


Abstract
The spatial folding of eukaryotic genome plays a key role in genome function. We report here that our recently developed method, Hi-TrAC, which specializes in detecting chromatin loops among accessible genomic regions, can detect active sub-TADs with a median size of 100 kb, most of which harbor one or two cell specifically expressed genes and regulatory elements such as super-enhancers organized into nested interaction domains. These active sub-TADs are characterized by highly enriched histone mark H3K4me1 and chromatin-binding proteins, including Cohesin complex. Deletion of selected sub-TAD boundaries have different impacts, such as decreased chromatin interaction and gene expression within the sub-TADs or compromised insulation between the sub-TADs, depending on the specific chromatin environment. We show that knocking down core subunit of the Cohesin complex using shRNAs in human cells or decreasing the H3K4me1 modification by deleting the H3K4 methyltransferase Mll4 gene in mouse Th17 cells disrupted the sub-TADs structure. Our data also suggest that super-enhancers exist as an equilibrium globule structure, while inaccessible chromatin regions exist as a fractal globule structure. In summary, Hi-TrAC serves as a highly sensitive and inexpensive approach to study dynamic changes of active sub-TADs, providing more explicit insights into delicate genome structures and functions.
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Multi-scale and proper spatial folding of chromatin is essential for eukaryotic genome packaging and biological processes such as DNA replication (1,2), immunoglobulin heavy chain V(D)J recombination (3–5), and cell differentiation (6–9). Disorganization of three-dimensional (3D) genome structure is associated with the pathogenesis of cancers and developmental diseases (10–13). The development of powerful new technologies, including super-resolution microscopy imaging methods and chromosome conformation capture (3C)-derived methods based on proximity ligation and high-throughput sequencing, has helped tremendously our understanding of the multilayer 3D genomic structure (14–16). The initial version of genome-wide 3C method, Hi-C, led to the discovery of large compartments at the megabase scale with a low resolution of 100 kb (17). With deeper sequencing, topologically associating domains (TADs) at the sub-megabase scale were identified at a better resolution of 40 kb (18–20). With the improved in situ Hi-C method and with much deeper sequencing, chromatin loops and hierarchically nested sub-TADs were identified at much higher 1–5 kb resolution (21). Another Hi-C derivative, Micro-C, was able to achieve a resolution of the single nucleosome level, although requiring billions of sequencing reads (22,23). Other sequencing-based methods with varying principles, such as ChIA-PET (24), GAM (25), SPRITE (26) and Trac-looping (27), have all been able to validate the existence of high-order structures of compartments, domains, and loops, advancing our understandings of dynamics of the 3D genome.
Since the initial identification of TADs, domain-centric analysis has remained essential in interpreting genome-wide sequencing data and elucidating 3D genome organization and regulation. Features of TADs, including their existence in a range of species (18,19,28,29), boundaries associated with CTCF and Cohesin (18,30–32), formation mechanism through loop-extrusion model (33), and regulatory role in gene expression (11,12,34), have been extensively documented with Hi-C data (35,36). Therefore, with a physical size of around several hundred nanometers (16,37), TADs are considered as the architectural units of chromatin, which define regulatory landscapes by framing the microenvironment for enhancer-promoter interactions. Our recently developed proximity ligation-independent technique, Hi-TrAC (38), provides high-resolution maps of interactions between accessibility sites as loops and generic chromatin interactions analogous to Hi-C and variants as domains (39). While the loop-centric analysis provided comprehensive catalogs of promoter–enhancer interactions (38), it is not clear how the Hi-TrAC data can be used for analysis of chromatin domains.
By explicitly examining the domain features revealed by modestly-sequenced Hi-TrAC data, we found that it can detect domains around 100 kb in size. These domains show cell specificity, harbor cell identity genes and super-enhancers; they contain enriched ChIP-seq signals of active histone modification marks and transcription factors including H3K4me1 and SMC3. We observed self-similar domain structures within the super-enhancers and their component enhancers. We found that super-enhancers interact with each other with a unique block-to-block pattern when they are in the same domain. Our data suggested that super-enhancers exist as an equilibrium globule structure, while inaccessible chromatin regions exist as a fractal globule structure. We further showed that knocking down RAD21 or deleting the H3K4 methyltransferase Mll4 gene severely compromised the active sub-TADs. Collectively, our results demonstrate that Hi-TrAC is a highly robust and compatible method for studying active domains at a high resolution, even with limited sequencing depth.
MATERIALS AND METHODS
Public data and pre-processing
Public data used in this study, including human Hi-TrAC, Hi-C, HiChIP, ChIA-PET and ChIP-seq, were summarized in our previous work (38). Hg38 alignment BAM files of ENCODE (40) ChIP- seq data were obtained from ENCODE website and summarized in Supplemental Table S1. Biological and technical replicates were merged for the same factor, and only unique reads were used for the following analyses. Human gene annotations from GENCODE (41) (gencode.v30.basic.annotation.gtf) were used in any gene-related analysis. Hg38 was used in this study. If human data processed in other genome versions were downloaded, they were always converted to hg38 for analysis. GM12878 and K562 super-enhancers were downloaded from dbSUPER (42) in hg19 and converted to hg38 by UCSC liftOver.
Segregation score calculation and domain calling from Hi-TrAC data
Domains from Hi-TrAC data were called based on segregation scores. For each bin at the assigned resolution (parameter) from the contact matrix, its upstream and downstream window size (parameter) region is used to construct the contact matrix. The contact matrix is further log2 transformed then calculated as a correlation matrix by calculating the Pearson correlation coefficient between all pairs of rows and columns in the transformed matrix. For the up-right corner sub-matrix of the correlation matrix, all values smaller than zero are assigned as zero, and the mean value of the matrix is assigned as a segregation score for the bin. After calculating segregation scores for all bins in one chromosome, z-score normalization was performed to the segregation scores, and bins with positive segregation scores were stitched together as candidate domains. These candidate domains further required an enrichment score (the number of PETs within the domain divided by the number of PETs with only one end within the domain) > = 1, and the interaction density is higher than the two folds of chromosome-wide density. The scheme of the algorithm was summarized in Supplementary Figure S1A and implemented as the cLoops2 callDomains module (39). For calling sub-TADs from Hi-TrAC data, bin size was set to 1 kb, and windows size was set to 50 kb, details parameters of -bs 1000 -ws 50000 -strict were used.
Domain aggregation analysis
For a domain with its 0.5-fold sized neighboring upstream and downstream regions, interacting PETs were grouped into a 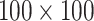 bins matrix. An individual enrichment score for a domain is calculated as the number of PETs with both ends located within the domain compared to the number of PETs with only one end located within the domain. The global enrichment score is the mean value of all enrichment scores for individual domains. Heatmap was plotted of the upper triangular matrix of the average matrix by default. The analysis was implemented in the cLoops2 agg module with the option of -domains (39), and default parameters were used in all related analyses in this manuscript.
bins matrix. An individual enrichment score for a domain is calculated as the number of PETs with both ends located within the domain compared to the number of PETs with only one end located within the domain. The global enrichment score is the mean value of all enrichment scores for individual domains. Heatmap was plotted of the upper triangular matrix of the average matrix by default. The analysis was implemented in the cLoops2 agg module with the option of -domains (39), and default parameters were used in all related analyses in this manuscript.
Deep-learning model for classification of Hi-TrAC active sub-TADs against background regions with ChIP-seq data
A deep-learning model was implemented based on 85 shared transcription factor or histone modification ChIP-seq datasets between GM12878 and K562 from ENCODE (40). For the classification of Hi-TrAC active sub-TADs against background regions, background regions were defined as the same sized flanking regions as active sub-TADs but not overlapping with any of them. ChIP-seq reads were quantified as RPKM values for active sub-TADs and background. All data were separated randomly into the training set, validation set, and test set, with a ratio of 0.8:0.1:0.1. The training set was used to train the model, and the model was selected by the lowest loss observed for the validation set. The test set was finally used to evaluate the performance of the model. Briefly, the model was built with the sequential connected Keras (43) neuron network layers of Dense, BatchNormalization, rectifier activation, Dropout, and finally, a Dense layer for outputting classification probability of being putative domain. The categorical cross-entropy loss and the Adam optimizer were used to train the model, with the settings of accuracy as metric and an early stop for avoiding overfitting. The scheme of the model was summarized in Supplementary Figure S5B. The high accuracy of the classification model should capture the important latent features for distinguishing Hi-TrAC active sub-TADs from the background with ChIP-seq data. Therefore, the estimation for feature importance was performed for each factor by shuffling the RPKM values for all regions 100 times. Then the mean value of decreased accuracy was used as the feature importance. The deep-learning-related functions were powered by the Keras module in TensorFlow (44), and ROC curves were powered by the scikit-learn (45). The scheme of the model-based feature importance evaluation was summarized in Supplementary Figure S5D. The code and data for the proposed model and feature importance evaluation method are deposited at GitHub and available with the link of https://github.com/YaqiangCao/Hi-TrAC_active_subTADs_supplemental.
Calling cell- or condition-specific active sub-TADs
Segregation scores of active sub-TADs were quantified both in GM12878 and K562 with the cLoops2 quant module (39), and segregation score difference >1 was used to get GM12878 specific active sub-TADs. K562-specific active sub-TADs were called in the same way. Significantly lost active sub-TADs after knockdown of CTCF or RAD21, or combined were called in the same way.
