

INTRODUCTION
Sickle cell disease (SCD) is a genetic hemoglobinopathy associated with extensive morbidity and early mortality. While there have been recent improvements in available disease-modifying therapies for SCD, cardiopulmonary complications remain a major risk factor for death in this population. Unfortunately, existing guidelines regarding screening for, diagnosis of, and management of cardiopulmonary complications are limited because of a lack of high-quality prospective longitudinal data in SCD populations.1,2 For example, we know that individuals with SCD have lower lung function compared with those without SCD but we still do not understand patterns of lung function progression from childhood to adulthood, the clinical significance of abnormal lung function, or if there are potential benefits of universal lung function screening in this population. While it is clear that pulmonary hypertension (PH) is a major risk factor for early death in SCD, the benefit of universal screening echocardiography for PH and the approach to management of abnormal screening echocardiography, especially in asymptomatic individuals, has not yet been determined. Herein we provide an overview of current knowledge regarding the presentation, clinical significance, and approach to management of acute and chronic cardiopulmonary complications of SCD.
ACUTE CHEST SYNDROME
The acute chest syndrome (ACS) in SCD is defined as a lung injury syndrome characterized by a new pulmonary infiltrate which: (1) is consistent with alveolar consolidation, rather than atelectasis; (2) involves at least one complete lung segment; and (3) is accompanied by chest pain, fever, tachypnea, wheezing, or cough.3 ACS is the second most common cause of hospitalization in patients with SCD, the leading cause of admission to an intensive care unit, and, together with PH, a leading cause of premature death, accounting for 25% of SCD-related mortality in earlier cohorts.4,5
ACS can occur in any of the sickle hemoglobinopathies, but it is more common in individuals with homozygous sickle cell disease (HbSS). Clinical parameters that appear to increase risk for, or are associated with, the development of ACS include: young age, active smoking, environmental smoke exposure,6 major surgical procedures, acute rib infarcts, avascular necrosis of the hips, pregnancy, use of opioids, acute anemic events, nocturnal or day-time hypoxemia, and previous pulmonary events.7,8 In children, a number of studies suggest that asthma is a risk factor for the development of ACS.9–11 An elevated white blood cell count, a higher steady-state level of hemoglobin, and a lower steady-state level of fetal hemoglobin are all laboratory parameters associated with an increased risk of developing ACS.7,8
The etiology of ACS is multifactorial. Although a specific cause is not identified in a substantial proportion of patients, three major pathogenetic mechanisms are involved, including infection, bone marrow fat embolization, and direct red cell intravascular sequestration, which cause lung injury and infarction (Table 1). As much as 80% of patients with ACS present with fever and 62% with cough; approximately 40% have chest pain, tachypnea, dyspnea, and abdominal, arm, leg, rib, or sternal pain.3 Some of the clinical features of ACS are age-dependent, which likely reflects the different disease etiologies in different age groups. Children have a higher proportion of underlying infectious etiologies in comparison with adults, who have fat embolization as a major cause. Most adult patients present with severe extremity or chest pain and develop ACS 24–72 hours later. Reactive airways disease is observed in 13% of cases and is much more common in children.3 Some patients manifest evidence of systemic fat embolization. In these cases, ACS is part of the spectrum of the systemic fat emboli syndrome, evident as acute multiorgan system failure (MOSF). Patients with MOSF experience acute hypoxic respiratory failure, acute cor pulmonale, renal and hepatic dysfunction, alterations in mental status, seizures, thrombocytopenia, and coagulopathy.12,13
Table 1.
Causes of the acute chest syndromea
| All Episodes (n = 670) | Age at Episode of Acute Chest Syndrome | |||
|---|---|---|---|---|
| Causes | No. of Episodes (%) | |||
| 0–9 y (n = 329) | 10–19 y (n = 188) | ≥ 20 y (n = 153) | ||
| Fat embolism, with or without infectionb | 59 (8.8) | 24 | 16 | 19 |
| Chlamydiac | 48 (7.2) | 19 | 15 | 14 |
| Mycoplasmad | 44 (6.6) | 29 | 7 | 8 |
| Virus | 43 (6.4) | 36 | 5 | 2 |
| Bacteria | 30 (4.5) | 13 | 15 | 12 |
| Mixed infections | 25 (3.7) | 16 | 6 | 3 |
| Legionella | 4 (0.6) | 3 | 0 | 1 |
| Miscellaneous infectionse | 3 (0.4) | 0 | 3 | 0 |
| Infarctionf | 108 (16.1) | 50 | 43 | 15 |
| Unknowng | 306 (45.7) | 139 | 88 | 79 |
Data on one episode were excluded because the patient’s birth date was not known.
Nineteen of the episodes of pulmonary fat embolism were associated with infectious pathogens.
This category included episodes in which Chlamydia alone was identified but not episodes involving mixed infections or pulmonary fat embolism.
This category included only episodes in which only Mycoplasma pneumoniae or M hominis was identified, but not episodes involving mixed infections, Mycobacterium tuberculosis or pulmonary fat embolism.
This category included two cases of tuberculosis and one case of M avium complex infection.
A pulmonary infarction was presumed to have occurred when the results of the analysis for pulmonary fat embolism, bacterial studies, viral isolation studies, and serologic tests were complete and were all negative.
The cause of episodes for which some or all of the diagnostic data were incomplete and no etiologic agent was identified was considered to be unknown.
(From Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, Dean D. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342.)
Since the triggers and risk factors for ACS are well known, preventive strategies in the outpatient setting, clinical surveillance, and aggressive and early therapy are likely to improve prognosis. Patients with multiple episodes of vaso-occlusive crises or a previous history of ACS should be treated with hydroxyurea in the outpatient setting because its use has been shown to reduce the risk of developing ACS by approximately 50%.14,15 A chronic transfusion regimen is also effective in reducing the incidence of ACS,16 as is preoperative blood transfusion in patients undergoing surgical procedures.17 In acutely ill patients, specific strategies, such as aggressive pain management and incentive spirometry, can minimize chest wall splinting, mitigating the development of atelectasis and alveolar hypoxia.18 We recommend use of empiric antimicrobial therapy in all patients with ACS, given the high prevalence of infectious etiologies. Coverage should include agents effective against atypical bacteria and encapsulated organisms. Although not tested in randomized trials, blood transfusion remains the mainstay of ACS therapy and is considered standard of care. Acute red cell transfusion increases PaO2 and hemoglobin oxygen saturation and may rapidly resolve the pulmonary event.3,19
ASTHMA AND AIRWAYS DISEASE IN INDIVIDUALS WITH SICKLE CELL DISEASE
Asthma is the most common chronic disease in childhood and disproportionately affects African Americans in terms of prevalence, morbidity, and mortality. Because SCD is the most common inherited disorder among African Americans, a certain number of individuals will be affected by both SCD and asthma. While a few small case-control studies have suggested that asthma occurs at a higher frequency among those with SCD compared with the general population,10 larger research cohort studies have not conclusively demonstrated this to be the case.20,21
Questions remain regarding whether asthma occurs as an independent comorbidity or whether the pathogenesis of SCD predisposes individuals to airway inflammation and asthma. Experimental mouse studies have shown that compared with wild type and heterozygous sickle mice, SCD mice develop exaggerated inflammatory responses in response to allergen challenges with ovalbumin and house dust mite sensitization.22–24 In addition to allergic inflammation, other mechanisms for airway disease have been proposed, including anemia-associated increased cardiac output and dilation of the pulmonary vasculature. Studies of adults and children with SCD have demonstrated associations between increased pulmonary capillary blood volume and measures of lower airway obstruction and airway resistance.25,26
Determining which individuals with SCD actually have asthma is not always straight-forward, as isolated features that are common in asthma are also common in SCD. Wheezing is common during episodes of ACS,3,8 and may be present at other times when there is acutely increased cardiac output, such as during febrile illnesses. Glassberg and colleagues27 noted that of 262 patients who presented to an emergency department for an SCD-related encounter, 49 (19%) had at least 1 documented episode of wheezing; fewer than half of those patients had been diagnosed with asthma. One research cohort study of 114 adults found that 34 (30%) reported 2 or more episodes of severe wheeze, leading to shortness of breath.28 Airway hyperresponsiveness is present in more than 50% of children with SCD, even in the absence of other features associated with asthma, including baseline airway obstruction, elevated blood eosinophil count, or aeroallergen sensitization.29
Physician-diagnosed asthma has been associated with an increased incidence of ACS in children with sickle cell anemia (ie, with HbSS) and seems to be an even bigger risk factor for ACS in children with HbSC.30 Prospective data from 291 children with HbSS in the Cooperative Study of SCD found that children with asthma were younger at the time of their first episode of ACS and had twice as many episodes of ACS as children without asthma.9 Children in the Sleep and Asthma Cohort (SAC) study with physician-diagnosed asthma also had higher rates of ACS episodes than children without asthma.31
Individuals with SCD should have careful screening for recurrent respiratory symptoms32 including wheezing, dyspnea, or exercise limitation, as well as a personal history of allergies/atopy and a family history of asthma.31 If positive, patients should be referred for evaluation and management by a pulmonologist that includes pulmonary function tests.33 Treatment with anti-inflammatory controller therapy according to the National Heart, Lung, and Blood Institute (NHLBI) and/or Global Initiative for Asthma (GINA) guidelines is recommended in patients who seem to have persistent asthma.34,35 Several groups have reported improved adherence to national evidence-based asthma guidelines and improved SCD-specific outcomes when patients have SCD and asthma care that is delivered in an integrative care model.33,36,37
LUNG FUNCTION IN SICKLE CELL DISEASE
Given the frequency of dyspnea and functional limitation among adults with SCD,38 gathering knowledge of the natural history of lung function in SCD should be a priority. Unfortunately, longitudinal studies of lung function across the lifespan for individuals with SCD are limited. While there are several cross-sectional studies of lung function among individuals with SCD, there are no large prospective studies that follow children with SCD into adulthood. Furthermore, interpretation of lung function across studies is not always consistent; while international guidelines recommend use of lower-limit-of-normal (LLN) criteria (which can vary by age, height, and gender) to define abnormal lung function patterns, several studies in SCD use arbitrary cut-offs (such as an FEV1/FVC < 0.70 or 0.80 to distinguish normal vs obstruction). Despite these limitations, when taken together, there are some general patterns that have emerged (Table 2).
