

Abstract
Dysregulation of histone deacetylases (HDACs) is closely related to tumor development and progression. As promising anticancer targets, HDACs have gained a great deal of research interests and two decades of effort has led to the approval of five HDAC inhibitors (HDACis). However, currently traditional HDACis, although effective in approved indications, exhibit severe off-target toxicities and low sensitivities against solid tumors, which have urged the development of next-generation of HDACi. This review investigates the biological functions of HDACs, the roles of HDACs in oncogenesis, the structural features of different HDAC isoforms, isoform-selective inhibitors, combination therapies, multitarget agents and HDAC PROTACs. We hope these data could inspire readers with new ideas to develop novel HDACi with good isoform selectivity, efficient anticancer effect, attenuated adverse effect and reduced drug resistance.
Key words: HDACs, Oncogenesis, Selective inhibitor, Combination therapy, Multitarget agent, PROTAC
Graphical abstract
The manuscript systematically reviews the biological functions of HDACs, the role of HDACs in oncogenesis, the structure features of different HDAC isoforms, isozyme-specific inhibitor, combination therapy, multitarget agent and HDAC PROTAC.

1. Epigenetics and histone deacetylation
Epigenetics refers to a reversible or inheritable process that regulates gene expression without altering DNA nucleotide sequence1. There are several primary epigenetic mechanisms: DNA/RNA methylation, RNA editing, noncoding RNA-mediated regulation, histone variant, histone modification and chromatin remodeling2, 3, 4. Histone modification, especially acetylation/deacetylation on histone mediated by histone deacetylases (HDACs) and histone acetyltransferases (HATs), is a major epigenetic mechanism. This dynamic process confers unique structural properties to histones and nucleosomes, thus regulating gene expression5,6.
As the subunit of eukaryotic chromatin, nucleosome consists of DNA coiled around a well-protected histone core7. HDACs and HATs modulate gene transcription by altering the structure of nucleosome. Briefly, HDACs deacetylate the ε-NH2 group of lysine in nucleosome histone, which tauts the charged association of histone tails with DNA and prevents the binding of transcription co-factors to DNA, thereby repressing gene transcription. Conversely, HATs acetylate the ε-NH2 group and relax the charged interaction, followed by greater accesses of transcription co-factors to DNA, thus promoting gene transcription (Fig. 1)6,8,9.
Figure 1.

The regulation of gene transcription mediated by HDACs and HATs.
Under physiological conditions, HDACs together with HATs regulate the dynamic equilibrium of deacetylation/acetylation on histones/nonhistone proteins and maintain gene expression, homeostasis and ontogeny. However, in pathological conditions, external stimuli or gene mutation may result in the overexpression of HDACs. Aberrant HDACs activity disrupts the balance of acetylation–deacetylation process and causes a more compact nucleosome structure, thus downregulating tumor suppressor genes, followed by tumorigenesis10,11. Aside from tumor, aberrant HDACs are also associated with a wide range of diseases (Table 1), which make HDACs promising targets.
Table 1.
Diseases involved in HDACs.
| HDACs | Diseases | Ref. |
|---|---|---|
| HDAC1 | Cancers, cardiovascular diseases, allergic diseases, memory and learning, neurodegenerative disorders, viral infections, osteoarthritis, allergic rhinitis. | 12, 13, 14, 15, 16, 17 |
| HDAC2 | Cancers, liver diseases, memory and learning, neurodegenerative disorders, viral infections, osteoarthritis, cognitive deficits, diabetes mellitus, airway diseases, chronic obstructive pulmonary disease, cardiac hypertrophy. | 13, 14, 15, 16,18, 19, 20, 21, 22, 23 |
| HDAC3 | Cancers, obesity, diabetes mellitus, neurodegenerative disorders, rheumatoid arthritis, inflammations, cardiovascular diseases, memory and learning, osteoarthritis, allergic rhinitis, cognitive deficits, lung and kidney diseases. | 16,17,20,24,25 |
| HDAC8 | Cancers, cornelia de lange syndrome (CdLS), X-linked disorders, infectious diseases, duchenne muscular dystrophy (DMD), cardiovascular diseases, liver fibrosis, pulmonary diseases, hepatic diseases, myopathy, renal fibrosis, neuroinflammation-mediated diseases. | 26,27 |
| HDAC4 | Cognitive disorders, neurodegenerative disorders. | 28,29 |
| HDAC5 | Vascular hypertrophy, diabetes mellitus, schizophrenia, neurodegenerative disorders. | 20,30 |
| HDAC7 | Cancers, osteoarthritis, autoimmune diseases, inflammatory bowel diseases, systemic sclerosis, peyronie's diseases, type 2 diabetes mellitus, hepatic steatosis, liver fibrosis, primary sclerosing cholangitis, respiratory diseases, cystic fibrosis, acute lung injury, huntington's diseases. | 31 |
| HDAC9 | Cerebrovascular diseases, osteoporosis, autoimmune diseases, cancers, obesity, liver fibrosis. | 32 |
| HDAC6 | Cancers, neurodegenerative disorders, cardiovascular diseases, inflammatory diseases, autoimmune diseases, viral infections, cognitive deficits. | 20,33, 34, 35 |
| HDAC10 | Cancers, schizophrenia, HIV infection, intracerebral hemorrhage and immunoglobulin A nephropathy. | 36 |
| HDAC11 | Allergic rhinitis, neurological and mental disorders, metabolic disorders, cancers, age-related macular degeneration. | 17,37,38 |
2. The classification and function of HDACs
2.1. Classification of HDACs
According to the cofactors and primary homologous structures, 18 HDAC members phylogenetically fall into Zn2+-dependent HDACs and NAD+-dependent HDACs. Zn2+-dependent HDACs are also known as classical HDACs (mainly containing class I, II, IV) and they catalyze the deacetylation of histones relying on zinc cation (Fig. 2). On the other hand, NAD+-dependent enzymes refer to class III (Sirt 1–7), which employ NAD + as a cofactor39. Notably, Sirt 1–7 are structurally and evolutionarily unrelated to the classical HDACs and the term “HDACs” usually refers to classical HDACs.
Figure 2.

Schematic depiction of HDAC isoforms. Catalytic domains are shown as bright blue column (zinc-dependent HDACs) and green column (NAD+-dependent HDACs).
Generally, class I HDACs share a large degree of similarities in structures as well as functions, and their superior reactivities confer them the major intracellular deacetylation functions40. Of note, class I HDACs are ubiquitously expressed by diverse cells, predominantly reside in the nucleus and participate in multiple biological events through their enzymic functions or formation of multiprotein corepressor complexes (Tables 2 and 3)41. In addition, many studies revealed that overexpression of class I HDACs is common in multiple malignancies and class I HDACs are closely involved in cancer progression42.
Table 2.
The key features of HDAC family members25.
| HDACs | Gene | Homology | Size (aa) | Catalytic domaina | Localization | Substrate |
|---|---|---|---|---|---|---|
| HDAC1 | 1p34 | Yeast Rpd3 | 482 | 9–321 | Nb | Histones, YY1, p53, SHP, DNMT1, E2F-1, ER, AR, ATM, STAT3, pRb, GATA, MeCP2, MEF2, MyoD, NF-κB. |
| HDAC2 | 6q21 | Yeast Rpd3 | 488 | 9–322 | N | Histones, YY1, STAT3, BRCA1, pRb, GATA2, NF-κB, Bcl-6, HOP. |
| HDAC3 | 5q31 | Yeast Rpd3 | 428 | 3–316 | C/N | Histones, pRb, SHP, MEF2D, YY-1, STAT3, NF-κB, GATA1, RelA. |
| HDAC8 | Xq13 | Yeast Rpd3 | 377 | 14–324 | C/N | Histones, SMC3, p53, ERRα, HSP70. |
| HDAC4 | 2q37.3 | Yeast HDA1 | 1084 | 655–1084 | C/N | Histones, p21, HP1, p53, HIF-1α, FOXO, GATA1, SRF, Runx 2, SUV39H1, GCMa. |
| HDAC5 | 17q21 | Yeast HDA1 | 1122 | 684–1028 | C/N | CaM, YY1, HP1, MEF2, GCMa, Runx 2, Smad 7. |
| HDAC7 | 12q13.1 | Yeast HDA1 | 952 | 518–865 | C/N | FLAG1, FLAG2. |
| HDAC9 | 7p21.1 | Yeast HDA1 | 1011 | 631–978 | C/N | Histones, CaM, MEF2, HIF-1α, PML, Runx 2. |
| HDAC6 | Xp11.23 | 1215 | 87–404, 482–800 |
Cc | α-Tubulin, Tau, SHP, HSP90, HSF-1, Runx 2, Smad 7, cortactin. | |
| HDAC10 | 22q13.31 | 669 | 1–323 | C | HSP90, LcoR. | |
| HDAC11 | 3p25.1 | Class I, II HDACs |
347 | 14–326 | N/C | Histones (longer chain acyllysine residues), Cdt 1. |
Catalytic domain: amino acid sequences.
N: Nuclear.
C: Cytoplasm.
Table 3.
| HDAC isoform | Partners | Functions |
|---|---|---|
| HDAC1 | Form complex with LSD1, CoREST 1 and HDAC2. Interact with HDAC2, NuRD, MTA1-3, RBBP4/7, CHD3-5, GATAD2A/B, CDK2AP1 and MBD2/3. Component of MiDAC complex including HDAC2, MIDEAS and DNTTIP1. Form a mSin3A multiprotein complex containing HDAC1-2, SUDS3/SAP45, ARID4B/SAP180, SIN3A and SAP130. Bind to APBB1 and TSHZ3. Form complex with HDAC2, RBBP4, 7. |
It regulates stem cell fate and mediates cell plasticity in cancer and vascular diseases. It modulates cell cycle progression, chromatin remodeling and gene expression. It works with MTA2 to regulate cell growth and apoptosis via p53. It suppresses circadian genes expression. |
| HDAC2 | Component of SMRT complex with HDAC3/4/7, SMRT, GPS2 and TBL1X. Found in a complex including HDAC1, HDAC2, RBBP4,7. Form a HDAC/KDM1A/GFI/RCOR complex. Form a BHC-HDAC heterodimer. Bind to unphosphorylated DAXX, DEK and histones. Interact with ATR and CHD4. Form mSin3A–HDAC complex. Form a HDAC1, 2, EHMT2 and TRIM28 complex. Bind to CDYL, MIER1 and MIER2. |
It is involved in lipid metabolism, fibroblast cell cycle, circadian rhythms, macrophage polarization and inflammation. It interacts with YY1 to form the transcriptional repressor complexes. It is associated with gene expression and ontogeny. HDAC2 knockout causes aberrant transcription. |
| HDAC3 | Forms a complex with YY1. Bind to NCOR1 and SMRT. Form N-CoR/HDAC3 co-repressor complex. Interacts with APEX1, BCL6, BCOR, BEND3, BTBD14B, CCAR2, DACH1, DAXX, GLIS2, HDAC7, HDAC9, HDAC10, INSM1, MAPK14, MEF2D, MJD2A/JHDM3A, NRIP1, NR2C1, PRDM6, SRY, SMRT/NCOR2, XBP1 isoform 1 and ZMYND15. |
It may take part in the modulation of transcription by binding to YY1. HDAC3 is considered a potential tumor suppressor gene. HDAC3 can repress the function of p53, thereby regulating cell growth and apoptosis. |
| HDAC8 | Interacts with alpha-SMA, CBFA2T3, PEPB2-MYH11 and phosphorylated SMG5/EST1B. | HDAC8 regulates the release of cohesion complexes from chromatin. |
| HDAC4 | Interacts with 14-3-3 chaperone protein, AHRR, ANKRA, BTBD14B, EP300, HDAC7, KDM5B, MEF2C, MYOCD, MORC2 and NR2C1. | HDAC4 binds to DNA indirectly via MEF2C and MEF2D. |
| HDAC5 | Interacts with HDAC7, HDAC9, 14-3-3 protein, AHRR, BAHD1, BCOR, CTBP1, EP300, GRK5, KDM5B, MEF2C, SMRT, NRIP1 and PHB2 at least. | HDAC5 binds to MEF2, leading to the repression of MEF2-dependent gene. It may form multicomplex with HDAC3. It is involved in tumor progression. |
| HDAC7 | Interacts with the 14-3-3 protein, EDNRA, FOXP3, HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, KAT5, KDM5B, MBD3, MEF2A, MEF2B, MEF2C, MTA1L1, NCOR1, NCOR2, PML, RBBP4, RBBP7, SIN3A, SIN3B, SAP30 YWHAE and ZMYND15. | In muscle differentiation, HDAC7 allows the expression of myocyte enhancer factors. Its gene encodes splicing transcript variants of different isoforms. |
| HDAC9 | Homodimer. Interacts with the 14-3-3 protein, BCL6, CTBP1, ETV6, HDAC1, HDAC3, FOXP3, MEF2, MAPK10 and NCOR1. | HDAC9 inhibits the activity of MEF2 via the formation of CtBP–HDACs complexes. It participates in hematopoiesis, skeletal myogenesis inhibition and heart development. It prevents neuronal apoptosis. |
| HDAC6 | Bind to LIMK1 and TPPP1. Form complex with APOBEC3G. Interact with FAT10. |
It promotes drug resistance. It is involved in HIV-1 infection. It is required for misfolded protein degradation. |
| HDAC10 | Bind to SMRT, HDAC2 and HDAC3. | It suppresses cell growth and also serves as prognostic factor. Regulates DNA mismatch repair and G2/M transition. HDAC10 upregulation may mediate autophagy and growth decline. |
| HDAC11 | Interact with HDAC6. | HDAC11 participates in immune regulation via IL-10 expression. It also mediates the stability of CDT1 protein. |
Class IIa HDACs and class IIb HDACs share high homology sequences respectively, but the additional catalytic domain of HDAC6 leads to an overall low homology similarity of the class II HDACs43. Although class IIa HDACs share 57% homology with class I HDACs in the catalytic domains, their reactivities are significantly lower than that of class I HDACs44. Moreover, class II HDACs are located in cytoplasm and nucleus, where they can deacetylate histone proteins and nonhistone proteins44,45.
As the sole member of class IV HDACs, HDAC11 is the smallest histone deacetylase, and the catalytic domain accounts for more than 80% of its sequence. HDAC11 was firstly characterized in 2002 and is somewhat more distantly related to other HDAC family members39. Interestingly, HDAC11 removes acetyl group from lysine with lower efficacy, but it is the most proficient fatty acid deacetylase in HDAC family46.
2.2. The biological functions of HDACs
HDACs are able to participate in multiple biological processes by their deacetylase activities or forming protein complexes with various partners. As shown in Fig. 3, HDACs are involved in following biological cascades: (1) Gene transcription. HDACs regulate transcription by altering the structure of nucleosome or the association of transcription factors (ETS, RUNX and SMAD7) to DNA47, 48, 49, 50. (2) Protein stability. HDACs and HATs can affect protein stability (such as that of p53 and CDT1) since lysine residues are subjected to both acetylation and ubiquitylation51,52. (3) Protein translocation. When Ku70 (a DNA damage-associated protein) is acetylated by HATs, Bax protein is released from the “Ku70–Bax” complex, followed by Bax mitochondrial localization53, 54, 55. Moreover, reversible acetylation also influences STAT3 subcellular localization. (4) Protein–protein interaction. For example, acetylation could intervene in the p53–MDM2 interaction56,57. (5) Protein import and export. HDACs together with HATs modulate importin protein acetylation, thereby regulating the nuclear import and export of HMGB1 or transcription factors58. (6) Immunomodulation. Class I HDACs participate in innate immunity by regulating the production of inflammatory cytokines, while class IIa HDACs take part in specific immunity by influencing antigen presentation59. (7) Cell motility and migration. HDAC6 removes the acetyl group from its substrates (α-tubulin, HSP90 and cortactin), and low acetylation promotes cell motility and migration60, 61, 62. Moreover, HDACs can also increase the expression of MMPs which degrade the intact basement membrane and extracellular matrix, followed by migration and invasion63,64. (8) Degradation of misfolded protein. HDAC6 can recruit misfolded protein to the aggresome, followed by degradation via ubiquitin–proteasome system65. (9) Apoptosis. HDACs promote cell survival and evade apoptosis by modulating the intrinsic or extrinsic apoptosis pathway66, 67, 68, 69. (10) Autophagy. Upon HDACs inhibition, autophagy is initiated by inactivating mTOR, upregulating ATG, Beclin-1 and LC370,71. (11) Cell cycle. The protein–protein interaction of cell-cycle regulator (CCR) modulates cell cycle progression and HDACs are able to change the expression or function of CCR to regulate cell cycle progression71. (12) Angiogenesis. HDACs not only increase the expression of pro-angiogenesis factors including VEGF, HIF-1α and CXCR4, but also downregulate p53 and VHL protein, thus stimulating angiogenesis72, 73, 74. (13) DNA synthesis. HDACs inhibition represses the expression of thymidylate synthetase and CTP synthase, which reduces DNA synthesis25. (14) DNA repair. HDACs are required for the function or gene expression of DNA repair proteins, such as Ku70, Ku86, BRCA1, BRCA2 and RAD5175, 76, 77, 78.
Figure 3.

A schematic view of HDACs biological functions. (a) Nuclear export of HMGB1; (b) Bax translocation and the intrinsic apoptosis; (c) Degradation of misfolded protein; (d) STAT3 nuclear translocation; (e) Cell motility and migration; (f) The acetylation and function of importin; (g) p53 protein stability; (h) Association of transcription factors with DNA.
In addition to abovementioned cascades, HDACs are able to complex with some proteins to achieve a number of biological functions. Table 3 illustrates the partners interacting with HDACs and related functions.
3. HDACs and cancer
3.1. The expression of HDACs in different tumors
A considerable number of studies have proven that HDAC overexpression is closely related to tumor progression and 13 out of 15 tumors (breast cancer, colon cancer, gastric cancer, kidney cancer, liver cancer, lung cancer, ovarian cancer, lymphoma, pancreatic cancer, prostate cancer, medulloblastoma, neuroblastoma and chronic lymphocytic leukemia) overexpress HDACs47,81,82. Accumulating evidence revealed that HDACs are required for tumor-related functions, such as cell growth/proliferation, cell differentiation, cell cycle, cell motility/migration, drug resistance, prognosis, angiogenesis, autophagy and apoptosis83, 84, 85, 86. Given the overexpression and tumor-related functions, HDACs therefore have emerged as promising anti-cancer targets.
3.2. HDACs in cancer pathogenesis
HDACs play multiple roles in tumor progression (Fig. 4). In tumorigenesis, overexpressed HDACs transcriptionally silence tumor suppressor genes and promote tumorigenesis. In tumor development, overexpressed HDACs (1) induce angiogenesis via hypoxia inducible factor-1 (HIF-1) and provide nutrients for tumor growth and metabolism; (2) disrupt the cell cycle progression and promote the proliferation of tumor cells; (3) improve cell infiltration and increase the metastasis as well as invasion of tumor cells; (4) reduce the sensitivity of tumor cells to apoptosis signals or chemical drugs, resulting in apoptosis escape or drug resistance.
Figure 4.

