

Abstract
Alzheimer’s disease (AD) and Parkinson’s disease (PD) represent the most prevalent neurodegenerative disorders severely impacting life expectancy and quality of life of millions of people worldwide. AD and PD exhibit both a very distinct pathophysiological disease pattern. Intriguingly, recent researches, however, implicate that overlapping mechanisms may underlie AD and PD. In AD and PD, novel cell death mechanisms, encompassing parthanatos, netosis, lysosome-dependent cell death, senescence and ferroptosis, apparently rely on the production of reactive oxygen species, and seem to be modulated by the well-known, “old” second messenger cAMP. Signaling of cAMP via PKA and Epac promotes parthanatos and induces lysosomal cell death, while signaling of cAMP via PKA inhibits netosis and cellular senescence. Additionally, PKA protects against ferroptosis, whereas Epac1 promotes ferroptosis. Here we review the most recent insights into the overlapping mechanisms between AD and PD, with a special focus on cAMP signaling and the pharmacology of cAMP signaling pathways.
Keywords: cAMP, oxidative stress, mitochondria, parthanatos, ferroptosis, Alzheimer’s disease, Parkinson disease
Introduction
Alzheimer’s disease (AD) and Parkinson’s disease (PD) represent two of the most prevalent neurodegenerative disorders worldwide, affecting over 50 million and 10 million people, respectively (Rocca, 2018; Nichols et al., 2022). AD primarily affects learning and memory processes, and is characterized by progressive memory loss, language difficulties, disorientation, mood and behavior changes, and difficulty with daily routine tasks (Long and Holtzman, 2019; Scheltens et al., 2021). PD mainly affects coordination of movements, causing rest tremor, rigidity, bradykinesia, and difficulty with balance and coordination (Tolosa et al., 2021). Although PD can also cause non-motor symptoms such as depression, anxiety, sleep problems, and cognitive impairment, these symptoms are generally less severe than those of AD (Tolosa et al., 2021). AD and PD are multifactorial disorders that seemingly involve multiple cellular pathways and mechanisms. In AD, aggregation of amyloid beta (Aβ) and hyperphosphorylated tau are important contributors to the disease, along with oxidative stress, neuroinflammation, loss of cholinergic neurons, and mitochondrial dysfunction. On the other hand, the development and progression of PD also involve several cellular changes, including accumulation of α-synuclein, dopaminergic neurodegeneration, oxidative stress, neuroinflammation and mitochondrial dysfunction (Hou et al., 2019). Interestingly, accumulated α-synuclein was found in more than 50% of autopsied AD brain while tau protein could also be detected in one-third of PD cases, which may suggest overlapping mechanisms between AD and PD (Roberts et al., 2017; Zhang et al., 2018; Smith et al., 2019; Twohig and Nielsen, 2019; Visanji et al., 2019).
Reactive oxygen species (ROS), such as superoxide anion (O2-), hydrogen peroxide (H2O2) and hydroxyl radical (-OH), are intermediates in the process of aerobic metabolism. Mitochondria are the main organelles to generate ROS within the cells, particularly during aerobic respiration involving electron transfer through respiratory chain complexes I and III. Low levels of ROS function as signaling molecules, regulating transcription, phosphorylation and other pathways (Mittler, 2017). Some studies suggest that a low level of ROS plays specific role in cellular proliferation, differentiation and immunity (Cairns et al., 2011; West et al., 2011; Mittal et al., 2014; Diebold and Chandel, 2016; Mittler, 2017). In AD and PD, however, most probably due to an insufficient elimination and/or overproduction of ROS aberrantly high cellular ROS levels initiate cellular alterations defined as oxidative stress. For example, under normal physiological conditions, the intracellular level of H2O2 is tightly controlled within the low nanomolar concentration of approximately 1–10 nM and serves as redox signaling, while concentrations above 100 nM are considered as oxidative stress (Sies and Jones, 2020) (Figure 1). Oxidative stress damages (large) molecules including lipids, proteins and DNA, and thereby impacts cellular activities such as synaptic function, calcium homeostasis, receptor trafficking, endocytosis and regulated cell death (RCD) (Halliwell, 2006; Tönnies and Trushina, 2017). Indeed, increasing evidence indicates that the presence of oxidative stress in brains is closely associated with pathological hallmarks of AD and PD (Sultana et al., 2009; Padurariu et al., 2010; Rajkumar et al., 2019; Trist et al., 2019; Oun et al., 2022). In AD, Aβ oligomers trigger a significant influx of Ca2+ into the mitochondria, subsequently stimulating oxidative phosphorylation, and ultimately resulting in dysfunctional mitochondria and increased ROS production. Interestingly, a recent study reviewing RNA sequencing data also indicates that dysfunctional mitochondria in late AD exhibit an alteration of the expression of ROS-related gens in neurons, such as oxidative phosphorylation and electron transport chain (Marmolejo-Garza et al., 2022). In addition, in PD models it has been hypothesized that binding of α-synuclein to mitochondria disrupts the mitochondrial membrane potential, subsequently leading to ROS generation and impairment of mitochondria protein import (Krabbendam et al., 2018). Apart from dysfunctional mitochondria, accumulation of bioactive metals and activated microglia are the other two sources of oxidative stress in AD and PD (Tönnies and Trushina, 2017). Bioactive metals including copper, iron, and zinc, are found to be accumulated in the brains of AD and PD and serve as catalysts in ROS production via Fenton and Haber-Weiss reactions. In addition, in AD and PD, disease-associated microglia (DAM) are activated via damage-associated molecular patterns (DAMPs) that are released in response to neuronal damage or protein aggregation. Activated DAM results in persistent inflammation and ROS production, and have been implicated in the exacerbation of neurodegeneration (Calsolaro and Edison, 2016; Simpson and Oliver, 2020).
FIGURE 1.
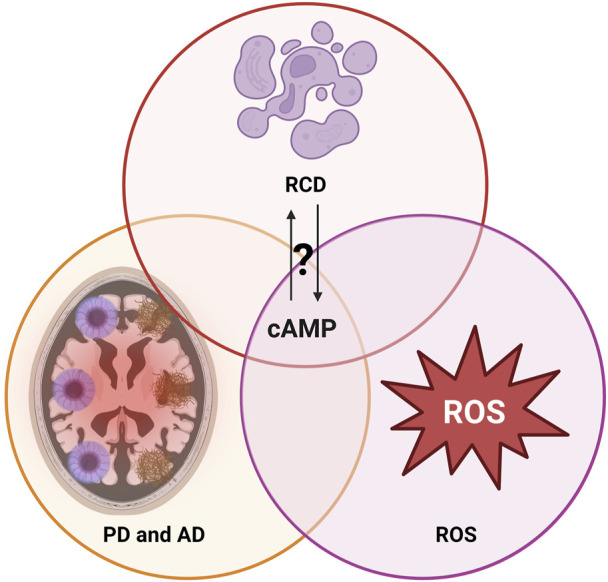
Interplay of regulated cell death (RCD) mechanisms, reactive oxygen species (ROS) and the neurodegenerative disorders Alzheimer’s disease (AD) and Parkinson disease (PD): Mechanisms potentially overlapping on the level of cAMP. For further details, see text.
Cyclic adenosine monophosphate (cAMP) is a key second messenger that mediates a variety of physiological processes, including memory and learning, myocardial relaxation and contraction, inflammation and immune function (Cheng et al., 2008; Serezani et al., 2008), and various cellular activities, such as calcium homeostasis, gene transcription, glutaminergic receptor trafficking, cell death, and neurotransmission (Schmidt et al., 2013; Liu et al., 2016). However, dysregulation of cAMP signaling has been linked to various diseases such as cancer, diabetes, and neurological disorders, highlighting the importance of tight regulation of intracellular cAMP levels. cAMP signaling is initiated by activation of G protein-coupled receptors (GPCRs), such as the Gs-coupled β-adrenoceptors. Subsequently, Gs activates transmembrane adenylate cyclases (tmACs), leading to an elevation of intracellular cAMP levels. In addition, soluble AC (sAC), known to be regulated by CO2/HCO−3/pH-, calcium and ATP, contributes to cAMP elevations (Wiggins et al., 2018). The duration and amplitude of cAMP signaling is tightly controlled by phosphodiesterases (PDEs), including cAMP specific PDE4, PDE7, and PDE8, and cAMP/cGMP dual substrate specific PDE1, PDE2, PDE3, PDE10 and PDE11 (Zuo et al., 2019). cAMP signaling pathway involves four main effectors, namely, protein kinase A (PKA), exchange protein directly activated by cAMP (Epac), cyclic nucleotide regulated ion channels and Popeye domain containing (POPDC) proteins, which all share a conserved cAMP binding domain except POPDC proteins (Brand, 2018; Robichaux and Xiaodong Cheng, 2018). As the most studied cAMP effector, PKA exerts its effects by either directly activating proteins or modulating other kinase pathways through phosphorylation. The multifunctional facets of PKA signaling are coordinated by the interaction with A-kinase anchoring proteins (AKAPs), tethering PKA to receptors (such as the Gs-coupled receptors, ion channels) and subcellular structures such as actin filaments, microtubules, mitochondria and nuclei (Poppinga et al., 2014; Han et al., 2015) (Figure 2). PKA also plays a role in regulating gene expression by phosphorylating cAMP response element-binding protein (CREB) in the nucleus. Epac, on the other hand, functions as a guanine nucleotide exchange factor (GEF) and modulates downstream targets by activating small GTPases such as Rap and Ras (Schmidt et al., 2013). Meanwhile, cAMP-gated ion channels, such as hyperpolarization-activated cyclic nucleotide-gated channels (HCNs), directly modulate ion influx and membrane depolarization, facilitating action potentials (Santoro and Shah, 2019). Unlike PKA and Epac, POPDC proteins are transmembrane proteins that interact with cAMP and downstream proteins via their highly conserved intracellular POPEYE domains (Tucker and Zorn, 2022). As mentioned above, AKAPs play a crucial role in cAMP signaling by coordinating G proteins, ACs, PDEs, PKA, and Epac (Nijholt et al., 2008; Schmidt et al., 2013) (Figure 2). This coordination leads to the formation of signalsomes, enabling efficient sensing and functioning of cAMP in various subcellular locations.
FIGURE 2.

The cAMP signaling pathways. The well-known, “old” second messenger cAMP is generated upon activation of Gs-coupled receptors (such as the β-adrenoceptors) and subsequent stimulation of a diverse subset of adenylyl cyclases (ACs), shown are the transmembrane ACs (tmACs) and the soluble ACs (sACs). cAMP exerts its effects through its primary effector protein kinase A (PKA), next to the exchange protein directly activated by cAMP (Epac) cyclic nucleotide-gated channels (HCN) and Popeye domain containing (POPDC) proteins (Brand, 2018; Robichaux and Xiaodong Cheng, 2018). The multifaceted signaling properties of cAMP are controlled by A-kinase anchoring proteins (AKAPs) and phosphodiesterases (PDEs), enabling cAMP signaling in space and time (Schmidt et al., 2013; Poppinga et al., 2014; Han et al., 2015). For further details, see text.
Many studies have investigated the effects of ROS on cAMP signaling pathway. For example, in rat adipocytes it was reported that micromolar concentrations of H2O2 inhibited type II PKA activity via the formation of disulfide bonds between Cys-97 in the regulatory subunit and Cys-199 in the catalytic subunit (De Piña et al., 2008). Similarly, a study in pancreatic islets also suggested that ROS reduced cAMP levels via ROS sensitive ACs and inhibit PKA via oxidizing specific residues (Li et al., 2012). In addition, it was found that ROS can bypass cAMP stimulation and directly modulate PKA activity. In cardiomyocytes, it was reported that ROS oxidized the regulatory subunits of type I PKA at Cys17 and Cys38, promoting disulfide bond formation between two regulatory subunits leaving PKA activity unchanged. The cytosolic disulfide bonds containing type I PKA was subsequently translocated to the membrane and activated independent of cAMP elevations (Brennan et al., 2006). In a model of ischemic heart failure, it was found that increased ROS activated PKA in the absence of cAMP elevations and consequently initiated substrate phosphorylation (Andre et al., 2013). A more recent study in mouse cardiomyocytes addressed that oxidized type I PKA preferentially localized to the lysosome via increased AKAP binding (Simon et al., 2021). On the other hand, the cAMP/PKA pathway can seemingly modulate ROS production by regulating enzyme activities involved in the respiration. It was reported that activation of PKA attenuated ROS production via phosphorylation on NADH ubiquinone oxidoreductase subunit S4 (NDUFS4) in complex I in different human and murine cells (Piccoli et al., 2006). Another study revealed that activation of PKA by forskolin increased complex I activity, leading to a reduction in ROS levels in cancer cells (Palorini et al., 2013). On the contrary, however, in cardiomyocytes, PKA activation triggered by a β-adrenergic agonist promoted ROS production, while pretreatment with a PKA inhibitor abrogated ROS production, suggesting that PKA exerts distinct, cell type specific functions in cellular ROS production (Andersson et al., 2011). In line with these findings, the other main cAMP effector, Epac has been proposed to negatively regulate ROS production. It was reported that Epac downregulated downstream PI3K/Akt pathway, leading to a reduced ROS levels in pancreatic β-cells (Mukai et al., 2011). In another study of ischemia-reperfusion injury, activation of Epac-Rap1 pathway reduced the production of mitochondrial superoxide and thereby prevented ROS production (Stokman et al., 2014). These studies demonstrate the intricate interplay between ROS and cAMP signaling, which depends on several factors, including ROS concentrations, specific cell types and signaling pathways (Figures 1, 2).