Table 2.
Summary of studies on lung function in sickle cell disease
| Studies (year) | Study Designs | Populations | Findings |
|---|---|---|---|
| Ivankovich et al39, 2019 | Cross sectional case-control | 22 infants with SCD, 37 healthy control infants aged 6–18 months | Normal lung function in 77% of infants with SCA but those w SCA had lower FVC, FEF0.5, FEF25–75 compared with controls |
| Koumbourlis et al40, 1997 | Cross sectional | 20 infants with SCD aged 3–30 months (12 with HbSS) | Elevated FRC, decreased maximum expiratory flow at FRC, and increased time to maximum expired flow/total expiratory time, suggesting hyperinflation and lower airway obstruction |
| Cohen et al41 2016 | Cross sectional and longitudinal cohort | 149 children aged 6–19 years with HbSS with spirometry and plethysmography; 139 had morbidity data from birth, 136 with prospective morbidity data (Sleep and Asthma Cohort Study) | 70% with normal lung function, 16%, 7%, and 6% with obstructive, restrictive, nonspecific, and mixed patterns, respectively. Lung function pattern not associated with prior or future pain or ACS episodes |
| Arteta et al42, 2014 | Cross sectional | 146 children with HbSS or HbSβ0 (PUSH study) | 19% had obstruction (44% of whom had asthma), 9% had restriction, 11% had nonspecific abnormal lung function, and 19% had reduced DLCO |
| MacLean et al46, 2008 | Single center longitudinal cohort study | 312 children with SCD aged 8–18 years | Average decline in FEV1: 2.93% per year for males, 2.95% per year for females; average decline in TLC: 2.15% per year for males, 2.43% per year for females |
| Lunt et al47, 2016 | Longitudinal case-control study in 2 cohorts |
Cohort 1: 47 children with SCD, 26 controls; mean age 8.8 (range 3–14) years, followed up for 2 years Cohort 2: 45 children with SCD, 24 controls; mean age 10.2 (range 4–17) years, followed up for 10 years |
Cohort 1: At baseline: 34% with obstruction, 2% with restriction. FEV1 decline 1.45% per year Cohort 2 at baseline: 24% with obstruction, 11% with restriction. FEV1 decline 0.9% per year. |
| Klings et al53, 2006 | Multicenter longitudinal cohort study | 310 adults with HbSS (Cooperative Study of SCD) | 74% had restriction, mean TLC 70% of predicted; 13% had isolated reduced DLCO. DLCO inversely associated with age. |
| Field etal52, 2008 | Single center longitudinal cohort study | 92 adults with SCD, lung function testing obtained for clinical reasons | 35.7% with restriction, 18% with obstruction. FEV1 decreased by 49 cc per year |
| Hodges et al51, 2022 | Single center longitudinal cohort study | 193 adults with SCD 309 adults with cystic fibrosis Age 18–65 years |
FEV1 declined 23 and 26 mL/year in those with SCD and cystic fibrosis, respectively, no difference between these two cohorts |
Abbreviations: ACS, acute chest syndrome; DLCO, diffusion capacity for carbon monoxide; FEF0.5, forced expiratory flow in 0; 5 seconds; FEF25–75, forced expiratory flow from the 25th to 75%ile of the maneuver; FEV1, forced expiratory volume in 1 second; FRC, functional residual capacity; FVC, forced vital capacity; SCA, sickle cell anemia; SCD, sickle cell disease; TLC, total lung capacity.
Children with SCD have, on average, reduced lung function compared with age, gender, and race-matched healthy control children, but generally have lung function in the normal range. This has been documented in two small studies of infants, both of which demonstrated some airflow limitation compared with healthy control children.39,40 While 70–80% of school-age children have lung function above the LLN, approximately 15–20% have lower airway obstruction and 7–9% have a restrictive defect.41,42 In adults, restriction is the more common abnormality (ranging from 27% to 74% across studies), but some adults do have lower airway obstruction (19% in one research cohort28). Reductions in pulmonary function parameters have been associated with abnormalities seen on high resolution computed tomography of the chest, including lobar volume loss, irregular linear opacities, and central vessel prominence.43 More recently, single breath washout and multiple breath washout techniques have been used to demonstrate ventilation heterogeneity in children and adults with sickle cell anemia,44,45 which may serve as an earlier marker of peripheral lung disease before abnormalities on spirometry and plethysmography are detected.
The longitudinal trajectory of lung function among individuals with SCD has not been well studied. A few studies have reported variable and inconsistent rates of progressive decline in FEV1, FVC, and TLC among children46–48 (ranging from 0.5% to 2–3% per year); but there are studies showing treatment with hydroxyurea may slow the rate of lung function decline.49,50 Data in adults have shown declines in FEV151,52 as well as reductions in diffusion capacity for carbon monoxide (DLCO) that correlate with age but are independent of the degree of anemia.53 Of note, one study suggests that FEV1 decline may be associated with an increased risk of mortality.54
Current NIH and American Society of Hematology guidelines recommend against routine screening pulmonary function tests (PFTs) for asymptomatic individuals with SCD. However, clinicians should have a low threshold for obtaining PFTs as part of a diagnostic evaluation of respiratory signs and symptoms including cough, dyspnea, chest pain, reduced exercise capacity, oxygen desaturation, etc.1 Pulmonary function abnormalities should be interpreted and addressed in the context of the clinical presentation; management may involve treating asthma and/or sleep-disordered breathing (SDB), optimizing SCD-modifying therapies (ie, hydroxyurea), and/or evaluation and management of PH. Although the clinical benefit of universal screening PFTs has not yet been demonstrated, there is a need for well-designed longitudinal prospective studies to evaluate (1) the natural history of lung function across the lifespan; (2) risk factors for lung function decline in SCD; (3) the relationship between lung function and morbidity and mortality in SCD patients; and (4) the impact of pulmonary function screening on changes in SCD management and outcomes.1
SLEEP-DISORDERED BREATHING AND NOCTURNAL HYPOXEMIA
SDB and hypoxemia appear to be common in SCD and may impact SCD morbidity and mortality. SDB includes obstructive sleep apnea (OSA), central sleep apnea, and nonobstructive hypoventilation. In addition, individuals may have nonobstructive sustained or intermittent nocturnal hypoxemia or a daytime oxygen requirement. Children with SCD appear to have an increased frequency of nocturnal desaturation compared with children without SCD. This can be partially explained by an increased risk of OSA in children with SCD,20 although children may have nocturnal desaturation in the absence of OSA.55 Some have attributed oxygen desaturation in individuals with SCD to a multifactorial rightward shift of the oxyhemoglobin dissociation curve and decreased oxyhemoglobin affinity (Fig. 1), because of a combination of increased production of 2,3 diphosphoglycerate (2,3 DPG) in the setting of severe anemia to prevent tissue hypoxia; and hypoventilation with resulting CO2 retention from pain, opioid use, and SDB.56
Fig. 1.
