

This genetic association study investigates the genetic cause for bicuspid aortic valve development.
Key Points
Question
What genes are associated with the development of bicuspid aortic valve (BAV)?
Findings
By integrating independent human genetic approaches starting with familial segregation and followed by rare and common variants analyses in this genetic association study of 938 patients, the MIB1 gene was identified; MIB1 is an essential regulator of NOTCH ligands signaling and represents a potential new gene in the development of BAV in humans. These data were further supported by mouse models; mice carrying the identified human MIB1 gene variants demonstrated BAV.
Meaning
These findings suggest a role for MIB1 in the pathophysiology of BAV and identify the NOTCH pathway as a potential target in the management of BAV.
Abstract
Importance
Nonsyndromic bicuspid aortic valve (nsBAV) is the most common congenital heart valve malformation. BAV has a heritable component, yet only a few causative genes have been identified; understanding BAV genetics is a key point in developing personalized medicine.
Objective
To identify a new gene for nsBAV.
Design, Setting, and Participants
This was a comprehensive, multicenter, genetic association study based on candidate gene prioritization in a familial cohort followed by rare and common association studies in replication cohorts. Further validation was done using in vivo mice models. Study data were analyzed from October 2019 to October 2022. Three cohorts of patients with BAV were included in the study: (1) the discovery cohort was a large cohort of inherited cases from 29 pedigrees of French and Israeli origin; (2) the replication cohort 1 for rare variants included unrelated sporadic cases from various European ancestries; and (3) replication cohort 2 was a second validation cohort for common variants in unrelated sporadic cases from Europe and the US.
Main Outcomes and Measures
To identify a candidate gene for nsBAV through analysis of familial cases exome sequencing and gene prioritization tools. Replication cohort 1 was searched for rare and predicted deleterious variants and genetic association. Replication cohort 2 was used to investigate the association of common variants with BAV.
Results
A total of 938 patients with BAV were included in this study: 69 (7.4%) in the discovery cohort, 417 (44.5%) in replication cohort 1, and 452 (48.2%) in replication cohort 2. A novel human nsBAV gene, MINDBOMB1 homologue MIB1, was identified. MINDBOMB1 homologue (MIB1) is an E3-ubiquitin ligase essential for NOTCH-signal activation during heart development. In approximately 2% of nsBAV index cases from the discovery and replication 1 cohorts, rare MIB1 variants were detected, predicted to be damaging, and were significantly enriched compared with population-based controls (2% cases vs 0.9% controls; P = .03). In replication cohort 2, MIB1 risk haplotypes significantly associated with nsBAV were identified (permutation test, 1000 repeats; P = .02). Two genetically modified mice models carrying Mib1 variants identified in our cohort showed BAV on a NOTCH1-sensitized genetic background.
Conclusions and Relevance
This genetic association study identified the MIB1 gene as associated with nsBAV. This underscores the crucial role of the NOTCH pathway in the pathophysiology of BAV and its potential as a target for future diagnostic and therapeutic intervention.
Introduction
Bicuspid aortic valve (BAV) is a heritable condition, affecting 1% to 2% of the population1,2 and is the most common congenital heart valve defect. BAV is heritable, with first-degree family members having a 10-fold increased risk of being affected.3,4,5,6,7 When seen in families, BAV mostly presents as a dominant inherited trait with incomplete penetrance and a higher prevalence in male individuals (sex ratio of 3:1).3,4,8,9,10 BAV genetics are complex and can be caused by a combination of mendelian, oligogenic, and polygenic inheritance.5 Several candidate genes, often identified in animal models, were reported.6,7,11,12,13 However, only few genes were shown to cause nonsyndromic BAV (nsBAV) in humans.14,15 These include NOTCH1, GATA6, and SMAD6, each accounting for 1% to 3% of cases.3,14,16
The NOTCH pathway, an evolutionarily conserved cell-cell communication signaling pathway involved in multiple developmental processes,17 is known to be related to BAV. NOTCH signaling regulates aortic valve morphogenesis, and its disruption causes aortic valve diseases in both humans and mice.18,19,20 MIB1 (MINDBOMB1) is an essential E3-ubiquitin ligase that induces NOTCH ligand ubiquitination and endocytosis, an essential pathway activation step.21
Methods
The study was approved by each institution’s review board according to local regulations, and written informed consent was obtained from all participants. Animal studies were approved by the CNIC Animal Experimentation Ethics Committee and the Madrid community. All animal procedures conformed to European Union Directive 2010/63EU and Recommendation 2007/526/EC regarding protection of animals used for experimental and other scientific purposes, enforced in Spanish law under Real Decreto 1201/2005. This study followed the Strengthening the Reporting of Genetic Association Studies (STREGA) reporting guidelines.
We combined 3 human genetic approaches and animal model functional studies. The discovery cohort was a large cohort of inherited cases from 28 pedigrees of French and Israeli origin. Replication cohort 1 for rare variants analysis included unrelated sporadic cases from various European ancestries. Replication cohort 2 was a second validation cohort for common variants analysis in unrelated sporadic cases from Europe and the US.
Study Cohort
This study did not address a specific race or ethnicity and was based on the population of origin. Data collected included self-reported ancestry to avoid subpopulation stratification error.
Discovery Cohort
In the discovery cohort, we included a large familial cohort from 2 tertiary medical centers. Index cases were included if they had BAV and if 1 or more relatives had BAV or thoracic aortic aneurysm. All patients underwent a comprehensive clinical examination and personal medical and family histories. Inclusion was based on BAV presence as shown by echocardiography, magnetic resonance imaging, or cardiac surgery. Dysmorphic features were evaluated medically, and syndromic conditions were excluded. Patients younger than 18 years were included only as part of a family, not as index cases.
Replication Cohort 1
Patients whose relatives were unaffected (a negative BAV or thoracic aortic aneurysm test) or unavailable were classified as sporadic cases and were included in replication cohort 1, along with additional cases from the Mechanistic Interrogation of BAV Aortopathy (MIBAVA)–Leducq consortium.
Replication Cohort 2
Replication cohort 2 included patients with BAV from Mass General Brigham Hospital who were of European ancestry. In addition, ethnically matched controls from the Framingham Heart Study were also included.6
Whole-Exome and Candidate Gene Sequencing
Discovery Cohort
DNA was extracted from the peripheral blood or saliva of study participants using standard protocols. Whole-exome sequencing was performed at the Genomics Core Facility in the Imagine Institute.22 DNA extraction using the QIAmp Blood DNA Mini Kit (Qiagen) was followed by exome capture using the SureSelect Human All Exon Kit (Agilent) and sequencing on a HiSeq2500 system (Illumina). The mean depth of coverage obtained for each sample was greater than 150×, with more than 97% of the exome covered at least 30×.
Replication Cohort 1
Sanger sequencing of the 21 exons and flanking intronic sequences of MIB1 (NM_020774) was performed on the sporadic Israeli and French cohort. Sequences were amplified by polymerase chain reaction with specific primers, sequenced using BigDye Terminator v3.1 cycle sequencing kits (Thermo Fisher Scientific), and run on an ABI Prism 3730XL DNA Analyzer (Life Technologies). DNA variants were identified using Sequencher software (Gene Codes Corporation).
Mechanistic Interrogation of BAV Aortopathy–Leducq Consortium
Whole blood–derived genomic DNA of patients was used for whole-exome sequencing with the Nimblegen SeqCap Exome Enrichment Kit (Roche). Samples were 2 × 100 base pair–end sequenced on a HiSeq1500 instrument (Illumina). Raw data were processed using an in-house developed pipeline,23 followed by variant calling with the Genome Analysis Toolkit Unified Genotyper (Broad Institute).24 Variants were annotated and filtered with VariantDB, an in-house developed tool.25 All rare variants of MIB1 identified by whole-exome sequencing were confirmed by targeted Sanger sequencing. When samples and phenotypes from relatives were available, segregation analysis of the selected variants was performed by Sanger sequencing as described previously.
