

Abstract
In humans, the NK cell marker CD161 identifies several subsets of T cells, including a polyclonal CD8 αβ T cell receptor expressing subset with characteristic specificity for tissue-localized viruses. This subset has also been reported to display enhanced cytotoxic and memory phenotypes. Here, we characterized the biology and functional properties of this unique T cell subset and determined its potential suitability for use in chimeric antigen receptor (CAR) T cell therapy. In mice, gene expression profiling among the CD161 equivalent CD8+ T cell populations (CD8+NK1.1+) revealed substantial upregulation of granzymes, perforin, killer lectin-like receptors and innate signaling molecules in comparison to CD8+NK1.1− T cells. Adoptive transfer of CD8+NK1.1+ cells from previously exposed animals offered substantially enhanced protection and improved survival against melanoma tumors and influenza infection compared to CD8+NK1.1− cells. Freshly isolated human CD8+CD61+ T cells exhibited heightened allogeneic killing activity in comparison to CD8+CD61− T cells or total peripheral blood mononuclear cells (PBMC). To determine if this subset might be utilized to improve the antitumor efficacy CAR T cell therapy in the solid tumor setting, we compared bulk PBMC, CD8+CD161−, and CD8+CD161+ T cells transduced with a HER2-specific CAR construct. In vitro, CD8+CD161+ CAR-transduced T cells killed HER2+ targets faster and with greater efficiency. Similarly, these cells mediated substantially enhanced in vivo anti-tumor efficacy and survival in xenograft models of HER2+ pancreatic ductal adenocarcinoma, exhibiting elevated expression of granzymes and reduced expression of exhaustion markers. These data suggest that this unique T cell subset presents an opportunity to improve CAR T cell therapy for the treatment of solid tumors.
One Sentence Summary:
A highly cytotoxic T cell memory subset demonstrates intrinsic advantages when utilized as a CAR T cell platform in a solid tumor model.
Introduction
Pancreatic Ductal Adenocarcinoma (PDAC) is a highly aggressive tumor with an abysmal five-year survival rate of < 9% (1) despite aggressive surgery, radiation, and high-dose chemotherapy. In recent years, adoptive chimeric antigen receptor (CAR) T cell therapy has shown great potential as a therapeutic modality in cancer, although this is currently limited to select CD19+ malignancies (2, 3). The common second-generation CAR design consists of a single-chain variable fragment (scFv) extracellular domain that targets a cell surface tumor antigen, a transmembrane domain, a hinge region, and the intracellular signaling domain of CD3ζ typically fused to the intracellular signaling domains of either the 4–1BB or CD28 costimulatory molecules (4). In a pilot phase I clinical trial, adoptive cell therapy using autologous, mesothelin-specific CAR T cells was shown to be safe and perhaps mildly efficacious against chemotherapy-refractory metastatic human PDAC in a small number of patients (5); however, CAR T cell therapy against pancreatic tumors is still poorly-evolved. Indeed, few CAR-based therapeutic trials have demonstrated efficacy in the solid tumor setting (6, 7) and none to the degree of success observed in CD19+ malignancies.
A critical characteristic of cell-mediated immunity to viral infection is the establishment of long-lived memory T cell populations that provide durable immunity to subsequent challenge through accelerated expansion and cytotoxicity kinetics (8). Several groups have previously identified an intriguing subset of such memory T cells (9–17) that may be identified by expression of the natural cytotoxicity receptor NK1.1 in mice or CD161 in humans. In contrast to T cell receptor (TCR) invariant or CD8αα+CD161+ cells, this polyclonal TCR αβ cell population exhibits a stem cell like capacity for self-renewal and differentiation, a distinct transcriptional profile with upregulated transcripts from the granzyme super family (15, 17), a characteristic anti-viral specificity (11, 14, 18, 19), and tissue-homing properties (14). Canonically, CD161 is known as an innate natural killer (NK) cell receptor, but may also be expressed on CD4, CD8, and NKT cells (16). Though also found in circulation, CD8+CD161+ cells contribute to tissue pathogenesis during chronic viral infection and autoimmune conditions due to tissue-resident properties or a propensity for extravasation (13, 14, 20). Further, high expression of CD161 in tumor resident immune infiltrates is associated with substantially improved clinical outcome and survival in non-small cell lung cancer (NSCLC) (21).
In previous work, we demonstrated that adoptively transferred, antigen experienced CD8+NK1.1+ cells could mediate durable protection in a model of disseminated murine PDAC. These cells were present nine months after initial exposure to antigen and were highly protective when adoptively transferred into naïve mice subsequently challenged with the parental PDAC cell line (22). Expanding upon these results, we sought to characterize additional biological and functional properties of these cells in a variety of in vivo model systems including severe combined immunodeficient (SCID) orthotopic and disseminated xenograft models of CAR T cell therapy for the treatment of PDAC. The results demonstrated that CD8+CD161+ T cells may comprise a superior platform for CAR T cell therapy when expanded in undifferentiated fashion.
Results
Gene expression profiling following chemo-immunotherapy of PDAC identifies innate-like and cytotoxic characteristics of CD3+CD8+NK1.1+ cells.
Previous work demonstrated that very small numbers (< 1,500 per mouse) of splenic CD8+NK1.1+ cells isolated nine months after chemo-immunotherapy for the treatment of orthotopic PDAC could still provide rapid and robust anti-tumor protection against the parent PDAC cell line in a model of disseminated disease (22). To gain insight into key functional characteristics of this NK1.1+ CD3+ CD8+ T cell subset, we orthotopically implanted cohorts of mice with KrasG12D/p53−/− PDAC tumors and subsequently treated them by means of the previously published (22) chemo/immunotherapy-based therapeutic protocol. Two months post-treatment, CD8+ splenocytes were negatively selected and subdivided into NK1.1+ and NK1.1− fractions. These fractions were then separately co-cultured overnight with PDAC-loaded matured dendritic cells (DC), and the PDAC antigen-specific cells were identified and isolated by upregulated CD69 expression (fig. S1). The results of the microarray indicated that 1642 genes were differentially regulated between CD8+NK1.1+ and CD8+NK1.1− cells upon antigenic stimulation (Fig. 1A). Although many different pathways were potentially impacted (tables S1 and S2), the most striking differences were found in the cytolytic granzyme serine proteases, particularly noncanonical granzyme isoforms F, D, G, and C, as well as among the innate-like cytotoxicity receptors (Fig. 1B). These results suggested CD8+ NK1.1+ cells represent a population of CD8+ T cells with enhanced cytolytic capabilities.
Figure 1. Gene expression analysis by microarray of antigen stimulated T cells indicates upregulated cytotoxic and innate-like characteristics among CD8+NK1.1+ cells.
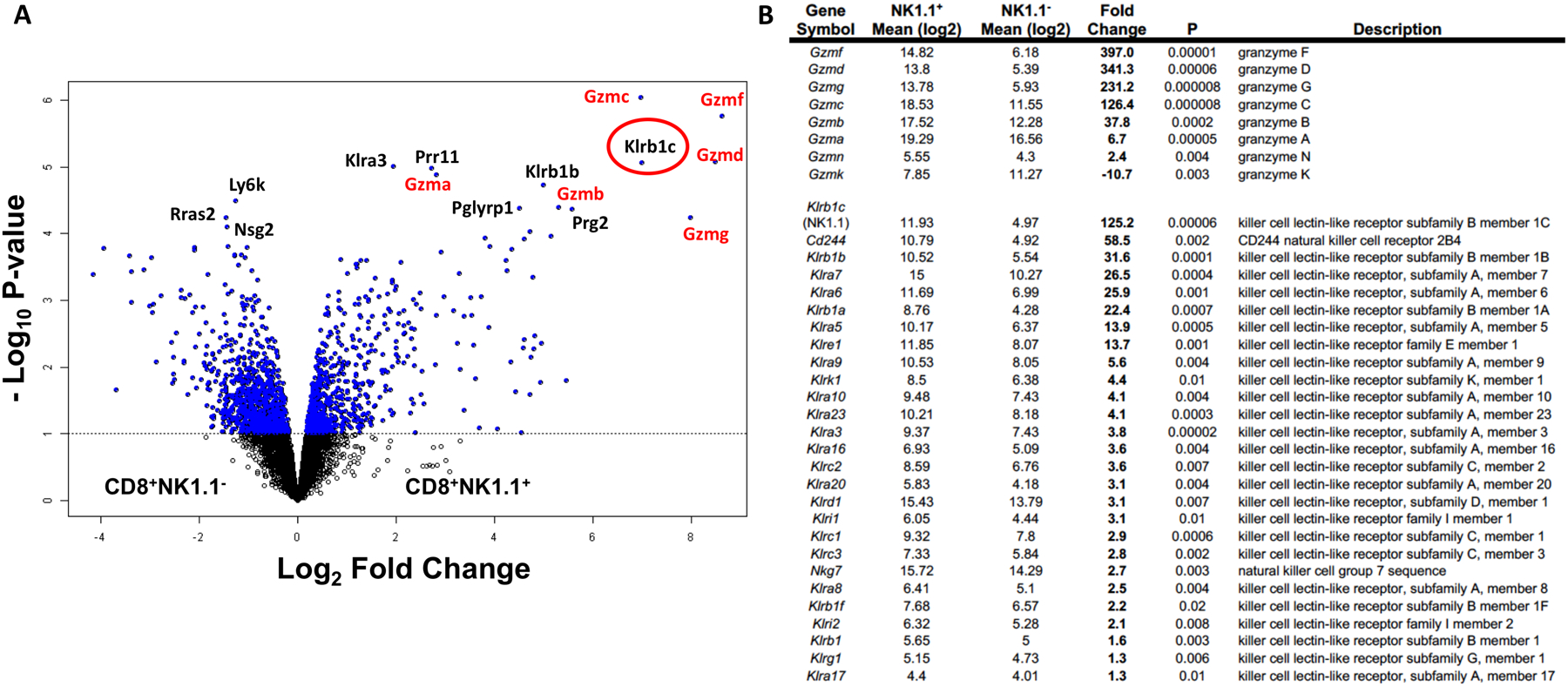
A cohort of 15 mice received dendritic cell-based vaccination in combination with chemotherapy against murine pancreatic ductal adenocarcinoma. 60 days post tumor inoculation, spleens were harvested, pooled into three groups of five each and activated overnight with dendritic cells loaded with tumor antigens. Stimulated cells were then sorted by flow cytometry into NK1.1− and NK1.1+ subsets by gating on CD8+CD69+ population. (A) Volcano plot shows 1642 genes that are differentially regulated between CD8+NK1.1− and CD8+NK1.1+ cells at a univariate degree of 0.1. Top 15 genes that are differentially regulated at an FDR of 0.05 are labeled on the plot with genes from the granzyme and killer-like lectin pathways are labeled in red. NK1.1 (Klrb1c) is circled in red. (B) Fold change and P values for the genes grouped into the granzyme pathway and killer cell like receptor subfamily pathway are indicated.
