

Abstract
Mapping the gene targets of chromatin-associated transcription regulators (TRs) is a major goal of genomics research. ChIP-seq of TRs and experiments that perturb a TR and measure the differential abundance of gene transcripts are a primary means by which direct relationships are tested on a genomic scale. It has been reported that there is a poor overlap in the evidence across gene regulation strategies, emphasizing the need for integrating results from multiple experiments. Although research consortia interested in gene regulation have produced a valuable trove of high-quality data, there is an even greater volume of TR-specific data throughout the literature. In this study, we show a workflow for the identification, uniform processing, and aggregation of ChIP-seq and TR perturbation experiments for the ultimate purpose of ranking human and mouse TR–target interactions. Focusing on an initial set of eight regulators (ASCL1, HES1, MECP2, MEF2C, NEUROD1, PAX6, RUNX1, and TCF4), we identified 497 experiments suitable for analysis. We used this corpus to examine data concordance, to identify systematic patterns of the two data types, and to identify putative orthologous interactions between human and mouse. We build upon commonly used strategies to forward a procedure for aggregating and combining these two genomic methodologies, assessing these rankings against independent literature-curated evidence. Beyond a framework extensible to other TRs, our work also provides empirically ranked TR–target listings, as well as transparent experiment-level gene summaries for community use.
Understanding the regulatory interactions underlying gene expression programs is of considerable interest to contemporary experimental and computational biology. A fundamental objective is to map the relationships between transcription regulators (TRs) and the sets of gene targets they functionally influence. TRs, which include DNA sequence-specific transcription factors and chromatin proteins like MECP2 that bind methylated DNA, are a large class of proteins generalized by their ability to promote or repress gene activity (Lambert et al. 2018; Serebreni and Stark 2021). Learning the regulatory range of TRs is essential to understanding development, the functional identities of cell types, and the origins of diseases (Arendt et al. 2016, Lambert et al. 2018). However, experimentally establishing TR–target interactions is laborious and expensive, especially for precious tissues like the human brain. Additionally, efforts to predictively model these interactions as networks remain a challenging task complicated by a lack of known interactions (Marbach et al. 2012; Rothenberg 2019; Nord and West 2020). Identifying high-confidence sets of experimentally supported regulatory relationships is beneficial to inform biology as well as predictive method optimization.
We recently curated the literature for low-throughput biochemical assays showing TR–target regulation (Chu et al. 2021), focusing on neurologically relevant TRs in mouse and human to expand upon existing resources like TRRUST (Han et al. 2018). These biochemical assays can provide strong evidence for direct regulation, but their coverage is limited relative to the potential number of TR–target interactions. Accordingly, there are currently multiple genome-scale assays that provide regulatory information, but many rely on inference to determine TR–target relationships (Hawe et al. 2019). The most prominent means for high-throughput experimental assessment of direct interactions are to sequence purified DNA bound by an immunologically selected TR (ChIP-seq) or to measure changes in gene transcript levels upon perturbation of the TR (differential expression [DE]).
ChIP-seq and TR perturbation each have biological and technical considerations that complicate the task of assigning direct targets (Cusanovich et al. 2014; Kang et al. 2020). For example, perturbation experiments may prioritize indirect targets that are regulated by processes downstream from the perturbed TR. Consequently, applying both methods to a TR and intersecting the resulting gene lists is a common approach to enrich for regulatory interactions. This can be as simple as binarizing the significantly DE genes affiliated with a proximal ChIP-seq binding event or using moderately more advanced strategies like the BETA algorithm, which combines the two gene lists into a single ordered ranking (Wang et al. 2013). However, efforts to evaluate the intersection of individual experimental pairs between these two genomic strategies in yeast (Hu et al. 2007; Gitter et al. 2009; Kang et al. 2020) and human (Cusanovich et al. 2014) have revealed that their evidence rarely converges.
Despite these complexities, ChIP-seq and perturbation experiments remain important strategies for biological discovery that continue to proliferate (Luo et al. 2020). These data sets (or their intersection) are also often used as the “gold standard” for evaluating computational strategies that predict TR–target interactions (Marbach et al. 2016; Miraldi et al. 2019; Pearl et al. 2019; Qin et al. 2020). In particular, two recent resources for human TRs have shown the importance of aggregating distinct lines of evidence for the purpose of gene target assignment (Garcia-Alonso et al. 2019; Keenan et al. 2019). However, both focused solely on human TRs and held out the perturbation data to be used as a benchmark for their gene target rankings. Given the relative abundance of mouse gene regulation data, there is a clear need for the parallel organization and analysis of human and mouse TR–target interactions.
Here we outline a framework to identify and rank experimentally derived TR–target relationships by aggregating ChIP-seq and perturbation data sets from both mouse and human. We describe the degree of similarity across experiments and note characteristics of each data type that can complicate target assignment. We provide a framework for the aggregation and integration of these data sets, evaluating these empirically derived rankings against independent experimental evidence. This study also shows how the collected information can potentiate further work, such as aligning bound regions to orthogonal genomic annotations, or identifying TR–target interactions with cross-species evidence.
Results
For the current study, we focused on an initial set of eight TRs to establish methodology and to calibrate expectations: ASCL1, HES1, MECP2, MEF2C, NEUROD1, PAX6, RUNX1, and TCF4 (not to be confused with TCF7L2, which is sometimes referred to as TCF4 in the literature). The selection of TRs was largely guided by our prior work on the curation of low-throughout experiments of neurologically relevant TRs (Chu et al. 2021). We emphasize that the data used in the current study were not required to be conducted in a brain-relevant system: Our goal was to identify all ChIP-seq and high-throughput TR perturbation expression data sets for these regulators. Experiments excluded from analysis are noted in Supplemental Table S3. Our workflow is outlined in Figure 1. In the subsequent sections, we give an overview of each genomic strategy before describing the aggregation and intersection approaches and their evaluation, leading to a consolidated ranking of candidate regulatory targets.
Figure 1.
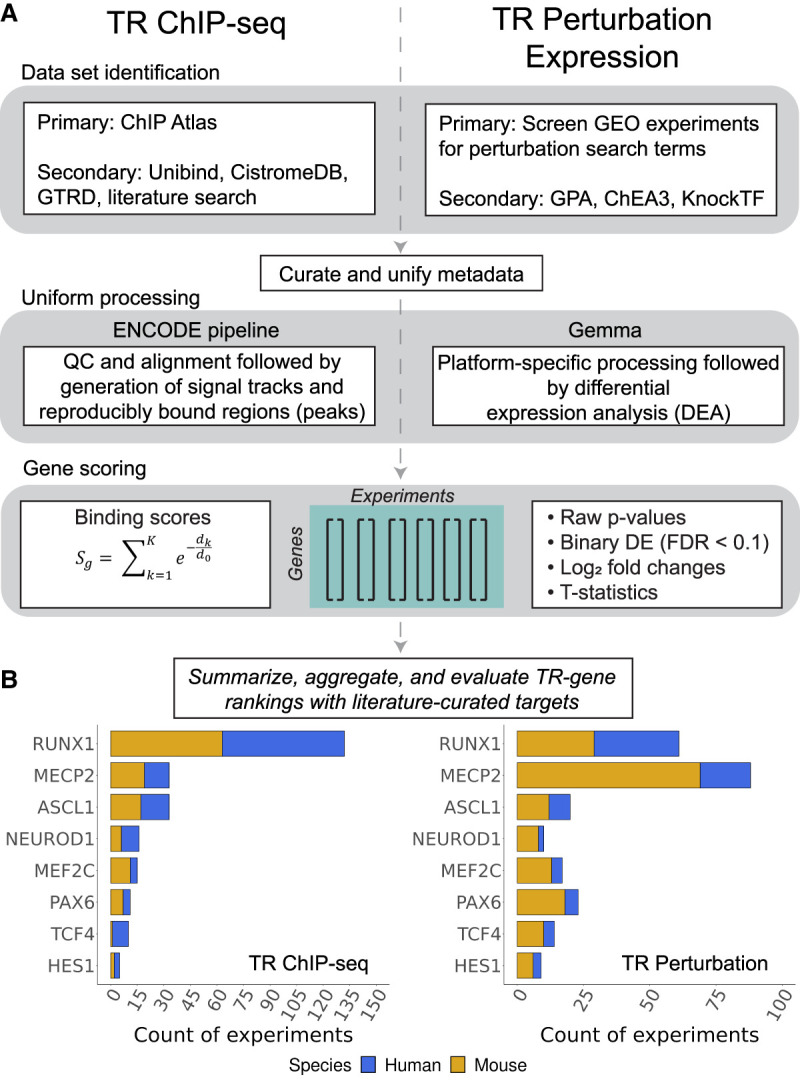
Study overview. (A) Workflow for TR ChIP-seq (left) and TR perturbation (right) data. (B) Counts of TR experiments considered for analysis.
Identification, summarization, and gene-scoring ChIP-seq data sets
ChIP-seq data were predominantly identified across existing resources (particularly ChIP Atlas) (Zou et al. 2022) and supplemented with literature curation (Methods) (Fig. 1A). All samples were curated into experimental units (based upon sample replication and presence of input controls) and uniformly processed using the ENCODE pipeline (Landt et al. 2012). A total of 255 experiments from 363 samples and 244 input controls was kept for analysis. Although there was approximately equal representation of experiments across species, this equality does not extend to individual regulators (Fig. 1B; Supplemental Table S1).