GO terms enrichment analysis
Gene Ontology (GO) terms enrichment analysis for genes was performed by findGO.pl in the HOMER package (46), requiring more than ten overlapping genes in the terms, and there are fewer than 1000 genes in the terms. Only top enriched terms sorted by ascending P-values were shown.
Establishing CTCF-AID and domain boundary deletion cells
CTCF-AID and active sub-TAD boundary deletion K562 cells were generated using the CRISPR/Cas9 system. The targeting sequences were cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 vector (Addgene, #62988). For CTCF-AID cells, K562 cells were co-transfected with CRISPR and donor plasmids, which contain Hygromycin selection marker. After 24 h, cells were treated with 2 μg/ml Puromycin and 200 μg/ml Hygromycin for 48 h to remove non-transfected cells. Surviving cells were selected further by treating with 200 μg/ml Hygromycin for two weeks. The survived cells were sorted into 96-well plate with one cell per well and cultured for two to three weeks, then genotyped by PCR, western blotting, and sequencing. For generating active sub-TAD boundary deletion cells, K562 cells were transfected with CRISPR plasmids. After 24 h, cells were treated with 2 μg/ml Puromycin for 48 h to remove non-transfected cells. Survived cells were sorted into 96-well plate with one cell per well, and further cultured for two to three weeks. Cell clones were genotyped by PCR and sequencing. Targeting sequences are: CTCF, AGAAGTCCTGGCGACGCACA; BHLHE40, 1-TACTATCTATAGTAACTCCC, 2-TACCAGACTTCCACCGTATC; GATA1, 1-GGGTCCTCCCGACAATCCTC, 2-TTCCGGGCACTCTGACTATA; LMO2, 1-TTTAAGTGGAAGGGCCATAG, 2-TCACAACATACCTCGATGAT.
Auxin inducible degradation of CTCF-AID and western blotting
CTCF-AID K562 cells were infected with empty or OsTIR1 retrovirus. After 72 h, cells were treated with 500 μM auxin IAA for 6 h, and then sorted for detecting the effects of depleting CTCF. The depletion efficiency was examined by western blotting. Primary antibodies used for detecting corresponding proteins were: anti-CTCF (Cell Signaling Technology, 3418S, dilution 1:1000), anti-OsTIR1 (MBL Life Science, PD048, dilution 1:1000), anti-β-Actin (Santa Cruz, sc-47778 HRP, dilution 1:5000).
Generation of mouse Th17 cells
Mll4 fl/fl CD4-Cre+ mice were described previously (47). Naive CD4+ T cells were purified from the lymph nodes of Mll4+/+CD4-Cre+ and Mll4fl/flCD4-Cre + mice with EasySep Mouse CD4 + T cell Isolation Kit (StemCell, #19852). Cells were cultured in Th17 differentiation medium (2 μg/ml anti-CD28, 10 μg/ml anti-IL4, 10 μg/ml anti-IFNg, 10 μg/ml anti-IL12, 10 ng/ml TGFβ, 20 ng/ml IL6, 10 ng/ml IL1β) on plates coated with anti-CD3 and anti-CD28. Cells were collected by cell sorting with DAPI on 24 and 72 h for RNA-seq, ChIP-seq and Hi-TrAC.
ChIP-seq, RNA-seq and Hi-TrAC assays
Hi-TrAC was performed as described previously (38). Briefly, cells were fixed with 1% Formaldehyde at room temperature for 10 min. The biotinylated linker for bridging interacting chromatin was inserted into the genome by Tn5. After reverse crosslinking, genomic DNA was purified, and with gaps repaired. After digesting with MluCI and NlaIII restriction enzymes, biotinylated DNA fragments were enriched with streptavidin beads. Universal adapters were ligated to DNA fragments, and the libraries were amplified by PCR with Illumina Multiplexing primers.
ChIP-seq assays were performed as described previously (48,49). In brief, cells were fixed with 1% formaldehyde at room temperature for 10 min. Chromatin was sonicated and immunoprecipitated with anti-H3K4me1(ab8895, Abcam), anti-H3K4me3 (17-614, Millipore) and anti-H3K27ac (ab4729, Abcam) antibodies. Purified ChIP DNA was repaired with End-It DNA End-Repair Kit (Epicentre). The library was then indexed and amplified and sequenced on an Illumina platform. For RNA-seq, RNA from 5000 cells were purified with QIAzol lysis reagent (QIAGEN) and RNeasy mini kit (QIAGEN). The libraries were then constructed following Smart-Seq2 method (50).
ChIP-seq, RNA-seq and Hi-TrAC data analysis
Mouse sequencing data were processed to the reference genome mm10, and human data were processed to hg38 in this manuscript.
ChIP-seq data were mapped by Bowtie2 (v.2.3.5) (51). Only non-redundant reads with MAPQ ≥10 were used for the following analysis. Sample-wise genome-wide correlation analysis was performed with multiBigwigSummary bins and parameter -bs 1000 in deepTools2 (v3.3.0) package (52). Peaks were called for each ChIP-seq library first with the cLoops2 callPeaks module (39); specific parameters of -eps 300,500 -minPts 10,20 -sen were used for H3K4me1 datasets, and -eps 150,300 -minPts 10,20 were used for H3K4me3 and H3K27ac datasets. Overlapped regions from the same histone modification's biological replicates and wild-type or Mll4 knockout conditions were compiled together as union sets (Supplemental Table S8). The overlapped peaks of H3K4me1, H3K4me3, and H3K27ac were assigned as putative genomic segments with the following criteria (Supplemental Table S8): (i) H3K4m3 overlapped H3K27ac peaks at the transcription start site (TSS) as active TSS first; (ii) H3K4me3 peaks without H3K27ac peaks at TSS as poised TSS; (iii) H3K27ac peaks at TSS distal region as active enhancers; (iv) H3K4me1 only peaks at TSS distal region as poised enhancers; (v) finally, other parts of H3K4me1 peaks overlapped with above segments but not totally covered as the flank regions. Super-enhancers were called based on wild-type H3K27ac ChIP-seq data and peaks with ROSE (53).
RNA-seq data were mapped with STAR (v2.7.3a) (54) and quantified with Cufflinks (v2.2.1) (55).
Hi-TrAC raw data were pre-processed into highly-quality non-redundant paired-end tags (PETs) with tracPre2.py in the cLoops2 package. The cLoops2 estSim module performed correlation analysis among replicates, and the cLoops2 estRes module carried out resolution estimations of pooled PETs. The cLoops2 callDomains module called active sub-TADs with parameters of -bs 1000 -ws 25000 -mcut 1000000 -strict. Significantly decreased domains comparing wild-type and Mll4 knockout cells were called in the same way as human data with the same segregation score difference threshold of 1.
Data visualizations
Most of the tracks were shown by the cLoops2 plot module. Other plots were generated by the matplotlib (56) and seaborn (57) with in-house code.
Code available
Segregation score based domain-calling algorithm was coded as the cLoops2 callDomains module, domain aggregation analysis was coded in the cLoops2 agg module, domain quantification was coded in the cLoops2 quant module, insulation score calculation was summarized as getIS.py in the cLoops2 package as a script, segregation score calculation was summarized as getSS.py in the cLoops2 package as a script for using data besides Hi-TrAC, all these codes are available at: https://github.com/YaqiangCao/cLoops2.
RESULTS
Segregation scores reveal TADs-like domains from hi-TrAC data
Insulation scores (28,58) are widely used to obtain domain boundaries from Hi-C and its variant data by transforming 2D contact matrix into one-dimensional signals. Based on the insulation scores, the resulting domains are dependent on the critical parameters of resolution, sliding window size, and the cutoff of insulation score to define the boundaries and combinations of boundaries to dictate domains. Especially when comparing different samples, parameter tuning may be further needed. In addition to these constraints, we found that the insulation scores calculated from the Hi-TrAC data failed to accurately indicate the boundaries with the local minimal values. To overcome such limitations, we proposed the segregation score to identify putative domains using Hi-TrAC data (Materials and Methods) (Supplementary Figure S1A). These two approaches shared the same idea of transforming two-dimensional matrix into one-dimensional signals. Unlike the insulation score, with which the local minimal score indicates the domain boundary, genomic regions with positive segregation scores were stitched together as the chromatin domains from the Hi-TrAC data (Supplementary Figure S1A and B). Two other metrics were required to define a putative Hi-TrAC domain: (1) more interacting paired-end tags (PETs) within the putative domain than the PETs with only one end located within the domain (enrichment score (ES) > 1); (2) two-folds higher interaction density than chromosome-wide interaction density.
