to the lead contact, Dr. Joseph Fraietta (jfrai@upenn.edu), who will fulfill these requests.
Materials availability
Requests for materials should be directed to the Penn Center for Innovation (pciinfo@pci.upenn.edu). All requests will be promptly reviewed to determine if they involve any intellectual property considerations. Materials eligible for sharing will be released following the execution of a material transfer agreement.
Experimental model and subject details
Cell lines
The NALM6 B-cell acute lymphoblastic leukemia cell line, pancreatic ductal adenocarcinoma cell lines Capan-2 and AsPC-1 expressing mesothelin, the PC3 prostatic adenocarcinoma cell line, and HEK 293T cells were acquired from the American Type Culture Collection (ATCC). PC3 cells were modified to express PSMA (PC3-PSMA), and both PC3-PSMA and NALM6 cells were engineered to express click beetle green luciferase and green fluorescent protein (CBG-GFP). A human mesothelioma cell line, EMP (parental), derived from a patient’s tumor in March 2010, was also used. EMP was altered to express human mesothelin (EMMESO) as it does not endogenously express it. Antigen expression was confirmed in these cell lines before functional assays. Capan-2, PC3-PSMA, and HEK 293T cells were cultured in D10 media with Dulbecco’s Modified Eagle Medium (DMEM, Gibco), supplemented with high glucose, GlutaMAX, 10% fetal bovine serum (FBS, Gibco), and 1% penicillin/streptomycin. NALM6, AsPC-1, and EMMESO cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 media containing 10% FBS and 1% streptomycin/penicillin (R10). The University of Arizona Genetics Core authenticated all cell lines used in this study, and mycoplasma contamination was tested using the MycoAlert Mycoplasma Detection kit (Lonza) according to the manufacturer’s protocol.
Primary human T-cells
For the generation of CAR T-cells, mononuclear cells from peripheral blood (PBMC) were acquired through leukapheresis from healthy volunteers. These cells were obtained from the Human Immunology Core at the University of Pennsylvania, with the approval of the university’s Institutional Review Board. Study participants provided written informed consent, in compliance with the Declaration of Helsinki, the International Conference on Harmonization’s Good Clinical Practice guidelines, and the United States Common Rule.
Mice
Animal studies were conducted using both male and female NOD/SCID/IL-2Rγ-null (NSG) mice aged between eight and twelve weeks, obtained from the University of Pennsylvania Stem Cell and Xenograft Core. These experiments were carried out under a protocol approved by the Institutional Animal Care and Use Committee. This ensured that all experiments involving the mice adhered to the highest ethical standards and guidelines for animal care and use in scientific research. The mice were housed in a pathogen-free environment, subjected to a 12-h light/dark cycle, and maintained at a consistent temperature of 21 ± 2°C with 60 ± 20% relative humidity. The animals were given unrestricted access to acidified water and sterilized food.
Method details
Lentivirus production
Lentivirus manufacturing was carried out previously described.42 Briefly, the pTRPE lentiviral transfer plasmid was modified to facilitate expression of the human RUNX3 transcription factor or second-generation CAR constructs comprised of extracellular anti-mesothelin (M5 and SS1), -PSMA or anti-CD19 single chain variable fragments, CD8a hinge/transmembrane domains, and 4-1BB and CD3ζ cytoplasmic signaling modules. At 70–80% confluency, low passage HEK 293T cells were transfected with lentiviral transfer plasmid and packaging vectors using Lipofectamine 2000 (ThermoFisher Scientific). At 24- and 48-h post-transfection, lentivirus supernatant was harvested, followed by concentration using high-speed ultracentrifugation. After removal of supernatant, R10 media was added for resuspension of viral pellets and stored at stored at −80°C.
T-cell culture and lentiviral transduction
PBMCs were isolated from buffy coats and cryopreserved at 4 × 107 cells per vial. Cryopreserved PBMCs were thawed, and T-cell purification was carried out using the Pan T Cell Isolation Kit according to the manufacturer’s instructions (Miltenyi Biotec). For CAR-TCONV manufacturing, T-cells were activated with CD3/CD28 Dynabeads (ThermoFisher Scientific) at a bead to cell ratio of 3:1 for three days in T-cell OpTmizer CTS SFM media (Gibco) supplemented with 5% human AB serum and 100U/mL human IL-2 (T-cell media). At 24-h post-T-cell activation, T-cells were transduced with the above lentiviral vectors at a multiplicity of infection (MOI) of three. T-cell media was added to cultures every two days, and on day nine, CAR T-cells were cryopreserved in T-cell media supplemented with 20% human AB serum and 10% dimethyl sulfoxide. For CAR-TRM production, T-cell culture media was supplemented with 2 ng/mL of TGF-β (R&D Systems or Cell Signaling Technology) in addition to IL-2, and the CAR T-cell engineering process was carried out as specified above.
Gene editing using CRISPR/Cas9
At three days following T-cell activation, CD3/CD28 Dynabeads were magnetically removed. T-cells were washed with phosphate-buffered saline (PBS), and electroporation was carried out using the P3 primary cell 4D-nucleofector kit (Lonza). Ribonucleoprotein (RNP) complexes were generated by preincubating 12 μg TrueCut S. Cas9 (ThermoFisher Scientific), 0.2 nmol Alt-R tracrRNA (trans-activating CRISPR RNA), and crRNA (crispr RNA) (Integrated DNA Technologies) for 10 min. Three million CAR T-cells resuspended in P3 buffer were added to the appropriate RNP complexes, followed by transfer to nucleocuvette strips for electroporation using the 4D-Nucleofactor (Lonza, protocol EO-115). After electroporation, CAR T-cells were maintained in T-cell media supplemented with or without 2 ng/mL of TGF-β. Three days post-electroporation, CAR T-cells were collected for genomic DNA isolation. Target regions flanking double-strand brake sites were amplified by polymerase chain reaction (PCR) for Sanger sequencing. Editing efficiencies were determined by Inference of CRISPR Edits (ICE) analysis.
