

Abstract
MicroRNAs (miRNAs) play a crucial role in regulating gene expression. MicroRNA expression levels fluctuate, and point mutations and methylation occur in cancer cells; however, to date, there have been no reports of carcinogenic point mutations in miRNAs. MicroRNA 142 (miR‐142) is frequently mutated in patients with follicular lymphoma, diffuse large B‐cell lymphoma, chronic lymphocytic leukemia (CLL), and acute myeloid leukemia/myelodysplastic syndrome (AML/MDS). To understand the role of miR‐142 mutation in blood cancers, the CRISPR‐Cas9 system was utilized to successfully generate miR‐142‐55A>G mutant knock‐in (Ki) mice, simulating the most frequent mutation in patients with miR‐142 mutated AML/MDS. Bone marrow cells from miR‐142 mutant heterozygous Ki mice were transplanted, and we found that the miR‐142 mutant/wild‐type cells were sufficient for the development of CD8+ T‐cell leukemia in mice post‐transplantation. RNA‐sequencing analysis in hematopoietic stem/progenitor cells and CD8+ T‐cells revealed that miR‐142‐Ki/+ cells had increased expression of the mTORC1 activator, a potential target of wild‐type miR‐142‐3p. Notably, the expression of genes involved in apoptosis, differentiation, and the inhibition of the Akt–mTOR pathway was suppressed in miR‐142‐55A>G heterozygous cells, indicating that these genes are repressed by the mutant miR‐142‐3p. Thus, in addition to the loss of function due to the halving of wild‐type miR‐142‐3p alleles, mutated miR‐142‐3p gained the function to suppress the expression of distinct target genes, sufficient to cause leukemogenesis in mice.
Keywords: acute myeloid leukemia, microRNA, miR‐142, model mice, single nucleotide mutation
Recently, single nucleotide mutations and the upregulation or downregulation of miRNA expression have been observed in various cancers, however, carcinogenesis caused by single nucleotide mutations in miRNAs has not yet been reported. To elucidate the role of the miR‐142 mutation in AML, the CRISPR‐Cas9 system was used to generate miR‐142‐55A>G mutation knock‐in mice (Ki), which were most frequently identified in patients. Bone marrow cells were transplanted from Ki heterozygous mice and miR‐142‐Ki heterozygous mice were found to develop CD8+ T‐cell leukemia. Evidence is provided for carcinogenesis due to a gain‐of‐function mutation in microRNA using an in vivo study.
Abbreviations
- HSPCs
hematopoietic stem/progenitor cells
- Ki
knock‐in
- miRNAs
microRNAs
- MDS
myelodysplastic syndrome
- TCGA
the Cancer Genome Atlas
1. INTRODUCTION
miRNAs are small noncoding RNAs that consist of 20–25 nucleotides that are not translated into proteins. miRNAs become mature single‐stranded RNAs in the cell, promote mRNA degradation, and inhibit protein translation. Thus, they are associated with various regulatory networks. Recent reports have indicated that single nucleotide mutations, 1 , 2 , 3 methylations, 4 , 5 and the upregulation or downregulation of miRNA expression have been observed in various cancers 6 , 7 , 8 , 9 ; however, no studies have reported carcinogenesis caused by single nucleotide mutations in miRNAs.
We focused on one of the miRNAs, microRNA 142 (miR‐142), which is a highly conserved miRNA, abundantly expressed in the hematopoietic system and reportedly regulates the differentiation and function of megakaryocytes, 10 erythrocytes, 11 dendritic cells, 12 and most frequently T‐cells. 13 , 14 , 15 , 16 Recently, miR‐142 has been reported to mutate frequently in hematologic cancers. Single nucleotide mutations within the miR‐142 gene, including the miR‐142 precursor, have been documented to occur throughout the gene in various forms of leukemia, such as follicular lymphoma, 17 , 18 chronic lymphocytic leukemia, 19 diffuse large B‐cell lymphoma, 20 , 21 , 22 , 23 and AML/MDS. 2 , 3 We focused on the miR‐142‐55A>G mutation, which is frequently found in patients with AML/MDS. 2 , 3 The miR‐142 sequence is 100% identical between humans and mice. In addition, homozygous knockout mice with the entire length of miR‐142 deleted show abnormal differentiation of their immune system cells, but there have been no reports on heterozygous mice. 14 , 24 However, most of the miR‐142 abnormalities in AML/MDS patients are single nucleotide substitutions and heterozygous. Therefore, we hypothesized that the heterozygosity of a single nucleotide mutation within the seed sequence of miR‐142‐3p is responsible for the development of leukemia.
In this study, to understand the role of the miR‐142 mutation in leukemia, we utilized the CRISPR‐Cas9 system to generate miR‐142 mutation Ki mice to simulate the most frequently identified mutation in patients. We analyzed miR‐142‐Ki heterozygous mice and showed that single nucleotide mutations in miRNAs are associated with carcinogenesis.
2. MATERIALS AND METHODS
2.1. Animal welfare
Animal experiments were carried out in accordance with the Regulations for Animal Experiments at our university and Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science, and Technology. Mice were maintained under specific‐pathogen‐free (SPF) conditions with ad libitum food and water. The light cycle was regulated to 7:00 AM lights on and 7:00 PM lights off. C57BL/6N and CD‐1 mice were purchased from Clea Japan (Tokyo, Japan) and Charles River Laboratories Japan (Kanagawa, Japan), respectively. The established mouse lines were backcrossed to C57BL/6N at least six times. Approval was obtained from the Institutional Animal Care and Use Committee and the DNA Experiment Committee of Kumamoto University (Approval Number for Animal Experiments: A2020‐016) (Approval Number for DNA Experiments: 29‐053).
2.2. Generation of the miR‐142‐55A>G mouse line
The first trial to produce the point mutation was performed using ES cells. pX330‐sgRNA9 was constructed by cloning a set of sgRNA oligos (5′‐caccGCACTACTAACAGCACTGGA‐3′ and 5′‐aaacTCCAGTGCTGTTAGTAGTGC‐3′) into a BbsI‐digested pX330 vector (#42230; Addgene). The 6NK‐7 ES cells, which were established from C57BL/6N mice in our laboratory, and were transfected with 2 μg of pX330‐sgRNA9 and 3 μg of ssODN with the mutated sequence (5′‐CAGACAGACAGTGCAGTCACCCATAAAGTAGAAAGCACTACTAACAGCACTGGAGGGTGTGGTGTTTCCTACTTTATGGATGAGTGCACTGTGGGCTTCGGAGACCACGCCACGCCGCGGC‐3′) using the Xfect mESC Transfection Reagent (Clontech Laboratories), following the manufacturer's protocol. After 2 days, the transfected ES cells were plated at clonal density (3000 pieces/dish) into a 10‐cm dish and cultured for 7 days. The culture conditions of the CO2 incubator were set at 6.2% (100% humidity). On day 8, colonies were picked and stocked. Subsequently, PCR was performed with primers for miR‐142‐S (5′‐GGGAAGAAGGTTACAAAGAGG‐3′) and miR‐142‐R (5′‐TGAGAGATGCTCACCTGTTTC‐3′) and the resulting product was sequenced. We could not obtain clones with the targeted point mutation but obtained clone #30, which had an 8‐bp deletion. Clone #30 ES cells were aggregated with morulae from ICR mice. Chimeric mice were mated with C57BL/6N females.
To introduce the miR‐142‐55A>G point mutation, in vitro fertilization was performed using females from line #30 and a wild‐type (WT) C57BL/6N male. Thereafter, fertilized eggs together with Cas9 protein (317–08441; Nippon Gene), tracrRNA (GE‐002; FASMAC), synthetic crRNA (FASMAC), and ssODN were introduced into C57BL/6N by electroporation, as previously described. 25 Synthetic crRNAs were designed for aagtagaagcactactaaca (GGG) at the junction of the 8‐bp deletion. The electroporation solution contained 10 μM of tracrRNA, 10 μM of synthetic crRNA, 0.1 μg/μL of Cas9 protein, and 1 μg/μL of ssODN in Opti‐MEM I Reduced Serum Medium (31,985,062; Thermo Fisher Scientific). DNA was extracted from each of the 14 pups born, after which PCR was performed with primers miR‐142‐S and miR‐142‐R, and the resulting product was sequenced.
