


Abstract
Drug discovery, which plays a vital role in maintaining human health, is a persistent challenge. Fragment-based drug discovery (FBDD) is one of the strategies for the discovery of novel candidate compounds. Computational tools in FBDD could help to identify potential drug leads in a cost-efficient and time-saving manner. The Auto Core Fragment in silico Screening (ACFIS) server is a well-established and effective online tool for FBDD. However, the accurate prediction of protein-fragment binding mode and affinity is still a major challenge for FBDD due to weak binding affinity. Here, we present an updated version (ACFIS 2.0), that incorporates a dynamic fragment growing strategy to consider protein flexibility. The major improvements of ACFIS 2.0 include (i) increased accuracy of hit compound identification (from 75.4% to 88.5% using the same test set), (ii) improved rationality of the protein-fragment binding mode, (iii) increased structural diversity due to expanded fragment libraries and (iv) inclusion of more comprehensive functionality for predicting molecular properties. Three successful cases of drug lead discovery using ACFIS 2.0 are described, including drugs leads to treat Parkinson's disease, cancer, and major depressive disorder. These cases demonstrate the utility of this web-based server. ACFIS 2.0 is freely available at http://chemyang.ccnu.edu.cn/ccb/server/ACFIS2/.
Graphical Abstract
Graphical Abstract.
ACFIS 2.0 is a one-stop platform for fragment-based drug design, aiming to help users obtain novel ligand structures for specific protein targets.
INTRODUCTION
Drugs save lives and improve health; thus, the discovery of novel drugs for various diseases is an important scientific issue. However, developing a new approved drug costs $1.5–2.0 billion and requires 10–15 years on average (1). Therefore, developing effective approaches to improve the success rate and reduce the cost for drug discovery is important (2). Lead discovery is a critical stage in drug research and development. The goal of this stage is to identify candidate molecules with satisfactory pharmacological activity. Lead discovery is accomplished through multiple iterations of compound design, synthesis, and bioassays. Surprisingly, it is estimated that approximate 100 000 compounds are synthesized at this stage for the development of a new drug (3). The poor efficiency of drug lead generation fundamentally restricts the progress of drug discovery. Therefore, the exploitation and application of helpful methodologies or tools for accelerating lead generation and optimization are pressing needs.
Fragment-based drug design (FBDD), a powerful methodology to identify quality leads starting from small sized molecules, has rapidly advanced in recent decades (4,5). Due to the weak binding of fragments to proteins, a variety of biophysical technologies, such as nuclear magnetic resonance (NMR) (6), X-ray diffraction (7), and surface plasmon resonance (SPR) (8), are more commonly employed in FBDD. However, fragment screening using these technologies is limited to only hundreds or thousands due to the expense, long experimental time, and high demand for purified raw materials (9,10). On the other hand, computational approaches have fewer limitations and large-scale screening (up to million) can be accomplished. In addition, computational approaches allow many molecular properties, such as synthetic accessibility, to be considered in advance. FBDD accelerated by computational tools has been successfully applied in the discovery of six approved drugs and over 40 candidates in clinical trials (11–13). For example, the discovery and development of vemurafenib took only 6 years from project initiation to approval (14). Overall, computational approaches exhibit great strengths in screening efficiency and economic cost.
A number of FBDD-derived computational methods or tools have been developed recently. According to the steps of FBDD they participate in, these computational tools can be divided into two categories: fragment screening tools and fragment optimization tools. Fragment optimization tools to determinate the core fragment include the fragment docking software like SEED (15,16) and the molecular decomposition tools like DAIM (17). Fragment optimization tools to generate lead compound include fragment growing tools like Frag_PELE (18) and DeepFrag (19) and fragment linking tools like DeLinker (20) and Syntalinker (21). These tools play important roles in expediting the FBDD process. Previously, we developed a comprehensive web server that facilitates FBDD named Auto Core Fragment in silico Screening (ACFIS) (22). A key innovation of ACFIS is the integration of multiple computation models to achieve a more integral workflow from fragment screening to lead generation. ACFIS is a useful tool that facilitates lead compound identification (23–26). However, protein flexibility was not fully estimated for the prediction of fragment binding and optimization in the previous version.
