

Abstract
Background
Adenoid cystic carcinoma (ACC) is a lethal malignancy of exocrine glands, characterized by the coexistence within tumor tissues of 2 distinct populations of cancer cells, phenotypically similar to the myoepithelial and ductal lineages of normal salivary epithelia. The developmental relationship linking these 2 cell types, and their differential vulnerability to antitumor treatments, remains unknown.
Methods
Using single-cell RNA sequencing, we identified cell-surface markers (CD49f, KIT) that enabled the differential purification of myoepithelial-like (CD49fhigh/KITneg) and ductal-like (CD49flow/KIT+) cells from patient-derived xenografts (PDXs) of human ACCs. Using prospective xenotransplantation experiments, we compared the tumor-initiating capacity of the 2 cell types and tested whether one could differentiate into the other. Finally, we searched for signaling pathways with differential activation between the 2 cell types and tested their role as lineage-specific therapeutic targets.
Results
Myoepithelial-like cells displayed higher tumorigenicity than ductal-like cells and acted as their progenitors. Myoepithelial-like and ductal-like cells displayed differential expression of genes encoding for suppressors and activators of retinoic acid signaling, respectively. Agonists of retinoic acid receptor (RAR) or retinoid X receptor (RXR) signaling (all-trans retinoic acid, bexarotene) promoted myoepithelial-to-ductal differentiation, whereas suppression of RAR/RXR signaling with a dominant-negative RAR construct abrogated it. Inverse agonists of RAR/RXR signaling (BMS493, AGN193109) displayed selective toxicity against ductal-like cells and in vivo antitumor activity against PDX models of human ACC.
Conclusions
In human ACCs, myoepithelial-like cells act as progenitors of ductal-like cells, and myoepithelial-to-ductal differentiation is promoted by RAR/RXR signaling. Suppression of RAR/RXR signaling is lethal to ductal-like cells and represents a new therapeutic approach against human ACCs.
Adenoid cystic carcinomas (ACCs) are malignant adenocarcinomas that originate in exocrine glands, most commonly the salivary glands (SGs) (1). ACCs display indolent growth, but their slow proliferation kinetics often belie an aggressive and relentless nature, characterized by perineural infiltration and early hematogenous spread (1-3). Current treatments for ACCs are limited to surgery and radiotherapy. Because ACCs usually arise within the craniofacial district, such treatments are often destructive and, in approximately 60% of cases, unable to prevent metastatic relapse and patient death (2-5). ACCs are usually refractory to chemotherapy, immunotherapy, and various types of targeted therapies (6-9). ACCs often associate with t(6; 9) MYB-NFIB chromosomal translocations (10-13), but no actionable treatments are currently available to suppress the oncogenic signaling that results from them (14).
Histologically, ACCs are characterized by a distinctive feature: the coexistence of 2 populations of malignant cells, termed ductal-like and myoepithelial-like, because of their phenotypic similarity to ductal and myoepithelial lineages of normal SG epithelia (15-21). The molecular causes of this feature are poorly understood and remain difficult to investigate because of the lack of experimental means to differentially isolate the 2 cell types. It remains unknown, for example, whether the 2 populations represent distinct genetic clones, arising from the divergent accumulation of distinct repertoires of somatic mutations, or distinct developmental lineages, arising from the retention by malignant tissues of normal differentiation programs (22-25). It also remains unclear how the 2 populations compare in terms of differential sensitivity to antitumor therapies.
In this study, we used single-cell RNA-sequencing (scRNA-seq) (26) to identify cell surface markers that enable, for the first time, the differential purification by fluorescence-activated cell sorting (FACS) of the 2 malignant cell types found in human ACCs. We then demonstrated that one can differentiate into the other and identified a signaling pathway that controls this process. Finally, we leveraged this knowledge to develop a new pharmacological approach for the treatment of human ACCs.
Methods
An expanded description of all materials and methods used in this study is provided in the Supplementary Methods (available online), together with relevant technical references.
Patient-derived xenograft (PDX) lines
PDX lines representative of human ACCs (Supplementary Table 1, available online) were obtained from the Adenoid Cystic Carcinoma Registry at the University of Virginia (27) and propagated subcutaneously in female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (Jackson Laboratory; stock #005557) (25).
Animal welfare
Animal experiments were approved by the institutional animal care and use committee (IACUC) of Columbia University (research protocols: AC-AAAL7751, AC-AAAW1466, AC-AABM9553).
.
Fluorescence-activated cell sorting
Solid tumors were dissociated into single-cell suspensions, and malignant cells isolated by FACS, following established protocols (Supplementary Figure 1, available online) (23,25). Monoclonal antibodies used to visualize different subtypes of malignant cells included mouse anti-human EpCAM (clone: 9C4) conjugated to fluorescein isothiocyanate (FITC), rat anti-human-and-mouse CD49f (clone: GoH3) conjugated to allophycocyanin (APC), and mouse anti-human KIT (clone: 104D2) conjugated to phycoerythrin (PE). Mouse cells were excluded using mouse anti-mouse H-2Kd (clone: SF1.1) conjugated to biotin, rat anti-mouse Cd45 (clone: 30-F11) conjugated to PE/Cyanine5, and streptavidin conjugated to PE/Cyanine5 (BD Biosciences). Cell-cycle distribution of sorted cells was evaluated using 4′,6-diamidino-2-phenylindole (DAPI), following permeabilization with BD Cytofix/Cytoperm (BD Biosciences).
RNA-sequencing
scRNA-seq experiments were performed using Chromium Single Cell 3’ Solution (10x Genomics) and NovaSeq-6000 (Illumina) platforms and analyzed using cellranger (v3.1.0) and Randomly (28). In conventional RNA-seq experiments, RNA was isolated using the NucleoSpin RNA XS kit (Takara) and cDNA libraries prepared using the TruSeq Stranded mRNA kit (Illumina). Conventional RNA-seq reactions were run on either HiSeq-4000 or NovaSeq-6000 platforms (Illumina), and results analyzed using DESeq2 (29) and STAR-fusion (30). Differentially expressed genes were identified based on false-discovery rates (FDR), calculated using the Benjamini–Hochberg method. RNA-seq datasets were deposited in the database of Genotypes and Phenotypes (dbGAP; https://www.ncbi.nlm.nih.gov/gap), under accession number: phs002764.
Immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded tissue blocks were stained with the following antibodies: mouse anti-human TP63 (clone: 4A4), rabbit anti-human KIT (clone: YR145), rabbit anti-human MKI67 (clone: 30-9).
In vivo tumorigenicity
Autologous pairs of CD49fhigh/KITneg and CD49flow/KIT+ cells were double sorted by FACS, resuspended in high-concentration matrigel (Corning), and injected subcutaneously, side by side, into opposite flanks (left vs. right) of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice. The frequency of tumor-initiating cells was calculated by Extreme Limiting Dilution Analysis (ELDA; http://bioinf.wehi.edu.au/software/elda) (31).
In vitro tissue cultures
ACC cells were cultured either as 3-dimensional organoids (32-34) or 2-dimensional monolayers (35) and treated with all-trans retinoic acid (ATRA; 0.1-10 µM), bexarotene (10 µM), BMS493 (1-10 µM), or AGN193109 (1-10 µM). Lentivirus vectors (36) were based on the pLL3.7 backbone (Addgene; #11795), re-engineered to drive constitutive expression of a dominant negative version of human retinoic acid receptor alpha (DNhRARα) (Addgene; #15153) (37) in tandem with the enhanced green fluorescent protein (EGFP), which was used as a fluorescent reporter. Cell viability was assessed using the alamarBlue HS Cell Viability Reagent (38).
In vivo therapeutic studies
Tumor-bearing animals were treated by intraperitoneal injection of BMS493 (1 mg x 3-4 days/week x 3 weeks) resuspended in 0.15 M hydroxypropyl-β-cyclodextrin (Cayman Chemicals).
Statistical analyses
A detailed explanation of all statistical tests used in this study is provided in the Supplementary Methods (available online).