2.3. Genotyping
Genotype was determined by PCR using primer sets to determine WT and mutant alleles. WT alleles were detected by PCR with 30 cycles of annealing at 65°C using miR‐142‐WT‐F (5′‐ TAACAGCACTGGAGGGTGTA‐3′) and miR‐142‐R (5′‐TGAGAGATGCTCACCTGTTTC‐3′). Mutant alleles were detected by PCR with 30 cycles of annealing at 66°C using miR‐142‐G‐F (5′‐TAACAGCACTGGAGGGTGTG‐3′) and miR‐142‐R.
2.4. Quantification of miRNA expression
Total RNA was extracted from mouse bone marrow (BM) samples using ISOGEN (Nippon Gene). cDNA synthesis was performed from 10 ng of total RNA using a TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems), according to the manufacturer's instructions. Thereafter, miRNA expression levels were quantified using a TaqMan MicroRNA Assay (Applied Biosystems) for miR‐142‐5p in a Thermal Cycler Dice TP800 Real‐Time PCR system (TaKaRa). Each assay was performed in technical triplicates. miR‐142 expression levels were normalized to Actb/β‐actin as an internal control. The average threshold cycle (Ct) for three replicates per sample was used to calculate ΔCt. Finally, the relative quantification for gene expression was calculated using the 2−ΔΔCt method.
2.5. Bone marrow transplantation
Whole BM (5 × 106 cells) from CD45.2+ donors was injected into the tail veins of irradiated CD45.1+ recipient mice. Irradiation was performed using an X‐ray irradiator delivering 8.5 Gy. For leukemia modeling, whole BM (1 × 106 cells) from moribund donors was injected into irradiated (5.5 Gy) CD45.1+ recipient mice. Animals that showed no engraftment of donor cells were excluded from further analysis. Mice exhibiting declining health status were sacrificed, and tissues were taken for analysis.
2.6. Flow cytometry and antibodies
Flow cytometry and cell sorting were performed using antihuman or antimurine antibodies, as indicated in Table S1. The lineage mixture solution contained biotin‐conjugated anti‐Gr1, B220, CD4, CD8α, Ter119, and IL‐7Rα antibodies. All flow cytometric analyses were performed on a FACSVerse or FACSCantoII cytometer (BD Biosciences), and cell sorting was performed on a FACSAria II cytometer (BD Biosciences).
2.7. Complete blood count and cell separation
A complete blood count (CBC) was performed using a Celltac Alpha MEK‐6450 hematology analyzer (Nihon Kohden, Tokyo, Japan), with 10 μL of peripheral blood (PB) collected from the tail. LSK (Lineage−Sca‐1+c‐Kit+) and CD8+ T‐cells were purified from BM by magnetic separation over a MACS LS Column (Miltenyi Biotec Inc., North Rhine‐Westphalia, Germany), following the manufacturer's instructions.
2.8. Cytospin and May–Grünwald–Giemsa staining
BM cells were cytospun using a Shandon Cytospin 3 Cytocentrifuge (Block Scientific) at 100 × g for 3 min. Next, air‐dried slides were stained in May–Grünwald solution (Sigma‐Aldrich, Tokyo, Japan) for 5 min. After a brief wash with water, the slides were stained in Giemsa solution (Sigma‐Aldrich) for 20 min. The slides were finally washed in distilled water and allowed to air dry before microscopic analysis.
2.9. Histological analysis
Mice were euthanized and organs were removed in PBS containing 2% FBS. Thereafter, organs were fixed with 15% formalin neutral buffer solution (Cat#: 067–02397; FUJIFILM Wako Pure Chemical Corporation), and 4‐μm thick paraffin‐embedded sections were prepared. Next, H&E staining was carried out using Mayer's hematoxylin solution (Cat #: 131–09665; FUJIFILM Wako Pure Chemical Corporation), eosin alcohol solution, and acid extract (Cat #: 050–06041; FUJIFILM Wako Pure Chemical Corporation). Finally, images were captured using a BX53 microscope (Olympus Corporation).
2.10. RNA sequencing
RNA was extracted from LSK and CD8+ T‐cells. Total RNA was extracted using ISOGEN (Nippon Gene). RNA sequencing was performed by the Liaison Laboratory Research Promotion Center (LILA), Kumamoto University. Library cDNAs were prepared using a NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs) and an Ultra II Directional RNA Library Prep Kit (Illumina) and sequenced using an Illumina NextSeq 500 system (Illumina) with a Nextseq 500/550 High Output v2.5 Kit (Illumina) to obtain single‐end 75‐nt reads. The resulting reads were aligned to the mouse genome UCSC mm10 using STAR ver.2.6.0a software after trimming to remove the adapter sequence and low‐quality ends using Trim Galore! V0.5.0 (cutadapt v1.16). Gene expression levels were measured as counts, and transcripts per million were determined using RSEM v1.3.1. A GTF file was derived from UCSC mm10. Gene Ontology analysis was performed using Gene Set Enrichment Analysis (GSEA) in the clusterProfiler package ver.4.1.0. GSEA software was downloaded from the websites of the Broad Institute (http://software.broadinstitute.org/gsea/downloads.jsp).
2.11. Quantitative RT‐PCR
Total RNA was extracted from mouse BM samples and hematopoietic cells using ISOGEN (Nippon Gene), and 500 ng of RNA was converted to cDNA using a PrimeScript 1st strand cDNA Synthesis Kit (6110A; TaKaRa). cDNA aliquots were used for quantitative RT‐PCR (RT‐qPCR). For RT‐qPCR, TB Green Premix Ex Taq II (RR820A; TaKaRa) was used as the Taq polymerase, and reactions were performed using the intercalator method in a Thermal Cycler Dice TP800 Real‐Time PCR system (TaKaRa). In experiments using relative quantification, expression levels were normalized to Actb/β‐actin expression. The sequences of all primers used in this study are listed in Table S2.
2.12. Statistical analysis
All statistical tests were performed using GraphPad Prism version 9 (GraphPad Software). The significance of differences was measured using an unpaired two‐tailed Student's t‐test, Mann–Whitney nonparametric test, or chi‐squared test. A p‐value <0.05 was considered significant.
3. RESULTS
3.1. Generation of miR‐142‐55A>G mice using the CRISPR‐Cas9 system
To determine whether a single nucleotide mutation in the miR‐142‐3p seed sequence (miR‐142‐3p‐55A>G), 2 , 3 which has been recurrently detected in patients with AML (Figure 1A), drives leukemogenesis, the same mutation was introduced into the murine Mir142 gene with the same sequence as the human gene. The first attempt at genome editing using 6NK‐7 ES cells (C57BL/6N) did not result in the desired 55A>G mice but resulted in 8‐bp‐deficient mice (C57BL/6N‐Mir142em1Card) (Figure 1B). For the second attempt at genome editing, fertilized eggs from the 8‐bp‐deficient mice were used and yielded the desired miR‐142‐55A>G (miR‐142‐Ki) mice (C57BL/6N‐Mir142em6Card) (Figure 1B). MicroRNAFold, a web server that predicts the hairpin structure of miRNAs in the genome using the ab initio method, revealed that the insertion of the miR‐142‐55A>G mutation forms the hairpin structure with equal efficiency (Figure S1a,b). 26 , 27 A TaqMan miRNA assay was performed to determine the actual miRNA expression levels. In this assay, the miR‐142‐5p kit was applied to determine the miR‐142 expression level because the miR‐142‐55A>G mutation prevents precise assessments with the miR‐142‐3p kit. miRNA expression levels were confirmed to be equal to those of the WT, even when replaced by miR‐142‐55A>G (Figure S1c). We designed forward primers for the position where the single nucleotide mutation was inserted. Genotyping confirmed that the mutated mice only had the designated single nucleotide mutation, which was confirmed by Sanger sequencing (Figure 1C,D).
FIGURE 1.