Exome Data Analyses and Prioritization in Discovery Cohort
A filtering pipeline was systematically applied for exome sequencing data.22 Variant calling was performed with the Genome Analysis Toolkit Unified Genotyper based on the ENSEMBL 72 database, using standard parameters. Variants that did not pass the quality filters were excluded (read depth <10 × and/or Phred score <30). Only rare variants (minor allele frequency [MAF] <0.1% according to gnomAD, version 2.1.1) with a combined annotation dependent depletion (CADD) score above 20 (using the CADD, version 1.6 framework26) and with coding impact were retained for any further analysis.
We assembled a list of 34 known BAV genes by thorough and critical literature review, identifying the most robust genes associated with syndromic and nsBAV (in humans and/or mice, eTable 1 in Supplement 1). We aimed to exclude cases with predicted deleterious variants in these 34 known genes. The remaining genes carrying variants in the discovery cohort were then subjected to a prioritization and ranking process, aimed to identify a leading candidate gene for further investigation via validation cohorts and functional work based on their roles in heart development pathophysiology (detailed in eFigure 2 and eTable 1 in Supplement 1 and eTable 2 in Supplement 2).24,25,26,27 The resulting candidate genes were prioritized by in-silico bioinformatics tools (VarElect and Endeavor), based on pathobiology, relevant pathways, and genetic and biological interactions. The top-rated genes were chosen for further qualitative analyses according to (1) biological relevance to BAV (genes were prioritized if known to have a role in the valve development), (2) frequency (the number of families and family members sharing pathogenic variants in the gene), and (3) variant weight (according to the variant type and its predicted deleteriousness).
Rare Variants Burden Testing in Discovery Cohort and Replication Cohort 1
Burden testing was performed for MIB1 rare variants with coding impact in index cases from the discovery cohort and replication cohort 1 against gnomAD, version 2.1.1, publicly available controls using the TRAPD software package (BioTools).28 Only gnomAD variants passing filters from exome data were retained, using the same filtering process as in the discovery step (MAF <0.1%, CADD score >20, read depth ≥10×). Similarly, burden testing was performed on presumably benign, silent variants used as an internal method control. Owing to the various ethnic backgrounds of patients, testing was performed in all patients with BAV vs all gnomAD controls, as well as by ethnically matched subpopulations. A 2 × 2 contingency table was constructed for each of the groups described previously. A 1-sided Fisher exact test was used to estimate the association P values. An AlphaFold model (DeepMind) of MIB1 with BAV variants mapped was constructed to demonstrate the sites of the identified variants.29,30
Common Single-Nucleotide Variations Genotyping Data Analyses in Replication Cohort 2
Patients of the second replication cohort were recruited from Mass General Brigham, Boston, Massachusetts.6 Genotypes were generated using the Omni2.5 chip (Illumina) for cases and the HumanOmni5.0 chip for controls (obtained from the Framingham Heart Study). The Illumina Omni2.5 chip is a subset of the Omni5.0 chip, with a similar set of primers. In the quality control process, only markers with MAF greater than 1% were included, and populations were stratified by principal components–based filtering (eFigure 3 in Supplement 1). After merging cases and controls, we had BAV cases and controls with a set of 24 single-nucleotide variations (SNVs) in MIB1 ([uc002ktp.3] hg19 chr18:19,284,918-19 450 912 region +/−100kb) common to both Illumina arrays,6 which were included for final analysis.
PLINK, version 1.9 (Harvard University) was used for SNV association tests in an additive logistic regression model for BAV cases and controls. The false discovery rate method was applied for multiple comparison correction. The coefficient of linkage disequilibrium (D′) between SNVs was calculated using LDlink (National Cancer Institute).31 PHASE, version 2.1 (University of Chicago)32 was used for haplotype reconstruction and for performing association tests between haplotypes and BAV status. We used a permutation test with 1000 repeats to test for differences in haplotype frequencies between BAV cases and controls, each time shuffling the case/control status of individuals.
Generation of Mib1K735R Mouse Line
Clustered regularly interspaced short palindromic repeats (CRISPR) RNA (crRNA) sequences were designed using the CRISPOR-TEFOR online tool (Tefor Paris-Saclay). The annealed 2-part synthetic crRNA (Alt-R CRISPR-Cas9 crRNA, 2 nmol [Integrated DNA Technologies]) and tracrRNA (Alt-R CRISPR-Cas9 tracrRNA, 5 nmol; Integrated DNA Technologies) molecules were diluted in microinjection buffer (1mM Tris hydrochloric acid, pH 7.5; 0.1mM EDTA) and incubated with Sp Cas9 nuclease (Integrated DNA Technologies 1081058). To generate the Mib1K735R line, a complementary and asymmetric single-stranded oligodeoxynucleotide (ssODN) was designed33 as custom synthetic genes (Megamer single-stranded Gene Fragments [Integrated DNA Technologies]) introducing this point variation. The final concentration of components was 0.61 pmol/μL of crRNA and tracrRNA, 30 ng/μL of Cas9 protein, and 10 ng/μL of ssODN. Microinjections were performed at 1-cell stage fertilized C57BL/6 mouse embryos.34 Pups were screened for the targeted gene variation or insertion by polymerase chain reaction analysis and sequencing, and the selected founders were backcrossed to the C57BL/6 background. Detailed information about the microinjection reagents, summary of results, genotyping, and tissue processing35,36 is given in eTable 3 in Supplement 1. We have described recently the generation of the mice harboring the Mib1V943F variant.37 Mice heterozygous for Notch38 (denoted as Notch1+/−) and Rbp39 (denoted as Rbp+/−) variant lines were used for genetic sensitization studies with Mib1 alleles.
Statistical Analysis
Study data were analyzed from October 2019 to October 2022. In addition to the analysis stated before, we performed the additional statistical analysis using R studio (RStudio). We reported categorical variables as number and percentage and compared variables using the χ2 or Fisher exact test. We reported continuous variables as mean and SD and compared variables using the t test or Mann-Whitney U test. All P values were 2-sided (unless otherwise specified), and P <.05 was considered statistically significant.
Results
A total of 938 patients with BAV were included in this study: 69 (7.4%) in the discovery cohort, 417 (44.5%) in replication cohort 1, and 452 (48.2%) in replication cohort 2. Mean (SD) overall age was 49.8 (14.2) years. A flowchart summarizing the study design and main results is displayed in eFigure 1 in Supplement 1. The composition of the study cohorts including participant age and sex data is presented in the Table.
Table. Nonsyndromic Bicuspid Aortic Valve (nsBAV) Discovery Cohort and Replication Cohorts (N = 938).
| Cohort | No./total No. of cases | Center | Location | No. of patients with BAV (No. of families) | Male sex, No. (%) | Female sex, No. (%) | Age, mean (SD), y |
|---|---|---|---|---|---|---|---|
| Discovery cohort | 69/938 Familial cases | APHP-Hôpital Européen Georges Pompidou | Paris, France | 46 (18) | 30 (65.2) | 16 (34.8) | 39 (16.5) |
| Hadassah Medical Center | Jerusalem, Israel | 23 (11) | 17 (73.9) | 6 (26.1) | 40 (22.7) | ||
| Replication cohort 1 | 417/938 Casesa | MIBAVA-Leducq | Europe, Canada, US | 195 | NA | NA | 52.2 (11.8) |
| APHP-Hôpital Européen Georges Pompidou | Paris, France | 164 | 127 (77.4) | 37 (22.6) | 51 (15.5) | ||
| Hadassah Medical Center | Jerusalem, Israel | 58 | 40 (68.9) | 18 (31.1) | 44.8 (18.8) | ||
| 62 874 Controls | Genome Aggregation Database | Mixed | NA | NA | NA | NA | |
| Replication cohort 2 | 452/938 Cases | Mass General Brigham Hospital | Boston, Massachusetts, US | 452 | 336 (74.3) | 116 (25.7) | 54.4 (11.8) |
| 1834 Controls | The Database of Genotypes and Phenotypes | Framingham Heart Study, US | NA | 713 (38.9) | 1121 (61.1) | NA |
Abbreviations: AP-HP, Assistance Publique–Hȏpitaux de Paris; MIBAVA, Mechanistic Interrogation of BAV Aortopathy; NA, not available.