In AD, cAMP levels are generally reduced, although sometimes elevated or unchanged, depending on the expression/activity of AC and PDE in specific brain regions, which may contribute to AD pathogenesis (Kelly, 2018; Di Benedetto et al., 2021). The basal, Gαs-mediated, and forskolin-induced AC activities are decreased in the hippocampus and temporal cortex, due to reduced expression of AC1 and AC2, rather than other AC isoforms. However, Gαs-mediated AC activity is only decreased in the cerebellum and occipital cortex (Kelly, 2018). On the other hand, in late AD the mRNA level of PDE4A is reduced in the frontal cortex and CA2, while PDE4D is increased in the putamen (Pérez-Torres et al., 2003; Ugarte et al., 2015). The mRNA level of PDE7A is decreased in the dentate gyrus (DG), whereas the mRNA level of PDE8B is increased in both the hippocampal DG and CA2 regions (Pérez-Torres et al., 2003). Notably, studies in in vivo AD models have demonstrated that hippocampal impairments can be mitigated by increasing cAMP levels through either forskolin-stimulated AC activity or reduced expression/activity of PDEs (Kelly, 2018). Furthermore, a number of PDE4 inhibitors have shown clinical potential to improve cognition and disease severity via compensating decreased cAMP in AD (Prickaerts et al., 2017). In addition, soluble Aβ was found to lead to mitochondrial Ca2+ overload (Calvo-Rodriguez et al., 2020), which may trigger activation of mitochondrial sAC in AD (Valsecchi et al., 2014). Studies in in vivo, in vitro models of AD and AD patients reported on reduced expression and/or activity of global PKA (Bonkale et al., 1996; Kelly, 2018). It is worth noting that localized PKA and AKAP5 are involved in hyperphosphorylation of tau pathology in AD (Jicha et al., 1999). Localized PKA signaling may provide an explanation for the puzzling finding that rather low concentrations of the PKA inhibitor H89 attenuated memory deficits, oxidative stress and NfkB signaling in the male Wistar rats (Eftekharzadeh et al., 2012; Amini et al., 2015). In PD, on the other hand, alterations in cAMP due to (gene and protein) changes in PDE4D, PDE8B and PDE10A correlates with dementia progression and symptom severity (Azuma et al., 2015; Niccolini et al., 2015; Kaut et al., 2017). Similarly, in the 6-hydroxydopamine (6-OHDA) model of PD a decreased PDE10A expression correlated with an increased cAMP level (Sancesario et al., 2014). Signorile et al. (2021) performed studies in fibroblasts from Parkin-mutants and reported on cAMP elevations due to reduced PDE4 activity, while treatment with resveratrol (known to inhibit several molecular targets next to PDE4) restored the cellular cAMP level pointing to the involvement of cAMP modulators distinct from PDE4. Indeed, Signorile et al. (2021) demonstrated that resveratrol inhibited transmembrane ACs (tmACs) and activated soluble ACs (sACs) via upregulation of cytosolic and mitochondrial Ca2+, resulting in a remodeling of the cAMP homeostasis. In addition, compartmentalized cAMP/PKA also play an important role in PD pathology. A study revealed that both AKAP1 overexpression and PKA activation restored dysfunctional mitochondrial respiration and lysosome function in PD cell models with deficient PTEN-induced kinase 1 (PINK1), indicating the role of compartmentalized cAMP/PKA signaling in mitochondrial pathology (Dagda et al., 2011).
Extensive evidence has already indicated that cell death can occur in either accidental ways or regulated ways. Compared with accidental cell death, regulated cell death (RCD) is tightly and precisely modulated by various molecular machineries, and it has been suggested that RCD can be inhibited genetically and/or pharmacologically (Galluzzi et al., 2018). The first identified RCD, apoptosis, was found by John Kerr in 1972 and had been used as the synonyms for all RCDs up until 2000 (Kerr et al., 1972). After 2000, many novel RCD mechanisms, such as parthanatos, NETosis, lysosome-dependent cell death, cellular senescence and ferroptosis, were gradually identified and studied (Tang et al., 2019).
Mature neurons in a healthy CNS are generally resistant to cell death and can persist throughout the individual lifespan (Chi et al., 2018). Neuronal cell death is generally observed as a final outcome of multiple stress accumulations, surpassing the ability of neurons to recover. In AD, cell death occurs in various regions, including the entorhinal cortex, nucleus basalis, and locus coeruleus, even before clinical symptoms (Arendt et al., 2013). As AD progresses, more severe neuronal loss occurs in the frontal cortex and other subcortical regions (Martínez-Pinilla et al., 2016). In PD, over 50% of neurons in the substantia nigra are lost prior to diagnosis (Ross et al., 2004). Until now, various RCDs are observed in AD and PD, and we provide here the most updated summary. However, it remains unclear whether RCDs are primary contributors or merely represent secondary responses due to the underlying pathophysiology of AD and PD. Oxidative stress is a well-established trigger for various RCDs, as comprehensively reviewed in (Tang et al., 2019). cAMP signaling, on the other hand, has been demonstrated to engage in reciprocal regulation with oxidative stress. In this review, we aim to elaborate several ROS related RCD mechanisms in AD and PD, and will emphasize the impact of cAMP signaling to those novel RCD mechanisms (Figures 1, 2).
Parthanatos
Parthanatos is a caspase-independent RCD that is triggered by hyperactivation of poly (ADP-ribose) polymerase (PARP-1) (Galluzzi et al., 2018). Unlike apoptosis, parthanatos occurs without the formation of an apoptotic body or small-size DNA fragmentation (Wang et al., 2009). PARP-1 is vital for repairing single or double strand DNA breaks induced by toxic stimuli such as oxidative stress, nitrosative stress, hypoxia, hypoglycemia, or inflammation (Galluzzi et al., 2018). Severe DNA damage causes PARP-1 overactivation, which subsequently catalyzes a variety of DNA repair proteins via transferring ADP-ribose (PAR) to itself or its target proteins (Andrabi et al., 2008; Narne et al., 2017). However, excessive poly (ADP-ribosyl)ation (PARlyation) causes nicotinamide adenine dinucleotide (NAD+) and ATP depletion, which in turn contributes to parthanatotic cell death (Figure 3). Furthermore, the accumulated PAR polymers translocate from the nucleus to mitochondria and induce the release of apoptosis-inducing factor (AIF) from mitochondria. AIF recruits macrophage migration inhibitory factor (MIF) into the nucleus and causes parthanatotic chromatinolysis and large-size DNA fragmentation inside the nucleus (Wang et al., 2011; Wang et al., 2016). In AD and PD, excessive release of the neurotransmitter glutamate activates N-Methyl-D-aspartic acid (NMDA) receptors or α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, further induces Ca2+ influx and activation of nitric oxide synthase (NOS) (Fatokun et al., 2013; Gerace et al., 2014). Nitric oxide (NO) reacts with superoxide anion (O2 −) which is a common ROS in neurodegenerative disorders, and forms into peroxynitrite (ONOO−). NO and ONOO− can potently induce DNA damage and consequently lead to parthanatotic neuronal death (Fatokun et al., 2013) (Figure 3; Table 1).
FIGURE 3.

ROS and other stimuli induce DNA damage subsequently leading to the hyperactivation of PARP-1 and PARylation (Andrabi et al., 2008; Narne et al., 2017). Excessive PAR polymers translocate from the nucleus to mitochondria and induce AIF release from mitochondria. AIF recruits MIF into the nucleus and causes chromatinolysis and large-size DNA fragmentation (Wang et al., 2011; Wang et al., 2016). In AD and PD, excessive glutamate activates N-Methyl-D-aspartic acid (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors that induce Ca2+ influx and NOS activation, leading to generation of NO and ONOO−, which can potentiate DNA damage and trigger parthanatos (Fatokun et al., 2013). Activation of cAMP/PKA axis promotes phosphorylation of mitochondrial PARP-1, thereby promoting parthanatos (Brunyanszki et al., 2014). Epac activates Rap and H-Ras, and upregulates PARP expression by inhibiting its cleavage (Grandoch et al., 2009b). These mechanisms highlight the role of cAMP signaling in promoting parthanatos via both PKA and Epac. For further details, see text.
TABLE 1.
Studies of cAMP modulators in in vitro models of regulated cell deaths.
| Research | Regulated cell deaths | cAMP modulators | Target | Model | Main research findings |
|---|---|---|---|---|---|
| Brunyanszki et al. (2014) | Parthanatos | Propranolol | β-receptor antagonist | U937 | β -adrenoceptor/cAMP/PKA axis phosphorylates PARP-1 subsequently leading to parthanatos |
| DDA | AC inhibitor | C2C12 | |||
| PKAi | PKA inhibitor | ||||
| Isoproterenol | β-receptor agonist | ||||
| Forskolin | AC activator | ||||
| 8-Br-cAMP | PKA&Epac activator | ||||
| Grandoch et al. (2009a) | Parthanatos | 8-pCPT-2′-O-Me-cAMP | Epac activator | U937 | Epac activation leads to inhibition of PARP cleavage |
| Grandoch et al. (2009b) | Parthanatos | 8-Br-cAMP | PKA&Epac activator | WEHI 231 | Epac upregulates PARP expression via Rap1 and H-Ras |
| dd-Ado | AC inhibitor | ||||
| SQ22536 | AC inhibitor | ||||
| H-89 | PKA inhibitor | ||||
| Forskolin | AC activator | ||||
| Rp-8CPT-cAMP | PKA inhibitor | ||||
| 8-pCPT-2′-O-MecAMP | Epac activator | ||||
| Bokoch et al. (2009) | Netosis | H-89 | PKA inhibitor | HEK293 | NOXA1 phosphorylation by PKA recruits 14-3-3 protein subsequently inhibiting Nox holoenzyme assembly and potentially preventing netosis |
| Forskolin | AC activator | HT-29 | |||
| Eby et al. (2014) | Netosis | Forskolin | AC activator pan-PDE inhibitor | Neutrophil | Elevated cAMP by AC inhibits NETs formation and oxidative burst |
| IBMX | |||||
| Stahl-Meyer et al. (202) | Lysosome dependent cell death | Forskolin | AC activator | MCF7 | Lysosphingolipid-induced lysosome-dependent cell death requires cAMP signaling seemingly sensitizing cells to lysosome-dependent cell death |
| Fibroblast | |||||
| Sung et al. (2020) | Senescence | H-89 | PKA inhibitor | Vascular smooth muscle cell | cAMP/PKA-SIRT1 seem to mediate antisenescence mechanisms |
| Forskolin | AC activator | ||||
| Wang et al. (2022) | Senescence | Forskolin | AC activator | mesenchymal stem cells | cAMP alterations impact senescence and senescence-related inflammatory phenotypes |
| SQ22536 | AC inhibitor | ||||
| Ishii et al. (2019) | Ferroptosis | 8-Br-cAMP | PKA&Epac activator | Yeast mitochondria | PKA suppresses mitochondria and indirectly inhibits Nrf2 |
| Musheshe et al. (2022) | Ferroptosis | CE3F4 | Epac1 inhibitor | HT-22 | Epac1 inhibition reduces ROS and lipid peroxidation subsequently preventing ferroptosis |
| ESI-05 | Epac2 inhibitor | ||||
| ESI-09 | Epac inhibitor | ||||
| 8-pCPT-2′-O-Me-cAMP | Epac activator |
AC, adenylyl cyclase; DDA, 2′,3′-dideoxyadenosine; PKA, protein kinase A; epac, exchange protein directly activated by cAMP; IBMX, 3-isobutyl-1-methylxanthine; PARP, Poly (ADP-ribose) polymerase; Nox, NADPH oxidase; NETs, neutrophil extracellular traps; SIRT1, Sirtuin 1; Nrf2, nuclear factor erythroid 2-related factor 2.
Many studies have indicated that parthanatos is associated with AD and PD. In AD, Aβ has been shown to activate PARP-1 in various cell models, including microglia, astrocytes, and neuronal/glial cell cultures (Martire et al., 2013; Wang and Ge, 2020). Love et al. (1999) reported that PARP-1 and PAR were more expressed in postmortem AD brains compared to controls. Interestingly, in those AD brains, PAR containing cells were mainly represented by neurons and rarely by astrocytes or microglia, suggesting that parthanatos mainly occurs in neurons in AD (Love et al., 1999). Additionally, Lee et al. (2012) found AIF was significantly elevated in hippocampal CA1 region at early stages of AD and CA2 region at advanced stages of AD. In PD, a recent study reported that parthanatos was triggered by α-synuclein pre-formed fibrils (α-syn PFFs) via NOS mediated DNA damage, while other RCDs including necroptosis and autophagy were not involved in α-synuclein induced cell death, which may imply that parthanatos is a prominent RCD in PD pathology. Furthermore, PAR promoted the fibrillization of α-synuclein into a new strain with 25-fold greater toxicity, which further activated PARP-1. The level of PAR was found to be increased in the cerebrospinal fluid (CSF) and substantia nigra of PD patients, further confirming that PARP-1 activation is involved in PD pathogenesis (Kam et al., 2018) (Table 1).
Several PARP inhibitors have been developed and approved for clinical trials of cancer treatment, but their precise effects on AD and PD still need to be determined (Zhou et al., 2021). On one hand, inhibition of PARP-1 prevents parthanatos via mitigating over-active PARP-1. For example, it was found that treatment with the PARP-1 specific inhibitor 3AB prevented ROS-mediated cell death parthanatos and downregulated parthanatotic pathway in human SH-SY5Y neuroblastoma cells (Wang et al., 2018). On the other hand, PARP-1 depletion also leads to dysfunctional DNA repair machinery, severe DNA damage, and consequently promoting cell death, which has been used as the strategy in cancer treatment (Zhou et al., 2021). Therefore, considering the unknown side effect of long-term PARP-1 inhibition, the use of PARP-1 inhibitors in treating long-term AD and PD should be applied with caution. Alternative approaches to prevent parthanatos include targeting AIF and MIF. Inhibition of AIF release from mitochondria, its interaction with binding proteins such as cyclosporin A, or its translocation into the nucleus with compounds such as N-phenylmaleimide can prevent AIF-mediated parthanatos (Fatokun et al., 2013). Similarly, adenoviral knockdown of MIF in a rat spinal cord model has been found to reduce oxidative stress-induced parthanatos in neurons (Yang et al., 2020), and MIF antagonists such as antibodies, inhibitors, and fragment peptides of MIF have been successfully used to diminish parthanatos driven cell death (Song et al., 2022). Recent studies have shown promising results using the MIF nuclease inhibitor PAANIB-1 in vivo to mitigate α-synuclein or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurodegeneration (Park et al., 2022). These studies offer potential usage of parthanatos associated therapeutics in the future treatment of AD and PD.
Intriguingly, it was demonstrated that activation of the cAMP/PKA axis promotes mitochondrial PARP-1 activation induced by ROS (Brunyanszki et al., 2014) (Figure 3). Stimulation of the Gs-coupled β-adrenoceptor increase the cellular cAMP levels subsequently leading to the activation of PKA. Activated PKA translocates to the mitochondria where it modulates parthanatos by phosphorylating PARP-1 (Brunyanszki et al., 2014). The activation of Epac has also been found to potentially contribute to parthanatotic cell death by inhibiting the cleavage of PARP (Grandoch et al., 2009a) (Figure 3). Additionally, their findings suggest that Epac upregulates PARP expression via Rap1 and H-Ras, as inhibition of these proteins has been observed to promote PARP cleavage (Grandoch et al., 2009b) (Table 1). Taken together modulation of parthanatos driven cell death might rely on the cAMP effectors PKA and Epac, and modulation of these cAMP signaling pathways might bear the potential for future treatment options in AD and PD.
Netosis
Netosis is a type of RCD dependent on neutrophil extracellular traps (NETs) in response to infection or injury (Galluzzi et al., 2018). Activated neutrophils undergo netosis and release NETs to limit the growth or kill various pathogenic microorganisms including bacteria, fungi, virus and parasite (Mutua and Gershwin, 2021). Netosis is initiated by activation of neutrophils via a variety of pattern recognition receptors (PPRs) including toll-like receptors, NOD-like receptors and C-type lectin receptors, followed by Ca2+ influx (Huang et al., 2022). This elevated Ca2+ triggers the activation of a subset of calcium-dependent protein kinase C (PKCs) and downstream Raf-MEK-ERK cascades, which phosphorylate NADPH oxidase (NOX) and enhance ROS production (Hakkim et al., 2011; Dahlgren et al., 2019) (Figure 4). Excessive ROS leads to the degradation of cytoplasmic granule and causes the release of elastase (NE) and myeloperoxidase (MPO) into the nucleus, together with peptidyl arginine deiminase 4 (PAD4), leads to chromatin decondensation, ultimately resulting in cell rupture and NETs release (Papayannopoulos et al., 2010) (Figure 4; Table 1).