Results
Identification of surface markers differentially expressed between myoepithelial-like and ductal-like cells
To identify surface markers differentially expressed between myoepithelial-like and ductal-like cells, we analyzed by scRNA-seq a bulk preparation of epithelial cancer cells (EpCAM+) purified by FACS from a PDX line representative of a human ACC with classic cribriform histology (Figure 1, A and B; Supplementary Figure 1, available online) (27). We used the Randomly (28) algorithm to remove stochastic contributions to the transcriptional variability observed between cells and then clustered cells based on systematic differences in transcriptional patterns, identifying an optimal clustering solution consisting of 3 subgroups (Figure 1, C; Supplementary Figure 2, available online). Of these 3 subgroups, the largest 2 displayed mutually exclusive expression of known myoepithelial (ACTA2, CNN1, TP63) and ductal (KRT7, KRT18, ELF5) cell markers (Supplementary Figure 3, available online), and a third appeared to represent a highly proliferating (MKI67high) subset of ductal-like cells (Figure 1, G and J; Supplementary Figure 3, C, available online). Among the differentially expressed genes, we identified those encoding for cell-surface markers CD49f (ITGA6) and KIT, also known as CD117 (KIT), which associated with myoepithelial-like and ductal-like cell types, respectively (Figure 1, D-I). We then tested whether CD49f and KIT could be leveraged to visualize myoepithelial-like and ductal-like cells by FACS. Indeed, staining with fluorophore-conjugated antibodies directed against the 2 markers enabled clear discrimination of 2 cell populations, cells expressing high levels of CD49f and no measurable levels of KIT (CD49fhigh/KITneg) and cells expressing low levels of CD49f and measurable levels of KIT (CD49flow/KIT+), across 5 independent PDX lines representative of biphenotypic ACCs (Figure 2, A). Analysis of the same tumors by IHC also confirmed that KIT expression was restricted to ductal-like cells, and mutually exclusive to expression of TP63, a myoepithelial marker (Figure 2, B), in agreement with previous IHC studies (39).
Figure 1.

Identification of surface markers for the differential purification of myoepithelial-like and ductal-like cell populations in human ACCs. A) Histological analysis of the ACCX22 human PDX line, confirming retention of a cribriform histology with pseudocyst formation, characteristic of well-differentiated (grade 1) ACCs. B) Magnification of the tissue area outlined in (A) (dashed box), demonstrating the presence of 1) ductal-like cells, characterized by abundant eosinophilic cytoplasm and arranged in ringlike structures (arrows); and 2) myoepithelial-like cells (arrowheads), characterized by spindle-shaped morphology and arranged to line pseudocysts. C) Visualization by Uniform Manifold Approximation and Projection (UMAP) of scRNA-seq data obtained from a purified preparation of human malignant cells (EpCAM+) sorted by FACS from the ACCX22 human PDX line. In the UMAP scatter plot, the 3 cell clusters identified as representing the most robust clustering solution (ie, as displaying the highest mean silhouette score following clustering based on the Leiden algorithm) were labeled with different colors and displayed clear visual separation. Based on differentially expressed genes (Supplementary Figure 3, available online), the 3 clusters were annotated as follows: cluster 1 = myoepithelial-like cells; cluster 2 = ductal-like cells; cluster 3 = proliferating ductal-like cells. D) List of genes identified as displaying a statistically significant difference in mean expression levels between cluster 1 (myoepithelial-like) and cluster 2 (ductal-like), based on a Student’s t test (2-tailed) adjusted for multiple comparisons (FDR < 0.001; Benjamini–Hochberg method). Among the differentially expressed genes are those encoding for 2 surface markers: ITGA6 (CD49f) and KIT (CD117). E-G) UMAP plots displaying gene-expression levels for ITGA6 (E), KIT (F), and the proliferation marker MKI67 (G); q values are based on a Student's t test (2-tailed), corrected for multiple comparisons (Benjamini–Hochberg method), as described in Supplementary Figure 3 (available online). H-J) Violin plots displaying the distribution of gene-expression levels for ITGA6 (H), KIT (I), and MKI67 (J) across the 3 cell clusters identified by scRNA-seq; P values are based on a Kruskal–Wallis H test. ACC = adenoid cystic carcinoma; FACS = fluorescence-activated cell sorting; FDR = false discovery rate; PDX = patient-derived xenograft; scRNA-seq = single-cell RNA sequencing; TPM = transcripts per million.
Figure 2.

Differential purification by flow cytometry of myoepithelial-like (CD49fhigh/KITneg) and ductal-like (CD49flow/KIT+) cells. A) Analysis by flow cytometry of CD49f and KIT surface expression in 5 human PDX lines representative of biphenotypic ACCs, enabling visual discrimination of 2 distinct populations of human malignant cells: CD49fhigh/KITneg (top-left gates) and CD49flow/KIT+ (bottom-right gates). B) Analysis by immunohistochemistry (IHC) of corresponding tumors, confirming mutually exclusive expression of TP63 (a myoepithelial marker) and KIT (a ductal marker). Scale bar: 50 µm. C) Principal component analysis (PCA) of RNA-seq data obtained from 5 autologous pairs of CD49fhigh/KITneg (CD49fhigh) and CD49flow/KIT+ (KIT+) cells, purified in parallel from 5 biphenotypic PDX lines of human ACCs (ACCX5M1, ACCX6, ACCX14, ACCX22, SGTX6). PCA was performed using the top 500 genes displaying the highest level of variance across the full 10-sample dataset. The 10 samples segregated into 2 distinct clusters, corresponding to their surface marker phenotype (CD49fhigh/KITneg vs CD49flow/KIT+) and separating along the first principal component (PC1). (D) Heatmap of the top 100 genes identified as differentially expressed (DE) between CD49fhigh/KITneg and CD49flow/KIT+ cells, after mean centering of gene-expression levels and hierarchical clustering of both genes and samples. Differentially expressed genes were defined as those with a more than twofold difference in mean expression levels between the 2 populations (log2 fold-change >1) that was considered to be statistically robust based on a Wald test corrected for multiple comparisons (FDR < 0.05; Benjamini–Hochberg method). Differentially expressed genes were ranked based on the P value from the Wald test. Asterisk labels indicate genes encoding for previously validated myoepithelial (*) and ductal (**) markers. ACC = adenoid cystic carcinoma; APC = allophycocyanin; DE = differentially expressed; FDR = false discovery rate; PC1 = first principal component; PC2 = second principal component; PDX = patient-derived xenograft; PE = phycoerythrin; RNA-seq = RNA sequencing.
Transcriptional profiling of CD49fhigh/KITneg and CD49flow/KIT+ cells
To understand whether CD49fhigh/KITneg and CD49flow/KIT+ cells isolated from different patients displayed similar gene-expression patterns, we sorted autologous pairs of the 2 cell types from 5 biphenotypic PDX lines and analyzed them by conventional RNA-seq. When plotted based on a principal component analysis (PCA) of their transcriptional profiles, the 10 samples segregated into 2 equal clusters (5 samples per cluster) that matched the original phenotypes of sorted cells (CD49fhigh/KITneg vs. CD49flow/KIT+). The 2 clusters separated along the first principal component (PC1), which accounted for a dominant fraction (58%) of the variability within the dataset (Figure 2, C). This observation revealed that the 2 cell types were defined by systematic differences in transcriptional profiles, strongly conserved across different tumors irrespective of patient-specific variables (eg, site of origin, sex, repertoire of genetic alterations) (Supplementary Table 1, available online) (27). We then used DESeq2 (29) to identify genes differentially expressed between the 2 cell types (Supplementary Table 2, available online) and observed that CD49fhigh/KITneg cells expressed markers of myoepithelial cells (eg, ACTA2, MYH11, PDPN, TP63) (15-20), and CD49flow/KIT+ cells expressed markers of the ductal and/or luminal lineages of exocrine glands (eg, ELF5, KIT, SLPI, ANXA8) (40-43) (Figure 2, D), thus confirming their myoepithelial-like and ductal-like identities. Finally, we used STAR-Fusion (30) to test whether CD49fhigh/KITneg and CD49flow/KIT+ cells, which are both known to carry t(6; 9) MYB-NFIB translocations (44), differed in expression of MYB-NFIB chimeric transcripts. Our analysis revealed that, in ACCs that harbored such translocations, both cell types expressed MYB-NFIB chimeric transcripts, without evidence of meaningful differences in terms of absolute levels or alternative splicing (Supplementary Figure 4, available online).