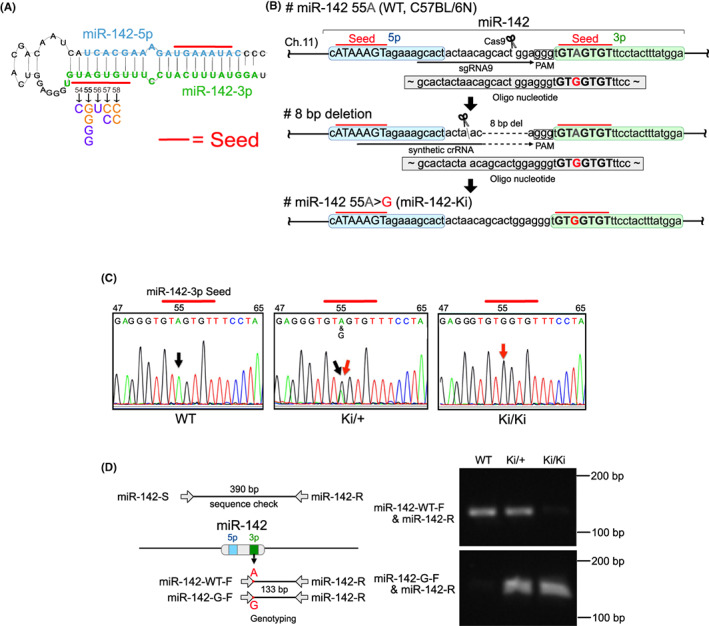
Generation of miR‐142‐55A>G mice using the CRISPR‐Cas9 system. (A) miR‐142 pre‐miR hairpin showing the position and nature of the individual single nucleotide variants identified in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) patients. Numbering is based on the pre‐miR sequence position. Mature miRNA sequences are shown in bold with seeds underlined. Wild‐type miR‐142‐3p is shown in green, with mutations identified by TCGA in orange and those identified by Thol et al. in purple. (B) Strategy for the CRISPR‐Cas9 system in conjunction with miR‐142‐3p. (C) Sequence analysis confirmed the mutation of miR‐142‐3p (nucleotides in the seed sequence are marked with red). A black arrow indicates the correct nucleotide, and a red arrow indicates the mutated nucleotide. (D) Verification of the introduced mutation using genotyping and sequence analysis. The position of each primer is shown. The primer sequences were designed for the wild‐type (A) and Ki (G) by aligning the right end of the forward primer (miR‐142‐WT‐F, miR‐142‐G‐F) with the position where the single nucleotide mutation was inserted.
3.2. miR‐142‐55A>G mutant mice show abnormal lymphocyte cell differentiation
According to Mendelian ratios, the number of miR‐142‐55A>G mutant Ki mice generated was significantly lower for homozygous mice (Figure 2A). To determine the phenotype of miR‐142‐55A>G mutant mice, we observed the survival of WT (+/+), miR‐142‐Ki heterozygous (Ki/+), and miR‐142‐Ki homozygous (Ki/Ki) mice. The results showed that Ki/Ki mice died within the first year of birth (median survival; 147 days versus undetermined, p < 0.0001), some Ki/+ mice began to die after 150 days, and only a few WT mice died during the observation period (Figure 2B). To elucidate the cause of death in the miR‐142‐55A>G mutant mice, we dissected and analyzed the 150‐day‐old mice, revealing that the spleen was enlarged in miR‐142‐Ki/+ and Ki/Ki mice (Figure 2C,D), showing phenotypic splenomegaly comparable with that previously reported for miR‐142‐KO mice. 14
FIGURE 2.
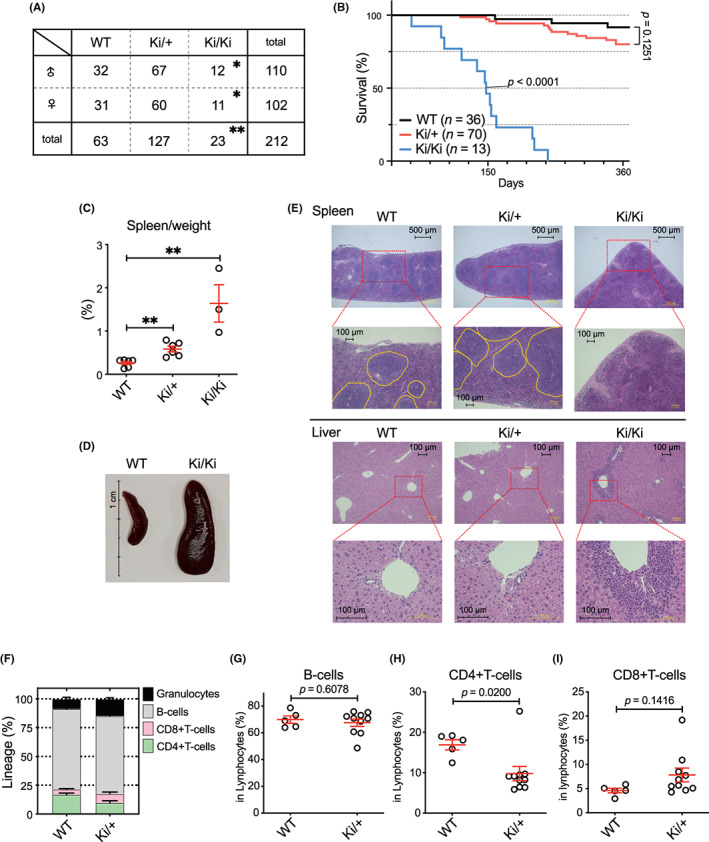
miR‐142‐55A>G mutant mice showed abnormal lymphocyte cell differentiation. (A) The genotypic ratio of miR‐142‐55A>G Ki mice. Genotype ratios for miR‐142‐55A>G Ki mice. *p < 0.05, and **p < 0.01 using the chi‐squared test. (B) Kaplan–Meier analysis of the survival of mice transplanted with wild‐type (WT; black, n = 36), Ki/+ (red, n = 70), or miR‐142 Ki/ Ki (blue, n = 13). A log‐rank test calculated statistical significance values between WT and the other groups. (C) Relative weight of the spleen of 150‐day‐old WT, Ki/+, and Ki/Ki mice (n = 3–6). (D) Spleens were removed from WT (left) and Ki/Ki (right) littermates after genotyping at 150 days. (E) Histologic features of the spleen (top panel) and liver (bottom panel) of 150‐day‐old WT, Ki/+, Ki/Ki mice. (F) Proportion of granulocytes, B‐cells, CD8+ T‐cells, and CD4+ T‐cells from FACS in the peripheral blood (PB) of WT (n = 5) and Ki/+ (n = 10) mice. (G–I) Proportion of B‐cells, CD4+ T‐cells, and CD8+ T‐cells in PB lymphoid cells.
Histological analysis of the grossly enlarged spleen revealed that the structure of the spleen was greatly altered in both Ki/+ and Ki/Ki mice (Figure 2E, top panel). The tissue structure was generally preserved in Ki/+ mice, but the white pulp was larger; however, the white pulp tissue was disrupted in Ki/Ki mice. Furthermore, abnormal cells infiltrated the liver in Ki/Ki mice (Figure 2E, bottom panel). Next, flow cytometry analysis of PB from miR‐142‐Ki/+ mice revealed an increase in granulocytes (Figure 2F), a decrease in CD4+ T‐cells, and a mild increase in CD8+ T‐cells within the lymphocyte lineage (Figure 2G,H). Taken together, T‐cell differentiation in PB was affected by the miR‐142‐55A>G mutation, even in heterozygous individuals.
3.3. miR‐142‐55A>G mutant mice develop CD8+ T‐cell leukemia
To determine whether the miR‐142‐55A>G mutation in blood cells drives the development of leukemia, we transplanted BM cells isolated from WT (+/+), miR‐142‐Ki heterozygous (Ki/+), and miR‐142‐Ki homozygous (Ki/Ki) mice into lethally irradiated Ly5.1+ recipient mice. Their hematopoietic phenotypes were then observed by collecting blood cell counts monthly (Figure 3A). Both the miR‐142‐Ki/+ and Ki/Ki mice died within a year of transplantation (median survival; 238 days versus undetermined, p < 0.0001, 147 days versus undetermined, p < 0.0001), and only a few WT mice died during this observation period (Figure 3B). CBCs showed lower white blood cell (WBC) levels in both the Ki/+ and Ki/Ki mice 2 months post‐transplantation, which showed impaired hematopoiesis phenotypically that was comparable with that previously reported for miR‐142‐KO mice. 24 , 28 Moribund Ki/+ and Ki/Ki mice showed increased WBC counts and decreased hemoglobin levels and platelet counts 5 months post‐transplantation (Figure 3C, Figure S2a,b). The Ki/+ (Pre) mice were referred to as Ki/+ mice at the pre‐disease stage 2 months post‐transplantation and Ki/+ (Leu) as moribund Ki/+ mice showing an expansion of leukemic cells. Flow cytometry analysis revealed that there was no significant change in hematopoietic differentiation in PB in the WT and Ki/+ (Pre) mice; however, Ki/+ (Leu) mice showed massive expansion of the CD4+/CD8+ T‐cells at the expense of B‐cells in the PB compared with those of WT mice (Figure 3D,E).