The CD161 mouse homologue, NK1.1, identifies a protective circulating memory T cell population in multiple mouse models of disease.
To validate that NK1.1 could identify a similar critical population of cytolytic memory cells in a model-independent fashion, we performed adoptive transfer experiments in an infectious disease model and in a second tumor model. We first inoculated a donor cohort of 6 to 8 week-old mice with a sub-lethal dose of H2N3 mouse-adapted A/HongKong/8/68 influenza virus by nebulized aerosol inoculation. Three weeks after inoculation and recovery from weight loss, splenocytes were harvested and CD8+ non-adherent cells were isolated by negative selection. After separation into NK1.1+ and NK1.1− fractions by positive selection, 5 × 105 NK1.1+ or NK1.1− cells were adoptively transferred into naïve cohorts that were lethally-challenged with the same influenza virus strain 24 hours after adoptive transfer (fig. S2A). Body weights were recorded as an index of recovery and survival was determined by Kaplan-Meier analysis. The cohort receiving donor CD8+NK1.1+ cells became convalescent, recovered body weights (p < 0.05, Fig. 2A) and survived the infection (p<0.05, Fig. 2B) whereas those adoptively transferred with CD8+NK1.1− cells lost weight and succumbed to infection at the same rate as controls adoptively transferred with naïve CD8+ splenocytes. Analysis of peripheral blood mononuclear cells (PBMC) at day 12 post infection showed a 40% increase in circulating CD3+CD8+IFN-γ+ cells among mice that received CD8+NK1.1+ cells in comparison to naïve and CD8+NK1.1− adoptively transferred cohorts (p<0.05, Fig. 2C).
Figure 2. CD8+NK1.1+ cells define a memory population that offers durable protection and improves survival against influenza infection or melanoma tumors.
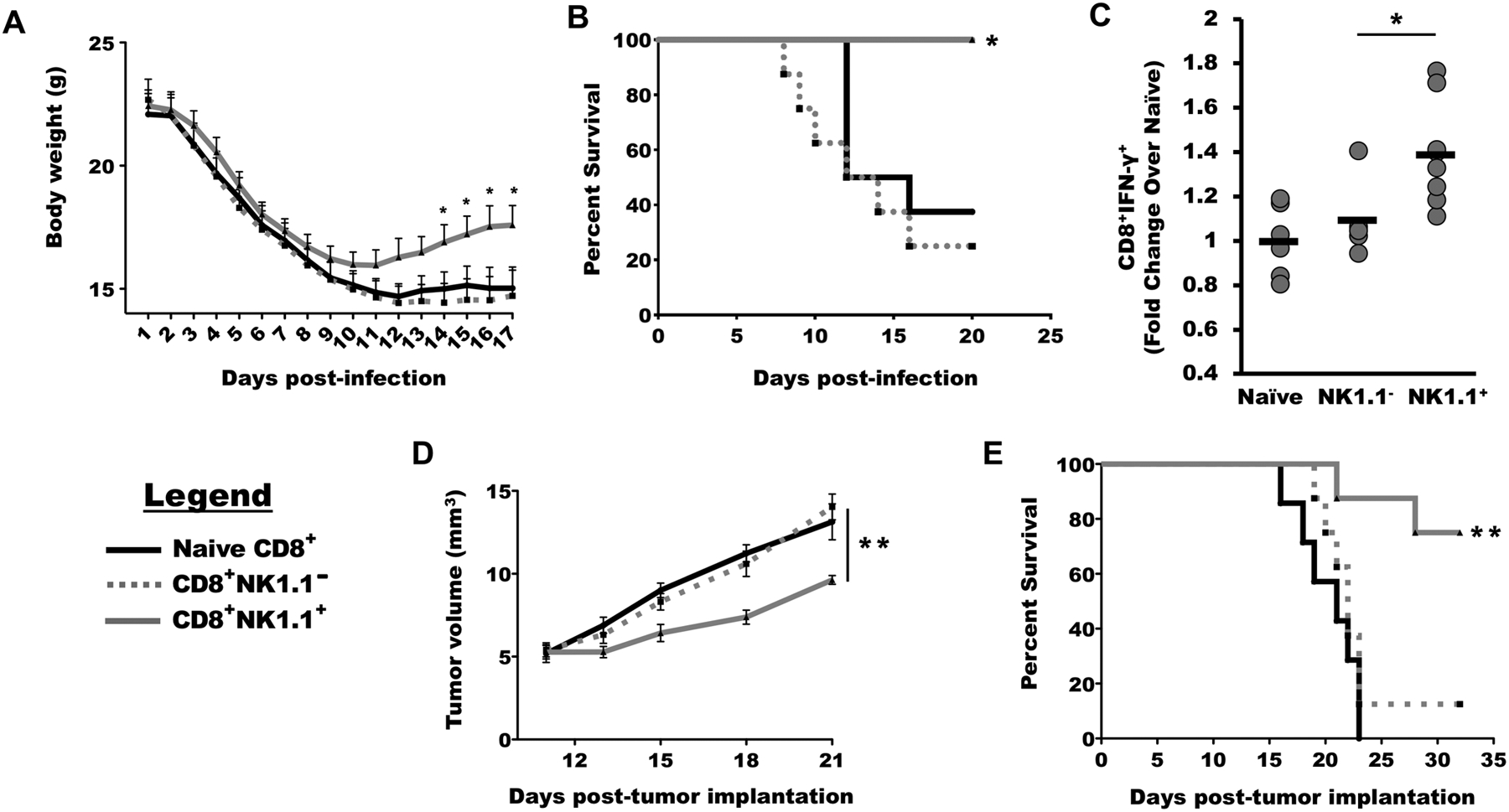
(A) Splenocytes were harvested from mice that recovered from influenza infection and sorted into CD8+NK1.1− and CD8+NK1.1+ cells. The sorted cells were adoptively transferred into naïve mice that were subsequently challenged with lethal influenza infection and weight was recorded beginning 1 day post infection (DPI). (B) Survival was measured in mice receiving adoptive transfers of CD8+NK1.1− cells, CD8+NK1.1+ cells, and naïve CD8+ cells. (C) The frequency of CD3+CD8+IFN-γ+ cells were analyzed in PBMCs of mice 12 days weeks post infection. (D and E) B16 melanoma-bearing mice were vaccinated with dendritic cells loaded with tumor antigens. Splenocytes were harvested after three weeks and sorted into CD8+NK1.1− and CD8+NK1.1+ cells and adoptively transferred into mice with palpable tumors, and tumor burden (D) and survival (E) were measured over time. For each experiment, n ≥ 7 mice per group. Data are presented as mean ± SEM, *p < 0.05, **p < 0.01, by one-way ANOVA with Tukey’s post-hoc analysis (A, C, D) or log-rank (Mantel-Cox) test (B, E).
In the second model system, a cohort of donor mice was inoculated subcutaneously with 2 × 105 B16 melanoma cells and vaccinated with a B16-loaded cell-based vaccine at 7 and 14 days post-inoculation. At day 21, the mice were euthanized, and splenocytes again harvested and sorted into CD8+NK1.1+ and CD8+NK1.1− cell populations. Naïve cohorts of mice were inoculated with palpable B16 tumors and then received 1.5 × 106 CD8+NK1.1+ or CD8+NK1.1− cells (fig. S2B). The mice that received the CD8+NK1.1+ cells exhibited a significant delay in tumor growth (p<0.01 on day 21, Fig. 2D) accompanied by significant survival benefit (p<0.01, Fig. 2E) whereas the cohort that received CD8+NK1.1− cells survived identically to the control cohort that received naïve splenocytes.
To determine any potential role of TCR specificity or variability in these results, pairs of mice were challenged with either sublethal influenza infection or melanoma inoculation and vaccination. At day 21, splenocytes from these animals were harvested and sorted CD8+ cells were cultured overnight with antigen-loaded DC. Subsequently, the antigen-specific CD69+ or CD25+ population was divided into NK1.1+ and NK1.1− subfractions (fig. S3), and the TCR sequences of these populations were determined for T cells isolated from each animal. Of note, 93% of TCRs were not shared at the amino acid level either between mice or between NK1.1+ and NK1.1− fractions derived from the same animal. Of the 7% that were shared, between 21% and 60% were shared between the oligoclonal NK1.1+ fraction and the highly polyclonal NK1.1− fraction among individual animals (table S3). Interestingly, the oligoclonal NK1.1+ cells expressed only a very small fraction of total unique TCRs sequenced. Among influenza-infected mice, these were 369 of 8,968 total TCRs (4.1%), and among melanoma inoculated and vaccinated mice, these represented 442 of 21,132 total TCRs (2.1%). Together these results suggest that the CD161 homolog NK1.1 defines a major CD8+ oligoclonal memory cell population in simple models of immunological challenge among adolescent mice.
The phenotype of the murine CD3+CD8+NK1.1+ cell population is conserved among the CD3+CD8+CD161+ human counterpart population.
Encouraged by the protective memory responses offered by the CD8+NK1.1+ cells in the mouse model systems studied, we next asked if the subset of memory CD8+ T cells defined by NK1.1 expression was phenotypically and transcriptionally conserved in human populations among the analogous CD3+CD8+CD161+ cell population in peripheral circulation. For this analysis, CD8+CD161+ and CD8+CD161− cells were differentially isolated from six different human donors. After verifying that the CD161+ cells were polyclonal by TCR-Vβ spectratyping (fig. S4), transcriptional profiling of each population was performed by microarray analysis with the most differentially regulated genes shown in tables S4 and S5. Despite the fact that these cells were not activated prior to analysis and in a homeostatic resting state, the profile of upregulated granzymes and natural cytotoxicity receptors was substantially recapitulated in these cells (Fig. 3A and B). Cross-species comparative analysis of genes between the activated mouse and unactivated human cells identified a conserved signature of 206 genes with common nomenclatures differentially regulated among the two populations (fig. S5). Reactome Pathway analysis (reactome.org) of the upregulated human genes identified signatures associated with differentiation and regulation (false discovery rate [FDR] < 5 × 10−4) including histone deacetylation, deoxyribonucleic acid (DNA) and histone methylation, nucleosome assembly, ribonucleic acid (RNA) polymerase I promoter escape, transcriptional regulation by small RNAs, and gene silencing by RNA (table S6).
Figure 3. The murine CD3+ CD8+ NK1.1+ cell population is phenotypically similar to the human CD3+ CD8+ CD161+ cell population.