Analyzed with 29 familial index cases.
Inherited nsBAV as a Highly Heterogeneous Trait
Sixty-nine familial cases from 29 pedigrees from various ethnic backgrounds were included in the discovery cohort, dedicated to new candidate gene identification (Table). Detailed description of the cohort demographics and clinical characteristics can be found in eTable 4 in Supplement 1 and eTable 5 and eTable 6 in Supplement 2. In most families, 2 members were affected (18 families, 62%); 10 families had 3 affected members (34.5%), and 1 family had 4 (3.5%). Patients with BAV were predominantly male (46 of 69 [66%]).
After conducting exome sequencing, we applied a step-by-step variant- and gene-filtering strategy detailed in eFigure 2 in Supplement 1. We retained only genes harboring rare and predicted damaging protein-altering variants, with MAF less than 1%, and CADD score greater than 20.
First, we excluded known or suspected syndromic and nsBAV genes (34 genes, including NOTCH1, ROBO4, and SMAD6) (eTable 1 in Supplement 1). We then excluded genes that are not expressed during cardiac development or involved in congenital heart valve defect (eTable 2 in Supplement 2).33,34,35 Following, we used in silico gene prioritization tools (Endeavor and VarElect)36,37 (eTable 7 in Supplement 2) to further prune the remaining genes. The top 10 ranked genes from each tool were chosen for further qualitative analysis based on population rarity, gene recurrence in families, and known or potential impact of each variant. Ultimately, we identified 4 candidate genes (eTable 8 in Supplement 1).
MIB1 was identified as the leading candidate gene. In addition to the genetic finding (Figure 1),40 our selection was based on literature data: (1) MIB1 role in NOTCH pathway, (2) mouse models of Mib1 inactivation have BAV,18,38 and (3) the identified variant was previously found in various congenital heart valve defect phenotypes including left ventricular noncompaction (LVNC).39,41,42The index case in our pedigree carrying the MIB1 variant had a complex phenotype combining BAV with a myocardial crypt. Myocardial crypts have been reported in increased prevalence among carriers of cardiomyopathy gene variants with otherwise normal phenotype.41
Figure 1. MIB1 Rare Variants in Nonsyndromic Bicuspid Aortic Valve (nsBAV).
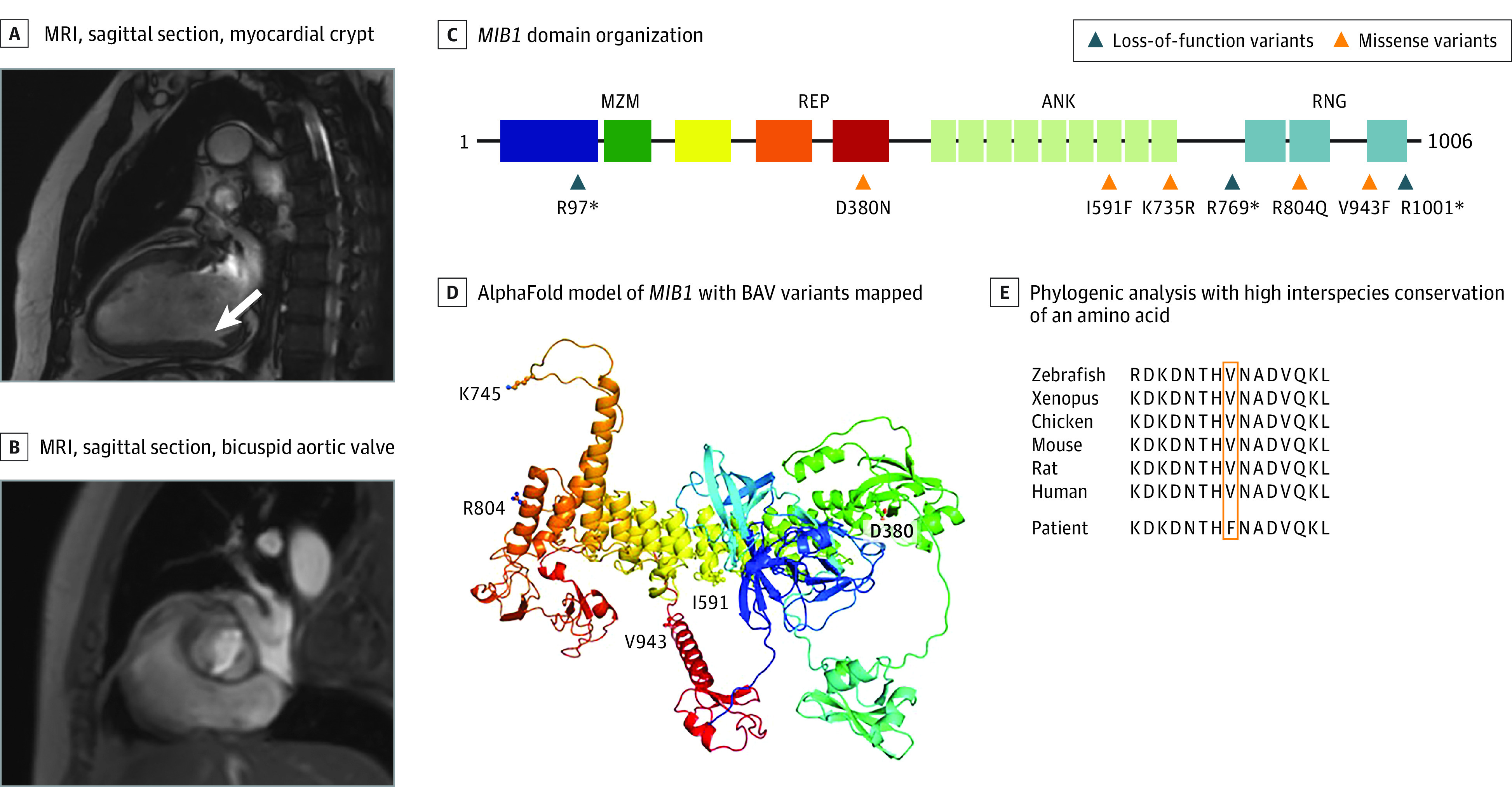
A, Cardiac magnetic resonance imaging (MRI) of a patient with the MIB1 p.V943F variant and a combined valve and muscle phenotype, sagittal section, showing a myocardial crypt in the left ventricle free wall. B, Sagittal section of the same study; showing BAV. C, A graphical representation of the MIB1 domain organization and the location of the identified MIB1 variants. The asterisk indicates a nonsense variant. D, An AlphaFold model (DeepMind) of MIB1 with BAV variants mapped. The model is rendered as a cartoon (colored on a rainbow scale from blue at the N-terminus to red at C-terminus) with sites of BAV variants indicated and rendered in ball-and-stick format. E, A phylogenetic analysis showing high interspecies conservation of the amino acid.40 ANK indicates ankyrin repeats 1 to 9; MZM, MIB-Herc2 domain 1 + ZZ finger domain + MIB-Herc2 domain 2; REP, MIB repeats 1 and 2; RNG, Ring domains 1 to 3.
Segregation analysis in this pedigree was complex as both parents of the index case had BAV (eFigure 4 in Supplement 1). The index case (III-1) and his affected mother (II-2) both harbored the MIB1 V943F variant. The affected father (II-1) was deceased at the time of the study. The affected brother of the index case (III-3) did not harbor the variant.