The role of HDACs in cancer pathogenesis.
3.2.1. HDACs in angiogenesis
Hypoxia is a significant feature of the tumor microenvironment (TME) and tumor cells can induce angiogenesis in TME via HIF-1α. In malignant cells, the activity of HIF-1α is regulated by HDACs through several pathways. Functionally, acetylation at Lys-532 leads to the ubiquitin-dependent degradation of HIF-1α, whereas HDAC1 and HDAC4 are capable of inducing the deacetylation of HIF-1α and repressing ubiquitination and degradation processes87,88. Moreover, HDAC5 and HDAC6 promote the maturation and stabilization of HIF-1α by regulating the deacetylation of the chaperones HSP70 and HSP90 rather than directly regulating the acetylation of HIF-1α. Upon HDAC5 and HDAC6 inhibition, hyperacetylation of HSP70 and HSP90 renders them a higher affinity for HIF-1α, which causes the attenuated nuclear accumulation and accelerated degradation of HIF-1α89. HDAC4, HDAC5 and HDAC7 increase the transcription of HIF-1α by triggering the association of HIF-1α with CBP/p30087,90,91. Aside from HIF-1α, HDAC9 hyperexpression results in endothelial cell germination and angiogenesis by blocking the antiangiogenic microRNA-17-92 cluster92.
Angiogenesis has great significance for TME and HDACs are able to regulate angiogenesis through multiple pathways. Therefore, HDACs can be potential anti-angiogenesis targets in tumor therapy93.
3.2.2. HDACs in cell cycle progression
Cell cycle is a complex and orchestrated cellular process, whose regulation involves in various cell-cycle regulators (CCRs), including cyclins, cyclin-dependent kinases (CDKs) and CDK inhibitors (CDKIs). In the network interactions of CCRs, the activities of CDKs are positively regulated by cyclins, but negatively modulated by CDKIs. The CIP/KIP family of CDKIs consist of p21WAF1/CIP1, p27KIP1 as well as p57, and HDACs can change their expression or function to regulate the cell cycle.
HDAC1 and HDAC2 are recruited to the promoters of p21WAF1/CIP1, p27KIP1 and p57KIP2 genes and repress CDKIs (p21, p27 and p57), thereby shortening the cell cycle and enhancing unrestricted proliferation94,95. HDAC3 directly deacetylates cyclin A and regulate the stability of cyclin A. HDAC3 inhibition induces PCAF/GCN5-mediated acetylation of cyclin A and leads to proteasome-dependent degradation of cyclin A, followed by S and G2/M phases arrest96. Knockdown of HDAC5 upregulates the CDK inhibitory protein p21 and simultaneously downregulates CDK2/4/6 as well as cyclin D1, causing G1 phase arrest97. Moreover, HDAC10 regulates the G2/M transition via a pathway mediated by let-7/HMGA2/cyclin A298.
3.2.3. HDACs in apoptosis
HDACs inhibition can directly regulate apoptosis through intrinsic and extrinsic apoptosis pathways. In extrinsic apoptosis pathway, HDACs inhibition can prime malignancies for TRAIL-induced apoptosis by upregulating cell surface death receptors, reducing c-FLIP and enhancing the formation of death inducing signal complex (DISC)99. Moreover, HDACs inhibition also directly induces the activation of TNFS10 gene promoter, followed by TRAIL expression and apoptosis99.
In intrinsic apoptotic pathways, the main characteristics of HDACs regulating apoptosis is elevated levels of antiapoptotic proteins and reduced expression of proapoptotic proteins (Fig. 5)100,101. For example, HDAC1 inhibition increases the ratio of Bax/Bcl-269. Knockdown of HDAC2 selectively upregulates proapoptotic proteins, including Bax, Apaf-1 and AIF, while downregulating antiapoptotic protein Bcl-2102. Additionally, inhibition of HDAC3 upregulates the expression of pro-apoptotic protein PUMA by promoting the binding of p53 to its promoter103. Moreover, HDAC8 together with HDAC1 interacts with the Bcl-2-modifying factor (BMF) gene and establishes a pro-survival scenario via the repression of BMF protein expression104. Notably, the knockout of HDAC8 can effectively activate the transcription of BMF and promote BMF-mediated apoptosis104. As for HDAC6, it interferes Bax/Ku70 interaction. In the absence of HDAC6, Ku70 is acetylated by CREB-binding protein (CBP) and then Bax is released from “Bax–Ku70” complex, followed by Bax conformation activation and intrinsic apoptosis initiation55.
Figure 5.

The intrinsic apoptotic pathway regulated by HDACs.
3.2.4. HDACs in cell metastasis and invasion
Downregulation of E-cadherin is a significant feature of epithelial-mesenchymal transition (EMT), and HDACs play a pivotal role in this process. In prostate cancer cells, the transcriptional repressor complex containing Snail, HDAC1 and HDAC2, can mediate the silencing of E-cadherin and accelerate EMT process, causing cell metastasis and invasion105.
On the other hand, an intact basement membrane and extracellular matrix are barriers that repress the metastasis and invasion of malignant cells. However, the upregulation of matrix metalloproteinases (MMPs) mediated by HDACs helps malignant cells to degrade and cross the barrier. For example, in breast cancer cells, overexpression of HDAC1, 6 and 8 increased cellular metastasis by upregulating the expression of MMP-963, while upregulation of MMP-2 mediated Sirt-1 promotes cellular metastasis and invasion in prostate cancer cells64.
3.2.5. HDACs in cell proliferation, differentiation and drug resistance
Many studies have revealed that HDACs control cell proliferation by regulating the cell cycle, whereas inhibition or knockout of HDACs induces apoptosis or cell cycle arrest. For example, class I HDACs can shorten the cell cycle of smooth muscle cells and accelerate cell proliferation. However, knockout of class I HDACs arrests the cell cycle in G1/S phase and significantly reduces cellular proliferative activity41. In glioma cells, HDAC4 promotes cell proliferation by downregulating CDK inhibitory proteins p27 and p21, whereas knockdown of HDAC4 induces G0/G1 cell cycle arrest and apoptosis106. Moreover, HDAC4 also represses the expression of proapoptotic genes, such as BIK and PMAIP1, thereby enhancing drug resistance107. HDAC5 is able to enhance cell proliferation by upregulating Six1 in human hepatoma cells and downregulating HDAC5 significantly inhibits the proliferation of human hepatoma cells108. In addition, both HDAC7 in HeLa cells and HDAC9 in retinoblastoma cells can shorten the cell cycle and promote cell proliferation109,110. For class IIb HDACs, HDAC6 overexpression promotes glioblastoma cell proliferation and induces temozolomide resistance. However, HDAC6 knockdown or HDAC6 isoform-selective inhibitor overcomes temozolomide resistance and significantly suppresses cell proliferation111.
Notably, HDACs have great significances for ontogeny and are widely involved in the differentiation of osteoblasts, nerve cells, skeletal muscle cells, colorectal cancer cells and glioma stem cells112, 113, 114, 115, 116, 117, 118.
Given the crucial role of HDACs in tumorigenesis, development, proliferation, apoptosis, metastasis, invasion and drug resistance, developing HDACi for the therapeutic intervention of aberrant HDACs is still an attractive field in medicinal chemistry.
4. Small molecular HDAC inhibitor (HDACi)
4.1. General pharmacophore model for HDACi
Zinc ion-dependent HDACs possess a highly conserved and homologous catalytic domain: a catalytic channel, a zinc cation and some secondary pockets. To achieve good inhibition of HDACs, various HDACis were designed according to the unique structure of the target enzymes. HDACi generally possesses three common pharmacophore models: surface binding region (cap), linker and zinc binding group (ZBG), whereas there are also a few HDACis with only two functional fragments of linker and ZBG119.
Cap group binds to the pockets in the rim of HDACs channel, which can increase the affinity of HDACi for target enzyme and block the entry of substrate into HDAC catalytic channel. The linker is responsible for delivering ZBG to chelate zinc cation. A suitable linker is capable of helping HDACi achieve good inhibitory activity. A short linker cannot allow ZBG to interact with zinc ion well, resulting in poor binding, while a long linker may affect the interaction of the cap group with the pocket in the rim of the catalytic channel and reduce the affinity of HDACi for target enzyme. ZBG, another key group for HDACi, can chelate zinc ion in the catalytic center and inhibit the catalytic activity of HDACs. Common ZBGs with strong chelating effect are hydroxamic acid and anilinobenzamide; weak ZBGs include trifluoromethyl ketone, thiocarbonate, hydrazide, carboxylic acid, boric acid, mercaptoacetamide, etc.120.
4.2. The indications and drawbacks of approved HDACis
4.2.1. Indications and clinical phases of HDACis
Up to date, two-decade efforts have led to the approval of five HDACis. Among them, vorinostat (SAHA), romidepsin (FK228), belinostat (PXD101) and panobinostat (LBH589) were approved by US Food and Drug Administration (FDA) or European Medicines Agency for the treatment of cutaneous T-cell lymphoma (CTCL), peripheral T-cell lymphoma (PTCL) and multiple myeloma (MM). Chidamide (CS055), approved by China Food and Drug Administration (CFDA), was used to treat PTCL and advanced (metastatic or locally recurrent, inoperable) breast cancer (Fig. 6).
Figure 6.

Chemical structures, HDACs inhibitory activities of approved HDACis and clinical candidate HDACis25,121, 122, 123, 124.
In addition to approved HDACis, some clinical candidate HDACis show good profiles concerning HDACs inhibitory activities and isoform selectivities. Herein, we investigated their chemical structures, HDACs inhibitory activities and clinical status (Fig. 6).
4.2.2. The drawbacks of approved HDACis
4.2.2.1. Adverse effects
Vorinostat (SAHA), a pan-HDACi with hydroxamic acid, was approved for CTCL therapy. In phase I trial, vorinostat was proven to be effective against a defined subset of hematological malignancies as intravenous and oral preparations, whereas it showed limited efficacy on solid tumors. Vorinostat displayed good efficacy with overall response rates of 24%–30% at a dosage of 400 mg/day in the treatment of refractory advanced CTCL in phase II development125. Despite the good efficacy toward hematological malignancies, a series of adverse effects have also been discovered. The common mild adverse effects are fatigue, nausea, diarrhea, anorexia, dysgeusia as well as thrombocytopenia, and the severe adverse effect is thrombosis125,126.
In 2009, romidepsin (FK-228) was approved for relapsed/refractory PTCL therapy. Romidepsin serves as a pan-HDACi but shows preferential inhibition toward class I HDACs and modest inhibition against class II HDACs. In a phase II clinical trial enrolling 131 patients with PTCL, romidepsin exhibited significant therapeutic effects with an objective response rate of 25%, a median duration of response of 28 months and the longest response of 56 months127,128. Notably, romidepsin is well tolerated in older patients, and the common adverse effects are similar, including anemia, fatigue, fever, thrombocytopenia, leukopenia and neutropenia129.
Belinostat (PXD-101) is also a pan-HDACi and was approved for relapsed/refractory PTCL therapy. In a phase II clinical study of belinostat in 129 patients with PTCL, the objective response rate is 25.8% and the median duration of response is 13.6 months130. Of note, 96.9% of patients treated with belinostat experienced moderate treatment-emergent adverse events, such as vomiting, nausea, pyrexia, fatigue, anemia, neutropenia, dyspnea, thrombocytopenia and pneumonia130,131.
Chidamide (CS055), an orally potent benzamide-derived HDACi, was first approved for PTCL therapy in 2014132. Unlike the aforementioned approved pan-HDACis, chidamide favors the inhibition of class I HDACs as well as IIb HDACs, and exhibits modest inhibition against class IV HDAC. Moreover, chidamide possesses the better properties in terms of anticancer effects and adverse effects relative to other approved HDACis. Upon treatment with chidamide, the objective response rate of patients was 28%. The good class I HDAC selectivity of chidamide seems to significantly decrease its clinical adverse events, and its frequent adverse events are thrombocytopenia, leukopenia and neutropenia133. Therefore, developing isoform-specific HDACi is supposed to be a promising approach to attenuate adverse effects or off-target toxicities.
Panobinostat (LBH-589) is a potent broad-spectrum HDACi developed by Novartis and it displays very good inhibitory activity against all HDAC enzymes. In 2015, panobinostat was approved in combination with dexamethasone and bortezomib for the treatment of relapsed MM, but its clinical toxicity and side effects are relatively severe. In the phase II clinical trial of panobinostat enrolling 35 patients, all patients experienced adverse events, with most (71.4%) reporting at least one grade 3 adverse events and 4 patients experiencing grade 4 toxicity134. The most common adverse events included fatigue (62.9%), nausea (51.4%), and thrombocytopenia (45.7%)135.
Currently conventional approved HDACis, although effective in specific indications, exhibit severe adverse effects and off-target toxicities due to the limited isoform selectivity136. Considering the tumor-related functions, class I HDACs selective inhibitor may be an alternative to pan-HDACi because of the good anticancer effect and less off-target toxicity137, 138, 139. Notably, HDAC6 selective inhibitors are well tolerated and they can be effective in the treatment of neurodegenerative diseases as monotherapy or various cancers in combination with other anti-cancer agents140,141.
4.2.2.2. Limited efficiency in solid tumors
Despite the great progress of approved HDACi in the therapy for a defined subset of hematological malignancies, their poor therapeutic efficacy as monotherapy in solid tumors has emerged as an obstacle to be overcome. As illustrated in Fig. 7, vorinostat was effective against a defined subset of hematological malignancies, whereas it displayed limited efficacy toward solid tumors142. Although some more potent HDACis (romidepsin, belinostat, chidamide and panobinostat) were subsequently developed, their efficacies toward solid tumors remain unsatisfactory, probably due to their ineffectively low concentrations143. Moreover, available data revealed that most of the known HDACis exhibit very modest benefits in almost all solid tumors, such as prostate cancer, breast cancer and renal cancer144.
Figure 7.

The therapeutic effects of vorinostat against solid tumors and hematomas142.
Accumulating evidence indicated that combining HDACi with anticancer drugs in synergy with HDACi is capable of providing breakthroughs in therapeutic effects and expansion of indications via synergistic or additive effects136. Therefore, combination therapy or multitarget agents-based HDACi opens the door to the effective therapy of solid tumor. Up to date, some combination regimens involved in HDACi141,145, 146, 147 and multitarget agents-based HDACi124,148, 149, 150 exhibit encouraging anticancer effects and superior safety profiles, which make one believe that combination therapy and multitarget agent can be effective approaches to sensitize solid tumors.
The drawbacks of conventional HDACis have motivated the development of next-generation HDACi. Isoform-specific inhibitors, combination therapies, multitarget agents and HDAC PROTACs have emerged as the trends of next-generation HDACi. In this manuscript, we will introduce the trends and challenges of next-generation HDACi.
5. The trends of next-generation HDACi
5.1. Isozyme-specific HDACi
A central theme on HDACi is isoform selectivity or lack of specificity because non-selective pan-HDACi is involved in a broad spectrum of adverse effects. An investigation showed that vorinostat, romidepsin, belinostat and panobinostat are non-selective HDACi that exhibit good inhibitory activities toward almost all HDAC isoforms (Fig. 6)121. Despite chidamide exhibits preferential inhibition of class I HDACs, its good inhibitory activities against HDAC10 and HDAC11 indicates that its isoform-selectivity remains to be further improved.
5.1.1. Advantages and challenges of isozyme-specific HDACi
It is well accepted that good isoform selectivity confers compound superior therapeutic efficacy and pivotal safety profile in the treatment of some diseases, especially for chronic diseases (obesity, fibrosis and inflammation)121. Moreover, isozyme-specific HDACi can be more effective anticancer agent or powerful tool to explore the biology as well as cellular distribution of individual HDAC isozyme in both physiological and pathological settings43. Therefore, HDAC isoform selective inhibitor can be an alternative to pan-HDACi and is a promising trend in the future80.
Despite some potent HDAC isoform selective inhibitors have been developed, there are some challenges for the development of isozyme-specific HDACi: (1) HDAC enzymes share a conserved tertiary structure with only minor differences, which makes it difficult to develop one compound targeting specific isoform; (2) The structures of several HDAC isoforms remain to be largely unknown due to lack of crystal structure; (3) We know relatively little about the enzymatic activities and biological functions of several HDAC isoforms, such as HDAC10 and HDAC11. Therefore, isozyme-specific HDACi has a long way to go.
5.1.2. Protein structure of different HDAC isoforms
The key to success in the rational design of HDAC isoform-selective inhibitor is comprehension and deeper knowledge of the structure of HDAC isoforms. Therefore, we investigated the structural features of different HDAC isoforms, and hope these data could benefit readers in the design of isozyme-specific HDACi.
The common tube-like binding channel of HDAC can be divided into rim, wall, and bottom regions (Fig. 8). In addition to above common structures, some HDAC isoforms also possess secondary pockets, such as side pocket, lower pocket, foot pocket and potential pocket (Supporting Information Fig. S1). These secondary pockets have great significance for the design of isoform-selective HDACi (see Figure 9, Figure 10, Figure 11, Figure 12, Figure 13, Figure 14).
Figure 8.

Schematic view of different HDAC isoform structures with selective inhibitors.
Figure 9.

Examples of HDAC1 and HDAC2 selective inhibitors.
Figure 10.

Examples of HDAC3 selective inhibitors.
Figure 11.

Examples of HDAC6 selective inhibitors.
Figure 12.

Examples of HDAC8 selective inhibitors.
Figure 13.

Examples of HDAC10 and HDAC11 selective inhibitors.
Figure 14.