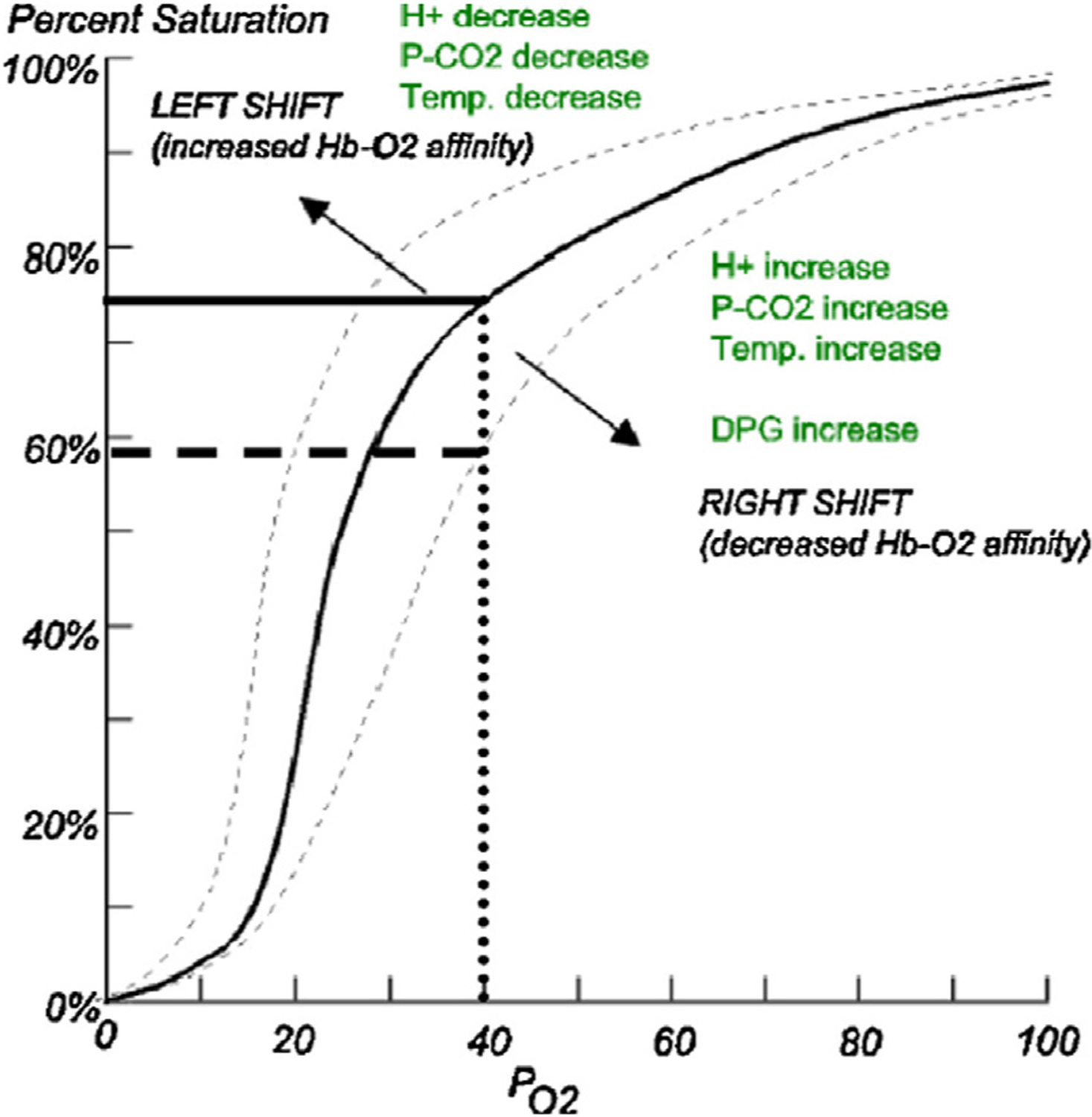
Oxyhemoglobin dissociation curve. The rightward shift, as seen in SCD, indicates that oxyhemoglobin saturation at a given arterial oxygen pressure is lower in patients with SCD because of decreased oxygen affinity of HbS, increased 2,3 diphosphoglycerate (2,3 DPG) in sickled erythrocytes, hypoventilation due to pain or opioid use, and as an adaptation to severe anemia to prevent tissue hypoxia. (From Caboot JB, Allen JL. Hypoxemia in sickle cell disease: significance and management. Paediatr Respir Rev. 2014 Mar;15(1):17–23. https://doi.org/10.1016/j.prrv.2013.12.004. Epub 2013 Dec 21. PMID: 24461342.)56
In the largest observational research cohort study of 243 children with sickle cell anemia, the prevalence of OSA [with an obstructive apnea hypopnea index (OAHI) of ≥1 event per hour] was 41%; 10% of children met criteria for moderate OSA (OAHI ≥5).20 While snoring was a significant risk factor for OSA in this cohort, 20% of children with moderate OSA did not have habitual snoring and only 32% had caretaker-witnessed apneas, suggesting that screening for common OSA symptoms in children with SCD may be insufficient in determining which patients warrant polysomnography. Additional features associated with OSA in that cohort included a daytime resting oxygen saturation of <96% on room air and nocturnal enuresis.20,57
Less is known about SDB and specifically OSA in adults with SCD. Of three small studies that utilize polysomnography rather than pulse oximetry alone,58–60 only one study enrolled young adults with SCD irrespective of symptoms.59 That said, the prevalence of OSA in all three studies was high (23%–50%). Given the logistical challenges, high cost, and limited insurance coverage of full-laboratory polysomnography for adults, Ayachi and colleagues examined use of the 3% oxygen desaturation index (ODI3%) to predict which adults with SCD have OSA. Using high-resolution pulse oximetry data recorded as part of a sleep study, they found that using an ODI3% had good agreement with AHI, suggesting that home nocturnal oximetry with a high-resolution pulse oximeter could be a useful tool to determine which patients warrant full -laboratory polysomnography.61
There are many outstanding questions about SDB and oxygen desaturation in SCD,62 including optimal approaches to evaluate SDB and oxygen saturation. Data are conflicting with regard to the extent to which OSA and sustained versus intermittent oxygen desaturation are associated with morbidity and mortality in this population. While some studies have found links between pain, ACS, cerebrovascular disease, and cardiovascular dysfunction,63–66 the results are inconsistent.67–70
While current SCD guidelines recommend against universal screening with formal polysomnography, a comprehensive sleep history and review of systems (besides simply asking about snoring) are essential for these patients.1 Individuals with snoring, witnessed apneas, and/or nonrestorative sleep; unexplained oxygen desaturation during sleep, while awake, or with exertion; nocturnal enuresis in an older child; recurrent priapism; PH; history of stroke; and cognitive and executive function issues, such as attention and concentration difficulties, confusion, and memory issues should strongly consider a sleep study. Several case series have shown that initiating treatment with hydroxyurea has the potential to improve OSA and nocturnal hypoxemia in patients with SCD.71–74 One large pediatric inpatient database study found that the increased risk of neurologic complications among children with SCD and OSA was mitigated by treatment with nocturnal noninvasive ventilation.75
PULMONARY VASCULAR DISEASE
Sustained intravascular hemolysis and anemia worsened by episodic vascular occlusion, inflammation, and organ ischemia, directly injure the vasculature.76–80 This leads to the development of PH, a major cause of death in patients with SCD.80,81 PH is currently defined as a mean pulmonary arterial pressure (mPAP) measured by right heart catheterization (RHC) of >20 mm Hg.82 Individuals with SCD may develop PH with primary vascular involvement [World Health Organization (WHO) group 1, or pulmonary arterial hypertension (PAH)] secondary to left heart disease (group 2 PH), secondary to interstitial lung disease or pulmonary fibrosis (group 3 PH), or due to chronic thromboembolic PH (CTEPH, or group 4 PH). Due to these myriad presentations, PH related to SCD is currently listed in group 5 in the clinical classification of PH81 (Fig. 2).
Fig. 2.