Consistent with a previous effort to identify literature-sourced ChIP-seq data (Marinov et al. 2014), we found appreciable heterogeneity in the structure of experimental designs. Factors like the presence of an input control or sequencing depth can introduce technical variation to the count of inferred bound regions (peaks) in an experiment (Supplemental Fig. S1; Landt et al. 2012). This is an unavoidable reality when aggregating literature-sourced data, motivating our use of the stringent approach for peak calling promoted by ENCODE. Peaks were assigned to genes using a continuous scoring metric (from here referred to as the binding score; see Methods), and the ChIP-seq data were thus represented and analyzed as gene-by-experiment matrices of binding scores.
ChIP-seq experiments targeting the same TR show moderately elevated similarity
As we aimed to aggregate TR data generated across distinct contexts, we first wanted to explore the similarity of binding profiles between the same TRs (intra-TR) and different TRs (inter-TR) across experiments. We examined both the Pearson's correlations (r) of binding scores, which provided a measure of similarity across all protein-coding genes, and multiple measures of overlap for the top 500 scoring genes, inspired by Sikora-Wohlfeld et al. (2013) and Keenan et al. (2019).
The results were consistent regardless of the approach or number of top genes selected, showing that, collectively, intra-TR experiments were moderately more similar than inter-TR pairs (Fig. 2A–C; Supplemental Figs. S2, S18, S20). Specifically, on average, intra-TR data sets shared 57/500 top genes, whereas inter-TR pairs shared 22/500. As expected, the most similar experiments were in comparable biological contexts from the same NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) submission (performed by the same laboratory), followed by experiments conducted in comparable contexts by distinct research groups. For example, the highest global correlation (r = 0.87, 307/500 top-scoring genes) was between two different ASCL1 constructs in the SH-SY5Y neuroblastoma cell line (GEO; GSE153823) (Ali et al. 2020), whereas the most correlated experiments from distinct groups both assayed mouse ASCL1 in the developing neural tube (r = 0.66; 251/500 top-scoring genes) (for GEO GSE43159, see Sun et al. 2013; for GEO GSE55840, see Borromeo et al. 2014).
Figure 2.

ChIP-seq experiment similarity and specifically bound genes. (A–C) Distribution of binding score correlations between ChIP-seq experiments targeting the same TR (intra-TR) versus different TRs (inter-TRs). (D) Log2 fold changes of binding scores for the top eight ranked genes (by P-value) for each TR in human experiments using a mixed effect linear model. As scoring was distance based, linearly proximal genes had similar ranks, and so for plotting, only the most significant genes (PCDHGA7 for MECP2 and KRTAP9-3 for ASCL1) are shown. (E) Distributions of binding scores for the most significant gene for each TR from the same model as in D.
Elevated intra-TR correlation was not universal to all comparisons (Supplemental Fig. S2), which in some instances may be attributable to cell type patterns. For example, the three human HES1 experiments had intra-correlations ranging from r = 0.16–0.19. A HES1 experiment from this trio conducted in the K562 cell line had inter-correlations ranging from 0.29–0.36 with five other K562 experiments targeting NEUROD1 or RUNX1. However, all intra-HES1 pairs had more genes in their top 500 overlap (range, 68–92) than any inter-HES1 comparison (range, two to 63). We also found instances in which there was less intra-TR similarity than might be expected (Supplemental Fig. S22A). Two ENCODE K562 RUNX1 experiments had an r = 0.32 (69/500 top scoring), despite targeting the same TR in the same cell type. These experiments used different RUNX1 antibodies and sequence library strategies, serving as a reminder of the considerable technical variation of the ChIP-seq methodology.
Using the same approach, we compared the similarity of mouse and human ChIP-seq experiments, based on orthology of TRs and of targets. For targets, we focused on a set of 16,686 high-confidence one-to-one mouse-human orthologs (Methods). The distribution of binding score correlations for the same TR but different species (intra-TR & cross-species) was shifted higher relative to both inter-TR & cross-species and inter-TR & within-species comparisons (collapsed as one group for simplicity in Fig. 2C; Supplemental Fig. S2C). The top of these intra-TR & cross-species rankings were dominated by RUNX1 experiments: Of the 2323 intra-TR & cross-species comparisons that had greater than average common top-scoring genes (more than 37/500), 2097 (90%) were associated with RUNX1. This is attributable to the relatively high abundance of data for this TR and the frequency in which RUNX1 experiments were conducted in blood contexts in both species, consistent with conserved regulatory profiles leading to cellular identities (Arendt et al. 2016). Although slightly attenuated, the shift in correlation distributions was held when excluding RUNX1 experiments (Supplemental Fig. S3).
Collectively, these observations support that consistent TR binding profiles may be identified across studies, albeit with an expected loss of highly context-dependent signals. Despite this potential for false negatives, this was nevertheless promising for our goal of aggregating data to uncover consistent evidence in support of specific TR–target relationships.
A mixed effect linear modeling framework identifies genes with TR-enriched binding
Finding evidence for intra-TR binding similarities, we looked to identify and rank the bound genes for each TR. Although we ultimately used the intra-TR mean binding score in our final aggregated rankings (Discussion), we found that certain regions had a propensity to be bound generically, consistent with prior observations (Discussion) (Supplemental Fig. S5). For example, the constitutively expressed GPAA1 was found to have a peak within 25 kbp of its transcription start site (TSS) in 104/129 (81%) of the human experiments, distributed across all TRs. We therefore developed a strategy to identify candidate TR targets that could address the concern of binding specificity (Methods). Briefly, we used a mixed effect linear modeling framework with TR identity as the main effect, accounting for high-level experimental factors and the heightened correlation among experiments generated by the same group. Although not without caveats (Discussion), this approach was designed to seek evidence for selective TR binding patterns despite the heterogeneity of contexts typically found in each comparator group.
Figure 2D shows the results of this approach, plotting the differential binding scores of the eight most significant genes for each human TR, whereas Figure 2E shows the boxplots of the binding scores of each TR's most significant gene (mouse in Supplemental Fig. S4). This yielded an average of 618 candidate target genes per TR (log2 fold change [FC] > 0 & false-discovery rate [FDR] < 0.05), ranging from three (mouse HES1) to 1402 (human RUNX1). This model revealed a number of previously characterized TR–target interactions. Well-described ASCL1 targets and Notch pathway effectors DLL1, DLL3, DLL4, and HES6 (Castro et al. 2006, 2011; Nelson et al. 2009) had elevated binding in both the human and mouse ASCL1 comparisons, whereas the HES1 target ATOH1 (Kazanjian and Shroyer 2011) was the most significant gene in the human HES1 comparison. Taken together, this analysis supported that aggregated ChIP-seq data can prioritize independently described regulatory interactions. We more formally evaluate this approach in a later section.
Identifying frequently bound loci relative to regulatory element annotations
The framework described thus far is gene-centric, relying on a scoring metric that sums the contribution of binding events around a TSS. Consequently, a gene may have a high binding score for a given TR, even if the individual binding events are dispersed around the TSS across experiments. We therefore identified the regions most commonly bound by a TR, providing discrete coordinates for future investigation (Supplemental Data S5).
To show an application of these bound loci, we examined their overlap with candidate cis-regulatory elements (cCREs) (Moore et al. 2020). These regions, which encompass predicted promoter- and enhancer-like regions, were previously defined through the integration of multiple genomic features across diverse cellular contexts. Genomic annotations like the cCREs aim to characterize the biological function of the underlying DNA sequences. To provide context before focusing on frequently bound loci, we first examined the proportion of each ChIP-seq experiment's peaks that overlapped each class of cCRE (Fig. 3).
Figure 3.

Overlap of ChIP-seq peaks with annotated regulatory elements. (A, left) The proportion of peaks for each human ChIP-seq experiment, grouped by TR, that overlapped with a candidate cis-regulatory element (cCRE). (PLS) Promoter-like sequence, (ELS) enhancer-like sequence, (p) TSS proximal, (d) TSS distal. (Right) The proportional breakdown of all cCRE groups. (B) Same as in A, except for mouse ChIP-seq experiments.
With the exception of MECP2, we found that 78% of the peaks in a typical human ChIP-seq experiment overlap with a cCRE (Fig. 3A); in mouse, 60% (Fig. 3B). We note that mouse cCRE groups were defined with less input data than those of human and are expected to have fewer discovered elements and thus less overlap. And although there was variation across TF experiments in how peaks were distributed across the cCRE groups (Supplemental Fig. S17), they generally followed the global cCRE group proportions, with most binding to regions characterized as enhancer-like sequences. MECP2 was the exception: The overlap of peaks dropped to an average of 19% for human and 3% for mouse, even though MECP2 experiments had a comparable number of peaks to the other experiments (Supplemental Fig. S1C). This lack of overlap may reflect MECP2's differential mode of binding relative to the TFs (Shah and Bird 2017). We also found that filtering peaks for cCRE overlap did not change the similarity structure between ChIP-seq experiments (Supplemental Fig. S21).
As discussed earlier, the concordance of binding across experiments for a TR was limited, but by focusing on areas of agreement, we hope to uncover meaningful biology. In the locus-specific analysis, there are many cases of highly reproducible sites. For example, for human ASCL1, we found 29 regions bound across all 16 experiments and 405 regions bound in at least 14, many of which were infrequently bound by the other TRs (Supplemental Fig. S6). A notable example is a ∼330-bp sequence in an intronic region of SHB, annotated as an enhancer-like cCRE, which had a peak called in all ASCL1 experiments but only twice in non-ASCL1 experiments (two of 113).