Association Between nsBAV and Rare MIB Protein-Altering Variants
We assembled replication cohort 1, including nsBAV sporadic cases (n = 417) from European, Canadian, and Israeli backgrounds (Table and eTable 4 and eTable 9 in Supplement 1) and performed next-generation sequencing or Sanger sequencing targeting MIB1. Using the same variant-filtering criteria as in the discovery cohort, we identified 7 additional rare, predicted deleterious, protein-altering MIB1 variants in 8 patients: 3 nonsense variants and 4 missense variants, of which 1 was found twice (eTable 10 in Supplement 1). Using Genomic Evolutionary Rate Profiling,42 all the identified variants were predicted to be under a selective constraint with high positive scores (range, 4.27-5.64). Segregation analysis could be done for the p.D380N missense variant, which was confirmed de novo (parents were healthy and did not harbor the variant). In an in vitro model, we previously showed that this variant induced impaired interaction between MIB1 and its ligand JAG1, leading to defective NOTCH pathway activation.43
The cumulative frequency of rare and predicted deleterious variants in sporadic BAV cases was 2% compared with 0.9% for silent variants. To determine if sporadic BAV index cases from the discovery cohort and replication cohort 1 present enrichment for rare and predicted deleterious variants, a variant association study was performed using TRAPD (Testing Rare Variants using Public Data) burden testing28 against either all controls or ethnically matched controls from gnomAD (Genome Aggregation Database). Enrichment analysis demonstrated association for all cases vs all gnomAD controls (1% vs 0.5% alleles; 2% vs 0.9% genotypes/cases and controls in a dominant model; P = .03) and the Israeli cases vs Ashkenazi Jewish and non-Finland European controls (1% vs 0.5% alleles; 2% vs 0.9% genotypes in a dominant model; P = .03). In contrast, silent variants were not enriched in our cases (eTable 11 in Supplement 1).
Association of MIB1 Locus With Sporadic nsBAV
Replication cohort 2 included 452 patients with BAV from the Mass General Brigham and Women’s Hospital and 1834 controls from the Framingham Heart Study. Demographics and clinical data of BAV cases and controls are detailed in eTable 12 Supplement 1 and eTable 13 in Supplement 2. We considered common SNVs at the MIB1 locus (Figure 2).44 Using a logistic regression model with a false discovery rate correction for multiple comparisons, we detected an association for 5 noncoding SNVs (eTable 14 in Supplement 1), 4 of them within coding regions or introns (Figure 2A).44 Linkage disequilibrium (LD) assessment demonstrated that all 5 SNVs were part of a delineated LD block, with D′ (coefficient of LD) of 1, indicating that the SNVs were in high LD. (Figure 2B).44 Two blocks were apparent from the LD analysis: one with SNVs 1, 2, and 5 (rs7241299, rs79023008, rs11083391) and the other with SNVs 3 and 4 (rs1893384, rs3017041). Haplotype reconstruction allowed us to identify significant differences in haplotype frequencies between cases and controls, identifying 2 risk haplotypes and 1 protective haplotype (permutation test, 1000 repeats; P = .02) (eTable 15 in Supplement 1). This was further supported by permutation tests in a randomly shuffled cohort of cases and controls, which resulted in nonsignificant differences.
Figure 2. MIB1 Common Single-Nucleotide Variations (SNVs) Associated With Bicuspid Aortic Valve (BAV).
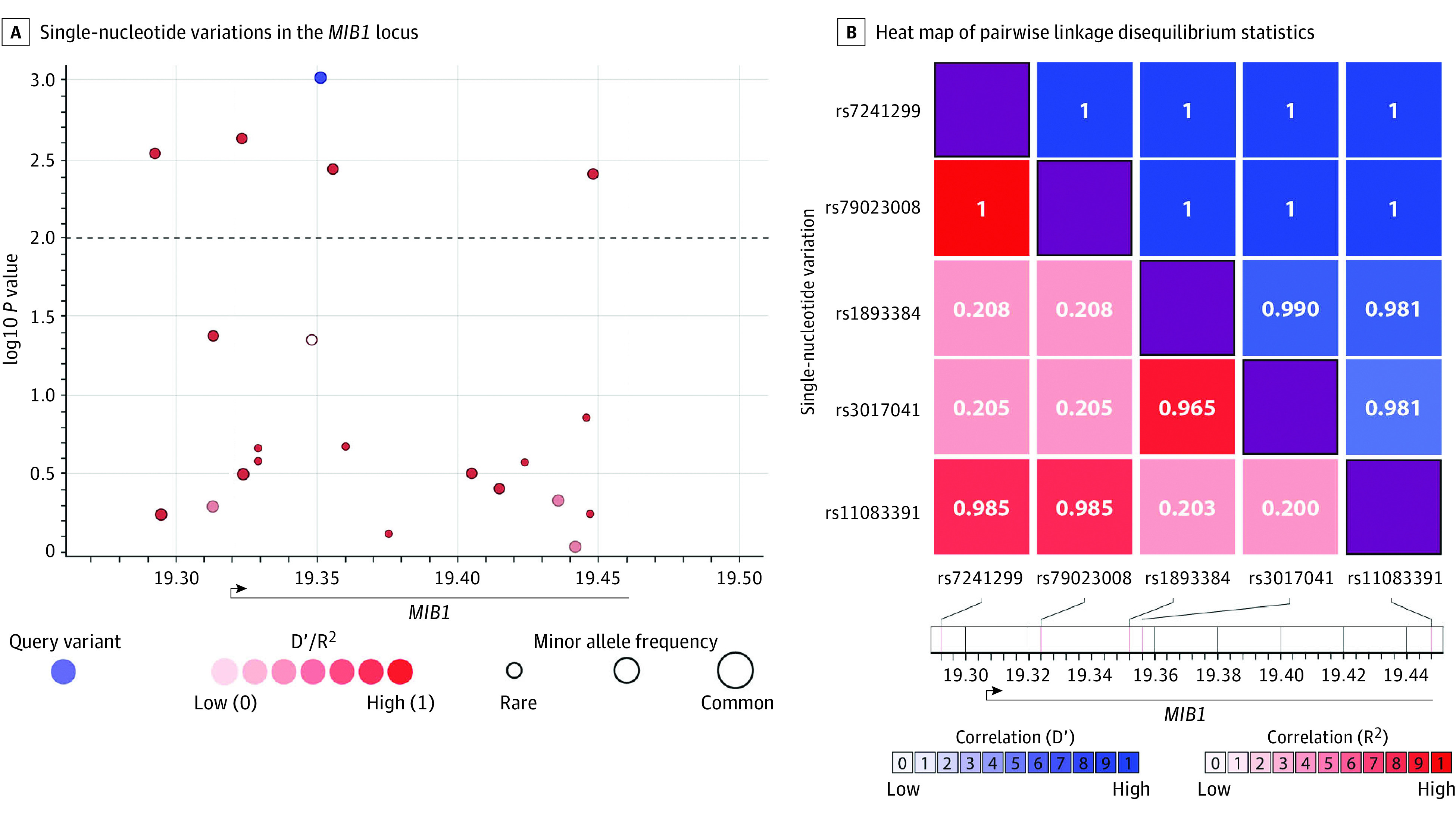
A, SNVs in the MIB1 locus. The x-axis represents the chromosomal coordinates; the y-axis represents the log10 P value (left) and the combined recombination rate from HapMap (right). The locus spans 166 Kb from chr18:19 284 918 to chr18:19 450 912 in GRCh37/hg19. Each point represents an SNV and is colored on the basis of D′ in relation to the most significant SNV, colored in blue. The dashed horizontal line marks the critical P value of .01, which was set by the false discovery rate multiple comparisons analysis.44 B, A heat map of pairwise linkage disequilibrium statistics for the statistically significant SNVs. The x and y dimensions represent the 5 significant SNVs identified, demonstrating linkage disequilibrium′ approaching 1 for all 5, implying high linkage disequilibrium, modified after being created by LDlink.31
Mice Harboring Mib1 Missense Variants Show BAV
We introduced the Mib1 p.K735R and the Mib1 p.V943F37 missense variants into the mouse genome using CRISPR-Cas9 genomic edition. Previously, only loss-of-function studies have been performed in mice based on conditional Mib1 inactivation in the heart.19,45 Genetic studies have suggested a dominant requirement of NOTCH signaling for aortic valve development; thus, NOTCH1 haploinsufficiency might predispose to cardiac outflow tract abnormalities, including BAV.46 We found that mice heterozygous or homozygous for the Mib1K735R variant (n=41 and n=28, respectively), or the Mib1V943F variant (n=50 and n=24, respectively) displayed normal aortic valves (Figure 3A [1, 4] and Figure 3C; eFigure 5A,B,G in Supplement 1 shows quantification of background controls). To test the sensitivity of the BAV phenotype to NOTCH gene dosage, we introduced Rbp or Notch1 loss-of-function alleles38,39 into the Mib1V943F and Mib1K735R backgrounds. Notch1+/− mice alone showed only a 9% BAV penetrance (n = 11; P ≤.05 by χ2 analysis) (Figure 3B [1, 2], Figure 3C, and eTable 16 in Supplement 1). In contrast, the combination of missense Mib1 variant alleles with Notch1 or Rbp loss-of-function gene variants revealed that Mib1K735R/+;Notch1+/− double-heterozygous mice developed BAV and associated valve defects with 44% penetrance (n = 9; P <.001 by χ2 analysis) (Figure 3B [3, 4], Figure 3C, and eTable 16 in Supplement 1). All Mib1KR/+; Notch1+/− mice showed 100% ventricular septal defect (VSD) and 44% BAV; thus, all BAV was accompanied by VSD, and there is no BAV without VSD in these double-heterozygous mice. Rbp+/− mice did not show BAV (n = 10) (Figure 3B [5, 6] and Figure 3C). Mib1V943F/+;Rbp+/− mice developed BAV with 6% frequency (n = 20) (Figure 3B [7, 8], Figure 3C, and eFigure 5E,F,G in Supplement 1 show background controls). These results suggest that NOTCH signaling attenuation in a double-heterozygous Mib1 and Notch1 variant background was associated with BAV development and valve defects in mice.