Flow cytometry
For flow cytometric characterizations, CAR T-cells were first stained with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (ThermoFisher Scientific) in PBS. Samples were subsequently washed and incubated with antibodies diluted in FACS buffer (PBS +2% FBS). Expression of anti-mesothelin CARs and the anti-CD19 CAR was measured using a PE-conjugated mesothelin protein and an APC-conjugated anti-FMC63 idiotype antibody, respectively. PSMA CAR expression was assessed using a biotinylated recombinant human PSMA protein sourced from Sino Biological, followed by a secondary stain with APC-conjugated streptavidin. Additional surface proteins were stained using the following antibodies: BV 421 anti-human CD279 (PD-1), BV 570 anti-human CD45RO, BV 650 anti-human CD8a, BV 785 anti-human CD4, BV 570 anti-human CD127 (IL-7Ralpha), PE anti-human CD366 (Tim-3), PE/Cyanine 5 anti-human CD62L, PerCP/Cyanine 5.5 anti-mouse CD45, FITC anti-human CD103 (Integrin alphaE), APC-H7 anti-human CD8, PE-CF594 anti-human CD197 (CCR7), APC-H7 anti-human CD45, APC anti-human CD25, APC/Fire 750 anti-human CD49a, PE/Cyanine 7 anti-human CD223 (LAG-3), and/or BV 711 anti-human CD39. For intracellular protein staining, cells were first fixed and permeabilized using the Foxp3 Transcription Factor Staining Buffer Set (ThermoFisher Scientific), followed by staining with the following antibodies: PE-CF594 anti-human IL-2, BV 570 anti-human IFN-gamma, AF 700 anti-human TNF-alpha, BV 421 anti-human Perforin, APC anti-human Perforin, PE anti-human RUNX3, PE/Cyanine 5.5 anti-human Granzyme B, and/or APC anti-human TOX. Fluorescence intensity for each sample was measured using an LSRII Fortessa cytometer (BD Biosciences), and analysis was performed with FlowJo software v10 (FlowJo, LLC).
In vitro restimulation assay
The resident memory phenotype of CAR T-cells was assessed using an in vitro restimulation assay. Briefly, 5 × 105 irradiated AsPC-1 cells were seeded a day before coincubation with CAR T-cells. M5 CAR T-cells were magnetically isolated using anti-mesothelin-coated beads (ACROBiosystems). The isolated 1 × 106 CAR T-cells were then added to AsPC-1 cells at an effector to target ratio of 2:1. After 18-h of coincubation, supernatant was collected for cytokine measurements using the 13-plex LEGENDplex human CD8 panel (BioLegend). On every fifth day following tumor cell challenge, CAR T-cells were harvested for immunophenotyping, and 1 × 106 viable CAR T-cells were transferred into plates containing AsPC-1 cells to facilitate chronic stimulation.
Cytotoxicity assay
The xCELLigence system (ACEA Biosciences Inc.) was used to monitor that cytolytic capacity of anti-M5 CAR-TCONV and CAR-TRM cells. For this assay, 4 × 104 AsPC-1 cells were seeded in E-Plate VIEW 96 PET microwell plates, and after 24-h, 2 × 104 M5 CAR T-cells or irrelevant CD19 CAR T-cells were added at an effector to target ratio of 1:2. Tween 20 was used as a positive control for full cell lysis. The real-time cell index was monitored in 20-min intervals over seven days, and the degree of CAR T-cell-mediated cytotoxic activity was calculated using the normalized cell index and percent cytolysis.
Mouse xenograft studies
To assess the in vivo antitumor potency of M5 CAR-TCONV and CAR-TRM cells in solid tumor indications, 4 × 106 AsPC-1 cells or 5 × 106 Capan-2 cells were premixed with Matrigel (Corning) and subcutaneously inoculated in the right flanks of 6- to 8-week-old NSG mice. For the AsPC-1 model, 3 × 105 M5 CAR T-cells were intravenously infused on day thirty-two following tumor engraftment. For Capan-2 xenograft experiments, 1 × 105 CAR T-cells were intravenously given to Capan-2-bearing mice on day twenty-nine. To examine the role of the resident memory population in mediating CAR-TRM cell antitumor responses, the CD103+ population in CAR-TRM products was magnetically depleted using biotinylated anti-human CD103 antibody and anti-biotin microbeads (Miltenyi Biotec). Following isolation, CD103+cell-depleted CAR-TRM cells were administered to mice. Longitudinal tumor growth was monitored using caliper measurements (tumor volume = (length × width2)/2) in AsPC-1 and Capan-2 models. To assess the trafficking of CAR T-cells, the M5 CAR-TCONV and CAR-TRM cells were labeled with 320 μg/mL IVISense DiR 750 fluorescent cell labeling dye (PerkinElmer) for 30 min. After washing the CAR T-cells twice with PBS, they were intravenously injected into the mice on day thirty-three post-Capan-2 engraftment. The migration of the CAR T-cells was monitored using the Xenogen IVIS Imaging System (PerkinElmer) with a 710 nm excitation and 760 nm emission filter.