We take such patterns of reproducibility and specificity as an indication of biological relevance. However, not all binding, even if reproducible, is expected to result in significant regulatory activity (Wasserman and Sandelin 2004; Teytelman et al. 2013; Cusanovich et al. 2014), hence the importance of considering orthogonal data (Garcia-Alonso et al. 2019). This brings us to the introduction of the TR perturbation experiments, which provides evidence of TR regulation at the RNA level. For example, the candidate ASCL1 target SHB we identified in the above binding analysis is differentially expressed (DE) in seven of eight human ASCL1 perturbation experiments, raising our confidence in its relevance. In the next sections, we describe the systematic analysis of TR perturbation experiments for integration with the ChIP data.
Acquisition and summarization of TR perturbation data sets
TR perturbation data were predominantly identified using a screen of GEO experiments and supplemented with existing resources (Methods) (Fig. 1A). All were processed in the Gemma database (Lim et al. 2021), and a total of 242 experiments were considered for downstream analysis (Supplemental Table S2). Sequencing platforms were slightly more represented than microarrays (Supplemental Fig. S7F). Unlike the ChIP-seq collection, in which there were similar amounts of mouse and human data, the TR perturbation corpus was heavily skewed toward the mouse, with just over twice as many experiments in the mouse than human (Fig. 1C), although the breakdown by TR was broadly similar. Gene knockouts were the most common perturbation strategy, making up nearly half of all experiments (Supplemental Fig. S7E). Overexpression and knockdowns followed with near equal representation. The remainder we classified as “mutants,” following the original investigators’ descriptions. These are somewhat distinct from knockouts, typically (but not always) involving loss of function-inducing point mutations rather than larger deletions.
TR perturbation experiments show modest DE effect sizes
We first explored the properties of the collected perturbation data before any aggregation. Consistent with the findings of Cusanovich et al. (2014), we found that the perturbation effect sizes tended to be modest. Given the distribution of FC across all experiments (Fig. 4A), we did not apply FC thresholding and classified genes as differentially expressed (DEGs) at a relaxed FDR < 0.1. This framework resulted in a median count of 216 DEGs per experiment (adding a constraint of a minimum absolute FC of one would result in a median of 26).
Figure 4.

Overview of TR perturbation experiments. (A) Distribution of gene log2 fold changes (FCs) across the 242 mouse and human perturbation experiments. FC was clipped at [−3, 3] for plotting. (B) Count (log10 scale) of differentially expressed genes grouped by TR; color denotes perturbation strategy, and shape denotes species. (C) Heatmap of correlation values of gene FC between experiments, grouped by TR. Note that only orthologous genes were calculated here to allow plotting of both species; the relatively minimal intra-TR correlation holds when considering mouse and human separately.
Also in line with the findings of Cusanovich et al. (2014), we found that the number of genes affected by a TR perturbation was poorly predicted by the FC magnitude of the perturbed TR (Supplemental Fig. S9B,C). For example, there was an appreciable number of experiments that had no DEGs despite substantial changes in the TR's expression level. Otherwise, there was a spread in DEG counts for each TR, with NEUROD1 having the most on average and HES1 the least (Fig. 4B). Although biological characteristics may explain these extremes (e.g., pioneering activity of NEUROD1 vs. a more repressive role for HES1), the variety of designs, contexts, and sample sizes complicate these comparisons. Still, we noted a difference in the count of DEGs associated with perturbation type, with knockdowns having the highest median (1975) and knockouts the lowest (73) (Supplemental Fig. S10A,B).
Despite select examples, intra-TR perturbation experiments show weak similarity
As with the ChIP-seq collection, we explored the similarity of intra-TR versus inter-TR perturbation experiments. Because there was unequal gene coverage across experiments owing to platform differences (Supplemental Fig. S7), we calculated Pearson's correlations of log2 FCs (as well as their absolute values) for the genes commonly measured between pairs of experiments. For the top 500 overlap comparison, we separately considered up- and down-regulated genes, as well as sorted by P-values from the DE analysis.
Consistent with expectations, the most similar experiments were intra-TR comparisons from the same research group, led by a pair of ASCL1 overexpression experiments (r = 0.95, 410/500 by up-regulated) (for GEO GSE153823, see Ali et al. 2020). As for ChIP-seq, experiments from different groups but comparable contexts showed elevated similarity, such as two experiments overexpressing mouse Ascl1 in astrocyte-to-neuron conversions (r = 0.60, 222/500 overlap of up-regulated genes) (for GEO GSE174238, see Kempf et al. 2021; for GEO GSE132674, see Rao et al. 2021). We also found examples of anticorrelative patterns that aligned with the opposing roles of the perturbations, typically involving a MECP2 overexpression versus a MECP2 loss of function (e.g., r = −0.66, 49/500 by P-value) (for GEO GSE126640, see Cholewa-Waclaw et al. 2019).
However, intra-TR similarities as a group were only marginally different from inter-TR comparisons (Fig. 4C; Supplemental Figs. S8, S18). Focusing on the top 500 overlaps by P-values, an average human intra-TR pair shared 32/500 genes to the 28/500 of inter-TR pairs; mouse intra-TR pairs had 29/500 compared with 21/500 in inter-TR pairs. These trends extended to the orthologous gene comparison between species. The strongest correlation was between NEUROD1 overexpression studies in the mouse and human that both looked to generate neuronal populations (r = 0.32, 135/500 by up-regulated) (for GEO GSE104435 see Matsuda et al. 2019; for GEO GSE149599, see Pomeshchik et al. 2020), whereas the strongest negative correlation was between a human MECP2 overexpression in a neuronal cell line and a mouse Mecp2 knockout in cortical neurons (r = −0.24, 39/500 by P-value) (for GEO GSE126640, see Cholewa-Waclaw et al. 2019; for GEO GSE124521, see Keidar et al. 2019). However, a typical intra-TR & cross-species comparison shared only three more genes in a top 500 comparison relative to the average inter-TR pair.
Aggregating perturbation data sets reveals repeatedly DE genes
We next used an aggregation approach to identify consistent patterns across perturbation experiments for each of the TRs. This is in keeping with our overall philosophy of taking advantage of commonalities while being cognizant of the issues noted above. To rank genes for each TR, we used a simple tally of the count of times a gene was DE across intra-TR experiments (Count DE), breaking ties with the average absolute FC.
Despite the weak experiment similarities, we identified many genes that were frequently DE for a given TR's set of perturbation experiments (Fig. 5C). The exception was HES1, which had fewer data overall and had few DEGs in both species (Fig. 4B). Mouse Mecp2 had the most perturbation data and, correspondingly, had the genes with the highest Count DE, led by Irak1 with 23/69 in mouse Mecp2 experiments and six of 19 in human. This NF-κB pathway gene has previously been associated with Mecp2 (Supplemental Material, “Overview of TR-targets”; Urdinguio et al. 2008; Kishi et al. 2016). However, we were unable to find literature support for other highly ranked Mecp2 candidates, such as the brain-enriched estrogen-related receptor gamma Esrrg (21/69 mouse, eight of 19 human) which also had strong MECP2 binding scores in our ChIP-seq analysis, suggesting that further investigation into this interaction is warranted.
Figure 5.

Demonstration of genes with recurrent differential expression (DE). (A) The x-axis is the count of times that a gene was DE across human experiments in the current study (n = 77), and the y-axis is the DE prior rank, where 1.0 represents the gene that was most likely found as DE across a large and diverse corpus of expression experiments. (B) Same as in A but for mouse experiments (n = 165). (C) Histograms of the count of times genes were DE across each group of TR experiments (top row, human; bottom row, mouse). (D) Demonstration of the top 15 genes by DE evidence for human ASCL1. FCs are clipped at [−2.5, 2.5] for plotting. The DE prior was binarized so that values above 0.9 (black squares) represent genes that are commonly DE regardless of design. (E) Same as in D but for mouse ASCL1.
We also observed many orthologous genes with recurring DE in both species, such as the neurogenic growth factor BMP7 for ASCL1 (six of eight human, six of 12 mouse). Human NEUROD1 had the fewest perturbation experiments (n = 2), yet we identified seven genes that were DE in both human studies as well as in seven of eight for mouse Neurod1 (nine of 10 of all NEUROD1 experiments): PTPRK, UGCG, TRIM9, SFT2D2, SLC35F1, ADGRL3, and SOGA1. SLC35F1 was recently identified as an understudied neurodevelopmental gene implicated in epileptic encephalopathies (Di Fede et al. 2021); our work thus connects NEUROD1 to the regulation of this presumed synaptic plasticity gene. On the other hand, there were also many examples of orthologous genes that were repeatedly measured as DE in one species but rarely in the other, such as MECP2 candidate targets SLC6A7 (15/69 mouse, zero of 19 human) and the proneural NRG2 (one of 69 mouse, eight of 19 human), but ruling these as species-specific targets requires further consideration beyond the scope of this study.