Examples of class IIa HDACs selective inhibitors.
HDAC1–3 have a deep and narrow binding channel, and there is an additional internal cavity domain called foot pocket at the bottom region, which is thought to be used to enter water and remove acetic acid80. Introducing a fragment into the ZBG group to accommodate the foot pocket of HDAC1-3 can improve the binding affinity and isoform selectivity (Fig. 8A)151. To be noted that the small Ser113 residues in HDAC1/2 confer them the large foot pocket while HDAC3 has a smaller foot pocket due to the presence of bulkier Tyr96 residue152. Additionally, HDACi with anilinobenzamide or trifluoromethyl ketone as ZBG appears to preferentially inhibit HDAC1 and HDAC2.
Moreover, HDAC8 possesses a side pocket consisting of L1 and L6 loops on the rim of channel and binding to the side pocket results in potent L-shaped HDAC8-selective inhibitor PCI-34051 (Fig. 8B)153.
The foot pocket and side pocket seem not to exist in class II HDACs, whereas class IIa HDACs have a lower pocket, and the occupation of the lower pocket with linker-aside phenyl leads to some potent U-shaped HDACis (Fig. 8C)154,155.
For class IIb HDACs, their shallow and wide binding channel prefers inhibitor with short and bulky linker. Of note, there are some potential binding pockets in the rim region (Fig. 8D) and the occupation of these potential pockets using large and extended cap groups is in favor of achieving high potency in class IIb HDACs inhibition and selectivity156.
To date, the structure of HDAC11 remains largely unknown due to a lack of crystal structure. However, some studies reveal that HDAC11 serves as a defattyacetylase instead of a deacetylase owing to its efficient defatty-acylation activity and low deacetylase activity46. According to its physiological function, an activity-guided design approach was applied in the development of HDAC11 selective inhibitors with long-chain fatty acyl groups157.
5.1.3. Observations/phenotypes of HDACs knockout
Apart from studying the structural features of HDAC isoforms, investigating their biological functions is also pivotal for the development of isozyme-specific HDACi. Therefore, this review summarizes the observations/phenotypes of HDACs knockout in mouse models (Table 4).
Table 4.
Observations/phenotypes of the different HDACs knockout.
| HDACs knockout | Observation/phenotype |
|---|---|
| HDAC1 | Lethal at E9.5–10.5 due to embryonic stem cell proliferation defects, apoptosis and cell cycle arrest. |
| HDAC2 | Perinatal lethal due to cardiac defects, cell cycle delay and apoptosis. |
| HDAC3 | Lethal at E9.5–10.5 due to gastrulation defects, cell cycle arrest, DNA damage and apoptosis. |
| HDAC8 | Perinatal lethal due to craniofacial defects. |
| HDAC4 | Segregation defects in p53-null carcinoma cell with no detectable effects in normal cells. |
| HDAC5 | Profoundly enlarged hearts. |
| HDAC7 | Embryonic lethality owing to blood vessels dilatation and rupture. |
| HDAC9 | Cardiac hypertrophy. |
| HDAC6 | Normal development with slight effects on bone density and immune response. |
| HDAC10 | Normal development with no defects. |
| HDAC11 | No significant effect on growth and differentiation. |
Global silence of HDAC1 results in embryonic lethality before E10.5 because of severe defects in proliferation as well as retardation in development, while upregulating HDAC2 and HDAC3 cannot compensate for HDAC1 loss158. HDAC2-deficient mice survive until the perinatal period, but they succumb to some cardiac defects, such as bradycardia, apoptosis of cardiomyocytes and obliteration of the lumen of the right ventricle159. Moreover, HDAC1/HDAC2 double knockout exhibited enhanced cell apoptosis and cell cycle arrest compared with HDAC1 or HDAC2 individual knockout160. Similarly, HDAC3 is required for early embryonic development and ablation of HDAC3 causes embryonic lethality at E9.5–10.5 due to gastrulation defects. In addition, cell cycle arrest, DNA breakage as well as apoptosis of mouse embryonic fibroblasts were observed in HDAC3-deficient cells161. Additionally, HDAC8 is associated with the morphogenesis of the skull by repressing a subset of transcription factors, and global deletion of HDAC8 results in skull instability, followed by perinatal lethality162.
Interestingly, disruption of HDAC4 selectively induces mitotic arrest and chromosome segregation defects, followed by caspase-dependent apoptosis in tumor cells, whereas it shows no detectable effect in embryonic fibroblasts, myelopoietic progenitors or normal human dermal fibroblasts (NHDFs). Of note, HDAC4 disruption influences the proliferation of both p53-wild-type and p53-null carcinoma cells but defect segregation was discovered only in p53-null cells163. Therefore, HDAC4 selective inhibitor may be such an agent with higher efficiency and less adverse effects in the treatment of p53-deficient tumors.
As suppressors of cardiac hypertrophy, HDAC5 and HDAC9 regulate a specific subset of cardiac stress signal pathways and play redundant roles in cardiac development164. Although mice lacking HDAC5 or HDAC9 can survive at birth with profoundly enlarged hearts, HDAC5/HDAC9 double deletion leads to death during embryogenesis or the perinatal period owing to ventricular septal defects (VSD) and thin ventricular walls164,165. HDAC7 is required for blood vessel development and the association of HDAC7 to myocyte enhancer factor-2 (MEF2) represses MMP-10 gene transcription, thus maintaining vascular integrity. Ablation of HDAC7 leads to embryonic lethality because of a failure in endothelial cell–cell adhesion and consequent blood vessels dilatation as well as rupture166.
Unlike the knockout of above HDAC isoforms, HDAC6 knockout does not lead to lethal or teratogenic effects. In the absence of HDAC6, mice develop normally with moderate impact on the bone mineral density and immune response167. Notably, a variety of HDAC6 selective inhibitors have been developed and applied in antitumor field, where they exhibit good antiproliferation activities with few adverse effects168, 169, 170. To date, several HDAC6 selective inhibitors including citarinostat (ACY-241), ricolinostat (ACY-1215), monohydrochloride (KA2507) and purinostat mesylate are under clinical development, and they achieve good anticancer effects with less toxicities compared with pan-HDACi when used alone or in combination (www.clinicaltrials.gov). These results indicated that HDAC6 selective inhibitors are well tolerated and could avoid the on-target adverse effects of some molecules.
Recently, Li et al.171 revealed that HDAC10 might serve as a presumed tumor suppressor and cells isolated from HDAC10 knockout mice showed highly tumorigenic and stem-like properties. Similar to HDAC6 disruption, HDAC10-deficient mice are viable and fertile without developmental defects.
In neural stem/precursor cells, inactivation of HDAC11 exhibits no significant effect on proliferation and differentiation172. Moreover, an additional study revealed that HDAC11 knockdown selectively affects malignant cells instead of normal cells173.
Class I HDACs are required for cell proliferation as well as differentiation, and disruption or inhibition of class I HDACs is able to significantly reduce cell proliferation rate via cell cycle retardation or apoptosis159. However, class I HDACs inhibition appears to cause a spectrum of adverse effects in clinical settings136.
Class II HDACs participate in development, where HDAC5/HDAC9 are associated with cardiac development and HDAC7 is involved in blood vessel development. Disruption of HDAC5/HDAC7/HDAC9 in mice causes dysplastic hearts or vessels, followed by perinatal lethality.
Conversely, deletion of HDAC4 and HDAC6 does not result in lethal or teratogenic effects, and mice are viable and fertile with no developmental defects. Notably, silence of HDAC4 induces segregation defects in p53-null carcinoma cells, and inactivation of HDAC6 suppresses the proliferation of tumor cells without harming normal-tissue cells. Therefore, the selective inhibitors of HDAC4 and HDAC6 may be efficient with attenuated adverse effects in the treatment of specific subsets of tumors.
Although the ablation of HDAC10 or HDAC11 shows no impact on development progression in mice, it does not affect cell proliferation. Thus, it seems to be an unwise choice to develop HDAC10 or HDAC11 isoform-selective inhibitors for cancer therapy in terms of anti-proliferation activity.
5.1.4. Known isozyme-specific HDACi and dual HDACi
There are several promising isoform selectivity patterns: (1) Target transcription-related nuclear enzymes (HDAC1, HDAC2 and HDAC3); (2) Focus on cytoplasmic HDAC6; (3) Selectively inhibit non-acetyllysine enzymes (HDAC8, HDAC10 and HDAC11); (4) Develop class IIa HDACs (HDAC4, HDAC5, HDAC7 and HDAC9) selective inhibitors121. This manuscript summarizes some representative selective inhibitors of aforementioned HDAC isoforms (see Figure 9, Figure 10, Figure 11, Figure 12, Figure 13, Figure 14, Figure 15).
Figure 15.

Examples of HDAC dual selective inhibitors.
5.1.4.1. HDAC1 selective inhibitors
Employing high throughput screening, Methot et al.152 discovered hydroxybiphenyl benzamide 1, which demonstrated preferential inhibition of HDAC1 in the absence of cap group. Further structural optimization of compound 1 led to a more potent and selective thiophene analogs 2, which is active against HDAC1/2 but is inactive toward other HDAC isoforms. Aside from enzymes inhibition, thiophene analog 2 also exhibits good anti-proliferation activity (HCT-116, GI50 = 0.11 μmol/L), druggability and pharmacokinetics152. In addition, Methot et al.174 subsequently identified spirocyclic carbamate nicotinamides 3 as HDAC1 selective inhibitor by targeting the foot pocket (an internal cavity domain) and it displays good profiles in terms of HDAC1 inhibition, HDAC1 selectivity and in vivo tumor growth inhibition.
5.1.4.2. HDAC2 selective inhibitors
β-Hydroxymethyl-chalcone 4, although lacks cap group, it preferentially inhibits HDAC2 (IC50 = 170 nmol/L) with moderate inhibition over HDAC1 (IC50 = 2740 nmol/L) and mild inhibition toward HDAC3 (IC50 = 50,000 nmol/L)175. QM/MM MD simulations revealed that the extended B-ring of compound 4 is able to occupy the large foot pocket of HDAC2 while the bulkier Tyr96 residue of HDAC3 blocks its access. The selective occupation of HDAC2 foot pocket may be responsible for HDAC2 selectivity. Despite the simple structure, α-hydroxy-ketone tropolone compound 5 not only well chelates zinc ion and inhibit enzymic reactivity, but also suppresses T cell lymphocytes growth176. Compound 6, whose phenyl ring attached to α-hydroxy-ketone tropolone, shares a similar structure to compound 5 but its HDAC2 selectivity is less than that of compound 5176.
5.1.4.3. HDAC3 selective inhibitors
Tacedinaline (CI-994) is a potent inhibitor against HDAC1/2/3, whereas addition of the C-4 fluorine gave a remarkable improvement in HDAC3 selectivity. BRD3308 (7), the 4-fluorine derivative of tacedinaline (CI-994), exhibits more than 17-fold HDAC3 selectivity compared with HDAC1/2 and it inhibits HDAC3 with an IC50 value of 64 nmol/L177. Conversely, 4-pyridyl ring substituents at C-5 of tacedinaline (CI-994) produced BRD2492, a potent HDAC1/2 selective inhibitor (HDAC1, IC50 = 2 nmol/L; HDAC2, IC50 = 19 nmol/L; HDAC3, IC50 = 2080 nmol/L)177. That BRD3308 (7) bearing C-4 fluorine substituent and BRD2492 with C-5 4-pyridyl substituent show preferential inhibition toward distinct HDAC isoform, further confirmed the different topography of the foot pocket in HDAC1, 2 and 3 (HDAC1/2 have a large foot pocket and HDAC3 possesses the small one).
Moreover, RGFP966 (8) shows high HDAC3 inhibitory activity and selectivity without inhibiting other isoforms13. As a molecular tool, RGFP966 (8) with the ability to cross the blood brain barrier is used to investigate the biological function of HDAC3 and is widely applied in various in vivo disease models.
Compound 9 is an hydroxamic acid-based HDAC3 selective inhibitor (IC50 = 5 nmol/L) and shows over 36-fold selectivity for HDAC3 compared with HDAC1 (IC50 = 181 nmol/L) and HDAC2 (IC50 = 298 nmol/L). SAR gathered to date suggested that removing the fluorine attached to the benzyl group or reducing the linker led to the decreased HDAC3 inhibitory activity178.
In addition to hydroxamic acid derivatives and ortho-aminoanilide derivatives, some hydrazide derivatives were also identified as HDAC3 selective inhibitors. Compound 10, a hydrazide-derived HDAC3 selective inhibitor, displays low nanomolar HDAC3 inhibitory activity (IC50 = 0.95 nmol/L) and 12-fold HDAC3-selectivity over HDAC1 (IC50 = 11.81 nmol/L) and HDAC2 (IC50 = 95.45 nmol/L)179. SAR analysis indicated that suitable alkyl (three to four carbon units) substitution at the hydrazide group is in favor of achieving HDAC3 inhibitory activity and HDAC3-selectivity, but shorter alkyl (two carbon units) or longer alkyl (more than four carbon units) impairs HDAC3 inhibitory activity and selectivity.
HDAC1 and HDAC2 have larger foot pocket, which prefers a ZBG group containing big fragment (phenyl or thienyl). However, there is a bulky Tyr96 residue in the foot pocket of HDAC3, which limits the access of big fragment. Therefore, introducing small substituent (alkyl or fluorine) into ZBG is a good choice to achieve HDAC3 selectivity.
5.1.4.4. HDAC6 selective inhibitors
Compared to HDAC1, 2 and 3, HDAC6 possesses the shallower and wider channel, which prefers inhibitor with shorter, bulkier linker and extended, larger cap. Considering the structure features of HDAC6, Butler et al.180 developed a potent HDAC6 selective inhibitor Tubastatin A (11), a tetrahydro-γ-carboline derivative with N-hydroxybenzamide. The short and bulky N-hydroxybenzamide of Tubastatin A can fit well into HDAC6 channel and its large cap is well tolerated by the potential binding pockets in the rim of channel. Therefore, Tubastatin A exhibits good potency in HDAC6 inhibition (IC50 = 15 nmol/L) and selectivity (more than 1000-fold selectivity over other HDACs excluding HDAC8)180. Despite the high potency in HDAC6 inhibition and selectivity, the modest ratio of brain to plasma levels of Tubastatin A (AUCbrain/AUCplasma = 0.18) would impair its therapeutic potency in central nervous system (CNS) disorders. Therefore, Kozikowski et al.181 subsequently identified the other potent HDAC6 selective inhibitors, SW-100 (12). Tetrahydroquinoline-based inhibitor SW-100 (12) demonstrates good brain penetrability, low-nanomolar HDAC6 inhibitory activity (IC50 = 2.3 nmol/L) and more than 1769-fold HDAC6 selectivity against other HDACs. In Fragile X Syndrome (FXS) mouse model, SW-100 (12) ameliorates several memory performances through its HDAC6 selective inhibition181.
Lee and co-workers identified a class of bicyclic arylamino/heteroarylamino hydroxamic acids as novel HDAC6 selective inhibitors168. As the most potent molecule in their study, MPT0G211 (13) displays remarkable potency in HDAC6 inhibition (IC50 = 0.29 nmol/L) and over 4000-fold selectivity against other HDAC isoforms168. It has been proved that MPT0G211 (13) is able to suppress the proliferation of NCI-H929, U266 and RPMI 8226 cells with no impact on normal cells. Moreover, MPT0G211 (13) also demonstrates significant in vivo antitumor activity when used alone (%TGI = 60.4%) or in combination with bortezomib (%TGI = 86.2%) in human multiple myeloma RPMI 8226 xenograft models.
KA2507 (14), a potent HDAC6 selective inhibitor under clinical investigation, delivers low nanomolar HDAC6 inhibitory activity (IC50 = 2.5 nmol/L) with little effect on other HDAC isoforms122. In preclinical models, KA2507 (14) play a crucial role in the antitumor response modulation. In a phase I study, KA2507 (14) exhibited good safety profiles and it was well tolerated at the oral dose of 800 mg. In the future clinical studies, KA2507 (14) may be used alone or combined with other immuno-oncology agents for the better antitumor efficacy122.
Nexturastat A (15) and HPOB (16) are examples of the “Y-shaped” HDAC6 selective inhibitor and they achieve high potency in HDAC6 inhibition and selectivity182,183. The cap groups of Nexturastat A (15) and HPOB (16) contain two fragments, which were thought to interact with the potential pockets in the rim of HDAC6 channel.
Ricolinostat/ACY-1215 (17) and citarinostat/ACY-241 (18) are well-known potent HDAC6 inhibitors and they are aliphatic hydroxamic acid derivatives140,184. Structural modification of ricolinostat gave an improvement in HDAC6 inhibitory activity and produced the more potent citarinostat/ACY-241 (18). Generally, ricolinostat and citarinostat share similar structures and prefer HDAC6 to other HDACs. Up to date, ricolinostat and citarinostat have undergone several clinical trials and they used as single agent or in combination achieve good anticancer therapeutic potency140,141.
5.1.4.5. HDAC8 selective inhibitors
PCI-34051 (19), a well-known HDAC8 specific inhibitor, was widely used as a tool molecule to explore the functions of HDAC8153. PCI-34051 (19) shows high potency in HDAC8 inhibition (IC50 = 10 nmol/L) and is free from significant class I, II as well as IV HDAC inhibitory activity. In subsequent biological evaluations, PCI-34051 (19) is able to induce caspase-dependent apoptosis in T-cell lymphomas or leukemias185.
WK2-16 (20) is another cinnamoyl terphenylhydroxamic acid-based HDAC8 specific inhibitor and it was developed by knowledge-based design combined with structural modeling techniques186. In addition to HDAC8 inhibitory activity comparable to PCI34051, WK2-16 (20) also exhibited good anti-proliferation activity against several human lung cancer cell lines with no cytotoxicity for normal IMR-90 cells.
Efforts of the structural optimization identified the most potent HDAC8 selective inhibitor OJI-1 (21) (IC50 = 0.8 nmol/L). In the future, OJI-1 (21) can be the best probe molecule to study the cellular function of HDAC8187.
5.1.4.6. HDAC10 selective inhibitors
Using a TR-FRET and BRET displacement assay, Géraldy and co-workers found that Tubastatin A, an HDAC6 inhibitor, strongly binds to HDAC10 instead of HDAC6. Subsequently, Géraldy et al.188 synthesized a library of Tubastatin A derivatives and SAR analysis revealed that a basic amine in the cap group was responsible for high HDAC10 potency. Docking study further confirmed that the cap group nitrogen forms a favorable hydrogen bond interaction with the gatekeeper residue Glu272. As the Tubastatin A analogues, compounds 22 and 23 displayed higher binding affinities and selectivities for HDAC10188.
5.1.4.7. HDAC11 selective inhibitors
Recently, a novel class of N-hydroxy-2-arylisoindoline-4-carboxamides were identified as the first-in-class HDAC11 selective inhibitors and FT895 (24) (HDAC11, IC50 = 3 nmol/L) exhibited the highest potency in HDAC11 inhibitory activity and selectivity189. Notably, the superior pharmacokinetic properties and cellular activities make FT895 (24) powerful tool to study the biological functions of HDAC11.
SIS17 (25), developed by an activity-guided rational design approach, was the most selective HDAC11 inhibitor, which is free from significant other HDACs inhibitory activity157. Interestingly, in HPLC-based assay with myristoyl-H3K9 peptide as substrate, SIS17 (25) are slightly less potent than FT895 (24). However, in cellular assay, SIS17 (25) performs better anti-proliferation activity compared with FT895 (24) because of the increased metabolic stability and permeability. Of course, SIS17 (25) can be a useful probe to investigate the biology and therapeutic potential of HDAC11.
5.1.4.8. HDAC4, 5, 7 and 9 selective inhibitors
Class IIa HDACs remains largely unknown due to lack of pharmacological tools to probe their biology, whereas TMP269 (26), a potent and selective class IIa inhibitor, fills this void. TMP269 (26) was firstly developed by Lobera et al.190 using trifluoromethyloxadiazole (TFMO) as ZBG. The crystal structure of TMP269 (26) bound to HDAC7 revealed that TMP269 (26) in a U-shaped conformation directly chelates zinc ion using TFMO group (fluorine and oxygen atoms) through weak electrostatic interactions. To be noted that TFMO play a critical role in class IIa HDACs selectivity of TMP269 (26) because the bulky TFMO is able to fit well into the roomier catalytic pockets of class IIa HDACs but cannot be accommodated by class I or IIb HDACs.
NVS-HD1 (27) shares the same ZBG (trifluoromethyloxadiazole, TFMO) with TMP269 (26), but it is more potent and selective than TMP269 (26). As the most potent and selective class IIa HDACs inhibitor, NVS-HD1 (27) displayed very high potency in class IIa HDACs inhibition (HDAC4, IC50 = 0.65 nmol/L; HDAC5, IC50 = 1.2 nmol/L and HDAC9, IC50 = 2.2 nmol/L) and showed over 200-fold selectivity for class IIa HDACs against class I HDACs (HDAC1, IC50 = 2400 nmol/L; HDAC3, IC50 = 1276 nmol/L; HDAC8, IC50 = 4383 nmol/L) and class IIb HDAC (HDAC6, IC50 = 7039 nmol/L)191. The good profiles concerning class IIa HDACs inhibition and selectivity make NVS-HD1 (27) a suitable tool molecule to investigate the enzymic activity and biological functions of class IIa HDACs.
Targeting the lower pocket, a unique structure of class IIa HDACs, is in favor of yielding class IIa HDACs inhibitory activity and selectivity. Recently, Burli et al.154 identified a class of trisubstituted diarylcyclopropanehydroxamic acid derivatives as class IIa HDACs inhibitors by exploiting the lower pocket and compound 28 exhibited the best profiles concerning the potency and selectivity against class IIa HDACs. The crystal structure of compound 28 bound to HDAC4 revealed that the linker-aside phenyl is well accommodated by the lower pocket and forms edge-to-face π-stacking interactions with Arg681 and Phe812. Subsequently, the structural optimization led to more potent as well as selective compound 29 and CHDI-390576 (30), which achieved improved pharmacokinetic profiles and CNS properties192,193.
5.1.4.9. Dual HDAC1/3, HDAC3/6 and HDAC6/8 selective inhibitors
Efforts of structural optimization resulted in a potent dual HDAC1/3 selective inhibitor 31, characterized by a N-hydroxycinnamamide linker, capped with an indole group194. Docking studies indicated that introducing p-chlorophenyl conferred compound 31 a boost potency in HDAC1/3 inhibition because the lipophilic p-chlorophenyl forms van der Waals interactions with HDACs protein. Further biological evaluations revealed the excellent carcinoma cells growth suppression activity and U937 xenograft tumor inhibition ability of compound 31. In addition, subsequent structural modification of compound 31 caused some other potent ortho-aminoanilide-based HDAC1/3 dual inhibitors and they showed good oral antitumor activity against HCT116 human tumor xenografts195.
Soumyanarayanan et al.196 employed a pharmacophore merging strategy to serendipitous identified a series of novel potent HDAC3/6 dual selective inhibitors. Among these molecules, compound 32 stands out due to its low nanomolar potency toward HDAC3/6 inhibition (34 and 2.6 nmol/L, respectively) and more than 26-fold HDAC3/6 selectivity over other HDAC isoforms. SAR studies indicated that the structure of compound 32 appears to fit well into HDAC6 binding pockets and any alterations in pharmacophore resulted in reduced HDAC6 inhibitory activity. Preliminary biological evaluations illustrated that these HDAC3/6 dual selective inhibitors exhibited low micromolar antiproliferation activity and potent compound 32 is able to induce apoptosis in MDA-MB-231 cells to exert antiproliferation activity.
CUDC-101 is a first-in-class EGFR/HER2/HDAC multitarget compound with good antitumor efficacy149. To yield a boost potency in HDACs inhibition, Yang et al.197 employed the bioisosterism principle to modify the structure of CUDC-101 by replacing the quinazoline scaffold of CUDC-101 with isoquinoline fragment, and developed a library of potent isoquinoline-based HDAC1/3/6 selective inhibitors, including compound 33. Moreover, aside from potency in HDACs inhibition, above soquinoline-based inhibitors demonstrated submicromolar anti-proliferation activity against several tumor cell lines (HCT-116, RPMI-8226 and HepG2 cells) with no effect on hERG channel.
The introduction of substituents into the linker of non-selective HDACs inhibitors seems to improve HDAC6/8 selectivity. SAHA is a well-known pan-HDAC inhibitor targeting most of 11 HDAC isoforms without HDAC6/8 selectivity. However, Negmeldin et al.198, 199, 200 modified the linker of SAHA at C2, C4 as well as C8 position and conferred SAHA-modified analogs HDAC6/8 selectivity. Briefly, modification at C2 and C5 position (compounds 34 and 35) led to decreased potency in HDAC inhibition with slight improvement in HDAC6/8 selectivity, whereas C4-modified SAHA analog (36) gave a boost in HDAC6/8 selectivity. The SAR analysis of SAHA analogues indicated that extended cap and short linker appear to fit well into the catalytic pockets of HDAC6 and 8.
Recently, Rodrigues et al.201 identified a novel series of N-acylhydrazone (NAH) derivatives as HDAC6/8 dual inhibitors based on the structure of trichostatin A (TSA). Briefly, they replaced CH moieties of TSA with nitrogen atom, replaced unsaturated double bond with N-hydroxybenzamide and explored the impact of alkylation at nitrogen atom. In their current study, compounds 37 and 38 exhibited the best potencies in HDAC6/8 inhibition and the modification gave an improvement in HDAC6/8 selectivity compared with TSA. Further evaluations revealed that compounds 37 and 38 are able to induce growth suppression, cell migration inhibition, apoptosis and cell cycle arrest. These results indicated that HDAC6/8 can be regarded as potential anti-tumor targets.
5.2. Combination therapies and multitarget agents
Clinical trials revealed that known HDACis are effective as monotherapies in a defined subset of hematological tumors, whereas they exhibit very modest effects against solid tumors144. Therefore, combination therapies with HDACi and HDACi-based multitarget agents came into being due to the poor therapeutic effect of HDACi in solid tumors202.
5.2.1. Combination therapies with HDACi
5.2.1.1. Advantages and challenges of combination therapies
HDACs are widely involved in various biological events by regulating the acetylation of substrates or interacting with various partners, which endows HDACi with the ability to synergize with a series of anticancer agents. Combining HDACi with anticancer drugs in synergy with HDACi can provide breakthroughs in therapeutic effects and expansion of indications via synergistic or additive effects, while reducing adverse effects or resistance by administering lower drug doses136. The development of combination therapy generally follows pragmatic guidelines: (i) both drugs should have antitumor activity and elicit synergistic effects; (ii) each drug should have different mechanisms of action or nonoverlapping patterns of resistance; (iii) the combination should be tolerable with reduced dosage203.
Combination therapies have been widely applied in a broad spectrum of dreadful disease therapies, especially acquired immunodeficiency syndrome (AIDS) and more difficult-to-treat solid tumors that are only infrequently cured at present, due to the flexible dosing schedules, improved efficiency profiles, reduced drug resistance and broad indications204, 205, 206. However, there are several issues to be addressed, such as poor patient compliance, complex clinical trial design and potential drug–drug interactions (DDIs). In addition, the different metabolic rates of each drug in combination therapy produce unpredictable pharmacokinetics (PK)/pharmacodynamic (PD) relationships, followed by necessitating extensive and expensive clinical studies207,208. Although cocktails or multicomponent drugs whereby multiple agents are coformulated in a single tablet improves patient compliance, other problems remain to be solved. Moreover, experience indicated that there may be more toxicities or adverse effects in drug combinations, since multiple drugs would be administered instead of a single compound208. Given the principal considerations above, multitarget agent, a single compound with multiple desired properties, may provide an appealing and cost-effective alternative to combination therapy209,210.
5.2.1.2. Known combination therapies with HDACi
Given the advantages of combination therapy, it is extensively explored and yields clinical benefits in the treatment of some dreadful diseases. Nowadays, the drugs in combination with HDACi include chemotherapy drugs, kinase inhibitors, protease inhibitors, immune checkpoint inhibitors (PD-1/PD-L1, CTLA-4), monoclonal antibodies as well as other agents (Fig. 16)140,211, 212, 213, 214, 215. For example, a phase III clinical trial (NCT02482753) of chidamide plus exemestane for patients with advanced, hormone receptor-positive breast cancer revealed that the combination regimen was well tolerated and significantly improved progression-free survival compared to exemestane plus placebo. Briefly, the median progression-free survival was 7.4 months in combination therapy group and 3.8 months in the placebo group145. In addition, chidamide plus carboplatin and paclitaxel (NCT01836679) in advanced non-small cell lung cancer146, citarinostat plus paclitaxel in advanced solid tumors141 and citarinostat plus nivolumab in advanced non-small cell lung cancer147 exhibit encouraging anti-solid tumor efficacy and superior safety profiles. Aforementioned clinical trials revealed that combination therapy is promising, beneficial and effective in the treatment of solid tumors, especially difficult-to-treat solid tumors and it opens the door to the effective therapy of solid tumor.
Figure 16.