Here, we present ACFIS 2.0, an updated version based on a dynamic fragment growing method that considers the influence of protein flexibly. Additionally, ACFIS 2.0 integrates molecular visualization and various molecular property prediction functions into the output interface. Tested on 122 reported cases covering 51 targets (Supplementary Table S1), ACFIS 2.0 achieved an accuracy of 88.5%. Compared to the previous version, the accuracy of ACFIS 2.0 is increased by more than 10% (Supplementary Figure S1). In addition, the prediction results produced by ACFIS 2.0 for potential bioactive compounds are more comprehensive, covering binding affinity, physicochemical properties, drug-likeness, and synthetic accessibility. It results in better identification of high-quality drug hits. To demonstrate the applicability of ACFIS 2.0, the case studies of applying this tool to design monoamine oxidase B (MAO-B), tropomyosin receptor kinase A (TRKA), and serotonin transporter (SERT) inhibitors as Parkinson's drug, anticarcinogen, and antidepressant are presented. The usability and performance of the upgraded ACFIS is significantly improved. ACFIS 2.0 could serve as a powerful platform for FBDD research to accelerate drug discovery.
PROGRAM DESCRIPTION
Workflow overview
The overall workflow for FBDD using ACFIS 2.0 can be divided into four steps with two modules (Figure 1A). The starting module, named CORE_GEN, is designed to generate a core fragment from a given ligand structure. This module is comprised of two steps: fragment deconstruction and core fragment identification. The purpose of the second module, named CAND_GEN, is to derive candidate compounds from the core fragment. The CAND_GEN module is comprised of two sequential steps: dynamic fragment growing based on the core fragment (identified in the first module) and molecular property evaluation for candidate selection. The core process of the ACFIS 2.0 pipeline is organized based on our previously developed pharmacophore-linked fragment virtual screening (PFVS) method (27). The details of the four steps are described in Supplementary Text S1.
Figure 1.

Overview of the ACFIS 2.0 workflow, input, and output. (A) The flow diagram of the CORE_GEN and CAND_GEN modules in ACFIS 2.0. (B) The panel for submission of a task on the Home page and a subsequent page for ligand selection. (C) The result page of a task in one-stop mode. The contents include a list of newly generated ligands, an interactive viewer of protein-ligand binding modes, and the predicted results of physicochemical properties, binding free energy, drug/pesticide-likeness evaluation and synthesis accessibility.
Server usage
Input
The input requirements of the one-stop mode on the ‘Home’ page are relatively simple (Figure 1B). First, a protein-ligand complex structure is required by uploading a PDB file or typing the 4-letter PDB code. The uploaded complex structure could be experimentally-determined, or derived from computational models (e.g. using molecular docking approach). In some cases where experimental protein 3D structures are not available, it is feasible to obtain docked complex structures using predicted protein structures by AlphaFold (28,29). Then, one of five fragment libraries is allowed to be specified for fragment growing. A fragment library derived from approved drugs is the default option. Optional inputs include an email address for notification about the job state, job name, and password to privatize the job. Once these initial inputs are submitted, a ‘Select Ligand’ page will appear. On this page, all ligands in the given complex structure are shown and the user is asked to choose a structure for virtual screening.
Inputs for the two advanced modes are slightly different from the one-stop mode. In CORE_GEN mode, only one protein-ligand complex structure is indispensable, and other inputs are optional, similar to in the one-stop mode. The ‘Select Ligand’ page appears after the initial submission in CORE_GEN. In CAND_GEN mode, the input requirements for the initial page and the ‘Select Ligand’ page are the same as the one-stop mode, but a protein-fragment complex structure is recommended. The subsequent page requires another input; all hydrogen atoms of the specified ligand are automatically labeled with numbers, and the user selects a hydrogen atom as the fragment growing point by entering its corresponding number.
Output
The outputs of ACFIS 2.0 include a list of compounds, a 3D visual presentation of protein-ligand binding, and three tables of molecular properties (Figure 1C). For tasks in one-stop mode, the results derived from CAND_GEN and CORE_GEN are separately displayed using two subpages. On the left side of the output (sub)page, a navigation list of all newly generated compounds is shown. The listed compounds can be potential drug hits derived from CAND_GEN or core fragments derived from CORE_GEN, and each compound is equipped with ‘VIEW’ and ‘DOWNLOAD’ buttons. On the right side of the output page, a presentation of prediction results for each compound, which varies with the click of a ‘VIEW’ button, is shown. The binding mode of the ligand with its surrounding protein residues is visualized via the 3D interactive viewer on the upper right of the page. The property prediction data, including physicochemical properties, binding free energy, drug/pesticide-likeness evaluation, is summarized in tables at the bottom right of the output page. All of compound structures and result tables can be downloaded.