To test this method, we performed the domain-calling algorithm based on the segregation score at a resolution of 10 kb in GM12878 Hi-TrAC data. The resulting domains were TAD-like and visually similar with the TADs from Hi-C data (21) (Supplementary Figure S1B and C), suggesting that the segregation score is reliable for detecting domains from Hi-TrAC data. We also noticed more explicit internal organizations than Hi-C data in these Hi-TrAC domains (Supplementary Figure S1B and C), demonstrating the ability of Hi-TrAC data to reveal fine-scale structures of smaller domains at higher resolution. Meanwhile, the segregation score and insulation score had a similar trend around domain boundaries both for Hi-TrAC and Hi-C data (Supplementary Figure S1B and C), but also had the different signs of values for some regions (Supplementary Figure S1B and C black arrows). Subtle differences of the two scores were observed at boundaries for Hi-TrAC data with a resolution of 10 kb (Supplementary Figure S1B, gray dash lines versus solid black lines for putative domain marked as c). To systematically evaluate the difference between the two scoring strategies, we further calculated the scores at the resolution of 1 kb in Hi-TrAC data to study subtle differences around boundaries. Genomic regions with positive segregation scores were stitched together as putative domains and boundaries were not included. Therefore, the upstream 1 kb bin of left boundaries and the downstream 1 kb bin of the right boundaries were aligned at the summit of chromatin accessibility peaks detected by ATAC-seq or boundary elements bound by CTCF and cohesin (Supplementary Figure S1D). Meanwhile, the minimum of aggregated segregation scores was closer to the aggregated summit of ATAC-seq or ChIP-seq peaks than insulation scores (Supplementary Figure S1D), and the minimum of aggregated insulation scores was nearly outside of the aggregated peaks, indicating insulation scores may not accurately mark the boundaries from Hi-TrAC data at high resolution. Even though the two scores showed a higher genome-wide correlation with Hi-TrAC data than with Hi-C data (Supplementary Figure S1E), the local minimal values were aligned at the valley for putative boundaries, both shown from the Hi-C example region (Supplementary Figure S1C) and globally (Supplementary Figure S1F), indicating the segregation scores can be used to call boundaries for Hi-C data.
We noticed that segregation scores calculated at high resolution as bin size set to 1 kb were highly consistent between Hi-TrAC biological replicate experiments both for GM12878 and K562 cells (Supplementary Figure S1G, Pearson correlation coefficient > 0.9). Further, the called putative domains were highly overlapped (88% for fewer domains called from the replicate experiments) (Supplementary Figure S1H), indicating the robustness of the proposed method for domain calling of Hi-TrAC data.
Hi-TrAC detects active sub-TADs
To check whether modestly sequenced Hi-TrAC data (GM12878: 116 million unique intra-chromosomal PETs; K562: 95 million unique intra-chromosomal PETs) can reveal smaller domains at an even higher resolution than Hi-C, we performed the domain-calling at a resolution of 1 kb. Among the top-ranked Hi-TrAC domains in GM12878 sorted by segregation scores (descending ranking), we found multiple intriguing features (Figure 1A and B): (i) domain sizes are smaller than 200 kb; (ii) top domains contain super-enhancers; (iii) a block-to-block interaction pattern exists within and between super-enhancers (in contrast to the typical dot-to-dot loop Hi-TrAC pattern) (38) and (iv) top-ranking domains harbor important functional genes in B cells. Since the GM12878 cell line is a B lymphocyte cell line transformed by the Epstein-Barr virus, these results suggested that domains may harbor the cell identity genes for GM12878, which were exemplified by the genomic locus of B cell proliferation- and differentiation-related genes such as BTG2 (59) and IKZF3 (60) (Figure 1A and B).
Figure 1.

Identification of active sub-TADs by Hi-TrAC. (A) Example of active sub-TAD containing the BTG2 gene locus detected by Hi-TrAC in GM12878 cells. The cLoops2 callDomains module called domains at 1kb resolution (Materials and Methods). The first exon of a gene and its name in the positive strand are indicated by blue color, and the first exon of a gene and its name in the negative strand are indicated by purple color. ENCODE (40) H3K27ac ChIP-seq profiles and the Hi-TrAC domain segregation scores are displayed below the genomic annotations. The filled black rectangle indicates the super-enhancers within the active sub-TADs. Hi-TrAC interaction matrix was shown at 500 bp resolution. The interaction domain is marked as blue dotted frames on the heatmap. The plot was generated by the cLoops2 plot module. (B) Example of active sub-TAD containing the IKZF3 gene locus detected by Hi-TrAC in GM12878 cells. (C) Aggregation analysis of 820 active sub-TADs identified from the Hi-TrAC data in GM12878 cells (Supplemental Table 2). Together with 0.5-fold neighboring upstream and downstream regions, all the Hi-TrAC active sub-TADs were aggregated for visualization (Materials and Methods). ES stands for enrichment scores of interacting PETs within the domains compared to the PETs with one end in the domain and the other outside the domain. The enrichments of various ChIP-seq signals were calculated based on ENCODE ChIP-seq data (40). The analysis was performed with the cLoops2 agg module. (D) Aggregation analysis of 1759 active sub-TADs identified from the K562 Hi-TrAC data (Supplemental Table 2). (E) Aggregation analysis of Hi-TrAC active sub-TADs with CTCF ChIA-PET data (24), RAD21 ChIA-PET data (68), H3K27ac HiChIP data (69) and in situ Hi-C data (21) in GM12878 cells.
In total, we identified 820 domains with a median size of 93 kb in GM12878 (Figure 1C and Supplemental Table S2) and 1759 domains with a median size of 87 kb in K562 (Figure 1D and Supplemental Table S2). As expected, both chromatin domain-boundary associated factors CTCF (18) and Cohesin complex core subunit SMC3 (61,62) were enriched at Hi-TrAC domain boundaries (Figure 1C and D), indicating their roles in domain establishment or maintenance. As these domains were identified in Tn5 accessible chromatin regions, they carried expected active chromatin features such as elevated H3K4me1, H3K4me2, H3K4me3 and H3K27ac signals inside the domain, as well as decreased H3K27me3 signals compared to flanking regions. Interestingly, H3K4me3 signals in both GM12878 and K562 were enriched at the boundaries, consistent with the observation that domain boundaries are enriched with promoters of actively transcribed genes, suggesting that certain promoters may also have boundary function (22,23,63,64). The median size of sub-TADs is typically around 180 kb (21), and insulated neighborhoods, one of its sub-classes, has a similar size of approximately186 kb (65). Previously identified supercoiling domains are around 100 kb (66), coinciding with the size of recently identified chromatin nanodomains through super-resolution microscopy in single cells (14,67). Even though the domain sizes (around 100 kb) detected by Hi-TrAC were smaller than the size of sub-TADs (180 kb) and were more similar to supercoiling domains and chromatin nanodomains, they still fell within the same size magnitude. Additionally, Hi-TrAC domains were identified in a similar way to sub-TADs via computational analysis of sequencing data, and therefore we coined them active sub-TADs.
We further validated Hi-TrAC active sub-TADs using the aggregation analysis of CTCF ChIA-PET data (24), RAD21 ChIA-PET data (68) and H3K27ac HiChIP data (69), but not the in situ Hi-C data (21) due to its limited resolution (Figure 1E). The aggregated dot-to-dot interaction patterns at the active sub-TAD boundaries observed from the CTCF and RAD21 ChIA-PET data indicate that Hi-TrAC active sub-TADs may also be formed through the loop extrusion mechanism (33,70), which is similar to the establishment of insulated neighborhoods (65,71), albeit at a smaller scale. We compared these data around a Hi-TrAC active sub-TAD containing the genomic locus of the TMSB4X gene in GM12878 (Supplementary Figure S2A and B), further validating the observation that other techniques may detect the active sub-TADs but not the fine structure within sub-TADs and their boundaries as revealed by Hi-TrAC. Together, our results indicate that Hi-TrAC is superior in detecting active sub-TADs and their fine structures in comparison with other methods.
Hi-TrAC reveals internal organizations of super-enhancers
To follow up on our observation that the active sub-TADs with the high segregation scores in GM12878 contained super-enhancers (Figure 1A and B), we performed more analysis for the association between Hi-TrAC active sub-TADs and super-enhancers. We found that around 80% (204 of a total 257) and 56% (413 of a total 731), respectively, super-enhancers in GM12878 and K562 cells overlapped with active sub-TADs (Figure 2A). Additionally, active sub-TADs with super-enhancers had higher segregation scores, enrichment scores (defined as Supplementary Figure S1A), and interaction densities than the active sub-TADs without super-enhancers (Figure 2B), indicating these domains are more insulated and interactive. Even within the same active sub-TAD, there were block-to-block interaction patterns between component enhancers from both the same and different super-enhancers (Figure 2C). Zooming in on the example domains at a 500 bp resolution revealed an internal structure within the individual component enhancers of each super-enhancer (Figure 2D). This internal structure has not been directly observed before through contact matrix heatmaps due to limitations of resolution with Hi-C, CTCF ChIA-PET or H3K27ac HiChIP (Supplementary Figure S3A). Binding of SMC3 was enriched at some boundaries of the internal structure and thus Cohesin may be important for the genomic folding within the composite enhancers of super-enhancers (Figure 2D).
Figure 2.

Hi-TrAC reveals the internal structure of super-enhancers. (A) Overlaps of active sub-TADs and super-enhancers in GM12878 and K562 cells. (B) Comparisons of segregation scores, enrichment scores, and interaction densities between active sub-TADs with and without super-enhancers. The box extends from the quartile to the third quartile of the data, with a line at the median. The whiskers extend from the box by 1.5× the inter-quartile range. Flier points past the end of the whiskers were not shown. All box plots shown in this study all follow the same definition. (C) Example of active sub-TAD containing the ENTPD1 gene locus detected by Hi-TrAC in GM12878 cells. Super-enhancer is marked with ‘a’ and its composite enhancers are marked with ‘b’ and ‘c’. (D) Zoom-in visualization of the internal structure of super-enhancer marked with ‘a’ in panel (C). There is also a clear internal structure within composite enhancer ‘c’. (E) Estimated scaling of interaction probability and genomic distance for the composite enhancer ‘c’ in panel (D) with internal Hi-TrAC PETs and linear regression model. The red dots represent observed data, and the black line shows the linear regression fitting results. The observed internal Hi-TrAC PETs grouped into 150 bp bins as frequencies and are shown as red dots. (F) Distribution of estimated scaling factors of various super enhancers and background control regions. The red dashed line indicates the theoretical scaling value of –1.0 if the chromatin chain is folding into the fractal globule model, and the blue dashed line indicates the theoretical scaling value of –1.5 if the chromatin conformation is the equilibrium globule (17,74). Composite enhancers of super-enhancers with a size longer than 5 kb were manually classified into enhancers with internal structure (EWIS) and enhancers without internal structure (EOIS) through 200 bp resolution Hi-TrAC interaction heatmap visualization one by one. The regions upstream and downstream of EWIS (10-fold size distance away from the EWIS) were defined as background (bg) for comparison. Individual estimated scaling factors were plotted and overlayed with the box plots as dots. The two-sided Mann–Whitney U rank test was used to draw P-values comparing different groups.