FIGURE 3.
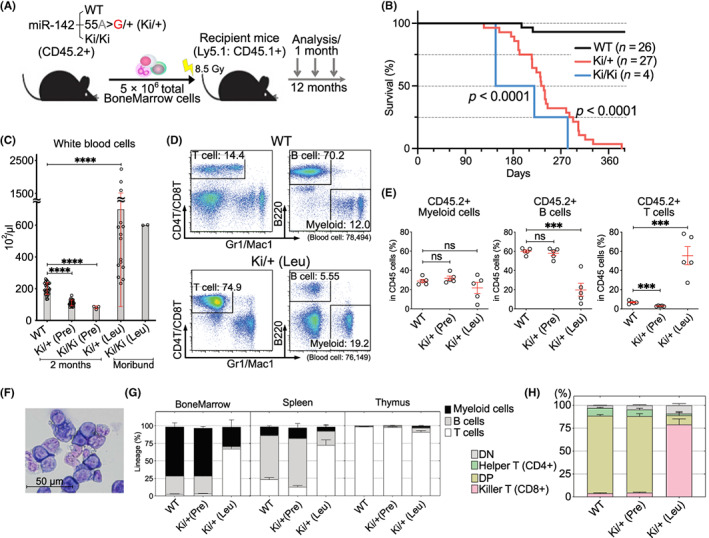
Development of miR‐142‐55A>G mutant mice with CD8+ T‐cell leukemia. (A) Diagram of bone marrow (BM) generation. Wild‐type (WT), miR‐142 55A>G/+ (Ki/+), or miR‐142 55A>G/55A>G (Ki/Ki) BM cells were transplanted into lethally irradiated WT (CD45.1+) hosts. The resulting mice were analyzed 12 months post‐transplantation. (B) Kaplan–Meier analysis of the survival of mice transplanted with WT (black, n = 26), Ki/+ (red, n = 27), or miR‐142 Ki/Ki (blue, n = 4). Statistically significant values between WT and other groups were calculated using a log‐rank test. (C) Complete blood counts (CBCs) in the peripheral blood (PB) of WT, Ki/+, and Ki/Ki mice (n = 4–27) 2 months post‐transplantation, moribund Ki/+ mice (n = 15), and moribund Ki/Ki mice (n = 2). (D) Representative FACS plots showing the proportion of CD4+/CD8+ T‐cells, B220+ B‐cells, and Gr1+/Mac1+ myeloid cells in the WT (top panels) or Ki/+ − Leukemia (Ki/+ [Leu], lower panels) from recipients of transplanted BM. (E) Proportion of CD45.2+ cells in myeloid cells, B‐cells, and CD4+/CD8+ T‐cells in the PB. Hematopoietic differentiation is biased toward T‐cells in Ki/+ (Leu). (F) Cytospin preparation of the BM from Ki/+ (Leu) mice, observed using May–Grünwald–Giemsa staining. (G) Myeloid, B‐cell, and T‐cell proportions from the FACS‐sorted BM, thymus, and spleen. Pooled data are shown from 3–5 independent experiments (WT, n = 3; Ki/+ [Pre], n = 3; Ki/+ [Leu], n = 5). (H) Percentage of CD4+ as helper T‐cells and CD8+ as killer T‐cells among the CD45.2+ cells in the thymus of WT, Ki/+ (Pre) mice (n = 3) 2 months post‐transplantation and moribund Ki/+ (Leu) mice (n = 4) at the time of sacrifice. Data are shown as the mean ± SEM.
miR‐142‐55A>G Ki/+ (Leu) mice showed expansion of immature CD8+ T‐cells in the PB and BM (Figure 3F), and this was also observed in the spleen (Figure 3G). Consistent with the expansion of T‐cells, the weight of the thymus in Ki/+ (Leu) mice was significantly increased compared with that of WT and Ki/+ (Pre) mice (Figure S2c). Furthermore, flow cytometry analysis of the thymus revealed a marked expansion of CD8+TCRb+ T‐cells (Figure 3H, Figure S2d,e), showing monoclonal expansion, which was confirmed by TCR repertoire analysis using RNA sequencing of the T‐cells in leukemic Ki/+ mice (Figure S3a,b). Collectively, miR‐142‐55A>G expression was sufficient to drive the malignant transformation of T‐cells after transplantation in mice.
3.4. miR‐142‐55A>G induces the transplantable capacity of T‐cell leukemia
The miR‐142‐55A>G heterozygous cells developed T‐cell leukemia in recipient mice. Thus, to assess the transplantable capacity of the leukemic cells, we performed secondary transplantation of the BM cells isolated from two Ki/+ leukemic mice into sublethally irradiated Ly5.1+ recipient mice (Figure 4A). All recipient mice in two cohorts died 60 days post‐transplantation (Figure 4B). Secondarily transplanted Ki/+ (Leu) mice showed a stronger increase in WBC counts and decreased hemoglobin levels and platelet counts at 1 month post‐transplantation compared with those in Ki/+ (Leu) mice at the time of transplantation (Figure S4a–c). Flow cytometry analysis revealed a marked expansion of T‐cell leukemia in the secondary recipient mice compared with that in the primary recipient mice (Figure 4C, Figure S4d–g). In addition, we observed that secondary recipients showed mild increases in thymic weight but significantly increased spleen and liver weights (Figure 4D–F). Thus, the miR‐142 mutant was able to induce the transplantable capacity of T‐cell leukemia, resulting in progressive leukemia in serial transplantation.
FIGURE 4.
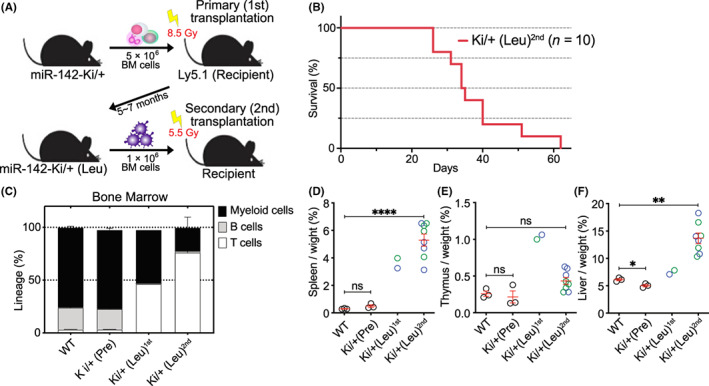
miR‐142‐55A>G enabled T‐cell leukemia to be transplanted. (A) Diagram of the second bone marrow (BM) transplantation. Ki/+ (Leu) (CD45.2+) BM cells were transplanted into antifractionally lethally irradiated Ly5.1 (CD45.1+) hosts. The resulting mice were analyzed 1–2 months post‐transplantation. (B) Kaplan–Meier analysis of the survival of second transplantation mice transplanted with Ki/+ (Leu) (red, n = 10). (C) Myeloid, B‐cell, and T‐cell proportions from the FACS‐sorted cells in the BM of wild‐type and Ki/+ (Pre) mice (n = 3) 2 months post‐transplantation, moribund primary transplantation Ki/+ (Leu) (1st, n = 2) mice, and moribund secondary transplantation Ki/+ (Leu) (2nd, n = 4) mice at the time of sacrifice. Data are shown as the mean ± SEM. (D–F) Relative weight of the spleen, thymus, and liver of +/+, Ki/+ (Pre) mice (n = 3) 2 months post‐transplantation, moribund primary transplantation Ki/+ (Leu) (1st, n = 2) mice, and moribund secondary transplantation Ki/+ (Leu) (2nd, n = 8) mice at the time of sacrifice.