Developmental relationship of CD49fhigh/KITneg and CD49flow/KIT+ cells
We next wanted to test whether the 2 cell populations represented different genetic clones that coexisted within the same tissue (Figure 3, A), or whether they were linked by a developmental relationship, whereby one population could differentiate into the other, in a process akin to those sustaining the normal morphogenesis of epithelial tissues (Figure 3, B). To explore this concept, we decided to perform prospective xenotransplantation studies with purified preparations of the 2 cell populations, to evaluate their tumor-initiating and multilineage differentiation capacity. Autologous pairs of CD49fhigh/KITneg and CD49flow/KIT+ cells were double sorted by FACS from 2 biphenotypic PDX lines (ACCX5M1, SGTX6) and injected, side by side, at progressively decreasing doses (10 000-250 cells per injection) in immune-deficient animals (Figure 3, C) (31). We observed that the frequency of tumor-initiating cells was higher in CD49fhigh/KITneg as compared with CD49flow/KIT+ cells (Figure 3, D and E; Supplementary Figure 5, A and B, available online), resulting in larger and faster-growing tumors (Supplementary Figure 5, C-F, available online) despite CD49fhigh/KITneg cells having a smaller fraction of actively proliferating cells (Supplementary Figure 5, G and H, available online). These results revealed that myoepithelial-like cells represent a biologically aggressive component of human ACCs, despite having a more quiescent phenotype. We then proceeded to analyze the cell composition of tumors originated from transplantation of sorted cells. Our results showed that tumors originated from sorted CD49fhigh/KITneg cells contained both cell types, at frequencies comparable with those observed in parent lines, irrespectively of the number of injected cells (Figure 3, F-H, L-N). This observation showed that CD49fhigh/KITneg cells can differentiate into CD49flow/KIT+ cells, thus excluding the clonal hypothesis. When we analyzed the few tumors originated from CD49flow/KIT+ cells, we also found them indistinguishable from parent lines (Figure 3, I-K, O-Q). In this specific case, however, given the high number of CD49flow/KIT+ cells required for tumor initiation, we could not exclude the possibility that such tumors arose from cross-contaminations of CD49fhigh/KITneg cells, despite the high purity achieved by double sorting.
Figure 3.

Tumorigenic properties of myoepithelial-like (CD49fhigh/KITneg) and ductal-like (CD49flow/KIT+) cells. A) Predicted outcomes of cell transplantation experiments under a clonal model, whereby different cell types give rise to distinct progenies, each retaining the phenotypic properties of the parent cells. B) Predicted outcomes of cell transplantation experiments under a differentiation model, whereby 1 or more cell types can serve as a progenitor of others, in a plastic and dynamic fashion. C) Experimental workflow of prospective xenotransplantation experiments aimed at comparing the tumor-initiating capacity of CD49fhigh/KITneg and CD49flow/KIT+ cells. The 2 populations were purified in parallel by FACS, starting from the same tumor lesion, double sorted to achieve high purity (>95%) and injected subcutaneously, side by side, into the opposite flanks of the same animal. D-E) Extreme limiting dilution analysis (ELDA) of xenotransplantation experiments using paired sets of CD49fhigh/KITneg and CD49flow/KIT+ cells sorted from ACCX5M1 (D) and SGTX6 (E) PDX lines. In both models, the frequency of tumor-initiating cells was higher in CD49fhigh/KITneg as compared with CD49flow/KIT+ cells. F-Q) Analysis by FACS and IHC of the cell composition of tumors originated from the xenotransplantation of purified preparations of either CD49fhigh/KITneg or CD49flow/KIT+ cells, sorted from either ACCX5M1 (F-K) or SGTX6 (L-Q) PDX lines. Analysis by FACS (F-G, I-J, L-M, O-P) showed that tumors originated from sorted cells contained both CD49fhigh/KITneg and CD49flow/KIT+ populations, irrespective of the original phenotype of sorted cells. In tumors originated from CD49fhigh/KITneg cells, the percentage of CD49fhigh/KITneg cells did not appear increased as compared with that observed in parent tumors but was lower than that observed in the purified preparations (G, M), indicating spontaneous in vivo differentiation (n.s. = not statistically significant; *P < .05; Mann–Whitney U test, 1-tailed). A symmetric scenario was observed in tumors originated from CD49flow/KIT+ cells (J, P). Analysis by IHC of tumors originated from sorted cells (H, K, N, Q) confirmed the reconstitution of a cribriform histology and of a biphenotypic cell composition, defined by the coexistence of 2 distinct subsets of cancer cells with mutually exclusive expression of myoepithelial-specific (TP63) and ductal-specific (KIT) biomarkers. Scale bars: 50 µm. APC = allophycocyanin; CI = confidence interval; FACS = fluorescence-activated cell sorting; IHC = immunohistochemistry; PDX = patient-derived xenograft; PE = phycoerythrin.
Differential expression of mechanistic regulators of retinoic acid (RA) signaling
To elucidate the molecular mechanisms that control the differentiation of CD49fhigh/KITneg cells into CD49flow/KIT+ cells, we searched for signaling pathways with differential activation in the 2 cell types. We tested whether CD49fhigh/KITneg and CD49flow/KIT+ cells differed in the expression of genes encoding for mechanistic regulators of RA signaling, such as enzymes involved in RA biosynthesis (45-47), RA binding proteins (48-50), retinoic acid receptors (RARs) and retinoid x receptors (RXRs) (51) (Figure 4, A), given that RA signaling plays a key role in the differentiation of SG epithelia (52-54) and antagonizes MYB signaling in human ACCs (55,56). We found that activators of RA signaling were overexpressed in CD49flow/KIT+ cells, whereas suppressors of RA signaling were overexpressed in CD49fhigh/KITneg cells, in a coordinated fashion (Figure 4, B and C).
Figure 4.

The role of retinoic acid (RA) signaling in controlling the cell composition of human ACC organoids. A) Schematic modeling of the RA signaling pathway. B) Comparison of the gene-expression levels for known mediators of RA signaling in CD49fhigh/KITneg (CD49f) and CD49flow/KIT+ (KIT) cells, as measured by RNA-seq on autologous pairs from biphenotypic ACCs. Genes identified as attenuators of RA signaling displayed preferential expression in CD49fhigh/KITneg cells (CD49f), whereas genes identified as potentiators of RA signaling displayed preferential expression in CD49flow/KIT+ cells (KIT). Error bars: mean +/- standard deviation (n.s. = not significant; *P < .10; **P < .05; ***P < .01; Student’s t test, paired samples). C) Heatmap displaying mean-centered z scores for the average expression levels of modulators of RA signaling in CD49fhigh/KITneg (CD49f) and CD49flow/KIT+ (KIT) cells. D-G) Analysis by microscopy and IHC of 3D organoids established from human ACCs. Organoids consisted in large adenoid structures (D-E) that recapitulated key elements of the histological architecture of primary tumors, such as the coexistence of 2 cell types with mutually exclusive expression of TP63 (F) and KIT (G). H-I) Analysis by flow cytometry of ACCX5M1 organoids treated for 1 week with either agonists (ATRA, 10 µM; bexarotene, 10 µM) or inhibitors (BMS493, 10 µM; AGN193109, 10 µM) of RAR/RXR signaling. Treatment with agonists induced an increase in the percentage of CD49flow/KIT+ cells, whereas treatment with inhibitors resulted in their reduction. J-M) Dose-response studies of the effects of agonists and inhibitors of RAR/RXR signaling on the cell composition of human ACC organoids. Treatment with increasing concentrations of ATRA (0.1-100 µM) resulted in a progressive increase of the percentage of CD49flow/KIT+ cells (ACCX5M1 [J-K]). The effects of ATRA were already detectable at low concentrations (0.1-1 µM; ACCX5M1 [J]). Treatment with inhibitors of RAR/RXR signaling (BMS493, AGN193109) resulted in a profound reduction of the percentage of CD49flow/KIT+ cells, even at low pharmacological doses (1 µM; ACCX5M1 [L]; SGTX6 [M]). Changes in the percentage of CD49flow/KIT+ cells were evaluated by FACS and tested for statistical significance using Welch’s 1-way ANOVA followed by Dunnett’s T3 test (n.s. = not significant; *P < .05; **P < .01; ***P < .001) assuming a normal distribution (Supplementary Figure 7, available online). 3D = 3-dimensional; ACC = adenoid cystic carcinoma; ANOVA = analysis of variance; APC = allophycocyanin; ATRA = all-trans retinoic acid; DMSO = dimethyl-sulfoxide; FACS = fluorescence-activated cell sorting; H&E = hematoxylin and eosin stain; IHC = immunohistochemistry; NT = untreated; PE = phycoerythrin; RA = retinoic acid; RAR = retinoic acid receptor; RNA-seq = RNA sequencing; RXR = retinoid X receptor.