To assess the immunophenotype of tumor-infiltrating CAR T-cells, Capan-2 tumors were harvested on day thirteen post-CAR-T cell injection and minced with a scalpel. Tumors were subsequently dissociated with 100 U/mL Collagenase Type IV (STEMCELL Technologies) and 0.25 mg/mL DNase I (STEMCELL Technologies) for 30 min at 37°C. Tumor-infiltrating CAR T-cells were then reactivated with 50 ng/mL of phorbol 12-myristate 13-acetate 1210 (PMA) and 1 μg/mL ionomycin in the presence of 5 μg/mL Brefeldin A for 6-h. Expression of effector cytokines and cytotoxic proteins were assessed by flow cytometry. To assess the antitumor efficacy of SS1 CAR T-cells, NSG mice were injected with EMMESO mesothelin-expressing mesothelioma cells. When tumor volume reached a size of ∼120 mm3, mice were infused with a single dose of 107 CAR T-cells.
For the NALM6 B-cell leukemia model, NSG mice were engrafted with 106 NALM6 cells intravenously. On day seven post-tumor injection, anti-CD19 CAR-TCONV, CAR-TRM cells, or irrelevant PSMA CAR T-cells (negative control), were infused. Longitudinal tumor growth was assessed weekly using bioluminescent imaging with the Xenogen IVIS Imaging System. On day nineteen after tumor injection, peripheral blood was collected from mice to evaluate CAR T-cell expansion capacity and differentiation phenotype. The number of human peripheral blood T-cells was measured using 123count eBeads (ThermoFisher Scientific), and surface memory markers were stained for flow cytometric analysis according to the above methods.
In the intraosseous PC3-PSMA model, 2 × 105 PC3-PSMA cells were intrafemorally transplanted into male NSG mice. After 21 days, the mice received an intravenous infusion of 1 × 105 anti-PSMA CAR-TCONV or -CAR-TRM cells. Tumor burden was assessed by administering an intraperitoneal injection of luciferin to the mice, followed by quantifying bioluminescence using the Xenogen IVIS Imaging System.
Bulk RNA-seq
Anti-mesothelin M5 CAR-TCONV and CAR-TRM cells were harvested after nine days of in vitro expansion. CD8+ CAR T-cells were magnetically isolated using anti-mesothelin-coated beads (ACROBiosystems) and anti-CD8 microbeads (Miltenyi Biotec), followed by resuspension in 500μL of TRIzol (Invitrogen). Total RNA was prepared using RNA Clean & Concentrator kits (Zymo Research) following the manufacturer’s instructions. Transcriptome sequencing was performed by Novogene using the NovaSeq 6000 system (Illumina, 150bp paired end reads). Paired-end reads were pseudoaligned to the human genome (GRCh38) using kallisto v0.46.0, and count estimates at the gene level were summarized using tximport 1.24.0.
Differential gene expression analyses between CAR-TRM and CAR-TCONV cells was conducted using edgeR v3.34.0. Briefly, filtered RNA-seq data was normalized using a TMM method (Trimmed Mean of M-values) and logarithmically transformed into counts per million (cpm). Generalized linear models were used to determine differential expression in a paired design using edgeR. Gene set enrichment analysis (GSEA) was conducted using enrichr and GSEABase on the following gene sets: resident-memory gene sets48 (Table S1: genes differentially expressed between CD69+CD103+ and CD69−CD103−CD8+ T-cells, adjusted p value <0.05 and log2FC ≥ 1), hypoxia and migration gene sets (MSigDB C2), and response to ICB signature.49
Single-cell RNA sequencing
For scRNA-seq, dead cells were eliminated using the Dead Cell Removal Kit (Miltenyi Biotec). Mesothelin-directed M5 CAR-TCONV and -TRM cells were magnetically isolated using anti-mesothelin-coated beads, as described above. After isolation, T-cell viability and quantity were evaluated using trypan blue exclusion and cells were resuspended in PBS containing 0.04% BSA. Approximately 20,000 cells were loaded per reaction to capture ∼10,000 cells for scRNA-seq library preparation using the Chromium Next GEM Single Cell 5′ Kit v2 (10X Genomics) according to manufacturer’s protocol. Sequencing was performed on a NovaSeq 6000 instrument at a depth of >20,000 reads per cell.
Raw read files were processed and aligned to the human reference genome (GRCh38) using Cell Ranger 6.1.2. Expression data were then analyzed in R using Seurat version 4.1.1. Poor quality cells with more than 10% mitochondrial reads were eliminated, and cells that express less than 1,000 genes or more than 7,000 genes were also eliminated. After cell filtration, 12,230 cells were obtained for subsequent analysis. Analyses were carried out using various additional packages of reproducible R code. Individual data sets were normalized via regularized negative binomial regression using SCTransform. Normalized expression datasets from CAR-TCONV and CAR-TRM samples were integrated with SelectIntegrationFeatures, PrepSCTIntegration, FindIntegrationAnchors, and IntegrateData commands. Principal component analysis was carried out on scaled data using RunPCA, followed by clustering of cells using FindNeighbors, and FindClusters. UMAP embedding was done with the runUMAP function. Differential gene expression analyses between CD8 clusters or CAR T-cell samples were performed using the FindMarkers and FindAllMarkers commands. We queried expression datasets for the expression of resident-memory48 and ‘stemness’-related (GSE83978) gene signatures and calculated module scores using AddModuleScore.
ATAC-seq analysis
Anti-mesothelin M5 CAR-TCONV and CAR-TRM cells were harvested after nine days of in vitro expansion. CAR T-cells were magnetically isolated according to the methods described above. After elimination of dead cells, 100,000 CD8+ CAR T-cells were cryopreserved. Library preparation was performed by Novogene as follows: a reaction mixture consisting of Tn5 transposase and equimolar adapter1 and adapter 2 was added to nuclei pellets and incubated at 37°C for 30 min. The library was enriched using PCR, and purification of final libraries was carried out using AMPure beads (Beckman Coulter). Paired-end sequencing (150bp reads) was conducted on a NovaSeq 6000 instrument.