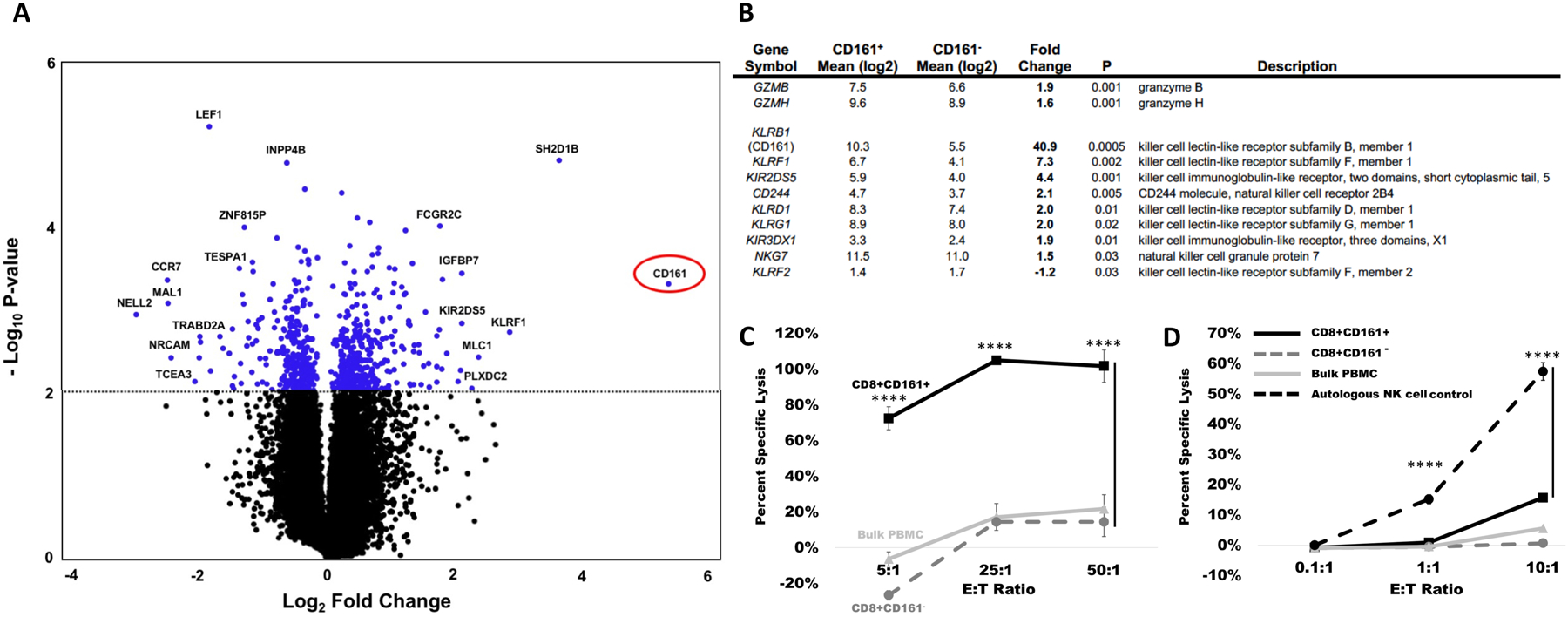
CD3+ CD8+ CD161+ and CD3+ CD8+ CD161− cells, the equivalent human population to the murine CD3+ CD8+ NK1.1+ cells and CD3+ CD8+ NK1.1− cells were magnetically sorted and evaluated by microarray. (A) A volcano plot is shown comparing the differential regulation of genes between CD8+ CD161+ and CD8+ CD161− cells, with the gene encoding CD161 circled in red. (B) The CD8+ CD161+ cell population exhibits elevated expression of granzymes and killer lectin like receptor genes. (C and D) Fresh buffy coat products were divided into bulk PBMC, CD8+CD161+, and CD8+CD161− fractions and tested in 51Cr killing assays against HEK293 allogeneic cell targets (C) or K562 targets (D). For each target, lysis was performed at the E:T ratio indicated on the X-axis. For all lysis assays, data are presented as mean ± standard deviation, ****p < 0.0001 by one-way ANOVA with Tukey’s post-hoc analysis.
To determine if human CD8+CD161+ cells could exhibit enhanced killing activity and to define the mechanism of cytotoxicity, freshly isolated CD3+CD8+CD161+ human buffy coat cells were co-cultured with allogeneic HEK293 or K562 cell targets. As shown, unsensitized CD3+CD8+CD161+ cells were robust allogeneic killers, exhibiting nearly 100% target lysis at E:T ratios as low as 25:1, whereas unsensitized autologous unmanipulated PMBC or CD3+CD8+CD161− counterparts did not exceed 20% allogeneic lysis at any E:T ratio (p<0.0001, Fig. 3C). In contrast, CD3+CD8+CD161+ cells exhibited no NK cell-like killing activity against K562 targets, except for slight activity observed at an effector-to-target ratio (E:T) of 10:1 in comparison to CD3+CD8+CD161− cells (p<0.0001, Fig. 3D). The data suggested that native CD3+CD8+CD161+ cells might possess substantially enhanced TCR-mediated killing capabilities.
The CD8+CD161+ T cell population can be used to generate CAR T cells for therapy of PDAC.
We hypothesized that the human CD8+CD161+ T cell subset might be able to offer more durable anti-tumor efficacy in the context of solid tumor CAR T cell therapy as compared to conventional bulk PBMC. To test this hypothesis, we specifically focused on the generation and characterization of systems to evaluate CAR T therapy of PDAC, an indication for which there exists an urgent and compelling medical need. Given that human epidermal growth factor receptor-2 (HER2) amplification and overexpression is reported in a large subset of pancreatic tumors (23), we chose a well-characterized second generation HER2-targeted CAR T construct that has been tested in early stage clinical trials (7, 24–27) for use in the model (Fig. 4A). We first characterized HER2 surface expression on seven common human pancreatic cancer cell lines (ASPCI, CAPAN1, CAPAN2, CFPAC1, HPAF1, PANC1, PL45) (Fig. 4B) to permit CAR T cell targeting at a variety of different surface antigen densities. We next isolated PBMC from three healthy donors, sorted them into bulk PBMC, CD8+CD161+cells and CD8+CD161− cells, transduced these populations with the HER2-CAR retroviral vector, and validated that the transduction efficiency (Fig. 4C), as well as the CAR surface density and proliferative capacity against HER2+ target (Fig. 4D), were comparable among all three populations. The ability of each transduced population to secrete interferon (IFN)-γ (Fig. 4E) and interleukin (IL)-2 (Fig. 4F) against each HER2+ target cell line was validated as well. At the time of selection, CD161 surface expression on positively-selected CD8+CD161+ cells measured 1–2 logs-fold higher than that of CD8+CD161− cells or unmanipulated PBMC, a phenotype that was maintained throughout transduction and optimized expansion (fig. S6).
Figure 4. Development of a CD8+CD161+CAR T cell therapy model for PDAC.
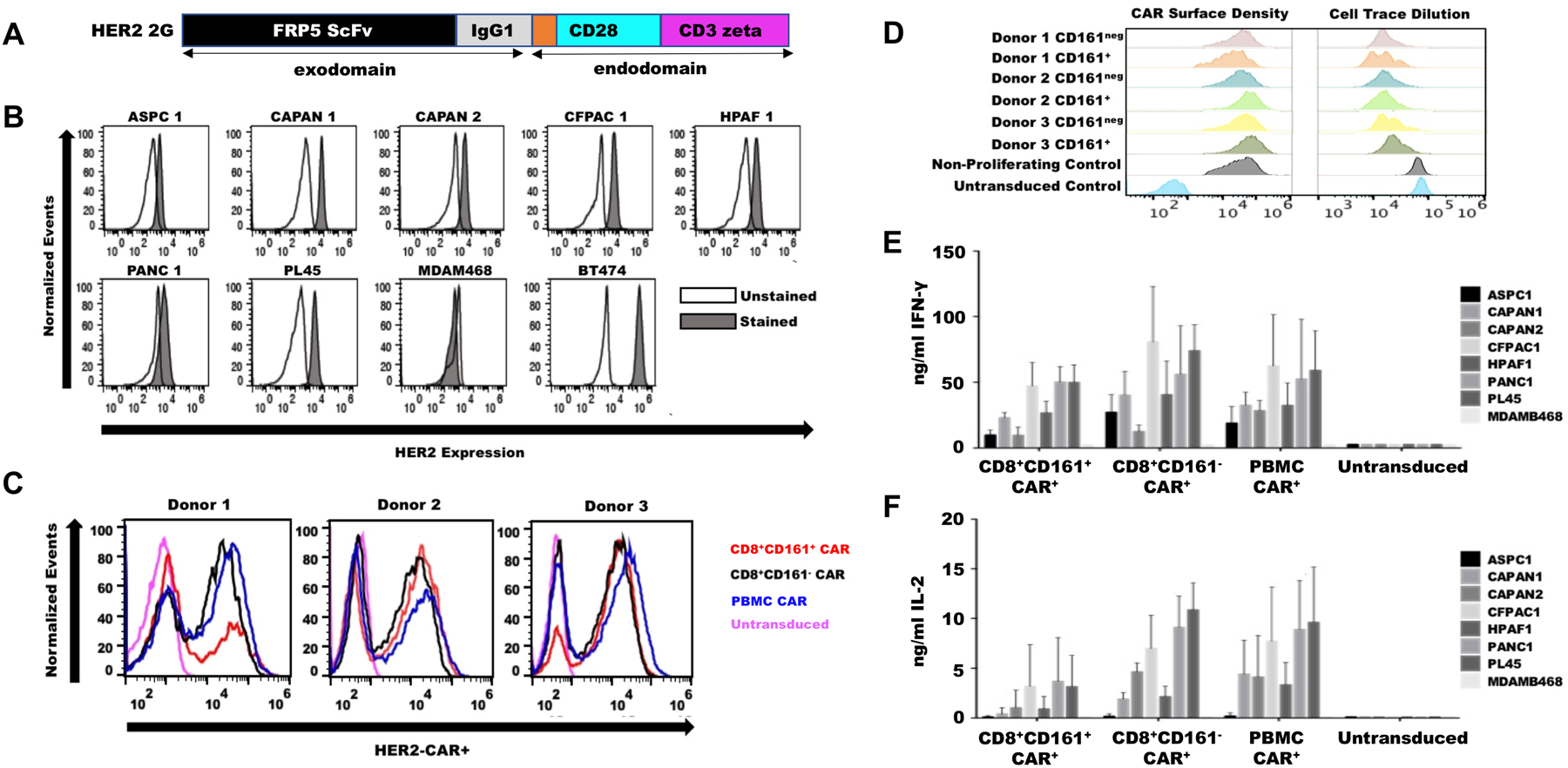
(A) A second generation anti-HER2 CAR construct was made with FRP5 ScFv exodomain and CD28, CD3zeta endodomains. (B) HER2 surface expression by flow cytometry was confirmed on seven common human pancreatic cell lines. MDAM486 served as HER2− and BT474 as HER2+ controls. (C) Bulk PBMC, CD8+161−, and CD8+161+ cells were sorted from normal human donors and transduced with HER2-CAR retroviral vector, and transduction efficiency was evaluated by flow cytometry. (D) CAR surface density and proliferative capacity against HER2+ targets were evaluated by flow cytometry. (E and F) 50,000 HER2+ and HER2− tumor cells were plated for 24 hours and cultured with 100,000 CAR+ cells or non-transduced controls for 48 hours. Culture media was assayed for IFN-γ (E) or IL-2 (F) secretion. Data are presented as mean ± SEM.