NEW FEATURES AND UPDATES
Expanded fragment libraries for increased structural diversity
In ACFIS 2.0, the original two libraries have been updated and two new libraries have been added to expand the structural diversity of generated ligands (Figure 2A). The first version of ACFIS contained only the FDA Drug Fragment Library and the Pesticide Fragment Library for fragment growing. In the current version, four fragment libraries are embedded, including the Approved Drug Fragment Library, the Approved Pesticide Fragment Library (30), the Hotspot Binding Fragment Library (31) and the Kinase Inhibitor Fragment Library (32).
Figure 2.
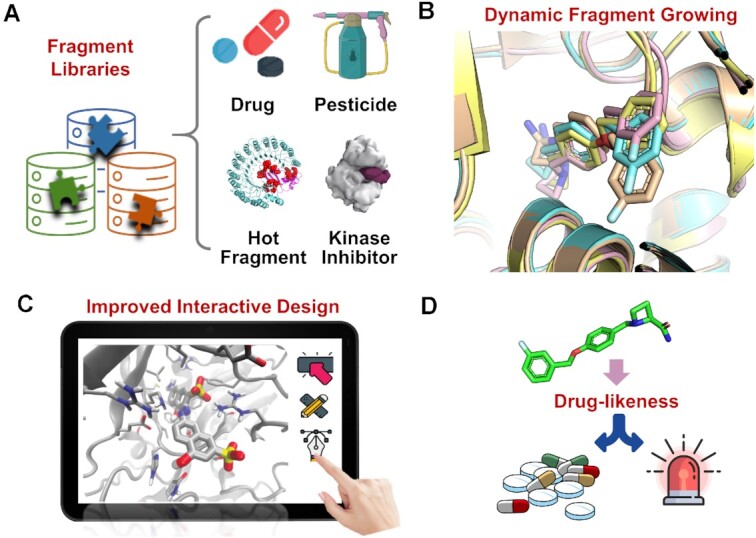
New features and updates of ACFIS 2.0, including (A) expanded fragment libraries, (B) a dynamic fragment strategy, (C) improved interactive design (C), and (D) drug likeness evaluation.
The main updates of deployed fragments in ACFIS 2.0 include: (i) the removal of highly similar fragments from the original two libraries based on a Tanimoto coefficient of 0.7, (ii) a supplementation with a series of novel fragments extracted from recently approved drugs and pesticides, (iii) the addition of 96 fragments that bind to the most preferred hotspots of popular drug targets (experimentally confirmed), (iv) the addition of 255 fragments that occur at a high frequency in kinase inhibitors and (v) the integration of the aforementioned four theme-specific fragment libraries.
Rational dynamic strategy for fragment growing
A new feature of ACFIS 2.0 is the dynamic fragment growing strategy that allows broad sampling of core fragment-bound protein conformations (Figure 2B). In the first version of ACFIS, only one receptor protein-core fragment complex conformation was used as a template to link fragments from the fragment library to the core fragment when constructing new compounds. This single protein-fragment complex conformation was originally deconstructed directly from the inputted protein-ligand complex structure without multiple sampling. This treatment ignored protein flexibility, probably resulting in a high probability of losing fragment-bound protein conformations at local energy minima and, thus, an unreasonable starting point for fragment growing. In ACFIS 2.0, molecular dynamics (MD) simulation of the protein-core fragment complex structure and conformation clustering of the MD trajectory are added before fragment growing (33), enabling an ensemble of complex structures at significantly different energy states (Supplementary Text S1). Therefore, the subsequent fragment growing is performed based on more than one protein-core fragment complex conformations, resulting in the generation of more protein-ligand conformations. This refinement of the fragment growing strategy rationalizes the previously proposed PFVS and may improve ACFIS 2.0 performance.