Additional considerations for perturbation data aggregation
We highlight two final considerations for aggregating the perturbation data. First, akin to the “generic” signals observed in the ChIP-seq collection, we observed genes that were frequently DE across TR studies (Supplemental Figs. S10B,C, S11). We compared these counts with a previous metric that ranks genes by their predictability of being DE (DE prior) (Crow et al. 2019), finding a weak but significant trend in both human (r = 0.09, P-value < 2.2 × 10−16) and mouse (r = 0.18, P-value < 2.2 × 10−16) (Fig. 5A,B). This trend could reflect biological underpinnings of the observations of Crow et al. (2019), but the weakness of the signal precluded making strong conclusions. Second, we observed genes could have discordant directions of change (up- vs. down-expressed) across the same type of perturbation for a given TR. We quantified this by adapting the metric of Purity (Supplemental Material), scoring the consistency of a gene's FC direction across loss- and gain-of-function experiments (Supplemental Fig. S12). As we do not assume that frequently DE genes are not true targets and that a given TR can be activating or repressive in different contexts (Lambert et al. 2018), we elected not to incorporate Purity or the DE prior into our final rankings (Fig. 5D,E). Still, we believe these metrics provide additional context, such as for identifying candidate interactions that are predominantly activating or repressive.
Combining aggregated ChIP-seq and perturbation evidence to prioritize gene targets
Having summarized each line of genomic evidence, we turned to our original goal of ranking gene targets by the combined evidence of the two strategies. We first simply examined the degree of overlap in their gene lists. Given the generally weak perturbation intra-TR experiment similarity, it was perhaps unsurprising to see the same trend when comparing across ChIP-seq and perturbation experiment pairs (Supplemental Figs. S13, S19). A typical intra-TR ChIP-seq and perturbation experiment pair shared 20/500 top-ranked genes versus 17/500 for inter-TR pairs.
However, we observed, much like the perturbation comparisons, many genes with frequent intra-TR overlaps between the two methods despite the overall weak group overlap. To reach a consolidated list for each TR, we extended a popular rank product approach previously used on individual ChIP-seq and perturbation experiment pairs (Methods) (Breitling et al. 2004; Tang et al. 2011; Wang et al. 2013). Rank products have been used extensively in genomics and other fields, representing a simplistic yet robust meta-analytic summarization of noisy data (Koziol 2010). Here, we used the aggregated perturbation and ChIP-seq gene lists as inputs, rather than individual experiments, to avoid biases for TRs with imbalanced data set counts between the two methods. Thus, for each TR, we obtained aggregated gene orderings for both genomic lines of evidence and a final combined ranking (integrated).
To evaluate the integrated rankings, we wished to use an orthogonal line of experimental evidence for comparison, similar to a recent benchmark of human TF–target interactions that used perturbation data as the gold standard (Garcia-Alonso et al. 2019). In contrast to Garcia-Alonso et al. (2019), we cannot use perturbation data as an evaluation set as we used it to generate the rankings. We reasoned that the information from TR perturbation data was more important to use for gene prioritization rather than to hold it out for assessment, particularly given that low overlap was already an expectation given our observations and prior work (Garcia-Alonso et al. 2019; Kang et al. 2020).
We thus turned to resources that curated low-throughput interactions (Supplemental Material, “Low-throughput curated target resources”; Supplemental Table S6; Han et al. 2018; Chu et al. 2021). These yielded 483 unique targets for the eight TRs, ranging from 11 for TCF4 to 156 for PAX6 (median, 51) (Supplemental Fig. S16A). This collection is not exhaustive, lacks annotation of negatives (nontarget genes), is diverse in contexts, and contains evidence from single-locus perturbation and binding experiments, so we do not consider it a true gold standard. However, we reasoned that it would still provide at least a sense of whether the integrated genomics data could help prioritize known (and, by extension, novel) interactions, relative to the performance of individual data sets or a single data modality.
First, we tested the difference in the aggregated gene rankings by presence in the curated set (Supplemental Figs. S14, S15). All mouse rankings showed evidence for prioritizing curated targets (Wilcoxon test P-value < 0.05), save for the TCF4 binding, integrated, and Neurod1 perturbation aggregations. This potentially may be explained by the size of the evaluation set, as these two TRs had the fewest curated targets (Supplemental Fig. S16). Some curated NEUROD1 targets like Insm1 (Breslin et al. 2003) were not highly ranked (perturbation rank, 1082nd) despite being DE in four of eight mouse NEUROD1 experiments. Thus, it is possible that the genes ranked higher than Insm1 by DE evidence include numerous real but unidentified targets. For example, Nova2 is a neurodevelopmental splicing regulator (Mattioli et al. 2020) that was DE six of eight times and had enriched mouse NEUROD1 binding, suggesting this may be a true interaction lacking low-throughput evidence. Although the human results were more mixed (none of the three MEF2C differential tests were significant, unlike mouse MEF2C), the majority of human comparisons still provided evidence that the rankings prioritized curated targets. In sum, this analysis supported that our aggregation strategy was able to assign heightened importance to genes with independent experimental evidence.
Aggregate rankings typically outperform null expectations and single experiments
The previous analysis considered the difference in median ranks by curation status. To more directly assess the ability of the aggregation strategies to preferentially rank known interactions, we performed a precision-recall analysis, calculating the area under the precision-recall curve (AUPRC) to summarize performance (Fig. 6A; Supplemental Fig. S16; Marbach et al. 2012). We first note the overall low performance of each of these rankings (e.g., ASCL1 in Fig. 6B,G), an unsurprising result given factors like the incomplete nature of the evaluation set (Discussion). The integrated rankings achieved a higher AUPRC than the single method aggregations for human ASCL1, mouse and human RUNX1, mouse Pax6, and human TCF4. However, the individual data type aggregations also sometimes outperformed the integrated ranking.
Figure 6.

Overview of literature curation evaluation framework. (A) Precision and recall were calculated at every step (k) of the aggregated rankings for presence in the curated target resource. (TP) True positives called at step k, (P) all curated targets for the current TR. (B) PR curve and the associated areas under the curves (AUPRC) for human ASCL1. (C) The distribution of AUPRCs when sampling random targets from the curated resource and using the integrated ASCL1 ranking to calculate the AUPRC. Bars refer to the observed performance when using the curated ASCL1 targets. (D) Distribution of AUPRCs when using the individual ASCL1 experiments or their rank product pairings to order genes. (E) Proportion of samples in C whose AUPRC exceeded the observed values. (F) The percentile of the aggregate AUPRCs relative to the distribution of all individual comparisons in D. (G) Example of ASCL1 targets with genomics and low-throughput evidence.
To better contextualize these relative differences in performance, we conducted two further comparisons using the precision-recall framework. First, we created a null distribution of AUPRCs for each TR, iteratively sampling curated targets from the entire literature resource and calculating the AUPRC with the aggregated rankings and sampled targets. This analysis revealed that, despite the low overall performances, almost every TR had an aggregate ranking that exceeded the null. Figure 6, C and E, shows that no null target set outperformed the human ASCL1 integrated rankings, a trend also seen for rankings like mouse PAX6 and RUNX1. Further, the human MEF2C perturbation and integrated rankings outperformed the null expectation, despite the lack of MEF2C differential ranking by curation status (Supplemental Figs. S14, S16). Thus, although aggregation for lesser-represented TRs may assign curated targets a similar rank to uncurated targets on average, the top of the aggregated ranking can still be enriched for curated targets relative to a null expectation.
Finally, we compared the aggregated AUPRCs to those obtained when calculated from individual ChIP-seq and perturbation experiments, or their individual rank–product pairings. This provided a direct comparison of the performance of the aggregated rankings to what is obtained from the individual experiments constituting the aggregation. Consistent with the previous analyses, most TRs had an aggregated ranking whose AUPRC exceeded the expected values from individual experiments, even if this was not always the integrated aggregation (Fig. 6D,F; Supplemental Fig. S16). The human ASCL1 integrated ranking, for example, outperformed every individual ASCL1 experiment, as well as the perturbation and binding aggregations, supporting the benefit of both data aggregation and cross-method integration.
Similarly, both the human and mouse RUNX1 integrated rankings were in the 98th percentile of RUNX1 AUPRCs, outperforming the perturbation and binding aggregations. Although the integrated ranking also outperformed the vast majority of individual experiments, the top human RUNX1 performances were typically rank–product pairings conducted in leukemic systems. As the most represented RUNX1 cell types in the curated resource were leukemia cell lines, the benchmark may be biased toward cell type–specific effects compared with the cell type–agnostic aggregations. This may also explain why mouse NEUROD1 had the least performant aggregations: The highest AUPRCs belonged to individual genomic experiments (and their rank–product pairings) conducted in pancreatic tissues. In concordance, pancreatic experiments were among the most represented from the relatively scarce collection of literature curated NEUROD1 targets, whereas the genomic NEUROD1 experiments covered a broader range of tissue (see Supplemental Material, “Commentary on aggregate ranking”).
In conclusion, although the integrated rankings were not always the most performant, the aggregation strategies presented are typically still more capable of prioritizing known targets than null expectations or the individual experiments composing the aggregation. All summarized rankings are found in Supplemental Data S1 and the contributing data in Supplemental Data S2. In the Supplemental Material, we show a context-specific analysis using RUNX1 K-562 experiments (Supplemental Fig. S22); we motivate an alternative ranking scheme that places greater importance on individual experiments (rankings provided in Supplemental Data S6); and we discuss a subset of the identified candidate targets for each TR.