CD8+CD161+CAR-transduced cells exhibit inherent advantages in killing, proliferation, and exhaustion in vitro.
To assess the cytotoxic capacity of CAR-modified CD8+CD161+ cells against that of CD8+CD161− cells and bulk PBMC, two independent cytotoxicity assays were performed in vitro, a chromium-based short-term assay and a cell impedance-based long-term killing assay. In the short-term assay using HER2+ BT474 cells and HER2− Raji cell controls, the CD8+CD161+ CAR-transduced cells exhibited roughly double the killing efficiency of either the transduced CD8+CD161− T cells or transduced bulk T cells at all E:T ratios tested from 1.25:1 to 40:1 (p<0.0001, Fig. 5A). To select an appropriate target cell line for longer term assays, we next determined the cytotoxic potential of the CD8+CD161+ CAR-transduced T cells against all seven HER2+ human pancreatic cancer cell lines in the impedance-based assay over 120 hours. Based on these results (fig. S7), we selected the CFPAC1 cell line for further experimentation given that this cell line was one of the most difficult to kill. Next, using CFPAC1 as the target in the long-term impedance-based cytotoxicity assay, we observed that the CD8+CD161+ CAR-transduced T cells exhibited significantly faster and more efficient tumor cell killing capacity as evidenced by CD8+CD161+ mean area under curve (AUC) was half that of the CD8+CD161− CAR-transduced cells and only a third that of CAR-transduced bulk PBMC (p<0.001, Fig. 5B). To further characterize the enhanced cytotoxic abilities of CD8+CD161+ CAR T cells, transduced CD8+CD161+ T cells, CD8+CD161− T cells, and bulk PBMC were incubated with either CAR target HER2-Fc protein or irrelevant CD19-Fc fusion protein and analyzed by flow cytometry. As shown, incubation of CD8+CD161+CAR+ T cells with target protein resulted in rapid and specific upregulation of perforin and granzyme B production (fig. S8A) whereas upregulation of IFN-γ and CD25 (fig. S8B) were of the same magnitude as that of CD8+CD161− or bulk PBMC T cells. To determine any advantages with regard to proliferative capacity or exhaustion, transduced bulk, CD8+CD161−, and CD8+CD161+ cells were stimulated four consecutive times in vitro with HER2-expressing CFPAC cell targets and analyzed. Following these repeated stimulations through the CAR, there were no statistical differences observed in cell count among these three lines; however, analysis of exhaustion markers indicated significant downregulation of the exhaustion markers T cell immunoglobulin and mucin-domain containing-3 (TIM3) (p<0.01) and programmed death-1 (PD-1) (p<0.05) among the CD8+CD161+ HER2CAR-transduced cells (fig. S8C and D) in comparison to CAR-transduced bulk PBMC and CD8+CD161− cells. Untransduced and CD19CAR-transduced controls did not proliferate at all in response to HER2-Fc target protein, and the few residual cells in these groups were not phenotyped. Together, the data indicated that CD8+CD161+ CAR T cells exhibit enhanced cytotoxicity kinetics and reduced expression of exhaustion markers.
Figure 5. CAR transduced CD8+CD161+ cells exhibit a killing advantage over CAR transduced bulk PBMC and CD8+CD161− cells.

(A) HER2+ BT474 (left) and HER2− Raji (right) tumor cells were labelled with 51chromium for 4 hours and co-cultured with indicated CAR T cells. Percent cell lysis was determined by chromium released into the media. NT, non-transduced. (B) Long term cytotoxicity was measured using an xCELLigence impedance-based assay. CFPAC1 tumor cells were seeded at 10,000 cells per well in a 96 well plate in triplicates and allowed to grow over 40 hours. T cells were then added at 2.5:1 tumor cell:T cell ratio and tumor growth or death, as indicated by cell index, was monitored over time. Data are presented as means ± SEM, ****p<0.0001 by one-way ANOVA with Tukey’s post-hoc analysis.
CD8+CD161+ CAR T cells substantially improve survival outcomes in vivo.
Building upon our in vitro results, we generated a luc2+ CFPAC1 line to model CAR T cell therapy of difficult-to-treat disseminated PDAC in vivo. In this model system, 6–8 week old male SCID mice were inoculated intraperitoneally (i.p.) with one million CFPAC1-luc2 cells. One week after tumor establishment (as determined by in vivo imaging [IVIS]), cohorts were generated by randomization and given a single i.p. administration of 2 × 106 in vitro-expanded CAR+ T cells. Disease burden was monitored by IVIS imaging. Disease burden among the cohorts treated with CD8+CD161+ CAR T cells was substantially lower over time than among cohorts treated with CD8+CD161− CAR T cells, bulk PBMC-derived CAR T cells, or non-transduced controls (Fig. 6 A and B). This prominent reduction in disease burden was reflected in the Kaplan-Meier survival estimate. 100% of mice in the cohort treated with CD8+CD161+ CAR T cells remained alive at day 80, significantly better than all other cohorts (p<0.01, Fig. 6C). A second series of independent experiments and analyses using a different T cell donor consistently replicated these results (fig. S9), with 100% of mice in the cohort treated with CD8+CD161+ CAR T cells remaining alive at day 120 (p<0.0001, fig. S9C). To determine the ability of CD8+CD161+ CAR T cells to address primary disease within the pancreas, we also developed an orthotopic model. In this model, pancreata were surgically exposed and subsequently injected with 5 × 105 human CFPAC-1-luc2+ PDAC cells resuspended in 50 μl phosphate buffered saline (PBS). Seven days later, tumor-bearing mice were randomized into cohorts of five and received adoptive transfers of a one-time bolus of 2 × 106 CAR T cells, and tumor progression was monitored by IVIS imaging (fig. S10A). Similarly to what was observed in the disseminated model, there was a substantial delay in tumor progression among mice treated with CD8+CD161+ CAR-transduced T cells (p<0.05, fig. S10B). By post-implantation day 74, nearly all mice in non-transduced, bulk PBMC, and CD8+CD161− groups had succumbed to disease whereas all mice in the CD8+CD161+ group remained alive (p<0.001, fig. S10C). Further, the addition of IL-21 to the culture medium prior to adoptive transfer, required for the maintenance of CD161 expression on CD8+CD161+ T cells during expansion, imparted no advantages or enhanced lytic properties to bulk PBMC-derived T cells (Fig. 5 and Fig. 6).
Figure 6. CD8+CD161+ CAR T cells improve survival outcomes in an in vivo disseminated model of PDAC.
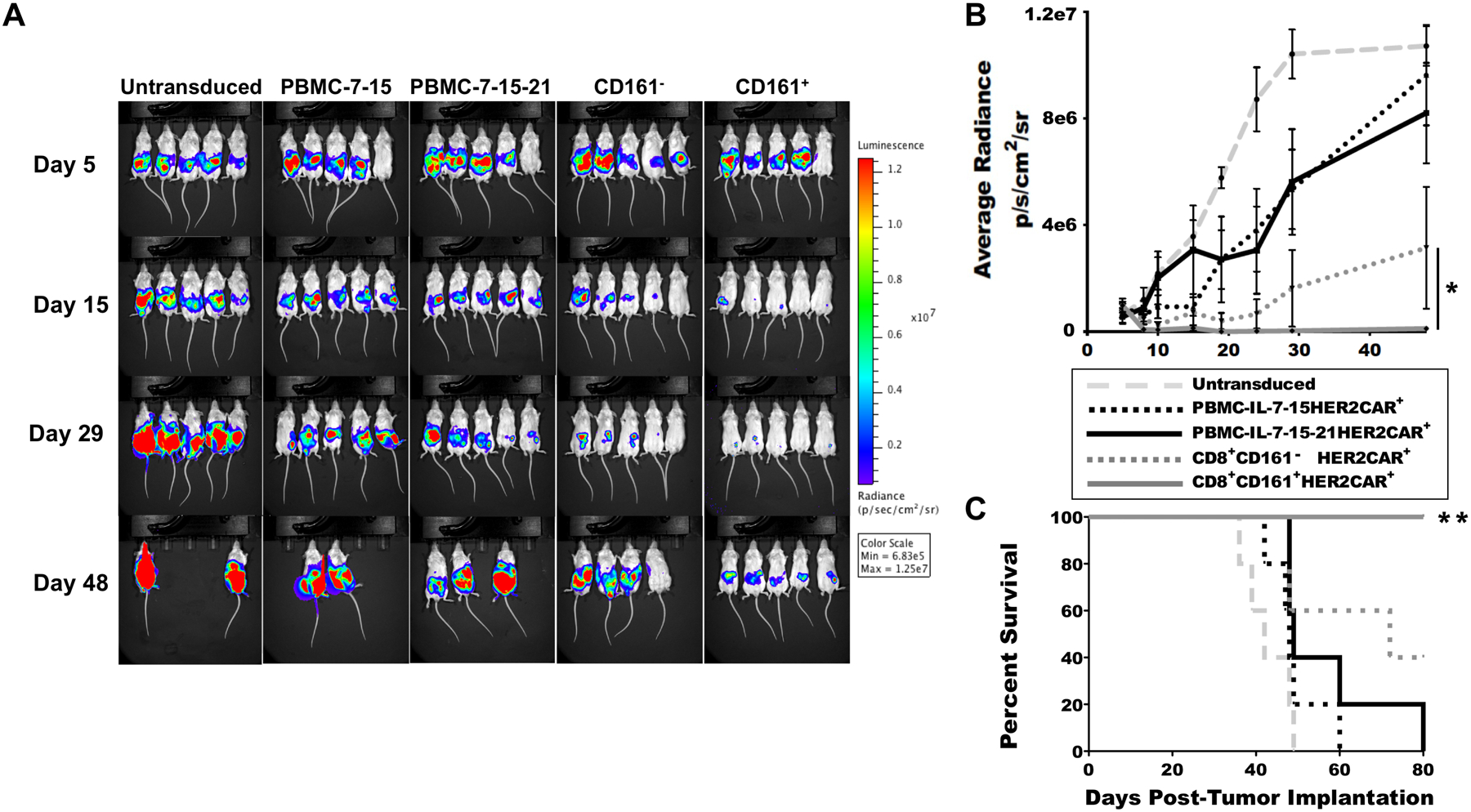
(A and B) Male SCID mice were inoculated i.p. with one million CFPAC1-luc2 cells. Five days after establishment of disease as determined by IVIS imaging, cohorts were generated by randomization and given a single administration of 2 × 106 in vitro-expanded CAR+ T cells on post-implantation day 7. Disease burden was monitored by IVIS imaging (A) and quantified (B). *p < 0.05 by one-way ANOVA with Fisher’s post-hoc analysis. C. Survival was measured over time. **p < 0.01 by log-rank (Mantel-Cox) test. One representative experiment shown. For each cohort, n=5 mice. Data are presented as means ± SEM.