3.5. miR‐142‐55A>G suppresses T‐cell differentiation regulators and promotes leukemic proliferation
To understand how the miR‐142 mutant induced the development of T‐cell leukemia, we performed RNA sequencing of lineage−Sca‐1+c‐Kit+ (LSK) HSPCs, CD8+ T‐cells (T‐cell) from two WT and Ki/+ (Pre) mice 2 months post‐transplantation, and three leukemic Ki/+ mice (Ki/+ (Leu)). Principal components analysis (PCA) revealed that HSPCs or T‐cells in the WT and Ki/+ (Pre) mice were proximally located, while both the HSPCs and T‐cells isolated from the three Ki/+ (Leu) mice were diversely located and apart from those of the WT and Ki/+ (Pre) mice (Figure 5A, Figure S5a,b). The results indicated that there were progressive transcriptional changes in the HSPCs and T‐cells during the development of T‐cell leukemia. In addition, GSEA revealed that leukemic T‐cells in Ki/+ (Leu) mice showed significant positive enrichment of the genes involved in cancer proliferation, such as the hallmark Myc and mTORC1 pathways, compared with T‐cells in WT mice (Figure 5B). In contrast, gene sets involved in immune function, such as the inflammatory response and apoptosis, were decreased in leukemic T‐cells in Ki/+ (Leu) mice. Notably, Ki/+ (Pre) HSPCs were inverted from Ki/+ (Leu) T‐cells, with downregulated gene sets such as Myc and mTOR target and upregulated gene sets associated with immune function (Figure 5B).
FIGURE 5.
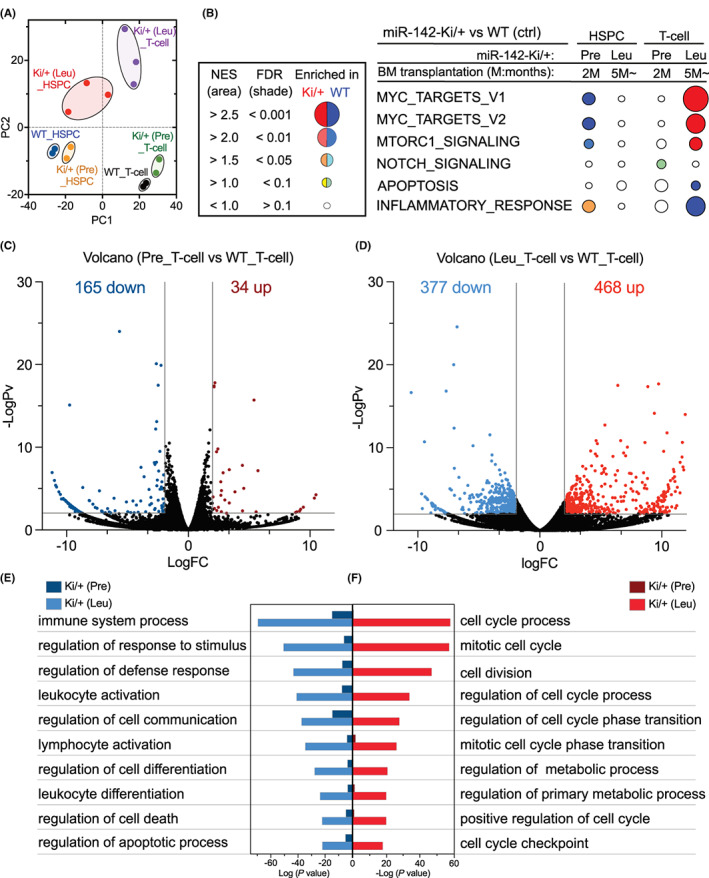
miR‐142‐55A>G suppressed T‐cell differentiation regulators and promoted leukemic proliferation. (A) Principal component analysis based on the gene expression of hematopoietic stem/progenitor cells (HSPCs) and T‐cells isolated from wild‐type (WT) (n = 2), Ki/+ (Pre) (n = 2), and Ki/+ (Leu) (n = 3) mice. (B) GSEA schematic of hallmark pathway genes that fluctuated in HSPCs and T‐cells of Ki/+ (Pre) mice and Ki/+ (Leu) mice. (C, D) Volcano plots showing differentially expressed genes (DEGs) in Ki/+ (Pre)_T‐cells versus the WT_T cells (C) and Ki/+ (Leu)_T‐cells versus the WT_T cells (D) (p < 0.05 colored dots, WT, n = 2; Ki/+ (Pre), n = 2; Ki/+ (Leu), n = 3). (E, F) GO analysis of the upregulated and downregulated genes in Ki/+ (Leu) T‐cells and Ki/+ (Pre) T‐cells detected in (C) and (D).
Additionally, we found 325, 258, 34, and 468 upregulated genes and 114, 103, 165, and 377 downregulated genes in the HSPCs in Ki/+ (Pre), HSPCs in Ki/+ (Leu), T‐cells in Ki/+ (Pre), and T‐cells in Ki/+ (Leu) mice, respectively, compared with their counterpart cells in WT mice (Figure 5C,D, Figure S5c,d; Data S1).
First, Gene Ontology (GO) analysis was performed using a group of genes whose expression varied significantly in Ki/+ (Pre) HSPCs and Ki/+ (Leu) HSPCs (Figure S5c,d). Compared with those of WT HSPCs, the upregulated genes were the same before and after leukemia and were mostly related to immune function and leukocyte activation terms (Figure S5c,d; right panel). Additionally, Ki/+ (Pre) HSPCs were associated with T‐cell activation and differentiation terms. However, most downregulated genes in Ki/+ (Pre) HSPCs were related to apoptosis (Figure S5c; left panel), but Ki/+ (Leu) HSPCs showed that the majority of downregulated genes were associated with metabolic processes, cell development, and differentiation (Figure S5d, left panel).
Next, to determine the characteristics of T‐cells from miR‐142‐55A>G mice, we concentrated on the clusters of genes whose expression fluctuated in T‐cells before and after leukemia relative to those in WT T‐cells. Pre‐leukemia had a greater number of downregulated than upregulated genes (Figure 5C); however, post‐leukemia, the manifestation of various gene clusters deviated significantly (Figure 5D). Evidently, GO analysis revealed that terms related to the immune system and apoptosis were enriched post‐leukemia, and these terms were enriched pre‐leukemia (Figure 5E).
Conversely, the upregulated genes were significantly enriched in GO terms associated with the cell cycle and metabolic processes. The gene expressions correlated with these terms were not enriched in pre‐leukemia T‐cells (Figure 5F), indicating that they are upregulated during leukemogenesis (Figure 5F).
Therefore, the miR‐142‐55A>G mutant induces the activation of a cluster of genes associated with immune function in HSPCs before and after leukemia, indicating that mutated HSPCs preferentially differentiated into lymphoid cells. Conversely, immune function and apoptosis were repressed in T‐cells before and after leukemia, and various factors implicated in cellular growth were manifested after leukemia, indicating that miR‐142‐55A>G suppressed immune function and differentiation in T‐cells but activated proliferation.
3.6. miR‐142‐55A>G mutant harbors gain and loss of function for repressing and derepressing target genes
miR‐142‐55A>G was found to suppress and activate the transcription of genes in leukemic cells directly and/or indirectly. To understand how WT and mutant forms of miR‐142 directly regulate the expression of genes, we utilized miRDB, an online database classifier, to predict potential miRNA‐target genes. 29 , 30 miR‐142‐55A>G was found to harbor distinct target genes from WT miR‐142 (Figure 6A); the individual target genes are listed in Data S3. Using those target gene sets of WT and mutant miR‐142, we performed hierarchical clustering of the genes in HSPCs and T‐cells (Figure 6B,C). Among 374 WT target genes, we found that WT T‐cell expression levels of genes were downregulated compared with those of WT HSPCs (Figure 6B), while for T‐cells in Ki/+ (Leu), a subset of target genes was upregulated (Leu‐Up; Figure 6B), forming a cluster that showed enrichments of the GO terms for metabolic processes and small GTPase signaling (Figure 6D; Data S4). This indicated a loss of function of the miR‐142 WT allele that activated the transcription of genes such as Rheb and Rras to drive signal transduction for cell growth. 31 , 32 , 33 , 34 Quantitative RT‐PCR confirmed that the expression levels of Rheb and Rras were increased in Ki/+ (Leu) HSPCs, compared with those in WT HSPCs (Figure 6E).