Improved function of design and analysis
Interactive webpage design
The interface of ACFIS 2.0 was redesigned to make job submission and the prediction result display more intuitive (Figure 2C). First, a function was added to show 2D molecular graphs for all ligands in the uploaded complex structure, allowing users to select a ligand of interest for a FBDD task. Second, when inputting CAND_GEN jobs, a 3D molecular graph of the core fragment labeled with hydrogen atom numbers was provided to aid users in specifying link points. Third, new compound navigation was implemented on the output page, allowing users to browse the prediction results of different compounds on the same page through mouse click. Furthermore, a web-based interactive viewer was implemented to provide 3D visualization of ligand-bound protein macromolecules, powered by NGL (34). In the viewer, users can interactively manipulate the complex structures, showing binding modes from different perspectives.
Physicochemical property analysis
New physicochemical property prediction functions for the generated compounds were added to ACFIS 2.0, providing basic structural information about the ligand/core fragment. The predicted physicochemical properties include SIMLES, molecular weight, number of heavy atoms, number of hydrogen bond donors, number of hydrogen bond acceptors, polar surface area, logP, number of rings, number of aromatic rings, number of rotated bonds and number of aromatic bonds.
Drug/pesticide-likeness evaluation
Another feature of ACFIS 2.0 is the inclusion of drug/pesticide-likeness evaluation to complement the original binding affinity evaluation (Figure 2D). This feature supports the identification of computationally generated compounds that are more likely to be drug/pesticide leads for subsequent synthesis. Three drug-likeness rules or scoring functions are used, including Lipinski's drug-likeness rule (35), the drug-likeness rule of Ghose et al. (36), and a quantitative estimate of drug-likeness (QED) scoring (37). The pesticide-likeness rule proposed by Hao et al. is employed (38). In addition, a synthetic accessibility score based on molecular complexity and fragment contributions is also provided on the output page.
RESULTS
Performance and comparison
The performance of ACFIS 2.0 in generating active ligands from core fragments was assessed by ranking predicted binding affinities. The detailed validation protocol was provided in Supplementary Text S2 and Figure S2. A test dataset of 122 cases covering 51 protein targets and a specific fragment library was used (Supplementary Table S1 and Figure S3). These cases were carefully collected from the literature. Sixty-one tested compounds that exhibited high experimental binding affinities or in vitro activities were considered positive samples; the remaining 61 compounds showed relatively low affinities or activities were considered negative samples. The input protein-core fragment complex structures were obtained by deconstructing the protein-ligand complexes reported in the PDBbind database (39). The CAND_GEN mode was used for testing. The output rank of the tested compound among all generated ligands was the criterion for estimating whether this prediction is true. As shown in Figure 3A, ACFIS 2.0 achieved an accuracy of 88.5%, a precision of 87.3%, a specificity of 86.8%, and a sensitivity value of 90.1%. Compared to previous version (Supplementary Figure S1), the accuracy of ACFIS 2.0 is increased by >10%. Thus, ACFIS 2.0 performed well in discovering fragment-derived compounds with potentially high bioactivity.
Figure 3.

Performance and key feature of ACFIS 2.0. (A) The predictive performance of ACFIS 2.0 validated on a test set of 122 cases. (B) Comparison of key features of ACFIS 2.0 with other computational tools for fragment-based drug discovery (FBDD). Compared to most of other tools, ACFIS 2.0 is one of the few tools that covers the global workflow of FBDD.
A comprehensive comparison of ACFIS 2.0 with other computational tools designed for FBDD was performed (Figure 3B and Supplementary Table S2). Most of tools only focus on a specific step of the FBDD workflow. SEED (15,16) aims to improve the fragment docking accuracy to obtain potent core fragments, while DAIM (17) and eMolFrag (40) were designed for structural fragmentation to extract core fragment from bioactive compounds. Tools for fragment-to-lead optimization are often organized according to the drug design strategies. For example, Frag_PELE (18) and DeepFrag (19) are used to perform fragment growing with a predetermined core fragment. DeLinker (20), SyntaLinker (21) and Ligbuilder (41) can link two or more different core fragments into a new lead compound. On the whole, ACFIS 2.0 is one of the few web-based tools to apply a one-stop workflow for FBDD, featuring pipeline presentation covering fragment library construction, core fragment identification and fragment-to-lead optimization.