FIGURE 4.

Netosis is initiated by activation of neutrophils via pattern recognition receptors (PRRs) and a subsequent influx of Ca2+ (Huang et al., 2022). This triggers a cascade of events involving Ca2+-dependent PKC, Raf-MEK-ERK pathway, NAPDH oxidase (NOX) phosphorylation, leading to ROS production (Hakkim et al., 2011; Dahlgren et al., 2019) (panel 4). The excessive ROS causes cytoplasmic granule degradation and release of elastase (NE) and myeloperoxidase (MPO) to the nucleus, along with peptidyl arginine deiminase 4 (PAD4), leading to chromatin decondensation and ultimately resulting in cell rupture and NETs release (Papayannopoulos et al., 2010) (panel 4). PKA phosphorylation of NOXA1 recruits 14-3-3 proteins, which block the assembly of NOX1 holoenzyme, ultimately preventing ROS production (Bokoch et al., 2009). Moreover, supraphysiological cAMP inhibits the formation of NETs and ROS burst (Eby et al., 2014). These findings highlight the role of cAMP signaling in inhibiting netosis via PKA. For further details, see text.
The blood-brain barrier (BBB) is a highly semipermeable border that insulates brain parenchyma from circulating leukocytes. Neutrophils infiltrated the BBB and migrated into parenchyma via adhesion to blood vessels, and released NETs and IL-17 in transgenic AD models (Zenaro et al., 2015). Furthermore, AD pathology and cognitive deficits were attenuated after temporary depletion of neutrophils or inhibition of neutrophil trafficking (Zenaro et al., 2015). In the same study, neutrophils and NETs were also found to accumulate around Aβ plagues in AD patients, while neutrophils from age-matched controls were remained in blood vessels. In a recent study, an increased adhesion of neutrophils to small blood vessels, along with NETs, were observed in an AD mouse model and AD patients, suggesting that netosis contributes to BBB breakdown in AD (Smyth et al., 2022). These available data suggest that netosis is involved in the pathogenesis of AD.
Inhibition of NET formation has already been used as a therapeutic approach against autoimmune and inflammatory diseases. NOX is a good target in netotic pathway and has entered several preclinical/clinical trials (Chamardani and Amiritavassoli, 2022). NOX inhibitors such as Apocynin and VAS2870, prevented ROS and netosis in human neutrophils (Vorobjeva et al., 2020). In addition to the regulation of NOX activity through the PKC pathway, NOX activity can also be regulated by PKA, which phosphorylates on NOXA1 and recruits regulatory14-3-3 proteins. The binding of 14-3-3 blocks the assembly of NOX1 holoenzyme, consequently preventing ROS production (Bokoch et al., 2009). In addition, it was reported that cAMP inhibited the formation of NETs and ROS burst (Eby et al., 2014) (Figure 4; Table 1). These studies suggest that activation of cAMP/PKA pathway may prevent netosis via NOX inhibition. Until now studies with focus on Epac and a potential link to PD are still missing and might be worthwhile to pursue in the future.
Lysosome-dependent cell death
Lysosome-dependent cell death is a type of RCD mediated by the lysosomal release of the proteolytic enzyme cathepsin. This cell death is induced by various stimuli including ROS, lysosomotropic detergents, dipeptide methyl esters and lipid metabolites (Tang et al., 2019) (Figure 5). After lysosomal membrane permeabilization (LMP), cytosolic cathepsins catalyze multiple substrates, such as Bid and anti-apoptotic proteins, and subsequently initiate caspase-dependent cell death (Taniguchi et al., 2015; Gómez-Sintes et al., 2016). It is worth noting that lysosome-dependent cell death can also trigger other forms of RCD, such as apoptosis and ferroptosis, which are respectively caused by the lysosomal leakage of Bid-specific cathepsins and iron-rich content (Kurz et al., 2008; Gao et al., 2018) (Figure 5; Table 1).
FIGURE 5.

Lysosome-dependent cell death is triggered by ROS or other stimuli, leading to LMP and a release of cathepsins into cytosol. Cathepsins catalyze multiple substrates, including Bid and anti-apoptotic proteins and initiate caspase-dependent cell death (Taniguchi et al., 2015; Gómez-Sintes et al., 2016). Additionally, lysosome-dependent cell death can activate other RCDs, such as apoptosis and ferroptosis, through the leakage of Bid-specific cathepsins and iron-rich content, respectively (Kurz et al., 2008; Gao et al., 2018). Lysosphingolipid induced lysosomal cell death in a process involving Ca2+-dependent adenylyl cyclase 1 (ADCY1), followed by an cAMP elevation, and finally inhibition of lysosomal acid sphingomyelinase (SMase) (Anand et al., 2019; Stahl-Meyer et al., 2022). These findings highlight the role of cAMP signaling in promoting lysosome-dependent cell death via ADCY1. For further details, see text.
It was reported that oligomeric Aβ42 induced LMP and subsequently released lysosomal enzymes, including cathepsins, resulting in neuronal death (Hook et al., 2020). In AD patients, cathepsin D and other lysosomal proteins were increased in blood exosome up to 10 years before clinical symptoms (Goetzl et al., 2015). Furthermore, active cathepsin D and cathepsin B were elevated and co-localized with senile plaques in the AD brains (Cataldo and Nixon, 1990). This can be attributed to the role of these cathepsins in Aβ degradation (Cermak et al., 2016). In a study of PD patients and a PD mouse model, reduced lysosomal proteins was found to be associated with increased Lewy body in nigral (Vila et al., 2011). LMP was also confirmed in 1-methyl-4-phenylpyridinium (MPP+)-induced PD cell model (Vila et al., 2011). These studies suggest that lysosome-dependent cell death is involved in pathogenesis of AD and PD.
A recent study found that lysosphingolipids induced lysosome-dependent cell death required activation of cAMP signaling pathways (Stahl-Meyer et al., 2022). Furthermore, elevated cAMP induced by AC activator forskolin, was found to sensitize cells to lysosomal cell death when co-treated with low concentrations of lysosphingolipids (Stahl-Meyer et al., 2022) (Table 1). This lysosomal cell death pathway probably started with a fast release of Ca2+ from lysosomes, followed by activation of Ca2+-dependent adenylyl cyclase 1 (ADCY1), which ultimately inhibited lysosomal acid sphingomyelinase (SMase) (Anand et al., 2019; Stahl-Meyer et al., 2022) (Figure 5; Table 1). However, it is important to note that further studies are required to fully understand how cAMP pathway is involved in lysosome-dependent cell death.
Cellular senescence
Cellular senescence refers to a pathophysiologial process that normal cells lose their proliferation abilities and undergo irreversible arrest in cell cycle when they reach their limits of replicative lifespan (Galluzzi et al., 2018). Cellular senescence was previously thought to be a type of RCD being induced by telomere shortening and other stressors, such as ROS, mitochondrial damage and tumor suppressor proteins (Campisi, 2013). ROS and other stressors can damage DNA and subsequently trigger DNA damage response (DDR) to phosphorylate a cell cycle repressor, p53 (Ramirez et al., 2001; Turenne et al., 2001; Nakamura et al., 2008). p53 further activates cyclin-dependent kinase 2 (CDK2) inhibitor p21 and results in decreased phosphorylation of retinoblastoma protein (Rb), which binds and inactivates transcription factor E2F, and subsequently arrests the cell cycle from G1 to S (Krenning et al., 2014). Additionally, p16INK4a can specifically inhibit CDK4 and CDK6 and thereby maintain the senescent suspension of cell cycle (Stein et al., 1999; Ruas et al., 2007). Senescent cells can release proinflammatory factors and ROS when they are turned into senescence-associated secretory phenotypes (SASPs) (Coppé et al., 2010). p38 mitogen-activated protein kinases (p38MAPK), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), cyclic GMP-AMP synthase (cGAS) and mammalian target of rapamycin (mTOR) are implicated in the SASP conversion process (Kritsilis et al., 2018) (Figure 6; Table 1).
FIGURE 6.

ROS or other stressors initiates cellular senescence via DNA damage and subsequent DNA damage response (DDR) activation. DDR results in p53 phosphorylation (Ramirez et al., 2001; Turenne et al., 2001; Nakamura et al., 2008) and p21 activation, leading to CDK2 inhibition and Rb/E2F-mediated G1 to S cell cycle arrest (Krenning et al., 2014). In addition, p16 can specifically inhibit CDK4/6 and maintain senescent cell cycle arrest (Stein et al., 1999; Ruas et al., 2007). Senescent cells produce proinflammatory factors and ROS when they turn into SASPs. cAMP/PKA/CREB pathway activates anti-senescence protein, Sirtuin 1 (SIRT1), and prevents cellular senescence (Sung et al., 2020; Zhu et al., 2022). These findings highlight the role of cAMP/PKA signaling in inhibiting cellular senescence via activating SIRT1. For further details, see text.
Cellular senescence was observed in wide range of cell types in central nervous system, including neurons, astrocytes, microglia, oligodendrocytes, oligodendrocyte progenitor cells and neural stem cells (NSCs) (Kritsilis et al., 2018). Senescent neurons and glia cells were found to be linked with AD and PD (Turnquist et al., 2016; Bussian et al., 2018; Chinta et al., 2018; Kritsilis et al., 2018), and pathological hallmarks of AD and PD, such as Aβ, tau, and α-synuclein, were reported to induce senescence in in vitro or in vivo models (Verma et al., 2021; Liu, 2022). In AD patients and mouse models, elevated senescent markers, p16INK4a, p21CIP1, p53 and senescence associated β-Galactosidase (SA-β-Gal) were observed (Arendt et al., 1996; Arendt et al., 1998; McShea et al., 1997; Lüth et al., 2000; Engidawork et al., 2001; Hochstrasser et al., 2011; Esteras et al., 2012; Esteras et al., 2013; Bhat et al., 2012; Campisi, 2013; Yates et al., 2015). Similarly, p16INK4a and SA-β-Gal were increased in PD patients (van Dijk et al., 2013; Chinta et al., 2018). Studies revealed that SASP factors, including interleukin-1β (IL-1β), interleukin-6 (IL-6), transforming growth factor-β (TGF-β), and tumor necrosis factor-α (TNF-α), were upregulated in AD, as evidenced by their elevation in the brains, CSF, and serum of AD patients (Blum-Degena et al., 1995; Dursun et al., 2015; Huell et al., 1995; Lai et al., 2017; Rea et al., 2018; Swardfager et al., 2010; Wood et al., 1993). Similarly, increased levels of IL-1β, IL-6, and TNF-α were observed in the striatum and CSF of PD patients (Blum-Degena et al., 1995; Hofmann et al., 2009; Scalzo et al., 2010; Lindqvist et al., 2012; Dursun et al., 2015; Chinta et al., 2018). Furthermore, hippocampal p38MAPK was reported to be activated during the early stage of AD (Sun et al., 2003). Taken together, these findings suggest that cellular senescence is very likely involved in the pathogenesis of AD and PD.
Cellular senescence has emerged as a potential target for the treatment of AD and PD. Senolytic treatment, which selectively induces death of senescent cells, has shown promise as a novel therapeutic strategy. However, it is still controversial which cell type should be the primary target of senolysis. A recent study found that oligodendrocyte progenitor cell (OPC) was the only glia cell type showing elevated senescent markers in AD patients and an Aβ mouse model. Selective elimination of senescent OPCs improved cognitive function, reduced neuroinflammation, and decreased Aβ load (Zhang et al., 2019). Another study correlated astrocytes and microglia with increased p16INK4a in P19 tau pathology mice, and demonstrated that genetic and pharmacological clearance of those cells could reduce gliosis, neurofibrillary tangles (NFTs), neuronal loss and cognitive impairment (Bussian et al., 2018). In contrast, the third study suggested that NFT-containing neurons were the primary target of senolysis in human and mouse AD brains. Treatment with senolytics reduced NFT density, neuronal loss and ventricular atrophy in a tau transgenic mouse model (Musi et al., 2018). Additionally, a PD study showed that senolytic treatment could reduce neuropathology and improve motor deficits (Chinta et al., 2018). Taken together, these studies provide compelling evidence for the potential of senolysis as a therapeutic approach for AD and PD. However, further research is needed to clarify the optimal cell types to target and the most effective senolytic treatment strategies.
Sirtuin 1 (SIRT1) is commonly accepted as an anti-senescence protein, as it possesses deacetylase activity that can be activated through cAMP signaling (Aslam and Ladilov, 2022). Sung et al. (2020) recently demonstrated that overexpression or activation of SIRT1 attenuated senescence via cAMP/PKA pathway in vascular smooth muscle cells. Another study revealed that senescence-accelerated mouse prone 8 (SAMP8) mice, which showed neurodegeneration induced by Aβ and accelerated senescence, expressed lower levels of cAMP, phosphorylated PKA, and phosphorylated CREB (Hou et al., 2022). Similarly, in a rat epithelial cell line, interleukin-13 (IL-13) induced senescence downregulated phosphorylated CREB (Zhu et al., 2022) (Figure 6). These studies suggest that activation of cAMP/PKA/CREB pathway prevents cellular senescence. However, in contrast to these findings, Wang et al. (2022) found that elevated cAMP induced by forskolin triggered cellular senescence in human-derived mesenchymal stem cells, while inhibition of cAMP by SQ22536 prevented against forskolin-induced senescence and senescence-related inflammatory phenotypes. These studies reveal differential roles of cAMP signaling in cellular senescence across various cells and tissues. Therefore, further research is necessary to fully understand the role of cAMP signaling in senescence and its potential therapeutic implications.
Ferroptosis
Dysregulation of trace metals is one of the pathological features in AD and PD. In AD, zinc, copper and iron accumulate in senile plaques, with zinc and copper inducing a conformational change in Aβ and resulting in non-fibrillar Aβ oligomerization, while iron is likely to be associated with plaques in a ferritin-bound form (Grundke-Iqbal et al., 1990; Lovell et al., 1998; Tiiman et al., 2013). Similarly, in PD, iron accumulation in Lewy bodies has been observed and implicated in accelerating α-synuclein aggregation (Hashimoto et al., 1999; Castellani et al., 2000; Uversky et al., 2001). Iron overload has been detected in specific brain regions including the parietal cortex, motor cortex, and hippocampus in AD patients (Bartzokis et al., 1994; Bartzokis and Tishler, 2000; Pfefferbaum et al., 2009; Langkammer et al., 2014; Tao et al., 2014), while increased iron levels have been observed in the substantia nigra in PD patients (Dexter et al., 1987). Furthermore, increased ferritin level in CSF is linked with the progression of AD (Ayton et al., 2018; 2015). The presence of reactive redox ions acts as catalysts to generate ROS via Fenton and Haber-Weiss reactions and further initiate lipid peroxidation, which have been detected in AD and PD patients (Tsang and Chung, 2009; Butterfield et al., 2010; Bradley-Whitman and Lovell, 2015). In addition, lipid peroxidation also causes breakdown of blood brain barrier and, therefore, exacerbates progression of early AD and PD (Chen et al., 2022).