Promising synergistic targets with HDACs.
Notably, this review also summarizes the drugs in combination with approved HDACis in clinical trials (Supporting Information Table S1). We hope readers can gain inspiration from these drug combination schedules and develop new drug combination regimens or multitarget drugs.
5.2.2. HDACi-based multitarget agents
5.2.2.1. Advantages and challenges of multitarget agents
Different from combination therapy, multitarget agent possesses a series of superior profiles, such as enhanced patient compliance, simple clinical trial design and predictable pharmacokinetics (PK)/pharmacodynamic (PD) relationships. Despite the advantages of multitarget agent, the rational design of multitarget agent with desired profiles is rather challenging because multitarget agent needs to match potencies at two completely different targets with a single chemical entity. Upon the design of multitarget agent, we need to consider all aspects of the factors, such as the structures of different targets, the structure-activity relationship of ligands bound to different targets and the ratio of activity level at the different targets. Fortunately, two main strategies for rationally designed multitarget ligands have been developed: (i) knowledge-based methods and (ii) screening methods210. Knowledge-based methods focus on existing data from the literature, classical molecules or clinical trials, whereas screening methods are based on computational chemistry.
5.2.2.2. Known HDACi-based multitarget agents
Accumulated efforts have identified various multitarget agents and the targets hybridizing to HDAC include kinases, receptors, DNA, enzymes, transcriptional regulators, apoptosis related proteins and some other proteins. Multitarget agents, the promising multifunctional molecules, exhibit superior properties concerning safety, growth suppression, anti-invasion, antiangiogenic activity, etc. This review investigates some known HDACi-based multitarget agents and related targets (Table 5, Table 6, Table 7, Table 8, Table 9, Table 10, Table 11). Readers who are interested in these multitarget agents can refer to the literatures in Table 6, Table 7, Table 8, Table 9, Table 10, Table 11, Table 12 for more details.
Table 5.
Kinase and HDAC dual inhibitors.
| Target | Name | Structure/Activity | Ref. |
|---|---|---|---|
| Abl/HDAC | Abl/HDAC-12b | 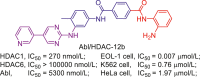 |
216 |
| ALK/HDAC | ALK/HDAC-10f | 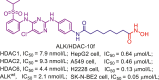 |
217 |
| BRAF/HDAC | BRAF/HDAC-14b |  |
218 |
| BRAF/HDAC-10e | 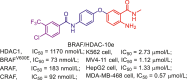 |
219 | |
| CDK/HDAC | CDK/HDAC-8b | 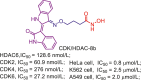 |
220 |
| CDK/HDAC-6d | 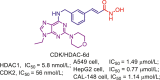 |
221 | |
| CK2/HDAC | CK2/HDAC-15c | 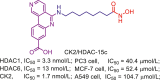 |
222 |
| c-Met/HDAC | c-Met/HDAC-2m | 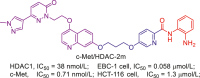 |
223 |
| c-Src/HDAC | c-Src/HDAC-4 |  |
224 |
| EphA2/HDAC | EphA2/HDAC-5a | 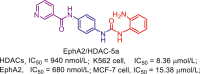 |
225 |
| FAK/HDAC | FAK/HDAC-6a |  |
226 |
| GSK-3β/HDAC | GSK-3β/HDAC-11 | 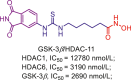 |
227 |
| JAK/HDAC | JAK/HDAC-24 |  |
228 |
| JAK/HDAC-51 | 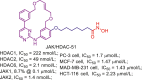 |
229 | |
| Mnk/HDAC | Mnk/HDAC-A12 | 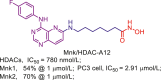 |
230 |
| mTOR/HDAC | mTOR/HDAC-50 | 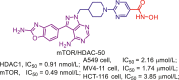 |
231 |
| PI3K/HDAC | CUDC-907 | 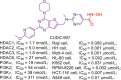 |
148 |
| PIM-1/HDAC | PIM-1/HDAC-4d |  |
232 |
Table 6.
Receptor and HDAC dual inhibitors.

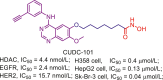

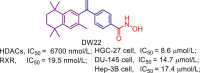
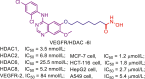
Table 7.
Transcriptional regulator and HDAC dual inhibitors.


Table 8.
Enzyme and HDAC dual inhibitors.
| Target | Name | Structure/Activity | Ref. |
|---|---|---|---|
| CYP51/HDAC | CYP51/HDAC-A5 | 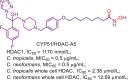 |
245 |
| DNMT/HDAC | C02S | 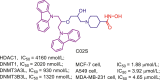 |
246 |
| DNMT/HDAC-204 | 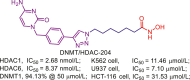 |
247 | |
| EZH2/HDAC | EZH2/HDAC-5 | 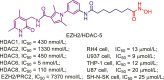 |
248 |
| GLP/HDAC | GLP/HDAC-D7 | 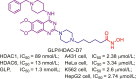 |
249 |
| HMGR/HDAC | JMF3086 | 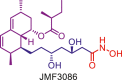 |
250 |
| IDO1/HDAC | IDO1/HDAC-10 | 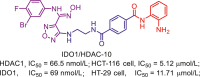 |
251 |
| LSD1/HDAC | 4SC-202 | 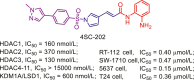 |
124 |
| MAO A/HDAC | MAO A/HDAC-15 | 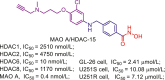 |
252 |
| MMP-2/HDAC | STOCK1N-46177 |  |
253 |
| NAMPT/HDAC | NAMPT/HDAC-7f | 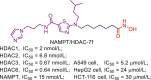 |
254 |
| PARP/HDAC | PARP/HDAC-P1 | 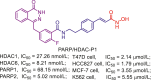 |
255 |
| PDE5/HDAC | PDE5/HDAC-29a | 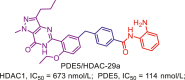 |
256 |
| PDE5/HDAC-44b |  |
257 | |
| Proteasome/HDAC | ZY-2 | 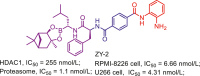 |
258 |
| SHP2/HDAC | SHP2/HDAC-8t | 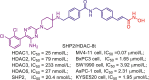 |
259 |
| Topo/HDAC | Topo/HDAC-3d | 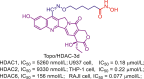 |
260 |
| Topo/HDAC-24d | 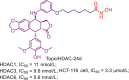 |
261 | |
| Topo/HDAC-8c | 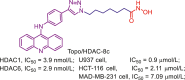 |
262 | |
| Topo/HDAC-7 | 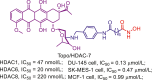 |
263 |
Table 9.
DNA/HDAC dual-targeting agents.
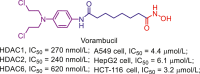
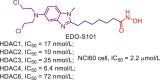



Table 10.
Multifunctional molecules targeting protein and HDAC.
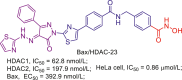
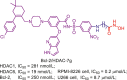
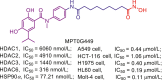
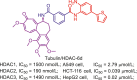
Table 11.
Other HDACi-based multitarget agents.
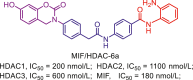
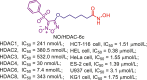
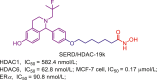
Table 12.
Clinical trials of HDACi-based multitarget agents.
| Compound | Indications | Phase | Trail numbera |
|---|---|---|---|
| EDO-S101 | Malignant melanoma. | I | NCT03903458 |
| Glioblastoma, GMT-unmethylated glioblastoma, gliosarcoma. | I | NCT03452930 | |
| Cutaneous T-cell lymphoma, hematological malignancies, hodgkin's lymphoma, multiple myeloma. | I | NCT02576496 | |
| Multiple myeloma in relapse, multiple myeloma progression, multiple myeloma with failed remission. | I, II | NCT03687125 | |
| Endometrial cancer, ovarian cancer, small-cell lung cancer, soft tissue sarcoma, triple-negative breast cancer. | I, II | NCT03345485 | |
| CUDC-907 | Brain tumor, lymphoma, neuroblastoma, solid tumor. | I | NCT02909777 |
| High-grade serous ovarian cancer, NUT midline carcinoma, triple-negative breast cancer. | I | NCT02307240 | |
| Diffuse intrinsic pontine glioma, recurrent anaplastic astrocytoma, recurrent glioblastoma, recurrent malignant glioma, recurrent medulloblastoma. | I | NCT03893487 | |
| Lymphoma, double-hit lymphoma, double-expressor lymphoma, high-grade B-cell lymphoma, relapsed/refractory lymphoma, relapsed/refractory diffuse large B-cell lymphoma, triple-hit lymphoma. | I | NCT01742988 | |
| Relapsed and/or refractory diffuse large B-cell lymphoma. | II | NCT02674750 | |
| Differentiated thyroid cancer, poorly differentiated and undifferentiated thyroid cancer, thyroid neoplasms. | II | NCT03002623 | |
| 4SC-202 | Advanced hematologic malignancies. | I | NCT01344707 |
| Malignant melanoma. | I, II | NCT03278665 | |
| Prostate cancer. | I, II | NCT02913131 | |
| Merkel cell carcinoma. | II | NCT04874831 | |
| GI cancer. | II | NCT03812796 | |
| CUDC-101 | Head and neck cancer. | I | NCT01384799 |
| Breast cancer; gastric cancer, head and neck cancer, liver cancer, non-small cell lung cancer. | I | NCT01171924 |
Data was obtained from www.clinicaltrials.gov.
Up to now, four HDACi-based multitarget agents (EDO-S101, CUDC-907, 4SC-202 and CUDC-101) are under clinical investigation and the results suggested that the above HDACi-based multitarget agents exhibit better properties in terms of antitumor efficacy, safety and indications compared with single drug124,148, 149, 150 (Table 12).
Although combination therapy is more extensively explored in the clinic than multitarget agents, both methods possess potential advantages in relation to monotherapy and are able to obtain enhanced efficacy, improved safety and reduced drug resistance. Therefore, combination therapy and multitarget agents will likely provide blueprints for the safe and effective treatment of diverse diseases, especially solid tumor. In either case, the key to success depends on the selection of appropriate targets or related signaling pathways that need to be concurrently regulated with the drug. Therefore, sufficient preliminary investigation is necessary for the development of combination therapy and multitarget agent.
5.3. PROTAC
5.3.1. General concept of PROTAC
Proteolysis-targeting chimera (PROTAC), a revolutionary technology in drug discovery, employs ubiquitin-proteasome system (UPS) to degrade target protein. As a heterobifunctional molecule, PROTAC consists of three pharmacophores: a target protein binding ligand, a E3 ligase binding ligand and a linker connecting above two ligands277. Different from traditional inhibitors inhibiting the activities of enzymes or blocking part of functions of target proteins at relatively high concentrations, PROTACs degrade target proteins and eliminate all the functions of target proteins (enzymatic and non-enzymatic functions) at low exposures due to the unique catalytic mode of action of PROTACs278. Nowadays, PROTAC has emerged as a promising approach in drug development, especially in “undruggable target” owing to its unique advantages.
5.3.2. Advantages and challenges of PROTAC
As a revolutionary technology, PROTACs have attracted great attentions because of their advantages: (1) Overcome drug resistance; (2) Eliminate non-enzymatic functions of kinase; (3) Target undruggable protein; (4) Reversible and fast protein knockdown; (5) Superior safety profiles; (6) Substoichiometric catalytic279. A library of advantages enables PROTACs to be powerful tools or agents in the treatment of cancer, inflammation/immune diseases and neurodegenerative disorders278,280. Aside from above advantages, there are some challenges: (1) Complicated evaluation of degradation activity; (2) Poor druggability and unpredictable pharmacokinetics (PK) and pharmacodynamics (PD) properties; (3) Few successes in undruggable targets; (4) Few E3 ligase ligands for PROTACs; (5) Unpredictable degradation of off-target proteins; (6) It is challenging to screen target protein ligands for PROTACs; (7) It is difficult to design PROTACs rationally278. Given the challenges, we therefore need to devote more efforts to PROTACs.
5.3.3. HDAC PROTACs
Up to date, PROTACs have been widely applied in the drug development and attempts to degrade HDACs with PROTACs have been made. This manuscript investigates some examples of HDAC PROTACs (see Figure 17, Figure 18, Figure 19, Figure 20).
Figure 17.