To determine the mechanism(s) by which CD8+CD161+ CAR-transduced T cells might be exerting enhanced cytolytic effects, we first analyzed bulk, CD8+CD161−, and CD8+CD161+ CAR-transduced T cells in a 51Cr lysis assay along with CD56+ NK cells controls to determine if CD8+CD161+ CAR-transduced T cells possessed any natural cytotoxicity lytic properties and observed no such activity (fig. S11). Only CD56+ autologous NK cell controls were able to lyse K562 targets whereas none of the CAR-transduced and expanded T cells, including the CD8+CD161+ CAR-transduced T cells, exhibited any lytic activity against MHC-deficient K562. To determine what other mechanisms might be mediating enhanced killing by CD8+CD161+ CAR-transduced T cells, we next performed a short-term in vivo study in which carboxyfluorescein succinimidyl ester (CFSE)-labeled, HER2CAR-transduced CD8+CD161− and CD8+CD161+ T cells were administered to mice bearing orthotopic CFPAC-1-luc2+ PDAC tumors. As a control, a cohort of CD19CAR-transduced CD8+CD161+ cells was included in addition to the non-transduced bulk T cell cohort. Consistent with all other experiments, the CD8+CD161+ HER2CAR-transduced T cells controlled the growth of tumor better than the CD8+CD161− HER2CAR-transduced T cells (p<0.05, Fig. 7A and B). Further, the uncontrolled tumor growth observed in the CD19CAR-transduced CD8+CD161+ control confirmed i) antigenic specificity of the antitumor response, ii) no impact of tonic CAR signaling, and iii) an absence of any non-specific CD8+CD161+ cell killing activity. Sixteen days after administration of the CAR-transduced T cells, animals were euthanized, tumors were harvested, and tumor infiltrating lymphocytes were collected for analysis. Numbers of human T cells present at the tumor site were indistinguishable between CD8+CD161− and CD8+CD161+ cohorts (Fig. 7C), and analysis of CFSE dilution and annexin V staining showed no differences in proliferation (Fig. 7D) or cell death (Fig. 7E). In contrast, CD8+CD161+ HER2CAR-transduced cells exhibited markedly lower expression of surface PD-1 than CD8+CD161− HER2CAR-transduced counterparts (p<0.05, Fig. 7F) as well as markedly higher expression of granzyme B (p<0.05, Fig. 7G). Although homing-related advantages could not be directly determined from antigen-specific CAR T cells that had undergone homeostatic expansion and contraction, such advantages could be inferred by comparing the numbers of untransduced bulk T cells in the tumor bed to the numbers of CD8+CD161+ CD19CAR-transduced T cells, as neither had recognized cognate antigen nor undergone subsequent proliferation and contraction. In this comparison, the CD8+CD161+ CD19CAR-transduced T cells were found in the tumor bed at double the frequency of the untransduced bulk T cell population, suggesting a potential homing advantage (p < 0.05, Fig. 7H).
Figure 7. CD8+CD161+ CAR T cell enhanced function may be mediated by elevated killing, enhanced homing, and reduced exhaustion.

(A) Male SCID mice were implanted orthotopically with 500,000 CFPAC1-luc2 cells. Five days after establishment of disease as determined by IVIS imaging, cohorts were generated by randomization and given a single administration of 2 × 106 in vitro-expanded HER2CAR T cells pre-stained with CFSE on post-implantation day 7, and post-treatment disease burden was monitored from day 9 to day 16 by IVIS imaging. (B) Tumor growth among the mice treated with CD8+CD161+ HER2CAR T cells was quantified over time following CAR T cell administration. (C-G) On day 16, tumors were excised and analyzed by flow cytometry for cell number (C), proliferation (CFSE dilution) (D), apoptosis (E), PD-1 expression (F), and granzyme B expression (G). (H) Presence of human CD3-expressing CD8+CD161+ CD19CAR-transduced T cells in the tumor bed was quantified by flow cytometry. Data are presented as mean ± SEM, n = 5 mice per group. *p < 0.05, ns = not significant by Student’s two-tailed t-test (F, G, H) or one-way ANOVA with Duncan’s post-hoc analysis (B).
Discussion
Although CAR T cell therapy has emerged as an exciting and effective treatment option for CD19+ B-cell malignancies (2, 3), its performance in the solid tumor setting has not yet approached a similar degree of efficacy. The inability of CAR T cells to effectively and durably control solid tumors has been attributed to a variety of factors, including the paucity of solid tumor-specific cell surface targets and tumor antigen heterogeneity (28), the immunosuppressive impact of the solid tumor microenvironment on T cell activation, exhaustion, and persistence (29), and the low efficiency by which ex vivo-expanded CAR T cells naturally traffic to tumor sites (30). To address these challenges, many investigators are employing a number of elegant strategies that seek to alter T cell biology through CAR-mediated expression of exogenous molecules or genome editing approaches (31). Here we have tested the hypothesis that TCRαβ CD8+CD161+ T lymphocytes, a memory T cell subset multiply identified and characterized in a variety of inflammatory contexts, might serve as an improved platform for CAR T cell therapy given that characteristics thought to be critical for CAR success in the solid tumor setting such as persistence, tissue extravasation, and serial killing appear to be inherent in this subset (14, 18, 19, 21, 32–36).
CD161 is known to be promiscuously expressed on NK cells, NKT cells, and a variety of T cell populations (15). It shares 47% homology with its murine homologue NK1.1 and is expressed by 5–25% of circulating peripheral T cells (12, 37). CD161 expression has previously been observed on a variety of different CD8+ cell types identified as CD8αα+ TCR mucosal-associated invariant T cells (MAIT cells) (9, 38), Tc 17 cells (11, 14), and memory stem cells (10). Transcriptional profile analysis of CD161-expressing cells identified a conserved transcriptional signature enriched in CD8+CD161+ T cells that can be extended to CD4+CD161+ T cells, TCRγδ+CD161+ T cells, and NK cells (17). Despite certain innate-like qualities of CD161 expressing cell types, the T cells identified here by us and by others exhibit an αβ polyclonal repertoire, a tendency toward anti-viral specificity, and no discernible natural cytotoxicity activity (11, 19, 32). We previously reported the functional importance of murine CD8+NK1.1+ cells, analogous to human CD8+CD161+ cells, and documented elevated numbers of these cells in mice following administration of a cell-based vaccine that mimics certain characteristics associated with viral infection (22). Microarray analysis of CD8+NK1.1+ cells revealed substantial upregulation of granzymes F, D, G, C, B, A, and N by these cells upon antigenic stimulation, as compared to activated CD8+NK1.1− counterparts. CD161+ human equivalents were also shown to upregulate granzymes B and H in the steady state and exhibited upregulated expression of the C-C Motif Chemokine Receptors CCR6, CCR5, and CCR2, with concomitant downregulation of CCR7, CCR4, CCR1, CCR3, and C-X-C Motif Chemokine Receptor 3 (CXCR3), suggesting altered homing characteristics of these cells. Similar expression patterns have been observed among circulating CD161+ cells in the peripheral blood of multiple sclerosis patients, potentiating their entry into the central nervous system and contributing to pathogenesis (20). In agreement with the genomic data, in vivo studies suggested that the mechanisms by which CD8+CD161+ CAR-transduced cells mediated improved antitumor activity were related to enhanced killing and, potentially, homing. Notably, lower expression of PD-1 after both in vitro and in vivo CAR stimulation suggested the additional mechanism of reduced exhaustion. Interestingly, among activated mouse CD8+NK1.1+ cells, multiple immunoglobulin heavy and light chains were among the most upregulated transcripts. We did not explore this phenomenon beyond verification that the CD8+NK1.1+ cell population analyzed was not contaminated with B cells; however, the unexpected yet fascinating phenomenon of B-cell receptor (BCR) and TCR co-expression in mouse T cells was recently reported in the context of autoimmune diabetes (39). It is possible that this population of “dual expressers” might overlap with NK1.1-expressing T cells to some extent.
The inclusion of IL-7 and IL-15 in the medium has been shown to benefit lymphocyte development, differentiation and homeostasis during ex vivo expansion of CAR T cells, resulting in higher survival after adoptive transfer in comparison to IL-2 expanded CAR T cells (40, 41). Others have shown that IL-21 promotes expansion of CD2+CD28+CD8+ T cells (42) and enhances potency of CD19-CAR T cell therapy (43). It has further been reported that addition of IL-15 and IL-21 helps enhance and maintain the memory potential of NKT cells (44). A combination of IL-7, IL-15 and IL-21-based ex vivo expansion of T-lymphocytes was reported to enhance memory cells, reduce metastasis and improve survival against murine melanoma (45). In the present study, we found that ex vivo culture and expansion of bulk PBMC-derived CAR T cells, CD8+CD161− CAR T cells, and CD8+CD161+ CAR T cells in a cocktail of IL-7, IL-15, and IL-21 benefitted exclusively the CD8+CD161+ cell population without altering the phenotypes of bulk PBMC or CD8+CD161− cells cultured without IL-21 or even in IL-2 alone. Under these conditions, CD8+CD161+ CAR-transduced cells functionally outperformed bulk T cell preparations and CD8+CD161− controls in lytic capability and persistence. Moreover, CD8+CD161+ CAR-transduced cells mediated substantially enhanced survival in orthotopic and disseminated models of PDAC disease in vivo in comparison to CAR-transduced bulk PBMC preparations, even if bulk PBMC were also expanded in IL-7, IL-15, and IL-21.
Some investigators have reported that optimal CAR T cell activity is achieved with a balanced population of CAR-transduced CD4+ and CD8+ T cells (46, 47). Given these reports, it is tempting to speculate that CD8+CD161+ CAR T cell therapy might be improved even further through the concomitant use of CD4+CD161+ CAR-transduced T cells. It is not clear, however, that such a strategy would be beneficial. Our results consistently demonstrate that CD8+CD161+ cells display enhanced cytotoxicity and durable anti-tumor responses in comparison to bulk PBMC made up of both CD4+ CAR-transduced T cells and CD8+ CAR-transduced T cells. Further, since most of CD161-expressing CD4+ cells are TH17 (IL-17 secreting) regulatory cells with low expression of cytotoxic molecules, it is not clear that a combination of CD161+ CD8+ CAR T cells and CD161+CD4+ CAR T cells would exhibit functional activity superior to that of CD8+CD161+ CAR T cells alone. Nonetheless, future experiments will explore this intriguing possibility.
There are certain potential limitations to our study. Because TCR sequencing indicated dissimilarities between NK1.1+ and NK1.1− T cell fractions (and indeed between identical fractions in different mice), we cannot rule out the possibility that variance in TCR affinity between these two subsets led to the differences in survival seen in the influenza and melanoma challenge models. Due to relatively small sample sizes, inherent variability in transcription, and a desire to avoid type II errors while still reporting potentially relevant biology, we can make few definitive claims regarding transcriptional differences between NK1.1/CD161+ and NK1.1/CD161− cells other than for genes which were subsequently validated prospectively through experimentation. Although genes with low q values identified through cross-species comparison are the most likely to correspond to true biological differences in transcription, the majority of these also require prospective validation. These are standard confounders in many genomics studies. Additionally, although CAR experiments were repeated many times, only three individual blood donors served as a source of starting materials. Therefore, the functional variability of this subset still requires validation more broadly. We also did not fully characterize the role that CD161/NK1.1 receptor ligation might play in the downstream functions of CD8+CD161+ cells. The determination of this variable will be particularly important in any future use of this subset in a therapeutic capacity.