Cutadapt was used to trim FASTQ files of adapter contamination. Reads were aligned to the hg19 reference genome using Bowtie2, retaining only properly aligned and paired reads from 10 to 1000 base pairs. Mitochondrial reads were then eliminated using removeChrom. Aligned reads were converted to BAM files and sorted with samtools, and Picard was used to remove PCR duplicates. BAM files were indexed using samtools to display tracks in the Integrative Genome Viewer (IGV). MACS2 was used to call peaks with an FDR q-value cutoff of 0.01. The R package Diffbind was used to remove ENCODE blacklisted regions, then to identify peaks differentially opened between the CAR-TCONV and -TRM populations. The findMotifsGenome script from HOMER was implemented to map differentially opened peaks for occurrences of known transcription factor motifs.
Statistical analyses
Statistical tests between two groups were carried out using a two-tailed unpaired/paired Student’s t t-test (for parametric data) or a Mann-Whitney U test (for non-parametric data). The Benjamini–Hochberg procedure was used to adjust p values for multiple comparisons testing following differential gene expression analyses. For statistic comparisons involving three or more groups, one- or two-way ANOVA tests were performed, followed by post-hoc testing for multiple comparisons. Mouse survival was assessed using the Log rank (Mantel-Cox) test. Statistical analyses were carried out using GraphPad Prism v9.1.0 (GraphPad), and p values <0.05 were considered significant. ∗p < 0.05, ∗p < 0.01, ∗∗∗p < 0.001, ns: not significant.
Acknowledgments
The authors gratefully acknowledge the Human Immunology Core for providing leukocytes, the Hospital of the University of Pennsylvania Apheresis Unit for peripheral blood mononuclear cell collections, and the Stem Cell and Xenograft Core for their valuable assistance with mouse studies, all at the University of Pennsylvania. We would like to extend our gratitude to Dr. Krishnendu Roy from the Georgia Institute of Technology for his valuable feedback and insightful suggestions, which greatly contributed to the improvement of our work. This project was supported by a National Science Foundation Engineering Research Center for Cell Manufacturing Technologies grant (EEC1648035; to B.L.L. and J.A.F.), the Bob Levis Funding Group (to B.L.L. and J.A.F.), a Prostate Cancer Foundation Tactical Award (to J.A.F.), an Alliance for Cancer Gene Therapy Investigator Award in Cell and Gene Therapy for Cancer (to J.A.F.), U54 CA244711 (to M.M.D. and J.A.F.), P01 CA214278 (to M.M.D. and J.A.F.), U01 AG066100 (to J.A.F.), a Samuel Waxman Cancer Research Foundation Grant (to J.A.F.), and an ACC P30 Core Grant P30 CA016520-42 (to J.A.F.). The graphical abstract was generated using BioRender.com.
Author contributions
Conceptualization, I.Y.J., E.K.M., S.M.A., and J.A.F.; methodology, I.Y.J., E.N.O., E.K.M., S.M.A., and J.A.F.; investigation, I.Y.J., E.N.O., R.B., S.M.C., E.W., M.D., J.K.J., G.P., D.L.S., A.C., B.L.L., S.L.B., E.K.M., S.M.A., and J.A.F.; writing – original draft, I.Y.J. and J.A.F.; writing – review and editing, I.Y.J., E.N.O., S.M.A., and J.A.F.; supervision, S.M.A. and J.A.F.; project administration, S.M.A. and J.A.F.; funding acquisition, J.A.F.
Declaration of interests
I.Y.J., D.L.S., M.M.D., A.C., B.L.L., E.K.M., S.M.A., and J.A.F. hold patents and intellectual property related to T cell-based cancer immunotherapy and have received royalties. M.M.D. has obtained research funding from Tmunity Therapeutics and is a member of the Scientific Advisory Board for Cellares Corporation. A.C. is a co-founder and has equity in Tmunity Therapeutics. B.L.L. serves as a consultant for Terumo and GSK; is on the Scientific Advisory Boards for Akron, Avectas, Immuneel, Immusoft, In8bio, Ori Biotech, Oxford Biomedica, and Vycellix; and holds equity as a co-founder in both Tmunity Therapeutics and Capstan Therapeutics. S.M.A. is a co-founder and equity holder in Capstan Therapeutics. J.A.F. receives research funding from Tmunity Therapeutics and Danaher Corporation, consults for Retro Biosciences, and is a member of the Scientific Advisory Boards for Cartography Bio and Shennon Biotechnologies Inc.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101053.
Supplemental information
Differentially expressed genes between CAR-TRM and CAR-TCONV cells. The table displays the following parameters: Gene ID, log fold change (log FC), log counts per million (log CPM), likelihood ratio (LR), p value, and false discovery rate (FDR).
Differential expression analysis of genes related to resident memory T cells, early memory T cells, and circulation-associated markers in CAR-TRM and CAR-TCONV cells as well as within CD8 subpopulations. The table is divided into three tabs: Tab 1, CD8 markers comparing TRM and TCONV cells; Tab 2, CD8 markers within distinct CD8 clusters; and Tab 3, TCF7CD8 markers comparing TRM and TCONV cells. Each tab displays the following parameters for the respective genes: p value (P_val), average log2 fold change (Avg_log2FC), percentage of cells expressing the gene in the first population (Pct.1), percentage of cells expressing the gene in the second population (Pct.2), adjusted p value (P_val_adj), cluster assignment, and gene symbol.
Sequences of CRISPR guide RNAs targeting AAVS1, IRF4, EGR2, and NR4A3 loci, as well as the corresponding forward and reverse sequencing primers for each target. The sequences are presented in the 5′-to-3′ orientation.
Data and code availability
-
•
The sequencing data reported in this paper is deposited in the Gene Expression Omnibus (GEO) database with accession number GSE224866. The data is available for public access and download from the GEO database, with no restrictions on its use or redistribution.