FIGURE 6.
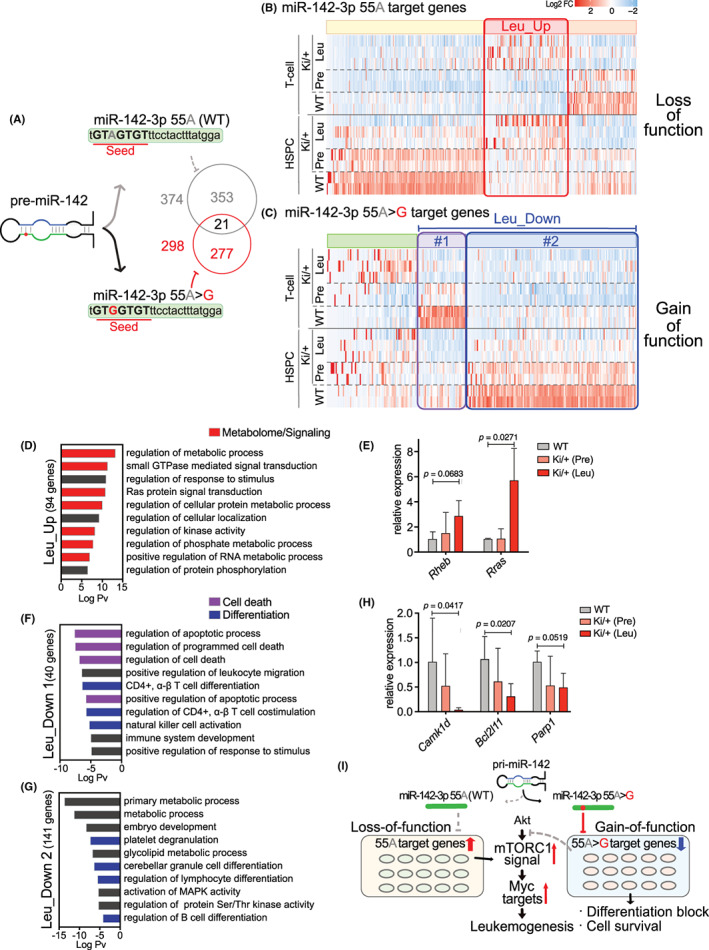
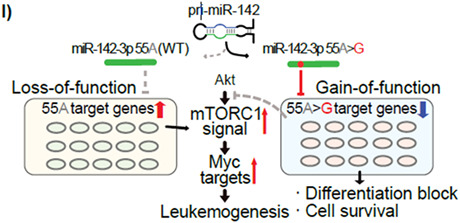
miR‐142‐55A>G mutant harbored gain‐of‐function and loss‐of‐function for the repression of target genes. (A) Diagram showing the variation in target genes due to single nucleotide mutations in miR‐142‐seeded sequences. (B, C) Heatmap showing expression changes of the representative genes with predicted miR‐142‐55A and miR‐142‐55A>G target sites. Red boxes in (B) show upregulated genes in Ki/+ (Leu)_ hematopoietic stem/progenitor cells (HSPCs) and T‐cells, purple boxes in (C) show gradually downregulated genes in HSPCs and T‐cells of Ki/+ (Pre) and Ki/+ (Leu), and indigo blue boxes in (C) show gradually downregulated genes, especially in HSPCs of Ki/+ (Pre) and Ki/+ (Leu). (D) GO analysis of the upregulated genes in the Ki/+ (Leu)_HSPCs and T‐cells detected in the red square in (B). (E) Expression levels of the representative genes in mTORC1 activators mRNA in HSPCs (n = 3–4). (F, G) GO analysis of the downregulated genes in the Ki/+ (Leu)_HSPCs and T‐cells; (F) detected in purple squares in (C), and Ki/+ (Leu)_HSPCs (G) detected in indigo blue squares in (C). (H) Expression levels of the representative genes in Akt–mTOR inhibitors mRNA in HSPCs (n = 3–4). (I) Schematic diagram for leukemia pathogenesis mechanism resulting from heterozygous miR‐142‐55A>G.
Moreover, among miR‐142‐55A>G target genes, T‐cells and HSPCs in Ki/+ (Leu) mice showed decreased gene expression of highly expressed genes in the T‐cells or HSPCs of WT mice, forming two clusters (Leu‐Down #1 and #2; Figure 6C). These genes were sequentially downregulated during the progression from pre‐leukemia to leukemia stages in Ki/+ mice. GO analysis revealed that, on the one hand, genes in cluster #1 were downregulated in Ki/+ T‐cells and enriched apoptosis and T‐cell functions (Figure 6F). On the other hand, genes in cluster #2 were downregulated in Ki/+ HSPCs and showed enrichment in differentiation and metabolic processes (Figure 6G; Data S4), indicating a gain of function for the miR‐142 mutant to directly repress the transcription of genes regulating T‐cell differentiation and apoptosis, such as Camk1d, Bcl2l11, and Parp1, repressing T‐cell leukemia development. 35 , 36 , 37 Quantitative RT‐PCR confirmed that the expression levels of Camk1d, Bcl2l11, and Parp1 were mildly decreased in Ki/+ (Leu) HSPCs relative to those of WT HSPCs (Figure 6H). Thus, the miR‐142‐55A>G mutation appeared to repress its newly acquired target genes and lose the function of WT miR‐142 to repress target genes, thereby driving the development of leukemia.
4. DISCUSSION
Single nucleotide mutations in miR‐142 are recurrently found in patients with various hematological malignancies. 2 , 3 , 17 , 18 , 19 , 20 , 21 , 22 , 23 miR‐142 may play a crucial role as an antitumor immunoregulator in various types of cancer, particularly in lymphoid neoplasms, 38 but miR‐142 knockouts decrease the proliferative capacity of hematopoietic cells that did not develop hematological malignancy in mice. The miR‐142 mutant has been analyzed in vitro in other studies but not in vivo. 39 Here, we generated a new miR‐142‐55A>G mutant mouse using a two‐step CRISPR‐Cas9 system and demonstrated that the miR‐142‐55A>G heterozygous mutation is sufficient for the development of T‐cell leukemia after BM transplantation.
To understand the molecular mechanism by which miR‐142 induces the onset of leukemia, we performed RNA‐sequence analysis of HSPCs and CD8+ T‐cells from miR‐142‐55A>G heterozygous mice at pre‐ and post‐leukemia stages. The results identified genes upregulated in leukemogenic T‐cells that are associated with cancer progression, including mTORC1 and MYC pathways driving proliferation and metabolic processes. The relationship between the mTOR complex and Myc has been the focus of various studies, 40 , 41 , 42 with some suggesting that mTORC1 activity is particularly important for leukemia differentiation and proliferation. 43 , 44 , 45 The miRDB is often used to identify candidates of specific miRNA‐target genes. By utilizing predicted genes for WT and mutant miR‐142‐3p, the transcriptomes of the HSPCs and T‐cells in mutant mice revealed that the expression levels of miR‐142‐3p WT target genes and 55A>G mutation target genes vary widely. The genes involved in activating mTORC1, such as Rras and Rheb, that targeted the genes of miR‐142‐3p WT, were upregulated in the HSPCs and T‐cells from leukemic mice. However, the miR‐142‐3p‐55A>G mutation suppressed new target genes, which may cause the inhibition of apoptosis and impaired differentiation into mature myeloid and lymphoid cells. Genes that inhibit the Akt–mTOR pathway, including Camk1d, Bcl2l11, and Parp1, were also downregulated in mutant mice. RT‐qPCR confirmed that subsets of target genes of both the WT and mutated miR‐142 were dysregulated in miR‐142‐55A>G heterozygous mice before they developed leukemia. Thus, introducing the miR‐142‐55A>G mutation in this study may result in both a loss of function, in which the miR‐142‐3p WT target gene is derepressed, and a gain of function, in which the miR‐142‐55A>G target gene is newly repressed (proposed model in Figure 6I).