Figure 3. Association of Heterozygous Mib1K735R and Mib1V943F Variants With Bicuspid Aortic Valve (BAV) in a NOTCH-Sensitized Mouse Genetic background.

A, Hematoxylin-eosin staining of aortic valves from (1) E16.5 Mib1K735R/+, (2) Mib1K735R/K735R, (3) Mib1V943F/+, and (4) Mib1V943F/V943F mice showing normal tricuspid morphology (asterisks). B, Aortic valves and transverse heart sections of E16.5 Mib1+/+;Notch1+/− (1, 2), Mib1K735R/+;Notch1+/− (3, 4), Mib1+/+;Rbp+/− (5, 6), and Mib1V943F/+;Rbp+/− (7, 8) embryos. The aortic valves leaflets are marked by an asterisk. BAVs are observed in the double heterozygotes (3, 7). Arrowheads mark the defective membranous ventricular septum, which is observed in the Notch1+/−;Mib1+/+ (1, 2) and Mib1K735R/+;Notch1+/− hearts (3, 4). C, Percentage of mice manifesting BAV and ventricular septal defect (VSD) phenotypes according to Mib1 variant and Notch1 and Rbp sensitization. (1) Mib1K735R/+ (n = 41), (2) Mib1K735R/K735R (n = 28), (3) Notch1+/− (n = 11), (4) Mib1K735R/+, Notch1+/− (n = 9), (5) Mib1V943F/+ (n = 50), (6) Mib1V943F/V943F (n = 24), (7) Rbp+/− (n = 10), and (8) Mib1V943F/+;Rbp+/− (n = 20). Scale bars, 100 μm for aortic valve sections and 200 μm for transverse heart sections.
aP ≤ .0001 by χ2.
Discussion
Using both human genetics and functional assays, we identified MIB1 as a novel gene associated with nsBAV in humans, with variants present in approximately 2% of BAV index cases in our cohorts. Our study implemented complementary human genetics approaches that allowed for the identification of MIB1 and was further supported by functional analysis (eFigure 1 in Supplement 1). The initial strategy was based on exome sequencing of a large BAV familial cohort (the discovery cohort), in which we identified an MIB1 germline variant (p.V943F) previously shown to cause LVNC.15 We used a first replication cohort of unrelated BAV cases, finding 7 additional rare variants with high predicted pathogenicity. A rare variant association study with burden testing39 showed enrichment in MIB1 variants among the BAV cohort. A common variant association study identified risk haplotypes in an additional independent cohort of 452 sporadic BAV cases compared with 1849 ethnically matched controls. Finally, we constructed functional models to confirm our findings, demonstrating that 2 genetically modified animal models, carrying the identified Mib1 missense variants in double heterozygous combination with Notch1 or Rbp deficiency, were associated with the development of BAV. These findings are entirely consistent with a mouse model of Mib1 inactivation in which mice developed cardiac abnormalities, including BAV.19
Very low penetrance and oligogenic architecture are distinct frameworks to interpret inheritance that is neither purely mendelian nor polygenic.3,8,9 Our data suggest that rare variants are associated with BAV, which evokes oligogenic inheritance involving MIB1. An oligogenic pattern has been demonstrated in mouse models with BAV.3,6,47,48,49,50 In the Slit/Robo pathway, the generation of double-variants’ progenies is necessary to increase the penetrance of BAV.49 Genetic interaction between NOS3 and NOTCH1 was also demonstrated to increase BAV penetrance.51 A similar oligogenic dose effect has been suggested for NOTCH pathway variations in outflow tract syndromes such as Alagille syndrome.52 Our mouse data show that the Mib1V943F and Mib1K735R variants were not associated with BAV in the heterozygous or homozygous condition, but rather when combined with Notch1 or Rbp heterozygous loss-of-function variants, indicating that the BAV phenotype is very sensitive to the combined insufficiency of NOTCH pathway genes (Figure 4). Our data are in full agreement with the results of a report showing that double heterozygous Mib1V943F/+;Notch1+/− mice show highly significant BAV.37 Previously, our group and others demonstrated that deleterious MIB1 variants associated with LVNC,40,53 atrial septal defects and VSDs, and patent ductus arteriosus.54 A complex phenotype of BAV and cardiac muscle malformation, as found in 1 case in the discovery cohort, has also been described.55,56 This phenotypic overlap between congenital heart valve defects, including BAV and other left heart anomalies, is common. The genes GATA4, GATA5, GATA6, ROBO4, and TBX20 show patterns of inheritance in multiple heart defects (such as atrial septal defect, VSD, BAV, patent ductus arteriosus, and mitral valve anomalies).57,58,59,60,61,62,63,64 The phenotypic overlap between LVNC and valve anomalies was also observed in a mouse model of Mib1 inactivation,40 as well as in a diversity of phenotypes induced by variations in other BAV-associated genes. Genetic sensitization experiments, in which variations in various genes are combined to uncover a dose-sensitive variant phenotype as found in our mice model, are typical of complex functional studies and are essential to identify the full implication of a given signaling pathway (ie, NOTCH) in a developmental process or disease.27
Figure 4. Schematic Presentation of the Main Players in the NOTCH Pathway.