To further study the internal organizations of super-enhancer components in GM12878 cells, we manually classified those component enhancers longer than 5 kb (n = 410) into enhancers with internal structure (EWIS, n = 175, 42.68%) and enhancers without internal structure (EOIS, n = 235) through one-by-one visualization of the Hi-TrAC heatmap at 200 bp resolution (Supplemental Table S3). We also defined the genomic regions of same sizes either upstream or downstream to the EWIS ten-folds of the size away as background for comparison. The polymer-based equilibrium globule (72,73) and fractal globule (17,74,75) models were proposed to understand and simulate the chromatin conformation. Simulation of the fractal globule model and equilibrium globule model of chromatin fiber folding reveal distinct scaling values of –1.0 and –1.5, respectively, which is the slope of linear fitting of the interaction probability as a function of genomic distance (17). The scaling of the initial Hi-C data at the range from 500 kb to 7 Mb fitted well the fractal globule model (17). We sought to explore the scaling for individual EWIS with the internal interacting PETs from Hi-TrAC data (Figure 2E) and compare the scaling distributions with EOIS and background (Figure 2F). Interestingly, the scaling property was significantly different between the three groups (Figure 2F). The scaling of the background regions is –1.009, close to a theoretical fractal globule structure as previously proposed based on the Hi-C data. Meanwhile, the scaling of EWIS regions is –1.390, significantly lower than that of EOIS (–1.243) and background (Figure 2F), indicating that EWIS has properties closer to the equilibrium model and EOIS may be an intermediate state between the fractal globule model and the equilibrium globule model. Even though Hi-C and H3K27ac HiChIP data may not show the internal organization as clearly as Hi-TrAC (Supplementary Figure S3A), they displayed the same trend and difference of scaling between EWIS compared with EOIS and EWIS compared with background (Figure 2F). The internal interaction densities measured by Hi-TrAC were significantly higher for EWIS compared with EOIS and background; however, the external interaction densities with other regions for EWIS were not higher than EOIS (Supplementary Figure S3B). Hi-C and H3K27ac HiChIP data also validated the significant difference between EWIS and EOIS for internal interaction densities and the small external difference (Supplementary Figure S3B).
In summary, our data show that there are internal organizations for component enhancers of some super-enhancers. The chromatin folding displayed distinct patterns at different functional regions. While the background regions (mostly inaccessible chromatin regions) may exist as a ‘fractal globule’ structure, super-enhancers with internal structures have properties closer to an ‘equilibrium globule’ structure and enhancers without internal structures may be at an intermediate state between the ‘fractal globule’ and ‘equilibrium globule’ structures.
Active sub-TADs associated epigenetic features
Over 60% of active sub-TADs contained fewer than two genes (Figure 3A), which exhibited significantly higher expression levels than other genes (Figure 3B). This implies that a substantial number of highly transcribed genes are regulated within their own gene-specific domains. Gene ontology (GO) analysis showed that the genes harbored in active sub-TADs were related to regulation of T cell activation, regulation of hemopoiesis, lymphocyte differentiation, and positive regulation of cytokine production, indicating that these domains may mark specific GM12878 cells’ cellular identity and functions as B cells (Figure 3C). Together, these results suggest that active sub-TADs may play important roles in regulating and determining cell function.
Figure 3.

Properties of Hi-TrAC detected active sub-TADs. (A) Distribution of numbers of genes in an active sub-TAD. (B) Distribution of expression levels for the genes located in the active sub-TADs and background regions. RNA-seq data were obtained from GSE30567 (98). Background regions were defined as the same sized regions as sub-TADs, being located either upstream or downstream and not overlapping with any active sub-TADs. Two-sided Wilcoxon rank-sum test was used to calculate P-values. (C) Gene ontology (GO) analysis for genes located within the Hi-TrAC active sub-TADs in GM12878 cells. The top 5 enriched GO terms are shown. (D) Chromatin interaction changes revealed by Hi-TrAC after deleting the left boundary of the BHLHE40 active sub-TAD in K562 cells. Two replicates of Hi-TrAC libraries (Supplemental Table 4) were pooled and down-sampled to 33 million cis unique PETs for both the CRISPR/Cas9 control and boundary deletion samples. Displayed are the Hi-TrAC pileup 1D profiles and interaction matrices. The region of the BHLHE40 active sub-TAD is indicated by the blue dashed line square, and the scissor symbol indicates the deleted left boundary region by the CRISPR/Cas9. The black circles indicate the loops originating from the left boundary, which are decreased after the boundary deletion. (E) Interaction changes within the BHLHE40 active sub-TAD resulting from the boundary deletion. The pileup 1D profiles and interaction matrix difference from the Hi-TrAC data are displayed. The heatmap was generated by the plotDiffHeatmap.py script in the cLoops2 package. The region with a clear decrease in interaction is indicated by the black line and red arrow. (F) Genome browser images of RNA-seq expression from the control and BHLHE40 active sub-TAD boundary deletion cells. Fold change (deletion/control) and P-value for BHLHE40 were generated by Cuffdiff (55). (G) Scheme and receiver operating characteristic (ROC) curves for the performance of classification model for the identification of important features of active sub-TADs using ENCODE ChIP-seq data for GM12878 and K562 (Materials and Methods). All data were separated into training, validation, and test sets (8:1:1), with training data used to train the model, validation data used to select the model, and test data used to evaluate the model's final performance. (H) Top ten most important features associated with the active sub-TADs. (I) Aggregation analysis for ChIP-seq signals of the top important features in the active sub-TADs and nearby background regions.
To evaluate the regulation of gene expression by active sub-TADs, we deleted the boundaries of domains containing the BHLHE40, GATA1 and LMO2 genes in K562 cells using CRISPR/Cas9 (Supplementary Figure S4A). These target boundaries are located distally from the target gene promoters and bound by CTCF and RAD21 (Supplementary Figure S4A). We generated Hi-TrAC libraries to examine the chromatin interaction alterations resulting from the boundary deletions (Supplemental Table S4). Remarkably, deleting the left boundary of the BHLHE40 active sub-TAD led to a decrease in both internal and external chromatin interactions anchored at the boundary (Figure 3D and E), particularly at proximal accessible sites within the domain (Figure 3E), accompanied with a significant decrease in BHLHE40 and upstream ITPR1 gene expression (Figure 3F and Supplementary Figure S4B), suggesting that this CTCF/Cohesin-bound boundary acts to facilitate nearby enhancer-promoter interactions as we proposed previously (76). By comparison, the deletion of the left boundary of the GATA1 active sub-TAD only led to a modest change in a proximal accessible site (Supplementary Figure S4C black arrow), accompanied with modest changes in interactions and no notable changes in gene expression (Supplementary Figure S4C). The deletion of the right boundary of the LMO2 active sub-TAD led to a decrease of the intra-domain interactions, which was accompanied by a decreased in the expression of LMO2 (Supplementary Figure S4D). However, increased interactions were detected in the adjacent domain containing the CAPRIN1 gene, which was accompanied by increased expression of CAPRIN1 (Supplementary Figure S4D), suggesting that this boundary acts as an insulator to restrain the regulatory elements to their appropriate chromatin domains. In conclusion, these results demonstrate that active sub-TADs play crucial roles in regulating proper gene expression by maintaining a suitable chromatin micro-environment and that alterations in their boundaries can lead to intricate changes in chromatin interactions and gene expressions.
To further study the epigenetic features associated with active sub-TADs, which may provide information on the potential regulators for these active sub-TADs, we quantified the enriched signals of ENCODE (40) ChIP-seq data of 85 factors (shared between GM12878 and K562) in active sub-TADs and flanking background regions (Supplementary Figure S5A) in both GM12878 and K562 cells (Supplemental Table S1). These 85 factors included histone modifications and transcription factors (TFs). We implemented a deep learning model to investigate whether epigenetic markers and TFs ChIP-seq signals have the classification power to distinguish the active sub-TADs from the flanking backgrounds (Supplementary Figure S5B) (Methods). If the model is accurate, it should be able to capture the important latent features and assign the high-value weights for them, which could then reveal important regulatory factors. After training, the model was able to classify active sub-TADs against background based on 1D ChIP-seq information (Supplementary Figure S5C and Figure 3G). Even for the test data (10% of all data) that had never been previously used in model training or validation, the classification accuracy remained reasonably high (0.911: the percentage of correctly classified items in the total item count) (Supplementary Figure S5C). The areas under the receiver operating characteristic curve (AUCs) were higher than 0.96 throughout training, validation, and test datasets, indicating that the model was properly trained, not overfitted, and highly reliable (Figure 3G). Equipped with high accuracy, the model can reliably reveal important features associated with active sub-TADs (Supplementary Figure S5D) (Materials and Methods). According to the classification model, H3K4me1 is the most important feature in classifying active sub-TADs against background (Figure 3H). SMC3 ranks top 2 as a known chromatin domain associated factor. Other top features including active histone marks H4K20me1(top 5), H3K9ac (top 10) and transcription factors RCOR1, YBX1, MTA2 and LARP7 exhibited elevated signals in the active sub-TADs and at the boundaries (Figure 3I). However, it is unclear whether the enrichment of these features in these active sub-TADs is causative or simply correlative (Figure 3I).