In summary, we report that CD8+CD161+ cells and their murine equivalent CD8+NK1.1+ cells exhibit high cytotoxic potential both in vitro and in vivo. Gene expression profile analysis by microarray revealed enhanced expression of granzyme, perforin, and innate-like receptors by these cells when activated compared to NK1.1− and CD161− counterparts. In vitro, CD8+CD161+ CAR-transduced T cells killed with greater speed and efficiency than bulk PBMC or CD8+CD161− CAR-transduced populations. In vivo, a single administration of CD8+CD161+ CAR-transduced T cells offered substantially improved anti-tumor protection and survival compared to CD8+CD161− and bulk PBMC CAR-transduced T cells. Use of this subset for CAR T cell therapy may offer an opportunity for effective treatment of solid tumors, including PDAC, and additional studies are warranted.
Materials and Methods
Study design.
The aim of this study was to characterize the biology and functional properties of a unique polyclonal CD8+ αβT cell subset that expresses the natural cytotoxicity receptor NK1.1 in the mouse and the homologous CD161 in humans. The study also aimed to determine the potential suitability of this subset for use in solid tumor CAR T cell therapy, specifically against HER2+ pancreatic ductal adenocarcinoma. Based on previously published work, we investigated the efficacy of the CD8+NK1.1+ subset against different disease models including influenza and melanoma. We performed microarray analysis of the antigen-specific CD8+NK1.1+ and CD8+NK1.1− cells, identifying substantial upregulation of cytotoxic granzymes and natural cytotoxicity receptors among CD8+NK1.1+ cells. We further extended these studies to CD161+ human T cells, characterizing the cytotoxic and memory potential of these cells through in vitro and in vivo experimentation. We next isolated bulk PBMC, CD8+CD161− and CD8+CD161+ cells from healthy donors and transduced the cells with a CD3ζ/CD28-HER2-CAR construct. We performed short term and long-term in vitro cytotoxicity analyses to test the efficacy of these CAR-transduced cells against human pancreatic cancer cell lines and then followed-up in vivo. We xenografted the HER2+ human PDAC cell line CFPAC1 into mice by both i.p. and orthotopic inoculation and subsequently treated the mice with CAR-transduced cells. CD19-CAR transduced CD8+CD161+ cells served as negative controls. We performed IVIS imaging, Kaplan-Meier survival analysis and phenotyped cells isolated from tumor microenvironment by flow cytometry to characterize proliferation, activation and exhaustion. All animal experiments were performed in compliance with institutional IACUC policies. All in vivo mouse experiments including xenografts were performed after randomization of the mice into treatment groups. The number of replicates for each experiment are described in the figure legends. Sample size number was determined by power calculation, the assumed variance (σ) of which was determined by previous investigator experience. Where possible, investigators were blinded during data collection.
Mouse microarray analysis.
CD8+NK1.1+ cells and CD8+NK1.1− cells were isolated from mice that previously received pancreatic tumors and were therapeutically treated with cell-based vaccine in conjunction with gemcitabine chemotherapy (22). Briefly, spleens from the treatment groups were excised separately, minced thoroughly in Roswell Park Memorial Institute (RPMI)-1640 medium (Invitrogen), strained through a 70 μm filter (Corning), washed in PBS, and pooled and activated with PDAC antigen-loaded autologous DC overnight. The activated cells were incubated with anti-CD3-Phycoerythrin-Cyanine5 (PE-Cy5) (Tonbo Biosciences), CD8-Allophycocyanin (APC) (BD Biosciences), CD69-Phycoerythrin (PE) (BD Biosciences) and NK1.1-Phycoerythrin-Cyanine7 (PE-Cy7) (BD Biosciences) at a dilution of 1:100 in FACS buffer (PBS supplemented with 1% fetal bovine serum and 0.01% sodium azide) for 20–30 min at 4° C for flow-based cell sorting. This FACS buffer was used for all flow-based cell sorting experiments. Cells were gated on the CD8+CD69+ population and sorted for NK1.1+ and NK1.1− subsets. Total RNA was isolated from the sorted cells using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Gene expression profiling of CD8+NK1.1+ and CD8+NK1.1− cells was performed with the Affymetrix Mouse Transcriptome Array 1.0 Chip (Affymetrix) by the Sequencing and Microarray Facility at the University of Texas MD Anderson Cancer Center. The mouse microarray data have been deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE167276 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc= GSE167276).
Influenza model.
To generate T cells for adoptive transfer, C57BL/6 mice were sublethally challenged with influenza A/HongKong/8/68 (H3N2) Swiss mouse lung-adapted strain of H3N2 influenza A virus graciously provided by Dr. Brian Gilbert as described previously (48). Animals were infected in a 20-minute nebulized aerosol exposure of influenza virus diluted in Cellgro MEM media (Corning, Inc) + 0.05% gelatin (Sigma-Aldrich) using the Aerotech II nebulizer at 10 L/min of room air generated from an Aridyne 2000 compressor. All mice infected in any given experiment were treated simultaneously in a single exposure chamber. Three-weeks post infection, mice were sacrificed, spleens harvested and CD8+ cells negatively selected using the CD8 T cell Isolation Kit (Miltenyi Biotec) per manufacturer’s instructions. Isolated CD8+ cells were further magnetically sorted into NK1.1+ and NK1.1− populations using the Anti-NK1.1 Biotin Kit (Miltenyi Biotec) per manufacturer’s instructions. 500,000 CD8+NK1.1+ and CD8+NK1.1− cells were subsequently adoptively transferred into two groups of eight naïve mice each that were then challenged with a lethal dose of influenza virus after 24 hours. Control group received naïve CD8+ splenocytes followed by influenza challenge.
Melanoma model.
To generate T cells for adoptive transfer, C57BL/6 mice were subcutaneously inoculated with 250,000 B16F10 melanoma tumor cells (American Type Culture Collection) suspended in 100 μl PBS. DC were loaded with melanoma tumor antigens as described (22). One week post tumor inoculation, 200,000 antigen-loaded DC suspended in 50 μl PBS were injected into the footpad with a booster vaccination given seven days later. Ten days post-boost, vaccinated mice were euthanized, and the CD8+ splenocytes were isolated by negative selection using the CD8 T cell Isolation Kit (Miltenyi Biotec). Isolated CD8+ cells were further magnetically sorted into NK1.1+ and NK1.1− populations using the Anti-NK1.1 Biotin Kit (Miltenyi Biotec) per the manufacturer’s instructions. Three cohorts of naïve mice were each given a subcutaneous injection of 250,000 B16F10 tumor cells. Tumor sizes were recorded and animals were randomized so that each group possessed similar average tumor sizes and standard errors. Seven days post tumor inoculation, mice in the treatment groups received 1.5 million each of CD8+NK1.1+ cells or CD8+NK1.1− cells by i.p. adoptive transfer. Control mice received naïve CD8+ splenocytes. Tumor size was determined by external caliper measurement and calculated by means of the formula (length × width2) × π/6. The mice were euthanized once the tumor burden reached 1.5 cm in any linear direction.
Analysis of murine PBMC.
Twelve days post influenza infection, PBMC were collected from adoptively-transferred mice by retro-orbital bleed. Red blood cells were lysed by treatment with ammonium chloride (Sigma-Aldrich) according to the manufacturer’s instructions. The white blood cell pellet was washed once with PBS and re-suspended in Adoptive Immunotherapy (AIM)-V medium (Invitrogen) with 10% mouse serum. The cells were incubated for 20–30 min at 4° C with anti-CD3 (Tonbo Biosciences), CD4 (BioLegend), CD8 (BD Biosciences), and IFN-γ (BD Biosciences) antibodies at a dilution of 1:100 or 1:50 (IFN-γ) in FACS buffer. All flow cytometric analysis was performed using an LSR II flow cytometer (BD Biosciences). Data analysis was conducted on ≥10, 000 events with FlowJo version 10.0.00003 (Tree Star Inc.) for OS-X.
Human microarray analysis.
CD8+CD161+ and CD8+CD161− cells were magnetically separated from peripheral blood derived from six human donors. The isolated cells were not activated. Total RNA from the cells was isolated by RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Gene expression profiling of CD8+CD161+, and CD8+CD161− cells was performed with the Human Transcriptome Array 1.0 Chip (Affymetrix) by the Sequencing and Microarray Facility, at the University of Texas MD Anderson Cancer Center. A detailed description of sample requirements and data pre-analysis is available on the facility’s website (https://www.mdanderson.org/research/research-resources/core-facilities/sequencing-and-microarray-facility-smf/services-and-fees/microarray-services-overview.html). Data were analyzed and visualized with Transcriptome Analysis Console v3.0 (Affymetrix). The human microarray data have been deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE167279 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167279).
TCR repertoire analysis.
CD8+NK1.1− cells and CD8+NK1.1+ cells were isolated from mice after (a) infection with a sublethal dose of influenza or (b) vaccination against B16 melanoma as described above. Three-weeks post infection or vaccination, mice were euthanized, splenocytes harvested, CD8+ cells negatively selected using the CD8 T cell Isolation Kit (Miltenyi Biotec) and activated overnight with antigen (influenza virus or B16 determinants) loaded DC. The following day, the cells were stained with CD3-PE-Cy5 (Tonbo Biosciences), CD8-APC (BD Biosciences), CD69-PE (BD Biosciences), CD25-APC-Cy7 (BD Biosciences) and NK1.1-PE-Cy7 (BD Biosciences) for flow based cell sorting. Antigen-experienced, activated cells were selected by gating on the CD8+CD69+ CD25+ population and segregated by NK1.1 expression. Total genomic DNA was isolated from the sorted cells using the QiaAmp DNA micro kit (Qiagen) according to the manufacturer’s instructions. TCRβ sequencing was performed using the TCRβ CDR3 Adaptive Biotechnologies sequencing technology (immunoSEQTM Kit, Adaptive Biotechnologies). Data were analyzed with the ImmunoSEQ analyzer toolset and are publicly available for viewing at https://clients.adaptivebiotech.com/pub/konduri-2021-stm.
TCR Vβ spectratyping.
CD8+CD161+ cells isolated from the peripheral blood of a normal donor were spectratyped in a PCR-based assay by the Mayo Clinic. Resulting images are clusters of fluorescent peaks with single-base-pair separation and different fluorescent intensities corresponding to the number of fragments of that size represented in the donor’s original RNA for any given Vβ primer pair. The number of individual peaks was compared to a reference range established from over 140 healthy donors equally represented by both genders and across the pediatric and adult age spectrum. The reporting units were normalized among the patient population by using a diversity ratio for each TCR Vβ family. The diversity ratio for each Vβ family was determined by the number of peaks in that specific family relative to all peaks within the donor’s sample expressed as a percentage.