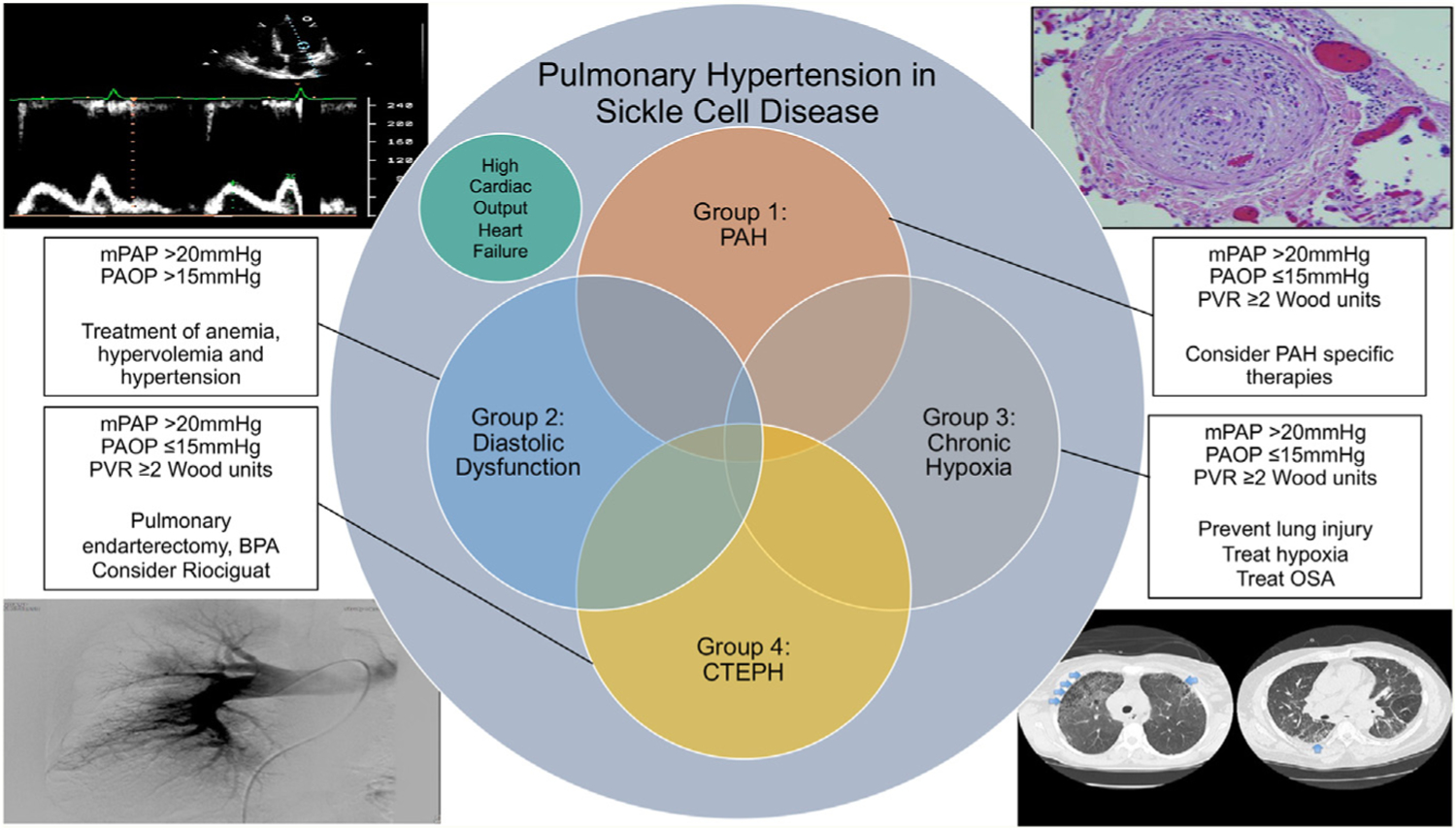
Overview of causes of PH in SCD. PH in SCD patients may be due to multiple different etiologies and often with multiple underlying causes. An overview of the different etiologies, their hemodynamics, and therapeutic options are listed. BPA, balloon pulmonary angioplasty; CTEPH, chronic thromboembolic PH; mPAP, mean pulmonary artery pressure; OSA, obstructive sleep apnea; PAOP, pulmonary artery occlusion pressure; PVR, pulmonary vascular resistance. (From Prohaska CC, Machado RF. The different facets of sickle cell disease-related pulmonary hypertension. Curr Opin Pulm Med. Sep 1 2021;27(5):319–328.)
Using the former definition of PH (mPAP ≥25 mm Hg) the prevalence of PH in SCD is 6%–11%83–85 and with the new definition, a significantly higher number of individuals with SCD could be classified as having SCD-related PH. From a hemodynamic perspective, half of patients have precapillary PH (defined as mPAP ≥ 25 mm Hg and PAOP ≤15 mm Hg), and half have post-capillary PH (defined as mPAP ≥25 mm Hg and PAOP >15 mm Hg).83–87 As such, most patients with SCD have hemodynamic profiles consistent with group 1 and group 2 disease. High-output heart failure may occur because of longstanding elevated cardiac output in the setting of anemia, although this is a rare etiology of PH. Advanced lung disease and pulmonary fibrosis leading to PH (group 3) are exceedingly rare, even if patients develop recurrent episodes of ACS over time. Given the increased risk of thromboembolism in SCD patients,88–92 they are at a higher risk of developing CTEPH (group 4), which is characterized by recurrent pulmonary thromboemboli followed by fibrotic remodeling of the occluded pulmonary vasculature.93 Scintigraphic evidence suggestive of CTEPH occurs in approximately 5% of patients with SCD and severe PH.94 Identification of CTEPH is important as these patients require lifelong anticoagulation, specific medications may be approved for therapeutic use,95 and surgical options may be available, including pulmonary artery balloon angioplasty or pulmonary endarterectomy.96–100
Regardless of the etiology, PH is a major threat to the well-being of individuals with SCD. Multiple studies have demonstrated that a high mPAP and other hemodynamic and laboratory parameters, such as elevated transpulmonary gradient, diastolic pulmonary gradient (pulmonary artery diastolic pressure—PAOP), pulmonary vascular resistance, NT-proBNP levels, 6-minute walk distance, and WHO functional class were all associated with increased mortality.87,101–103
Typical symptoms of PH include dyspnea on exertion and signs of right heart failure, such as elevated jugular venous pressure, a loud P2, and peripheral edema.93,104 These symptoms may also be attributable to anemia, left ventricular dysfunction, pulmonary fibrosis, or cirrhosis, all of which commonly develop in individuals with SCD. Diagnostic evaluation should include assessment for conditions that may also contribute to the development of PH [including iron overload, chronic liver disease, human immunodeficiency virus (HIV) infection, nocturnal hypoxemia, and pulmonary thromboembolism] and should follow established guidelines.82,92,103 In particular, assessment of functional capacity with a 6-minute walk test, measurement of brain natriuretic peptide (BNP) or N-terminal probrain natriuretic peptide (NT-proBNP) levels (a hormone released by the myocardium under pressure stress), as well as assessments of right ventricular systolic pressure via echocardiogram, should be used to identify patients with SCD at higher risk of PH, lower exercise capacity, and increased mortality risk.102,105,106
Mainstays of therapy include maximization of SCD-specific therapy (eg, hydroxyurea, transfusion therapy, stem cell transplantation), treatment of hypoxia with supplemental oxygen therapy, and treatment of associated cardiopulmonary conditions. Chronic red blood cell transfusions have been shown to reduce pulmonary pressures and increase 6-minute walk distance and functional classification in patients with SCD and PH.107 Bone marrow and lung transplantation have been shown to normalize pulmonary pressures and improve short-term outcome measures.108,109 In patients with PAH, namely, patients with an mPAP ≥25 mm Hg, PAOP <15 mm Hg and relatively high pulmonary vascular resistance (PVR) (≥160 dyn s/cm5), PAH-specific therapy may be considered. There is limited data on specific treatment of PH in SCD, and the choice of agent is empirical and based on the safety profile of the medication and physician preference.86,95,101,110–113 If these therapies should be considered, patients should be referred to a center specialized in the management of PH.
SICKLE CELL DISEASE CARDIOMYOPATHY
Recognizing sickle cell cardiomyopathy (SCC): Since its origins, cardiac pathophysiology in SCD has largely been attributed to anemia and the resulting high-output heart failure (HOHF).114 Because of reduced oxygen carrying capacity from anemia, cardiac output and stroke volume are increased with dilation of the peripheral vasculature, resulting in low systemic vascular resistance. Subsequent activation of the renin–angiotensin–aldosterone system results in volume overload and increased wall stress. Chronic volume overload contributes to the development of cardiac chamber dilation, hypertrophy, elevated filling pressures, and HOHF.
Growing evidence over the past two decades, however, has raised questions about the validity of this reflexive association alone. In fact, through a series of published works, the concept of SCC has emerged. Beyond HOHF, this entity further incorporates diastolic dysfunction,76,84 cardiac fibrosis,115 relatively prolonged repolarization (PR),116 and autonomic nervous system abnormalities, including reduced heart rate variability (HRV).117 These traits can contribute to the development of heart failure with preserved ejection fraction (HFpEF) in patients with SCD. Moreover, these factors with PH independently contribute to a reduced life expectancy in the 5th decade,76,84,91,92,118,119 a profound health disparity in African Americans.
SCC outcomes: With improvements in screening and management of acute complications, cardiopulmonary etiologies have emerged as the top risk factors associated with mortality and poor outcomes in SCD patients.76 Using a large publicly available database, prior work has reported on cardiovascular causes as the largest source of deaths, accounting for over 32% of total death certificates evaluated, followed by 28% associated with respiratory causes and 16% for renal etiologies.120 And yet, determining causes of death remains challenging in patients with SCD as the reported causes on death certificates do not always reflect the burden of disease. The paucity of autopsy studies provides additional, albeit limited, insight into this inquiry, but they remain poorly powered due to sample sizes. Nonetheless, autopsy studies have suggested that sudden death is a frequent outcome in patients with SCD, with one report attributing over 40% of the cases analyzed.91 While animal data121,122 and small Holter monitoring studies123 suggest a possible propensity for cardiac arrhythmias, the link from sudden death to sudden cardiac death remains uncharacterized in SCD.