Cell-specific active sub-TADs harbor cell identity genes
To study the cell specificity of active sub-TADs, we compared active sub-TADs detected in GM12878 and K562 cells. We identified 538 (65% of a total 820) GM12878 specific active sub-TADs, and 818 (46% of a total 1759) K562 specific active sub-TADs based on the difference in segregation scores (Figure 4A, Supplemental Table S5, Materials and Methods). Cell-specific active sub-TADs in their respective cells showed higher signals of the top ten important features identified from the classification model (Figure 4A). The identification of these cell-specific sub-TADs was validated using differential aggregation analysis of the unbiased Hi-C data, and feature-selective H3K27ac HiChIP and RAD21 ChIA-PET data (Figure 4B). Our results also showed that genes located in cell-specific active sub-TADs have higher expression levels in their corresponding cell type (Figure 4C). Consistent with a B cell transformed cell line, the top enriched KEGG terms for the genes in the cell-specific active sub-TADs in GM12878 cells contained genes involved in the B cell receptor signaling pathway (log(P-value) = –18.2). For K562, a chronic myelogenous leukemia cell line, one of the most enriched GO terms was myeloid leukocyte mediated immunity (log(P-value) = –17.7). This was further exemplified by PLCG2, a gene critical for B cell signaling (77), which had a super-enhancer and was specifically expressed in an active sub-TAD only detectable in GM12878 cells (Figure 4D). The leukemia-associated gene PIM1 (78), expressed only in K562, was localized to a cell-specific active sub-TAD containing a super-enhancer spanning almost the entire sub-TAD (Figure 4D). Although the data from other methods, such as Hi-C and H3K27ac HiChIP, could validate the cell specificity of sub-TADs using aggregate analysis (Figure 4B), they were unable to achieve high resolution in interaction heatmaps of specific genomic loci as shown by Hi-TrAC (Supplementary Figure S6). In summary, our results demonstrated that the specific cell-specific active sub-TADs detected by Hi-TrAC are chromatin structures critical to regulating cell identity and activity.
Figure 4.

Hi-TrAC detects cell-specific active sub-TADs. (A) Aggregation analysis of cell-specific active sub-TADs in GM12878 and K562 cells (Supplemental Table 5). Also shown are the top 10 most important features identified from the classification model. (B) Aggregated domain analyses for Hi-TrAC detected cell-specific active sub-TADs using in situ Hi-C (top panel), H3K27ac HiChIP (middle panel), and RAD21 ChIA-PET data (lower panel). (C) Distribution of expression levels for the genes located within the cell-specific sub-TADs identified by Hi-TrAC. The blue box shows the genes of GM12878 cell-specific sub-TADs, and the green box shows that of K562, respectively. (D) Examples of GM12878 and K562 specific active sub-TADs.
Active sub-TADs are disrupted by knocking down RAD21
To test whether CTCF or Cohesin contributes to the maintenance of active sub-TADs, we further analyzed the Hi-TrAC and Hi-C data from K562 cells knocked down of CTCF or RAD21, a major cohesin subunit, using shRNAs. The results from the Hi-TrAC and Hi-C samples showed that knocking down RAD21 substantially impaired chromatin interactions up to hundreds of kb (Figure 5A and B), suggesting that the changes occur mainly at domain levels, including TADs and sub-TADs. Additionally, the changes in interaction distance were consistent with previous results from Cohesin-depleted Hi-C data in which loop domains were eliminated but compartment domains remained (79). However, knocking down CTCF did not show the impairment of loop domains as caused by knocking down RAD21 (Figure 5A and B). Taken together, the changes in aggregated segregation scores from Hi-TrAC data (Figure 5C) and aggregated differential interaction contact matrix from both Hi-TrAC and Hi-C data (Figure 5D) indicated that knocking down RAD21 leads to global disruption across Hi-TrAC sub-TADs. Analysis of segregation scores and interaction matrix from Hi-TrAC data revealed that knocking down RAD21 resulted in decreased intra-domain interactions (Figure 5C) and noticeably blurred domain boundaries (Figure 5D). Compared to the shRNA control sample, significantly impaired active sub-TADs by knocking down CTCF, RAD21 or both CTCF and RAD21 were called (Supplemental Table S6) and checked for overlaps with each other (Figure 5E) and overlaps with super-enhancers (Figure 5F). This showed that RAD21 plays a much more critical role than CTCF in maintaining the active sub-TADs and in the potential role of super-enhancers. As exemplified by the leukemia oncogene LMO2 (80) gene locus (Figure 5G), Hi-TrAC was able to detect changes in active sub-TADs caused by the different knockdowns. Additionally, the resulting five-fold decrease in LMO2 expression indicated that active sub-TADs may also play an important role in regulating gene expression (control RPKM: 13.5 versus RAD21 KD RPKM:2.3, averages of two replicates, data included in the supplemental table of (38)). In T cell acute lymphoblastic leukemia, the proto-oncogenes LMO2 was found to be silent and insulated from active enhancers, and removal of the boundary not only enhanced its chromatin interactions with distal region, but also activated its expression in HEK-293T cells (13). As low levels of CTCF are thought sufficient to maintain TADs (81,82), we further tagged CTCF with the auxin-inducible degron (AID) (83) system. Expression of osTIR1 and auxin treatment of the resulting cells rapidly depleted the CTCF-AID protein (Figure 5H and I). Hi-TrAC libraries were generated from these cells (Supplemental Table S4), and analysis of the Hi-TrAC data showed no significant changes in the genomic distance distribution of interacting PETs (Figure 5J), in the aggregation of segregation scores of the active sub-TADs (Figure 5K), or at the active sub-TAD containing the LMO2 gene (Figure 5L). These results were consistently with the shRNA results. In summary, our results collectively demonstrated that RAD21 but not CTCF plays a more important role in maintaining the active sub-TADs detected by Hi-TrAC. Additionally, it is also possible that Cohesin plays a role in maintaining enhancer-promoter looping for proper gene expression control (68) and thereby preserving the active sub-TAD structures.
Figure 5.

Active sub-TADs are disrupted by knockdown of RAD21. (A) Distribution of frequencies of interacting PETs against genomic distances of Hi-TrAC data in wild-type and various knockdown K562 cells. KD: knockdown; dKD: double knockdown. (B) Distribution of frequencies of interacting PETs against genomic distances of Hi-C data in wild-type and various knockdown K562 cells. (C) Aggregation analyses of segregations scores of sub-TADs identified from Hi-TrAC data after knocking down CTCF or RAD21 either alone or in combination in K562 cells. (D) Aggregation analyses of interaction matrix of sub-TADs identified from Hi-TrAC data after knocking down CTCF or RAD21 either alone or in combination in K562 cells with Hi-TrAC (upper panel) or Hi-C data (lower panel). (E) Overlaps of significantly changed K562 Hi-TrAC active sub-TADs after knocking down CTCF or RAD21 alone or in combination. (F) Overlaps of significant changed K562 Hi-TrAC active sub-TADs with super-enhancers. (G) Example of disrupted active sub-TADs in K562 by knocking down RAD21 at the LMO2 gene locus. SEs stands for super-enhancers. (H) CTCF protein was rapidly degraded by expression of osTIR1 and auxin treatment in K562 cells as measured by Western blotting. (I) Genome browser images of RNA-seq expression from the control and CTCF-AID K562 cells. Fold change (AID/control) and P-value for CTCF were reported by Cuffdiff (55). CTCF RNA level was not affected by tagging with AID. (J) The frequency distribution of interacting PETs against genomic distances in Hi-TrAC data shows no difference between control and CTCF-depleted cells as the two lines overlap. Two biological replicates, each with three technological replicates of Hi-TrAC PETs (Supplemental Table 4) were combined and down-sampled to 54 million cis unique PETs each for both the control and CTCF-AID cells. (K) The aggregation analysis of the segregation scores of K562 active sub-TADs in control and CTCF-depleted cells showed that the two lines overlapped, and no significant difference was detected. (L) An example of preservation of active sub-TADs at the LMO2 gene locus in the CTCF-depleted cells.
Interactions of active sub-TADs are decreased by deleting mll4 in mouse Th17 cells
Our prediction model suggested that H3K4me1 is the most enriched feature associated with the Hi-TrAC active sub-TADs (Figure 3H), and thus we decided to further investigate whether H3K4me1 contributes these structures. MLL4, also known as KMT2D, is a primary H4K4 mono- and di-methyltransferase in mammalian cells and its deletion significantly decreases H3K4me1 levels on enhancers in T cells (47). Thus, we performed RNA-seq, ChIP-seq for H3K4me1, H3K4me3 and H3K27ac, and Hi-TrAC analyses of T helper 17 (Th17) cells from wild-type and Mll4 conditional knockout mice (Supplemental Table S7). The deletion of Mll4’s exons was validated by the RNA-seq data (Supplementary Figure S7A). While the global patterns of the histone marks were similar between wild-type and Mll4 deletion cells (Supplementary Figure S7B), enhancers showed significantly decreased histone modification signals in the Mll4 deleted cells (Supplementary Figure S7CSupplemental Table S8). Based on the Hi-TrAC interaction signals, wild-type and Mll4 deleted cells were clustered into different groups with either 1Kb or 5Kb resolution (Supplementary Figure S7D). With the limited sequencing depth, both pooled wild-type and Mll4 KO Hi-TrAC data showed the highest estimated genome-wide resolution as 1 kb (Supplementary Figure S7E). By examining different regulatory regions, we found that chromatin interactions showed the highest decreases at putative enhancers (Supplementary Figure S7F). By randomly checking the genomic regions, we noticed that H3K4me1 and H3K4me3 marks aligned well at TAD-like regions, and decreased interaction densities may happen at around 200 kb scale (Supplementary Figure S7G). The size coincided with the size of active sub-TADs, indicating that Hi-TrAC has the detection sensitivity for individual active sub-TAD with subtle interaction changes.