Ferroptosis is a type of RCD that is characterized by severe lipid peroxidation due to ROS generation and iron overload, both of which are also the pathological features in AD and PD (Galluzzi et al., 2018) (Figure 7). The lipid hydroperoxides are originated from polyunsaturated fatty acids (PUFA), such as arachidonoyl (AA) and adrenoyl (AdA) phospholipids, which are stepwise oxidized into PUFA-OOHs by long-chain-fatty-acid-CoA ligase 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3) and 15-lipoxygenase (15-LOX), and eventually act as ferroptotic signals and execute ferroptosis (Kagan et al., 2017; Li and Li, 2020). Glutathione peroxidase 4 (GPX4) is the only enzyme that catalyzes lipid hydroperoxides into hydroxy derivatives within biological membranes (Conrad and Friedmann Angeli, 2017). GPX4 oxidizes its substrate glutathione (GSH) into glutathione disulfide (GSSG) to clear lipid hydroperoxides (Li and Li, 2020). GSH is synthesized from a nonessential amino acid cysteine, which limits the rate of GSH synthesis (Lewerenz et al., 2013). Cysteine is directly transported into cells or in its oxidative form, cystine, via the cystine/glutamate antiporter System Xc − (Lewerenz et al., 2013). Inhibition of System Xc − by its inhibitors or high concentration of glutamate results in decreased intracellular cysteine pool and further leads to GSH depletion (Bannai and Kitamura, 1980). GSH depletion attenuates the activity of GPX4 and activates 15-LOX, and further trigger lipid peroxidation and ferroptosis (Lewerenz et al., 2018). Iron overload also contributes to lipid peroxidation since ferrous ion is a cofactor of 15-LOX (Yang et al., 2016). Additionally, the major metabolite of 15-LOX, 15-hydroxyeicosatetraenoic acid (15-HETE), disrupts the tight junctions of endothelial cells in blood brain barrier and results in BBB dysfunction (Chattopadhyay et al., 2014) (Figure 7).
FIGURE 7.

PUFA are stepwise oxidized into lipid hydroperoxides, PUFA-OOH by ACSL4, LPCAT3, and 15-LOX (Kagan et al., 2017; Li and Li, 2020). GPX4 uses GSH as a substrate to catalyze lipid hydroperoxides into hydroxy derivatives, limiting lipid peroxidation (Lewerenz et al., 2013). Excessive glutamate or System Xc-inhibition causes GSH depletion and attenuates GPX4 activity, leading to lipid peroxidation and ferroptosis (Bannai and Kitamura, 1980). Moreover, iron overload generates hydroxyl radicals via Fenton reaction, which also contributes to lipid peroxidation (Yang et al., 2016). Epac1 inhibition reduces ROS levels and lipid peroxidation by decreasing mitochondrial superoxide (Musheshe et al., 2022), while PKA suppresses mitochondrial activation and ROS production via inhibiting protein import (Ishii et al., 2019). In the meantime, PKA indirectly inhibits Nrf2 via outcompeting with Nrf2 regulator CK2. In addition, CREB regulates GPX4 transcription to prevent ferroptosis (Wang et al., 2021). These findings highlight the role of cAMP signaling in preventing ferroptosis via both PKA and mitochondrial Epac. For further details, see text.
It has been demonstrated a correlation between the cognitive decline in AD and PD and the reduction of GSH in hippocampus and frontal cortex (Mandal et al., 2015). Iron and copper have been found to promote GSH loss and cell death in HT22 hippocampal neurons (Maher, 2018), and decreased GSH levels have been observed in dopaminergic neurons in PD brains (Pearce et al., 1997). A conditional ablation of GPX4 in mouse forebrain neurons exhibited AD-like dysfunctions in learning and memory, as well as neurodegeneration in the hippocampus (Hambright et al., 2017). A recent RNA-seq analysis of AD patients reported that ferroptosis related genes, such as GPX4 and glutathione synthase (GSS), were downregulated at early AD and upregulated at late AD, suggesting that neuronal cells gradually compensate ferroptotic cell death by increasing antioxidant proteins with the progression of AD (Majerníková et al., 2021) (Figure 7). Interestingly, neurons expressed more ferroptosis related genes than glia cells in AD patients, probably revealing that ferroptosis initially affects neurons and subsequently spread to glia cells (Majerníková et al., 2021). This could be due to the fact that glial cells produce and transfer antioxidants to neurons, while neurons express antioxidant genes to a lesser extent (Ishii et al., 2019) (Table 1). These studies highlight the role of ferroptosis as a major type of RCD in AD and PD.
Inhibition of ferroptosis could be a strategy to treat AD and PD. The major anti-ferroptosis drugs are divided into three categories, 1) iron chelators, 2) antioxidants, 3) modulators of GSH synthesis (Lane et al., 2018; Majerníková et al., 2021). Examples of iron chelators include deferoxamine (DFO) and clioquinol, which have shown promise in delaying AD and PD progression in preclinical models (Grossi et al., 2009; Lei et al., 2012; Sripetchwandee et al., 2016; Finkelstein et al., 2017). However, those iron chelators still face limitations before clinical trials, including inability to specifically target at iron-overloaded brain regions and inconsistent dose regimen across different clinical trials (Derry et al., 2020; Farr and Xiong, 2021). Antioxidants attenuate ROS production via either trapping free radicals or inhibiting 15-LOX (Mabalirajan et al., 2009; Kagan et al., 2017; Zilka et al., 2017). Ferrostatin-1 and vitamin E are two examples of antioxidants that have been studied in the context of AD and PD. Ferrostatin-1 traps radicals and has been shown to decrease neurodegeneration in GPX4 knockout mice (Hambright et al., 2017), while vitamin E inhibits LOX and reduces lipid peroxidation (Mabalirajan et al., 2009; Ju et al., 2021) (Figure 7). A study of GPX4 ablation mice reported that Vitamin E deficient diets accelerated hippocampal degeneration and behavioral dysfunction (Hambright et al., 2017). Clinical trials also reported that vitamin E delayed AD progression by 6 months in mild to moderate AD patients and improve symptoms and GSH levels in PD patients (Epperly et al., 2017; Taghizadeh et al., 2017). However, there is still no consensus on the effectiveness of vitamin E, as another study reported that vitamin E non-respondent patients, showed sharp cognitive decline compared with placebo group (Lloret et al., 2009). Modulation of GSH synthesis related proteins, such as GPX4, is another potential therapy against ferroptosis. It was reported that α-Lipoic acid (LA) upregulated GPX4 expression and thereby reduced lipid peroxidation in a Tau mouse model (Zhang et al., 2018). In addition, the transcription of GPX4 and other antioxidant response element (ARE)-containing genes such as GSH S-transferase, GSH reductase and GSS are regulated by a redox induced transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2) (Song and Long, 2020). Activation of Nrf2 by its activator, sulforaphane, results in increased GSH level, decreased ROS production and improving memory deficits in various preclinical AD models (Kim, 2021) (Figure 7). Overall, these various approaches to inhibiting ferroptosis offer potential methods for treating AD and PD.
In astrocytes, Nrf2 upregulates antioxidant proteins during mitochondrial activation, while PKA suppresses mitochondrial activation and ROS production via blocking protein import into mitochondria, and indirectly inhibits Nrf2 via outcompeting with Nrf2 regulator, casein kinase 2 (CK2) (Ishii et al., 2019). Besides, it was reported that CREB mitigated ferroptosis via regulating GPX4 transcription in a lung cell line (Wang et al., 2021). In a recent study it was demonstrated that inhibition of Epac1 in the immortalized hippocampal (HT22) neuronal cells led to a reduction in ROS levels and lipid peroxidation by decreasing mitochondrial superoxide levels and subsequently preventing ferroptosis (Musheshe et al., 2022) (Figure 7; Table 1). These studies collectively suggest that the cAMP pathway may suppress mitochondrial activity and thereby prevent ferroptosis and shed light on the potential therapeutic targets for preventing ferroptosis by regulating the cAMP pathway.
Conclusion and outlook
Both AD and PD severely impact life expectancy and quality of life of millions of people worldwide. Unfortunate, despite the global burden, the search after novel drugs able to diminish or even prevent the pathophysiological disease patterns is still characterized by a very limited progress in the last decades. Intriguingly, recent evidence indicates that cAMP-linked drugs - already approved by both FDA (Food and Drug Administration, U.S.) and EMA (European Medicines Agency, Europe)–such as β-adrenoceptor agonists (e.g., salbutamol, albuterol) and antagonists (e.g., propranolol, sotalol) seem to enjoy some clinical success (Cepeda et al., 2019; Crotty and Schwarzschild, 2022; Fella et al., 2022; Lu’O’Ng and Nguyen, 2013; Mittal et al., 2014; Ramos et al., 2008; Wang et al., 2009; Wang et al., 2011; Andersson et al., 2013; Bond et al., 2007). However, patient benefit, potential underlying mechanisms and prevalence of unwanted treatment outcomes are still a matter of debate.
Global elevators and/or inhibitors of cAMP will have numerous (side) effects based on the widespread expression of the effectors of the cAMP signaling pathway in many cells and tissues including the brain. Therapeutics that specifically activate and/or inhibit effectors of cAMP signaling will likely enjoy greater clinical benefits with fewer side effects. In addition, gaining increasing knowledge about the impact of cAMP effectors distinct from β-adrenoceptors may open new avenues for combination therapies–allowing reduction in the concentration of each active drug component thereby most likely limiting unwanted treatment effects. Such combination therapies are already operational to treat chronic lung disorders (e.g., asthma and chronic obstructive pulmonary disease) and cancer. Neurodegenerative disorders may alter also cAMP effector expression and/or localization, causing dysfunctioning of cAMP signaling. Such mechanisms may profoundly contribute to oxidative stress triggered-RCD mechanisms known to result in neuronal death in AD and PD. Therefore, more research into how specific cAMP signaling play key roles in distinct subcellular compartments is needed.
For this purpose, we provided in this review a comprehensive overview about mechanisms underlying ROS-related novel cell death mechanisms in AD and PD. Notably, cAMP signaling appears to play differential roles in these RCDs. Signaling of cAMP via PKA and Epac promotes parthanatos and induces lysosomal cell death, while signaling of cAMP via PKA inhibits netosis and cellular senescence. Additionally, PKA protects against ferroptosis, whereas Epac1 promotes ferroptosis. Specifically, in parthanatos, PKA and Epac have been implicated in promoting cell death through phosphorylation of PARP-1 and inhibition of PARP cleavage, respectively (Grandoch et al., 2009a; Brunyanszki et al., 2014). In lysosomal cell death, elevated cAMP is required for sensitizing cells to lysosphingolipid-induced lysosomal cell death (Stahl-Meyer et al., 2022). Conversely, cAMP signaling has been shown to prevent netosis by inhibiting NOX activity and ROS production through cAMP/PKA pathway (Bokoch et al., 2009), and to prevent cellular senescence via regulation of SIRT1 through cAMP/PKA/CREB pathway (Sung et al., 2020; Zhu et al., 2022). In ferroptosis, PKA activation suppresses mitochondrial activation and upregulates GPX4 via CREB to reduce ROS production and ferroptosis (Ishii et al., 2019), while Epac1 inhibition decreases mitochondrial superoxide levels and subsequently prevents ferroptosis (Musheshe et al., 2022). Therefore, modulation of cAMP signaling pathways bears the potential for targeting various RCDs in AD and PD.
Pharmacologically modulating PKA can result in unintended adverse effects due to its ubiquitous expression in various tissues and cells. To address this issue, alternative strategies that increase the specificity of PKA signaling have been proposed (Insel et al., 2012). Recently, it was found that the type I regulatory subunit of PKA drives the formation of cAMP and PKA condensate, which facilitates cAMP subcellular compartmentation (Zhang et al., 2020). This finding suggests that selectively targeting PKA regulatory or catalytic subunits that are expressed in a specific tissue or subcellular compartment could be a potential strategy to minimize the side effects of PKA activation. The type I-beta and type II-beta regulatory subunits of PKA are promising candidates for modulation due to their selective expression patterns in the brain and lower expression levels in other tissues, and may provide new therapeutic opportunities against RCDs in AD and PD. Additionally, targeting AKAPs such as brain specific AKAP5, or specific phosphorylation targets of PKA can also increase the specificity of PKA signaling. Interestingly, it has been reported that AKAP5 is upregulated in AD patients (Jicha et al., 1999). In addition, AKAP5 has been reported to diminish β-adrenoceptor-induced cAMP signaling (Cong et al., 2001). Targeting of AKAP5 signaling has successfully applied to diminish cardiomyocyte dysfunctioning (Musheshe et al., 2018a; 2018b). By employing these approaches, we hypothesize that it might be possible in the future to achieve targeted activation of PKA signaling while minimizing the potential for off-target effects.
Modulating Epac signaling could be another potential therapeutic strategy against RCDs in neurodegenerative diseases. Indeed, it was reported that Epac1 gene was upregulated whereas Epac2 gene was downregulated in hippocampal and frontal cortex samples of AD patients compared to age-matched controls (Mcphee et al., 2005). Likewise, inhibition of Epac1 largely diminished ROS-related ferroptosis in HT22 hippocampal cells (Musheshe et al., 2022). It is tempting to speculate that an sophisticated interplay between Epac1 and Epac2 may of central importance in the regulation of ROS-driven RCDs in AD and PD. Unlike PKA, Epac2 is specifically expressed in the brain and adrenal glands, and has been implicated in neuronal differentiation and regeneration, thereby potentially reducing side effects in other tissues (Schmidt et al., 2013). Recently, a specific Epac2 activator Sp-8-BnT-cAMPS (“S220”) was developed using structure-guided chemistry, exhibiting 10 times higher potency and 7 times efficacy for Epac2 compared to cAMP (Schwede et al., 2015). Furthermore, a membrane-penetrating version of S220 called S223-AM has been developed recently expected to cross BBB and being therefore more suitable for future treatment opportunities in AD and PD (Xu et al., 2019). Definitely, further research is needed to elucidate the specific mechanisms involved in Epac2-mediated signaling, and its potential interplay with Epac1 against RCDs.