Examples of PROTACs 39–41.
Figure 18.

Examples of PROTACs 42–44.
Figure 19.

Examples of PROTACs 45–49.
Figure 20.

Examples of PROTACs 50–51.
Smalley et al.281 developed four HDAC PROTACs by introducing the linker from the terminal acetyl group of CI-994, a class I HDACs selective inhibitor. In their PROTACs development, they explored the length of linker (six carbon and twelve carbon) and different E3 ligase ligands (VHL ligand and CRBN ligand). Interestingly, PROTACs with longer linker including JSP004 (39) are less potent but they are more effective in histone acetylation induction. The poor HDACs binding affinity and the increased histone acetylation indicated that these PROTACs with longer linker are likely to degrade rather than inhibit HDACs. Quantitative Western blot revealed that VHL-based JSP004 (39) demonstrated better degradation efficacy in relation to CRBN-based PROTACs and it is able to degrade approximately 50% HDAC1, 2 at 1000 nmol/L281. Subsequently, Smalley et al.282 explored the impacts of linker length (8–15 atoms) and linker composition (alkyl, alkoxy, alkyl incorporating piperazine and poly ethylene glycol (PEG)) on the HDACs degradation ability of PROTACs. In this study, they identified a more potent degrader JSP016 (40) (HDAC1, DC50 = 550 nmol/L; HDAC2, DC50 = 530 nmol/L), which showed the highest potency in sub-G1 phase arrest among their PROTACs. Then they further developed two additional PROTACs (JPS026 (41) and JPS027), which employ IAP as E3 ligases ligand283. In addition to HDAC1/2 degradation, JSP026 (41) induced apoptosis as well as DNA replication repression and exhibited higher potency in cell death induction compared with JSP004 (39).
Recently, SR-3558, a potent class I HDACs selective inhibitor, was converted into HDAC3-specific PROTAC XZ9002 (42) using VH032 (VHL) as E3 ligase ligand284. Docking studies revealed that the terminal phenyl of SR-3558 was exposed to solvent and the occupation of foot pocket using the alkyl tail in the hydrazide was responsible for its class I HDACs selectivity. Considering HDACs binding affinity, the linker was attached to the terminal phenyl moiety. Upon treatment with XZ9002 (42) for 14 h, HDAC3 was specifically degraded with a DC50 value of 42 nmol/L without affecting HDACs 1, 2 and 6 in MDA-MB-468 cells284. Notably, the use of VHL ligand or a proteasome inhibitor blocking XZ9002 (42)-mediated HDAC3 degradation further confirmed that the HDAC3 degradation mediated by XZ9002 (42) is required for VHL E3 ligase and UPS. In the future, compound 42 can be a powerful tool to explore the biology of HDAC3.
Yang et al.285 firstly developed zinc-dependent HDAC degraders by connecting CRBN ligand to a non-selective HDAC inhibitor crebinostat. Surprisingly, dHDAC6 (43) derived from crebinostat preferentially degrades HDAC6 (DC50 = 34 nmol/L) instead of all HDAC enzymes. Upon treatment with 10 μmol/L dHDAC6 (43), 70.5% HDAC6 degradation was observed without “Hook effect”. Subsequently, they employed a general cell-based target engagement assay to identify a cell-permeable CRBN E3 ligase ligand, which was used to develop two series of potent PROTACs, including the most potent molecule YZ167 (44) (HDAC6, DC50 = 1.94 nmol/L)286.
Employing solid-phase synthesis, Sinatra et al.287 synthesized two classes of HDAC6 PROTACs. SAHA-based degrader A6 (45) exhibited the highest potency in HDAC6 degradation (DC50 = 3.46 nmol/L) with no effects on HDAC1 and HDAC4. Biological evaluations revealed the promising anti-tumor therapeutic potential of degrader A6 (45) and it was able to induce caspase-dependent apoptosis and sub-G1 phase arrest. In addition, a library of potent HDAC6 PROTACs derived from Nexturastat A have been developed, such as NP8 (46)288, compounds 47289, 48290 and 49291. They all selectively degrade HDAC6 with nanomolar DC50 values.
Recently, PROTAC 50292 and SZUH280 (51)293 were identified as potent HDAC8 selective PROTACs. PROTAC 50 with CRBN as E3 ligase ligand was derived from HDAC8 selective inhibitor NCC-149 and it showed good growth inhibition against Jurkat cells292. Different from PROTAC 50, SZUH280 (51) was PCI-34051-based degrader, which induced rapid HDAC8 degradation. Notably, SZUH280 (51) blocked DNA damage repair and demonstrated good in vivo antitumor activity against solid tumor xenograft model (A549)293. This study further confirmed the anticancer therapeutic potential of HDAC PROTACs.
Xiong et al.294 constructed a library of 48 HDAC PROTACs to systematically map the degradability of different HDAC isoforms. In their current study, SAHA (a pan-HDACs inhibitor), dacinostat (class I and IIb HDACs selective inhibitor), TMP269 (class IIa selective inhibitor) and NVS-HD1 (class IIa selective inhibitor) were chosen to target HDACs. Pomalidomide (CRBN ligand), VHL ligand, VH032 analog and LCL161(IAP ligand) were used to recruit E3 ligases. Alkyl or alkoxy linkers with diverse lengths were employed to conjugate the HDAC inhibitor and E3 ligase ligand. Their study identified HDAC3/6/8 as the most degradable HDACs, HDAC1/2/4 as infrequently degraded HDACs, HDAC9 as the least degraded HDAC and these results explain why non-selective HDAC inhibitor-based PROTACs are able to selectively degrade a defined subset of HDACs294. Notably, their studies also revealed that CRBN-based PROTACs prefer to degrade HDAC6/8, IAP recruiting PROTACs preferentially degrade HDAC6 and PROTACs with VHL exhibit preference for HDAC3 in KELLY cells294. Aside from E3 ligase ligand, both linker and cell identity affect the degradability of HDAC family proteins.
Different from traditional HDAC inhibitors, the catalytic mode of action confers PROTACs a series of advantages and it can be an effective approach to overcome drug resistance, does limited toxicity, “undruggable” target protein or the isoform selectivity limitation of traditional inhibitor. Some degraders under clinical investigation strongly proved that PROTACs is a promising technology in drug development295,296. However, keep in mind that there are still some problems and challenges alongside with PROTACs and we still need to devote more efforts to address these issues.
6. Conclusions
HDAC enzymes, as critical modulators, regulate the deacetylation/acetylation of histones/nonhistone proteins and maintain homeostasis as well as ontogeny. However, available evidence revealed that the overexpression of HDACs has been purchased as an oncogenesis biomarker and therapeutic intervention of aberrant HDACs with inhibitor is able to yield good antitumor effects. Such multifaceted features of HDACs make one believe that HDACs are promising anticancer targets. To date, decades of efforts have caused five approved HDACis, including vorinostat, romidepsin, belinostat, panobinostat and chidamide. Currently traditional HDACis, although effective in approved indications, display severe adverse effects, off-target toxicities and poor therapeutic effects on solid tumors, which hamper their clinical applications. Therefore, it is urgent to develop the next-generation novel HDACi, including isozyme-specific HDACi, combination therapy, multitarget agent and HDAC PROTAC. Isozyme-specific HDACi, an alternative to pan-HDACi, will turn into very valuable drugs since they can be used as probes for the functional investigation of individual HDAC isozymes or as effective anticancer agents with superior therapeutic efficacy and safety profile. However, it is challenging for the development of isozyme-specific HDACi due to the highly conserved catalytic domain, lack of crystal structure and little known about the enzymatic activities, biological functions of several HDAC isoforms. Combination therapy and multitarget agent are able to well address the poor therapeutic efficacy on solid tumors and drug resistance via synergistic or additive effects, which make them the promising trends for solid tumor therapy, especially difficult-to-treat solid tumors. Moreover, PROTAC, a novel technology in drug discovery, exhibits some advantages, whereas there are still several challenges and problems alongside with PROTAC. In summary, isozyme-specific HDACi, combination therapy, multitarget agent and HDAC PROTAC will likely be the trends of next-generation HDACi, whereas more efforts need to be devoted to addressing current challenges.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81874288, 82003590 and 92053105), the Natural Science Foundation of Shandong Province (ZR2020QH342, China), the Key Project of Natural Science Foundation of Anhui Province for College Scholar (2022AH051216, China) and Scientific Research Project of Anhui Provincial Health Commission (AHWJ2022b005, China).
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2023.02.007.
Contributor Information
Wengang Chen, Email: 13505532539@163.com.
Hao Fang, Email: haofangcn@sdu.edu.cn.
Xuben Hou, Email: hxb@sdu.edu.cn.
Author contributions
Tao Liang conceptualized and wrote the manuscript. Reham M Elhassan and Xiaolei Tang revised the manuscript. Fengli Wang and Yongmei Cheng drew figures. Wengang Chen, Xuben Hou and Hao Fang reviewed the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
References
- 1.Peixoto P., Cartron P.F., Serandour A.A., Hervouet E. From 1957 to nowadays: a brief history of epigenetics. Int J Mol Sci. 2020;21:7571. doi: 10.3390/ijms21207571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nadhamuni V.S., Korbonits M. Novel insights into pituitary tumorigenesis: genetic and epigenetic mechanisms. Endocr Rev. 2020;41:821–846. doi: 10.1210/endrev/bnaa006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skinner M.K., Ben Maamar M., Sadler-Riggleman I., Beck D., Nilsson E., McBirney M., et al. Alterations in sperm DNA methylation, non-coding RNA and histone retention associate with DDT-induced epigenetic transgenerational inheritance of disease. Epigenet Chromatin. 2018;11:8. doi: 10.1186/s13072-018-0178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei J.W., Huang K., Yang C., Kang C.S. Non-coding RNAs as regulators in epigenetics. Oncol Rep. 2017;37:3–9. doi: 10.3892/or.2016.5236. [DOI] [PubMed] [Google Scholar]
- 5.Hai R.H., Yang D.Y., Zheng F.F., Wang W.Q., Han X., Bode A.M., et al. The emerging roles of HDACs and their therapeutic implications in cancer. Eur J Pharmacol. 2022;931 doi: 10.1016/j.ejphar.2022.175216. [DOI] [PubMed] [Google Scholar]
- 6.Verza F.A., Das U., Fachin A.L., Dimmock J.R., Marins M. Roles of histone deacetylases and inhibitors in anticancer therapy. Cancers. 2020;12:1664. doi: 10.3390/cancers12061664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsunaka Y., Furukawa A., Nishimura Y. Histone tail network and modulation in a nucleosome. Curr Opin Struct. 2022;75 doi: 10.1016/j.sbi.2022.102436. [DOI] [PubMed] [Google Scholar]
- 8.Giannopoulou A.F., Velentzas A.D., Konstantakou E.G., Avgeris M., Katarachia S.A., Papandreou N.C., et al. Revisiting histone deacetylases in human tumorigenesis: the paradigm of urothelial bladder cancer. Int J Mol Sci. 2019;20:1291. doi: 10.3390/ijms20061291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng X.P., Sun Z.Q., Kuang P.H., Chen J.J. Recent progress on HDAC inhibitors with dual targeting capabilities for cancer treatment. Eur J Med Chem. 2020;208 doi: 10.1016/j.ejmech.2020.112831. [DOI] [PubMed] [Google Scholar]
- 10.Eckschlager T., Plch J., Stiborova M., Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang M.L., Zhang J., Yan C.J., Li X.H., Zhang J.L., Ling R. Small molecule HDAC inhibitors: promising agents for breast cancer treatment. Bioorg Chem. 2019;91 doi: 10.1016/j.bioorg.2019.103184. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y.F., Wang H.Y. The emerging role of histone deacetylase 1 in allergic diseases. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.1027403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowers M.E., Xia B., Carreiro S., Ressler K.J. The class I HDAC inhibitor RGFP963 enhances consolidation of cued fear extinction. Learn Mem. 2015;22:225–231. doi: 10.1101/lm.036699.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar V., Kundu S., Singh A., Singh S. Understanding the role of histone deacetylase and their inhibitors in neurodegenerative disorders: current targets and future perspective. Curr Neuropharmacol. 2022;20:158–178. doi: 10.2174/1570159X19666210609160017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roizman B. The checkpoints of viral gene expression in productive and latent infection: the role of the HDAC/CoREST/LSD1/REST repressor complex. J Virol. 2011;85:7474–7482. doi: 10.1128/JVI.00180-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H., Ji L., Yang Y., Zhang X.N., Gang Y., Bai L.H. The role of HDACs and HDACi in cartilage and osteoarthritis. Front Cell Dev Biol. 2020;8 doi: 10.3389/fcell.2020.560117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J.P., Zhong W., Xue K., Wang Z.G. Epigenetic changes: an emerging potential pharmacological target in allergic rhinitis. Int Immunopharm. 2019;71:76–83. doi: 10.1016/j.intimp.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y.R., Wang J.Q., Huang Z.G., Chen R.N., Cao X., Zhu D.C., et al. Histone deacetylase-2: a potential regulator and therapeutic target in liver disease (Review) Int J Mol Med. 2021;48:131. doi: 10.3892/ijmm.2021.4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gediya P., Parikh P.K., Vyas V.K., Ghate M.D. Histone deacetylase 2: a potential therapeutic target for cancer and neurodegenerative disorders. Eur J Med Chem. 2021;216 doi: 10.1016/j.ejmech.2021.113332. [DOI] [PubMed] [Google Scholar]
- 20.Weiwer M., Lewis M.C., Wagner F.F., Holson E.B. Therapeutic potential of isoform selective HDAC inhibitors for the treatment of schizophrenia. Future Med Chem. 2013;5:1491–1508. doi: 10.4155/fmc.13.141. [DOI] [PubMed] [Google Scholar]
- 21.Christensen D.P., Dahllof M., Lundh M., Rasmussen D.N., Nielsen M.D., Billestrup N., et al. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med. 2011;17:378–390. doi: 10.2119/molmed.2011.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barnes P.J. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol. 2009;71:451–464. doi: 10.1146/annurev.physiol.010908.163257. [DOI] [PubMed] [Google Scholar]
- 23.Eom G.H., Kook H. Role of histone deacetylase 2 and its posttranslational modifications in cardiac hypertrophy. Bmb Rep. 2015;48:131–138. doi: 10.5483/BMBRep.2015.48.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarkar R., Banerjee S., Amin S.A., Adhikari N., Jha T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: a review. Eur J Med Chem. 2020;192 doi: 10.1016/j.ejmech.2020.112171. [DOI] [PubMed] [Google Scholar]
- 25.Adhikari N., Jha T., Ghosh B. Dissecting histone deacetylase 3 in multiple disease conditions: selective inhibition as a promising therapeutic strategy. J Med Chem. 2021;64:8827–8869. doi: 10.1021/acs.jmedchem.0c01676. [DOI] [PubMed] [Google Scholar]
- 26.Kim J.Y., Cho H., Yoo J., Kim G.W., Jeon Y.H., Lee S.W., et al. Pathological role of HDAC8: cancer and beyond. Cells. 2022;11:3161. doi: 10.3390/cells11193161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fontana A., Cursaro I., Carullo G., Gemma S., Butini S., Campiani G. A therapeutic perspective of HDAC8 in different diseases: an overview of selective inhibitors. Int J Mol Sci. 2022;23 doi: 10.3390/ijms231710014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y.L., Hou F., Wang X., Kong Q.S., Han X.L., Bai B. Aberrant expression of histone deacetylases 4 in cognitive disorders: molecular mechanisms and a potential target. Front Mol Neurosci. 2016;9:114. doi: 10.3389/fnmol.2016.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mielcarek M., Zielonka D., Carnemolla A., Marcinkowski J.T., Guidez F. HDAC4 as a potential therapeutic target in neurodegenerative diseases: a summary of recent achievements. Front Cell Neurosci. 2015;9:42. doi: 10.3389/fncel.2015.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathias R.A., Guise A.J., Cristea I.M. Post-translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease. Mol Cell Proteomics. 2015;14:456–470. doi: 10.1074/mcp.O114.046565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y.Z., Abrol R., Mak J., Gupta K.D., Ramnath D., Karunakaran D., et al. Histone deacetylase 7: a signalling hub controlling development, inflammation, metabolism and disease. FEBS J. 2023;290:2805–2832. doi: 10.1111/febs.16437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu S.Q., Cho E.H., Lee J.Y. Histone deacetylase 9: its role in the pathogenesis of diabetes and other chronic diseases. Diabetes Metab J. 2020;44:234–244. doi: 10.4093/dmj.2019.0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang T., Fang H. Structure, functions and selective inhibitors of HDAC6. Curr Top Med Chem. 2018;18:2429–2447. doi: 10.2174/1568026619666181129141822. [DOI] [PubMed] [Google Scholar]
- 34.Seidel C., Schnekenburger M., Dicato M., Diederich M. Histone deacetylase 6 in health and disease. Epigenomics. 2015;7:103–118. doi: 10.2217/epi.14.69. [DOI] [PubMed] [Google Scholar]
- 35.Pulya S., Amin S.A., Adhikari N., Biswas S., Jha T., Ghosh B. HDAC6 as privileged target in drug discovery: a perspective. Pharmacol Res. 2021;163 doi: 10.1016/j.phrs.2020.105274. [DOI] [PubMed] [Google Scholar]
- 36.Cheng F.J., Zheng B., Wang J.W., Zhao G.T., Yao Z.S., Niu Z.H., et al. Histone deacetylase 10, a potential epigenetic target for therapy. Biosci Rep. 2021;41 doi: 10.1042/BSR20210462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu S.S., Wu F., Jin Y.M., Chang W.Q., Xu T.M. HDAC11: a rising star in epigenetics. Biomed Pharmacother. 2020;131 doi: 10.1016/j.biopha.2020.110607. [DOI] [PubMed] [Google Scholar]