Blood donors, human cell lines, and culture conditions.
Normal donor apheresis products were obtained from the University of Texas MD Anderson Cancer Center according to an IRB-approved research protocol. Pancreatic cancer cell lines (ASPC1, CAPAN1, CAPAN2, CFPAC1, HPAF1, PANC1 and PL45) were a gracious gift from Drs. Cathy Yao and Sujita Sukumaran (Baylor College of Medicine). All pancreatic cancer cell lines were grown in Dulbecco’s modified Eagle’s medium (Invitrogen) with 20% fetal calf serum (FCS, GE Life Sciences) and 2mmol/l GlutaMAX (Invitrogen). Raji cells were cultured in RPMI- 1640, 10% FCS and 2mmol/l GlutaMAX (Invitrogen). All T cells were maintained in T cell media defined as 250 ml RPMI-1640 (Invitrogen), 200ml Click’s medium (EHAA, Irvine Scientific), 10% FCS, and 2mmol/l GlutaMAX (Invitrogen).
Retrovirus production.
The FRP5.CD28.CD3-zeta retroviral CAR construct used here has been previously described (49). This CAR construct was assembled, synthesized and sequence-verified as previously described (7, 24–26). A retroviral vector encoding the fusion protein eGFP-Firefly Luciferase (eGFP.FFluc) previously described (50) was used to generate firefly enhanced GFP-luciferase expressing tumor cells for in vivo experiments. To produce retroviral supernatant, HEK293T cells were co-transfected and supernatants harvested as previously described (24).
Flow cytometry analysis of the CAR transduced cells
Samples were analyzed on the Gallios flow cytometer (Beckman Coulter), Accuri C6 flow cytometer (BD Biosciences) or LSRII flow cytometer (BD Biosciences). For HER2 testing on pancreatic cancer lines, 500, 000 cells were stained with IgG1 PE isotype antibody (2μl, BD 340761) or HER2-PE (20 μl, BD 340552) according to the manufacturer’s instructions. For CAR T cell staining, cells were incubated for 30–60 minutes at 4° C with recombinant human HER2 Fc chimera protein (3 μl @ 0.1 μg/μl, R&D systems 1129-ER-050) and then stained with goat anti-human IgG1 Fc-PE (0.5 μl, eBioscience 12–4998-82), anti-human CD161-APC (5μl, BioLegend, 339912, Clone HP-3G10), CD3-Flourescein isothiocyanate (FITC) (20 μl, BD Biosciences 56180) or CD8-Phytoplankton (PerCP) (20 μl, BD Biosciences 347314) according to the manufacturer’s instructions. To ensure proper compensation for the panels, Versacomp antibody capture beads (Beckman Coulter) were stained with the same antibodies (51). Data analysis was conducted on ≥10, 000 events with FlowJo version 10.0.00003 (Tree Star Inc.) for iOS.
T cell proliferation assay.
Cells were incubated with CellTrace Violet at 1μM according to the manufacturer’s instructions (Invitrogen). 1 × 106 Violet dye labeled CAR+ T cells were added per well to 250,000 HER2+ pancreatic cancer cells and cultured together for 3 days. Cells were collected and stained for the CAR as described (26).
Cytokine Release assay.
50,000 tumor cell targets were plated for 24 hours and then cultured with 100,000 CAR+ cells or non-transduced (NT) control T cells. After 48 hours, conditioned culture media was collected. IFN-γ and IL-2 release were measured by ELISA (R&D systems) according to the manufacturer’s instructions.
Cytotoxicity assays.
For short-term cytotoxicity assays, T cells or NK cells were incubated with targets radioactively labeled with 51Chromium (Perkin Elmer) for 4 hours at 50:1–0.1:1 effector:target cell ratios. Cell lysis was determined by chromium released into the media, read using a Wizard2 gamma counter (Perkin Elmer) or a MicroBeta TriLux scintillation counter (Perkin Elmer). Long-term cytotoxicity was measured using the xCELLigence (ACEA bioscience) impedance-based assay. Briefly, gold electrodes at the bottom of the plate measure the impedance of the signal as a measure of tumor cell growth. Tumor cells were seeded at 10,000 cells per well of a 96 well plate and allowed to grow for 20 to 40 hours. T cells were then added at 2.5:1 tumor cell:T cell ratio and tumor growth or death as indicated by cell index was monitored over time.
Retroviral transduction of isolated PBMC, CD8+CD161− and CD8+CD161+ cells.
CD8+CD161+ cells, CD8+CD161− cells, and bulk PBMC isolated from normal donor apheresis products were stimulated with plate bound anti-CD3/CD28 (eBioscience) and recombinant Clec2d (R&D Systems) and expanded in RPMI-1640, 10% fetal bovine serum, and 2 mmol/L GlutaMAX (Invitrogen) in a cytokine cocktail comprised of 10 ng/ml IL-7, 5 ng/ml IL-15, and 30 ng/ml IL-21 (all from Peprotech), given that IL-21 was important for maintenance of CD161 expression and phenotype. In some experiments, a second bulk PBMC control was expanded using the standard expansion protocol of only 10 ng/ml IL-7 and 5 ng/ml IL-15 to demonstrate that the presence of IL-21 during expansion made no functional impact at all upon the function of CAR-transduced bulk PBMC. After 48 hours, T cell blasts were harvested and added to retronectin (Takara Bio USA) coated plates onto which retroviral supernatants had previously been affixed by centrifugation at 2,000 × G for 60 minutes. After an additional 48 hours, transduced blasts were moved to tissues culture plates and expanded an additional 10 to 15 days in the IL-7/15/21 cocktail.
Characterization of CAR transduced T cell cytotoxic potential.
To evaluate the stimulation conditions required by the different CAR T cell populations for cytotoxicity and cytokine release, 100,000 CAR+ T cells from each group were plated in a 96 well plate in triplicate against the CAR target HER2-Fc protein (R&D Systems), and irrelevant protein CD19-Fc (R&D Systems), each at a final concentration of 1 μg/ml. After 48 hours, cytokine production and cytotoxicity of the effector cells were evaluated by flow cytometry analysis for CD3, CD8, CD161, granzyme, CD25 (all from BioLegend), Perforin (BD Biosciences) and IFN-γ (Invitrogen), as described above. For repeated antigenic stimulation, CAR transduced cells were plated against HER2+ CFPAC target cells at a ratio of 1:4 (E:T) every 2 to 3 days. After four consecutive, repeated antigenic stimulations, additional analyses were performed with anti-TIM3 and anti-PD-1 (also from BioLegend).
In vivo PDAC tumor models.
In the disseminated peritoneal model, male SCID mice at 6–8 weeks of age were i.p. inoculated with 1 × 106 human CFPAC-1-luc2+ PDAC cells. In the orthotopic model, 5 × 105 human CFPAC-1-luc2+ PDAC cells were resuspended in 50 μl PBS or Matrigel (Corning) and injected directly into the surgically-exposed pancreas. One-week post tumor implantation, mice were randomized into cohorts of five and adoptively transferred with 2 × 106 unlabeled or CFSE (Invitrogen) labeled CAR+ T cells by i.p. injection. Tumor growth was monitored by IVIS imaging and survival differences calculated by Kaplan-Meier analysis.
Analysis of tumor infiltrating lymphocytes.
Sixteen days post tumor cell inoculation, mice were euthanized and tumors were harvested. Tumors were digested into single-cell suspensions by treatment with collagenase type 1 at 1mg/ml (Worthington) for 30 min on a shaking incubator at 37°C. Cells were then washed with RPMI-1640 media, filtered through a 70 μM filter, and resuspended in FACS buffer. Cells were incubated with anti-human CD3, CD8, CD161, Annexin V, granzyme B, and PD-1 antibodies (all from BioLegend) at a dilution of 1:100 dilution or 1:50 (granzyme B) in FACS buffer for 20–30 min at 4°C. Flow cytometry analysis was performed on LSR II Flow cytometer (BD Biosciences). Data analysis was conducted on ≥10, 000 events using FlowJo version 10.0.00003 (Tree Star Inc.) for OS-X.
Statistical Analysis.
Significance of differences, unless stated otherwise, was determined by one-way analysis of variance (ANOVA) using Tukey’s honestly significant difference (HSD), Fisher’s (protected) least significant difference (LSD), or Duncan’s multiple range (MRT) post hoc tests as statistically appropriate. Pairwise comparisons were performed by Student’s two-tailed t-test. Kaplan–Meier survival significance was determined by the log rank (Mantel-Cox) test. All data are displayed as the mean ± SEM unless stated otherwise, and all analyses were performed using Prism software version 8.3 for iOS (GraphPad Software) or Microsoft Excel for Mac version 16.30. Statistical significance was defined as p ≤ 0.05. The normality of all data was assessed by the Shapiro-Wilk test and Q-Q plot.
Supplementary Material
Supplementary Materials
Fig. S1. Flow cytometry gating, sorting, and collection strategy for splenocytes subjected to microarray analysis.
Fig. S2. CD8+NK1.1+ cells are identified as critical circulating memory cells in multiple mouse models of disease.
Fig. S3. Flow cytometry gating, sorting, and collection strategy for splenocytes subjected to T cell receptor sequencing analysis.
Fig. S4. TCR-Vβ spectratyping indicated CD3+CD8+CD161+ cells as polyclonal in nature.
Fig. S5. Cross-species comparative gene analysis reveals a conserved gene signature of 206 genes differentially regulated between the two populations.
Fig. S6. CD8+CD161+ T cells exhibit logs-fold higher expression of CD161 than CD8+CD161- T cells or unmanipulated PBMC post-selection.
Fig. S7. Human PDAC cell line CFPAC1 selected as target cell for in vitro and in vivo assays.
Fig. S8. CAR transduced CD8+CD161+ cells exhibit enhanced cytotoxicity and reduced exhaustion following stimulation with target proteins.
Fig. S9. CD8+CD161+ CAR T cells substantially improve survival outcomes in an in vivo model of PDAC.
Fig. S10. CD8+CD161+ CAR T cells substantially improve survival outcomes in an in vivo orthotopic model of PDAC.
Fig. S11. CD8+CD161+ CAR T cells do not possess natural cytotoxicity activity.
Table S1. Top 25 upregulated genes among mouse CD8+NK1.1+ cells.
Table S2. Top 25 downregulated genes among mouse CD8+NK1.1+ cells.
Table S3. TCR sharing among NK1.1+ and NK1.1- cells derived from the same animal.
Table S4. Top 25 upregulated genes among human CD8+CD161+ cells.
Table S5. Top 25 downregulated genes among human CD8+CD161+ cells.
Table S6. Top 25 congruently regulated genes in the interspecies common gene signature.