In addition to traditional low molecular weight modulators, PROteolysis TArgeting Chimeras (PROTACs) have shown promising effect in selective degradation of target proteins opening new avenues to specifically inhibit the cAMP pathway. PROTACs are bifunctional molecules with specific ligands for protein of interest and E3 ubiquitin ligase, connected by a linker (Sun et al., 2019). The target protein forms a ternary complex with E3 ubiquitin ligase, and subsequently are ubiquitinated and degraded by ubiquitin proteasome system (Sun et al., 2019). Due to its unique mechanism, PROTACs exhibit higher sensitivity towards drug-resistant targets, which is often induced by exposure to high concentrations of small molecule inhibitors. Furthermore, PROTACs allow direct protein-level knockdown without potential genetic mutations and upregulations compared to other gene-editing techniques (Sun et al., 2019). In the context of AD and PD, PROTACs have reduced pathology and improved cognitive deficits (Konstantinidou et al., 2021; He et al., 2022). In addition, targeting a coactivator of CREB, CREB-binding protein (CBP) that forms a CREB/CBP complex and initiates gene transcription, exhibited a higher antiproliferative effect in myeloma cells compared to traditional inhibitors (Vannam et al., 2021). Next to PROTACs, one might envision to apply the CRISPR/Cas-9 technology to manipulate cAMP pathway. In a recent study, a PDE3A mutant introduced by CRISPR/Cas-9 was shown to provide long-term cardioprotection compared to nonselective PDE3 inhibitors in cardiomyocytes (Ercu et al., 2022). Intriguingly, recent studies point to the usage of molecular glues acting as small molecule mediators that bring two proteins together, regardless of their natural affinity or binding capabilities (Soini et al., 2022). The linker of PROTACs that connects E3 ligase and target proteins is an example of molecular glue. A class of molecular glue are 14-3-3 stabilizers, such as fusicoccin-A and pyrrolidone1, which seem to stabilize the interaction between 14-3-3 and its binding partners (Soini et al., 2022). As outlined in detail above, PKA phosphorylates NOXA1 and recruits 14-3-3, inhibiting the assembly of the NOX1 holoenzyme, ROS production, and ultimately netosis (Bokoch et al., 2009) (Table 1). It is tempting to speculate that 14-3-3 stabilizers strengthen the 14-3-3/NOXA1 complex, and thereby potentially preventing netosis. The inclusion of novel techniques such as PROTACs, CRISPR/Cas-9 and molecular glues will certainly support our future research to further unravel the distinct roles of PKA, AKAPs and Epac’s, their cell-cell type specific interplay to regulate the distinct features of RCDs.
Acknowledgments
We thank the Department of Molecular Pharmacology and Molecular Neurobiology for the support.
Funding Statement
AD is the recipient of an Alzheimer Nederland grant (WE.03-2018-04, Netherlands), Parkinson Fonds (Netherlands) and a Rosalind Franklin Fellowship co-funded by the European Union and the University of Groningen. UE and MS received support by Alzheimer Nederland grant WE.03-2019-05. MS was supported by the Deutsche Forschungsgemeinschaft (IRTG1874DIAMICOM-SP2) and Novartis unrestricted grant 50199468.
Author contributions
TZ and MS wrote the manuscript; ML created the figures with Biorender.com; UE, AD, and MS performed supervision and critical proof-reading. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- Amini E., Nassireslami E., Payandemehr B., Khodagholi F., Foolad F., Khalaj S., et al. (2015). Paradoxical role of PKA inhibitor on amyloidβ-induced memory deficit. Physiol. Behav. 149, 76–85. 10.1016/j.physbeh.2015.05.029 [DOI] [PubMed] [Google Scholar]
- Anand A., Liu B., Giacobini J. D., Maeda K., Rohde M., Jaattela M. (2019). Cell death induced by cationic amphiphilic drugs depends on lysosomal Ca2+ release and cyclic AMP. Mol. Cancer Ther. 18, 1602–1614. 10.1158/1535-7163.MCT-18-1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson D. C., Fauconnier J., Yamada T., Lacampagne A., Zhang S. J., Katz A., et al. (2011). Mitochondrial production of reactive oxygen species contributes to the β-adrenergic stimulation of mouse cardiomycytes. J. Physiology 589, 1791–1801. 10.1113/jphysiol.2010.202838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson K. E., Martin N., Nitti V. (2013). Selective β₃-adrenoceptor agonists for the treatment of overactive bladder. J. Urology 190, 1173–1180. 10.1016/j.juro.2013.02.104 [DOI] [PubMed] [Google Scholar]
- Andrabi S. A., Dawson T. M., Dawson V. L. (2008). Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann. N. Y. Acad. Sci. 1147, 233–241. 10.1196/annals.1427.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre L., Fauconnier J., Reboul C., Feillet-Coudray C., Meschin P., Farah C., et al. (2013). Subendocardial increase in reactive oxygen species production affects regional contractile function in ischemic heart failure. Antioxid. Redox Signal 18, 1009–1020. 10.1089/ars.2012.4534 [DOI] [PubMed] [Google Scholar]
- Arendt T., Holzer M., Gärtner U. (1998). Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J. Neural Transm. 105, 949–960. 10.1007/s007020050104 [DOI] [PubMed] [Google Scholar]
- Arendt T., Rödel L., Gärtner U., Holzer M. (1996). Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. Neuroreport 7, 3047–3049. 10.1097/00001756-199611250-00050 [DOI] [PubMed] [Google Scholar]
- Aslam M., Ladilov Y. (2022). Targeting the sAC-dependent cAMP pool to prevent SARS-cov-2 infection. Cells 9, 1962. 10.3390/cells9091962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton S., Diouf I., Bush A. I. Alzheimer’s disease Neuroimaging Initiative (2018). Evidence that iron accelerates alzheimer’s pathology: A CSF biomarker study. J. Neurol. Neurosurg. Psychiatry 89, 456–460. 10.1136/jnnp-2017-316551 [DOI] [PubMed] [Google Scholar]
- Ayton S., Faux N. G., Bush A. I., Weiner M. W., Aisen P., Petersen R., et al. (2015). Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 6, 6760. 10.1038/ncomms7760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma R., Ishikawa K., Hirata K., Hashimoto Y., Takahashi M., Ishii K., et al. (2015). A novel mutation of PDE8B Gene in a Japanese family with autosomal-dominant striatal degeneration. Mov. Disord. 30, 1964–1967. 10.1002/mds.26345 [DOI] [PubMed] [Google Scholar]
- Bannai S., Kitamura E. (1980). Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J. Biol. Chem. 255, 2372–2376. 10.1016/s0021-9258(19)85901-x [DOI] [PubMed] [Google Scholar]
- Bartzokis G., Sultzer D., Mintz J., Holt L. E., Marx P., Kelly Phelan C., et al. (1994). In vivo evaluation of brain iron in Alzheimer’s disease and normal subjects using MRI. Biol. Psychiatry 35, 480–487. 10.1016/0006-3223(94)90047-7 [DOI] [PubMed] [Google Scholar]
- Bartzokis G., Tishler T. A. (2000). MRI evaluation of basal ganglia ferritin iron and neurotoxicity in Alzheimer’s and Huntingon’s disease. Cell. Mol. Biol. 46, 821–833. [PubMed] [Google Scholar]
- Bhat R., Crowe E. P., Bitto A., Moh M., Katsetos C. D., Garcia F. U., et al. (2012). Astrocyte senescence as a component of alzheimer’s disease. PLoS One 7, e45069. 10.1371/journal.pone.0045069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum-Degena D., Müller T., Kuhn W., Gerlach M., Przuntek H., Riederer P. (1995). Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 202, 17–20. 10.1016/0304-3940(95)12192-7 [DOI] [PubMed] [Google Scholar]
- Bokoch G. M., Diebold B., Kim J. S., Gianni D. (2009). Emerging evidence for the importance of phosphorylation in the regulation of NADPH oxidases. Antioxid. Redox Signal 11 (10), 2429–2441. 10.1089/ars.2009.2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond R. A., Spina D., Parra S., Page C. P. (2007). Getting to the heart of asthma: Can “β blockers” be useful to treat asthma? Pharmacol. Ther. 115, 360–374. 10.1016/j.pharmthera.2007.04.009 [DOI] [PubMed] [Google Scholar]
- Bonkale W. L., Fastbom J., Wiehager ’ B., Ravid R., Winblad ’ B., Cowburn ’ R. F. (1996). Impaired G-protein-stimulated adenylyl cyclase activity in Alzheimer’s disease brain is not accompanied by reduced cyclic-AMP-dependent protein kinase A activity. Brain Research 737. Elsevier. [DOI] [PubMed] [Google Scholar]
- Bradley-Whitman M. A., Lovell M. A. (2015). Biomarkers of lipid peroxidation in alzheimer disease (AD): An update. Arch. Toxicol. 89, 1035–1044. 10.1007/s00204-015-1517-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand T. (2018). The Popeye domain containing genes and their function as cAMP effector proteins in striated muscle. J. Cardiovasc Dev. Dis. 5, 18. 10.3390/jcdd5010018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan J. P., Bardswell S. C., Burgoyne J. R., Fuller W., Schröder E., Wait R., et al. (2006). Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 281, 21827–21836. 10.1074/jbc.M603952200 [DOI] [PubMed] [Google Scholar]
- Brunyanszki A., Olah G., Coletta C., Szczesny B., Szabo C. (2014). Regulation of mitochondrial poly(ADP-Ribose) polymerase activation by the β-adrenoceptor/cAMP/protein kinase A axis during oxidative stress. Mol. Pharmacol. 86, 450–462. 10.1124/mol.114.094318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian T. J., Aziz A., Meyer C. F., Swenson B. L., van Deursen J. M., Baker D. J. (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. 10.1038/s41586-018-0543-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield D. A., Bader Lange M. L., Sultana R. (2010). Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1801, 924–929. 10.1016/j.bbalip.2010.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns R. A., Harris I. S., Mak T. W. (2011). Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95. 10.1038/nrc2981 [DOI] [PubMed] [Google Scholar]
- Calsolaro V., Edison P. (2016). Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dementia 12, 719–732. 10.1016/j.jalz.2016.02.010 [DOI] [PubMed] [Google Scholar]
- Calvo-Rodriguez M., Hou S. S., Snyder A. C., Kharitonova E. K., Russ A. N., Das S., et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 11, 2146. 10.1038/s41467-020-16074-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. (2013). Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705. 10.1146/annurev-physiol-030212-183653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani R. J., Siedlak S. L., Perry G., Smith M. A. (2000). Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 100, 111–114. 10.1007/s004010050001 [DOI] [PubMed] [Google Scholar]
- Cataldo A. M., Nixon R. A. (1990). Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc. Natl. Acad. Sci. U. S. A. 87, 3861–3865. 10.1073/pnas.87.10.3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda M. S., Kern D. M., Seabrook G. R., Lovestone S. (2019). Comprehensive real-world assessment of marketed medications to guide Parkinson’s grug discovery. Clinical Drug Investigation 39 (11), 1067–1075. 10.1007/s40261-019-00830-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak S., Kosicek M., Mladenovic-Djordjevic A., Smiljanic K., Kanazir S., Hecimovic S. (2016). Loss of cathepsin B and L leads to lysosomal dysfunction, NPC-like cholesterol sequestration and accumulation of the key Alzheimer’s proteins. PLoS One 11, 167428. 10.1371/journal.pone.0167428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamardani T. M., Amiritavassoli S. (2022). Inhibition of NETosis for treatment purposes: Friend or foe? Mol. Cell. Biochem. 477, 673–688. 10.1007/s11010-021-04315-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay R., Dyukova E., Singh N. K., Ohba M., Mobley J. A. (2014). Vascular endothelial tight junctions and barrier function are disrupted by 15(S)- hydroxyeicosatetraenoic acid partly via protein kinase C e-mediated zona occludens-1 phosphorylation at threonine 770/772. J. Biol. Chem. 289 (6), 3148–3163. 10.1074/jbc.M113.528190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Pang X., Yeo A. J., Xie S., Xiang M., Shi B., et al. (2022). The molecular mechanisms of ferroptosis and its role in blood-brain barrier dysfunction. Front. Cell. Neurosci. 16, 889765. 10.3389/fncel.2022.889765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Ji Z., Tsalkova T., Mei F. (2008). Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. (Shanghai) 40, 651–662. 10.1111/j.1745-7270.2008.00438.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H., Chang H. Y., Sang T. K. (2014). Neuronal cell death mechanisms in major neurodegenerative diseases. Int. J. Mol. Sci. 10.3390/ijms19103082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta S. J., Woods G., Demaria M., Rane A., Zou Y., McQuade A., et al. (2018). Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell. Rep. 22, 930–940. 10.1016/j.celrep.2017.12.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong M., Perry S. J., Lin F. T., Fraser I. D., Hu L. A., Chen W., et al. (2001). Regulation of membrane targeting of the G protein-coupled receptor kinase 2 by protein kinase A and its anchoring protein AKAP79. J. Biol. Chem. 276, 15192–15199. 10.1074/jbc.M009130200 [DOI] [PubMed] [Google Scholar]
- Conrad M., Friedmann Angeli J. P. (2017). Glutathione peroxidases. Compr. Toxicol. 2017, 260–276. 10.1016/B978-0-12-801238-3.95621-6 [DOI] [Google Scholar]
- Coppé J. P., Desprez P. Y., Krtolica A., Campisi J. (2010). The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathology Mech. Dis. 5, 99–118. 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty G. F., Schwarzschild M. A. (2022). What to Test in Parkinson Disease Prevention Trials?: Repurposed, Low-Risk, and Gene-Targeted Drugs. Neurology 99 (7), S34–S41. 10.1212/WNL.0000000000200238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda R. K., Gusdon A. M., Pien I., Strack S., Green S., Li C., et al. (2011). Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell. Death Differ. 18, 1914–1923. 10.1038/cdd.2011.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren C., Karlsson A., Bylund J. (2019). Intracellular neutrophil oxidants: From laboratory curiosity to clinical reality. J. Immunol. 202, 3127–3134. 10.4049/jimmunol.1900235 [DOI] [PubMed] [Google Scholar]
- De Piña M. Z., Vázquez-Meza H., Pardo J. P., Rendón J. L., Villalobos-Molina R., Riveros-Rosas H., et al. (2008). Signaling the signal, cyclic AMP-dependent protein kinase inhibition by insulin-formed H2O2 and reactivation by thioredoxin. J. Biol. Chem. 283, 12373–12386. 10.1074/jbc.M706832200 [DOI] [PubMed] [Google Scholar]
- Derry P. J., Hegde M. L., Jackson G. R., Kayed R., Tour J. M., Tsai A. L., et al. (2020). Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog. Neurobiol. 184, 101716. 10.1016/j.pneurobio.2019.101716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter D. T., Wells F. R., Agid F., Agid Y., Lees A. J., Jenner P., et al. (1987). Increased nigral iron content in postmortem parkinsonian brain. Lancet 2, 1219–1220. 10.1016/S0140-6736(87)91361-4 [DOI] [PubMed] [Google Scholar]