We called active sub-TADs from Th17 cells based on the wild-type Hi-TrAC data (Materials and Methods and Supplemental Table S9). The Rorc locus, encoding the Th17 master transcription factor RAR-related orphan receptor gamma (RORγt) (84), was located in a ∼100 kb active sub-TAD (Figure 6A), which is consistent with our observation that active sub-TADs harbor cell identity genes in human cells. In total, we identified 1427 active sub-TADs with a median size of 82 kb (Figure 6B). KEGG enrichment analysis for the genes contained within these active sub-TADs revealed interesting terms including Systemic lupus erythematosus (SLE), Th17 cell differentiation, and T cell receptor signaling pathway. These terms are related to Th17 cell differentiation, function, or potential diseases (85–87), further supporting our observation in human cells that active sub-TADs may play a regulatory role in cell functions.
Figure 6.

Interactions within active sub-TADs are decreased by deletion of Mll4 in mouse Th17 cells. (A) The Rorc genomic locus, encoding a master transcription factor in Th17 cells, is located in an active sub-TADs. ChIP-seq and Hi-TrAC data were generated in this study (Supplemental Table 7). ChIP-seq signals were the average values from two replicates, and Hi-TrAC data were pooled from three biological replicates (Supplemental Table 7). (B) Aggregation analysis of 1427 active sub-TADs identified from Hi-TrAC data in mouse Th17 cells (Supplemental Table 9). (C) KEGG terms enrichment analysis for genes located within the Hi-TrAC active sub-TADs in mouse Th17 cells. Only the top 5 enriched terms were shown. (D) Overlaps of active sub-TADs and super-enhancers in mouse Th17 cells. (E) Example of active sub-TAD containing the Irf4 gene locus detected by Hi-TrAC showing block-to-block interaction pattern between super-enhancers. Two super-enhancers were marked as ‘a’ and ‘b’. Super-enhancer ‘a’ was highlighted with a gray box to show its internal structure. (F) Correlation analysis between H3K4me1 ChIP-seq signal intensities and Hi-TrAC interaction densities for wild-type Th17 cells active sub-TADs. PCC stands for Pearson Correlation Coefficient. (G) Correlation analysis between the changes in H3K4me1 ChIP-seq signal intensity and Hi-TrAC interaction density in active sub-TADs after deletion of Mll4 Th17 cells. (H) Distribution of expression levels for the genes located within the active sub-TADs. (I) Aggregation analysis of significantly decreased active sub-TADs based on segregation scores in Th17 cells comparing wild-type and Mll4 knockout mice (Supplemental Table 9). (J) Example of a significantly decreased active sub-TAD harboring the Il6ra gene important for Th17 function.
We further called super-enhancers based on H3K27ac ChIP-seq data (Materials and Methods and Supplemental Table S8) and found 495 (68.18%) Th17 super-enhancers overlapped with active sub-TADs (Figure 6D), consistent with the result that more than half super-enhancers in human GM12878 and K562 cells were overlapped with active sub-TADs. We also observed block-to-block interactions between the two super-enhancers of the Irf4 gene locus (Figure 6E) and the internal structures within an individual super-enhancer (Figure 6E, gray box). The interactions measured by Hi-TrAC in active sub-TADs showed a high correlation with the level of H3K4me1 as the Pearson Correlation Coefficient (PCC) was 0.687 (Figure 6F). The decreases in H3K4me1 levels and interactions in the active sub-TADs resulting from Mll4 deletion were also highly correlated (PCC = 0.762, Figure 6G). The expression levels of genes in active sub-TADs were decreased by Mll4 deletion (Figure 6H). We further obtained 200 significantly decreased active sub-TADs based on changes of segregation scores (Figure 6I, Methods, and Supplemental Table S9). The H3K4me1 and H3K4me3 levels were decreased in these active sub-TADs but not the H3K27ac (Figure 6I). The global pattern was exemplified by the genomic locus of Il6ra (Figure 6J), whose decreased expression correlates with reduced Th17 response (88) and affects Th17 maintenance (89).
In summary, the Hi-TrAC data from mouse Th17 cells showed consistent results with the data from human cells regarding the size of the active sub-TADs, the high overlap between active sub-TADs with super-enhancers, and enrichment of cell function and identity genes within active sub-TADs. Our results strongly suggest that H3K4me1 is important for maintaining the interaction densities within active sub-TADs.
DISCUSSION
Since TADs were first identified, domain-centric analyses of Hi-C data have been widely performed in uncovering the relationship between TADs and the 3D genome folding (35,36). Here we demonstrated that the domain-centric analysis with modestly sequenced Hi-TrAC data is a sensitive yet robust method for accurately identifying and studying dynamic changes of active sub-TADs around 100 kb scale with a resolution of 1 kb. The results from our analyses revealed internal domain structures within multiple super-enhancers, which could not be explicitly observed from the interaction matrix heatmaps from current methods such as Hi-C, CTCF ChIA-PET, or H3K27ac HiChIP data. Thus, Hi-TrAC holds a considerable advantage in profiling genome organizations, especially in highly accessible regions at a fine-scale resolution. However, the exact mechanism of why and how the super-enhancers maintain such internal organizations remains elusive. It is currently being debated whether TADs reflect probabilistic preferential interactions from bulk cells or stable domains (90–92), and this debate itself could be directly linked to a parallel debate surrounding the existence of super-enhancer internal structures. Future studies of single-cell level data may prove to be a worthwhile approach to address these hypotheses.
Our integration of the ENCODE ChIP-seq data and active sub-TADs using a classification model revealed potentially important factors associated with active sub-TADs. Among these factors, Cohesin and active histone marks of H3K4me1 and H3K27ac were top-ranked, while CTCF was not on the top 10 list. Further, our knockdown experiments supported that Cohesin playa s more important role than CTCF in the maintenance of the active sub-TAD structure, partially validating the model. The model was further validated by deletion of the Mll4 gene in mouse Th17 cells, which simultaneously decreased the H3K4me1 signals and the interaction density within the active sub-TADs. We also believe that this model will serve as an excellent framework for identifying more factors associated with active sub-TADs and will improve as the ENCODE consortium expands its high-quality datasets (93). However, there are two important caveats in this study to note: (i) It is still the correlation analysis for the signal enrichment at active sub-TADs, which does not indicate any causality. (ii) Potential cell-specific factors were excluded as only factors shared between GM12878 and K562 were used to improve the robustness of the model. A potential solution to the first issue is to perform experimental perturbation and validation. For the second issue, a more detailed analysis focused solely on the K562 dataset may prove worthwhile as it has much rich public data.
We used a mouse strain with conditional deletion of Mll4, which is a prominent writer of the H3K4me1 modification, to study the potential role of H3K4me1 in the maintenance of active sub-TADs. In Th17 cells derived from Mll4 deletion mice, we observed decreases of H3K4me1 signals and compromised active sub-TADs structure, suggesting that H3K4me1 may contribute to the maintenance of active sub-TADs. However, we cannot rule out the possibility that the MLL4-regulated H3K4me1 does not play a direct role in maintaining the 3D chromatin organization since MLL4 was found to have methyltransferase-independent functions for regulating enhancer activity or gene expression (94,95). Thus, the deletion of Mll4 could diminish enhancer activity and gene expression, independently of the H3K4me1 modification, which may in turn lead to the observed loss of sub-TADs. Furthermore, loss of H3K4me1 reduced binding of chromatin remodelers at a subset of enhancers (96), which could also in turn lead to the loss of sub-TADs. MLL3, which has redundant activity with MLL4 in regulating H3K4me1 (97), may not compensate for the loss of MLL4 as its gene expression decreased substantially (from 5.06 RPKM to 1.48 RPKM) in the Mll4-deleted Th17 cells. Further studies are needed to understand the correlative or causative effects of H3K4me1 or Mll4/Mll3 in regulating the active sub-TADs in the future.
With its compatibility and sensitivity in this study, Hi-TrAC has been shown to be an effective method in studying chromatin domains covering regulatory elements at high resolutions, even with modest sequencing depths. We hope that Hi-TrAC will serve as a valuable tool for the 3D genome research community and further our understanding of chromatin domain dynamics in developmental and disease processes.
DATA AVAILABILITY
Sequencing data generated in this study have been deposited to the Gene Expression Omnibus database with the accession of GSE208085. Visualization tracks are available through the WashU Epigenome Browser with session bundle id: 1321f050-fec0-11ec-882d-4588547110d7 (mouse Th17 ChIP-seq, RNA-seq and Hi-TrAC data) and 6c5ce1e0-ac24-11ed-9669-31cbb10ec594 (human K562 boundary deletion and CTCF-AID Hi-TrAC data).