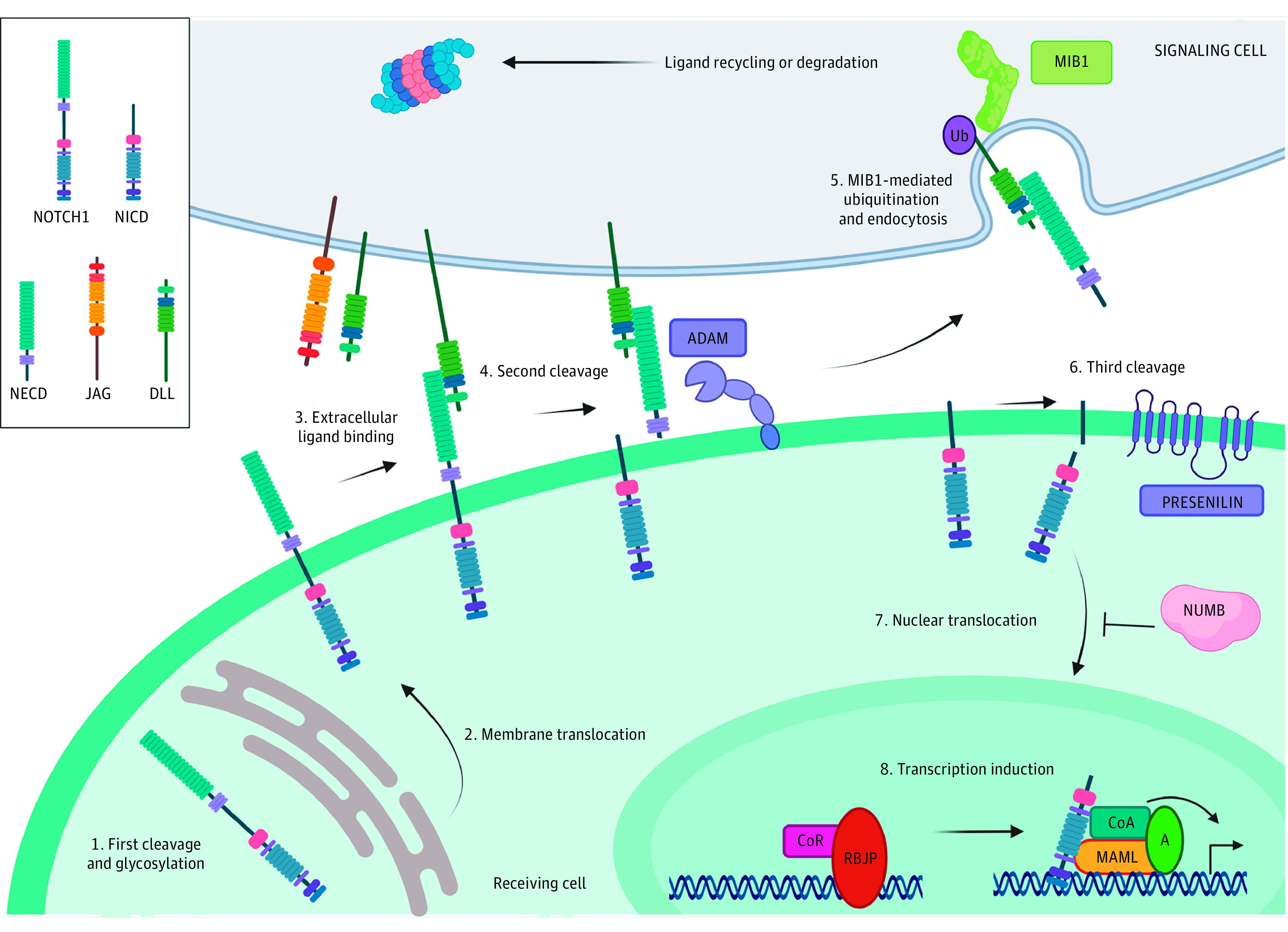
NOTCH is a local signaling mechanism in which cells are in a close position. Ligand-receptor interaction leads to a series of cleavage events that ultimately lead to the generation of the NOTCH intracellular domain (NICD), which is able to activate gene expression when bound to the appropriate factors. MIB1 and the DELTA and JAGGED ligands are expressed in the signaling cell (gray). The receiving cell (green) expresses receptors from the NOTCH family that are cleaved and glycosylated in the Golgi apparatus (step 1). Once at the membrane (step 2), NOTCH receptor binds to the ligands expressed in a neighboring cell (step 3). After this interaction, the exposed S2 cleavage site in membrane-bound NOTCH is recognized by ADAM metalloproteinases and cleaved (step 4), while MIB1 ubiquitinates the intracellular domain of the ligand in the signaling cell (step 5), eliciting ligand-receptor complex endocytosis and degradation or recycling. This induces the γ-secretase cleavage (step 6) that releases NICD, which is able to translocate to the nucleus (step 7). Once in the nucleus, NICD binds to the repressor RBPJ/RBP/CSL/Su(H), releasing its corepressors and recruiting coactivators as MAML, leading to the activation of a tissue-specific transcriptional program (step 8). MIB1 is essential for NOTCH pathway activation. This image was created using BioRender image creation software.
Ligand ubiquitination by MIB1 is essential for NOTCH pathway activation, although the downstream mechanisms are not fully understood. The key notion is that ubiquitination in the signaling cell of the ligand bound to the extracellular domain of NOTCH drives its endocytosis,65 eliciting in the receiving cell further NOTCH receptor processing and signaling activation (Figure 4).66
Other promising candidates were identified during our analysis (eTable 7 in Supplement 2). One of the leading candidates is JAG1, another NOTCH1 pathway gene and a crucial substrate of MIB1 during cardiogenesis.19,67 Interestingly, both Dll4 and Jag1 are expressed in valve endocardium during early valve development (E9.5), whereas only Jag1 is expressed in aortic valve endocardium at later stages (E12.5 onward).19,67 These 3 ligands depend on MIB1 for their normal function as NOTCH signaling activating ligands68 (Figure 3). In our discovery cohort, we identified rare and predicted deleterious missense variants in JAG1 in 2 pedigrees (eTable 7 in Supplement 2). Previous mice models of a cardiac-specific JAG1 variant resulted in BAV in 7 of 15 mice (46.7%).19 Further studies and collaboration may facilitate the discovery of its role in BAV.
Limitations
Several limitations to our study should be considered. By using a referral center–based cohort, there is a possibility for bias as the hospital clinic patients are characterized by a more severe form of disease compared with the general BAV population. Therefore, the prevalence of the MIB1 variant in the general population of patients with BAV may be lower than that in our cohort. In both replication cohorts, we used public databases for controls, and these lack validated phenotyping and may also include BAV cases, as in the general population. Finally, here we studied only 1 gene in the NOTCH pathway, but further investigation of the entire pathway is warranted, as several genes of the pathway are involved in valve development in mice.69
Conclusions
In conclusion, results of our genetic association study combining various human analyses and in vivo functional studies suggest the involvement of MIB1 in the development of nsBAV, highlighting the NOTCH pathway as a potential significant contributor to nsBAV inheritance and pathophysiology. This work also underscores the need for further investigation of NOTCH pathway components as additional candidate genes for nsBAV and as a future therapeutic target.