Prevalence of SCC and associated risk factors: The “true” prevalence of heart failure (HF) and the possibility of ventricular arrhythmias remain unknown in SCD. Part of this absence can be explained by the under-recognition of SCC. In addition, most studies are limited by small sample sizes. Furthermore, the combination of health disparities and gaps in care, as well as challenges in accessing advanced cardiac imaging (in particular, cardiac or CMRI), all result in a paucity of reliable epidemiologic data.
Despite these limitations, significant advances in the understanding of the scope of cardiac pathologies have been made. While prospective large sickle cohort studies are sparse, Parent and colleagues found at least ~2.5%–3% overall prevalence of elevated filling pressures during right heart catheterization (RHC). Importantly, RHC was only performed in those subjects with an elevated TRV, which would imply that this prevalence may be underestimated.124 These data would suggest a potentially doubling of risk for the development of HFpEF compared with its overall prevalence in the general population, which is ~1.5%.125
Underlying diastolic dysfunction is a critical component of HFpEF. Most published sickle studies reference criteria guided by the American Society of Echocardiography. Thresholds and definitions of diastolic dysfunction, however, remain unclear in the setting of chronic anemia, dilated myocardium, and high output. Nonetheless, Niss and colleagues126 reported that lateral and septal ratios of mitral velocity to early diastolic velocity of the mitral annulus (E/e’) were severely abnormal in 8% and 14% of SCD patients, respectively, suggesting impaired diastolic function. In the same study, the prevalence of pulmonary venous hypertension, a complication of HFpEF, was seen in ~61% of patients with an elevated TRV, much higher than the estimates from the Parent and colleagues84 study. In contrast to HFpEF, HF with reduced EF (HFrEF) seems to be uncommon in patients with SCD with preservation of left and right ventricular systolic function.115 In parallel with this finding, obstructive atherosclerotic coronary artery disease is infrequent in SCD, likely a reflection of the younger age of the population and relatively low cholesterol levels.127,128
Fibrosis was also reported in patients with SCD. Depending on the technique, CMRI-based observations suggest a significant proportion of patients with cardiac fibrosis, ranging from 25% to nearly 100% with diffuse patterns.115,129 Mirroring these patterns, prolonged repolarization has also been reported in a large subset of patients with SCD. For example, Indik and colleagues116 reported QTc prolongation was present in 39% of males and 27% of females with SCD. While this study was not adjusted for confounders, such as methadone use, future studies will also need to evaluate the potential simultaneous presence of cardiac fibrosis, diastolic dysfunction, elevated filling pressures, and repolarization abnormalities to determine the actual prevalence of SCC.
Understanding the basis of SCC: HOHF contributes to many of the findings associated with SCC, including cardiac chamber dilation, hypertrophy, and, in late stages, impaired diastolic function and filling pressures. Beyond HOHF, the etiologies of diastolic dysfunction, cardiac fibrosis, and prolonged PR are likely multifactorial in origin. While hemolysis, vaso-occlusion (VOC), ischemia-reperfusion (I/R), and systemic inflammation are hallmarks of SCD, their links to SCC remain unclear. Similarly, while transfusion burden and iron overload are common in SCD, several studies using CMRI have confirmed the absence of significant myocardial iron deposition in the majority of patients.115
Based on a series of articles, a diffuse pattern of cardiac fibrosis115 seems to be associated with both the degree of hemolysis and diastolic dysfunction.129 Similarly, prolonged repolarization also seems to be associated with hemolysis burden in patients.116 However, mechanisms that contribute to prolonged QTc and cardiac fibrosis, changes to the action potential, and arrhythmogenic propensity are unknown. In animal models of and patients with SCD, inflammatory cytokines seem to partly address this gap and stratify those with and without diastolic dysfunction, prolonged QTc, and cardiac fibrosis. In fact, in animal models, evidence suggests interleukin-18 may be causally associated with these traits.121
Microvascular occlusion contributes to organ damage in SCD. Measurement of microvascular blood flow in patients using CMRI or contrast-enhanced echocardiography has detected abnormal microvascular blood flow in the hearts of patients with SCD.115
Additional studies have suggested genetic contributions to the development of SCC traits, including diastolic dysfunction and QTc intervals.121,130 More investigative studies are required to link these associations with causality in SCD.
SCC and propensity for ventricular tachycardia: Notably, animal models of SCC have identified a significant propensity for the development of ventricular tachycardia (VT).121,122 Moreover, the origin of the VT was predominantly from the right ventricle (RV) despite no discernible differences in fibrosis patterns between the RV and the left ventricle (LV). While differences in the electrophysiological and molecular remodeling between the RV and LV that may help explain this susceptibility have not been characterized, Gupta and colleagues speculated whether PH and increased RV afterload may be contributing factors. Previous reports show RV as the source of pacing-induced VT and spontaneous ventricular fibrillation in animal models of PH and RV failure with attenuation of VT after treating PH. PH is a well-established risk factor for mortality in patients with SCD,131 while SCD mice develop spontaneous PH with aging.132 These findings suggest that PH may play a role in SCC and VT susceptibility in SCD.
Management of SCC: In contrast to PH, there are no current guidelines for screening, diagnosis, or management of SCC based on the paucity of epidemiologic and treatment data. While the defining traits, including diastolic dysfunction, cardiac fibrosis, relatively prolonged repolarization (PR), and autonomic nervous system abnormalities, have all been reported by several studies to represent potential bio-markers for poor outcomes, whether they reflect modifiable risk factors remains unknown. Nonetheless, decompensated heart failure protocols have been well defined for the broader population, including those patients with SCD.
Noninvasive imaging studies including echocardiography with doppler, CMRI with gadolinium, and T1* mapping have started to be recognized and proliferate in the evaluation and management protocols of SCC. Consideration for additional cardiac workup, including the potential roles for right and left heart catheterization, is critical when working with a cardiologist.
Volume management via the use of diuretics has to be carefully tailored as patients typically need to remain in a relatively tight euvolemic range; intravascular volume depletion can represent a risk factor for VOC, while volume overload can result in decompensated HF. Treatment of any underlying risk factor, including hypertension, is also prioritized. While no approved therapies exist for HFpEF, angiotensin converting enzyme inhibitors or mineralocorticoid receptor antagonists is generally considered for relative hypertension and SCC.
KEY POINTS.
Cardiopulmonary complications are a major cause of morbidity and early mortality in sickle cell disease.
Acute chest syndrome is multifactorial, and treatment strategies need to address these multiple potential etiologies.
Respiratory symptoms and lung function abnormalities are common among children and adults with sickle cell disease.
Pulmonary hypertension is multifactorial in etiology and a major cause of morbidity and early mortality in sickle cell disease.
Sickle cell cardiomyopathy refers to a range of abnormalities, including diastolic dysfunction, cardiac fibrosis, prolonged repolarization, and autonomic system abnormalities that contribute to the development of heart damage and dysfunction.
CLINICS CARE POINTS.
Individuals with sickle cell disease (SCD) should be asked about presence of daytime and nighttime respiratory symptoms; clinicians should have a low threshold to refer symptomatic patients for evaluation by a pulmonologist – ideally with expertise in the cardiopulmonary manifestations of sickle cell disease.
Acute chest syndrome (ACS) has multiple causes. Clinicians should be aware of these multiple etiologies and address them accordingly.
Transfusion therapy should be considered for the treatment of patients with moderate to severe ACS.
Current NIH and American Society of Hematology guidelines recommend against routine screening pulmonary function tests (PFTs) for asymptomatic individuals with SCD; however, clinicians should have a low threshold to obtain PFT’s as part of a diagnostic evaluation of respiratory signs and symptoms including cough, dyspnea, chest pain, reduced exercise capacity, oxygen desaturation, etc.
Sleep disordered breathing is common among individuals with SCD; polysomnography should be considered for patients with snoring, unexplained oxygen desaturation at rest and/or with physical activity, nonrestorative sleep, excessive daytime sleepiness, cognitive and attentional difficulties, nocturnal enuresis that persists past middle childhood, recurrent nocturnal sickle cell pain, stroke, and pulmonary hypertension.
Pulmonary hypertension (PH) is a syndrome of multiple etiologies and the etiology of PH is patients with SCD is multifactorial. As such, a structured and thorough diagnostic approach to these patients is critical.
Mainstays of PH therapy include maximization of SCD-specific therapy, treatment of hypoxia with supplemental oxygen therapy and treatment of associated cardiopulmonary conditions. In patients with pulmonary arterial hypertension (PAH), PAH-specific therapy may be considered.