Discussion
In this study, we build upon existing strategies (Tang et al. 2011; Wang et al. 2013) to create a framework for aggregating genomics data to rank gene regulatory relationships. We deliberately focused on methodologies that directly assess TR activity rather than those based on inference (e.g., coexpression or DNA motif footprinting). Prior studies have similarly ranked TR–target interactions using orthogonal lines of evidence (Garcia-Alonso et al. 2019) or aggregated TR–target libraries (Keenan et al. 2019). Although these studies are more comprehensive in their TR coverage, they focus only on human interactions and, in particular, hold out TR perturbation evidence for benchmarking rather than for gene prioritization. Here, we did a detailed analysis of a subset of TRs to better understand the properties of the contributing genomics experiments and to identify potential biases of aggregation. Toward this end, our work also represents a novel meta-analysis of TRs with shown importance in mammals.
Data reuse and aggregation across diverse studies presents many challenges. Extending prior observations, we find generally weak similarities when comparing experiments targeting the same TR (Hu et al. 2007; Cusanovich et al. 2014; Garcia-Alonso et al. 2019; Kang et al. 2020). Further, the genomics evidence (aggregated or not) was not highly performant when evaluated using literature-curated targets. This is unsurprising, given (1) the incomplete nature of the evaluation set (true targets lacking curation would be treated as negatives), (2) the heterogeneity of contexts and experimental/technical factors in all considered data, and (3) the inherent difficulty in benchmarking gene regulation (Marbach et al. 2012; Garcia-Alonso et al. 2019). The first reason in particular guided our use of the broadly applicable rank product for data integration (Breitling et al. 2004; Wang et al. 2013). Here, this is a two-parameter model that gives equal importance to each genomic line of evidence, whereas training TR-specific models would require a robust gold standard for each TR. Nevertheless, our data aggregation revealed numerous candidate TR–target interactions supported by extensive convergent evidence. As we (generally) see that the aggregated data prioritized curated targets, our explicit assumption is that the rankings will also be enriched for unexplored direct interactions.
It is important to discuss caveats of our framework, as some of these complexities extend to general difficulties in studying gene regulation. First and foremost, our rankings must be interpreted as a sorting of existing genomic evidence, rather than one of absolute biological importance. If a developmentally-critical interaction is not assayed in the appropriate biological or temporal context of the included experiments, it cannot be expected to be highly ranked, if it can even be captured by the considered strategies. Similarly, a highly context-specific interaction will not be as highly prioritized as one common to a more abundantly represented context, because of our approach of aggregating data. Therefore, researchers interested in benchmarking predictions may wish to use the aggregated rankings (Supplemental Data S1), whereas those interested in a specific context may refer to the organized experiment matrices (Supplemental Data S2) or to the alternative ranking scheme presented in the Supplemental Material that prioritizes genes with a single positive finding in any experiment (Supplemental Data S6).
For the binding evidence, our gene-scoring method aligns with other contemporary studies analyzing ChIP-seq data at scale (Methods). Although providing more granularity than binarizing binding events, these formulations still rely on genomic distance as a measure of relevance to a gene (Chen et al. 2020). Current evidence suggests this to be a useful approximation (Yoshida et al. 2019), but future efforts may benefit from incorporating evidence from 3D chromosomal interactions, such as in the “activity by contact” model (Fulco et al. 2019). Additionally, the TSS-based logic of the binding score is likely better suited for TFs than other classes of regulators like MECP2. Although we were still able to capture curated MECP2 targets, researchers may choose to prioritize the perturbation rankings or focus on specific MECP2-bound coordinates (Supplemental Data S3–S5).
In line with prior reports, we also find that certain loci are frequently associated with a ChIP-seq signal across assays (Supplemental Fig. S5). The biological-versus-technical nature of these regions has been debated (Teytelman et al. 2013; Wreczycka et al. 2019; Partridge et al. 2020; Ramaker et al. 2020); regardless of its origin, this phenomenon motivated our analysis of differential binding activities (Fig. 2). For example, Li et al. (2019a) subtracted a gene-wise background signal when gene scoring a single ChIP-seq experiment. However, our study was based on a small and biased selection of TRs; thus, the comparator groups may participate in common regulatory pathways. For this reason, we based the final binding rankings on the mean binding score rather than on differential binding statistics. Nevertheless, we find evidence for specific binding that aligns with prior described interactions (e.g., HES1-ATOH1, ASCL1-DLL1). We believe that the binding score-based linear modeling framework has intriguing potential for forming more sophisticated TR group comparisons, such as for cobinding partners or TR families.
For the perturbation evidence, we tallied independent significance tests. Finding that genes commonly had variable changes in FC direction, we ultimately used the absolute FC as a tie-break. Together, this means that the final rankings are agnostic to the change of direction, although our inclusion of FC Purity in the summarized results allows researchers to identify interactions predominantly measured as activating or repressive. Ideally, a single model would jointly consider all TR perturbation experiments. However, this is greatly complicated by the diversity of technologies, gene coverage (non-uniformity of missing observations), sample sizes, and designs; thus, we elected for simplicity in this study. We used the DE prior (Fig. 5A,B) to provide additional context of a gene's behavior with respect to DE testing, as it can help identify “generically” DE genes.
Genes with frequent DE evidence showed a range of binding evidence across TRs. Although our primary interest was finding targets corroborated by both strategies, genes well supported by TR perturbation alone may warrant further investigation. These possibly reflect instances in which the assembled ChIP-seq contexts did not capture the binding event or in which it occurred at a genomic distance missed by our scoring scheme. Alternatively, a common interpretation for individual experiment pairs is that DE genes lacking in binding are indirectly regulated through an intermediate regulator. If an absence of binding can be confirmed, it is strongly suggestive that, even if indirect, the perturbed TR and frequently DEG participate in a tightly controlled regulatory pathway.
Although TRs are expected to have cell type–specific targets, our results nominate genes that may be regulated by a TR across contexts (Gertz et al. 2013; Lambert et al. 2018). While an ultimate goal of gene regulation research is to establish the specificity of TR–target interactions, it would still be desirable to characterize the extent by which each TR can be represented by a set of “core” targets. ASCL1, for example, appears to commonly regulate Notch pathway effectors like DLL1, DLL3, and HES6 (Castro et al. 2006, 2011; Nelson et al. 2009). Further examination of these frequent targets is warranted: if the same regions are bound or if there is more distributed enhancer usage, the degree to which these interactions are coexpressed across systems/conditions, conservation of the associated sequence, and the consistency of chromatin features or cobinding partners. Our examination of frequently bound loci is a step in this direction (Supplemental Fig. S6), but a more comprehensive exploration building on these observations requires further study. Similarly, a more comprehensive examination of TR–target interactions conserved across species is warranted, which can be potentiated by the examples provided by our work.
Consistent with our low-throughput curation resource (Chu et al. 2021), we found that the literature coverage of the studied regulators was greatly uneven, with MECP2 and RUNX1 receiving the most attention across low-throughput and high-throughput investigations. This is not surprising but highlights the need for investigation of less-studied, yet important regulators. Similarly, we identified multiple examples of genes that had convergent lines of evidence but are sparsely represented in the literature (Supplemental Material, “Overview of TR-targets”). Given that the assembled data are biased toward regulators deemed of interest by the broader research community, our work suggests that these understudied candidate targets could be prioritized by gene functionality studies.
In sum, we believe this study will be a useful resource for researchers interested in gene regulation. We present a large collection of transparently summarized information that catalogs the current state of the literature while also potentiating novel biological discovery. We also have documented many practical issues and limitations of the considered data and present an analytical framework that is readily extensible to the ever-growing collection of TR experimentation.
Methods
Except where noted, analyses were performed in the R statistical computing environment (R Core Team 2022).
Genomic feature tables
Gene annotations were based on NCBI RefSeq Select (mm10 and hg38), which assigns one TSS to each gene (https://www.ncbi.nlm.nih.gov/refseq/refseq_select/). ENCODE cCREs were obtained from https://screen.encodeproject.org/ (V3) (Moore et al. 2020). The DE prior rank information was an updated version for human and newly generated for mouse, using the same strategy as that of Crow et al. (2019) but expanded to a greater number of expression platforms and data sets (Supplemental Tables S4, S5). High-confidence one-to-one orthologous genes were accessed via the DIOPT resource (V8) (Hu et al. 2011), keeping only genes with a score of at least five that were also reciprocally the best score between mouse and human and excluding genes with more than one match.
Identification of ChIP-seq data sets
Identification of ChIP-seq data was predominantly facilitated using the ChIP Atlas database (Zou et al. 2022) owing to its breadth of mouse and human data and the organization of the associated metadata. Additional experiments were identified in the literature and other ChIP-seq resources: GTRD (Kolmykov et al. 2021), UniBind (Puig et al. 2021), and Cistromedb (Zheng et al. 2019). Experiments were curated and matched to their input controls, applying and extending ChIP Atlas's metadata framework such that each row corresponds to a unique SRX ID (https://www.ncbi.nlm.nih.gov/sra).