As a single miRNA is thought to be involved in the regulation of more than 100 genes depending on the sequence of its seeded regions, the results of this study suggest that a mutation in the seeded region of miR‐142‐3p may result in a new gain of function. As this experiment analyzed miR‐142‐55A>G heterozygous, it is difficult to say whether all expected miR‐142 target genes were altered, and further similar analysis of miR‐142‐55A>G homozygous will be required. However, as all single nucleotide mutations found in AML/MDS patients are heterozygous, we expect this finding to also apply to humans.
A limitation of this study is that a single nucleotide mutation obtained from an AML/MDS patient was introduced but resulted in acute lymphoid leukemia (ALL). Two factors can be considered as possible causes. The first is that tumorigenesis is caused solely by single nucleotide mutations in miRNAs. Here, we aimed to examine how single nucleotide mutations in miR‐142 found in AML/MDS patients are associated with cancer development. Notably, AML/MDS patients develop an accumulation of 5–10 or more gene mutations. Further work is needed to examine the effect of other genetic mutations. A paper from TCGA reported that three of four patients with miR‐142 mutations also had IDH2‐R140Q mutations, 2 and other researchers have reported that both miR‐142‐KO and IDH2‐R140Q cause AML. 24 , 41 The second is the difference between humans and mice. The miR‐142 mutations found in humans are more common in lymphoid cancers. There is also a significant difference in the pattern of hematopoietic differentiation in PB between humans and mice. In the C57BL/6N mice used in this study, hematopoietic differentiation was particularly biased toward the lymphocyte lineage B‐cells. Here, we introduced a mutation found in AML/MDS that may have affected the lymphoid lineage in mice. Although miR‐142 mutations have not been identified to date, several studies have reported the abnormal expression of miR‐142‐3p in ALL. 46 , 47 Additionally, the miRDB‐predicted miR‐142 target genes showed that 418 and 374 WT miR‐142‐3p target genes were found in humans and mice, respectively, and ~40%–45% of them were shared. However, the miR‐142‐55A>G mutation targets 294 genes in humans and 298 genes in mice, sharing only ~14% of the same genes, suggesting that the miR‐142 single nucleotide mutation may result in different phenotypes in humans and mice relative to WT miR‐142 (Data S5). The fact that the miR‐142‐55A>G target gene cluster focused on in this study was detected only in mice may have resulted in ALL rather than AML/MDS. As the miR‐142‐3p seed sequence contains various single nucleotide mutations other than 55A>G, further analysis is required to determine the relationship between single nucleotide mutations in the miRNA seed sequence and its target genes.
Abnormal expression of miRNAs, regulators of gene expression, has been observed in many diseases, including cancer. A miRNA gain‐of‐function mutation in human disease can be used to identify single miR‐140 seed sequence mutations detected in bone system disease. 48 However, single nucleotide mutations in miRNAs have widely been studied as loss‐of‐function mutations. 49 , 50 , 51 , 52 , 53 Here, we demonstrated a causal relationship between the presence of a single nucleotide mutation in miRNA‐142 and leukemia due to the combined effects of gain and loss of function through the monoallelic mutation of miRNA‐142‐55A>G. A single nucleotide mutation of a cancer‐promoting factor could also control other miRNAs to inhibit their functions, thereby promoting cancer development. 54 , 55 We believe that the results of this report provide new insights into cancer biology, and further research on miRNAs is warranted in the future.
AUTHOR CONTRIBUTIONS
K.A., M.A., R.N., and G.S. conceived the project and designed the study. S.K. performed the experiments and analysis involving mutant mice. J.B., I.F., K.T., K.Y., M.Y., S.T., T.K., and K.A. aided in the experiments. S.U. performed RNA sequencing. K.A. established mutant mice. S.K., G.S., and M.A. wrote the paper. All authors approved the final version of the manuscript.
FUNDING INFORMATION
This work was supported by the Japan Society for the Promotion of Science (JSPS KAKENHI, Grant number 21H02391 to K.A. and 21K05999 to M.A.).
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Reviewer Board: N/A
Informed Consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: This research was approved by the Institutional Animal Care and Use Committee and the DNA Experiment Committee of Kumamoto University (Approval Numbers for Animal Experiments: A2020‐016; Approval Number for DNA Experiments: 29–053).
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Data S1
Data S2
Data S3
Data S4
Data S5
Table S1
Table S2
ACKNOWLEDGMENTS
We thank Narumi Koga, Takako Keida, Arisa Igarashi, Sho Kubota, Mariko Morii‐Kubota, Mayumi Muta, Yoko Mine, and Kumiko Murakami for their technical assistance. We would also like to thank Editage (www.editage.com) for English language editing.
Kawano S, Araki K, Bai J, et al. A gain‐of‐function mutation in microRNA 142 is sufficient to cause the development of T‐cell leukemia in mice. Cancer Sci. 2023;114:2821‐2834. doi: 10.1111/cas.15794
Contributor Information
Goro Sashida, Email: sashidag@kumamoto-u.ac.jp.
Masatake Araki, Email: maraki@gpo.kumamoto-u.ac.jp.
REFERENCES
- 1. Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre‐miR‐146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci USA. 2008;105(20):7269‐7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. The Cancer Genome Atlas Research Network , Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thol F, Scherr M, Kirchner A, et al. Clinical and functional implications of microRNA mutations in a cohort of 935 patients with myelodysplastic syndromes and acute myeloid leukemia. Haematologica. 2015;100(4):e122‐e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Toyota M, Suzuki H, Sasaki Y, et al. Epigenetic silencing of microRNA‐34b/c and B‐cell translocation gene 4 is associated with CpG Island methylation in colorectal cancer. Cancer Res. 2008;68(11):4123‐4132. [DOI] [PubMed] [Google Scholar]
- 5. Lujambio A, Ropero S, Ballestar E, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67(4):1424‐1429. [DOI] [PubMed] [Google Scholar]
- 6. Markou A, Tsaroucha EG, Kaklamanis L, Fotinou M, Georgoulias V, Lianidou ES. Prognostic value of mature microRNA‐21 and microRNA‐205 overexpression in non‐small cell lung cancer by quantitative real‐time RT‐PCR. Clin Chem. 2008;54(10):1696‐1704. [DOI] [PubMed] [Google Scholar]
- 7. Schepeler T, Reinert JT, Ostenfeld MS, et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008;68(15):6416‐6424. [DOI] [PubMed] [Google Scholar]
- 8. Lu L, Katsaros D, de la Longrais IA, Sochirca O, Yu H. Hypermethylation of let‐7a‐3 in epithelial ovarian cancer is associated with low insulin‐like growth factor‐II expression and favorable prognosis. Cancer Res. 2007;67(21):10117‐10122. [DOI] [PubMed] [Google Scholar]
- 9. Yu SL, Chen HY, Chang GC, et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell. 2008;13(1):48‐57. [DOI] [PubMed] [Google Scholar]
- 10. Chapnik E, Rivkin N, Mildner A, et al. Mir‐142 orchestrates a network of Actin cytoskeleton regulators during megakaryopoiesis. Elife. 2014;3:e01964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rivkin N, Chapnik E, Mildner A, et al. Erythrocyte survival is controlled by microRNA‐142. Haematologica. 2017;102(4):676‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rivkin N, Chapnik E, Mildner A, et al. Mononuclear phagocyte miRNome analysis identifies miR‐142 as critical regulator of murine dendritic cell homeostasis. Blood. 2013;121(6):1016‐1027. [DOI] [PubMed] [Google Scholar]
- 13. Sun Y, Oravecz‐Wilson K, Mathewson N, et al. Mature T cell responses are controlled by microRNA‐142. J Clin Invest. 2015;125(7):2825‐2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kramer NJ, Wang WL, Reyes EY, et al. Altered lymphopoiesis and immunodeficiency in miR‐142 null mice. Blood. 2015;125(24):3720‐3730. [DOI] [PubMed] [Google Scholar]
- 15. Mildner A, Chapnik E, Varol D, et al. MicroRNA‐142 controls thymocyte proliferation. Eur J Immunol. 2017;47(7):1142‐1152. [DOI] [PubMed] [Google Scholar]