High-output heart failure combined with diastolic dysfunction, cardiac fibrosis, prolonged repolarization and autonomic nervous system abnormalities all can contribute to sickle cell cardiomyopathy (SCC).
Transthoracic echocardiography, cardiac MRI, and right heart catheterization are useful tests for the diagnosis of SCC.
Iron overload in the heart is rare in SCC.Volume management via the use of diuretics has to be carefully tailored as patients typically need to remain in a relatively tight euvolemic range.
Footnotes
DISCLOSURE
A.A. Desai: R01HL136603. R.F. Machado: NIH (R01HL127342); (R01HL158108); and (R01HL111656). R.T. Cohen: has no commercial or financial conflicts of interests.
REFERENCES
- 1.Liem RI, Lanzkron S, Coates TC, et al. American society of hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease. Blood Adv 2019;3(23):3867–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312(10):1033–48. [DOI] [PubMed] [Google Scholar]
- 3.Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med 2000;342(25):1855–65. [DOI] [PubMed] [Google Scholar]
- 4.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330(23):1639–44. [DOI] [PubMed] [Google Scholar]
- 5.Powars D, Weidman JA, Odom-Maryon T, et al. Sickle cell chronic lung disease: prior morbidity and the risk of pulmonary failure. Medicine (Baltimore) 1988; 67(1):66–76. [PubMed] [Google Scholar]
- 6.Cohen RT, DeBaun MR, Blinder MA, et al. Smoking is associated with an increased risk of acute chest syndrome and pain among adults with sickle cell disease. Blood 2010;115(18):3852–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994;84(2):643–9. [PubMed] [Google Scholar]
- 8.Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood 1997;89(5):1787–92. [PubMed] [Google Scholar]
- 9.Boyd JH, Macklin EA, Strunk RC, et al. Asthma is associated with acute chest syndrome and pain in children with sickle cell anemia. Blood 2006;108(9): 2923–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight-Madden JM, Forrester TS, Lewis NA, et al. Asthma in children with sickle cell disease and its association with acute chest syndrome. Thorax 2005;60(3): 206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nordness ME, Lynn J, Zacharisen MC, et al. Asthma is a risk factor for acute chest syndrome and cerebral vascular accidents in children with sickle cell disease. Clin Mol Allergy 2005;3(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vichinsky E, Williams R, Das M, et al. Pulmonary fat embolism: a distinct cause of severe acute chest syndrome in sickle cell anemia. Blood 1994;83(11): 3107–12. [PubMed] [Google Scholar]
- 13.Castro O. Systemic fat embolism and pulmonary hypertension in sickle cell disease. Hematol Oncol Clin North Am Dec 1996;10(6):1289–303. [DOI] [PubMed] [Google Scholar]
- 14.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332(20):1317–22. [DOI] [PubMed] [Google Scholar]
- 15.Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med 2008;358(13):1362–9. [DOI] [PubMed] [Google Scholar]
- 16.Miller ST, Wright E, Abboud M, et al. Impact of chronic transfusion on incidence of pain and acute chest syndrome during the Stroke Prevention Trial (STOP) in sickle-cell anemia. J Pediatr Dec 2001;139(6):785–9. [DOI] [PubMed] [Google Scholar]
- 17.Vichinsky EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med 1995;333(4):206–13. [DOI] [PubMed] [Google Scholar]
- 18.Bellet PS, Kalinyak KA, Shukla R, et al. Incentive spirometry to prevent acute pulmonary complications in sickle cell diseases. N Engl J Med 1995;333(11): 699–703. [DOI] [PubMed] [Google Scholar]
- 19.Styles LA, Vichinsky E. Effects of a long-term transfusion regimen on sickle cell-related illnesses. J Pediatr 1994;125(6 Pt 1):909–11. [DOI] [PubMed] [Google Scholar]
- 20.Rosen CL, Debaun MR, Strunk RC, et al. Obstructive sleep apnea and sickle cell anemia. Pediatrics 2014;134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen RT, Klings ES, Strunk RC. Sickle cell disease: wheeze or asthma? Asthma Res Pract 2015;1:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nandedkar SD, Feroah TR, Hutchins W, et al. Histopathology of experimentally induced asthma in a murine model of sickle cell disease. Blood 2008;112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pritchard KA, Feroah TR, Nandedkar SD, et al. Effects of experimental asthma on inflammation and lung mechanics in sickle cell mice. Am J Respir Cell Mol Biol 2012;46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andemariam B, Adami AJ, Singh A, et al. The sickle cell mouse lung: proinflammatory and primed for allergic inflammation. Translational Res 2015;166(3): 254–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wedderburn CJ, Rees D, Height S, et al. Airways obstruction and pulmonary capillary blood volume in children with sickle cell disease. Pediatr Pulmonol 2014;49(7):716–22. [DOI] [PubMed] [Google Scholar]
- 26.Lunt A, McGhee E, Robinson P, et al. Lung function, transfusion, pulmonary capillary blood volume and sickle cell disease. Respir Physiolo Neurobiol 2016;222:6–10. [DOI] [PubMed] [Google Scholar]
- 27.Glassberg JA, Chow A, Wisnivesky J, et al. Wheezing and asthma are independent risk factors for increased sickle cell disease morbidity. Br J Haematol 2012; 159(4):472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen RT, Madadi A, Blinder MA, et al. Recurrent, severe wheezing is associated with morbidity and mortality in adults with sickle cell disease. Am J Hematol 2011;86(9):756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Field JJ, Stocks J, Kirkham FJ, et al. Airway hyperresponsiveness in children with sickle cell anemia. Chest 2011;139(3):563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poulter EY, Truszkowski P, Thompson AA, et al. Acute chest syndrome is associated with history of asthma in hemoglobin SC disease. Pediatr Blood Cancer 2011;57(2):289–93. [DOI] [PubMed] [Google Scholar]
- 31.Strunk RC, Cohen RT, Cooper BP, et al. Wheezing symptoms and parental asthma are associated with a physician diagnosis of asthma in children with sickle cell anemia. J Pediatr 2014;164(4):821–826 e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sadreameli SC, Alade RO, Mogayzel PJ Jr, et al. Asthma Screening in Pediatric Sickle Cell Disease: A Clinic-Based Program Using Questionnaires and Spirometry. Pediatr Allergy Immunol Pulmonology 2017;30(4):232–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saxena S, Afolabi-Brown O, Ballester L, et al. Benefit of pulmonary subspecialty care for children with sickle cell disease and asthma. Pediatr Pulmonol 2022; 57(4):885–93. [DOI] [PubMed] [Google Scholar]
- 34.Expert Panel Working Group of the National Heart, Lung, and Blood Institute (NHLBI) administered and coordinated National Asthma Education and Prevention Program Coordinating Committee (NAEPPCC), Cloutier MM, Baptist AP, Blake KV, et al. 2020 focused updates to the asthma management guidelines: a report from the national asthma education and prevention program coordinating committee expert panel working group. J Allergy Clin Immunol 2020; 146(6):1217–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddel HK, Bacharier LB, Bateman ED, et al. Global initiative for asthma strategy 2021: executive summary and rationale for key changes. Am J Respir Crit Care Med 2022;205(1):17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McClain BL, Ivy ZK, Bryant V, et al. Improved guideline adherence with integrated sickle cell disease and asthma care. Am J Prev Med 2016;51(1 Suppl 1):S62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Black LV, Ezmigna D, Wallace-Farquharson T, et al. Feasibility and preliminary outcomes of an integrated pediatric sickle cell disease and pulmonary care clinic for children with sickle cell disease. Pediatr Blood Cancer 2020;67(11): e28672. [DOI] [PubMed] [Google Scholar]
- 38.Delclaux C, Zerah-Lancner F, Bachir D, et al. Factors associated with dyspnea in adult patients with sickle cell disease. Chest 2005;128(5):3336–44. [DOI] [PubMed] [Google Scholar]
- 39.Ivankovich DT, Braga JAP, Lanza FC, et al. Lung Function in Infants with Sickle Cell Anemia. J Pediatr 2019;207:252–4. [DOI] [PubMed] [Google Scholar]
- 40.Koumbourlis AC, Hurlet-Jensen A, Bye MR. Lung function in infants with sickle cell disease. Pediatr Pulmonol 1997;24(4):277–81. [DOI] [PubMed] [Google Scholar]
- 41.Cohen R, Strunk RC, Rodeghier M, et al. Pattern of lung function is not associated with prior or future morbidity in children with sickle cell anemia. Ann Am Thorac Soc 2016;13(8):1314–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arteta M, Campbell A, Nouraie M, et al. Abnormal pulmonary function and associated risk factors in children and adolescents with sickle cell anemia. J Pediatr Hematol Oncol 2014;36(3):185–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sylvester KP, Desai SR, Wells AU, et al. Computed tomography and pulmonary function abnormalities in sickle cell disease. Eur Respir J 2006;28(4):832–8. [DOI] [PubMed] [Google Scholar]