Case study 1: optimization of a highly potent MAO-B inhibitor for treating parkinson's disease
Inhibition of MAO-B is an effective treatment for Parkinson's disease (PD) (42). The MAO-B inhibitor, safinamide, is the only one approved Parkinson's drug. To discover more potent MAO-B inhibitors, Jin et al. used ACFIS 2.0 for structural optimization starting from safinamide structure (Figure 4A) (43). First, the crystal structure of the MAO-B and safinamide complex (PDB ID: 2v5z) was inputted into the CORE_GEN mode to obtain a list of computational core fragments. Fragment 1, which had the highest ligand efficiency (LE) among all output fragments, it was selected as the core fragment to start the CAND_GEN job. The complex structure of MAO-B with fragment 1 was uploaded and the Comprehensive Fragment Library was specified for fragment growing. Then, ACFIS 2.0 provided a series of fragment 1-derived compounds as potential new MAO-B ligands. Among them, compound C3 exhibited a lower calculated binding free energy than safinamide, which was inferred to be bioactive toward MAO-B. Eventually, compound C3 was synthesized and assayed. The experimentally-derived MAO-B inhibitory activity of compound C3 (IC50 = 21nM) was 10-fold higher than the inhibitory activity of safinamide (IC50 = 249 nM), demonstrating the dependability of ACFIS 2.0.
Figure 4.

Three cases of fragment-based drug discovery applications using ACFIS 2.0. (A) Design of a monoamine oxidas-B inhibitor for treating Parkinson's disease based on safinamide. (B) Design of a tropomyosin receptor kinase inhibitor for cancer therapies based on larotrectinib. (C) Design of a serotonin transporter inhibitor for treating depression based on vilazodone.
Case study 2: design of a less drug-resistant TRK inhibitor for cancer therapies
Tropomyosin receptor kinases (TRKs) are an important class of anti-tumor targets (44). Larotrectinib was the first approved TRK drug (45), but patients develop resistance problem induced by protein mutations, such as TRKAG595R (46). To seek for less drug-resistant TRK inhibitors, Wang et al. performed FBDD using ACFIS 2.0 (Figure 4B) (32). First, the structure of the TRKAG595R mutant protein was constructed by computationally mutating key residues on the holo-TRK crystal structure (PDB ID: 4aoj). The complex structure of Larotrectinib and TRKAG595R, which built through molecular docking, was submitted for a CORE_GEN job. Fragment 2 computationally exhibited high binding affinity and the highest LE value. Thus, fragment 2 was used as the core fragment. CAND_GEN was then employed to generate compounds based on fragment 2 and the Comprehensive Fragment Library. Dozens of compounds were generated, and the YT3 structure had the highest binding affinity score. Experimental results demonstrated that YT3 inhibited both wild-type TRKA (IC50 = 4.4 nM) and TRKAG595R (IC50 = 1.02 nM).
Case study 3: discovery of a novel SERT inhibitor with improved drug bioavailability for treating depression
The serotonin transporter (SERT) is an important pharmacological target for treating major depressive disorder (47). The SERT inhibitor, vilazodone, is a commercially available anti-depressant drug with insufficient oral bioavailability (F = 47.45%). To design novel anti-depressants with improved bioavailability, Wang et al. performed SERT-based drug design with the aid of ACFIS 2.0 (Figure 4C) (48). The crystal structure of the vilazodone-SERT complex (PDB ID: 7lwd) was uploaded to launch a CORE_GEN operation. Fragment 3 had an acceptable binding free energy and drug-likeness score for fragment optimization. To optimize the interaction between the core fragment and SERT, the adipose chain of fragment 3 was shortened to obtain fragment 4 as the final core fragment. Fragment growing was performed for fragment 4 using CAND_GEN, and the newly generated compound DH4 with a high binding affinity was identified for subsequent structural modification. Compound DH4 exhibits similar binding affinity and significantly improved oral bioavailability (F = 83.28%) compared with vilazodone, proving the successful performance of ACFIS 2.0 in this design.
LIMITATIONS
Despite the enhanced features of ACFIS 2.0, several limitations need to be considered. For instance, the current ACFIS version relies on protein-ligand complex structures to implement core fragment or compound generation. Thus, ACFIS 2.0 cannot be used fail to work when the protein structure of interest is unknown or not available. In addition, the performance of a job may be negatively affected if the starting complex structures have low accurate binding modes. Further, the workflow of ACFIS 2.0 relies on the assumption that adding a new fragment did not affect the binding mode of the core fragment, but sometimes the opposite is true.