- 38.Chen H.Z., Xie C.G., Chen Q., Zhuang S.G. HDAC11, an emerging therapeutic target for metabolic disorders. Front Endocrinol. 2022;13 doi: 10.3389/fendo.2022.989305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao L., Cueto M.A., Asselbergs F., Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. 2002;277:25748–25755. doi: 10.1074/jbc.M111871200. [DOI] [PubMed] [Google Scholar]
- 40.Schultz B.E., Misialek S., Wu J., Tang J., Conn M.T., Tahilramani R., et al. Kinetics and comparative reactivity of human class I and class IIb histone deacetylases. Biochemistry. 2004;43:11083–11091. doi: 10.1021/bi0494471. [DOI] [PubMed] [Google Scholar]
- 41.Reichert N., Choukrallah M.A., Matthias P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell Mol Life Sci. 2012;69:2173–2187. doi: 10.1007/s00018-012-0921-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinke G., Yamada D., Eguchi H., Iwagami Y., Asaoka T., Noda T., et al. Role of histone deacetylase 1 in distant metastasis of pancreatic ductal cancer. Cancer Sci. 2018;109:2520–2531. doi: 10.1111/cas.13700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruijter A.J.M.D., Gennip A.H.V., Caron H.N., Kemp S., Kuilenburg ABPv. Histone deacetylases (HDACs), characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Asfaha Y., Schrenk C., Alves Avelar L.A., Hamacher A., Pflieger M., Kassack M.U., et al. Recent advances in class IIa histone deacetylases research. Bioorg Med Chem. 2019;27 doi: 10.1016/j.bmc.2019.115087. [DOI] [PubMed] [Google Scholar]
- 45.Clocchiatti A., Florean C., Brancolini C. Class IIa HDACs: from important roles in differentiation to possible implications in tumourigenesis. J Cell Mol Med. 2011;15:1833–1846. doi: 10.1111/j.1582-4934.2011.01321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kutil Z., Novakova Z., Meleshin M., Mikesova J., Schutkowski M., Barinka C. Histone deacetylase 11 is a fatty-acid deacylase. ACS Chem Biol. 2018;13:685–693. doi: 10.1021/acschembio.7b00942. [DOI] [PubMed] [Google Scholar]
- 47.Chun P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res (Seoul) 2015;38:933–949. doi: 10.1007/s12272-015-0571-1. [DOI] [PubMed] [Google Scholar]
- 48.Li Y.X., Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6:a026831. doi: 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pahl M.C., Erdman R., Kuivaniemi H., Lillvis J.H., Elmore J.R., Tromp G. Transcriptional (ChIP-Chip) analysis of ELF1, ETS2, RUNX1 and STAT5 in human abdominal aortic aneurysm. Int J Mol Sci. 2015;16:11229–11258. doi: 10.3390/ijms160511229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simonsson M., Heldin C.H., Ericsson J., Gronroos E. The balance between acetylation and deacetylation controls Smad 7 stability. J Biol Chem. 2005;280:21797–21803. doi: 10.1074/jbc.M503134200. [DOI] [PubMed] [Google Scholar]
- 51.Caron C., Boyault C., Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays. 2005;27:408–415. doi: 10.1002/bies.20210. [DOI] [PubMed] [Google Scholar]
- 52.Glozak M.A., Seto E. Acetylation/deacetylation modulates the stability of DNA replication licensing factor Cdt 1. J Biol Chem. 2009;284:11446–11453. doi: 10.1074/jbc.M809394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen H.Y., Lavu S., Bitterman K.J., Hekking B., Imahiyerobo T.A., Miller C. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 54.Hada M., Kwok R.P. Regulation of ku70-bax complex in cells. J Cell Death. 2014;7:11–13. doi: 10.4137/JCD.S13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian C., Jarzembowski J.A., Opipari A.W., Jr., Castle V.P., Kwok R.P. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia. 2011;13:726–734. doi: 10.1593/neo.11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bode A.M., Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 57.Yu S.Y., Cai X.X., Wu C.X., Liu Y., Zhang J., Gong X., et al. Targeting HSP90-HDAC6 regulating network implicates precision treatment of breast cancer. Int J Biol Sci. 2017;13:505–517. doi: 10.7150/ijbs.18834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andersson U., Yang H. HMGB1 is a critical molecule in the pathogenesis of gram-negative sepsis. J Intensive Care. 2022;2:156–166. doi: 10.1016/j.jointm.2022.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Falkenberg K.J., Johnstone R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 60.Li D.W., Xie S.B., Ren Y., Huo L.H., Gao J.M., Cui D.D., et al. Microtubule-associated deacetylase HDAC6 promotes angiogenesis by regulating cell migration in an EB1-dependent manner. Protein Cell. 2011;2:150–160. doi: 10.1007/s13238-011-1015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park J.H., Kim S.H., Choi M.C., Lee J., Oh D.Y., Im S.A., et al. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem Biophys Res Commun. 2008;368:318–322. doi: 10.1016/j.bbrc.2008.01.056. [DOI] [PubMed] [Google Scholar]
- 62.Zhang X.H., Yuan Z.G., Zhang Y.T., Yong S., Salas-Burgos A., Koomen J., et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park S.Y., Jun J.A., Jeong K.J., Heo H.J., Kang J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol Rep. 2011;25:1677–1681. doi: 10.3892/or.2011.1236. [DOI] [PubMed] [Google Scholar]
- 64.Lovaas J.D., Zhu L., Chiao C.Y., Byles V., Faller D.V., Dai Y. SIRT1 enhances matrix metalloproteinase-2 expression and tumor cell invasion in prostate cancer cells. Prostate. 2013;73:522–530. doi: 10.1002/pros.22592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kawaguchi Y., Kovacs J.J., McLaurin A., Vance J.M., Ito A., Yao T. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 66.Fulda S. Histone deacetylase (HDAC) inhibitors and regulation of TRAIL-induced apoptosis. Exp Cell Res. 2012;318:1208–1212. doi: 10.1016/j.yexcr.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 67.Mohana Kumar B., Song H.J., Cho S.K., Balasubramanian S., Choe S.Y., Rho G.J. Effect of histone acetylation modification with sodium butyrate, a histone deacetylase inhibitor, on cell cycle, apoptosis, ploidy and gene expression in porcine fetal fibroblasts. J Reprod Dev. 2007;53:903–913. doi: 10.1262/jrd.18180. [DOI] [PubMed] [Google Scholar]
- 68.Wang J., Kim T.H., Ahn M.Y., Lee J., Jung J.H., Choi W.S., et al. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int J Oncol. 2012;41:1101–1109. doi: 10.3892/ijo.2012.1534. [DOI] [PubMed] [Google Scholar]
- 69.Zhao B., He T.L. Chidamide, a histone deacetylase inhibitor, functions as a tumor inhibitor by modulating the ratio of Bax/Bcl-2 and P21 in pancreatic cancer. Oncol Rep. 2015;33:304–310. doi: 10.3892/or.2014.3595. [DOI] [PubMed] [Google Scholar]
- 70.Mrakovcic M., Kleinheinz J., Frohlich L.F. Histone deacetylase inhibitor-induced autophagy in tumor cells: implications for p53. Int J Mol Sci. 2017;18:1883. doi: 10.3390/ijms18091883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manal M., Chandrasekar M.J., Gomathi Priya J., Nanjan M.J. Inhibitors of histone deacetylase as antitumor agents: a critical review. Bioorg Chem. 2016;67:18–42. doi: 10.1016/j.bioorg.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 72.Deng B., Luo Q., Halim A., Liu Q.P., Zhang B.Y., Song G.B. The antiangiogenesis role of histone deacetylase inhibitors: their potential application to tumor therapy and tissue repair. DNA Cell Biol. 2020;39:167–176. doi: 10.1089/dna.2019.4877. [DOI] [PubMed] [Google Scholar]
- 73.Qian D.Z. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12:634–642. doi: 10.1158/1078-0432.CCR-05-1132. [DOI] [PubMed] [Google Scholar]
- 74.Kim M.S., Kwon H.J., Lee Y.M., Baek J.H., Jang J.E., Lee S.W., et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7:437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 75.Al Emam A., Arbon D., Jeeves M., Kysela B. Ku70 N-terminal lysines acetylation/deacetylation is required for radiation-induced DNA-double strand breaks repair. Neoplasma. 2018;65:708–719. doi: 10.4149/neo_2018_171020N673. [DOI] [PubMed] [Google Scholar]
- 76.Munshi A., Kurland J.F., Nishikawa T., Tanaka T., Hobbs M.L., Tucker S.L., et al. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res. 2005;11:4912–4922. doi: 10.1158/1078-0432.CCR-04-2088. [DOI] [PubMed] [Google Scholar]
- 77.Adimoolam S., Sirisawad M., Chen J., Thiemann P., Ford J.M., Buggy J.J. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci U S A. 2007;104:19482–19487. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chao O.S., Goodman O.B., Jr. Synergistic loss of prostate cancer cell viability by coinhibition of HDAC and PARP. Mol Cancer Res. 2014;12:1755–1766. doi: 10.1158/1541-7786.MCR-14-0173. [DOI] [PubMed] [Google Scholar]
- 79.Lee K.W., Whedon S.D., Wang Z.A., Cole P.A. Distinct biochemical properties of the class I histone deacetylase complexes. Curr Opin Chem Biol. 2022;70 doi: 10.1016/j.cbpa.2022.102179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roche J., Bertrand P. Inside HDACs with more selective HDAC inhibitors. Eur J Med Chem. 2016;121:451–483. doi: 10.1016/j.ejmech.2016.05.047. [DOI] [PubMed] [Google Scholar]
- 81.Poyet C., Jentsch B., Hermanns T., Schweckendiek D., Seifert H.H., Schmidtpeter M., et al. Expression of histone deacetylases 1, 2 and 3 in urothelial bladder cancer. BMC Clin Pathol. 2014;14:10. doi: 10.1186/1472-6890-14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fritzsche F.R., Weichert W., Roske A., Gekeler V., Beckers T., Stephan C., et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer. 2008;8:381. doi: 10.1186/1471-2407-8-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Draney C., Austin M.C., Leifer A.H., Smith C.J., Kener K.B., Aitken T.J., et al. HDAC1 overexpression enhances beta-cell proliferation by down-regulating Cdkn1b/p27. Biochem J. 2018;475:3997–4010. doi: 10.1042/BCJ20180465. [DOI] [PubMed] [Google Scholar]
- 84.Hassell K.N. Histone deacetylases and their inhibitors in cancer epigenetics. Diseases. 2019;7:57. doi: 10.3390/diseases7040057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hayashi A., Horiuchi A., Kikuchi N., Hayashi T., Fuseya C., Suzuki A., et al. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. Int J Cancer. 2010;127:1332–1346. doi: 10.1002/ijc.25151. [DOI] [PubMed] [Google Scholar]
- 86.Zhao H.S., Yu Z.J., Zhao L., He M., Ren J., Wu H.Z., et al. HDAC2 overexpression is a poor prognostic factor of breast cancer patients with increased multidrug resistance-associated protein expression who received anthracyclines therapy. Jpn J Clin Oncol. 2016;46:893–902. doi: 10.1093/jjco/hyw096. [DOI] [PubMed] [Google Scholar]
- 87.Geng H., Harvey C.T., Pittsenbarger J., Liu Q., Beer T.M., Xue C., et al. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem. 2011;286:38095–38102. doi: 10.1074/jbc.M111.257055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yoo Y.G., Kong G., Lee M.O. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1 alpha protein by recruiting histone deacetylase 1. EMBO J. 2006;25:1231–1241. doi: 10.1038/sj.emboj.7601025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen S.Y., Yin C.Q., Lao T.T., Liang D.M., He D., Wang C.G., et al. AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1 alpha and functional activation of HIF-1 by deacetylating Hsp 70 in the cytosol. Cell Cycle. 2015;14:2520–2536. doi: 10.1080/15384101.2015.1055426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kato H., Tamamizu-Kato S., Shibasaki F. Histone deacetylase 7 associates with hypoxia-inducible factor 1 alpha and increases transcriptional activity. J Biol Chem. 2004;279:41966–41974. doi: 10.1074/jbc.M406320200. [DOI] [PubMed] [Google Scholar]
- 91.Seo H.W., Kim E.J., Na H., Lee M.O. Transcriptional activation of hypoxia-inducible factor-1 alpha by HDAC4 and HDAC5 involves differential recruitment of p300 and FIH-1. FEBS Lett. 2009;583:55–60. doi: 10.1016/j.febslet.2008.11.044. [DOI] [PubMed] [Google Scholar]
- 92.Kaluza D., Kroll J., Gesierich S., Manavski Y., Boeckel J.N., Doebele C., et al. Histone deacetylase 9 promotes angiogenesis by targeting the antiangiogenic microRNA-17-92 cluster in endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:533–543. doi: 10.1161/ATVBAHA.112.300415. [DOI] [PubMed] [Google Scholar]
- 93.Kong X.G., Lin Z., Liang D.M., Fath D., Sang N., Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2006;26:2019–2028. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yamaguchi T., Cubizolles F., Zhang Y., Reichert N., Kohler H., Seiser C., et al. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Gene Dev. 2010;24:455–469. doi: 10.1101/gad.552310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zupkovitz G., Grausenburger R., Brunmeir R., Senese S., Tischler J., Jurkin J., et al. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol. 2010;30:1171–1181. doi: 10.1128/MCB.01500-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vidal-Laliena M., Gallastegui E., Mateo F., Martinez-Balbas M., Pujol M.J., Bachs O. Histone deacetylase 3 regulates cyclin A stability. J Biol Chem. 2013;288:21096–21104. doi: 10.1074/jbc.M113.458323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fan J., Lou B., Chen W., Zhang J., Lin S., Lv F.F., et al. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumour Biol. 2014;35:11523–11532. doi: 10.1007/s13277-014-2358-2. [DOI] [PubMed] [Google Scholar]
- 98.Li Y.X., Peng L.R., Seto E. Histone deacetylase 10 regulates the cell cycle G2/M phase transition via a novel Let-7-HMGA2-Cyclin A2 pathway. Mol Cell Biol. 2015;35:3547–3565. doi: 10.1128/MCB.00400-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang J., Zhong Q. Histone deacetylase inhibitors and cell death. Cell Mol Life Sci. 2014;71:3885–3901. doi: 10.1007/s00018-014-1656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Duan H., Heckman C.A., Boxer L.M. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005;25:1608–1619. doi: 10.1128/MCB.25.5.1608-1619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schneller A., Zojer N., Bolomsky A., Ludwig H. Synergistic interaction between HDAC and MCL-1 inhibitors through downregulation of BCL-X-L in multiple myeloma. Haematologica. 2021;106:2516–2521. doi: 10.3324/haematol.2020.277152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim J.K., Noh J.H., Eun J.W., Jung K.H., Bae H.J., Shen Q., et al. Targeted inactivation of HDAC2 restores p16INK4a activity and exerts antitumor effects on human gastric cancer. Mol Cancer Res. 2013;11:62–73. doi: 10.1158/1541-7786.MCR-12-0332. [DOI] [PubMed] [Google Scholar]
- 103.Feng L.F., Pan M., Sun J., Lu H.Q., Shen Q., Zhang S.J., et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J Mol Med (Berl) 2013;91:49–58. doi: 10.1007/s00109-012-0932-x. [DOI] [PubMed] [Google Scholar]
- 104.Kang Y., Nian H., Rajendran P., Kim E., Dashwood W.M., Pinto J.T., et al. HDAC8 and STAT3 repress BMF gene activity in colon cancer cells. Cell Death Dis. 2014;5:e1476. doi: 10.1038/cddis.2014.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wawruszak A., Kalafut J., Okon E., Czapinski J., Halasa M., Przybyszewska A., et al. Histone deacetylase inhibitors and phenotypical transformation of cancer cells. Cancers. 2019;11:148. doi: 10.3390/cancers11020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cai J.Y., Xu T.T., Wang Y., Chang J.J., Li J., Chen X.Y., et al. Histone deacetylase HDAC4 promotes the proliferation and invasion of glioma cells. Int J Oncol. 2018;53:2758–2768. doi: 10.3892/ijo.2018.4564. [DOI] [PubMed] [Google Scholar]
- 107.Spaety M.E., Gries A., Badie A., Venkatasamy A., Romain B., Orvain C., et al. HDAC4 levels control sensibility toward cisplatin in gastric cancer via the p53-p73/BIK pathway. Cancers. 2019;11:1747. doi: 10.3390/cancers11111747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Feng G.W., Dong L.D., Shang W.J., Pang X.L., Wang Y. HDAC5 promotes cell proliferation in human hepatocellular carcinoma by up-regulating Six 1 expression. Eur Rev Med Pharmaco. 2014;18:811–816. [PubMed] [Google Scholar]
- 109.Zhang Y.T., Wu D., Xia F.J., Xian H.Y., Zhu X.Y., Cui H.J., et al. Downregulation of HDAC9 inhibits cell proliferation and tumor formation by inducing cell cycle arrest in retinoblastoma. Biochem Biophys Res Commun. 2016;473:600–606. doi: 10.1016/j.bbrc.2016.03.129. [DOI] [PubMed] [Google Scholar]
- 110.Zhu C.H., Chen Q., Xie Z.Q., Ai J., Tong L.J., Ding J., et al. The role of histone deacetylase 7 (HDAC7) in cancer cell proliferation: regulation on c-Myc. J Mol Med (Berl) 2011;89:279–289. doi: 10.1007/s00109-010-0701-7. [DOI] [PubMed] [Google Scholar]
- 111.Wang Z., Hu P., Tang F., Lian H., Chen X., Zhang Y., et al. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016;379:134–142. doi: 10.1016/j.canlet.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 112.Blixt N.C., Faulkner B.K., Astleford K., Lelich R., Schering J., Spencer E., et al. Class II and IV HDACs function as inhibitors of osteoclast differentiation. PLoS One. 2017;12 doi: 10.1371/journal.pone.0185441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dressel U., Bailey P.J., Wang S.C., Downes M., Evans R.M., Muscat G.E. A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. J Biol Chem. 2001;276:17007–17013. doi: 10.1074/jbc.M101508200. [DOI] [PubMed] [Google Scholar]
- 114.Ferrari L., Bragato C., Brioschi L., Spreafico M., Esposito S., Pezzotta A., et al. HDAC8 regulates canonical Wnt pathway to promote differentiation in skeletal muscles. J Cell Physiol. 2019;234:6067–6076. doi: 10.1002/jcp.27341. [DOI] [PubMed] [Google Scholar]