Uniform processing of ChIP-seq data
As there was heterogeneity across each resource's processing pipelines and how sets of samples were organized within an experiment unit (e.g., the pooling of replicates or inputs), we uniformly reprocessed all data. Using SRX IDs, sample library information was obtained using NCBI's ESearch utility (version 13.8), and FASTQ files were downloaded using fasterq-dump (version 2.10.8) before read trimming and quality control were performed using Trim Galore! (version 0.6.6) (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), keeping reads that were at least 30 bp. Processed FASTQ files were then submitted for processing in the comparatively stringent ENCODE ChIP-seq pipeline (version 1.3.6) (Landt et al. 2012; https://github.com/ENCODE-DCC/chip-seq-pipeline2) with the following considerations: First, samples were grouped into units based upon the replicate status of the experimental design, and the identified input controls for each experimental unit were pooled. We note that many experiments consisted of a single replicate (meaning that the reproducibility process operated on pseudoreplicates rather than true replicates) and that input controls could be shared across distinct experimental units. Second, we fixed the peak caller to MACS2 (Zhang et al. 2008), as the ENCODE default method SPP cannot be used for runs with no input controls. Finally, all samples were submitted to the TF pipeline procedure, save for MECP2 experiments, which were submitted to both the TF and histone procedures. Given MECP2's binding profile, prior studies have used broad peak-calling parameterization (Gabel et al. 2015; Ito-Ishida et al. 2018; Xiang et al. 2020). Correspondingly, more MECP2 experiments succeeded processing with the histone parameterization, and thus, we used this strategy for all MECP2 samples. We followed the advice of Marinov et al. (2014) to avoid applying flat QC cut-offs for heterogeneous ChIP-seq collections, with two exceptions: Experiments marked as “fail” in the reproducibility analysis (IDR for TF, Overlap for MECP2) in the generated QC report were excluded, and only experiments with at least 100 peaks were retained to avoid overly sparse binding vectors during analysis. We further note that these excluded experiments typically also had outlier ENCODE QC metrics.
ChIP-seq gene binding scores and normalization
The ENCODE pipeline produces a set of output files, among which is a single “optimal reproducible” peak set table of genomic coordinates (via the IDR procedure for TF and Overlap procedure for histone/MECP2) for each experimental unit; we focused on these tables for most analyses. We considered multiple approaches to score gene binding for each experiment. The most common strategy is binary assignment, in which genes are scored as one if a peak summit is found within a distance threshold to the TSS, and zero otherwise. Following the advice of Sikora-Wohlfeld et al. (2013), we focused on quantitative binding scores, in particular a slight modification to the exponential decay function introduced by Ouyang et al. (2009):
| (1) |
where S is the binding score for a gene (g) in one TR experiment, K is the number of peak summits within 1 Mbp of the gene TSS, dk represents the absolute distance in base pairs between the TSS and the peak summit, and d0 is the decay constant, set to 5000 as in the original publication. The original formulation scaled each element by the MACS2 intensity score, which we omitted as these scores no longer retained their original interpretation after the ENCODE reproducibility process. The omission of this scaling factor is consistent with other work that adopts an exponential decay formulation to score genes using ChIP-seq peaks (Wang et al. 2013; Garcia-Alonso et al. 2019; Chen et al. 2020). Thus, all ChIP-seq experimental units can be represented as a gene-by-experiment matrix of binding scores. We added one and applied a log10 transformation followed by quantile normalization (preprocessCore R package version 1.48) to these bind score matrices, finding that this strategy helped alleviate batch/technical considerations, and used these matrices for the similarity analyses and the final gene rankings.
Binding specificity analysis
To find genes with enriched binding scores for the same TR, we adopted the limma-voom framework (version 3.42.2) (Law et al. 2014), a common strategy for applying linear models to genomics data with a positive mean-variance relationship, as observed here. The raw binding score matrices were submitted to the voom transformation with quantile normalization specified, using limma's duplicateCorrelation function with laboratory identity as a blocking variable to account for expected elevated correlations among experiments submitted by the same research group. The following model was then fit to every gene:
| (2) |
where S is the same as in Equation 1, j indexes TR experiments, the main effect TR represents the ChIP'd protein, binary variables I and R capture if experiment j has at least one input control and at least one replicate, and C is the count of peaks for experiment j, with the residual (ε) having the covariance matrix as estimated by duplicateCorrelation, as well as regression weights provided by voom. Finally, for each TR, a “one-versus-rest” contrast was extracted from this model, which estimates for each gene the difference in mean binding scores (S) for the current TR's set of experiments relative to all other experiments, after using information from all experiments to account for the specified experimental structure/technical variables.
ChIP-seq peak region overlap
All overlap procedures were performed using the GenomicRanges R package (version 1.38) (Lawrence et al. 2013). For the cCRE region analysis, only the peak summit was used to detect overlap across any part of a cCRE region (which ranged from 150–350 bp). For the frequently bound region analysis, as the original peaks were variable in length, we resized each such that 150 bp was added in each direction from the summit and then merged those that overlapped. Our main conclusions were robust to minor variations of these processing steps.
Identification of TF perturbation high-throughput expression data sets
Perturbation strategies can be coarsely grouped by if they reduce the available pool of TR gene transcripts (knockdowns), if one or both TR alleles are functionally eliminated (knockouts), if a transgenic construct results in elevated levels of the TR (overexpression), or if sequence variations critically disrupt the function of the TR (mutants). We first queried existing resources that have aggregated TF perturbation experiments: Gene Perturbation Atlas (Xiao et al. 2015), ChEA3 (Keenan et al. 2019), and KnockTF (Feng et al. 2020). Most experiments were identified by extending strategies used by our group for the Gemma database (Lim et al. 2021). Briefly, this involves human curation of experiment suitability after programmatically searching the GEO database for co-occurrence of TR gene symbols and a list of perturbation terms (e.g., “siRNA,” “overexpression”). All identified experiments were checked for accurate curation by at least two individuals. Selected experiments were required to have at least two control and treatment samples and to have samples that perturbed only the single TR of interest and were of an appropriate technical strategy (single-cell sequencing, sorting by expression, and run-on sequencing were excluded).
We found that multiple experiments showed minimal or even “unexpected” expression changes in the perturbed TF (e.g., overexpression of TR yielding an apparent decrease in RNA levels for the TR) (Supplemental Fig. S9A). We inspected all such examples, finding that the perturbed TR was often among the top-ranked genes by absolute FC (median, 81st percentile). Although we cannot exclude the possibility of sample mislabeling on GEO, it is possible that temporal or biological factors such as proposed genetic compensatory mechanisms (El-Brolosy and Stainier 2017; El-Brolosy et al. 2019) may explain the measured TR transcript levels postperturbation. Given that the associated studies typically validated the perturbation independent of the microarray or RNA sequencing experiment, we chose to keep all such experiments.
Obtaining summarized TR perturbation results from Gemma
The selected expression studies were submitted to the Gemma framework for uniform processing (Lim et al. 2021). This entails human curation of the experimental design using controlled terminology, paired with automated handling of batch information, platform-specific support, and differential expression analysis (DEA). Gemma fits generalized linear models using the curated experimental factors, producing a table of summarized results for each factor (t-statistics, log2 FCs, and P-values). We note that some studies had experimental factors beyond the TF perturbation (e.g., “± LPS treatment”). When distinct cell types/tissues were one of these factors (e.g., a knockout in hypothalamus as well as cerebellum), a separate perturbation DEA was performed for each cell type/tissue. Otherwise, a single model was fit for all experimental factors, and the perturbation contrast effect sizes (controlling for the other factors) were extracted. Microarray probes that did not map to a single gene were excluded, and when multiple probes or sequencing elements mapped to a single gene, only the element with the maximum absolute t-statistic was kept. The FDR was controlled using the Benjamini–Hochberg procedure (p.adjust R, version 4.2.1) on the P-values after this filtering. Genes were binarized as DE at FDR < 0.1. In this manner, all experiments can be represented as gene-by-experiment matrices of the various effect sizes. Because microarrays vary in which genes are assayed, their inclusion resulted in variable gene coverage in the final corpus (Supplemental Fig. S7).
Ranking gene targets
For perturbation data, we ranked genes by the count of times that they were DE across experiments (Count DE), using the absolute log2 FC as a tie-break (D), and for ChIP-seq, we used the mean binding score (S). We considered multiple strategies to reach a single ranking from both data types. A popular approach for individual experiment pairs is the BETA algorithm, which takes the rank product (RP) of a DE gene list and binding scores from an exponential decay function based on distance from the TSS (as in this study) (Wang et al. 2013). However, we wished to rank targets using all the intra-TR data sets, not just pairs. Two options are to tally the count of times a gene was in a “top overlap” or to average a gene's RP across every intra-TR comparison. However, each requires making numerous nonindependent comparisons (each experiment is paired multiple times). Alternatively, rank aggregation strategies have a long history of use in genomics (e.g., Keenan et al. 2019 averages rankings across different TR–target libraries), with strategies extended to cases involving unevenly sized rankings (Kolde et al. 2012; Li et al. 2019b). Yet, the final ranking will be influenced if there are imbalanced experiment counts between the two genomic methods, and adjusting for this would require calibration against a known standard. Consequently, we applied a simplistic hybrid approach, calculating the RP, as in the BETA algorithm, but using the aggregated rankings as inputs:
| (3) |
where N are the number of genes for the respective lists. The RP returns a unitless value, which we in turn convert to a rank such that one represents the most prioritized gene g for TR j.
Data access
The code used for analysis in this study can be found at GitHub (https://github.com/PavlidisLab/TR_aggregation) and as Supplemental Code. The metadata of the analyzed experiments and their associated GEO accession identifiers can be found in Supplemental Tables S1 (ChIP-seq) and S2 (perturbation). The summarized gene rankings (Supplemental Data S1), data matrices (Supplemental Data S2), and bound regions (Supplemental Data S3–S5) used for analysis can additionally be found as RDS objects in the Borealis data repository (https://doi.org/10.5683/SP3/MAFGFL).