eTable 1. List of 34 Genes Known or Suspected to Cause Syndromic and Nonsyndromic BAV
eTable 3. Detailed Information About the Microinjection Reagents and Genotyping of the Mice Model
eTable 4. Phenotyping of French and Israeli Cases From Discovery and Replication I Cohorts
eTable 8. Candidate Genes Identified After Multistep Variant and Gene Filtering
eTable 9. MIBAVA-Leducq Cohort (Belonging to Replication Cohort I) Characteristics
eTable 10. MIB1 Identified Coding Variants
eTable 11. MIB1 Association Study by Burden Testing Using gnomAD Control Population
eTable 12. Replication Cohort II—Summary of Demographics and Clinical Data of BAV Cases
eTable 14. The 5 Most Significant SNVs in the Common Variants Analysis
eTable 15. Risk Haplotypes at the MIB1 Locus: a Comparison Between BAV Cases and Controls
eTable 16. Results From the Combination of Missense Mib1 K735R Mutant Alleles With Notch1 Loss of Function mutations
eFigure 1. A Flow Chart Summarizing the Design of the Study
eFigure 2. Flow Chart Displaying the Prioritization Process for Candidate Genes and Variants Analysis
eFigure 3. Multidimensional Scaling (MDS) Plot for 452 BAV Cases and 1911 FHS Controls
eFigure 4. Pedigree of Family BAV-003 With MIB1 p.V943F Variant and Bicuspid Aortic Valve (BAV) Phenotype
eFigure 5. Histological Analysis of Mib1+/+, Mib1+/+;Notch1+/+ and Mib1+/+;Rbp+/+ Control Mice
eTable 2. List of Known Candidate Genes
eTable 5. French Cohort Main Demographics and Clinical Characteristics
eTable 6. Israeli Cohort Main Demographics and Clinical Characteristics
eTable 7. Candidate Genes Used in the In Silico Analyses
eTable 13. Replication Cohort II Individuals Demographics and Clinical Data
Data Sharing Statement
References
- 1.Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890-1900. doi: 10.1016/S0735-1097(02)01886-7 [DOI] [PubMed] [Google Scholar]
- 2.Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J. 2005;150(3):513-515. doi: 10.1016/j.ahj.2004.10.036 [DOI] [PubMed] [Google Scholar]
- 3.Gillis E, Kumar AA, Luyckx I, et al. ; Mibava Leducq Consortium . Candidate gene resequencing in a large bicuspid aortic valve–associated thoracic aortic aneurysm cohort: SMAD6 as an important contributor. Front Physiol. 2017;8:400. doi: 10.3389/fphys.2017.00400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tessler I, Leshno M, Shmueli A, Shpitzen S, Durst R, Gilon D. Cost-effectiveness analysis of screening for first-degree relatives of patients with bicuspid aortic valve. Eur Heart J Qual Care Clin Outcomes. 2021;7(5):447-457. doi: 10.1093/ehjqcco/qcab047 [DOI] [PubMed] [Google Scholar]
- 5.Teekakirikul P, Zhu W, Gabriel GC, et al. Common deletion variants causing protocadherin-α deficiency contribute to the complex genetics of BAV and left-sided congenital heart disease. HGG Adv. 2021;2(3):2. doi: 10.1016/j.xhgg.2021.100037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gharibeh L, Komati H, Bossé Y, et al. ; Bicuspid Aortic Valve Consortium . GATA6 regulates aortic valve remodeling, and its haploinsufficiency leads to right-left type bicuspid aortic valve. Circulation. 2018;138(10):1025-1038. doi: 10.1161/CIRCULATIONAHA.117.029506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi L-M, Tao JW, Qiu XB, et al. GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int J Mol Med. 2014;33(5):1219-1226. doi: 10.3892/ijmm.2014.1700 [DOI] [PubMed] [Google Scholar]
- 8.Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004;44(1):138-143. doi: 10.1016/j.jacc.2004.03.050 [DOI] [PubMed] [Google Scholar]
- 9.Dargis N, Lamontagne M, Gaudreault N, et al. Identification of gender-specific genetic variants in patients with bicuspid aortic valve. Am J Cardiol. 2016;117(3):420-426. doi: 10.1016/j.amjcard.2015.10.058 [DOI] [PubMed] [Google Scholar]
- 10.Tessler I, Goudot G, Albuisson J, et al. Is bicuspid aortic valve morphology genetically determined? a family-based study. Am J Cardiol. 2022;163:85-90. doi: 10.1016/j.amjcard.2021.09.051 [DOI] [PubMed] [Google Scholar]
- 11.Li R-G, Xu YJ, Wang J, et al. GATA4 loss-of-function mutation and the congenitally bicuspid aortic valve. Am J Cardiol. 2018;121(4):469-474. doi: 10.1016/j.amjcard.2017.11.012 [DOI] [PubMed] [Google Scholar]
- 12.Qu X-K, Qiu XB, Yuan F, et al. A novel NKX2.5 loss-of-function mutation associated with congenital bicuspid aortic valve. Am J Cardiol. 2014;114(12):1891-1895. doi: 10.1016/j.amjcard.2014.09.028 [DOI] [PubMed] [Google Scholar]
- 13.Luyckx I, Kumar AA, Reyniers E, et al. ; MIBAVA Leducq Consortium . Copy number variation analysis in bicuspid aortic valve-related aortopathy identifies TBX20 as a contributing gene. Eur J Hum Genet. 2019;27(7):1033-1043. doi: 10.1038/s41431-019-0364-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gehlen J, Stundl A, Debiec R, et al. Elucidation of the genetic causes of bicuspid aortic valve disease. Cardiovasc Res. 2022;cvac099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thériault S, Gaudreault N, Lamontagne M, et al. A transcriptome-wide association study identifies PALMD as a susceptibility gene for calcific aortic valve stenosis. Nat Commun. 2018;9(1):988. doi: 10.1038/s41467-018-03260-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tessler I, Albuisson J, Goudot G, et al. Bicuspid aortic valve: genetic and clinical insights. Aorta (Stamford). 2021;9(4):139-146. doi: 10.1055/s-0041-1730294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284(5415):770-776. doi: 10.1126/science.284.5415.770 [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Wu B, Farrar E, et al. Notch-Tnf signaling is required for development and homeostasis of arterial valves. Eur Heart J. 2017;38(9):675-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacGrogan D, D’Amato G, Travisano S, et al. Sequential ligand-dependent notch signaling activation regulates valve primordium formation and morphogenesis. Circ Res. 2016;118(10):1480-1497. doi: 10.1161/CIRCRESAHA.115.308077 [DOI] [PubMed] [Google Scholar]
- 20.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. 2009;47(6):828-834. doi: 10.1016/j.yjmcc.2009.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Musse AA, Meloty-Kapella L, Weinmaster G. Notch ligand endocytosis: mechanistic basis of signaling activity. Semin Cell Dev Biol. 2012;23(4):429-436. doi: 10.1016/j.semcdb.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Megahed H, Nicouleau M, Barcia G, et al. Utility of whole exome sequencing for the early diagnosis of pediatric-onset cerebellar atrophy associated with developmental delay in an inbred population. Orphanet J Rare Dis. 2016;11(1):57. doi: 10.1186/s13023-016-0436-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giardine B, Riemer C, Hardison RC, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451-1455. doi: 10.1101/gr.4086505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491-498. doi: 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vandeweyer G, Van Laer L, Loeys B, Van den Bulcke T, Kooy RF. VariantDB: a flexible annotation and filtering portal for next generation sequencing data. Genome Med. 2014;6(10):74. doi: 10.1186/s13073-014-0074-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310-315. doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muskavitch MA. Delta-notch signaling and Drosophila cell fate choice. Dev Biol. 1994;166(2):415-430. doi: 10.1006/dbio.1994.1326 [DOI] [PubMed] [Google Scholar]
- 28.Guo MH, Plummer L, Chan Y-M, Hirschhorn JN, Lippincott MF. Burden testing of rare variants identified through exome sequencing via publicly available control data. Am J Hum Genet. 2018;103(4):522-534. doi: 10.1016/j.ajhg.2018.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583-589. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varadi M, Anyango S, Deshpande M, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50(D1):D439-D444. doi: 10.1093/nar/gkab1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555-3557. doi: 10.1093/bioinformatics/btv402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978-989. doi: 10.1086/319501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richardson CD, Ray GJ, Bray NL, Corn JE. Non-homologous DNA increases gene disruption efficiency by altering DNA repair outcomes. Nat Commun. 2016;7:12463. doi: 10.1038/ncomms12463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harms DW, Quadros RM, Seruggia D, et al. Mouse genome editing using the CRISPR/Cas system. Curr Protoc Hum Genet. 2014;83:15.7.1-27. doi: 10.1002/0471142905.hg1507s83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de la Pompa JL, Wakeham A, Correia KM, et al. Conservation of the Notch signalling pathway in mammalian neurogenesis. Development. 1997;124(6):1139-1148. doi: 10.1242/dev.124.6.1139 [DOI] [PubMed] [Google Scholar]
- 36.Kanzler B, Kuschert SJ, Liu YH, Mallo M. Hoxa-2 restricts the chondrogenic domain and inhibits bone formation during development of the branchial area. Development. 1998;125(14):2587-2597. doi: 10.1242/dev.125.14.2587 [DOI] [PubMed] [Google Scholar]