CONCLUSION AND OUTLOOK
In summary, ACFIS 2.0 is an upgraded web server that provides a computational pipeline to implement FBDD. While based on the core functionality of ligand generation from the original version, ACFIS 2.0 was optimized by incorporating improved dynamic fragment growing functionality. In addition, the expanded fragment libraries, comprehensive molecular properties analysis, and user-friendly visualization enable users to investigate generated compounds as potential hits, facilitating more comprehensive drug/pesticide molecular exploration. ACFIS 2.0 was 88.5% accurate in generating expected compounds with appropriate binding affinity rankings when tested on 122 cases. Three typical cases of ACFIS 2.0 applications, improving the designs of MAO-B, SERT and TRK inhibitors, demonstrated the applicability of the server. The new upgrades make ACFIS 2.0 more powerful and user-friendly for FBDD. In the future, further improvements to ACFIS 2.0 will be made, including the addition of a protein-fragment binding scoring function to improve fragment screening performance and the addition of a fragment linking strategy to generate more diverse compounds. ACFIS 2.0 benefited from users’ feedback and our upgrade efforts provide better service.
DATA AVAILABILITY
ACFIS 2.0 is freely available at http://chemyang.ccnu.edu.cn/ccb/server/ACFIS2/. The datasets used for the performance validation are provided in the Supplementary data.
Supplementary Material
Contributor Information
Xing-Xing Shi, National Key Laboratory of Green Pesticide, Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, Central China Normal University, Wuhan, 430079, P.R. China.
Zhi-Zheng Wang, National Key Laboratory of Green Pesticide, Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, Central China Normal University, Wuhan, 430079, P.R. China.
Fan Wang, National Key Laboratory of Green Pesticide, Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, Central China Normal University, Wuhan, 430079, P.R. China.
Ge-Fei Hao, National Key Laboratory of Green Pesticide, Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, Central China Normal University, Wuhan, 430079, P.R. China.
Guang-Fu Yang, National Key Laboratory of Green Pesticide, Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, Central China Normal University, Wuhan, 430079, P.R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [32125033, 21837001]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Harrer S., Shah P., Antony B., Hu J.. Artificial intelligence for clinical trial design. Trends Pharmacol. Sci. 2019; 40:577–591. [DOI] [PubMed] [Google Scholar]
- 2. Berdigaliyev N., Aljofan M.. An overview of drug discovery and development. Future Med. Chem. 2020; 12:939–947. [DOI] [PubMed] [Google Scholar]
- 3. Hughes J.P., Rees S., Kalindjian S.B., Philpott K.L.. Principles of early drug discovery. Br. J. Pharmacol. 2011; 162:1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Erlanson D.A., Davis B.J., Jahnke W.. Fragment-based drug discovery: advancing fragments in the absence of crystal structures. Cell Chem. Biol. 2019; 26:9–15. [DOI] [PubMed] [Google Scholar]
- 5. Murray C.W., Rees D.C.. The rise of fragment-based drug discovery. Nat. Chem. 2009; 1:187–192. [DOI] [PubMed] [Google Scholar]
- 6. Harner M.J., Frank A.O., Fesik S.W.. Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR. 2013; 56:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y.S., Strickland C., Voigt J.H., Kennedy M.E., Beyer B.M., Senior M.M., Smith E.M., Nechuta T.L., Madison V.S., Czarniecki M.et al.. Application of fragment-based NMR screening, X-ray crystallography, structure-based design, and focused chemical library design to identify novel microM leads for the development of nM BACE-1 (beta-site APP cleaving enzyme 1) inhibitors. J. Med. Chem. 2010; 53:942–950. [DOI] [PubMed] [Google Scholar]
- 8. Navratilova I., Hopkins A.L.. Fragment screening by surface plasmon resonance. ACS Med. Chem. Lett. 2010; 1:44–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumar A., Voet A., Zhang K.Y.. Fragment based drug design: from experimental to computational approaches. Curr. Med. Chem. 2012; 19:5128–5147. [DOI] [PubMed] [Google Scholar]