- Di Benedetto G., Iannucci L. F., Surdo N. C., Zanin S., Conca F., Grisan F., et al. (2021). Compartmentalized signaling in aging and neurodegeneration. Cells 10, 464. 10.3390/cells10020464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold L., Chandel N. S. (2016). Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 100, 86–93. 10.1016/j.freeradbiomed.2016.04.198 [DOI] [PubMed] [Google Scholar]
- Dursun E., Gezen-Ak D., Hanağasi H., Bilgiç B., Lohmann E., Ertan S., et al. (2015). The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 283, 50–57. 10.1016/j.jneuroim.2015.04.014 [DOI] [PubMed] [Google Scholar]
- Eby J. C., Gray M. C., Hewlett E. L. (2014). Cyclic AMP-mediated suppression of neutrophil extracellular trap formation and apoptosis by the Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 82, 5256–5269. 10.1128/IAI.02487-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekharzadeh B., Ramin M., Khodagholi F., Moradi S., Tabrizian K., Sharif R., et al. (2012). Inhibition of PKA attenuates memory deficits induced by β-amyloid (1-42), and decreases oxidative stress and NF-κB transcription factors. Behav. Brain Res. 226, 301–308. 10.1016/j.bbr.2011.08.015 [DOI] [PubMed] [Google Scholar]
- Engidawork E., Gulesserian T., Seidl R., Cairns N., Lubec G. (2001). Expression of apoptosis related proteins in brains of patients with Alzheimer’s disease. Neurosci. Lett. 303, 79–82. 10.1016/S0304-3940(01)01618-4 [DOI] [PubMed] [Google Scholar]
- Epperly T., Dunay M. A., Boice J. L. (2017). Alzheimer disease: Pharmacologic and nonpharmacologic therapies for cognitive and functional symptoms. Am. Fam. Physician 95, 771–778. [PubMed] [Google Scholar]
- Ercu M., Mücke M. B., Pallien T., Markó L., Sholokh A., Schächterle C., et al. (2022). Mutant phosphodiesterase 3A protects from hypertension-induced cardiac damage. Circulation 146, 1758–1778. 10.1161/CIRCULATIONAHA.122.060210 [DOI] [PubMed] [Google Scholar]
- Esteras N., Alquézar C., Bermejo-Pareja F., Bialopiotrowicz E., Wojda U., Martín-Requero A. (2013). Downregulation of extracellular signal-regulated kinase 1/2 activity by calmodulin KII modulates p21Cip1 levels and survival of immortalized lymphocytes from Alzheimer’s disease patients. Neurobiol. Aging 34, 1090–1100. 10.1016/j.neurobiolaging.2012.10.014 [DOI] [PubMed] [Google Scholar]
- Esteras N., Bartolomé F., Alquézar C., Antequera D., Muñoz Ú., Carro E., et al. (2012). Altered cell cycle-related gene expression in brain and lymphocytes from a transgenic mouse model of Alzheimer’s disease [amyloid precursor protein/presenilin 1 (PS1)]. Eur. J. Neurosci. 36, 2609–2618. 10.1111/j.1460-9568.2012.08178.x [DOI] [PubMed] [Google Scholar]
- Farr A. C., Xiong M. P. (2021). Challenges and opportunities of deferoxamine delivery for treatment of alzheimer’s disease, Parkinson’s disease, and intracerebral hemorrhage. Mol. Pharm. 18, 593–609. 10.1021/acs.molpharmaceut.0c00474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatokun A. A., Dawson V. L., Dawson T. M. (2013). Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016. 10.1111/bph.2014.171.issue-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fella E., Papacharalambous R., Kynigopoulos D., Ioannou M., Derua R., Christodoulou C., et al. (2022). Pharmacological activation of the C5a receptor leads to stimulation of the β-adrenergic receptor and alleviates cognitive impairment in a murine model of familial Alzheimer’s disease. Front. Immunol. 13. 10.3389/fimmu.2022.947071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein D. I., Billings J. L., Adlard P. A., Ayton S., Sedjahtera A., Masters C. L., et al. (2017). The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 5, 53. 10.1186/s40478-017-0456-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L., Vitale I., Aaronson S. A., Abrams J. M., Adam D., Agostinis P., et al. (2018). Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell. Death Differ. 25, 486–541. 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H., Bai Y., Jia Y., Zhao Y., Kang R., Tang D., et al. (2018). Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 503, 1550–1556. 10.1016/j.bbrc.2018.07.078 [DOI] [PubMed] [Google Scholar]
- Gerace E., Masi A., Resta F., Felici R., Landucci E., Mello T., et al. (2014). PARP-1 activation causes neuronal death in the hippocampal CA1 region by increasing the expression of Ca2+-permeable AMPA receptors. Neurobiol. Dis. 70, 43–52. 10.1016/j.nbd.2014.05.023 [DOI] [PubMed] [Google Scholar]
- Goetzl E. J., Boxer A., Schwartz J. B., Abner E. L., Petersen R. C., Miller B. L., et al. (2015). Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 85, 40–47. 10.1212/WNL.0000000000001702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Sintes R., Ledesma M. D., Boya P. (2016). Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 32, 150–168. 10.1016/j.arr.2016.02.009 [DOI] [PubMed] [Google Scholar]
- Grandoch M., López de Jesús M., Oude Weernink P. A., Weber A. A., Jakobs K. H., Schmidt M. (2009b). B cell receptor-induced growth arrest and apoptosis in WEHI-231 immature B lymphoma cells involve cyclic AMP and Epac proteins. Cell. Signal 21, 609–621. 10.1016/j.cellsig.2009.01.002 [DOI] [PubMed] [Google Scholar]
- Grandoch M., Bujok V., Fleckenstein D., Schmidt M., Fischer J. W., Weber A. A. (2009a). Epac inhibits apoptosis of human leukocytes. J. Leukoc. Biol. 86, 847–849. 10.1189/jlb.0109048 [DOI] [PubMed] [Google Scholar]
- Grossi C., Francese S., Casini A., Rosi M. C., Luccarini I., Fiorentini A., et al. (2009). Clioquinol decreases amyloid-β burden and reduces working memory impairment in a transgenic mouse model of alzheimer’s disease. J. Alzheimer’s Dis. 17, 423–440. 10.3233/JAD-2009-1063 [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I., Fleming J., Tung Y. C., Lassmann H., Iqbal K., Joshi J. G. (1990). Ferritin is a component of the neuritic (senile) plaque in Alzheimer dementia. Acta Neuropathol. 81, 105–110. 10.1007/BF00334497 [DOI] [PubMed] [Google Scholar]
- Hakkim A., Fuchs T. A., Martinez N. E., Hess S., Prinz H., Zychlinsky A., et al. (2011). Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 7, 75–77. 10.1038/nchembio.496 [DOI] [PubMed] [Google Scholar]
- Halliwell B. (2006). Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 97, 1634–1658. 10.1111/j.1471-4159.2006.03907.x [DOI] [PubMed] [Google Scholar]
- Hambright W. S., Fonseca R. S., Chen L., Na R., Ran Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. 10.1016/j.redox.2017.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B., Poppinga W. J., Schmidt M. (2015). Scaffolding during the cell cycle by A-kinase anchoring proteins. Pflugers Arch. 467, 2401–2411. 10.1007/s00424-015-1718-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M., Hsu L. J., Xia Y., Takeda A., Sisk A., Sundsmo M., et al. (1999). Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro . Neuroreport 10, 717–721. 10.1097/00001756-199903170-00011 [DOI] [PubMed] [Google Scholar]
- He M., Cao C., Ni Z., Liu Y., Song P., Hao S., et al. (2022). PROTACs: Great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target Ther. 7, 181. 10.1038/s41392-022-00999-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser T., Marksteiner J., Defrancesco M., Deisenhammer E. A., Kemmler G., Humpel C. (2011). Two blood monocytic biomarkers (CCL15 and p21) combined with the mini-mental state examination discriminate alzheimer’s disease patients from healthy subjects. Dement. Geriatr. Cogn. Dis. Extra 1, 297–309. 10.1159/000330468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K. W., Schuh A. F. S., Saute J., Townsend R., Fricke D., Leke R., et al. (2009). Interleukin-6 serum levels in patients with Parkinson’s disease. Neurochem. Res. 34, 1401–1404. 10.1007/s11064-009-9921-z [DOI] [PubMed] [Google Scholar]
- Hook V., Yoon M., Mosier C., Ito G., Podvin S., Head B. P., et al. (2020). Cathepsin B in neurodegeneration of Alzheimer’s disease, traumatic brain injury, and related brain disorders. Biochim. Biophys. Acta Proteins Proteom 1868, 140428. 10.1016/j.bbapap.2020.140428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y., Dan X., Babbar M., Wei Y., Hasselbalch S. G., Croteau D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. 10.1038/s41582-019-0244-7 [DOI] [PubMed] [Google Scholar]
- Hou Z., Yang X., Li Y., Chen J., Shang H. (2022). Electroacupuncture enhances neuroplasticity by regulating the orexin A-mediated cAMP/PKA/CREB signaling pathway in senescence-accelerated mouse prone 8 (SAMP8) mice. Oxid. Med. Cell. Longev. 2022, 8694462. 10.1155/2022/8694462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huell M., Strauss S., Volk B., Bergeru M., Bauer J., Huell M., et al. (1995). Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta. Neuropathol. 89 Springer-Verlag; [DOI] [PubMed] [Google Scholar]
- Huang J., Hong W., Wan M., Zheng L. (2022). Molecular mechanisms and therapeutic target of NETosis in diseases. MedComm (Beijing) 3, e162. 10.1002/mco2.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel P. A., Zhang L., Murray F., Yokouchi H., Zambon A. C. (2012). Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol. 204, 277–287. 10.1111/j.1748-1716.2011.02273.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T., Warabi E., Mann G. E. (2019). Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic. Biol. Med. 133, 169–178. 10.1016/j.freeradbiomed.2018.09.002 [DOI] [PubMed] [Google Scholar]
- Jicha G. A., Weaver C., Lane E., Vianna C., Kress Y., Rockwood J., et al. (1999). cAMP-dependent protein kinase phosphorylations on tau in alzheimer’s disease. J. Neurosci. 19, 7486–7494. 10.1523/JNEUROSCI.19-17-07486.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju J., Song Y. N., Wang K. (2021). Mechanism of ferroptosis: A potential target for cardiovascular diseases treatment. Aging Dis. 12, 261–276. 10.14336/AD.2020.0323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan V. E., Mao G., Qu F., Angeli J. P. F., Doll S., Croix C. S., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. 10.1038/nchembio.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam T. I., Mao X., Park H., Chou S. C., Karuppagounder S. S., Umanah G. E., et al. (2018). Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson's disease. Science 362, 8407. 10.1126/science.aat8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaut O., Schmitt I., Tost J., Busato F., Liu Y., Hofmann P., et al. (2017). Epigenome-wide DNA methylation analysis in siblings and monozygotic twins discordant for sporadic Parkinson’s disease revealed different epigenetic patterns in peripheral blood mononuclear cells. Neurogenetics 18, 7–22. 10.1007/s10048-016-0497-x [DOI] [PubMed] [Google Scholar]
- Kelly M. P. (2018). Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal 42, 281–291. 10.1016/j.cellsig.2017.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. F. R., Wyllie A. H., Currie A. R. (1972). Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. 10.1038/bjc.1972.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. (2021). Pre-clinical neuroprotective evidences and plausible mechanisms of sulforaphane in alzheimer’s disease. Int. J. Mol. Sci. 22, 2929. 10.3390/ijms22062929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidou M., Oun A., Pathak P., Zhang B., Wang Z., ter Brake F., et al. (2021). The tale of proteolysis targeting chimeras (PROTACs) for Leucine-Rich Repeat Kinase 2 (LRRK2). ChemMedChem 16, 959–965. 10.1002/cmdc.202000872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbendam I. E., Honrath B., Culmsee C., Dolga A. M. (2018). Mitochondrial Ca2+-activated K+ channels and their role in cell life and death pathways. Cell. Calcium 69, 101–111. 10.1016/j.ceca.2017.07.005 [DOI] [PubMed] [Google Scholar]
- Krenning L., Feringa F. M., Shaltiel I. A., vandenBerg J., Medema R. H. (2014). Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol. Cell. 55, 59–72. 10.1016/j.molcel.2014.05.007 [DOI] [PubMed] [Google Scholar]
- Kritsilis M., Rizou S. V., Koutsoudaki P. N., Evangelou K., Gorgoulis V. G., Papadopoulos D. (2018). Ageing, cellular senescence and neurodegenerative disease. Int. J. Mol. Sci. 19, 2937. 10.3390/ijms19102937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T., Terman A., Gustafsson B., Brunk U. T. (2008). Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell. Biol. 129, 389–406. 10.1007/s00418-008-0394-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane D. J. R., Ayton S., Bush A. I. (2018). Iron and alzheimer’s disease: An update on emerging mechanisms. J. Alzheimer’s Dis. 64, S379–S395. 10.3233/JAD-179944 [DOI] [PubMed] [Google Scholar]
- Lai K. P., Liu C. S., Rau A., Lanctôt, K. L., Köhler C. A., Pakosh M., et al. (2017). Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. Journal of Neurology, Neurosurgery and Psychiatry 88 (10), 876–882. 10.1136/jnnp-2017-316201 [DOI] [PubMed] [Google Scholar]
- Langkammer C., Ropele S., Pirpamer L., Fazekas F., Schmidt R. (2014). MRI for iron mapping in Alzheimer’s disease. Neurodegener. Dis. 13, 189–191. 10.1159/000353756 [DOI] [PubMed] [Google Scholar]
- Lee J. H., Cheon Y. H., Woo R. S., Song D. Y., Moon C., Baik T. K. (2012). Evidence of early involvement of apoptosis inducing factor-induced neuronal death in Alzheimer brain. Anat. Cell. Biol. 45, 26–37. 10.5115/acb.2012.45.1.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei P., Ayton S., Finkelstein D. I., Spoerri L., Ciccotosto G. D., Wright D. K., et al. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295. 10.1038/nm.2613 [DOI] [PubMed] [Google Scholar]
- Lewerenz J., Ates G., Methner A., Conrad M., Maher P. (2018). Oxytosis/ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 12, 214. 10.3389/fnins.2018.00214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J., Hewett S. J., Huang Y., Lambros M., Gout P. W., Kalivas P. W., et al. (2013). The cystine/glutamate antiporter system xc-in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 18, 522–555. 10.1089/ars.2011.4391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Li Y. (2020). The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target Ther. 5, 108. 10.1038/s41392-020-00216-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Li B., Brun T., Deffert-Delbouille C., Mahiout Z., Daali Y., et al. (2012). NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes 61, 2842–2850. 10.2337/db12-0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist D., Kaufman E., Brundin L., Hall S., Surova Y., Hansson O. (2012). Non-motor symptoms in patients with Parkinson’s disease - correlations with inflammatory cytokines in serum. PLoS One 7, 47387. 10.1371/journal.pone.0047387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R. M. (2022). Aging, cellular senescence, and alzheimer’s disease. Int. J. Mol. Sci. 23, 1989. 10.3390/ijms23041989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Chen Y., Tong J., Reynolds A. M., Proudfoot S. C., Qi J., et al. (2016). Epac signaling is required for cocaine-induced change in ampa receptor subunit composition in the ventral tegmental area. J. Neurosci. 36, 4802–4815. 10.1523/JNEUROSCI.3186-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloret A., Badía M. C., Mora N. J., Pallardó F. V., Alonso M. D., Viña J. (2009). Vitamin e paradox in alzheimer’s disease: It does not prevent loss of cognition and may even be detrimental. J. Alzheimer’s Dis. 17, 143–149. 10.3233/JAD-2009-1033 [DOI] [PubMed] [Google Scholar]