Supplementary Material
ACKNOWLEDGEMENTS
This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Contributor Information
Yaqiang Cao, Laboratory of Epigenome Biology, Systems Biology Center, Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
Shuai Liu, Laboratory of Epigenome Biology, Systems Biology Center, Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
Kairong Cui, Laboratory of Epigenome Biology, Systems Biology Center, Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
Qingsong Tang, Laboratory of Epigenome Biology, Systems Biology Center, Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
Keji Zhao, Laboratory of Epigenome Biology, Systems Biology Center, Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Author contributions: Y.C. and K.Z. conceived the project. Y.C. analyzed the data. S.L., K.C. contributed to the experiments and Q.T. contributed to the experimental design. Y.C., S.L. and K.Z. wrote the paper. All authors contributed to data interpretation.
FUNDING
Division of Intramural Research of the NHLBI. Funding for open access charge: Division of Intramural Research, National Heart, Lung, and Blood Institute; 4DN Transformative Collaborative Project Award [A-0066 to K.Z.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Marchal C., Sima J., Gilbert D.M.. Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol. 2019; 20:721–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pope B.D., Ryba T., Dileep V., Yue F., Wu W., Denas O., Vera D.L., Wang Y., Hansen R.S., Canfield T.K.et al.. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014; 515:402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y., Zhang X., Ba Z., Liang Z., Dring E.W., Hu H., Lou J., Kyritsis N., Zurita J., Shamim M.S.et al.. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature. 2019; 573:600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qiu X., Ma F., Zhao M., Cao Y., Shipp L., Liu A., Dutta A., Singh A., Braikia F.Z., De S.et al.. Altered 3D chromatin structure permits inversional recombination at the IgH locus. Sci. Adv. 2020; 6:eaaz8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hill L., Ebert A., Jaritz M., Wutz G., Nagasaka K., Tagoh H., Kostanova-Poliakova D., Schindler K., Sun Q., Bonelt P.et al.. Wapl repression by Pax5 promotes V gene recombination by Igh loop extrusion. Nature. 2020; 584:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zheng H., Xie W.. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019; 20:535–550. [DOI] [PubMed] [Google Scholar]
- 7. Stadhouders R., Filion G.J., Graf T.. Transcription factors and 3D genome conformation in cell-fate decisions. Nature. 2019; 569:345–354. [DOI] [PubMed] [Google Scholar]
- 8. Du Z., Zheng H., Huang B., Ma R., Wu J., Zhang X., He J., Xiang Y., Wang Q., Li Y.et al.. Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature. 2017; 547:232–235. [DOI] [PubMed] [Google Scholar]
- 9. Ke Y., Xu Y., Chen X., Feng S., Liu Z., Sun Y., Yao X., Li F., Zhu W., Gao L.et al.. 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell. 2017; 170:367–381. [DOI] [PubMed] [Google Scholar]
- 10. Wang H., Han M., Qi L.S.. Engineering 3D genome organization. Nat. Rev. Genet. 2021; 22:343–360. [DOI] [PubMed] [Google Scholar]
- 11. Lupianez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R.et al.. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015; 161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flavahan W.A., Drier Y., Liau B.B., Gillespie S.M., Venteicher A.S., Stemmer-Rachamimov A.O., Suva M.L., Bernstein B.E.. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016; 529:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hnisz D., Weintraub A.S., Day D.S., Valton A.L., Bak R.O., Li C.H., Goldmann J., Lajoie B.R., Fan Z.P., Sigova A.A.et al.. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016; 351:1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jerkovic I., Cavalli G.. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 2021; 22:511–528. [DOI] [PubMed] [Google Scholar]
- 15. Kempfer R., Pombo A.. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 2020; 21:207–226. [DOI] [PubMed] [Google Scholar]
- 16. Lakadamyali M., Cosma M.P.. Visualizing the genome in high resolution challenges our textbook understanding. Nat. Methods. 2020; 17:371–379. [DOI] [PubMed] [Google Scholar]
- 17. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O.et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G.. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012; 148:458–472. [DOI] [PubMed] [Google Scholar]
- 20. Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J.et al.. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012; 485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S.et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsieh T.S., Cattoglio C., Slobodyanyuk E., Hansen A.S., Rando O.J., Tjian R., Darzacq X.. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Mol. Cell. 2020; 78:539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krietenstein N., Abraham S., Venev S.V., Abdennur N., Gibcus J., Hsieh T.S., Parsi K.M., Yang L., Maehr R., Mirny L.A.et al.. Ultrastructural details of mammalian chromosome architecture. Mol. Cell. 2020; 78:554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang Z., Luo O.J., Li X., Zheng M., Zhu J.J., Szalaj P., Trzaskoma P., Magalska A., Wlodarczyk J., Ruszczycki B.et al.. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015; 163:1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beagrie R.A., Scialdone A., Schueler M., Kraemer D.C., Chotalia M., Xie S.Q., Barbieri M., de Santiago I., Lavitas L.M., Branco M.R.et al.. Complex multi-enhancer contacts captured by genome architecture mapping. Nature. 2017; 543:519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quinodoz S.A., Ollikainen N., Tabak B., Palla A., Schmidt J.M., Detmar E., Lai M.M., Shishkin A.A., Bhat P., Takei Y.et al.. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell. 2018; 174:744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lai B., Tang Q., Jin W., Hu G., Wangsa D., Cui K., Stanton B.Z., Ren G., Ding Y., Zhao M.et al.. Trac-looping measures genome structure and chromatin accessibility. Nat. Methods. 2018; 15:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crane E., Bian Q., McCord R.P., Lajoie B.R., Wheeler B.S., Ralston E.J., Uzawa S., Dekker J., Meyer B.J.. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015; 523:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu C., Cheng Y.J., Wang J.W., Weigel D. Prominent topologically associated domains differentiate global chromatin packing in rice from Arabidopsis. Nat. Plants. 2017; 3:742–748. [DOI] [PubMed] [Google Scholar]
- 30. Phillips-Cremins J.E., Sauria M.E., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S., Ong C.T., Hookway T.A., Guo C., Sun Y.et al.. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013; 153:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwarzer W., Abdennur N., Goloborodko A., Pekowska A., Fudenberg G., Loe-Mie Y., Fonseca N.A., Huber W., Haering C.H., Mirny L.et al.. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017; 551:51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haarhuis J.H.I., van der Weide R.H., Blomen V.A., Yanez-Cuna J.O., Amendola M., van Ruiten M.S., Krijger P.H.L., Teunissen H., Medema R.H., van Steensel B.et al.. The cohesin release factor WAPL restricts chromatin loop extension. Cell. 2017; 169:693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davidson I.F., Peters J.M.. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 2021; 22:445–464. [DOI] [PubMed] [Google Scholar]
- 34. Fang C., Wang Z., Han C., Safgren S.L., Helmin K.A., Adelman E.R., Serafin V., Basso G., Eagen K.P., Gaspar-Maia A.et al.. Cancer-specific CTCF binding facilitates oncogenic transcriptional dysregulation. Genome Biol. 2020; 21:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Szabo Q., Bantignies F., Cavalli G.. Principles of genome folding into topologically associating domains. Sci. Adv. 2019; 5:eaaw1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beagan J.A., Phillips-Cremins J.E.. On the existence and functionality of topologically associating domains. Nat. Genet. 2020; 52:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ou H.D., Phan S., Deerinck T.J., Thor A., Ellisman M.H., O'Shea C.C.. ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017; 357:eaag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu S., Cao Y., Cui K., Tang Q., Zhao K.. Hi-TrAC reveals division of labor of transcription factors in organizing chromatin loops. Nat. Commun. 2022; 13:6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cao Y., Liu S., Ren G., Tang Q., Zhao K.. cLoops2: a full-stack comprehensive analytical tool for chromatin interactions. Nucleic Acids Res. 2022; 50:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khan A., Zhang X.. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016; 44:D164–D171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gulli A., Pal S.. Deep Learning with Keras. 2017; Packt Publishing Ltd. [Google Scholar]
- 44. Abadi M., Agarwal A., Barham P., Brevdo E., Chen Z., Citro C., Corrado G.S., Davis A., Dean J., Devin M.. Tensorflow: large-scale machine learning on heterogeneous distributed systems. 2016; arXiv doi:16 March 2016, preprint: not peer reviewedhttps://arxiv.org/abs/1603.04467.