Supplementary Material
Acknowledgments
We thank Dr. Nathaniel Lim for the expansion of the Differential Expression (DE) prior ranking (manuscript in preparation), first generated by Crow et al. (2019). We also thank Dr. Marine Louarn, who has managed the update of the resource of curated regulatory interactions (Chu et al. 2021). This work was supported by National Institutes of Health grant MH111099 (https://www.nih.gov/) and Natural Sciences and Engineering Research Council of Canada (NSERC) grant RGPIN-2016-05991 (https://www.nserc-crsng.gc.ca/), both held by P.P. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. A.M. had funding support from the Canadian Institutes of Health Research Canada Graduate Scholarship (CIHR-CGS), Natural Sciences and Engineering Research Council of Canada - Collaborative Research and Training Experience (NSERC-CREATE), and UBC Institute of Mental Health (IMH) Marshall Scholars programs.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277273.122.
Competing interest statement
The authors declare no competing interests.
References
- Ali FR, Marcos D, Chernukhin I, Woods LM, Parkinson LM, Wylie LA, Papkovskaia TD, Davies JD, Carroll JS, Philpott A. 2020. Dephosphorylation of the proneural transcription factor ASCL1 re-engages a latent post-mitotic differentiation program in neuroblastoma. Mol Cancer Res 18: 1759–1766. 10.1158/1541-7786.MCR-20-0693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt D, Musser JM, Baker CVH, Bergman A, Cepko C, Erwin DH, Pavlicev M, Schlosser G, Widder S, Laubichler MD, et al. 2016. The origin and evolution of cell types. Nat Rev Genet 17: 744–757. 10.1038/nrg.2016.127 [DOI] [PubMed] [Google Scholar]
- Borromeo MD, Meredith DM, Castro DS, Chang JC, Tung K-C, Guillemot F, Johnson JE. 2014. A transcription factor network specifying inhibitory versus excitatory neurons in the dorsal spinal cord. Development 141: 2803–2812. 10.1242/dev.105866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling R, Armengaud P, Amtmann A, Herzyk P. 2004. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett 573: 83–92. 10.1016/j.febslet.2004.07.055 [DOI] [PubMed] [Google Scholar]
- Breslin MB, Zhu M, Lan MS. 2003. Neurod1/E47 regulates the E-box element of a novel zinc finger transcription factor, IA-1, in developing nervous system. J Biol Chem 278: 38991–38997. 10.1074/jbc.M306795200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro DS, Skowronska-Krawczyk D, Armant O, Donaldson IJ, Parras C, Hunt C, Critchley JA, Nguyen L, Gossler A, Göttgens B, et al. 2006. Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev Cell 11: 831–844. 10.1016/j.devcel.2006.10.006 [DOI] [PubMed] [Google Scholar]
- Castro DS, Martynoga B, Parras C, Ramesh V, Pacary E, Johnston C, Drechsel D, Lebel-Potter M, Garcia LG, Hunt C, et al. 2011. A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes Dev 25: 930–945. 10.1101/gad.627811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Zheng R, Tokheim C, Dong X, Fan J, Wan C, Tang Q, Brown M, Liu JS, Meyer CA, et al. 2020. Determinants of transcription factor regulatory range. Nat Commun 11: 2472. 10.1038/s41467-020-16106-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholewa-Waclaw J, Shah R, Webb S, Chhatbar K, Ramsahoye B, Pusch O, Yu M, Greulich P, Waclaw B, Bird AP. 2019. Quantitative modelling predicts the impact of DNA methylation on RNA polymerase II traffic. Proc Natl Acad Sci 116: 14995–15000. 10.1073/pnas.1903549116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu ECP, Morin A, Chang THC, Nguyen T, Tsai Y-C, Sharma A, Liu CC, Pavlidis P. 2021. Experiment level curation of transcriptional regulatory interactions in neurodevelopment. PLoS Comput Biol 17: e1009484. 10.1371/journal.pcbi.1009484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow M, Lim N, Ballouz S, Pavlidis P, Gillis J. 2019. Predictability of human differential gene expression. Proc Natl Acad Sci 116: 6491–6500. 10.1073/pnas.1802973116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Pavlovic B, Pritchard JK, Gilad Y. 2014. The functional consequences of variation in transcription factor binding. PLoS Genet 10: e1004226. 10.1371/journal.pgen.1004226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fede E, Peron A, Colombo EA, Gervasini C, Vignoli A. 2021. SLC35F1 as a candidate gene for neurodevelopmental disorders resembling Rett syndrome. Am J Med Genet A 185: 2238–2240. 10.1002/ajmg.a.62203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy MA, Stainier DYR. 2017. Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet 13: e1006780. 10.1371/journal.pgen.1006780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Günther S, Fukuda N, Kikhi K, Boezio GLM, Takacs CM, Lai S-L, et al. 2019. Genetic compensation triggered by mutant mRNA degradation. Nature 568: 193–197. 10.1038/s41586-019-1064-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Song C, Liu Y, Qian F, Gao Y, Ning Z, Wang Q, Jiang Y, Li Y, Li M, et al. 2020. KnockTF: a comprehensive human gene expression profile database with knockdown/knockout of transcription factors. Nucleic Acids Res 48: D93–D100. 10.1093/nar/gkz881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. 2019. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat Genet 51: 1664–1669. 10.1038/s41588-019-0538-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME. 2015. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522: 89–93. 10.1038/nature14319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alonso L, Holland CH, Ibrahim MM, Turei D, Saez-Rodriguez J. 2019. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res 29: 1363–1375. 10.1101/gr.240663.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J, Savic D, Varley KE, Partridge EC, Safi A, Jain P, Cooper GM, Reddy TE, Crawford GE, Myers RM. 2013. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol Cell 52: 25–36. 10.1016/j.molcel.2013.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitter A, Siegfried Z, Klutstein M, Fornes O, Oliva B, Simon I, Bar-Joseph Z. 2009. Backup in gene regulatory networks explains differences between binding and knockout results. Mol Syst Biol 5: 276. 10.1038/msb.2009.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E, et al. 2018. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res 46: D380–D386. 10.1093/nar/gkx1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawe JS, Theis FJ, Heinig M. 2019. Inferring interaction networks from multi-omics data. Front Genet 10: 535. 10.3389/fgene.2019.00535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Killion PJ, Iyer VR. 2007. Genetic reconstruction of a functional transcriptional regulatory network. Nat Genet 39: 683–687. 10.1038/ng2012 [DOI] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, Mohr SE. 2011. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12: 357. 10.1186/1471-2105-12-357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito-Ishida A, Yamalanchili HK, Shao Y, Baker SA, Heckman LD, Lavery LA, Kim J-Y, Lombardi LM, Sun Y, Liu Z, et al. 2018. Genome-wide distribution of linker histone H1.0 is independent of MeCP2. Nat Neurosci 21: 794–798. 10.1038/s41593-018-0155-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Patel NR, Shively C, Recio PS, Chen X, Wranik BJ, Kim G, McIsaac RS, Mitra R, Brent MR. 2020. Dual threshold optimization and network inference reveal convergent evidence from TF binding locations and TF perturbation responses. Genome Res 30: 459–471. 10.1101/gr.259655.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanjian A, Shroyer NF. 2011. NOTCH signaling and ATOH1 in colorectal cancers. Curr Colorectal Cancer Rep 7: 121–127. 10.1007/s11888-011-0090-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, Jagodnik KM, Kropiwnicki E, Wang Z, Ma'ayan A. 2019. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res 47: W212–W224. 10.1093/nar/gkz446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keidar L, Gerlitz G, Kshirsagar A, Tsoory M, Olender T, Wang X, Yang Y, Chen Y-S, Yang Y-G, Voineagu I, et al. 2019. Interplay of LIS1 and MeCP2: interactions and implications with the neurodevelopmental disorders lissencephaly and Rett syndrome. Front Cell Neurosci 13: 370. 10.3389/fncel.2019.00370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf J, Knelles K, Hersbach BA, Petrik D, Riedemann T, Bednarova V, Janjic A, Simon-Ebert T, Enard W, Smialowski P, et al. 2021. Heterogeneity of neurons reprogrammed from spinal cord astrocytes by the proneural factors Ascl1 and Neurogenin2. Cell Rep 36: 109409. 10.1016/j.celrep.2021.109409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi N, MacDonald JL, Ye J, Molyneaux BJ, Azim E, Macklis JD. 2016. Reduction of aberrant NF-κB signalling ameliorates Rett syndrome phenotypes in Mecp2-null mice. Nat Commun 7: 10520. 10.1038/ncomms10520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R, Laur S, Adler P, Vilo J. 2012. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 28: 573–580. 10.1093/bioinformatics/btr709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmykov S, Yevshin I, Kulyashov M, Sharipov R, Kondrakhin Y, Makeev VJ, Kulakovskiy IV, Kel A, Kolpakov F. 2021. GTRD: an integrated view of transcription regulation. Nucleic Acids Res 49: D104–D111. 10.1093/nar/gkaa1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziol JA. 2010. Comments on the rank product method for analyzing replicated experiments. FEBS Lett 584: 941–944. 10.1016/j.febslet.2010.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT. 2018. The human transcription factors. Cell 172: 650–665. 10.1016/j.cell.2018.01.029 [DOI] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22: 1813–1831. 10.1101/gr.136184.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. 2014. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: R29. 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. 2013. Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118. 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wan C, Zheng R, Fan J, Dong X, Meyer CA, Liu XS. 2019a. Cistrome-GO: a web server for functional enrichment analysis of transcription factor ChIP-seq peaks. Nucleic Acids Res 47: W206–W211. 10.1093/nar/gkz332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang X, Xiao G. 2019b. A comparative study of rank aggregation methods for partial and top ranked lists in genomic applications. Brief Bioinform 20: 178–189. 10.1093/bib/bbx101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim N, Tesar S, Belmadani M, Poirier-Morency G, Mancarci BO, Sicherman J, Jacobson M, Leong J, Tan P, Pavlidis P. 2021. Curation of over 10,000 transcriptomic studies to enable data reuse. Database 2021: baab006. 10.1093/database/baab006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Hitz BC, Gabdank I, Hilton JA, Kagda MS, Lam B, Myers Z, Sud P, Jou J, Lin K, et al. 2020. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res 48: D882–D889. 10.1093/nar/gkz1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Costello JC, Küffner R, Vega NM, Prill RJ, Camacho DM, Allison KR, Kellis M, Collins JJ, Stolovitzky G. 2012. Wisdom of crowds for robust gene network inference. Nature Methods 9: 796–804. 10.1038/nmeth.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Lamparter D, Quon G, Kellis M, Kutalik Z, Bergmann S. 2016. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat Methods 13: 366–370. 10.1038/nmeth.3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinov GK, Kundaje A, Park PJ, Wold BJ. 2014. Large-scale quality analysis of published ChIP-seq data. G3 (Bethesda) 4: 209–223. 10.1534/g3.113.008680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Irie T, Katsurabayashi S, Hayashi Y, Nagai T, Hamazaki N, Adefuin AMD, Miura F, Ito T, Kimura H, et al. 2019. Pioneer factor NeuroD1 rearranges transcriptional and epigenetic profiles to execute microglia-neuron conversion. Neuron 101: 472–485.e7. 10.1016/j.neuron.2018.12.010 [DOI] [PubMed] [Google Scholar]
- Mattioli F, Hayot G, Drouot N, Isidor B, Courraud J, Hinckelmann M-V, Mau-Them FT, Sellier C, Goldman A, Telegrafi A, et al. 2020. De novo frameshift variants in the neuronal splicing factor NOVA2 result in a common C-terminal extension and cause a severe form of neurodevelopmental disorder. Am J Hum Genet 106: 438–452. 10.1016/j.ajhg.2020.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraldi ER, Pokrovskii M, Watters A, Castro DM, De Veaux N, Hall JA, Lee J-Y, Ciofani M, Madar A, Carriero N, et al. 2019. Leveraging chromatin accessibility for transcriptional regulatory network inference in T Helper 17 Cells. Genome Res 29: 449–463. 10.1101/gr.238253.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JE, Purcaro MJ, Pratt HE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R. 2020. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583: 699–710. 10.1038/s41586-020-2493-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BR, Hartman BH, Ray CA, Hayashi T, Bermingham-McDonogh O, Reh TA. 2009. Acheate-scute like 1 (Ascl1) is required for normal δ-like (Dll) gene expression and notch signaling during retinal development. Dev Dyn 238: 2163–2178. 10.1002/dvdy.21848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, West AE. 2020. Neurobiological functions of transcriptional enhancers. Nat Neurosci 23: 5–14. 10.1038/s41593-019-0538-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, Zhou Q, Wong WH. 2009. ChIP-seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc Natl Acad Sci 106: 21521–21526. 10.1073/pnas.0904863106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge EC, Chhetri SB, Prokop JW, Ramaker RC, Jansen CS, Goh S-T, Mackiewicz M, Newberry KM, Brandsmeier LA, Meadows SK, et al. 2020. Occupancy maps of 208 chromatin-associated proteins in one human cell type. Nature 583: 720–728. 10.1038/s41586-020-2023-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl JR, Colantuoni C, Bergey DE, Funk CC, Shannon P, Basu B, Casella AM, Oshone RT, Hood L, Price ND, et al. 2019. Genome-scale transcriptional regulatory network models of psychiatric and neurodegenerative disorders. Cell Syst 8: 122–135.e7. 10.1016/j.cels.2019.01.002 [DOI] [PubMed] [Google Scholar]
- Pomeshchik Y, Klementieva O, Gil J, Martinsson I, Hansen MG, de Vries T, Sancho-Balsells A, Russ K, Savchenko E, Collin A, et al. 2020. Human iPSC-derived hippocampal spheroids: an innovative tool for stratifying Alzheimer disease patient-specific cellular phenotypes and developing therapies. Stem Cell Reports 15: 256–273. 10.1016/j.stemcr.2020.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig RR, Boddie P, Khan A, Castro-Mondragon JA, Mathelier A. 2021. UniBind: maps of high-confidence direct TF-DNA interactions across nine species. BMC Genomics 22: 482. 10.1186/s12864-021-07760-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Fan J, Zheng R, Wan C, Mei S, Wu Q, Sun H, Brown M, Zhang J, Meyer CA, et al. 2020. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol 21: 32. 10.1186/s13059-020-1934-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaker RC, Hardigan AA, Goh ST, Partridge EC, Wold B, Cooper SJ, Myers RM. 2020. Dissecting the regulatory activity and sequence content of loci with exceptional numbers of transcription factor associations. Genome Res 30: 939–950. 10.1101/gr.260463.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Z, Wang R, Li S, Shi Y, Mo L, Han S, Yuan J, Jing N, Cheng L. 2021. Molecular mechanisms underlying Ascl1-mediated astrocyte-to-neuron conversion. Stem Cell Reports 16: 534–547. 10.1016/j.stemcr.2021.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2022. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www-R-project.org/. [Google Scholar]
- Rothenberg EV. 2019. Causal gene regulatory network modeling and genomics: second-generation challenges. J Comput Biol 26: 703–718. 10.1089/cmb.2019.0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebreni L, Stark A. 2021. Insights into gene regulation: from regulatory genomic elements to DNA-protein and protein-protein interactions. Curr Opin Cell Biol 70: 58–66. 10.1016/j.ceb.2020.11.009 [DOI] [PubMed] [Google Scholar]
- Shah RR, Bird AP. 2017. MeCP2 mutations: progress towards understanding and treating Rett syndrome. Genome Med 9: 17. 10.1186/s13073-017-0411-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora-Wohlfeld W, Ackermann M, Christodoulou EG, Singaravelu K, Beyer A. 2013. Assessing computational methods for transcription factor target gene identification based on ChIP-seq data. PLoS Comput Biol 9: e1003342. 10.1371/journal.pcbi.1003342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Hu X, Lim MHK, Ng CKL, Choo SH, Castro DS, Drechsel D, Guillemot F, Kolatkar PR, Jauch R, et al. 2013. TherMos: estimating protein–DNA binding energies from in vivo binding profiles. Nucleic Acids Res 41: 5555–5568. 10.1093/nar/gkt250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Chen Y, Meyer C, Geistlinger T, Lupien M, Wang Q, Liu T, Zhang Y, Brown M, Liu XS. 2011. A comprehensive view of nuclear receptor cancer cistromes. Cancer Res 71: 6940–6947. 10.1158/0008-5472.CAN-11-2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teytelman L, Thurtle DM, Rine J, van Oudenaarden A. 2013. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Natl Acad Sci 110: 18602–18607. 10.1073/pnas.1316064110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdinguio RG, Lopez-Serra L, Lopez-Nieva P, Alaminos M, Diaz-Uriarte R, Fernandez AF, Esteller M. 2008. Mecp2-null mice provide new neuronal targets for Rett syndrome. PLoS One 3: e3669. 10.1371/journal.pone.0003669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, Liu XS. 2013. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 8: 2502–2515. 10.1038/nprot.2013.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Sandelin A. 2004. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet 5: 276–287. 10.1038/nrg1315 [DOI] [PubMed] [Google Scholar]
- Wreczycka K, Franke V, Uyar B, Wurmus R, Bulut S, Tursun B, Akalin A. 2019. HOT or not: examining the basis of high-occupancy target regions. Nucleic Acids Res 47: 5735–5745. 10.1093/nar/gkz460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Tanaka Y, Patterson B, Hwang S-M, Hysolli E, Cakir B, Kim K-Y, Wang W, Kang Y-J, Clement EM, et al. 2020. Dysregulation of BRD4 function underlies the functional abnormalities of MeCP2 mutant neurons. Mol Cell 79: 84–98.e9. 10.1016/j.molcel.2020.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Gong Y, Lv Y, Lan Y, Hu J, Li F, Xu J, Bai J, Deng Y, Liu L, et al. 2015. Gene Perturbation Atlas (GPA): a single-gene perturbation repository for characterizing functional mechanisms of coding and non-coding genes. Sci Rep 5: 10889. 10.1038/srep10889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, Desland F, Chudnovskiy A, Mortha A, Dominguez C, et al. 2019. The cis-regulatory atlas of the mouse immune system. Cell 176: 897–912.e20. 10.1016/j.cell.2018.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9: R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, Chen C-H, Brown M, Zhang X, Meyer CA, et al. 2019. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res 47: D729–D735. 10.1093/nar/gky1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, Ohta T, Miura F, Oki S. 2022. ChIP-Atlas 2021 update: a data-mining suite for exploring epigenomic landscapes by fully integrating ChIP-seq, ATAC-seq and Bisulfite-seq data. Nucleic Acids Res 50: W175–W182. 10.1093/nar/gkac199 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.