-
•
No original code is featured in this paper.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E., et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 4.Locke F.L., Miklos D.B., Jacobson C.A., Perales M.A., Kersten M.J., Oluwole O.O., Ghobadi A., Rapoport A.P., McGuirk J., Pagel J.M., et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N. Engl. J. Med. 2022;386:640–654. doi: 10.1056/NEJMoa2116133. [DOI] [PubMed] [Google Scholar]
- 5.Wang M., Munoz J., Goy A., Locke F.L., Jacobson C.A., Hill B.T., Timmerman J.M., Holmes H., Jaglowski S., Flinn I.W., et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020;382:1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raje N., Berdeja J., Lin Y., Siegel D., Jagannath S., Madduri D., Liedtke M., Rosenblatt J., Maus M.V., Turka A., et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019;380:1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munshi N.C., Anderson L.D., Jr., Shah N., Madduri D., Berdeja J., Lonial S., Raje N., Lin Y., Siegel D., Oriol A., et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021;384:705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 8.Mailankody S., Devlin S.M., Landa J., Nath K., Diamonte C., Carstens E.J., Russo D., Auclair R., Fitzgerald L., Cadzin B., et al. GPRC5D-Targeted CAR T cells for myeloma. N. Engl. J. Med. 2022;387:1196–1206. doi: 10.1056/NEJMoa2209900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2022. CA. Cancer J. Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen D.T., Ogando-Rivas E., Liu R., Wang T., Rubin J., Jin L., Tao H., Sawyer W.W., Mendez-Gomez H.R., Cascio M., et al. CAR T cell locomotion in solid tumor microenvironment. Cells. 2022;11 doi: 10.3390/cells11121974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez M., Moon E.K. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Craddock J.A., Lu A., Bear A., Pule M., Brenner M.K., Rooney C.M., Foster A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moon E.K., Carpenito C., Sun J., Wang L.C.S., Kapoor V., Predina J., Powell D.J., Jr., Riley J.L., June C.H., Albelda S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Y.J., Dougan M., Jailkhani N., Ingram J., Fang T., Kummer L., Momin N., Pishesha N., Rickelt S., Hynes R.O., Ploegh H. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA. 2019;116:7624–7631. doi: 10.1073/pnas.1817147116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Majzner R.G., Mackall C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 16.O'Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A., et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majzner R.G., Ramakrishna S., Yeom K.W., Patel S., Chinnasamy H., Schultz L.M., Richards R.M., Jiang L., Barsan V., Mancusi R., et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603:934–941. doi: 10.1038/s41586-022-04489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang O.Y., Tian L., Yoder T., Xu R., Kulikovskaya I., Gupta M., Melenhorst J.J., Lacey S.F., O'Rourke D.M., Binder Z.A. PD1 expression in EGFRvIII-directed CAR T cell infusion product for glioblastoma is associated with clinical response. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.872756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung I.Y., Narayan V., McDonald S., Rech A.J., Bartoszek R., Hong G., Davis M.M., Xu J., Boesteanu A.C., Barber-Rotenberg J.S., et al. BLIMP1 and NR4A3 transcription factors reciprocally regulate antitumor CAR T-cell stemness and exhaustion science. Trans Med. 2022;14:eabn7336. doi: 10.1126/scitranslmed.abn7336. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo A., Huang H., Zhu Z., Chen M.J., Shi H., Yuan S., Sharma P., Connelly J.P., Liedmann S., Dhungana Y., et al. cBAF complex components and MYC cooperate early in CD8(+) T cell fate. Nature. 2022;607:135–141. doi: 10.1038/s41586-022-04849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C.M., Quigley M.F., Almeida J.R., Gostick E., Yu Z., Carpenito C., et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masopust D., Vezys V., Marzo A.L., Lefrançois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 23.Hogan R.J., Zhong W., Usherwood E.J., Cookenham T., Roberts A.D., Woodland D.L. Protection from respiratory virus infections can be mediated by antigen-specific CD4(+) T cells that persist in the lungs. J. Exp. Med. 2001;193:981–986. doi: 10.1084/jem.193.8.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gebhardt T., Wakim L.M., Eidsmo L., Reading P.C., Heath W.R., Carbone F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009;10:524–530. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- 25.Wakim L.M., Waithman J., van Rooijen N., Heath W.R., Carbone F.R. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science. 2008;319:198–202. doi: 10.1126/science.1151869. [DOI] [PubMed] [Google Scholar]
- 26.Zheng L., Qin S., Si W., Wang A., Xing B., Gao R., Ren X., Wang L., Wu X., Zhang J., et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 2021;374:abe6474. doi: 10.1126/science.abe6474. [DOI] [PubMed] [Google Scholar]
- 27.Edwards J., Wilmott J.S., Madore J., Gide T.N., Quek C., Tasker A., Ferguson A., Chen J., Hewavisenti R., Hersey P., et al. CD103(+) tumor-resident CD8(+) T cells are associated with improved survival in immunotherapy-naive melanoma patients and expand significantly during anti-PD-1 treatment. Clin. Cancer Res. 2018;24:3036–3045. doi: 10.1158/1078-0432.CCR-17-2257. [DOI] [PubMed] [Google Scholar]
- 28.Bösmüller H.C., Wagner P., Peper J.K., Schuster H., Pham D.L., Greif K., Beschorner C., Rammensee H.G., Stevanović S., Fend F., Staebler A. Combined immunoscore of CD103 and CD3 identifies long-term survivors in high-grade serous ovarian cancer. Int. J. Gynecol. Cancer. 2016;26:671–679. doi: 10.1097/IGC.0000000000000672. [DOI] [PubMed] [Google Scholar]