In vitro effects of RAR/RXR activation and inhibition
To elucidate the role played by RA signaling in regulating cell differentiation, we leveraged a 3-dimensional in vitro organoid tissue-culture system (32-34) that recapitulated the biphenotypic composition of primary tissues (Figure 4, D-G), as well as key elements of their histological architecture (Supplementary Figure 6, available online). We observed that stimulation of organoid cultures with agonists of RARs (ATRA) or RXRs (bexarotene) caused an increase in the percentage of CD49flow/KIT+ cells, whereas suppression of the signaling mediated by RAR and RXR heterodimers (RAR/RXR) with inverse RAR agonists (BMS493, AGN193109) resulted in selective loss of CD49flow/KIT+ cells (Figure 4, H and I). These effects were observed at concentrations (0.1-10 µM) that spanned the drugs’ known median effective dose (ED50) (Figure 4, J-M) and were reproduced across 3 biphenotypic PDX lines (ACCX5M1, SGTX6, ACCX6) (Figure 5, A-F). To clarify the mechanism causing such changes in cell composition, we first tested whether ATRA or BMS493 induced preferential proliferation of one cell type. Analysis by IHC and FACS showed no increases in the frequency of MKI67+ cells (Figure 5, G-R; Supplementary Figure 8, A-O, available online) or of cells in either the G2 or M phases of the cell cycle (Supplementary Figure 8, P-S) in either cell type. The IHC analysis also confirmed that cells with a ductal-like phenotype, as defined by expression of KIT and lack of expression of TP63 (KIT+/TP63neg) became more abundant following treatment with ATRA, and were undetectable after treatment with BMS493 (Figure 5, G-R; Supplementary Figure 8, A-O, available online). Remarkably, organoids treated with BMS493 displayed a striking change in morphology, with areas occupied by KIT+ cells undergoing nuclear fragmentation, suggesting selective cytotoxicity toward ductal-like cells (Figure 5, O-R; Supplementary Figure 8, L, available online). We thus hypothesized that agonism and suppression of RAR/RXR signaling might have lineage-specific effects on the 2 cell populations (Figure 5, S). To formally test this hypothesis, we purified CD49fhigh/KITneg and CD49flow/KIT+ cells and treated them individually with ATRA (10 µM) or BMS493 (10 µM) using 2-dimensional monolayer cultures (35) (Figure 6, A-F). The experiment revealed that stimulation with ATRA did not impact the viability of CD49fhigh/KITneg cells (Figure 6, B) but changed their phenotype, with a majority of cells becoming CD49flow/KIT+ (Figure 6, C-D), suggesting myoepithelial-to-ductal differentiation. Conversely, treatment of purified CD49flow/KIT+ cells with BMS493 resulted in a substantial decrease in cell viability, indicating selective toxicity against ductal-like cells (Figure 6, E and F). To provide orthogonal evidence in support of RAR/RXR signaling as a key mediator of myoepithelial-to-ductal differentiation, we decided to test whether the effects of RAR/RXR inhibitors could be phenocopied by overexpression of a dominant-negative version of human RARα (DNhRARα), known to suppress the transcriptional activity of all 3 members of the human RAR family (RARα, RARβ, RARγ) (37). Indeed, infection of CD49fhigh/KITneg cells with a lentivirus driving constitutive expression of the DNhRARα construct resulted in complete abrogation of their spontaneous differentiation into CD49flow/KIT+ cells (Figure 6, G-N).
Figure 5.

Pharmacological perturbation of RAR/RXR signaling across different PDX models and analysis of its effects. A-F) Analysis and quantification by flow cytometry of the relative percentage of CD49flow/KIT+ cells in ACC organoids established from 3 independent PDX lines (ACCX5M1, SGTX6, ACCX6), following 1 week of treatment with either ATRA (10 µM) or BMS493 (10 µM). Treatment with ATRA was associated with an increase in the percentage of CD49flow/KIT+ cells, whereas treatment with BMS493 was associated with its reduction, as compared with control organoids treated only with DMSO, the solvent used to resuspend the 2 drugs. Differences in the mean percentage of CD49flow/KIT+ cells were tested for statistical significance using Welch’s 1-way ANOVA followed by Dunnett’s T3 test (*P < .05; **P< .01; ***P < .001) assuming a normal distribution (Supplementary Figure 7, available online). Box plots report the results of at least 2 independent experiments (with a minimum of 3 replicates for each condition). G-R) Analysis by IHC of 3D organoids established from the ACCX6 PDX line and treated with DMSO, ATRA (10 µM), or BMS493 (10 µM). Treatment with ATRA resulted in a visual expansion of KIT+ cells (M) as compared with treatment with DMSO alone (I), whereas treatment with BMS493 resulted in a complete loss of KIT expression (Q) and was associated with a dramatic change in the organoids’ morphology, characterized by the appearance of amorphous, eosin-rich deposits at their center (O). Neither ATRA nor BMS493 appeared to upregulate MKI67 expression in either cell population (H, L, P). Scale bars: 100 µm. S) Schematic modeling of the effects produced by agonism and inhibition of RAR/RXR signaling on the cell composition of human ACCs, as hypothesized based on the observations conducted on whole 3D organoids: stimulation of RAR/RXR signaling induces the differentiation of myoepithelial-like cells into ductal-like cells, whereas inhibition of RAR/RXR causes selective death of ductal-like cells. 3D = 3-dimensional; ACC = adenoid cystic carcinoma; ANOVA = analysis of variance; APC = allophycocyanin; ATRA = all-trans retinoic acid; DMSO = dimethyl-sulfoxide; H&E = hematoxylin and eosin stain; IHC = immunohistochemistry; PDX = patient-derived xenograft; PE = phycoerythrin; RAR = retinoic acid receptor; RXR = retinoid X receptor.
Figure 6.

Effects of RAR/RXR signaling on the differentiation of myoepithelial-like cells into ductal-like cells and the survival of ductal-like cells. A) Schematic workflow of experiments aimed at elucidating the population-specific effects of pharmacological manipulations of RAR/RXR signaling. Paired sets of CD49fhigh/KITneg and CD49flow/KIT+ cells were sorted in parallel from the same tumor (ACCX5M1) and cultured for 1 week as 2D monolayers, in the presence of either ATRA (10 µM) or BMS493 (10 µM), respectively. B-D) Evaluation of the effects of ATRA on sorted CD49fhigh/KITneg cells. Treatment with ATRA did not affect the viability of CD49fhigh/KITneg cells ([B] alamarBlue assay) but caused CD49fhigh/KITneg cells to change phenotype and become CD49flow/KIT+ ([C-D] FACS). E-F) Evaluation of the effects of BMS493 on sorted CD49flow/KIT+ cells. Treatment with BMS493 caused the death of the majority CD49flow/KIT+ cells ([E] alamarBlue assay). On visual inspection by conventional microscopy, CD49flow/KIT+ cells treated with BMS493 appeared fragmented as compared with control cells treated with DMSO alone (F). Scale bar: 100 µm. G) Schematic workflow of the experiment aimed at testing the effects of a DNhRARα construct on the capacity of CD49fhigh/KITneg cells to undergo myoepithelial-to-ductal differentiation. CD49fhigh/KITneg cells were sorted from ACCX5M1 tumors, cultured for 6 days as 2D monolayers, and infected with lentivirus vectors encoding for either a DNhRARα-EGFP construct or a control EGFP reporter. Infected cells were then cultured for 1 additional week and analyzed by FACS for CD49f and KIT expression, restricting the analysis to infected (EGFP+) cells. H-J) Analysis by FACS of CD49fhigh/KITneg cells purified form ACCX5M1 tumors and infected with lentivirus vectors encoding for either a DNhRARα-EGFP construct or a control EGFP reporter. Forced DNhRARα expression completely abrogated the capacity of CD49fhigh/KITneg cells to produce a CD49flow/KIT+ progeny, whereas forced expression of EGFP alone did not. K-N) Evaluation of the role of DNhRARα as a suppressor of myoepithelial-to-ductal differentiation in a second, independent PDX model (SGTX6). Error bars: mean +/- standard deviation; P values: Student’s t test, 2-tailed (n.s. = not significant; *P < .05; ***P < .001). 2D = 2-dimensional; APC = allophycocyanin; ATRA = all-trans retinoic acid; DMSO = dimethyl-sulfoxide; DNhRARα = dominant negative human retinoic acid receptor alpha; EGFP = enhanced green fluorescent protein; FACS = fluorescence-activated cell sorting; PDX = patient-derived xenograft; PE = phycoerythrin; RAR = retinoic acid receptor; RXR = retinoid X receptor.