- 44.Arigliani M, Kirkham FJ, Sahota S, et al. Lung Clearance Index May Detect Early Peripheral Lung Disease in Sickle Cell Anemia. Ann Am Thorac Soc 2022. [DOI] [PubMed] [Google Scholar]
- 45.Machogu EM, Khurana M, Kaericher J, et al. Lung clearance index in children with sickle cell disease. Pediatr Pulmonol 2021;56(5):1165–72. [DOI] [PubMed] [Google Scholar]
- 46.MacLean JE, Atenafu E, Kirby-Allen M, et al. Longitudinal decline in lung volume in a population of children with sickle cell disease. Am J Respir Crit Care Med 2008;178(10):1055–9. [DOI] [PubMed] [Google Scholar]
- 47.Lunt A, McGhee E, Sylvester K, et al. Longitudinal assessment of lung function in children with sickle cell disease. Pediatr Pulmonol 2016;51(7):717–23. [DOI] [PubMed] [Google Scholar]
- 48.Willen SM, Cohen R, Rodeghier M, et al. Age is the only predictor of small decrease in lung function in children with sickle cell anemia. Am J Hematol 2018;93(3):408–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kotwal N, Pillai DK, Darbari DS, et al. Spirometric changes after initiation of hydroxyurea in children with sickle cell anemia. J Pediatr Hematol Oncol 2022; 44(6):e923–5. [DOI] [PubMed] [Google Scholar]
- 50.McLaren A, Klingel M, Behera S, et al. Effect of Hydroxyurea Therapy on Pulmonary Function in Children with Sickle Cell Anemia. Am J Respir Crit Care Med 2017;195(5):689–91. [DOI] [PubMed] [Google Scholar]
- 51.Hodges B, Ivy ZK, Cronin RM, et al. Annual decline in lung function in adults with sickle cell disease is similar to that observed in adults with cystic fibrosis. Blood Adv 2022;6(6):1937–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Field JJ, Glassberg J, Gilmore A, et al. Longitudinal analysis of pulmonary function in adults with sickle cell disease. Am J Hematol 2008;83(7):574–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klings ES, Wyszynski DF, Nolan VG, et al. Abnormal pulmonary function in adults with sickle cell anemia. Am J Respir Crit Care Med 2006;173(11):1264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chaturvedi S, Labib Ghafuri D, Kassim A, et al. Elevated tricuspid regurgitant jet velocity, reduced forced expiratory volume in 1 second, and mortality in adults with sickle cell disease. Am J Hematol 2017;92(2):125–30. [DOI] [PubMed] [Google Scholar]
- 55.Needleman JP, Franco ME, Varlotta L, et al. Mechanisms of nocturnal oxyhemoglobin desaturation in children and adolescents with sickle cell disease. Pediatr Pulmonol 1999;28(6):418–22. [DOI] [PubMed] [Google Scholar]
- 56.Caboot JB, Allen JL. Hypoxemia in sickle cell disease: significance and management. Paediatr Respir Rev 2014;15(1):17–23. [DOI] [PubMed] [Google Scholar]
- 57.Lehmann GC, Bell TR, Kirkham FJ, et al. Enuresis associated with sleep disordered breathing in children with sickle cell anemia. J Urol 2012;188(4 Suppl): 1572–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma S, Efird JT, Knupp C, et al. Sleep disorders in adult sickle cell patients. J Clin Sleep Med 2015;11(3):219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whitesell PL, Owoyemi O, Oneal P, et al. Sleep-disordered breathing and nocturnal hypoxemia in young adults with sickle cell disease. Sleep Med 2016;22:47–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Al-Saqqaf R, Merdad R, Wali SO, et al. The prevalence of obstructive sleep apnea in adult patients with sickle cell disease. J Appl Hematol 2017;8(1):16–22. [Google Scholar]
- 61.Ayache M, Rosen CL, Chiang A, et al. Overnight oxygen desaturation index in sickle cell disease: prediction of sleep apnea. Sleep Med 2019;64:12–4. [DOI] [PubMed] [Google Scholar]
- 62.Ruhl AP, Sadreameli SC, Allen JL, et al. Identifying clinical and research priorities in sickle cell lung disease. An official american thoracic society workshop report. Ann Am Thorac Soc 2019;16(9):e17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Katz T, Schatz J, Roberts CW. Comorbid obstructive sleep apnea and increased risk for sickle cell disease morbidity. Sleep Breath 2018;22(3):797–804. [DOI] [PubMed] [Google Scholar]
- 64.Kirkham FJ, Hewes DK, Prengler M, et al. Nocturnal hypoxaemia and central-nervous-system events in sickle-cell disease. Lancet 2001;357(9269):1656–9. [DOI] [PubMed] [Google Scholar]
- 65.Johnson MC, Kirkham FJ, Redline S, et al. Left ventricular hypertrophy and diastolic dysfunction in children with sickle cell disease are related to asleep and waking oxygen desaturation. Blood 2010;116(1):16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mondal P, Stefek B, Sinharoy A, et al. The association of nocturnal hypoxia and an echocardiographic measure of pulmonary hypertension in children with sickle cell disease. Pediatr Res Mar 2019;85(4):506–10. [DOI] [PubMed] [Google Scholar]
- 67.Willen SM, Rodeghier M, Rosen CL, et al. Sleep disordered breathing does not predict acute severe pain episodes in children with sickle cell anemia. Am J Hematol 2018;93(4):478–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goldstein NA, Keller R, Rey K, et al. Sleep-disordered breathing and transcranial Dopplers in sickle cell disease. Arch Otolaryngol Head Neck Surg 2011; 137(12):1263–8. [DOI] [PubMed] [Google Scholar]
- 69.Worsham CM, Martin ST, Nouraie SM, et al. Clinical and laboratory findings associated with sleep disordered breathing in sickle cell disease. Am J Hematol 2017;92(12):E649–51. [DOI] [PubMed] [Google Scholar]
- 70.Abulhamail A, Selati S, Alasqah R. The association between obstructive sleep apnea and stroke in sickle-cell disease children. Eur Arch Otorhinolaryngol 2022;279(2):843–51. [DOI] [PubMed] [Google Scholar]
- 71.Grady AJ, Hankins JS, Haberman B, et al. Hydroxyurea treatment effect on children with sickle cell disease and obstructive sleep apnea. Sleep Breath 2017; 21(3):697–701. [DOI] [PubMed] [Google Scholar]
- 72.Narang I, Kadmon G, Lai D, et al. Higher nocturnal and awake oxygen saturations in children with sickle cell disease receiving hydroxyurea therapy. Ann Am Thorac Soc 2015;12(7):1044–9. [DOI] [PubMed] [Google Scholar]
- 73.Singh SA, Koumbourlis AC, Aygun B. Resolution of chronic hypoxemia in pediatric sickle cell patients after treatment with hydroxyurea. Pediatr Blood Cancer 2008;50(6):1258–60. [DOI] [PubMed] [Google Scholar]
- 74.van Geyzel L, Arigliani M, Inusa B, et al. Higher oxygen saturation with hydroxyurea in paediatric sickle cell disease. Arch Dis Child 2020;105(6):575–9. [DOI] [PubMed] [Google Scholar]
- 75.Tsou PY, Cielo CM, Xanthopoulos MS, et al. The burden of obstructive sleep apnea in pediatric sickle cell disease: a Kids’ inpatient database study. Sleep 2021;44(2):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gladwin MT, Sachdev V. Cardiovascular abnormalities in sickle cell disease. J Am Coll Cardiol 2012;59(13):1123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Detterich JA, Kato RM, Rabai M, et al. Chronic transfusion therapy improves but does not normalize systemic and pulmonary vasculopathy in sickle cell disease. Blood 2015;126(6):703–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gladwin MT. Cardiovascular complications and risk of death in sickle-cell disease. Lancet 2016;387(10037):2565–74. [DOI] [PubMed] [Google Scholar]
- 79.Sundd P, Gladwin MT, Novelli EM. Pathophysiology of Sickle Cell Disease. Annu Rev Pathol 2019;14:263–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Saraf SL, Zhang X, Kanias T, et al. Haemoglobinuria is associated with chronic kidney disease and its progression in patients with sickle cell anaemia. Br J Haematol 2014;164(5):729–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Prohaska CC, Machado RF. The different facets of sickle cell disease-related pulmonary hypertension. Curr Opin Pulm Med 2021;27(5):319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mehari A, Gladwin MT, Tian X, et al. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA 2012;307(12):1254–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Parent F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med 2011;365(1):44–53. [DOI] [PubMed] [Google Scholar]
- 85.Fonseca GH, Souza R, Salemi VM, et al. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J 2012;39(1):112–8. [DOI] [PubMed] [Google Scholar]
- 86.Machado RF, Barst RJ, Yovetich NA, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 2011;118(4):855–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mehari A, Alam S, Tian X, et al. Hemodynamic predictors of mortality in adults with sickle cell disease. Am J Respir Crit Care Med 2013;187(8):840–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hagger D, Wolff S, Owen J, et al. Changes in coagulation and fibrinolysis in patients with sickle cell disease compared with healthy black controls. Blood Coagul Fibrinolysis 1995;6(2):93–9. [DOI] [PubMed] [Google Scholar]