- 29.Workel H.H., Komdeur F.L., Wouters M.C.A., Plat A., Klip H.G., Eggink F.A., Wisman G.B.A., Arts H.J.G., Oonk M.H.M., Mourits M.J.E., et al. CD103 defines intraepithelial CD8+ PD1+ tumour-infiltrating lymphocytes of prognostic significance in endometrial adenocarcinoma. Eur. J. Cancer. 2016;60:1–11. doi: 10.1016/j.ejca.2016.02.026. [DOI] [PubMed] [Google Scholar]
- 30.Skon C.N., Lee J.Y., Anderson K.G., Masopust D., Hogquist K.A., Jameson S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013;14:1285–1293. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milner J.J., Toma C., Yu B., Zhang K., Omilusik K., Phan A.T., Wang D., Getzler A.J., Nguyen T., Crotty S., et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature. 2017;552:253–257. doi: 10.1038/nature24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holz L.E., Prier J.E., Freestone D., Steiner T.M., English K., Johnson D.N., Mollard V., Cozijnsen A., Davey G.M., Godfrey D.I., et al. CD8(+) T cell activation leads to constitutive formation of liver tissue-resident memory T cells that seed a large and flexible niche in the liver. Cell Rep. 2018;25:68–79.e4. doi: 10.1016/j.celrep.2018.08.094. [DOI] [PubMed] [Google Scholar]
- 33.Szabo P.A., Miron M., Farber D.L. Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol. 2019;4 doi: 10.1126/sciimmunol.aas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milner J.J., Toma C., He Z., Kurd N.S., Nguyen Q.P., McDonald B., Quezada L., Widjaja C.E., Witherden D.A., Crowl J.T., et al. Heterogenous populations of tissue-resident CD8(+) T cells are generated in response to infection and malignancy. Immunity. 2020;52:808–824.e7. doi: 10.1016/j.immuni.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li G., Srinivasan S., Wang L., Ma C., Guo K., Xiao W., Liao W., Mishra S., Zhang X., Qiu Y., et al. TGF-beta-dependent lymphoid tissue residency of stem-like T cells limits response to tumor vaccine. Nat. Commun. 2022;13:6043. doi: 10.1038/s41467-022-33768-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurtulus S., Madi A., Escobar G., Klapholz M., Nyman J., Christian E., Pawlak M., Dionne D., Xia J., Rozenblatt-Rosen O., et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1(-)CD8(+) tumor-infiltrating T cells. Immunity. 2019;50:181–194.e6. doi: 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casey K.A., Fraser K.A., Schenkel J.M., Moran A., Abt M.C., Beura L.K., Lucas P.J., Artis D., Wherry E.J., Hogquist K., et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012;188:4866–4875. doi: 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mackay L.K., Rahimpour A., Ma J.Z., Collins N., Stock A.T., Hafon M.L., Vega-Ramos J., Lauzurica P., Mueller S.N., Stefanovic T., et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 39.Zhang N., Bevan M.J. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity. 2013;39:687–696. doi: 10.1016/j.immuni.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ganesan A.P., Clarke J., Wood O., Garrido-Martin E.M., Chee S.J., Mellows T., Samaniego-Castruita D., Singh D., Seumois G., Alzetani A., et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017;18:940–950. doi: 10.1038/ni.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hombrink P., Helbig C., Backer R.A., Piet B., Oja A.E., Stark R., Brasser G., Jongejan A., Jonkers R.E., Nota B., et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat. Immunol. 2016;17:1467–1478. doi: 10.1038/ni.3589. [DOI] [PubMed] [Google Scholar]
- 42.Kloss C.C., Lee J., Zhang A., Chen F., Melenhorst J.J., Lacey S.F., Maus M.V., Fraietta J.A., Zhao Y., June C.H. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 2018;26:1855–1866. doi: 10.1016/j.ymthe.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dahmani A., Delisle J.S. TGF-beta in T Cell biology: implications for cancer immunotherapy. Cancers. 2018;10 doi: 10.3390/cancers10060194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liikanen I., Lauhan C., Quon S., Omilusik K., Phan A.T., Bartrolí L.B., Ferry A., Goulding J., Chen J., Scott-Browne J.P., et al. Hypoxia-inducible factor activity promotes antitumor effector function and tissue residency by CD8+ T cells. J. Clin. Invest. 2021;131 doi: 10.1172/JCI143729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Good Z., Spiegel J.Y., Sahaf B., Malipatlolla M.B., Ehlinger Z.J., Kurra S., Desai M.H., Reynolds W.D., Wong Lin A., Vandris P., et al. Post-infusion CAR T(Reg) cells identify patients resistant to CD19-CAR therapy. Nat. Med. 2022;28:1860–1871. doi: 10.1038/s41591-022-01960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haradhvala N.J., Leick M.B., Maurer K., Gohil S.H., Larson R.C., Yao N., Gallagher K.M.E., Katsis K., Frigault M.J., Southard J., et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat. Med. 2022;28:1848–1859. doi: 10.1038/s41591-022-01959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hou A.J., Chang Z.L., Lorenzini M.H., Zah E., Chen Y.Y. TGF-beta-responsive CAR-T cells promote anti-tumor immune function. Bioeng. Transl. Med. 2018;3:75–86. doi: 10.1002/btm2.10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hasan F., Chiu Y., Shaw R.M., Wang J., Yee C. Hypoxia acts as an environmental cue for the human tissue-resident memory T cell differentiation program. JCI Insight. 2021;6 doi: 10.1172/jci.insight.138970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luoma A.M., Suo S., Wang Y., Gunasti L., Porter C.B.M., Nabilsi N., Tadros J., Ferretti A.P., Liao S., Gurer C., et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell. 2022;185:2918–2935.e29. doi: 10.1016/j.cell.2022.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gabriel S.S., Tsui C., Chisanga D., Weber F., Llano-León M., Gubser P.M., Bartholin L., Souza-Fonseca-Guimaraes F., Huntington N.D., Shi W., et al. Transforming growth factor-beta-regulated mTOR activity preserves cellular metabolism to maintain long-term T cell responses in chronic infection. Immunity. 2021;54:1698–1714.e5. doi: 10.1016/j.immuni.2021.06.007. [DOI] [PubMed] [Google Scholar]