In vivo antitumor activity of BMS493
We next aimed to elucidate whether the selective toxicity displayed by BMS493 against ductal-like cells in vitro could be leveraged for the in vivo therapy of ACCs. We hypothesized that, among ACCs, those enriched in ductal-like cells would represent the most susceptible targets. Although most ACCs display biphenotypic histology, over the course of the disease, a subgroup progresses to a solid histological pattern, consisting predominantly of KIT+ cells (20). Progression to solid histology associates with NOTCH1-activating mutations, increased proliferation kinetics, and worse clinical outcomes (57-63). To understand whether ACCs with solid histology represented monophenotypic expansions of ductal-like cells, we analyzed 2 PDX models representative of this specific subtype (ACCX9, ACCX11) (20) and confirmed that they consisted of a single KIT+/TP63neg population (Figure 7, A-F). We then performed RNA-seq on KIT+ cells purified by FACS from these 2 models and repeated the principal component analysis (PCA), combining the new data with those from purified pairs of myoepithelial-like and ductal-like cells from biphenotypic ACCs. Indeed, KIT+ cells from solid ACCs clustered with CD49flow/KIT+ cells from biphenotypic ACCs (Figure 7, G), indicating retention of a ductal-like transcriptional profile (Figure 7, H). Furthermore, when treated with BMS493 (10 µM), organoids established from solid PDX lines displayed loss of structural integrity and decreased viability, indicating retention of sensitivity to suppression of RAR/RXR signaling (Figure 7, I-K). As a final step, we tested whether in vivo administration of BMS493 (40 mg/kg, intraperitoneal) could be leveraged for the treatment of PDX lines with either solid (ACCX9, ACCX11) or cribriform (ACCX5M1) histology (Figure 8). We used a more intense regimen for the cribriform model (4 times/week x 3 weeks; Figure 8, F) as compared with the solid models (3 times/week x 3 weeks; Figure 8, A), assuming lower sensitivity of the former. Treatment with BMS493 was associated with side effects reminiscent of vitamin A deficiency in mice (eg, encrusted eyelids, rough coat, scaling of skin) (64). Of 18 tumor-bearing animals treated with BMS493, 33% (6 of 18) experienced tumor shrinkage (Supplementary Figure 9, available online). Four (22%) animals were prematurely euthanized because of abrupt deterioration of general health conditions. In 3 of these animals, health deterioration occurred immediately following tumor shrinkage, suggesting acute toxicity due to tumor lysis (Supplementary Figure 9, available online). Overall, treatment with BMS493 led to a statistically significant reduction in tumor growth across all 3 models, even after removal of animals that underwent premature euthanasia (Figure 8, B-E, G-H; Supplementary Figure 9, available online).
Figure 7.

Transcriptional profile and drug sensitivity of ACCs with solid histology. A-B) Analysis by flow cytometry of 2 PDX lines representative of human ACCs with solid histology ([A] ACCX9; [B] ACCX11) revealing a monophenotypic, ductal-like (CD49flow/KIT+) cell composition. C-F) Analysis by IHC of KIT and TP63 expression in ACCX9 and ACCX11 tumors, showing ubiquitous expression of the ductal-specific marker KIT (C, E) and complete loss of the myoepithelial-specific marker TP63 (D, F). Scale bars: 50 µm. G) Principal component analysis (PCA) of RNA-seq data from human ACCs, in which data from the 2 PDX lines with solid histology (ACCX9, ACCX11) are combined with those from the 5 autologous pairs of CD49fhigh/KITneg and CD49flow/KIT+ cells isolated from biphenotypic PDX lines (ACCX5M1, ACCX6, ACCX14, ACCX22, SGTX6). PCA was performed using the top 500 genes that displayed the highest level of variance across the full 12-sample dataset. H) Hierarchical clustering of RNA-seq data from human ACCs, based on the expression levels of the same list of 100 genes identified as differentially expressed (DE) between CD49fhigh/KITneg and CD49flow/KIT+ cells and reported in Figure 2, D. Solid ACCs clustered with CD49flow/KIT+ cells from biphenotypic tumors, irrespective of the method used to analyze their transcriptional profile. I-J) On visual inspection by conventional microscopy, ACCX9 organoids cultured for 1 week in the presence of BMS493 (10 µM) displayed widespread cell fragmentation, in contrast to organoids cultured with DMSO alone. K) Quantification of organoid viability using the alamarBlue assay, confirming the cytotoxic activity of BMS493 (10 µM) against PDX lines with solid histology (ACCX9, ACCX11). Error bars: mean +/- standard deviation; P values: Student’s t test (2-tailed; **P < .01). ACC = adenoid cystic carcinoma; APC = allophycocyanin; DMSO = dimethyl-sulfoxide; IHC = immunohistochemistry; PC1 = first principal component; PC2 = second principal component; PDX = patient-derived xenograft; PE = phycoerythrin; RNA-seq = RNA sequencing.
Figure 8.
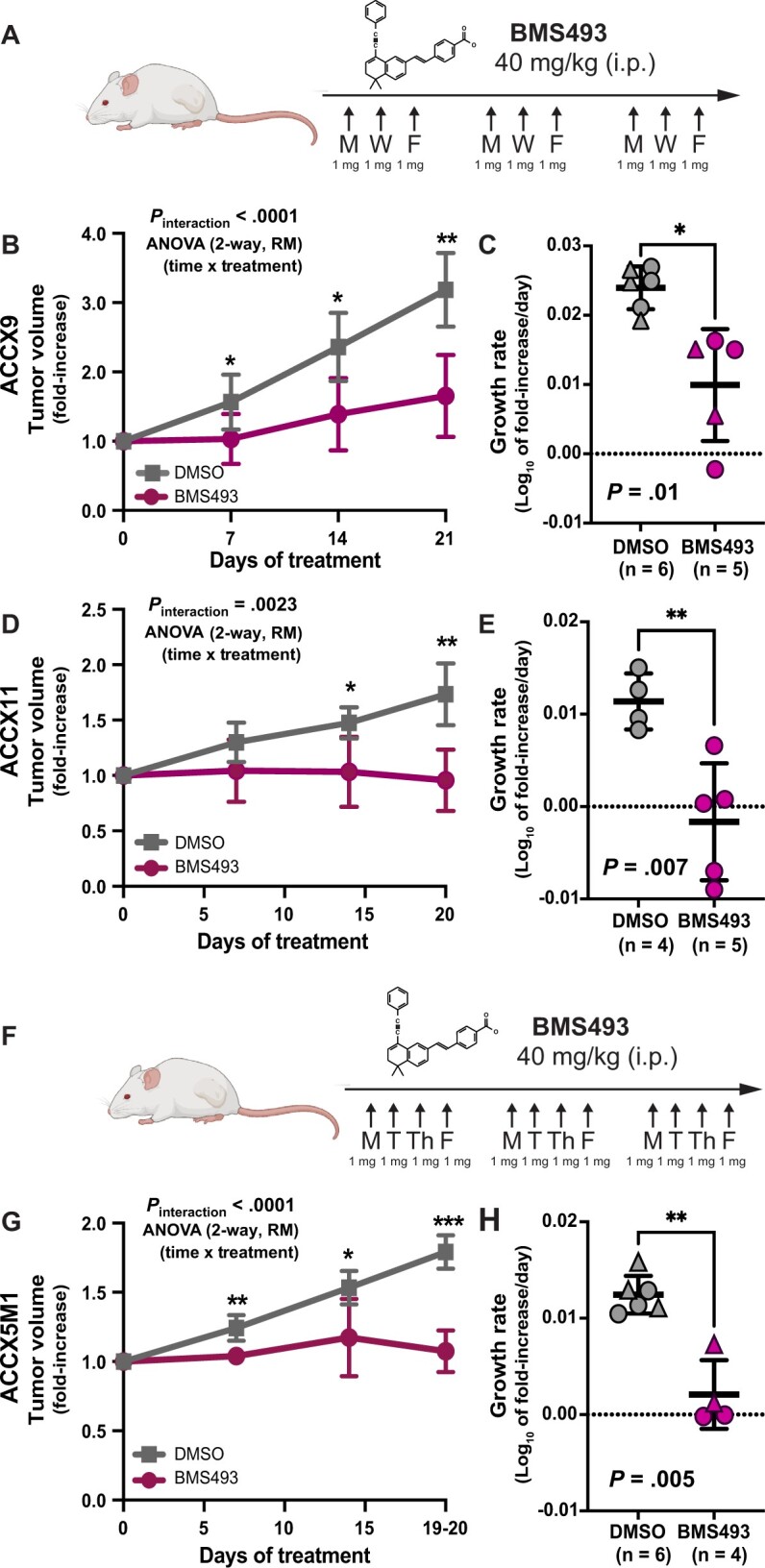
In vivo antitumor activity of BMS493. A) Schematic description of the BMS493 dosing regimen used for the in vivo treatment of solid ACC models (40 mg/kg doses, intraperitoneal, 3 times/week x 3 weeks). B-E) Comparison of tumor growth kinetics between mice treated with BMS493 and mice treated with the drug’s vehicle alone (DMSO), following subcutaneous engraftment of 2 solid ACC models (ACCX9 [B-C]; ACCX11 [D-E]). F) Schematic of the BMS493 dosing regimen used for the in vivo treatment of the biphenotypic ACC model (40 mg/kg doses, intraperitoneal, 4 times/week x 3 weeks). G-H) Comparison of tumor growth kinetics between mice treated with BMS493 and mice treated with the drug’s vehicle alone (DMSO), following subcutaneous engraftment of a biphenotypic ACC model (ACCX5M1). Differences in tumor growth kinetics were quantified by comparing either mean fold increases in tumor volume (B, D, G) or mean growth rates (C, E, H). Differences in mean fold increases in tumor volume (B, D, G) were tested for statistical significance using 2 approaches: 1) at each time point, using a 2-tailed Student’s t test (*P < .05; **P < .01; ***P < .001); and 2) across the full experimental dataset, using a 2-way ANOVA for repeated measures (RM), where measurements performed on the same mouse at different time points were treated as repeated measures (Pinteraction = time x treatment). Growth rates were calculated assuming exponential kinetics. Differences between mean growth rates (C, E, H) were tested for statistical significance using a 2-tailed Welch’s t test. Error bars: mean +/- standard deviation. Schematic descriptions of dosing regimens were created using BioRender.com. ACC = adenoid cystic carcinoma; ANOVA = analysis of variance; DMSO = dimethyl-sulfoxide.