- Long J. M., Holtzman D. M. (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cell. 179, 312–339. 10.1016/j.cell.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S., Barber R., Wilcock G. K. (1999). Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 122, 247–253. 10.1093/brain/122.2.247 [DOI] [PubMed] [Google Scholar]
- Lovell M. A., Robertson J. D., Teesdale W. J., Campbell J. L., Markesbery W. R. (1998). Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158, 47–52. 10.1016/S0022-510X(98)00092-6 [DOI] [PubMed] [Google Scholar]
- Lu’O’Ng K. V. Q., Nguyen L. T. H. (2013). The role of betaAdrenergic receptor blockers in Alzheimer’s disease: potential genetic and cellular signaling mechanisms. American Journal of Alzheimer’s Disease and Other Dementias. SAGE Publications Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüth H. J., Holzer M., Gertz H. J., Arendt T. (2000). Aberrant expression of nNOS in pyramidal neurons in Alzheimer’s disease is highly co-localized with p21(ras) and p16(INK4a). Brain Res. 852, 45–55. 10.1016/S0006-8993(99)02178-2 [DOI] [PubMed] [Google Scholar]
- Mabalirajan U., Aich J., Leishangthem G. D., Sharma S. K., Kumar Dinda A., Ghosh B. (2009). Effects of vitamin E on mitochondrial dysfunction and asthma features in an experimental allergic murine model. J. Appl. Physiol. 107, 1285–1292. 10.1152/japplphysiol.00459.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P. (2018). Potentiation of glutathione loss and nerve cell death by the transition metals iron and copper: Implications for age-related neurodegenerative diseases. Free Radic. Biol. Med. 115, 92–104. 10.1016/j.freeradbiomed.2017.11.015 [DOI] [PubMed] [Google Scholar]
- Majerníková N., den Dunnen W. F. A., Dolga A. M. (2021). The potential of ferroptosis-targeting therapies for alzheimer’s disease: From mechanism to Transcriptomic analysis. Front. Aging Neurosci. 13, 745046. 10.3389/fnagi.2021.745046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P. K., Saharan S., Tripathi M., Murari G. (2015). Brain glutathione levels - a novel biomarker for mild cognitive impairment and alzheimer’s disease. Biol. Psychiatry 78, 702–710. 10.1016/j.biopsych.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Marmolejo-Garza A., Medeiros-Furquim T., Rao R., Eggen B. J. L., Boddeke E., Dolga A. M. (2022). Transcriptomic and epigenomic landscapes of Alzheimer’s disease evidence mitochondrial-related pathways. Biochim. Biophys. Acta Mol. Cell. Res. 1869, 119326. 10.1016/j.bbamcr.2022.119326 [DOI] [PubMed] [Google Scholar]
- Martínez-Pinilla E., Ordóñez C., Valle E. V., Navarro A., Tolivia J. (2016). Regional and gender study of neuronal density in brain during aging and in Alzheimer’s disease. Front. Aging Neurosci. 8. 10.3389/fnagi.2016.00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire S., Fuso A., Rotili D., Tempera I., Giordano C., De Zottis I., et al. (2013). PARP-1 modulates amyloid beta peptide-induced neuronal damage. PLoS One 8, e72169. 10.1371/journal.pone.0072169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcphee I., Gibson L. C. D., Kewney J., Darroch C., Stevens P. A., Spinks D., et al. (2005). Cyclic nucleotide signalling: A molecular approach to drug discovery for alzheimer’s disease. Biochem. Soc. Trans. 33, 1330–1332. 10.1042/BST20051330 [DOI] [PubMed] [Google Scholar]
- McShea A., Harris P. L. R., Webster K. R., Wahl A. F., Smith M. A. (1997). Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathology 150, 1933–1939. [PMC free article] [PubMed] [Google Scholar]
- Mittal M., Siddiqui M. R., Tran K., Reddy S. P., Malik A. B. (2014). Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal 20, 1126–1167. 10.1089/ars.2012.5149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittler R. (2017). ROS are good. Trends Plant Sci. 22, 11–19. 10.1016/j.tplants.2016.08.002 [DOI] [PubMed] [Google Scholar]
- Mukai E., Fujimoto S., Sato H., Oneyama C., Kominato R., Sato Y., et al. (2011). Exendin-4 suppresses Src activation and reactive oxygen species production in diabetic Goto-Kakizaki rat islets in an Epac-dependent manner. Diabetes 60, 218–226. 10.2337/db10-0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musheshe N., Lobo M. J., Schmidt M., Zaccolo M. (2018a). Targeting fret-based reporters for cAMP and PKA activity using AKAP79. Sensors Switz. 18, 2164. 10.3390/s18072164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musheshe N., Oun A., Sabogal-Guáqueta A. M., Trombetta-Lima M., Mitchel S. C., Adzemovic A., et al. (2022). Pharmacological inhibition of Epac1 averts ferroptosis cell death by preserving mitochondrial integrity. Antioxidants 11, 314. 10.3390/antiox11020314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musheshe N., Schmidt M., Zaccolo M. (2018b). cAMP: From long-range second messenger to nanodomain signalling. Trends Pharmacol. Sci. 39, 209–222. 10.1016/j.tips.2017.11.006 [DOI] [PubMed] [Google Scholar]
- Musi N., Valentine J. M., Sickora K. R., Baeuerle E., Thompson C. S., Shen Q., et al. (2018). Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 17, 12840. 10.1111/acel.12840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutua V., Gershwin L. J. (2021). A review of neutrophil extracellular traps (NETs) in disease: Potential anti-NETs therapeutics. Clin. Rev. Allergy Immunol. 61, 194–211. 10.1007/s12016-020-08804-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A. J., Chiang Y. J., Hathcock K. S., Horikawa I., Sedelnikova O. A., Hodes R. J., et al. (2008). Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin 1, 6. 10.1186/1756-8935-1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narne P., Pandey V., Simhadri P. K., Phanithi P. B. (2017). Poly(ADP-ribose)polymerase-1 hyperactivation in neurodegenerative diseases: The death knell tolls for neurons. Semin. Cell. Dev. Biol. 63, 154–166. 10.1016/j.semcdb.2016.11.007 [DOI] [PubMed] [Google Scholar]
- Niccolini F., Foltynie T., Reis Marques T., Muhlert N., Tziortzi A. C., Searle G. E., et al. (2015). Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson’s disease. Brain 138, 3003–3015. 10.1093/brain/awv219 [DOI] [PubMed] [Google Scholar]
- Nichols E., Steinmetz J. D., Vollset S. E., Fukutaki K., Chalek J., Abd-Allah F., et al. (2022). Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the global burden of disease study 2019. Lancet Public Health 7, 105–e125. 10.1016/S2468-2667(21)00249-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijholt I. M., Dolga A. M., Ostroveanu A., Luiten P. G. M., Schmidt M., Eisel U. L. M. (2008). Neuronal AKAP150 coordinates PKA and Epac-mediated PKB/Akt phosphorylation. Cell. Signal 20, 1715–1724. 10.1016/j.cellsig.2008.05.001 [DOI] [PubMed] [Google Scholar]
- Oun A., Soliman A., Trombetta-Lima M., Tzepapadaki A., Tsagkari D., Kortholt A., et al. (2022). LRRK2 protects immune cells against erastin-induced ferroptosis. Neurobiol. Dis. 175, 105917. 10.1016/j.nbd.2022.105917 [DOI] [PubMed] [Google Scholar]
- Padurariu M., Ciobica A., Hritcu L., Stoica B., Bild W., Stefanescu C. (2010). Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 469, 6–10. 10.1016/j.neulet.2009.11.033 [DOI] [PubMed] [Google Scholar]
- Palorini R., De Rasmo D., Gaviraghi M., Danna L. S., Signorile A., Cirulli C., et al. (2013). Oncogenic K-ras expression is associated with derangement of the cAMP/PKA pathway and forskolin-reversible alterations of mitochondrial dynamics and respiration. Oncogene 32, 352–362. 10.1038/onc.2012.50 [DOI] [PubMed] [Google Scholar]
- Papayannopoulos V., Metzler K. D., Hakkim A., Zychlinsky A. (2010). Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell. Biol. 191, 677–691. 10.1083/jcb.201006052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H., Kam T. I., Peng H., Chou S. C., Mehrabani-Tabari A. A., Song J. J., et al. (2022). PAAN/MIF nuclease inhibition prevents neurodegeneration in Parkinson's diseasee. Cell. 185, 1943–1959. 10.1016/j.cell.2022.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce R. K. B., Owen A., Daniel S., Jenner P., Marsden C. D. (1997). Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 104, 661–677. 10.1007/BF01291884 [DOI] [PubMed] [Google Scholar]
- Pérez-Torres S., Cortés R., Tolnay M., Probst A., Palacios J. M., Mengod G. (2003). Alterations on phosphodiesterase type 7 and 8 isozyme mRNA expression in Alzheimer’s disease brains examined by in situ hybridization. Exp. Neurol. 182, 322–334. 10.1016/S0014-4886(03)00042-6 [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A., Adalsteinsson E., Rohlfing T., Sullivan E. V. (2009). MRI estimates of brain iron concentration in normal aging: Comparison of field-dependent (FDRI) and phase (SWI) methods. Neuroimage 47, 493–500. 10.1016/j.neuroimage.2009.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli C., Scacco S., Bellomo F., Signorile A., Iuso A., Boffoli D., et al. (2006). cAMP controls oxygen metabolism in mammalian cells. FEBS Lett. 580, 4539–4543. 10.1016/j.febslet.2006.06.085 [DOI] [PubMed] [Google Scholar]
- Poppinga W. J., Muñoz-Llancao P., González-Billault C., Schmidt M. (2014). A-Kinase anchoring proteins: CAMP compartmentalization in neurodegenerative and obstructive pulmonary diseases. Br. J. Pharmacol. 171, 5603–5623. 10.1111/bph.12882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickaerts J., Heckman P. R. A., Blokland A. (2017). Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin. Investig. Drugs 26, 1033–1048. 10.1080/13543784.2017.1364360 [DOI] [PubMed] [Google Scholar]
- Rajkumar A. P., Bidkhori G., Shoaie S., Clarke E., Morrin H., Hye A., et al. (2019). Postmortem cortical Transcriptomics of Lewy body dementia reveal mitochondrial dysfunction and lack of neuroinflammation. Am. J. Geriatric Psychiatry 28, 75–86. 10.1016/j.jagp.2019.06.007 [DOI] [PubMed] [Google Scholar]
- Ramirez R. D., Morales C. P., Herbert B. S., Rohde J. M., Passons C., Shay J. W., et al. (2001). Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes. Dev. 15, 398–403. 10.1101/gad.859201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos B. P., Colgan L. A., Nou E., Arnsten A. F. T. (2008). B2 Adrenergic Agonist, Clenbuterol, Enhances Working Memory Performance in Aging Animals. Neurobiology of Aging 8 (7), 1060–1069. 10.1016/j.neurobiolaging.2007.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea I. M., Gibson D. S., McGilligan V., McNerlan S. E., Alexander H. D., Ross O. A. (2018). Age and age related diseases: role of inflammation triggers and cytokines. Front. Immunol. Frontiers Media S.A. 10.3389/fimmu.2018.00586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts H. L., Schneider B. L., Brown D. R. (2017). α-Synuclein increases β-amyloid secretion by promoting β-/γ-secretase processing of APP. PLoS One 12, 1925. 10.1371/journal.pone.0171925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux W. G., Xiaodong Cheng X. (2018). Intracellular cAMP sensor EPAC: Physiology, pathophys-iology, and therapeutics development. Physiol. Rev. 98, 919–1053. 10.1152/physrev.00025.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca W. A. (2018). The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol. 17, 928–929. 10.1016/S1474-4422(18)30355-7 [DOI] [PubMed] [Google Scholar]
- Ross G. W., Petrovitch H., Abbott R. D., Nelson J., Markesbery W., Davis D. (2004). Parkinsonian signs and Substantia Nigra neuron density in decendents elders without PD. Annals of Neurology 56 (4), 532–539. 10.1002/ana.20226 [DOI] [PubMed] [Google Scholar]
- Ruas M., Gregory F., Jones R., Poolman R., Starborg M., Rowe J., et al. (2007). CDK4 and CDK6 delay senescence by kinase-dependent and p16INK4a-independent mechanisms. Mol. Cell. Biol. 27, 4273–4282. 10.1128/mcb.02286-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancesario G., Morrone L. A., D’Angelo V., Castelli V., Ferrazzoli D., Sica F., et al. (2014). Levodopa-induced dyskinesias are associated with transient down-regulation of cAMP and cGMP in the caudate-putamen of hemiparkinsonian rats: Reduced synthesis or increased catabolism? Neurochem. Int. 79, 44–56. 10.1016/j.neuint.2014.10.004 [DOI] [PubMed] [Google Scholar]
- Santoro B., Shah M. M. (2019). Hyperpolarization-activated cyclic nucleotide-gated channels as drug targets for neurological disorders. Annu. Rev. Pharmacol. Toxicol. 60, 109–131. 10.1146/annurev-pharmtox-010919-023356 [DOI] [PubMed] [Google Scholar]
- Scalzo P., Kümmer A., Cardoso F., Teixeira A. L. (2010). Serum levels of interleukin-6 are elevated in patients with Parkinson’s disease and correlate with physical performance. Neurosci. Lett. 468, 56–58. 10.1016/j.neulet.2009.10.062 [DOI] [PubMed] [Google Scholar]
- Scheltens P., De Strooper B., Kivipelto M., Holstege H., Chételat G., Teunissen C. E., et al. (2021). Alzheimer’s disease. Lancet 397, 1577–1590. 10.1016/S0140-6736(20)32205-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M., Dekker F. J., Maarsingh H. (2013). Exchange protein directly activated by cAMP (epac): A multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol. Rev. 65, 670–709. 10.1124/pr.110.003707 [DOI] [PubMed] [Google Scholar]
- Schwede F., Bertinetti D., Langerijs C. N., Hadders M. A., Wienk H., Ellenbroek J. H., et al. (2015). Structure-guided design of selective Epac1 and Epac2 agonists. PLoS Biol. 13, e1002038. 10.1371/journal.pbio.1002038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serezani C. H., Ballinger M. N., Aronoff D. M., Peters-Golden M. (2008). Cyclic AMP: Master regulator of innate immune cell function. Am. J. Respir. Cell. Mol. Biol. 39, 127–132. 10.1165/rcmb.2008-0091TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H., Jones D. P. (2020). Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 21, 363–383. 10.1038/s41580-020-0230-3 [DOI] [PubMed] [Google Scholar]
- Signorile A., Ferretta A., Pacelli C., Capitanio N., Tanzarella P., Matrella M. L., et al. (2021). Resveratrol treatment in human parkin-mutant fibroblasts modulates camp and calcium homeostasis regulating the expression of mitochondria-associated membranes resident proteins. Biomolecules 11, 1511. 10.3390/biom11101511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J. N., Vrellaku B., Monterisi S., Chu S. M., Rawlings N., Lomas O., et al. (2021). Oxidation of protein kinase A regulatory subunit PKARIα protects against myocardial ischemia-reperfusion injury by inhibiting lysosomal-triggered calcium release. Circulation 143, 449–465. 10.1161/CIRCULATIONAHA.120.046761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson D. S. A., Oliver P. L. (2020). Ros generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 9, 743. 10.3390/antiox9080743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C., Malek N., Grosset K., Cullen B., Gentleman S., Grosset D. G. (2019). Neuropathology of dementia in patients with Parkinson’s disease: A systematic review of autopsy studies. J. Neurol. Neurosurg. Psychiatry 90, 1234–1243. 10.1136/jnnp-2019-321111 [DOI] [PubMed] [Google Scholar]