- 51.Im S.J., Hashimoto M., Gerner M.Y., Lee J., Kissick H.T., Burger M.C., Shan Q., Hale J.S., Lee J., Nasti T.H., et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caushi J.X., Zhang J., Ji Z., Vaghasia A., Zhang B., Hsiue E.H.C., Mog B.J., Hou W., Justesen S., Blosser R., et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature. 2021;596:126–132. doi: 10.1038/s41586-021-03752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pauken K.E., Sammons M.A., Odorizzi P.M., Manne S., Godec J., Khan O., Drake A.M., Chen Z., Sen D.R., Kurachi M., et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Araki Y., Fann M., Wersto R., Weng N.P. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B) J. Immunol. 2008;180:8102–8108. doi: 10.4049/jimmunol.180.12.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Araki Y., Wang Z., Zang C., Wood W.H., 3rd, Schones D., Cui K., Roh T.Y., Lhotsky B., Wersto R.P., Peng W., et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912–925. doi: 10.1016/j.immuni.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petrie H.T., Zúñiga-Pflücker J.C. Zoned out: functional mapping of stromal signaling microenvironments in the thymus. Annu. Rev. Immunol. 2007;25:649–679. doi: 10.1146/annurev.immunol.23.021704.115715. [DOI] [PubMed] [Google Scholar]
- 57.Wherry E.J. T cell exhaustion. Nat. Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 58.Doering T.A., Crawford A., Angelosanto J.M., Paley M.A., Ziegler C.G., Wherry E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schietinger A., Greenberg P.D. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35:51–60. doi: 10.1016/j.it.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schøller A.S., Nazerai L., Christensen J.P., Thomsen A.R. Functionally competent, PD-1(+) CD8(+) trm cells populate the brain following local antigen encounter. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.595707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lynn R.C., Weber E.W., Sotillo E., Gennert D., Xu P., Good Z., Anbunathan H., Lattin J., Jones R., Tieu V., et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293–300. doi: 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dai X., Park J.J., Du Y., Na Z., Lam S.Z., Chow R.D., Renauer P.A., Gu J., Xin S., Chu Z., et al. Massively parallel knock-in engineering of human T cells. Nat. Biotechnol. 2023 doi: 10.1038/s41587-022-01639-x. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshikawa T., Wu Z., Inoue S., Kasuya H., Matsushita H., Takahashi Y., Kuroda H., Hosoda W., Suzuki S., Kagoya Y. Genetic ablation of PRDM1 in antitumor T cells enhances therapeutic efficacy of adoptive immunotherapy. Blood. 2022;139:2156–2172. doi: 10.1182/blood.2021012714. [DOI] [PubMed] [Google Scholar]
- 64.Omilusik K.D., Nadjsombati M.S., Shaw L.A., Yu B., Milner J.J., Goldrath A.W. Sustained Id2 regulation of E proteins is required for terminal differentiation of effector CD8(+) T cells. J. Exp. Med. 2018;215:773–783. doi: 10.1084/jem.20171584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Masson F., Minnich M., Olshansky M., Bilic I., Mount A.M., Kallies A., Speed T.P., Busslinger M., Nutt S.L., Belz G.T. Id2-mediated inhibition of E2A represses memory CD8+ T cell differentiation. J. Immunol. 2013;190:4585–4594. doi: 10.4049/jimmunol.1300099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seo H., Chen J., González-Avalos E., Samaniego-Castruita D., Das A., Wang Y.H., López-Moyado I.F., Georges R.O., Zhang W., Onodera A., et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA. 2019;116:12410–12415. doi: 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Utzschneider D.T., Gabriel S.S., Chisanga D., Gloury R., Gubser P.M., Vasanthakumar A., Shi W., Kallies A. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 2020;21:1256–1266. doi: 10.1038/s41590-020-0760-z. [DOI] [PubMed] [Google Scholar]
- 68.Du N., Kwon H., Li P., West E.E., Oh J., Liao W., Yu Z., Ren M., Leonard W.J. EGR2 is critical for peripheral naive T-cell differentiation and the T-cell response to influenza. Proc. Natl. Acad. Sci. USA. 2014;111:16484–16489. doi: 10.1073/pnas.1417215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kurd N.S., He Z., Louis T.L., Milner J.J., Omilusik K.D., Jin W., Tsai M.S., Widjaja C.E., Kanbar J.N., Olvera J.G., et al. Early precursors and molecular determinants of tissue-resident memory CD8(+) T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.aaz6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harberts A., Schmidt C., Schmid J., Reimers D., Koch-Nolte F., Mittrücker H.W., Raczkowski F. Interferon regulatory factor 4 controls effector functions of CD8(+) memory T cells. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2014553118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frigault M.J., Lee J., Basil M.C., Carpenito C., Motohashi S., Scholler J., Kawalekar O.U., Guedan S., McGettigan S.E., Posey A.D., Jr., et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015;3:356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moon E.K., Wang L.C., Dolfi D.V., Wilson C.B., Ranganathan R., Sun J., Kapoor V., Scholler J., Puré E., Milone M.C., et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014;20:4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haas A.R., Tanyi J.L., O'Hara M.H., Gladney W.L., Lacey S.F., Torigian D.A., Soulen M.C., Tian L., McGarvey M., Nelson A.M., et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol. Ther. 2019;27:1919–1929. doi: 10.1016/j.ymthe.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oh S.A., Li M.O. TGF-beta: guardian of T cell function. J. Immunol. 2013;191:3973–3979. doi: 10.4049/jimmunol.1301843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee Y.T., Suarez-Ramirez J.E., Wu T., Redman J.M., Bouchard K., Hadley G.A., Cauley L.S. Environmental and antigen receptor-derived signals support sustained surveillance of the lungs by pathogen-specific cytotoxic T lymphocytes. J. Virol. 2011;85:4085–4094. doi: 10.1128/JVI.02493-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cretney E., Xin A., Shi W., Minnich M., Masson F., Miasari M., Belz G.T., Smyth G.K., Busslinger M., Nutt S.L., Kallies A. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 2011;12:304–311. doi: 10.1038/ni.2006. [DOI] [PubMed] [Google Scholar]