- 10. de Souza Neto L.R., Moreira-Filho J.T., Neves B.J., Maidana R., Guimarães A.C.R., Furnham N., Andrade C.H., Silva F.P. Jr. In silico strategies to support fragment-to-lead optimization in drug discovery. Front. Chem. 2020; 8:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Esch I.J.P., Erlanson D.A., Jahnke W., Johnson C.N., Walsh L.. Fragment-to-lead medicinal chemistry publications in 2020. J. Med. Chem. 2022; 65:84–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walsh L., Erlanson D.A., de Esch I.J.P., Jahnke W., Woodhead A., Wren E.. Fragment-to-lead medicinal chemistry publications in 2021. J. Med. Chem. 2023; 66:1137–1156. [DOI] [PubMed] [Google Scholar]
- 13. Wang Z.Z., Shi X.X., Huang G.Y., Hao G.F., Yang G.F.. Fragment-based drug discovery supports drugging ‘undruggable’ protein-protein interactions. Trends Biochem. Sci. 2023; 10.1016/j.tibs.2023.01.008. [DOI] [PubMed] [Google Scholar]
- 14. Bollag G., Tsai J., Zhang J., Zhang C., Ibrahim P., Nolop K., Hirth P.. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012; 11:873–886. [DOI] [PubMed] [Google Scholar]
- 15. Marchand J.R., Caflisch A.. In silico fragment-based drug design with SEED. Eur. J. Med. Chem. 2018; 156:907–917. [DOI] [PubMed] [Google Scholar]
- 16. Majeux N., Scarsi M., Apostolakis J., Ehrhardt C., Caflisch A.. Exhaustive docking of molecular fragments with electrostatic solvation. Proteins. 1999; 37:88–105. [PubMed] [Google Scholar]
- 17. Kolb P., Caflisch A.. Automatic and efficient decomposition of two-dimensional structures of small molecules for fragment-based high-throughput docking. J. Med. Chem. 2006; 49:7384–7392. [DOI] [PubMed] [Google Scholar]
- 18. Perez C., Soler D., Soliva R., Guallar V.. FragPELE: dynamic Ligand Growing within a Binding Site. A Novel Tool for Hit-To-Lead Drug Design. J. Chem. Inf. Model. 2020; 60:1728–1736. [DOI] [PubMed] [Google Scholar]
- 19. Green H., Koes D.R., Durrant J.D.. DeepFrag: a deep convolutional neural network for fragment-based lead optimization. Chem. Sci. 2021; 12:8036–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Imrie F., Bradley A.R., van der Schaar M., Deane C.M.. Deep generative models for 3D linker design. J. Chem. Inf. Model. 2020; 60:1983–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang Y., Zheng S., Su S., Zhao C., Xu J., Chen H.. SyntaLinker: automatic fragment linking with deep conditional transformer neural networks. Chem. Sci. 2020; 11:8312–8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hao G.F., Jiang W., Ye Y.N., Wu F.X., Zhu X.L., Guo F.B., Yang G.F.. ACFIS: a web server for fragment-based drug discovery. Nucleic Acids Res. 2016; 44:W550–W556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yan Y.C., Wu W., Huang G.Y., Yang W.C., Chen Q., Qu R.Y., Lin H.Y., Yang G.F.. Pharmacophore-oriented discovery of novel 1,2,3-benzotriazine-4-one derivatives as potent 4-hydroxyphenylpyruvate dioxygenase inhibitors. J. Agric. Food Chem. 2022; 70:6644–6657. [DOI] [PubMed] [Google Scholar]
- 24. Yang J.F., Chen W.J., Zhou L.M., Hewage K.A.H., Fu Y.X., Chen M.X., He B., Pei R.J., Song K., Zhang J.H.et al.. Real-time fluorescence imaging of the abscisic acid receptor allows nondestructive visualization of plant stress. ACS Appl. Mater. Interfaces. 2022; 14:28489–28500. [DOI] [PubMed] [Google Scholar]
- 25. Liu J., Dai H., Wang B., Liu H., Tian Z., Zhang Y.. Exploring disordered loops in DprE1 provides a functional site to combat drug-resistance in Mycobacterium strains. Eur. J. Med. Chem. 2022; 227:113932. [DOI] [PubMed] [Google Scholar]
- 26. Wang E., Sun H., Wang J., Wang Z., Liu H., Zhang J.Z.H., Hou T.. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 2019; 119:9478–9508. [DOI] [PubMed] [Google Scholar]
- 27. Hao G.F., Wang F., Li H., Zhu X.L., Yang W.C., Huang L.S., Wu J.W., Berry E.A., Yang G.F.. Computational discovery of picomolar Q(o) site inhibitors of cytochrome bc1 complex. J. Am. Chem. Soc. 2012; 134:11168–11176. [DOI] [PubMed] [Google Scholar]