- 115.Li H., Xie H., Liu W., Hu R., Huang B., Tan Y.F., et al. A novel microRNA targeting HDAC5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J Clin Invest. 2009;119:3666–3677. doi: 10.1172/JCI39832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Spurling C.C., Godman C.A., Noonan E.J., Rasmussen T.P., Rosenberg D.W., Giardina C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol Carcinog. 2008;47:137–147. doi: 10.1002/mc.20373. [DOI] [PubMed] [Google Scholar]
- 117.Yang W., Liu Y.Y., Gao R.L., Yu H.Q., Sun T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli 1 signaling pathway. Cancer Lett. 2018;415:164–176. doi: 10.1016/j.canlet.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 118.Ye F., Chen Y., Hoang T., Montgomery R.L., Zhao X.H., Bu H., et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wagner F.F., Wewer M., Lewis M.C., Holson E.B. Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics. 2013;10:589–604. doi: 10.1007/s13311-013-0226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li Y., Wang F., Chen X.X., Wang J., Zhao Y.L., Li Y.J., et al. Zinc-dependent deacetylase (HDAC) inhibitors with different zinc binding groups. Curr Top Med Chem. 2019;19:223–241. doi: 10.2174/1568026619666190122144949. [DOI] [PubMed] [Google Scholar]
- 121.Ho T.C.S., Chan A.H.Y., Ganesan A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J Med Chem. 2020;63:12460–12484. doi: 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- 122.Tsimberidou A.M., Beer P.A., Cartwright C.A., Haymaker C., Vo H.H., Kiany S., et al. Preclinical development and first-in-human study of KA2507, a selective and potent inhibitor of histone deacetylase 6, for patients with refractory solid tumors. Clin Cancer Res. 2021;27:3584–3594. doi: 10.1158/1078-0432.CCR-21-0238. [DOI] [PubMed] [Google Scholar]
- 123.Moffat D., Patel S., Day F., Belfield A., Donald A., Rowlands M., et al. Discovery of 2-(6-{(6-fluoroquinolin-2-yl)methyl amino}bicyclo[3.1.0]hex-3-yl)-N-hydroxypyrimidine-5-carboxamide (CHR-3996), a class I selective orally active histone deacetylase inhibitor. J Med Chem. 2010;53:8663–8678. doi: 10.1021/jm101177s. [DOI] [PubMed] [Google Scholar]
- 124.Pinkerneil M., Hoffmann M.J., Kohlhof H., Schulz W.A., Niegisch G. Evaluation of the therapeutic potential of the novel isotype specific HDAC inhibitor 4SC-202 in urothelial carcinoma cell lines. Targeted Oncol. 2016;11:783–798. doi: 10.1007/s11523-016-0444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Duvic M., Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expet Opin Invest Drugs. 2007;16:1111–1120. doi: 10.1517/13543784.16.7.1111. [DOI] [PubMed] [Google Scholar]
- 126.Mann B.S., Johnson J.R., He K., Sridhara R., Abraham S., Booth B.P., et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin Cancer Res. 2007;13:2318–2322. doi: 10.1158/1078-0432.CCR-06-2672. [DOI] [PubMed] [Google Scholar]
- 127.Moskowitz A.J., Horwitz S.M. Targeting histone deacetylases in T-cell lymphoma. Leuk Lymphoma. 2017;58:1306–1319. doi: 10.1080/10428194.2016.1247956. [DOI] [PubMed] [Google Scholar]
- 128.Shustov A., Coiffier B., Horwitz S., Sokol L., Pro B., Wolfson J., et al. Romidepsin is effective and well tolerated in older patients with peripheral T-cell lymphoma: analysis of two phase II trials. Leuk Lymphoma. 2017;58:2335–2341. doi: 10.1080/10428194.2017.1295143. [DOI] [PubMed] [Google Scholar]
- 129.Whittaker S.J., Demierre M.F., Kim E.J., Rook A.H., Lerner A., Duvic M., et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 130.O'Connor O.A., Horwitz S., Masszi T., Van Hoof A., Brown P., Doorduijn J., et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33:2492–2499. doi: 10.1200/JCO.2014.59.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Poole R.M. Belinostat: first global approval. Drugs. 2014;74:1543–1554. doi: 10.1007/s40265-014-0275-8. [DOI] [PubMed] [Google Scholar]
- 132.Ning Z.Q., Li Z.B., Newman M.J., Shan S., Wang X.H., Pan D.S., et al. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol. 2012;69:901–909. doi: 10.1007/s00280-011-1766-x. [DOI] [PubMed] [Google Scholar]
- 133.Shi Y., Dong M., Hong X., Zhang W., Feng J., Zhu J., et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol. 2015;26:1766–1771. doi: 10.1093/annonc/mdv237. [DOI] [PubMed] [Google Scholar]
- 134.Rathkopf D.E., Picus J., Hussain A., Ellard S., Chi K.N., Nydam T., et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2013;72:537–544. doi: 10.1007/s00280-013-2224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.San-Miguel J.F., Hungria V.T.M., Yoon S.-S., Beksac M., Dimopoulos M.A., Elghandour A., et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15:1195–1206. doi: 10.1016/S1470-2045(14)70440-1. [DOI] [PubMed] [Google Scholar]
- 136.Shah R.R. Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Saf. 2019;42:235–245. doi: 10.1007/s40264-018-0773-9. [DOI] [PubMed] [Google Scholar]
- 137.Chia K.M., Beamish H., Jafferi K., Gabrielli B. Abstract #4612: selective inhibition of type I histone deacetylase improves tumor selective cytotoxicity over conventional pan specific inhibitors. Cancer Res. 2009;69:4612. [Google Scholar]
- 138.Luo Y.X., Li H.L. Structure-based inhibitor discovery of class I histone deacetylases (HDACs) Int J Mol Sci. 2020;21:8828. doi: 10.3390/ijms21228828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stubbs M.C., Kim W., Bariteau M., Davis T., Vempati S., Minehart J., et al. Selective inhibition of HDAC1 and HDAC2 as a potential therapeutic option for B-ALL. Clin Cancer Res. 2015;21:2348–2358. doi: 10.1158/1078-0432.CCR-14-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Santo L., Hideshima T., Kung A.L., Tseng J.C., Tamang D., Yang M., et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gordon M.S., Shapiro G., Sarantopoulos J., Juric D., Lu B., Chen P. A phase 1b study of the safety, pharmacokinetics, and preliminary antitumor activity of citarinostat (ACY-241) in combination with paclitaxel (Pac) in patients (pts) with advanced solid tumors (AST) J Clin Oncol. 2018;36:2547. [Google Scholar]
- 142.Zeng H.L., Qu J., Jin N., Xu J., Lin C.C., Chen Y., et al. Feedback activation of leukemia inhibitory factor receptor limits response to histone deacetylase inhibitors in breast cancer. Cancer Cell. 2016;30:459–473. doi: 10.1016/j.ccell.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 143.Gryder B.E., Sodji Q.H., Oyelere A.K. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012;4:505–524. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huang Z., Zhou W., Li Y.T., Cao M., Wang T.Q., Ma Y.K., et al. Novel hybrid molecule overcomes the limited response of solid tumours to HDAC inhibitors via suppressing JAK1-STAT3-BCL2 signalling. Theranostics. 2018;8:4995–5011. doi: 10.7150/thno.26627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jiang Z.F., Li W., Hu X.C., Zhang Q.Y., Sun T., Cui S.D., et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20:806–815. doi: 10.1016/S1470-2045(19)30164-0. [DOI] [PubMed] [Google Scholar]
- 146.Hu X.S., Wang L., Lin L., Han X.H., Dou G.F., Meng Z.Y., et al. A phase I trial of an oral subtype-selective histone deacetylase inhibitor, chidamide, in combination with paclitaxel and carboplatin in patients with advanced non-small cell lung cancer. Chin J Cancer Res. 2016;28:444–451. doi: 10.21147/j.issn.1000-9604.2016.04.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Awad M.M., Le Bruchec Y., Lu B., Ye J., Miller J., Lizotte P.H., et al. Selective histone deacetylase inhibitor ACY-241 (Citarinostat) plus nivolumab in advanced non-small cell lung cancer: results from a phase Ib study. Front Oncol. 2021;11 doi: 10.3389/fonc.2021.696512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Qian C., Lai C.J., Bao R., Wang D.G., Wang J., Xu G.X., et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin Cancer Res. 2012;18:4104–4113. doi: 10.1158/1078-0432.CCR-12-0055. [DOI] [PubMed] [Google Scholar]
- 149.Cai X., Zhai H.X., Wang J., Forrester J., Qu H., Yin L., et al. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDc-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J Med Chem. 2010;53:2000–2009. doi: 10.1021/jm901453q. [DOI] [PubMed] [Google Scholar]
- 150.Liu C., Ding H.Y., Li X.X., Pallasch C.P., Hong L.Y., Guo D.W., et al. A DNA/HDAC dual-targeting drug CY190602 with significantly enhanced anticancer potency. EMBO Mol Med. 2015;7:438–449. doi: 10.15252/emmm.201404580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li X.Y., Jiang Y.Q., Peterson Y.K., Xu T.Q., Himes R.A., Luo X., et al. Design of hydrazide-bearing HDACi based on panobinostat and their p53 and FLT3-ITD dependency in antileukemia activity. J Med Chem. 2020;63:5501–5525. doi: 10.1021/acs.jmedchem.0c00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Methot J.L., Chakravarty P.K., Chenard M., Close J., Cruz J.C., Dahlberg W.K., et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2008;18:973–978. doi: 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 153.Marek M., Shaik T.B., Heimburg T., Chakrabarti A., Lancelot J., Ramos-Morales E., et al. Characterization of histone deacetylase 8 (HDAC8) selective inhibition reveals specific active site structural and functional determinants. J Med Chem. 2018;61:10000–10016. doi: 10.1021/acs.jmedchem.8b01087. [DOI] [PubMed] [Google Scholar]
- 154.Burli R.W., Luckhurst C.A., Aziz O., Matthews K.L., Yates D., Lyons K.A., et al. Design, synthesis, and biological evaluation of potent and selective class IIa histone deacetylase (HDAC) inhibitors as a potential therapy for huntington's disease. J Med Chem. 2013;56:9934–9954. doi: 10.1021/jm4011884. [DOI] [PubMed] [Google Scholar]
- 155.Mak J.Y.W., Wu K.C., Gupta P.K., Barbero S., McLaughlin M.G., Lucke A.J., et al. HDAC7 inhibition by phenacetyl and phenylbenzoyl hydroxamates. J Med Chem. 2021;64:2186–2204. doi: 10.1021/acs.jmedchem.0c01967. [DOI] [PubMed] [Google Scholar]
- 156.Liang T., Hou X.B., Zhou Y., Yang X.Y., Fang H. Design, synthesis, and biological evaluation of 2,4-imidazolinedione derivatives as HDAC6 isoform-selective inhibitors. ACS Med Chem Lett. 2019;10:1122–1127. doi: 10.1021/acsmedchemlett.9b00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Son S.I., Cao J., Zhu C.L., Miller S.P., Lin H. Activity-auided design of HDAC11-specific inhibitors. ACS Chem Biol. 2019;14:1393–1397. doi: 10.1021/acschembio.9b00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lagger G., Rembold M., Tischler J., Schuettengruber B., Brunmeir R., Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Montgomery R.L., Davis C.A., Potthoff M.J., Haberland M., Fielitz J., Qi X., et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lin C.L., Tsai M.L., Lin C.Y., Hsu K.W., Hsieh W.S., Chi W.M., et al. HDAC1 and HDAC2 double knockout triggers cell apoptosis in advanced thyroid cancer. Int J Mol Sci. 2019;20:454. doi: 10.3390/ijms20020454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bhaskara S., Chyla B.J., Amann J.M., Knutson S.K., Cortez D., Sun Z.W., et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Haberland M., Mokalled M.H., Montgomery R.L., Olson E.N. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009;23:1625–1630. doi: 10.1101/gad.1809209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cadot B., Brunetti M., Coppari S., Fedeli S., de Rinaldis E., Dello Russo C., et al. Loss of histone deacetylase 4 causes segregation defects during mitosis of p53-deficient human tumor cells. Cancer Res. 2009;69:6074–6082. doi: 10.1158/0008-5472.CAN-08-2796. [DOI] [PubMed] [Google Scholar]
- 164.Chang S., McKinsey T.A., Zhang C.L., Richardson J.A., Hill J.A., Olson E.N. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhang C.L., Mckinsey T.A., Chang S., Antos C.L., Hill J.A., Olson E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chang S., Young B.D., Li S., Qi X., Richardson J.A., Olson E.N. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006;126:321–334. doi: 10.1016/j.cell.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 167.Zhang Y., Kwon S., Yamaguchi T., Cubizolles F., Rousseaux S., Kneissel M., et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee H.Y., Nepali K., Huang F.I., Chang C.Y., Lai M.J., Li Y.H., et al. (N-Hydroxycarbonylbenylamino)quinolines as selective histone deacetylase 6 inhibitors suppress growth of multiple myeloma in vitro and in vivo. J Med Chem. 2018;61:905–917. doi: 10.1021/acs.jmedchem.7b01404. [DOI] [PubMed] [Google Scholar]
- 169.Santo L., Cirstea D., Wang B., Yao T.P., Wu J.Y., Waterman P., et al. Role of selective HDAC6 inhibition on multiple myeloma bone disease. Blood. 2012;120 328-28. [Google Scholar]
- 170.Yang Z., Wang T.J., Wang F., Niu T., Liu Z.W., Chen X.X., et al. Discovery of selective histone deacetylase 6 inhibitors using the quinazoline as the cap for the treatment of cancer. J Med Chem. 2016;59:1455–1470. doi: 10.1021/acs.jmedchem.5b01342. [DOI] [PubMed] [Google Scholar]
- 171.Li Y.X., Zhang X.Y., Zhu S.Q., Dejene E.A., Peng W.Q., Sepulveda A., et al. HDAC10 regulates cancer stem-like cell properties in KRAS-driven lung adenocarcinoma. Cancer Res. 2020;80:3265–3278. doi: 10.1158/0008-5472.CAN-19-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bryant D.T., Landles C., Papadopoulou A.S., Benjamin A.C., Duckworth J.K., Rosahl T., et al. Disruption to schizophrenia-associated gene Fez 1 in the hippocampus of HDAC11 knockout mice. Sci Rep. 2017;7 doi: 10.1038/s41598-017-11630-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Deubzer H.E., Schier M.C., Oehme I., Lodrini M., Haendler B., Sommer A., et al. HDAC11 is a novel drug target in carcinomas. Int J Cancer. 2013;132:2200–2208. doi: 10.1002/ijc.27876. [DOI] [PubMed] [Google Scholar]
- 174.Methot J.L., Hoffman D.M., Witter D.J., Stanton M.G., Harrington P., Hamblett C., et al. Delayed and prolonged histone hyperacetylation with a selective HDAC1/HDAC2 inhibitor. ACS Med Chem Lett. 2014;5:340–345. doi: 10.1021/ml4004233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhou J., Li M., Chen N., Wang S., Luo H.B., Zhang Y., et al. Computational design of a time-dependent histone deacetylase 2 selective inhibitor. ACS Chem Biol. 2015;10:687–692. doi: 10.1021/cb500767c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ononye S.N., VanHeyst M.D., Oblak E.Z., Zhou W., Ammar M., Anderson A.C., et al. Tropolones as lead-like natural products: the development of potent and selective histone deacetylase inhibitors. ACS Med Chem Lett. 2013;4:757–761. doi: 10.1021/ml400158k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wagner F.F., Lundh M., Kaya T., McCarren P., Zhang Y.L., Chattopadhyay S., et al. An isochemogenic set of inhibitors to define the therapeutic potential of histone deacetylases in beta-cell protection. ACS Chem Biol. 2016;11:363–374. doi: 10.1021/acschembio.5b00640. [DOI] [PubMed] [Google Scholar]
- 178.Cheng G.L., Wang Z., Yang J.Y., Bao Y., Xu Q.H., Zhao L.X., et al. Design, synthesis and biological evaluation of novel indole derivatives as potential HDAC/BRD4 dual inhibitors and anti-leukemia agents. Bioorg Chem. 2019;84:410–417. doi: 10.1016/j.bioorg.2018.12.011. [DOI] [PubMed] [Google Scholar]
- 179.McClure J.J., Zhang C., Inks E.S., Peterson Y.K., Li J., Chou C.J. Development of allosteric hydrazide-containing class I histone deacetylase inhibitors for use in acute myeloid leukemia. J Med Chem. 2016;59:9942–9959. doi: 10.1021/acs.jmedchem.6b01385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Butler K.V., Kalin J., Brochier C., Vistoli G., Langley B., Kozikowski A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kozikowski A.P., Shen S., Pardo M., Tavares M.T., Szarics D., Benoy V., et al. Brain penetrable histone deacetylase 6 inhibitor SW-100 ameliorates memory and learning impairments in a mouse model of fragile x syndrome. ACS Chem Neurosci. 2019;10:1679–1695. doi: 10.1021/acschemneuro.8b00600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bergman J.A., Woan K., Perez-Villarroel P., Villagra A., Sotomayor E.M., Kozikowski A.P. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J Med Chem. 2012;55:9891–9899. doi: 10.1021/jm301098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lee J.H., Mahendran A., Yao Y., Ngo L., Venta-Perez G., Choy M.L., et al. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc Natl Acad Sci U S A. 2013;110:15704–15709. doi: 10.1073/pnas.1313893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Awad M.M., Le Bruchec Y., Lu B., Ye J., Miller J.A., Lizotte P.H., et al. Corrigendum: selective histone deacetylase inhibitor ACY-241 (citarinostat) plus nivolumab in advanced non-small cell lung cancer: results from a phase Ib study. Front Oncol. 2022;12 doi: 10.3389/fonc.2022.889996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Balasubramanian S., Ramos J., Luo W., Sirisawad M., Verner E., Buggy J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 186.Huang W.J., Wang Y.C., Chao S.W., Yang C.Y., Chen L.C., Lin M.H., et al. Synthesis and biological evaluation of ortho-aryl N-hydroxycinnamides as potent histone deacetylase (HDAC) 8 isoform-selective inhibitors. ChemMedChem. 2012;7:1815–1824. doi: 10.1002/cmdc.201200300. [DOI] [PubMed] [Google Scholar]
- 187.Ingham O.J., Paranal R.M., Smith W.B., Escobar R.A., Yueh H., Snyder T., et al. Development of a potent and selective HDAC8 inhibitor. ACS Med Chem Lett. 2016;7:929–932. doi: 10.1021/acsmedchemlett.6b00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Géraldy M., Morgen M., Sehr P., Steimbach R.R., Moi D., Ridinger J., et al. Selective inhibition of histone deacetylase 10: hydrogen bonding to the gatekeeper residue is implicated. J Med Chem. 2019;62:4426–4443. doi: 10.1021/acs.jmedchem.8b01936. [DOI] [PubMed] [Google Scholar]