Discussion
In this study, we identified 2 cell-surface markers (CD49f, KIT) that, for the first time, enabled the differential purification and comparative study of the 2 subtypes of malignant cells (myoepithelial-like and ductal-like) that are known to coexist in human ACCs. Our data reveal that the 2 cell types do not represent distinct genetic clones but rather distinct developmental lineages (ie, distinct cell types that originate as a result of multilineage differentiation processes, akin to those that enable stem-cell populations to sustain the homeostatic turnover of normal tissues). Our data also reveal that myoepithelial-like cells are highly tumorigenic upon xenotransplantation in immune-deficient animals, despite their low proliferation rates. In tumors originated from exocrine glands (eg, breast cancer), myoepithelial-like cells are often considered tumor suppressive (65,66). Our findings caution against this interpretation in ACCs and indicate that, to be curative, treatment strategies will need to eradicate myoepithelial-like components. Furthermore, our data show that, in ACCs, myoepithelial-like cells act as progenitors of ductal-like cells and that myoepithelial-to-ductal differentiation is promoted by RAR/RXR signaling. These findings provide a mechanistic explanation for the conflicting results that have been recently obtained in studies that tested ATRA’s antitumor activity in human ACCs. ATRA displayed marked antiproliferative activity against PDX models (55,56) but appeared to provide limited benefit when administered to patients (67). We hypothesize that, in ACC patients, the therapeutic benefit of ATRA might be short lived because of the cytostatic nature of its effect, which consists in a transient perturbation of the tumor tissues’ cell composition.
Perhaps more importantly, our data also revealed that suppression of RAR/RXR signaling induces selective death of ductal-like cells. This finding provides an opportunity for the selective pharmacological targeting of ACCs, especially of cases with solid histology, which are characterized by monophenotypic expansions of ductal-like cells. These tumors often originate during the natural progression of ACCs, following the acquisition of NOTCH1-activating mutations, in a scenario that is reminiscent of the “blast crisis” observed in chronic myelogenous leukemias, whereby a population of more differentiated, yet highly proliferative cells becomes dominant, because of mutations that aberrantly activate self-renewal (22,68,69). Our data indicate that, in solid ACCs, treatment with an inverse agonist of RAR/RXR signaling (BMS493) can have robust antitumor activity. Agonists of RAR/RXR signaling have been extensively explored as antitumor agents in humans (70-74), whereas inverse agonists, to the best of our knowledge, have not. Our findings advocate in support of the clinical development of inverse agonists of RAR/RXR signaling as antitumor agents, to assess their efficacy and toxicity in the treatment of selected subtypes of epithelial cancer, especially those characterized by ductal and/or luminal phenotypes.
Finally, our study demonstrates that, by “hacking” the signaling pathways that control multilineage differentiation in epithelial tissues, it is possible to discover novel pharmacological manipulations with selective toxicity on specific cellular lineages. Because multilineage differentiation programs are retained in many forms of cancer (22-24), this study could provide a methodological template to guide the discovery of novel antitumor drugs against a large and diverse repertoire of human malignancies.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Erin C. Bush (J.P. Sulzberger Columbia Genome Center, Columbia University) for technical guidance regarding the execution of single-cell RNA-seq experiments, as well as Jeffrey Kaufman and Nicole Spardy Burr (ACCRF) for sharing critical insights regarding the biology of human ACCs.
The funders of the study did not play any role in the design of the study, the collection, analysis, and interpretation of the data, the writing of the manuscript or the decision to submit the manuscript for publication.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. National Institutes of Health.
Part of the data reported in this manuscript has been previously presented during research conferences and invited seminars at academic institutions, such as: 1) the Keystone Symposium on Organoids as Tools for Fundamental Discovery and Translation (April 3-6, 2022); 2) the Rare Cancers: Unmet Medical Needs conference (April 26, 2022) organized by the Jed Ian Taxel Foundation; 3) the Gordon Research Conference on Epithelial Stem Cells and Niches (June 5-10, 2022); and 4) a research seminar held at the Department of Pathology of the University of Virginia (April 19, 2022). Part of the data reported in this manuscript has been included in a preprint that was uploaded onto the bioRxiv repository (January 21, 2022): https://www.biorxiv.org/content/10.1101/2022.01.19.476843v1.
Contributor Information
Sara Viragova, Department of Pathology and Cell Biology, Columbia University Medical Center, New York, NY, USA; Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, NY, USA; Columbia Stem Cell Initiative, Columbia University Medical Center, New York, NY, USA; Integrated Program in Cellular, Molecular and Biomedical Studies, Columbia University, New York, NY, USA.
Luis Aparicio, Program for Mathematical Genomics, Department of Systems Biology, Columbia University, New York, NY, USA; Department of Biomedical Informatics, Columbia University, New York, NY, USA.
Pierangela Palmerini, Department of Pathology and Cell Biology, Columbia University Medical Center, New York, NY, USA; Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, NY, USA; Columbia Stem Cell Initiative, Columbia University Medical Center, New York, NY, USA.
Junfei Zhao, Program for Mathematical Genomics, Department of Systems Biology, Columbia University, New York, NY, USA; Department of Biomedical Informatics, Columbia University, New York, NY, USA.
Luis E Valencia Salazar, Department of Pathology and Cell Biology, Columbia University Medical Center, New York, NY, USA; Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, NY, USA; Columbia Stem Cell Initiative, Columbia University Medical Center, New York, NY, USA.
Alexandra Schurer, Department of Pathology and Cell Biology, Columbia University Medical Center, New York, NY, USA; Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, NY, USA.
Anika Dhuri, Department of Pathology and Cell Biology, Columbia University Medical Center, New York, NY, USA.
Debashis Sahoo, Department of Pediatrics, University of California San Diego, San Diego, CA, USA; Department of Computer Science and Engineering, University of California San Diego, San Diego, CA, USA; Rebecca and John Moores Comprehensive Cancer Center, University of California San Diego, San Diego, CA, USA.
Christopher A Moskaluk, Department of Pathology, University of Virginia School of Medicine, Charlottesville, VA, USA.
Raul Rabadan, Program for Mathematical Genomics, Department of Systems Biology, Columbia University, New York, NY, USA; Department of Biomedical Informatics, Columbia University, New York, NY, USA.
Piero Dalerba, Department of Pathology and Cell Biology, Columbia University Medical Center, New York, NY, USA; Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, NY, USA; Columbia Stem Cell Initiative, Columbia University Medical Center, New York, NY, USA; Department of Medicine, Columbia University Medical Center, New York, NY, USA; Digestive and Liver Disease Research Center, Columbia University Medical Center, New York, NY, USA.
Data availability
All software used in this study is either publicly or commercially available, including Randomly (https://github.com/RabadanLab/randomly). The RNA-seq data generated as part of this study are available in the database of Genotypes and Phenotypes (dbGAP; https://www.ncbi.nlm.nih.gov/gap), using the following accession number: phs002764.