- 28. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A.et al.. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022; 50:D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang J.F., Wang F., Jiang W., Zhou G.Y., Li C.Z., Zhu X.L., Hao G.F., Yang G.F.. PADFrag: a database built for the exploration of bioactive fragment space for drug discovery. J. Chem. Inf. Model. 2018; 58:1725–1730. [DOI] [PubMed] [Google Scholar]
- 31. Bajusz D., Wade W.S., Satała G., Bojarski A.J., Ilaš J., Ebner J., Grebien F., Papp H., Jakab F., Douangamath A.et al.. Exploring protein hotspots by optimized fragment pharmacophores. Nat. Commun. 2021; 12:3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Z.Z., Wang M.S., Wang F., Shi X.X., Huang W., Hao G.F., Yang G.F.. Exploring the kinase-inhibitor fragment interaction space facilitates the discovery of kinase inhibitor overcoming resistance by mutations. Brief. Bioinf. 2022; 23:bbac203. [DOI] [PubMed] [Google Scholar]
- 33. Shi X.X., Wang Z.Z., Wang Y.L., Huang G.Y., Yang J.F., Wang F., Hao G.F., Yang G.F.. PTMdyna: exploring the influence of post-translation modifications on protein conformational dynamics. Brief. Bioinf. 2022; 23:bbab424. [DOI] [PubMed] [Google Scholar]
- 34. Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlic A., Rose P.W.. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018; 34:3755–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J.. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 2001; 46:3–26. [DOI] [PubMed] [Google Scholar]
- 36. Ghose A.K., Viswanadhan V.N., Wendoloski J.J.. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999; 1:55–68. [DOI] [PubMed] [Google Scholar]
- 37. Bickerton G.R., Paolini G.V., Besnard J., Muresan S., Hopkins A.L.. Quantifying the chemical beauty of drugs. Nat. Chem. 2012; 4:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hao G., Dong Q., Yang G.. A comparative study on the constitutive properties of marketed pesticides. Mol. Inf. 2011; 30:614–622. [DOI] [PubMed] [Google Scholar]
- 39. Wang R., Fang X., Lu Y., Wang S.. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 2004; 47:2977–2980. [DOI] [PubMed] [Google Scholar]
- 40. Liu T., Naderi M., Alvin C., Mukhopadhyay S., Brylinski M.. Break down in order to build up: decomposing small molecules for fragment-based drug design with eMolFrag. J. Chem. Inf. Model. 2017; 57:627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yuan Y., Pei J., Lai L.. LigBuilder 2: a practical de novo drug design approach. J. Chem. Inf. Model. 2011; 51:1083–1091. [DOI] [PubMed] [Google Scholar]
- 42. Tan Y.Y., Jenner P., Chen S.D.. Monoamine oxidase-B inhibitors for the treatment of Parkinson's disease: past, present, and future. J. Parkinsons Dis. 2022; 12:477–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jin C.F., Wang Z.Z., Chen K.Z., Xu T.F., Hao G.F.. Computational fragment-based design facilitates discovery of potent and selective monoamine oxidase-B (MAO-B) inhibitor. J. Med. Chem. 2020; 63:15021–15036. [DOI] [PubMed] [Google Scholar]
- 44. Cocco E., Scaltriti M., Drilon A.. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018; 15:731–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Scott L.J. Larotrectinib: first global approval. Drugs. 2019; 79:201–206. [DOI] [PubMed] [Google Scholar]
- 46. Fuse M.J., Okada K., Oh-Hara T., Ogura H., Fujita N., Katayama R.. Mechanisms of resistance to NTRK inhibitors and therapeutic strategies in NTRK1-rearranged cancers. Mol. Cancer Ther. 2017; 16:2130–2143. [DOI] [PubMed] [Google Scholar]
- 47. Cowen P.J. Serotonin and depression: pathophysiological mechanism or marketing myth?. Trends Pharmacol. Sci. 2008; 29:433–436. [DOI] [PubMed] [Google Scholar]
- 48. Wang Z.Z., Yi C., Huang J.J., Xu T.F., Chen K.Z., Wang Z.S., Xue Y.P., Lu J.L., Nie B., Zhang Y.J.et al.. Deciphering nonbioavailable substructures improves the bioavailability of antidepressants by serotonin transporter. J. Med. Chem. 2023; 66:371–383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ACFIS 2.0 is freely available at http://chemyang.ccnu.edu.cn/ccb/server/ACFIS2/. The datasets used for the performance validation are provided in the Supplementary data.