Acknowledgements
On January 2, 2021, author Jonathan M. Levitt, Ph.D. passed away suddenly and unexpectedly after a short illness. The remaining authors dedicate this work in his honor and to the memory of his unparalleled scientific career, to his widow Kelly, and to his sons Ryan and Christopher. Dad loved you all so much. This project was supported in part by the Cytometry and Cell Sorting Core at Baylor College of Medicine with the expert assistance of Joel M. Sederstrom.
Funding
This work was supported in part by Cancer Cures 4 Kids foundation (to WKD) and NIH R01 AI127387 (to WKD). The authors also received funding from the St. Baldrick’s-Stand Up to Cancer Dream Team Translational Research Grants 5SU2C-AACR-DT-11-13 (to NMA and MH) and SU2C-AACR-IRG-17-17 (to MH) as well as US NIH PHS grant U54-CA23256 (to NMA and MH). Stand Up to Cancer is a division of the Entertainment Industry Foundation. Research Grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. MH is also supported by Alex’s Lemonade Stand Foundation for Childhood Cancer. This project was also supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (AI036211, CA125123, and RR024574). The sequencing and microarray facility at the University of Texas MD Anderson Cancer Center is supported by core grant CA016672.
Footnotes
Competing Interests
Institutional policy requires WKD, MMH, and VK to declare their ownership stakes in Diakonos Research, Ltd. VK, WKD, MMH, SJ, MH, and NMA are inventors on patent application 62/931,670 “Method for Producing Cytotoxic Effector Memory T-Cells for CAR T-Cell Treatment of Cancer” held by Baylor College of Medicine. This patent covers the methods by which CD8+CD161+ cells may be transduced and expanded in an undifferentiated fashion. NMA, MH and TTB also hold additional intellectual property interests related to other aspects of CAR T cell therapy. WKD performs paid consulting work for Leerink Partners and the Gerson Lehrman Group. MMH is a paid consultant for Medterra CBD, Inc. at the time this work was performed. All other authors declare no competing interests.
Data Availability
Mouse microarray data were deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE167276 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167276). Human microarray data were deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE167279 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167279). Mouse TCRβ sequencing was performed by Adaptive Biotechnologies and is publicly available for viewing at https://clients.adaptivebiotech.com/pub/konduri-2021-stm.
References
- 1.Ansari D, Gustafsson A, Andersson R, Update on the management of pancreatic cancer: surgery is not enough. World J Gastroenterol 21, 3157–3165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL et al. , Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neelapu SS et al. , Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Stegen SJ, Hamieh M, Sadelain M, The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov 14, 499–509 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beatty GL et al. , Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 155, 29–32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmed N et al. , HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol 3, 1094–1101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmed N et al. , Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33, 1688–1696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seaman MS et al. , Subsets of memory cytotoxic T lymphocytes elicited by vaccination influence the efficiency of secondary expansion in vivo. J Virol 78, 206–215 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin E et al. , Stepwise development of MAIT cells in mouse and human. PLoS Biol 7, e54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turtle CJ, Swanson HM, Fujii N, Estey EH, Riddell SR, A distinct subset of self-renewing human memory CD8+ T cells survives cytotoxic chemotherapy. Immunity 31, 834–844 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Northfield JW et al. , CD161 expression on hepatitis C virus-specific CD8+ T cells suggests a distinct pathway of T cell differentiation. Hepatology 47, 396–406 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Takahashi T, Dejbakhsh-Jones S, Strober S, Expression of CD161 (NKR-P1A) defines subsets of human CD4 and CD8 T cells with different functional activities. J Immunol 176, 211–216 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Assarsson E et al. , CD8+ T cells rapidly acquire NK1.1 and NK cell-associated molecules upon stimulation in vitro and in vivo. J Immunol 165, 3673–3679 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Billerbeck E et al. , Analysis of CD161 expression on human CD8+ T cells defines a distinct functional subset with tissue-homing properties. Proc Natl Acad Sci U S A 107, 3006–3011 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fergusson JR, Fleming VM, Klenerman P, CD161-expressing human T cells. Front Immunol 2, 36 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fergusson JR et al. , CD161(int)CD8+ T cells: a novel population of highly functional, memory CD8+ T cells enriched within the gut. Mucosal Immunol 9, 401–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fergusson JR et al. , CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell Rep 9, 1075–1088 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Havenith SH et al. , Analysis of stem-cell-like properties of human CD161++IL-18Ralpha+ memory CD8+ T cells. Int Immunol 24, 625–636 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Poon K, Montamat-Sicotte D, Cumberbatch N, McMichael AJ, Callan MF, Expression of leukocyte immunoglobulin-like receptors and natural killer receptors on virus-specific CD8+ T cells during the evolution of Epstein-Barr virus-specific immune responses in vivo. Viral Immunol 18, 513–522 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Annibali V et al. , CD161(high)CD8+T cells bear pathogenetic potential in multiple sclerosis. Brain 134, 542–554 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Braud VM et al. , Expression of LLT1 and its receptor CD161 in lung cancer is associated with better clinical outcome. Oncoimmunology 7, e1423184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konduri V et al. , Chemo-immunotherapy mediates durable cure of orthotopic Kras(G12D)/p53(−/−) pancreatic ductal adenocarcinoma. Oncoimmunology 5, e1213933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart J et al. , Analysis of wntless (WLS) expression in gastric, ovarian, and breast cancers reveals a strong association with HER2 overexpression. Mod Pathol 28, 428–436 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Ahmed N et al. , HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res 16, 474–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grada Z et al. , TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids 2, e105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hegde M et al. , Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther 21, 2087–2101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hegde M et al. , Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat Commun 11, 3549 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majzner RG, Mackall CL, Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 25, 1341–1355 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Mardiana S, Solomon BJ, Darcy PK, Beavis PA, Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Sci Transl Med 11, (2019). [DOI] [PubMed] [Google Scholar]
- 30.Schmidts A, Maus MV, Making CAR T Cells a Solid Option for Solid Tumors. Front Immunol 9, 2593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guedan S, Ruella M, June CH, Emerging Cellular Therapies for Cancer. Annu Rev Immunol 37, 145–171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dominguez-Molina B et al. , Immune Correlates of Natural HIV Elite Control and Simultaneous HCV Clearance-Supercontrollers. Front Immunol 9, 2897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez Y et al. , CD161 Expression Defines a Th1/Th17 Polyfunctional Subset of Resident Memory T Lymphocytes in Bronchoalveolar Cells. PLoS One 10, e0123591 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SE et al. , CD161(+) T cells as predictive markers for acute graft-versus-host disease. Biol Blood Marrow Transplant 21, 421–428 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Nicol B et al. , An intermediate level of CD161 expression defines a novel activated, inflammatory, and pathogenic subset of CD8(+) T cells involved in multiple sclerosis. J Autoimmun 88, 61–74 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Steel KJA et al. , Synovial IL-17A+ CD8+ T cells display a polyfunctional, pro-inflammatory and tissue-resident memory phenotype and function in psoriatic arthritis. Arthritis Rheumatol, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lanier LL, Chang C, Phillips JH, Human NKR-P1A. A disulfide-linked homodimer of the C-type lectin superfamily expressed by a subset of NK and T lymphocytes. J Immunol 153, 2417–2428 (1994). [PubMed] [Google Scholar]
- 38.Goldfinch N et al. , Conservation of mucosal associated invariant T (MAIT) cells and the MR1 restriction element in ruminants, and abundance of MAIT cells in spleen. Vet Res 41, 62 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmed R et al. , A Public BCR Present in a Unique Dual-Receptor-Expressing Lymphocyte from Type 1 Diabetes Patients Encodes a Potent T Cell Autoantigen. Cell 177, 1583–1599 e1516 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Y et al. , Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123, 3750–3759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rochman Y, Spolski R, Leonard WJ, New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol 9, 480–490 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santegoets SJ et al. , IL-21 promotes the expansion of CD27+ CD28+ tumor infiltrating lymphocytes with high cytotoxic potential and low collateral expansion of regulatory T cells. J Transl Med 11, 37 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh H et al. , Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res 71, 3516–3527 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ngai H et al. , IL-21 Selectively Protects CD62L(+) NKT Cells and Enhances Their Effector Functions for Adoptive Immunotherapy. J Immunol 201, 2141–2153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zoon CK, Wan W, Graham L, Bear HD, Addition of interleukin-21 for expansion of T-cells for adoptive immunotherapy of murine melanoma. Int J Mol Sci 16, 8744–8760 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sommermeyer D et al. , Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turtle CJ et al. , CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 126, 2123–2138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang D et al. , AIMp1 Potentiates TH1 Polarization and Is Critical for Effective Antitumor and Antiviral Immunity. Front Immunol 8, 1801 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmed N et al. , Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res 67, 5957–5964 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Vera J et al. , T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood 108, 3890–3897 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Byrd T et al. , Polystyrene microspheres enable 10-color compensation for immunophenotyping of primary human leukocytes. Cytometry A 87, 1038–1046 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials
Fig. S1. Flow cytometry gating, sorting, and collection strategy for splenocytes subjected to microarray analysis.
Fig. S2. CD8+NK1.1+ cells are identified as critical circulating memory cells in multiple mouse models of disease.
Fig. S3. Flow cytometry gating, sorting, and collection strategy for splenocytes subjected to T cell receptor sequencing analysis.
Fig. S4. TCR-Vβ spectratyping indicated CD3+CD8+CD161+ cells as polyclonal in nature.
Fig. S5. Cross-species comparative gene analysis reveals a conserved gene signature of 206 genes differentially regulated between the two populations.
Fig. S6. CD8+CD161+ T cells exhibit logs-fold higher expression of CD161 than CD8+CD161- T cells or unmanipulated PBMC post-selection.
Fig. S7. Human PDAC cell line CFPAC1 selected as target cell for in vitro and in vivo assays.
Fig. S8. CAR transduced CD8+CD161+ cells exhibit enhanced cytotoxicity and reduced exhaustion following stimulation with target proteins.
Fig. S9. CD8+CD161+ CAR T cells substantially improve survival outcomes in an in vivo model of PDAC.
Fig. S10. CD8+CD161+ CAR T cells substantially improve survival outcomes in an in vivo orthotopic model of PDAC.
Fig. S11. CD8+CD161+ CAR T cells do not possess natural cytotoxicity activity.
Table S1. Top 25 upregulated genes among mouse CD8+NK1.1+ cells.
Table S2. Top 25 downregulated genes among mouse CD8+NK1.1+ cells.
Table S3. TCR sharing among NK1.1+ and NK1.1- cells derived from the same animal.
Table S4. Top 25 upregulated genes among human CD8+CD161+ cells.
Table S5. Top 25 downregulated genes among human CD8+CD161+ cells.
Table S6. Top 25 congruently regulated genes in the interspecies common gene signature.
Data Availability Statement
Mouse microarray data were deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE167276 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167276). Human microarray data were deposited in the NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE167279 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167279). Mouse TCRβ sequencing was performed by Adaptive Biotechnologies and is publicly available for viewing at https://clients.adaptivebiotech.com/pub/konduri-2021-stm.