- 77.Li C., Zhu B., Son Y.M., Wang Z., Jiang L., Xiang M., Ye Z., Beckermann K.E., Wu Y., Jenkins J.W., et al. The transcription factor Bhlhe40 programs mitochondrial regulation of resident CD8(+) T cell fitness and functionality. Immunity. 2019;51:491–507.e7. doi: 10.1016/j.immuni.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zaid A., Mackay L.K., Rahimpour A., Braun A., Veldhoen M., Carbone F.R., Manton J.H., Heath W.R., Mueller S.N. Persistence of skin-resident memory T cells within an epidermal niche. Proc. Natl. Acad. Sci. USA. 2014;111:5307–5312. doi: 10.1073/pnas.1322292111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mackay L.K., Wynne-Jones E., Freestone D., Pellicci D.G., Mielke L.A., Newman D.M., Braun A., Masson F., Kallies A., Belz G.T., Carbone F.R. T-Box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity. 2015;43:1101–1111. doi: 10.1016/j.immuni.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 80.Fonseca R., Burn T.N., Gandolfo L.C., Devi S., Park S.L., Obers A., Evrard M., Christo S.N., Buquicchio F.A., Lareau C.A., et al. Runx3 drives a CD8(+) T cell tissue residency program that is absent in CD4(+) T cells. Nat. Immunol. 2022;23:1236–1245. doi: 10.1038/s41590-022-01273-4. [DOI] [PubMed] [Google Scholar]
- 81.Pallett L.J., Davies J., Colbeck E.J., Robertson F., Hansi N., Easom N.J.W., Burton A.R., Stegmann K.A., Schurich A., Swadling L., et al. IL-2(high) tissue-resident T cells in the human liver: sentinels for hepatotropic infection. J. Exp. Med. 2017;214:1567–1580. doi: 10.1084/jem.20162115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar B.V., Ma W., Miron M., Granot T., Guyer R.S., Carpenter D.J., Senda T., Sun X., Ho S.H., Lerner H., et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 2017;20:2921–2934. doi: 10.1016/j.celrep.2017.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mackay L.K., Minnich M., Kragten N.A.M., Liao Y., Nota B., Seillet C., Zaid A., Man K., Preston S., Freestone D., et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459–463. doi: 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 84.Freitas K.A., Belk J.A., Sotillo E., Quinn P.J., Ramello M.C., Malipatlolla M., Daniel B., Sandor K., Klysz D., Bjelajac J., et al. Enhanced T cell effector activity by targeting the Mediator kinase module. Science. 2022;378 doi: 10.1126/science.abn5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y., Kadel E.E., III, Koeppen H., Astarita J.L., Cubas R., et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tauriello D.V.F., Palomo-Ponce S., Stork D., Berenguer-Llergo A., Badia-Ramentol J., Iglesias M., Sevillano M., Ibiza S., Cañellas A., Hernando-Momblona X., et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 87.Tang N., Cheng C., Zhang X., Qiao M., Li N., Mu W., Wei X.F., Han W., Wang H. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5 doi: 10.1172/jci.insight.133977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Narayan V., Barber-Rotenberg J.S., Jung I.Y., Lacey S.F., Rech A.J., Davis M.M., Hwang W.T., Lal P., Carpenter E.L., Maude S.L., et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat. Med. 2022;28:724–734. doi: 10.1038/s41591-022-01726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stüber T., Monjezi R., Wallstabe L., Kühnemundt J., Nietzer S.L., Dandekar G., Wöckel A., Einsele H., Wischhusen J., Hudecek M. Inhibition of TGF-beta-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differentially expressed genes between CAR-TRM and CAR-TCONV cells. The table displays the following parameters: Gene ID, log fold change (log FC), log counts per million (log CPM), likelihood ratio (LR), p value, and false discovery rate (FDR).
Differential expression analysis of genes related to resident memory T cells, early memory T cells, and circulation-associated markers in CAR-TRM and CAR-TCONV cells as well as within CD8 subpopulations. The table is divided into three tabs: Tab 1, CD8 markers comparing TRM and TCONV cells; Tab 2, CD8 markers within distinct CD8 clusters; and Tab 3, TCF7CD8 markers comparing TRM and TCONV cells. Each tab displays the following parameters for the respective genes: p value (P_val), average log2 fold change (Avg_log2FC), percentage of cells expressing the gene in the first population (Pct.1), percentage of cells expressing the gene in the second population (Pct.2), adjusted p value (P_val_adj), cluster assignment, and gene symbol.
Sequences of CRISPR guide RNAs targeting AAVS1, IRF4, EGR2, and NR4A3 loci, as well as the corresponding forward and reverse sequencing primers for each target. The sequences are presented in the 5′-to-3′ orientation.
Data Availability Statement
-
•
The sequencing data reported in this paper is deposited in the Gene Expression Omnibus (GEO) database with accession number GSE224866. The data is available for public access and download from the GEO database, with no restrictions on its use or redistribution.
-
•
No original code is featured in this paper.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.