Author contributions
Sara Viragova, PhD (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Validation; Visualization; Writing—original draft; Writing—review & editing), Luis Aparicio, PhD (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Software; Visualization; Writing—review & editing), Pierangela Palmerini, PhD (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Visualization; Writing—review & editing), Junfei Zhao, PhD (Data curation; Formal analysis; Software; Visualization), Luis E. Valencia Salazar, BA (Investigation; Validation), Alexandra Schurer, MS (Investigation; Validation) Anika Dhuri (Investigation; Validation), Debashis Sahoo, PhD (Data curation; Formal analysis; Software), Christopher A Moskaluk, MD, PhD (Methodology; Resources), Raul Rabadan, PhD (Funding acquisition; Resources; Supervision) and, Piero Dalerba, MD (Conceptualization; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing—original draft; Writing—review & editing).
Funding
This study was supported by the U.S. National Institutes of Health (grant numbers: R01-DE028961 to PD, RC1-DE020687 to C.A.M, TL1-TR001875 to SV, T32-GM008224 to SV, R01-GM138385 to DS, UG3-TR003355 to DS, R01-AI155696 to DS, R35-CA253126 to RR, U01-CA261822 to RR); the Adenoid Cystic Carcinoma Research Foundation (“Kara Gelb” Memorial Grant to PD); the Damon Runyon Cancer Research Foundation (grant number: DRR-44-16, 2016 Runyon-Rachleff Innovator Award to PD); the National Organization of Rare Disorders (to CAM); and the Prostate Cancer Foundation (grant number 19CHAL02 to RR). This study used the shared resources of Columbia University’s Herbert Irving Comprehensive Cancer Center (HICCC), such as the Flow Cytometry Shared Resource, the Molecular Pathology Shared Resource and the Genomics and High Throughput Screening Shared Resource, funded in part through the U.S. National Institutes of Health/National Cancer Institute Cancer Center Support Grant P30-CA013696. This study used the shared resources of the Irving Institute for Clinical and Translational Research (IICTR), such as the J.P. Sulzberger Columbia Genome Center, funded in part through the U.S. National Institutes of Health/National Center for Advancing Translational Sciences Cooperative Agreement UL1-TR001873.
Conflicts of interest
Sara Viragova and Piero Dalerba are acknowledged as co-inventors on a patent application (US-20220031644-A1), describing the use of inverse agonists of retinoic acid receptors as antitumor agents for the treatment of ACCs. Piero Dalerba received royalties from Oncomed Pharmaceuticals, Quanticel Pharmaceuticals and Forty Seven Inc, as a results of his acknowledgment as a co-inventor on patents granted to the University of Michigan (US-07723112) and Stanford University (US-09329170, US-09850483, US-10344094, US-11130813) and related to 1) the discovery of surface markers for the differential purification of cancer stem cell populations from human malignancies; 2) the use of single-cell genomics technologies for the identification of pharmacological targets expressed in cancer stem cell populations; 3) the combination of anti-CD47 and anti-EGFR monoclonal antibodies for the treatment of human colon cancer. Piero Dalerba recently owned stock of Eli Lilly and Company. Raul Rabadan is a founder of Genotwin, a consultant for Arquimea Research and a member of the scientific advisory board of AimedBio, in activities unrelated to the current study. Piero Dalerba’s spouse is employed by Regeneron and owns (or recently owned) stock of the following pharmaceutical companies: AbbVie, Amgen, AstraZeneca, Eli Lilly and Company, GlaxoSmithKline, Johnson & Johnson, Merck & Co, Novartis, Organon, Pfizer, Teva Pharmaceutical Industries Ltd and Viatris. All other authors have no competing interests to disclose.
References
- 1. Moskaluk CA. Adenoid cystic carcinoma: clinical and molecular features. Head Neck Pathol. 2013;7(1):17-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee RJ, Tan AP, Tong EL, et al. Epidemiology, prognostic factors, and treatment of malignant submandibular gland tumors: a population-based cohort analysis. JAMA Otolaryngol Head Neck Surg. 2015;141(10):905-912. [DOI] [PubMed] [Google Scholar]
- 3. Gao M, Hao Y, Huang MX, et al. Clinicopathological study of distant metastases of salivary adenoid cystic carcinoma. Int J Oral Maxillofac Surg. 2013;42(8):923-928. [DOI] [PubMed] [Google Scholar]
- 4. Bjorndal K, Krogdahl A, Therkildsen MH, et al. Salivary gland carcinoma in Denmark 1990-2005: outcome and prognostic factors. Results of the Danish Head and Neck Cancer Group (DAHANCA). Oral Oncol. 2012;48(2):179-185. [DOI] [PubMed] [Google Scholar]
- 5. Lloyd S, Yu JB, Wilson LD, et al. Determinants and patterns of survival in adenoid cystic carcinoma of the head and neck, including an analysis of adjuvant radiation therapy. Am J Clin Oncol. 2011;34(1):76-81. [DOI] [PubMed] [Google Scholar]
- 6. Laurie SA, Licitra L.. Systemic therapy in the palliative management of advanced salivary gland cancers. J Clin Oncol 2006;24(17):2673-2678. [DOI] [PubMed] [Google Scholar]
- 7. Dodd RL, Slevin NJ.. Salivary gland adenoid cystic carcinoma: a review of chemotherapy and molecular therapies. Oral Oncol. 2006;42(8):759-769. [DOI] [PubMed] [Google Scholar]
- 8. Laurie SA, Ho AL, Fury MG, et al. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: a systematic review. Lancet Oncol. 2011;12(8):815-824. [DOI] [PubMed] [Google Scholar]
- 9. Di Villeneuve L, Souza IL, Tolentino FDS, et al. Salivary gland carcinoma: novel targets to overcome treatment resistance in advanced disease. Front Oncol. 2020;10:580141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Persson M, Andrén Y, Mark J, et al. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci. 2009;106(44):18740-18744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitani Y, Li J, Rao PH, et al. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: incidence, variability, and clinicopathologic significance. Clin Cancer Res. 2010;16(19):4722-4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ho AS, Kannan K, Roy DM, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45(7):791-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rettig EM, Talbot CC Jr, Sausen M, et al. Whole-genome sequencing of salivary gland adenoid cystic carcinoma. Cancer Prev Res (Phila). 2016;9(4):265-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang Y, Gao R, Cao C, et al. MYB-activated models for testing therapeutic agents in adenoid cystic carcinoma. Oral Oncol. 2019;98:147-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Du F, Zhou CX, Gao Y.. Myoepithelial differentiation in cribriform, tubular and solid pattern of adenoid cystic carcinoma: a potential involvement in histological grading and prognosis. Ann Diagn Pathol. 2016;22:12-17. [DOI] [PubMed] [Google Scholar]
- 16. Jaso J, Malhotra R.. Adenoid cystic carcinoma. Arch Pathol Lab Med. 2011;135(4):511-515. [DOI] [PubMed] [Google Scholar]
- 17. Azumi N, Battifora H.. The cellular composition of adenoid cystic carcinoma. An immunohistochemical study. Cancer. 1987;60(7):1589-1598. [DOI] [PubMed] [Google Scholar]
- 18. Prasad ML, Barbacioru CC, Rawal YB, et al. Hierarchical cluster analysis of myoepithelial/basal cell markers in adenoid cystic carcinoma and polymorphous low-grade adenocarcinoma. Mod Pathol. 2008;21(2):105-114. [DOI] [PubMed] [Google Scholar]
- 19. Wu HM, Ren GX, Wang LZ, et al. Expression of podoplanin in salivary gland adenoid cystic carcinoma and its association with distant metastasis and clinical outcomes. Mol Med Rep. 2012;6(2):271-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Drier Y, Cotton MJ, Williamson KE, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016;48(3):265-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parikh AS, Wizel A, Davis D, et al. Single-cell RNA sequencing identifies a paracrine interaction that may drive oncogenic notch signaling in human adenoid cystic carcinoma. Cell Rep. 2022;41(9):111743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dalerba P, Cho RW, Clarke MF.. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58(1):267-284. [DOI] [PubMed] [Google Scholar]
- 23. Dalerba P, Kalisky T, Sahoo D, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol. 2011;29(12):1120-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105-111. [DOI] [PubMed] [Google Scholar]
- 25. Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104(24):10158-10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu AR, Neff NF, Kalisky T, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11(1):41-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moskaluk CA, Baras AS, Mancuso SA, et al. Development and characterization of xenograft model systems for adenoid cystic carcinoma. Lab Invest. 2011;91(10):1480-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aparicio L, Bordyuh M, Blumberg AJ, et al. A random matrix theory approach to denoise single-cell data. Patterns. 2020;1(3):100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Love MI, Huber W, Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haas BJ, Dobin A, Li B, et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019;20(1):1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu Y, Smyth GK.. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1-2):70-78. [DOI] [PubMed] [Google Scholar]