- 37.Siguero-Álvarez M, Salguero-Jiménez A, Grego-Bessa J, et al. Human hereditary cardiomyopathy shares a genetic substrate with bicuspid aortic valve. Circulation. 2023;147(1):47-65. doi: 10.1161/CIRCULATIONAHA.121.058767 [DOI] [PubMed] [Google Scholar]
- 38.Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121(5):1533-1545. doi: 10.1242/dev.121.5.1533 [DOI] [PubMed] [Google Scholar]
- 39.Oka C, Nakano T, Wakeham A, et al. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 1995;121(10):3291-3301. doi: 10.1242/dev.121.10.3291 [DOI] [PubMed] [Google Scholar]
- 40.Luxán G, Casanova JC, Martínez-Poveda B, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. 2013;19(2):193-201. doi: 10.1038/nm.3046 [DOI] [PubMed] [Google Scholar]
- 41.Child N, Muhr T, Sammut E, et al. Prevalence of myocardial crypts in a large retrospective cohort study by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2014;16(1):66. doi: 10.1186/s12968-014-0066-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A; NISC Comparative Sequencing Program . Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15(7):901-913. doi: 10.1101/gr.3577405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMillan BJ, Schnute B, Ohlenhard N, et al. A tail of two sites: a bipartite mechanism for recognition of notch ligands by mind bomb E3 ligases. Mol Cell. 2015;57(5):912-924. doi: 10.1016/j.molcel.2015.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Machiela MJ, Chanock SJ. LDassoc: an online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics. 2018;34(5):887-889. doi: 10.1093/bioinformatics/btx561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Captur G, Wilson R, Bennett MF, et al. Morphogenesis of myocardial trabeculae in the mouse embryo. J Anat. 2016;229(2):314-325. doi: 10.1111/joa.12465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koenig SN, LaHaye S, Feller JD, et al. Notch1 haploinsufficiency causes ascending aortic aneurysms in mice. JCI Insight. 2017;2(21):2. doi: 10.1172/jci.insight.91353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quintero-Rivera F, Xi QJ, Keppler-Noreuil KM, et al. MATR3 disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Hum Mol Genet. 2015;24(8):2375-2389. doi: 10.1093/hmg/ddv004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biben C, Weber R, Kesteven S, et al. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ Res. 2000;87(10):888-895. doi: 10.1161/01.RES.87.10.888 [DOI] [PubMed] [Google Scholar]
- 49.Mommersteeg MTM, Yeh ML, Parnavelas JG, Andrews WD. Disrupted Slit-Robo signalling results in membranous ventricular septum defects and bicuspid aortic valves. Cardiovasc Res. 2015;106(1):55-66. doi: 10.1093/cvr/cvv040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas PS, Sridurongrit S, Ruiz-Lozano P, Kaartinen V. Deficient signaling via Alk2 (Acvr1) leads to bicuspid aortic valve development. PLoS One. 2012;7(4):e35539. doi: 10.1371/journal.pone.0035539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bosse K, Hans CP, Zhao N, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol. 2013;60:27-35. doi: 10.1016/j.yjmcc.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z, Chen S, Boyle S, et al. The extracellular domain of Notch2 increases its cell-surface abundance and ligand responsiveness during kidney development. Dev Cell. 2013;25(6):585-598. doi: 10.1016/j.devcel.2013.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Waning JI, Caliskan K, Hoedemaekers YM, et al. Genetics, clinical features, and long-term outcome of noncompaction cardiomyopathy. J Am Coll Cardiol. 2018;71(7):711-722. doi: 10.1016/j.jacc.2017.12.019 [DOI] [PubMed] [Google Scholar]
- 54.Li B, Yu L, Liu D, et al. MIB1 mutations reduce Notch signaling activation and contribute to congenital heart disease. Clin Sci (Lond). 2018;132(23):2483-2491. doi: 10.1042/CS20180732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karatza A, Mylonas KS, Tzifa A. Left ventricular noncompaction in a child with bicuspid aortic valve and aortic coarctation. Cardiol Young. 2019;29(9):1208-1210. doi: 10.1017/S1047951119001707 [DOI] [PubMed] [Google Scholar]
- 56.Stähli BE, Gebhard C, Biaggi P, et al. Left ventricular non-compaction: prevalence in congenital heart disease. Int J Cardiol. 2013;167(6):2477-2481. doi: 10.1016/j.ijcard.2012.05.095 [DOI] [PubMed] [Google Scholar]
- 57.Yang B, Zhou W, Jiao J, et al. Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nat Commun. 2017;8:15481. doi: 10.1038/ncomms15481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garg V, Kathiriya IS, Barnes R, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424(6947):443-447. doi: 10.1038/nature01827 [DOI] [PubMed] [Google Scholar]
- 59.Jiang J-Q, Li RG, Wang J, et al. Prevalence and spectrum of GATA5 mutations associated with congenital heart disease. Int J Cardiol. 2013;165(3):570-573. doi: 10.1016/j.ijcard.2012.09.039 [DOI] [PubMed] [Google Scholar]
- 60.Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E. GATA4 sequence variants in patients with congenital heart disease. J Med Genet. 2007;44(12):779-783. doi: 10.1136/jmg.2007.052183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maitra M, Koenig SN, Srivastava D, Garg V. Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatr Res. 2010;68(4):281-285. doi: 10.1203/PDR.0b013e3181ed17e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang R-T, Wang J, Xue S, et al. TBX20 loss-of-function mutation responsible for familial tetralogy of Fallot or sporadic persistent truncus arteriosus. Int J Med Sci. 2017;14(4):323-332. doi: 10.7150/ijms.17834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu C, Shen A, Li X, Jiao W, Zhang X, Li Z. T-box transcription factor TBX20 mutations in Chinese patients with congenital heart disease. Eur J Med Genet. 2008;51(6):580-587. doi: 10.1016/j.ejmg.2008.09.001 [DOI] [PubMed] [Google Scholar]
- 64.Posch MG, Gramlich M, Sunde M, et al. A gain-of-function TBX20 mutation causes congenital atrial septal defects, patent foramen ovale, and cardiac valve defects. J Med Genet. 2010;47(4):230-235. doi: 10.1136/jmg.2009.069997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nichols JT, Miyamoto A, Olsen SL, D’Souza B, Yao C, Weinmaster G. DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J Cell Biol. 2007;176(4):445-458. doi: 10.1083/jcb.200609014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kovall RA, Gebelein B, Sprinzak D, Kopan R. The canonical Notch signaling pathway: structural and biochemical insights into shape, sugar, and force. Dev Cell. 2017;41(3):228-241. doi: 10.1016/j.devcel.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.D’Amato G, Luxán G, del Monte-Nieto G, et al. Sequential Notch activation regulates ventricular chamber development. Nat Cell Biol. 2016;18(1):7-20. doi: 10.1038/ncb3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo B, McMillan BJ, Blacklow SC. Structure and function of the Mind bomb E3 ligase in the context of Notch signal transduction. Curr Opin Struct Biol. 2016;41:38-45. doi: 10.1016/j.sbi.2016.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, Fang Y, Lu P, Wu B, Zhou B. NOTCH signaling in aortic valve development and calcific aortic valve disease. Front Cardiovasc Med. 2021;8:682298. doi: 10.3389/fcvm.2021.682298 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. List of 34 Genes Known or Suspected to Cause Syndromic and Nonsyndromic BAV
eTable 3. Detailed Information About the Microinjection Reagents and Genotyping of the Mice Model
eTable 4. Phenotyping of French and Israeli Cases From Discovery and Replication I Cohorts
eTable 8. Candidate Genes Identified After Multistep Variant and Gene Filtering
eTable 9. MIBAVA-Leducq Cohort (Belonging to Replication Cohort I) Characteristics
eTable 10. MIB1 Identified Coding Variants
eTable 11. MIB1 Association Study by Burden Testing Using gnomAD Control Population
eTable 12. Replication Cohort II—Summary of Demographics and Clinical Data of BAV Cases
eTable 14. The 5 Most Significant SNVs in the Common Variants Analysis
eTable 15. Risk Haplotypes at the MIB1 Locus: a Comparison Between BAV Cases and Controls
eTable 16. Results From the Combination of Missense Mib1 K735R Mutant Alleles With Notch1 Loss of Function mutations
eFigure 1. A Flow Chart Summarizing the Design of the Study
eFigure 2. Flow Chart Displaying the Prioritization Process for Candidate Genes and Variants Analysis
eFigure 3. Multidimensional Scaling (MDS) Plot for 452 BAV Cases and 1911 FHS Controls
eFigure 4. Pedigree of Family BAV-003 With MIB1 p.V943F Variant and Bicuspid Aortic Valve (BAV) Phenotype
eFigure 5. Histological Analysis of Mib1+/+, Mib1+/+;Notch1+/+ and Mib1+/+;Rbp+/+ Control Mice
eTable 2. List of Known Candidate Genes
eTable 5. French Cohort Main Demographics and Clinical Characteristics
eTable 6. Israeli Cohort Main Demographics and Clinical Characteristics
eTable 7. Candidate Genes Used in the In Silico Analyses
eTable 13. Replication Cohort II Individuals Demographics and Clinical Data
Data Sharing Statement