- 45. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V.et al.. Scikit-learn: machine Learning in Python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 46. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Placek K., Hu G., Cui K., Zhang D., Ding Y., Lee J.E., Jang Y., Wang C., Konkel J.E., Song J.et al.. MLL4 prepares the enhancer landscape for Foxp3 induction via chromatin looping. Nat. Immunol. 2017; 18:1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K.. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–837. [DOI] [PubMed] [Google Scholar]
- 49. Wei G., Abraham B.J., Yagi R., Jothi R., Cui K., Sharma S., Narlikar L., Northrup D.L., Tang Q., Paul W.E.et al.. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. 2011; 35:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Picelli S., Faridani O.R., Bjorklund A.K., Winberg G., Sagasser S., Sandberg R.. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014; 9:171–181. [DOI] [PubMed] [Google Scholar]
- 51. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L.. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013; 31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007; 9:90–95. [Google Scholar]
- 57. Waskom M.L. Seaborn: statistical data visualization. J. Open Source Softw. 2021; 6:3021. [Google Scholar]
- 58. Olivares-Chauvet P., Mukamel Z., Lifshitz A., Schwartzman O., Elkayam N.O., Lubling Y., Deikus G., Sebra R.P., Tanay A.. Capturing pairwise and multi-way chromosomal conformations using chromosomal walks. Nature. 2016; 540:296–300. [DOI] [PubMed] [Google Scholar]
- 59. Rouault J.P., Falette N., Guehenneux F., Guillot C., Rimokh R., Wang Q., Berthet C., Moyret-Lalle C., Savatier P., Pain B.et al.. Identification of BTG2, an antiproliferative p53-dependent component of the DNA damage cellular response pathway. Nat. Genet. 1996; 14:482–486. [DOI] [PubMed] [Google Scholar]
- 60. Wang J.H., Avitahl N., Cariappa A., Friedrich C., Ikeda T., Renold A., Andrikopoulos K., Liang L., Pillai S., Morgan B.A.et al.. Aiolos regulates B cell activation and maturation to effector state. Immunity. 1998; 9:543–553. [DOI] [PubMed] [Google Scholar]
- 61. Parelho V., Hadjur S., Spivakov M., Leleu M., Sauer S., Gregson H.C., Jarmuz A., Canzonetta C., Webster Z., Nesterova T.et al.. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008; 132:422–433. [DOI] [PubMed] [Google Scholar]
- 62. Wendt K.S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T.et al.. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008; 451:796–801. [DOI] [PubMed] [Google Scholar]
- 63. Harrold C.L., Gosden M.E., Hanssen L.L., Stolper R.J., Downes D.J., Telenius J.M., Biggs D., Preece C., Alghadban S., Sharpe J.A.. A functional overlap between actively transcribed genes and chromatin boundary elements. 2020; bioRxiv doi:01 July 2020, preprint: not peer reviewed 10.1101/2020.07.01.182089. [DOI]
- 64. Oudelaar A.M., Higgs D.R.. The relationship between genome structure and function. Nat. Rev. Genet. 2021; 22:154–168. [DOI] [PubMed] [Google Scholar]
- 65. Hnisz D., Day D.S., Young R.A.. Insulated neighborhoods: structural and functional units of mammalian gene control. Cell. 2016; 167:1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Naughton C., Avlonitis N., Corless S., Prendergast J.G., Mati I.K., Eijk P.P., Cockroft S.L., Bradley M., Ylstra B., Gilbert N.. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 2013; 20:387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Szabo Q., Donjon A., Jerkovic I., Papadopoulos G.L., Cheutin T., Bonev B., Nora E.P., Bruneau B.G., Bantignies F., Cavalli G.. Regulation of single-cell genome organization into TADs and chromatin nanodomains. Nat. Genet. 2020; 52:1151–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Grubert F., Srivas R., Spacek D.V., Kasowski M., Ruiz-Velasco M., Sinnott-Armstrong N., Greenside P., Narasimha A., Liu Q., Geller B.et al.. Landscape of cohesin-mediated chromatin loops in the human genome. Nature. 2020; 583:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mumbach M.R., Satpathy A.T., Boyle E.A., Dai C., Gowen B.G., Cho S.W., Nguyen M.L., Rubin A.J., Granja J.M., Kazane K.R.et al.. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 2017; 49:1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Alipour E., Marko J.F.. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res. 2012; 40:11202–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schujiers J., Lee T.I., Zhao K.et al.. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014; 159:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Munkel C., Langowski J.. Chromosome structure predicted by a polymer model. Phys. Rev. E. 1998; 57:5888–5896. [Google Scholar]
- 73. Mateos-Langerak J., Bohn M., de Leeuw W., Giromus O., Manders E.M., Verschure P.J., Indemans M.H., Gierman H.J., Heermann D.W., van Driel R.et al.. Spatially confined folding of chromatin in the interphase nucleus. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:3812–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mirny L.A. The fractal globule as a model of chromatin architecture in the cell. Chromosome Res. 2011; 19:37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grosberg A.Y., Nechaev S.K., Shakhnovich E.I.. The role of topological constraints in the kinetics of collapse of macromolecules. J. Phys-Paris. 1988; 49:2095–2100. [Google Scholar]
- 76. Ren G., Jin W., Cui K., Rodrigez J., Hu G., Zhang Z., Larson D.R., Zhao K.. CTCF-mediated enhancer-promoter interaction is a critical regulator of cell-to-cell variation of gene expression. Mol. Cell. 2017; 67:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kim Y.J., Sekiya F., Poulin B., Bae Y.S., Rhee S.G.. Mechanism of B-cell receptor-induced phosphorylation and activation of phospholipase C-gamma2. Mol. Cell. Biol. 2004; 24:9986–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lilly M., Le T., Holland P., Hendrickson S.L.. Sustained expression of the Pim-1 kinase is specifically induced in myeloid cells by cytokines whose receptors are structurally related. Oncogene. 1992; 7:727–732. [PubMed] [Google Scholar]
- 79. Rao S.S.P., Huang S.C., Glenn St Hilaire B., Engreitz J.M., Perez E.M., Kieffer-Kwon K.R., Sanborn A.L., Johnstone S.E., Bascom G.D., Bochkov I.D.et al.. Cohesin loss eliminates all loop domains. Cell. 2017; 171:305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. McCormack M.P., Young L.F., Vasudevan S., de Graaf C.A., Codrington R., Rabbitts T.H., Jane S.M., Curtis D.J.. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science. 2010; 327:879–883. [DOI] [PubMed] [Google Scholar]
- 81. Nora E.P., Goloborodko A., Valton A.L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G.. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017; 169:930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kubo N., Ishii H., Xiong X., Bianco S., Meitinger F., Hu R., Hocker J.D., Conte M., Gorkin D., Yu M.et al.. Promoter-proximal CTCF binding promotes distal enhancer-dependent gene activation. Nat. Struct. Mol. Biol. 2021; 28:152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nishimura K., Fukagawa T., Takisawa H., Kakimoto T., Kanemaki M.. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods. 2009; 6:917–922. [DOI] [PubMed] [Google Scholar]
- 84. Dong C. Cytokine regulation and function in T cells. Annu. Rev. Immunol. 2021; 39:51–76. [DOI] [PubMed] [Google Scholar]
- 85. Alunno A., Bartoloni E., Bistoni O., Nocentini G., Ronchetti S., Caterbi S., Valentini V., Riccardi C., Gerli R.. Balance between regulatory T and Th17 cells in systemic lupus erythematosus: the old and the new. Clin. Dev. Immunol. 2012; 2012:823085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shah K., Lee W.W., Lee S.H., Kim S.H., Kang S.W., Craft J., Kang I.. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res. Ther. 2010; 12:R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dolff S., Bijl M., Huitema M.G., Limburg P.C., Kallenberg C.G., Abdulahad W.H.. Disturbed Th1, Th2, Th17 and T(reg) balance in patients with systemic lupus erythematosus. Clin. Immunol. 2011; 141:197–204. [DOI] [PubMed] [Google Scholar]
- 88. Chen X., Zhang M., Liao M., Graner M.W., Wu C., Yang Q., Liu H., Zhou B.. Reduced Th17 response in patients with tuberculosis correlates with IL-6R expression on CD4+ T Cells. Am. J. Respir. Crit. Care Med. 2010; 181:734–742. [DOI] [PubMed] [Google Scholar]
- 89. Jones G.W., McLoughlin R.M., Hammond V.J., Parker C.R., Williams J.D., Malhotra R., Scheller J., Williams A.S., Rose-John S., Topley N.et al.. Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J. Immunol. 2010; 184:2130–2139. [DOI] [PubMed] [Google Scholar]
- 90. Flyamer I.M., Gassler J., Imakaev M., Brandao H.B., Ulianov S.V., Abdennur N., Razin S.V., Mirny L.A., Tachibana-Konwalski K.. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature. 2017; 544:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nagano T., Lubling Y., Varnai C., Dudley C., Leung W., Baran Y., Mendelson Cohen N., Wingett S., Fraser P., Tanay A.. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 2017; 547:61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stevens T.J., Lando D., Basu S., Atkinson L.P., Cao Y., Lee S.F., Leeb M., Wohlfahrt K.J., Boucher W., O'Shaughnessy-Kirwan A.et al.. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature. 2017; 544:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Consortium E.P., Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A.et al.. Author correction: expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2022; 605:E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dorighi K.M., Swigut T., Henriques T., Bhanu N.V., Scruggs B.S., Nady N., Still C.D. 2nd, Garcia B.A., Adelman K., Wysocka J.. Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol. Cell. 2017; 66:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rickels R., Herz H.M., Sze C.C., Cao K., Morgan M.A., Collings C.K., Gause M., Takahashi Y.H., Wang L., Rendleman E.J.et al.. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet. 2017; 49:1647–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Local A., Huang H., Albuquerque C.P., Singh N., Lee A.Y., Wang W., Wang C., Hsia J.E., Shiau A.K., Ge K.et al.. Identification of H3K4me1-associated proteins at mammalian enhancers. Nat. Genet. 2018; 50:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang C., Lee J.E., Lai B., Macfarlan T.S., Xu S., Zhuang L., Liu C., Peng W., Ge K.. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:11871–11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F.et al.. Landscape of transcription in human cells. Nature. 2012; 489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data generated in this study have been deposited to the Gene Expression Omnibus database with the accession of GSE208085. Visualization tracks are available through the WashU Epigenome Browser with session bundle id: 1321f050-fec0-11ec-882d-4588547110d7 (mouse Th17 ChIP-seq, RNA-seq and Hi-TrAC data) and 6c5ce1e0-ac24-11ed-9669-31cbb10ec594 (human K562 boundary deletion and CTCF-AID Hi-TrAC data).