- 89.Villagra J, Shiva S, Hunter LA, et al. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 2007;110(6):2166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lim MY, Ataga KI, Key NS. Hemostatic abnormalities in sickle cell disease. Curr Opin Hematol 2013;20(5):472–7. [DOI] [PubMed] [Google Scholar]
- 91.Manci EA, Culberson DE, Yang YM, et al. Causes of death in sickle cell disease: an autopsy study. Br J Haematol 2003;123(2):359–65. [DOI] [PubMed] [Google Scholar]
- 92.Graham JK, Mosunjac M, Hanzlick RL. Sickle cell lung disease and sudden death: a retrospective/prospective study of 21 autopsy cases and literature review. Am J Forensic Med Pathol 2007;28(2):168–72. [DOI] [PubMed] [Google Scholar]
- 93.Lai YC, Potoka KC, Champion HC, et al. Pulmonary arterial hypertension: the clinical syndrome. Circ Res 2014;115(1):115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Anthi A, Machado RF, Jison ML, et al. Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Respir Crit Care Med 2007;175(12):1272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weir NA, Conrey A, Lewis D, et al. Riociguat use in sickle cell related chronic thromboembolic pulmonary hypertension: A case series. Pulm Circ 2018;8(4). 2045894018791802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ghofrani HA, D’Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med Jul 25 2013; 369(4):319–29. [DOI] [PubMed] [Google Scholar]
- 97.Naik RP, Streiff MB, Haywood C Jr, et al. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. J Thromb Haemost 2014;12(12): 2010–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yung GL, Channick RN, Fedullo PF, et al. Successful pulmonary thromboendarterectomy in two patients with sickle cell disease. Am J Respir Crit Care Med 1998;157(5 Pt 1):1690–3. [DOI] [PubMed] [Google Scholar]
- 99.Agrawal H, Petit CJ, Miro J, et al. Contralateral pulmonary hypertension following resuscitation of unilateral ductal origin of a pulmonary artery: a multi-institutional review. Pediatr Cardiol 2018;39(1):71–8. [DOI] [PubMed] [Google Scholar]
- 100.Mahesh B, Besser M, Ravaglioli A, et al. Pulmonary endarterectomy is effective and safe in patients with haemoglobinopathies and abnormal red blood cells: the Papworth experience. Eur J Cardio Thorac Surg 2016;50(3):537–41. [DOI] [PubMed] [Google Scholar]
- 101.Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood 2003;101(4):1257–61. [DOI] [PubMed] [Google Scholar]
- 102.Machado RF, Anthi A, Steinberg MH, et al. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA 2006;296(3):310–8. [DOI] [PubMed] [Google Scholar]
- 103.Ataga KI, Moore CG, Jones S, et al. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. Br J Haematol 2006;134(1):109–15. [DOI] [PubMed] [Google Scholar]
- 104.Klings ES, Machado RF, Barst RJ, et al. An official American Thoracic Society clinical practice guideline: diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am J Respir Crit Care Med 2014; 189(6):727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gladwin MT, Barst RJ, Gibbs JS, et al. Risk factors for death in 632 patients with sickle cell disease in the United States and United Kingdom. PloS one 2014; 9(7):e99489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Machado RF, Hildesheim M, Mendelsohn L, et al. NT-pro brain natriuretic peptide levels and the risk of death in the cooperative study of sickle cell disease. Br J Haematol 2011;154(4):512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Turpin M, Chantalat-Auger C, Parent F, et al. Chronic blood exchange transfusions in the management of pre-capillary pulmonary hypertension complicating sickle cell disease. Eur Respir J 2018;52(4). [DOI] [PubMed] [Google Scholar]
- 108.George MP, Novelli EM, Shigemura N, et al. First successful lung transplantation for sickle cell disease with severe pulmonary arterial hypertension and pulmonary veno-occlusive disease. Pulm Circ 2013;3(4):952–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pittman C, Hsieh MM, Coles W, et al. Reversal of pre-capillary pulmonary hypertension in a patient with sickle cell anemia who underwent haploidentical peripheral blood stem cell transplantation. Bone Marrow Transplant 2017;52(4):641–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Machado RF, Martyr S, Kato GJ, et al. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol 2005;130(3):445–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Minniti CP, Machado RF, Coles WA, et al. Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. Br J Haematol 2009;147(5):737–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Barst RJ, Mubarak KK, Machado RF, et al. Exercise capacity and haemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: results of the ASSET studies. Br J Haematol 2010;149(3):426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weir NA, Saiyed R, Alam S, et al. Prostacyclin-analog therapy in sickle cell pulmonary hypertension. Haematologica 2017;102(5):e163–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Reddy YNV, Borlaug BA. High-output heart failure in sickle cell anemia. JACC Cardiovasc Imaging 2016;9(9):1122–3. [DOI] [PubMed] [Google Scholar]
- 115.Desai AA, Patel AR, Ahmad H, et al. Mechanistic insights and characterization of sickle cell disease-associated cardiomyopathy. Circ Cardiovasc Imaging 2014;7(3):430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Indik JH, Nair V, Rafikov R, et al. Associations of Prolonged QTc in Sickle Cell Disease. PLoS One 2016;11(10):e0164526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sangkatumvong S, Coates TD, Khoo MC. Abnormal autonomic cardiac response to transient hypoxia in sickle cell anemia. Physiol Meas 2008;29(5): 655–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Darbari DS, Kple-Faget P, Kwagyan J, et al. Circumstances of death in adult sickle cell disease patients. Am J Hematol 2006;81(11):858–63. [DOI] [PubMed] [Google Scholar]
- 119.Fitzhugh CD, Lauder N, Jonassaint JC, et al. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am J Hematol 2010;85(1):36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hamideh D, Alvarez O. Sickle cell disease related mortality in the united states (1999–2009). Pediatr Blood Cancer 2013;60(9):1482–6. [DOI] [PubMed] [Google Scholar]
- 121.Gupta A, Fei YD, Kim TY, et al. IL-18 mediates sickle cell cardiomyopathy and ventricular arrhythmias. Blood 2021;137(9):1208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bakeer N, James J, Roy S, et al. Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci U S A 2016;113(35): E5182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maisel A, Friedman H, Flint L, et al. Continuous electrocardiographic monitoring in patients with sickle-cell anemia during pain crisis. Clin Cardiol 1983;6(7): 339–44. [DOI] [PubMed] [Google Scholar]
- 124.Anderson TJ, Charbonneau F, Title LM, et al. Microvascular function predicts cardiovascular events in primary prevention: long-term results from the Fire-fighters and Their Endothelium (FATE) study. Circulation 2011;123(2):163–9. [DOI] [PubMed] [Google Scholar]
- 125.McDonagh TA, Metra M, Adamo M, et al. Corrigendum to: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2021;42(48):4901. [DOI] [PubMed] [Google Scholar]
- 126.Niss O, Quinn CT, Lane A, et al. Cardiomyopathy With Restrictive Physiology in Sickle Cell Disease. JACC Cardiovasc Imaging 2016;9(3):243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zorca S, Freeman L, Hildesheim M, et al. Lipid levels in sickle-cell disease associated with haemolytic severity, vascular dysfunction and pulmonary hypertension. Br J Haematol 2010;149(3):436–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.el-Hazmi MA, Jabbar FA, Warsy AS. Cholesterol and triglyceride level in patients with sickle cell anaemia. Scand J Clin Lab Invest 1987;47(4):351–4. [PubMed] [Google Scholar]
- 129.Niss O, Fleck R, Makue F, et al. Association between diffuse myocardial fibrosis and diastolic dysfunction in sickle cell anemia. Blood 2017;130(2):205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Duarte JD, Desai AA, Sysol JR, et al. Genome-wide analysis identifies IL-18 and FUCA2 as novel genes associated with diastolic function in african americans with sickle cell disease. PLoS One 2016;11(9):e0163013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350(9):886–95. [DOI] [PubMed] [Google Scholar]
- 132.Gladwin MT, Barst RJ, Castro OL, et al. Pulmonary hypertension and NO in sickle cell. Blood 2010;116(5):852–4. [DOI] [PMC free article] [PubMed] [Google Scholar]