- Smyth L. C. D., Murray H. C., Hill M., van Leeuwen E., Highet B., Magon N. J., et al. (2022). Neutrophil-vascular interactions drive myeloperoxidase accumulation in the brain in Alzheimer’s disease. Acta Neuropathol. Commun. 10, 38. 10.1186/s40478-022-01347-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soini L., Leysen S., Davis J., Ottmann C. (2022). Molecular glues to stabilise protein–protein interactions. Curr. Opin. Chem. Biol. 69, 102169. 10.1016/j.cbpa.2022.102169 [DOI] [PubMed] [Google Scholar]
- Song S., Xiao Z., Dekker F. J., Poelarends G. J., Melgert B. N. (2022). Macrophage migration inhibitory factor family proteins are multitasking cytokines in tissue injury. Cell. Mol. Life Sci. 79, 105. 10.1007/s00018-021-04038-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X., Long D. (2020). Nrf2 and ferroptosis: A new research direction for neurodegenerative diseases. Front. Neurosci. 14, 267. 10.3389/fnins.2020.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripetchwandee J., Wongjaikam S., Krintratun W., Chattipakorn N., Chattipakorn S. C. (2016). A combination of an iron chelator with an antioxidant effectively diminishes the dendritic loss, tau-hyperphosphorylation, amyloids-β accumulation and brain mitochondrial dynamic disruption in rats with chronic iron-overload. Neuroscience 332, 191–202. 10.1016/j.neuroscience.2016.07.003 [DOI] [PubMed] [Google Scholar]
- Stahl-Meyer K., Bilgin M., Holland L. K. K., Stahl-Meyer J., Kirkegaard T., Petersen N. H. T., et al. (2022). Galactosyl- and glucosylsphingosine induce lysosomal membrane permeabilization and cell death in cancer cells. PLoS One 17, e0277058. 10.1371/journal.pone.0277058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein G. H., Drullinger L. F., Soulard A., Dulić V. (1999). Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 19, 2109–2117. 10.1128/mcb.19.3.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokman G., Qin Y., Booij T. H., Ramaiahgari S., Lacombe M., Dolman M. E. M., et al. (2014). Epac-rap signaling reduces oxidative stress in the tubular epithelium. J. Am. Soc. Nephrol. 25, 1474–1485. 10.1681/ASN.2013070679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R., Perluigi M., Butterfield D. A. (2009). Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: Role of Abeta in pathogenesis. Acta Neuropathol. 118, 131–150. 10.1007/s00401-009-0517-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A., Liu M., Nguyen X. V., Bing G. (2003). p38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 183, 394–405. 10.1016/S0014-4886(03)00180-8 [DOI] [PubMed] [Google Scholar]
- Sun X., Gao H., Yang Y., He M., Wu Y., Song Y., et al. (2019). Protacs: Great opportunities for academia and industry. Signal Transduct. Target Ther. 4, 64. 10.1038/s41392-019-0101-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung J. Y., Kim S. G., Cho D. H., Kim J. R., Choi H. C. (2020). SRT1720-induced activation of SIRT1 alleviates vascular smooth muscle cell senescence through PKA-dependent phosphorylation of AMPKα at Ser485. FEBS Open Bio 10, 1316–1325. 10.1002/2211-5463.12895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swardfager W., Lanctt K., Rothenburg L., Rothenburg A., Cappell J., Herrmann N. (2010). A meta-analysis of cytokines in Alzheimer’s disease. Biological Psychiatry 68 (10), 930–941. 10.1016/j.biopsych.2010.06.012 [DOI] [PubMed] [Google Scholar]
- Taghizadeh M., Tamtaji O. R., Dadgostar E., Daneshvar Kakhaki R., Bahmani F., Abolhassani J., et al. (2017). The effects of omega-3 fatty acids and vitamin E co-supplementation on clinical and metabolic status in patients with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Neurochem. Int. 108, 183–189. 10.1016/j.neuint.2017.03.014 [DOI] [PubMed] [Google Scholar]
- Tang D., Kang R., Berghe T. V., Vandenabeele P., Kroemer G. (2019). The molecular machinery of regulated cell death. Cell. Res. 29, 347–364. 10.1038/s41422-019-0164-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M., Ogiso H., Takeuchi T., Kitatani K., Umehara H., Okazaki T. (2015). Lysosomal ceramide generated by acid sphingomyelinase triggers cytosolic cathepsin B-mediated degradation of X-linked inhibitor of apoptosis protein in natural killer/T lymphoma cell apoptosis. Cell. Death Dis. 6, 1717. 10.1038/cddis.2015.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y., Wang Y., Rogers J. T., Wang F. (2014). Perturbed iron distribution in alzheimer’s disease serum, cerebrospinal fluid, and selected brain regions: A systematic review and meta-analysis. J. Alzheimer’s Dis. 42, 679–690. 10.3233/JAD-140396 [DOI] [PubMed] [Google Scholar]
- Tiiman A., Palumaa P., Tõugu V. (2013). The missing link in the amyloid cascade of Alzheimer’s disease-Metal ions. Neurochem. Int. 62, 367–378. 10.1016/j.neuint.2013.01.023 [DOI] [PubMed] [Google Scholar]
- Tolosa E., Garrido A., Scholz S. W., Poewe W. (2021). Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 20, 385–397. 10.1016/S1474-4422(21)00030-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tönnies E., Trushina E. (2017). Oxidative stress, synaptic dysfunction, and alzheimer’s disease. J. Alzheimer’s Dis. 57, 1105–1121. 10.3233/JAD-161088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trist B. G., Hare D. J., Double K. L. (2019). Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell. 18, 13031. 10.1111/acel.13031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang A. H. K., Chung K. K. K. (2009). Oxidative and nitrosative stress in Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 1792, 643–650. 10.1016/j.bbadis.2008.12.006 [DOI] [PubMed] [Google Scholar]
- Tucker S. J., Zorn A. J. (2022). The role of Popeye domain-containing protein 1 (POPDC1) in the progression of the malignant phenotype. Br. J. Pharmacol. 179, 2829–2843. 10.1111/bph.15403 [DOI] [PubMed] [Google Scholar]
- Turenne G. A., Paul P., Laflair L., Price B. D. (2001). Activation of p53 transcriptional activity requires ATM’s kinase domain and multiple N-terminal serine residues of p53. Oncogene 20, 5100–5110. 10.1038/sj.onc.1204665 [DOI] [PubMed] [Google Scholar]
- Turnquist C., Horikawa I., Foran E., Major E. O., Vojtesek B., Lane D. P., et al. (2016). P53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell. Death Differ. 23, 1515–1528. 10.1038/cdd.2016.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twohig D., Nielsen H. M. (2019). α-synuclein in the pathophysiology of Alzheimer's disease. Mol. Neurodegener. 14, 23. 10.1186/s13024-019-0320-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte A., Gil-Bea F., García-Barroso C., Cedazo-Minguez Á., Ramírez M. J., Franco R., et al. (2015). Decreased levels of guanosine 3’, 5’-monophosphate (cGMP) in cerebrospinal fluid (CSF) are associated with cognitive decline and amyloid pathology in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 41, 471–482. 10.1111/nan.12203 [DOI] [PubMed] [Google Scholar]
- Uversky V. N., Li J., Fink A. L. (2001). Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein: A possible molecular link between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296. 10.1074/jbc.M105343200 [DOI] [PubMed] [Google Scholar]
- Valsecchi F., Konrad C., Manfredi G. (2014). Role of soluble adenylyl cyclase in mitochondria. Biochim. Biophys. Acta Mol. Basis Dis. 1842, 2555–2560. 10.1016/j.bbadis.2014.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk K. D., Persichetti E., Chiasserini D., Eusebi P., Beccari T., Calabresi P., et al. (2013). Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 28, 747–754. 10.1002/mds.25495 [DOI] [PubMed] [Google Scholar]
- Vannam R., Sayilgan J., Ojeda S., Karakyriakou B., Hu E., Kreuzer J., et al. (2021). Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell. Chem. Biol. 28, 503–514. 10.1016/j.chembiol.2020.12.004 [DOI] [PubMed] [Google Scholar]
- Verma D. K., Seo B. A., Ghosh A., Ma S. X., Hernandez-Quijada K., Andersen J. K., et al. (2021). Alpha-synuclein preformed fibrils induce cellular senescence in Parkinson’s disease models. Cells 10, 1694. 10.3390/cells10071694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M., Bové J., Dehay B., Rodríguez-Muela N., Boya P. (2011). Lysosomal membrane permeabilization in Parkinson disease. Autophagy 7, 98–100. 10.4161/auto.7.1.13933 [DOI] [PubMed] [Google Scholar]
- Visanji N. P., Lang A. E., Kovacs G. G. (2019). Beyond the synucleinopathies: Alpha synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener. 8, 28. 10.1186/s40035-019-0172-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobjeva N., Galkin I., Pletjushkina O., Golyshev S., Zinovkin R., Prikhodko A., et al. (2020). Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165664. 10.1016/j.bbadis.2020.165664 [DOI] [PubMed] [Google Scholar]
- Wang H. F., Wang Z. Q., Ding Y., Piao M. H., Feng C. S., Chi G. F., et al. (2018). Endoplasmic reticulum stress regulates oxygen-glucose deprivation-induced parthanatos in human SH-SY5Y cells via improvement of intracellular ROS. CNS Neurosci. Ther. 24, 29–38. 10.1111/cns.12771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Su X., Zhu R., Zhao R. C. (2022). cAMP agonist forskolin disrupts mitochondrial metabolism and induces senescence in human mesenchymal cells. Stem Cells Dev. 32, 87–98. 10.1089/scd.2022.0180 [DOI] [PubMed] [Google Scholar]
- Wang X., Ge P. (2020). Parthanatos in the pathogenesis of nervous system diseases. Neuroscience 449, 241–250. 10.1016/j.neuroscience.2020.09.049 [DOI] [PubMed] [Google Scholar]
- Wang Y., An R., Umanah G. K., Park H., Nambiar K., Eacker S. M., et al. (2016). A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354, 6872. 10.1126/science.aad6872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Dawson V. L., Dawson T. M. (2009). Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp. Neurol. 218, 193–202. 10.1016/j.expneurol.2009.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Kim N. S., Haince J. F., Kang H. C., David K. K., Andrabi S. A., et al. (2011). Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal 4, ra20. 10.1126/scisignal.2000902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Zhang X., Tian X., Yang Y., Ma L., Wang J., et al. (2021). CREB stimulates GPX4 transcription to inhibit ferroptosis in lung adenocarcinoma. Oncol. Rep. 45, 88. 10.3892/OR.2021.8039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. P., Shadel G. S., Ghosh S. (2011). Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402. 10.1038/nri2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins S. V., Steegborn C., Levin L. R., Buck J. (2018). Pharmacological modulation of the CO 2/HCO 3−/pH-calcium-and ATP-sensing soluble adenylyl cyclase. Pharmacol. Ther. 190, 173–186. 10.1016/j.pharmthera.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood J. A., Wood P. L., Ryan R., Graff-Radford N. R., Pilapil C., Robitaille Y. (1993). Cytokine indices in Alzheimer’s temporal cortex: no changes in mature IL-1/3 or IL-1RA but increases in the associated acute phase proteins IL-6, A2- macroglobulin and C-reactive Protein. Brain Research 629. [DOI] [PubMed] [Google Scholar]
- Xu Y., Schwede F., Wienk H., Tengholm A., Rehmann H. (2019). A membrane permeable prodrug of S223 for selective epac2 activation in living cells. Cells 8, 1589. 10.3390/cells8121589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Shu T., Zhao H., Sun Y., Xu W., Tu G. (2020). Knockdown of macrophage migration inhibitory factor (MIF), a novel target to protect neurons from parthanatos induced by simulated post-spinal cord injury oxidative stress. Biochem. Biophys. Res. Commun. 523, 719–725. 10.1016/j.bbrc.2019.12.115 [DOI] [PubMed] [Google Scholar]
- Yang W. S., Kim K. J., Gaschler M. M., Patel M., Shchepinov M. S., Stockwell B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U. S. A. 113, E4966–E4975. 10.1073/pnas.1603244113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates S. C., Zafar A., Rabai E. M., Foxall J. B., Nagy S., Morrison K. E., et al. (2015). The effects of two polymorphisms on p21cip1 function and their association with Alzheimer’s disease in a population of European descent. PLoS One 10, 0114050. 10.1371/journal.pone.0114050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenaro E., Pietronigro E., Bianca V. D., Piacentino G., Marongiu L., Budui S., et al. (2015). Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886. 10.1038/nm.3913 [DOI] [PubMed] [Google Scholar]
- Zhang J. Z., Lu T. W., Stolerman L. M., Tenner B., Yang J. R., Zhang J. F., et al. (2020). Phase separation of a PKA regulatory subunit controls cAMP compartmentation and oncogenic signaling. Cell. 182, 1531–1544. 10.1016/j.cell.2020.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Kishimoto Y., Grammatikakis I., Gottimukkala K., Cutler R. G., Zhang S., et al. (2019). Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22, 719–728. 10.1038/s41593-019-0372-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Gao F., Wang D., Li C., Fu Y., He W., et al. (2018a). Tau pathology in Parkinson’s disease. Front. Neurol. 9, 809. 10.3389/fneur.2018.00809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. H., Wang D. W., Xu S. F., Zhang S., Fan Y. G., Yang Y. Y., et al. (2018b). α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 14, 535–548. 10.1016/j.redox.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Liu L., Tao S., Yao Y., Wang Y., Wei Q., et al. (2021). Parthanatos and its associated components: Promising therapeutic targets for cancer. Pharmacol. Res. 163, 105299. 10.1016/j.phrs.2020.105299 [DOI] [PubMed] [Google Scholar]
- Zhu M., Min S., Mao X., Zhou Y., Zhang Y., Li W., et al. (2022). Interleukin-13 promotes cellular senescence through inducing mitochondrial dysfunction in IgG4-related sialadenitis. Int. J. Oral Sci. 14, 29. 10.1038/s41368-022-00180-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilka O., Shah R., Li B., Friedmann Angeli J. P., Griesser M., Conrad M., et al. (2017). On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 3, 232–243. 10.1021/acscentsci.7b00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo H., Cattani-Cavalieri I., Musheshe N., Nikolaev V. O., Schmidt M. (2019). Phosphodiesterases as therapeutic targets for respiratory diseases. Pharmacol. Ther. 197, 225–242. 10.1016/j.pharmthera.2019.02.002 [DOI] [PubMed] [Google Scholar]