- 16. Anandagoda N, Willis JC, Hertweck A, et al. microRNA‐142‐mediated repression of phosphodiesterase 3B critically regulates peripheral immune tolerance. J Clin Invest. 2019;129(3):1257‐1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bouska A, Zhang W, Gong Q, et al. Combined copy number and mutation analysis identifies oncogenic pathways associated with transformation of follicular lymphoma. Leukemia. 2017;31(1):83‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rheinbay E, Nielsen MM, Abascal F, et al. Analyses of non‐coding somatic drivers in 2,658 cancer whole genomes. Nature. 2020;578(7793):102‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Puente XS, Beà S, Valdés‐Mas R, et al. Non‐coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519‐524. [DOI] [PubMed] [Google Scholar]
- 20. Morin RD, Assouline S, Alcaide M, et al. Genetic landscapes of relapsed and refractory diffuse large B‐cell lymphomas. Clin Cancer Res. 2016;22(9):2290‐2300. [DOI] [PubMed] [Google Scholar]
- 21. Hornshøj H, Nielsen MM, Sinnott‐Armstrong NA, et al. Pan‐cancer screen for mutations in non‐coding elements with conservation and cancer specificity reveals correlations with expression and survival. NPJ Genom Med. 2018;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hezaveh K, Kloetgen A, Bernhart SH, et al. Alterations of microRNA and microRNA‐regulated messenger RNA expression in germinal center B‐cell lymphomas determined by integrative sequencing analysis. Haematologica. 2016;101(11):1380‐1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwanhian W, Lenze D, Alles J, et al. MicroRNA‐142 is mutated in about 20% of diffuse large B‐cell lymphoma. Cancer Med. 2012;1(2):141‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marshall A, Kasturiarachchi J, Datta P, et al. Mir142 loss unlocks IDH2R140‐dependent leukemogenesis through antagonistic regulation of HOX genes. Sci Rep. 2020;10(1):19390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oka K, Fujioka S, Kawamura Y, et al. Resistance to chemical carcinogenesis induction via a dampened inflammatory response in naked mole‐rats. Commun Biol. 2022;5(1):287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tempel S, Tahi F. A fast ab‐initio method for predicting miRNA precursors in genomes. Nucleic Acids Res. 2012;40(11):e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tav C, Tempel S, Poligny L, Tahi F. miRNAFold: a web server for fast miRNA precursor prediction in genomes. Nucleic Acids Res. 2016;44(W1):W181‐W184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shrestha A, Carraro G, El Agha E, et al. Generation and validation of miR‐142 knock out mice. PLoS One. 2015;10(9):e0136913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127‐D131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu W, Wang X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019;20(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Long X, Lin Y, Ortiz‐Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15(8):702‐713. [DOI] [PubMed] [Google Scholar]
- 32. Babcock JT, Quilliam LA. Rheb/mTOR activation and regulation in cancer: novel treatment strategies beyond rapamycin. Curr Drug Targets. 2011;12(8):1223‐1231. [DOI] [PubMed] [Google Scholar]
- 33. Deng L, Chen L, Zhao L, et al. Ubiquitination of Rheb governs growth factor‐induced mTORC1 activation. Cell Res. 2019;29(2):136‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alcover‐Sanchez B, Garcia‐Martin G, Wandosell F, Cubelos B. R‐Ras GTPases signaling role in myelin neurodegenerative diseases. Int J Mol Sci. 2020;21(16):5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jin Q, Zhao J, Zhao Z, et al. CAMK1D inhibits glioma through the PI3K/AKT/mTOR signaling pathway. Front Oncol. 2022;12:845036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tapodi A, Bognar Z, Szabo C, Gallyas F, Sumegi B, Hocsak E. PARP inhibition induces Akt‐mediated cytoprotective effects through the formation of a mitochondria‐targeted phospho‐ATM‐NEMO‐Akt‐mTOR signalosome. Biochem Pharmacol. 2019;162:98‐108. [DOI] [PubMed] [Google Scholar]
- 37. Wood CD, Veenstra H, Khasnis S, et al. MYC activation and BCL2L11 silencing by a tumor virus through the large‐scale reconfiguration of enhancer‐promoter hubs. Elife. 2016;5:e18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Galka‐Marciniak P, Kanduła Z, Tire A, et al. Mutations in the miR‐142 gene are not common in myeloproliferative neoplasms. Sci Rep. 2022;12(1):10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trissal MC, Wong TN, Yao JC, et al. MIR142 loss‐of‐function mutations derepress ASH1L to increase HOXA gene expression and promote leukemogenesis. Cancer Res. 2018;78(13):3510‐3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ben‐Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. 2017;45:72‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hsin IL, Shen HP, Chang HY, Jiunn‐Liang Ko JL, Wang PH. Suppression of PI3K/Akt/mTOR/c‐Myc/mtp53 by dual PI3K/mTOR inhibitor PQR309 in endometrial cancer cell lines. Cell. 2021;10(11):2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu P, Ge M, Hu J, et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c‐Myc‐driven hepatocarcinogenesis. Hepatology. 2017;66(1):167‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoshii T, Tadokoro Y, Naka K, et al. mTORC1 is essential for leukemia propagation but not stem cell self‐renewal. J Clin Invest. 2012;122(6):2114‐2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Simioni C, Martelli AM, Zauli G, Melloni E, Neri LM. Targeting mTOR in acute lymphoblastic leukemia. Cell. 2019;8(2):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishio T, Kumar S, Shimono J, et al. Genome‐wide CRISPR screen identifies CDK6 as a therapeutic target in adult T‐cell leukemia/lymphoma. Blood. 2022;139(10):1541‐1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bellon M, Lepelletier Y, Hermine O, Nicot C. Deregulation of microRNA involved in hematopoiesis and the immune response in HTLV‐I adult T‐cell leukemia. Blood. 2009;113(20):4914‐4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lv M, Zhang X, Jia H, et al. An oncogenic role of miR‐142‐3p in human T‐cell acute lymphoblastic leukemia (T‐ALL) by targeting glucocorticoid receptor‐α and cAMP/PKA pathways. Leukemia. 2012;26(4):769‐777. [DOI] [PubMed] [Google Scholar]
- 48. Grigelioniene G, Suzuki HI, Taylan F, et al. Gain‐of‐function mutation of microRNA‐140 in human skeletal dysplasia. Nat Med. 2019;25(4):583‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mencía A, Modamio‐Høybjør S, Redshaw N, et al. Mutations in the seed region of human miR‐96 are responsible for nonsyndromic progressive hearing loss. Nat Genet. 2009;41(5):609‐613. [DOI] [PubMed] [Google Scholar]
- 50. Soldà G, Robusto M, Primignani P, et al. A novel mutation within the MIR96 gene causes non‐syndromic inherited hearing loss in an Italian family by altering pre‐miRNA processing. Hum Mol Genet. 2012;21(3):577‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hughes AE, Bradley DT, Campbell M, et al. Mutation altering the miR‐184 seed region causes familial keratoconus with cataract. Am J Hum Genet. 2011;89(5):628‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Iliff BW, Riazuddin SA, Gottsch JD. A single‐base substitution in the seed region of miR‐184 causes EDICT syndrome. Invest Ophthalmol Vis Sci. 2012;53(1):348‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lechner J, Bae HA, Guduric‐Fuchs J, et al. Mutational analysis of MIR184 in sporadic keratoconus and myopia. Invest Ophthalmol Vis Sci. 2013;54(8):5266‐5272. [DOI] [PubMed] [Google Scholar]
- 54. Bagheri F, Mesrian Tanha H, Mojtabavi Naeini M, Ghaedi K, Azadeh M. Tumor‐promoting function of single nucleotide polymorphism rs1836724 (C3388T) alters multiple potential legitimate microRNA binding sites at the 3′‐untranslated region of ErbB4 in breast cancer. Mol Med Rep. 2016;13(5):4494‐4498. [DOI] [PubMed] [Google Scholar]
- 55. Shasttiri A, Rostamian Delavar M, Baghi M, Dehghani Ashkezari M, Ghaedi K. SNP rs10800708 within the KIF14 miRNA binding site is linked with breast cancer. Br J Biomed Sci. 2019;76(1):46‐48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Data S1
Data S2
Data S3
Data S4
Data S5
Table S1
Table S2