- 32. Shimono Y, Mukohyama J, Isobe T, et al. Organoid culture of human cancer stem cells. Methods Mol Biol. 2019;1576:23-31. [DOI] [PubMed] [Google Scholar]
- 33. Sato T, Stange DE, Ferrante M, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141(5):1762-1772. [DOI] [PubMed] [Google Scholar]
- 34. Nanduri LS, Baanstra M, Faber H, et al. Purification and ex vivo expansion of fully functional salivary gland stem cells. Stem Cell Rep. 2014;3(6):957-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Panaccione A, Chang MT, Carbone BE, et al. NOTCH1 and SOX10 are essential for proliferation and radiation resistance of cancer stem–like cells in adenoid cystic carcinoma. Clin Cancer Res. 2016;22(8):2083-2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tiscornia G, Singer O, Verma IM.. Production and purification of lentiviral vectors. Nat Protoc. 2006;1(1):241-245. [DOI] [PubMed] [Google Scholar]
- 37. Damm K, Heyman RA, Umesono K, et al. Functional inhibition of retinoic acid response by dominant negative retinoic acid receptor mutants. Proc Natl Acad Sci USA. 1993;90(7):2989-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eilenberger C, Kratz SRA, Rothbauer M, et al. Optimized alamarBlue assay protocol for drug dose-response determination of 3D tumor spheroids. MethodsX. 2018;5:781-787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bell D, Roberts D, Kies M, et al. Cell type-dependent biomarker expression in adenoid cystic carcinoma: biologic and therapeutic implications. Cancer. 2010;116(24):5749-5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iglesias JM, Cairney CJ, Ferrier RK, et al. Annexin A8 identifies a subpopulation of transiently quiescent c-kit positive luminal progenitor cells of the ductal mammary epithelium. PLoS One. 2015;10(3):e0119718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lim E, Vaillant F, Wu D, et al. ; for the kConFab. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907-913. [DOI] [PubMed] [Google Scholar]
- 42. Nguyen QH, Pervolarakis N, Blake K, et al. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat Commun. 2018;9(1):2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oakes SR, Naylor MJ, Asselin-Labat ML, et al. The Ets transcription factor Elf5 specifies mammary alveolar cell fate. Genes Dev. 2008;22(5):581-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. West RB, Kong C, Clarke N, et al. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am J Surg Pathol. 2011;35(1):92-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghyselinck NB, Duester G.. Retinoic acid signaling pathways. Development. 2019;146(13):dev167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shannon SR, Moise AR, Trainor PA.. New insights and changing paradigms in the regulation of vitamin A metabolism in development. Wiley Interdiscip Rev Dev Biol. 2017;6(3):e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Metzler MA, Sandell LL.. Enzymatic metabolism of vitamin A in developing vertebrate embryos. Nutrients. 2016;8(12):812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dong D, Ruuska SE, Levinthal DJ, et al. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J Biol Chem. 1999;274(34):23695-23698. [DOI] [PubMed] [Google Scholar]
- 49. Schug TT, Berry DC, Shaw NS, et al. Dual transcriptional activities underlie opposing effects of retinoic acid on cell survival. Cell. 2007;129(4):723-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Napoli JL. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol Ther. 2017;173:19-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bastien J, Rochette-Egly C.. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1-16. [DOI] [PubMed] [Google Scholar]
- 52. Wright DM, Buenger DE, Abashev TM, et al. Retinoic acid regulates embryonic development of mammalian submandibular salivary glands. Dev Biol. 2015;407(1):57-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abashev TM, Metzler MA, Wright DM, et al. Retinoic acid signaling regulates Krt5 and Krt14 independently of stem cell markers in submandibular salivary gland epithelium. Dev Dyn. 2017;246(2):135-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Metzler MA, Raja S, Elliott KH, et al. RDH10-mediated retinol metabolism and RARα-mediated retinoic acid signaling are required for submandibular salivary gland initiation. Development. 2018;145(15):dev164822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mandelbaum J, Shestopalov IA, Henderson RE, et al. Zebrafish blastomere screen identifies retinoic acid suppression of MYB in adenoid cystic carcinoma. J Exp Med. 2018;215(10):2673-2685. doi: 10.1084/jem.20180939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun B, Wang Y, Sun J, et al. Establishment of patient-derived xenograft models of adenoid cystic carcinoma to assess pre-clinical efficacy of combination therapy of a PI3K inhibitor and retinoic acid. Am J Cancer Res. 2021;11(3):773. [PMC free article] [PubMed] [Google Scholar]
- 57. Ho AS, Ochoa A, Jayakumaran G, et al. Genetic hallmarks of recurrent/metastatic adenoid cystic carcinoma. J Clin Invest. 2019;129(10):4276-4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stephens PJ, Davies HR, Mitani Y, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013;123(7):2965-2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ferrarotto R, Mitani Y, Diao L, et al. Activating NOTCH1 mutations define a distinct subgroup of patients with adenoid cystic carcinoma who have poor prognosis, propensity to bone and liver metastasis, and potential responsiveness to notch1 inhibitors. J Clin Oncol 2017;35(3):352-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ferrarotto R, Mitani Y, McGrail DJ, et al. Proteogenomic analysis of salivary adenoid cystic carcinomas defines molecular subtypes and identifies therapeutic targets. Clin Cancer Res. 2021;27(3):852-864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fordice J, Kershaw C, El-Naggar A, et al. Adenoid cystic carcinoma of the head and neck: predictors of morbidity and mortality. Arch Otolaryngol Head Neck Surg. 1999;125(2):149-152. [DOI] [PubMed] [Google Scholar]
- 62. van Weert S, van der Waal I, Witte BI, et al. Histopathological grading of adenoid cystic carcinoma of the head and neck: analysis of currently used grading systems and proposal for a simplified grading scheme. Oral Oncol. 2015;51(1):71-76. [DOI] [PubMed] [Google Scholar]
- 63. Romani C, Lorini L, Bozzola A, et al. Functional profiles of curatively treated adenoid cystic carcinoma unveil prognostic features and potentially targetable pathways. Sci Rep. 2023;13(1):1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wolbach SB, Howe PR.. Tissue changes following deprivation of fat-soluble A vitamin. J Exp Med. 1925;42(6):753-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Adriance MC, Inman JL, Petersen OW, et al. Myoepithelial cells: good fences make good neighbors. Breast Cancer Res. 2005;7(5):1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sirka OK, Shamir ER, Ewald AJ.. Myoepithelial cells are a dynamic barrier to epithelial dissemination. J Cell Biol. 2018;217(10):3368-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hanna GJ, ONeill A, Cutler JM, et al. A phase II trial of all-trans retinoic acid (ATRA) in advanced adenoid cystic carcinoma. Oral Oncol. 2021;119:105366. [DOI] [PubMed] [Google Scholar]
- 68. Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte–macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. New Engl J Med. 2004;351(7):657-667. [DOI] [PubMed] [Google Scholar]
- 69. Akala OO, Park IK, Qian D, et al. Long-term haematopoietic reconstitution by Trp53-/-p16Ink4a-/-p19Arf-/- multipotent progenitors. Nature. 2008;453(7192):228-232. [DOI] [PubMed] [Google Scholar]
- 70. Duvic M, Hymes K, Heald P, et al. ; for Members of the Bexarotene Worldwide Study Group. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol 2001;19(9):2456-2471. [DOI] [PubMed] [Google Scholar]
- 71. Matthay KK, Villablanca JG, Seeger RC, et al. ; for the Children’s Cancer Group. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. N Engl J Med. 1999;341(16):1165-1173. [DOI] [PubMed] [Google Scholar]
- 72. Tallman MS, Andersen JW, Schiffer CA, et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337(15):1021-1028. [DOI] [PubMed] [Google Scholar]
- 73. Lo-Coco F, Avvisati G, Vignetti M, et al. ; for the Study Alliance Leukemia. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111-121. [DOI] [PubMed] [Google Scholar]
- 74. Walmsley S, Northfelt DW, Melosky B, et al. ; for the Panretin Gel North American Study Group. Treatment of AIDS-related cutaneous Kaposi’s sarcoma with topical alitretinoin (9-cis-retinoic acid) gel. J Acquir Immune Defic Syndr. 1999;22(3):235-246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All software used in this study is either publicly or commercially available, including Randomly (https://github.com/RabadanLab/randomly). The RNA-seq data generated as part of this study are available in the database of Genotypes and Phenotypes (dbGAP; https://www.ncbi.nlm.nih.gov/gap), using the following accession number: phs002764.