- 189.Martin M.W., Lee J.Y., Lancia D.R., Jr., Ng P.Y., Han B., Thomason J.R., et al. Discovery of novel N-hydroxy-2-arylisoindoline-4-carboxamides as potent and selective inhibitors of HDAC11. Bioorg Med Chem Lett. 2018;28:2143–2147. doi: 10.1016/j.bmcl.2018.05.021. [DOI] [PubMed] [Google Scholar]
- 190.Lobera M., Madauss K.P., Pohlhaus D.T., Wright Q.G., Trocha M., Schmidt D.R., et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013;9:319–325. doi: 10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- 191.Luo L., Martin S.C., Parkington J., Cadena S.M., Zhu J., Ibebunjo C., et al. HDAC4 controls muscle homeostasis through deacetylation of myosin heavy chain, PGC-1 alpha, and Hsc70. Cell Rep. 2019;29:749–763 e12. doi: 10.1016/j.celrep.2019.09.023. [DOI] [PubMed] [Google Scholar]
- 192.Luckhurst C.A., Breccia P., Stott A.J., Aziz O., Birch H.L., Burli R.W., et al. Potent, selective, and CNS-penetrant tetrasubstituted cyclopropane class IIa histone deacetylase (HDAC) inhibitors. ACS Med Chem Lett. 2016;7:34–39. doi: 10.1021/acsmedchemlett.5b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Luckhurst C.A., Aziz O., Beaumont V., Burli R.W., Breccia P., Maillard M.C., et al. Development and characterization of a CNS-penetrant benzhydryl hydroxamic acid class IIa histone deacetylase inhibitor. Bioorg Med Chem Lett. 2019;29:83–88. doi: 10.1016/j.bmcl.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 194.Li X.Y., Inks E.S., Li X.G., Hou J.N., Chou C.J., Zhang J., et al. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J Med Chem. 2014;57:3324–3341. doi: 10.1021/jm401877m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Li X.Y., Zhang Y.J., Jiang Y.Q., Wu J.D., Inks E.S., Chou C.J., et al. Selective HDAC inhibitors with potent oral activity against leukemia and colorectal cancer: design, structure-activity relationship and anti-tumor activity study. Eur J Med Chem. 2017;134:185–206. doi: 10.1016/j.ejmech.2017.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Soumyanarayanan U., Ramanujulu P.M., Mustafa N., Haider S., Fang Nee A.H., Tong J.X., et al. Discovery of a potent histone deacetylase (HDAC) 3/6 selective dual inhibitor. Eur J Med Chem. 2019;184 doi: 10.1016/j.ejmech.2019.111755. [DOI] [PubMed] [Google Scholar]
- 197.Yang W., Li L.X., Wang Y.L., Wu X.W., Li T.T., Yang N., et al. Design, synthesis and biological evaluation of isoquinoline-based derivatives as novel histone deacetylase inhibitors. Bioorg Med Chem. 2015;23:5881–5890. doi: 10.1016/j.bmc.2015.06.071. [DOI] [PubMed] [Google Scholar]
- 198.Negmeldin A.T., Padige G., Bieliauskas A.V., Pflum M.K. Structural requirements of HDAC inhibitors: SAHA analogues modified at the C2 position display HDAC6/8 selectivity. ACS Med Chem Lett. 2017;8:281–286. doi: 10.1021/acsmedchemlett.6b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Negmeldin A.T., Knoff J.R., Pflum M.K.H. The structural requirements of histone deacetylase inhibitors: C4-modified SAHA analogs display dual HDAC6/HDAC8 selectivity. Eur J Med Chem. 2018;143:1790–1806. doi: 10.1016/j.ejmech.2017.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Negmeldin A.T., Pflum M.K.H. The structural requirements of histone deacetylase inhibitors: SAHA analogs modified at the C5 position display dual HDAC6/8 selectivity. Bioorg Med Chem Lett. 2017;27:3254–3258. doi: 10.1016/j.bmcl.2017.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rodrigues D.A., Ferreira-Silva G.A., Ferreira A.C., Fernandes R.A., Kwee J.K., Sant'Anna C.M., et al. Design, synthesis, and pharmacological evaluation of novel N-acylhydrazone derivatives as potent histone deacetylase 6/8 dual inhibitors. J Med Chem. 2016;59:655–670. doi: 10.1021/acs.jmedchem.5b01525. [DOI] [PubMed] [Google Scholar]
- 202.Arrighetti N., Corno C., Gatti L. Drug combinations with HDAC inhibitors in antitumor therapy. Crit Rev Oncog. 2015;20:83–117. doi: 10.1615/critrevoncog.2014012378. [DOI] [PubMed] [Google Scholar]
- 203.Pomeroy A.E., Schmidt E.V., Sorger P.K., Palmer A.C. Drug independence and the curability of cancer by combination chemotherapy. Trends Cancer. 2022;8:915–929. doi: 10.1016/j.trecan.2022.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tonekaboni S.A.M., Ghoraie L.S., Manem V.S.K., Haibe-Kains B. Predictive approaches for drug combination discovery in cancer. Briefings Bioinf. 2018;19:263–276. doi: 10.1093/bib/bbw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yuan S.Q., Wang F., Chen G., Zhang H., Feng L., Wang L., et al. Effective elimination of cancer stem cells by a novel drug combination strategy. Stem Cell. 2013;31:23–34. doi: 10.1002/stem.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zahedi P., De Souza R., Huynh L., Piquette-Miller M., Allen C. Combination drug delivery strategy for the treatment of multidrug resistant ovarian cancer. Mol Pharm. 2011;8:260–269. doi: 10.1021/mp100323z. [DOI] [PubMed] [Google Scholar]
- 207.Al-Lazikani B., Banerji U., Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30:679–692. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 208.Anighoro A., Bajorath J., Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem. 2014;57:7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- 209.Fortin S., Bérubé G. Advances in the development of hybrid anticancer drugs. Expet Opin Drug Discov. 2013;8:1029–1047. doi: 10.1517/17460441.2013.798296. [DOI] [PubMed] [Google Scholar]
- 210.Morphy R., Rankovic Z. Designed multiple ligands. an emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 211.Chao M.W., Chang L.H., Tu H.J., Chang C.D., Lai M.J., Chen Y.Y., et al. Combination treatment strategy for pancreatic cancer involving the novel HDAC inhibitor MPT0E028 with a MEK inhibitor beyond K-Ras status. Clin Epigenet. 2019;11:85. doi: 10.1186/s13148-019-0681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kyaw M.T.H., Yamaguchi Y., Choijookhuu N., Yano K., Takagi H., Takahashi N., et al. The HDAC inhibitor, SAHA, combined with cisplatin synergistically induces apoptosis in alpha-fetoprotein-producing hepatoid adenocarcinoma cells. Acta Histochem Cytoc. 2019;52:1–8. doi: 10.1267/ahc.18044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ngamphaiboon N., Dy G.K., Ma W.W., Zhao Y., Reungwetwattana T., DePaolo D., et al. A phase I study of the histone deacetylase (HDAC) inhibitor entinostat, in combination with sorafenib in patients with advanced solid tumors. Invest N Drugs. 2015;33:225–232. doi: 10.1007/s10637-014-0174-6. [DOI] [PubMed] [Google Scholar]
- 214.Novotny-Diermayr V., Hart S., Goh K.C., Cheong A., Ong L.C., Hentze H., et al. The oral HDAC inhibitor pracinostat (SB939) is efficacious and synergistic with the JAK2 inhibitor pacritinib (SB1518) in preclinical models of AML. Blood Cancer J. 2012;2:e69. doi: 10.1038/bcj.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Que Y., Zhang X.L., Liu Z.X., Zhao J.J., Pan Q.Z., Wen X.Z., et al. Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J Immunother Cancer. 2021;9 doi: 10.1136/jitc-2020-001696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Mahboobi S., Dove S., Sellmer R., Winkler M., Eichhorn E., Pongratz H., et al. Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: a series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rβ, and histone deacetylases. J Med Chem. 2009;52:2265–2279. doi: 10.1021/jm800988r. [DOI] [PubMed] [Google Scholar]
- 217.Pan T., Dan Y.R., Guo D.F., Jiang J.H., Ran D.Z., Zhang L., et al. Discovery of 2,4-pyrimidinediamine derivatives as potent dual inhibitors of ALK and HDAC. Eur J Med Chem. 2021;224 doi: 10.1016/j.ejmech.2021.113672. [DOI] [PubMed] [Google Scholar]
- 218.Li Y.J., Huang Y.J., Cheng H.M., Xu F., Qi R.X., Dai B.T., et al. Discovery of BRAF/HDAC dual inhibitors suppressing proliferation of human colorectal cancer cells. Front Chem. 2022;10 doi: 10.3389/fchem.2022.910353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Geng A.X., Cui H., Zhang L.Y., Chen X., Li H.M., Lu T., et al. Discovery of novel phenoxybenzamide analogues as Raf/HDAC dual inhibitors. Bioorg Med Chem Lett. 2019;29:1605–1608. doi: 10.1016/j.bmcl.2019.04.047. [DOI] [PubMed] [Google Scholar]
- 220.Cao Z.X., Yang F.F., Wang J., Gu Z.C., Lin S.X., Wang P., et al. Indirubin derivatives as dual inhibitors targeting cyclin-dependent kinase and histone deacetylase for treating cancer. J Med Chem. 2021;64:15280–15296. doi: 10.1021/acs.jmedchem.1c01311. [DOI] [PubMed] [Google Scholar]
- 221.Yu Y., Ran D., Jiang J., Pan T., Dan Y., Tang Q., et al. Discovery of novel 9H-purin derivatives as dual inhibitors of HDAC1 and CDK2. Bioorg Med Chem Lett. 2019;29:2136–2140. doi: 10.1016/j.bmcl.2019.06.059. [DOI] [PubMed] [Google Scholar]
- 222.Rangasamy L., Ortín I., Zapico J.M., Coderch C., Ramos A., de Pascual-Teresa B. New dual CK2/HDAC1 inhibitors with nanomolar inhibitory activity against both Enzymes. ACS Med Chem Lett. 2020;11:713–719. doi: 10.1021/acsmedchemlett.9b00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lu D., Yan J., Wang L., Liu H., Zeng L., Zhang M., et al. Design, synthesis, and biological evaluation of the first c-Met/HDAC inhibitors based on pyridazinone derivatives. ACS Med Chem Lett. 2017;8:830–834. doi: 10.1021/acsmedchemlett.7b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ko K.S., Steffey M.E., Brandvold K.R., Soellner M.B. Development of a chimeric c-Src kinase and HDAC inhibitor. ACS Med Chem Lett. 2013;4:779–783. doi: 10.1021/ml400175d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zhu Y., Ran T., Chen X., Niu J.Q., Zhao S., Lu T., et al. Synthesis and biological evaluation of 1-(2-aminophenyl)-3-arylurea derivatives as potential EphA2 and HDAC dual inhibitors. Chem Pharm Bull. 2016;64:1136–1141. doi: 10.1248/cpb.c16-00154. [DOI] [PubMed] [Google Scholar]
- 226.Mustafa M., Abd El-Hafeez A.A., Abdelhamid D., Katkar G.D., Mostafa Y.A., Ghosh P., et al. A first-in-class anticancer dual HDAC2/FAK inhibitors bearing hydroxamates/benzamides capped by pyridinyl-1,2,4-triazoles. Eur J Med Chem. 2021;222 doi: 10.1016/j.ejmech.2021.113569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.De Simone A., La Pietra V., Betari N., Petragnani N., Conte M., Daniele S., et al. Discovery of the first-in-class GSK-3 beta/HDAC dual inhibitor as disease-modifying agent to combat alzheimer's disease. ACS Med Chem Lett. 2019;10:469–474. doi: 10.1021/acsmedchemlett.8b00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Yao L., Mustafa N., Tan E.C., Poulsen A., Singh P., Duong-Thi M.D., et al. Design and synthesis of ligand efficient dual inhibitors of janus kinase (JAK) and histone deacetylase (HDAC) based on ruxolitinib and vorinostat. J Med Chem. 2017;60:8336–8357. doi: 10.1021/acs.jmedchem.7b00678. [DOI] [PubMed] [Google Scholar]
- 229.Yang E.G., Mustafa N., Tan E.C., Poulsen A., Ramanujulu P.M., Chng W.J., et al. Design and synthesis of janus kinase 2 (JAK2) and histone deacetlyase (HDAC) bispecific inhibitors based on pacritinib and evidence of dual pathway inhibition in hematological cell lines. J Med Chem. 2016;59:8233–8262. doi: 10.1021/acs.jmedchem.6b00157. [DOI] [PubMed] [Google Scholar]
- 230.Xing K., Zhang J., Han Y., Tong T., Liu D., Zhao L.X. Design, synthesis and bioactivity evaluation of 4,6-disubstituted pyrido[3,2-d]pyrimidine derivatives as Mnk and HDAC inhibitors. Molecules. 2020;25:4318. doi: 10.3390/molecules25184318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Zhang M.M., Wei W., Peng C.J., Ma X.D., He X., Zhang H., et al. Discovery of novel pyrazolopyrimidine derivatives as potent mTOR/HDAC bi-functional inhibitors via pharmacophore-merging strategy. Bioorg Med Chem Lett. 2021;49 doi: 10.1016/j.bmcl.2021.128286. [DOI] [PubMed] [Google Scholar]
- 232.Bass A.K.A., Nageeb E.M., El-Zoghbi M.S., Mohamed M.F.A., Badr M., Abuo-Rahma G.E.A. Utilization of cyanopyridine in design and synthesis of first-in-class anticancer dual acting PIM-1 kinase/HDAC inhibitors. Bioorg Chem. 2022;119 doi: 10.1016/j.bioorg.2021.105564. [DOI] [PubMed] [Google Scholar]
- 233.Yan W.Z., Ling L.J., Wu Y.R., Yang K.X., Liu R.Q., Zhang J.F., et al. Structure-based design of dual-acting compounds targeting adenosine A (2A) receptor and histone deacetylase as novel tumor immunotherapeutic agents. J Med Chem. 2021;64:16573–16597. doi: 10.1021/acs.jmedchem.1c01155. [DOI] [PubMed] [Google Scholar]
- 234.Tang C., Li C., Zhang S., Hu Z., Wu J., Dong C., et al. Novel bioactive hybrid compound dual targeting estrogen receptor and histone deacetylase for the treatment of breast cancer. J Med Chem. 2015;58:4550–4572. doi: 10.1021/acs.jmedchem.5b00099. [DOI] [PubMed] [Google Scholar]
- 235.Chen G.L., Wang L.H., Wang J., Chen K., Zhao M., Sun Z.Z., et al. Discovery of a small molecular compound simultaneously targeting RXR and HADC: design, synthesis, molecular docking and bioassay. Bioorg Med Chem Lett. 2013;23:3891–3895. doi: 10.1016/j.bmcl.2013.04.067. [DOI] [PubMed] [Google Scholar]
- 236.Peng F.W., Wu T.T., Ren Z.W., Xue J.Y., Shi L. Hybrids from 4-anilinoquinazoline and hydroxamic acid as dual inhibitors of vascular endothelial growth factor receptor-2 and histone deacetylase. Bioorg Med Chem Lett. 2015;25:5137–5141. doi: 10.1016/j.bmcl.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 237.Atkinson S.J., Soden P.E., Angell D.C., Bantscheff M., Chung C.W., Giblin K.A., et al. The structure based design of dual HDAC/BET inhibitors as novel epigenetic probes. Medchemcomm. 2014;5:342–351. [Google Scholar]
- 238.Zhang Z.M., Hou S.H., Chen H.L., Ran T., Jiang F., Bian Y.Y., et al. Targeting epigenetic reader and eraser: rational design, synthesis and in vitro evaluation of dimethylisoxazoles derivatives as BRD4/HDAC dual inhibitors. Bioorg Med Chem Lett. 2016;26:2931–2935. doi: 10.1016/j.bmcl.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 239.Amemiya S., Yamaguchi T., Hashimoto Y., Noguchi-Yachide T. Synthesis and evaluation of novel dual BRD4/HDAC inhibitors. Bioorg Med Chem Lett. 2017;25:3677–3684. doi: 10.1016/j.bmc.2017.04.043. [DOI] [PubMed] [Google Scholar]
- 240.Pan Z.P., Li X., Wang Y.J., Jiang Q.L., Jiang L., Zhang M., et al. Discovery of thieno[2,3-d]pyrimidine-based hydroxamic acid derivatives as bromodomain-containing protein 4/histone deacetylase dual inhibitors induce autophagic cell death in colorectal carcinoma cells. J Med Chem. 2020;63:3678–3700. doi: 10.1021/acs.jmedchem.9b02178. [DOI] [PubMed] [Google Scholar]
- 241.Orlikova B., Schnekenburger M., Zloh M., Golais F., Diederich M., Tasdemir D. Natural chalcones as dual inhibitors of HDACs and NF-kappaB. Oncol Rep. 2012;28:797–805. doi: 10.3892/or.2012.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.He S., Dong G., Wu S., Fang K., Miao Z., Wang W., et al. Small molecules simultaneously inhibiting p53-murine double minute 2 (MDM2) interaction and histone deacetylases (HDACs): discovery of novel multitargeting antitumor agents. J Med Chem. 2018;61:7245–7260. doi: 10.1021/acs.jmedchem.8b00664. [DOI] [PubMed] [Google Scholar]
- 243.Cui H., Huang J.K., Lei Y., Chen Q.W., Hu Z., Niu J.Q., et al. Design and synthesis of dual inhibitors targeting snail and histone deacetylase for the treatment of solid tumour cancer. Eur J Med Chem. 2022;229 doi: 10.1016/j.ejmech.2021.114082. [DOI] [PubMed] [Google Scholar]
- 244.Ren Y.H., Li S.S., Zhu R., Wan C.Y., Song D.M., Zhu J.W., et al. Discovery of STAT3 and histone deacetylase (HDAC) dual-pathway inhibitors for the treatment of solid cancer. J Med Chem. 2021;64:7468–7482. doi: 10.1021/acs.jmedchem.1c00136. [DOI] [PubMed] [Google Scholar]
- 245.Zhu T.B., Chen X., Li C.L., Tu J., Liu N., Xu D.F., et al. Lanosterol 14 alpha-demethylase (CYP51)/histone deacetylase (HDAC) dual inhibitors for treatment of candida tropicalis and cryptococcus neoformans infections. Eur J Med Chem. 2021;221 doi: 10.1016/j.ejmech.2021.113524. [DOI] [PubMed] [Google Scholar]
- 246.Yuan Z.G., Chen S.P., Gao C.M., Dai Q.Z., Zhang C.L., Sun Q.S., et al. Development of a versatile DNMT and HDAC inhibitor C02S modulating multiple cancer hallmarks for breast cancer therapy. Bioorg Chem. 2019;87:200–208. doi: 10.1016/j.bioorg.2019.03.027. [DOI] [PubMed] [Google Scholar]
- 247.Sun Q.S., Dai Q.Z., Zhang C.L., Chen Y., Zhao L., Yuan Z.B., et al. Design, synthesis and antitumor evaluations of nucleoside base hydroxamic acid derivatives as DNMT and HDAC dual inhibitors. Chin Chem Lett. 2021;32:2479–2483. [Google Scholar]
- 248.Romanelli A., Stazi G., Fioravanti R., Zwergel C., Di Bello E., Pomella S., et al. Design of first-in-class dual EZH2/HDAC inhibitor: biochemical activity and biological evaluation in cancer cells. ACS Med Chem Lett. 2020;11:977–983. doi: 10.1021/acsmedchemlett.0c00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zheng H.T., Dai Q.Z., Yuan Z.G., Fan T.T., Zhang C.L., Liu Z.J., et al. Quinazoline-based hydroxamic acid derivatives as dual histone methylation and deacetylation inhibitors for potential anticancer agents. Bioorg Med Chem. 2022;53 doi: 10.1016/j.bmc.2021.116524. [DOI] [PubMed] [Google Scholar]
- 250.Wei T.T., Lin Y.T., Chen W.S., Luo P., Lin Y.C., Shun C.T., et al. Dual targeting of 3-hydroxy-3-methylglutaryl coenzyme a reductase and histone deacetylase as a therapy for colorectal cancer. EBioMedicine. 2016;10:124–136. doi: 10.1016/j.ebiom.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Fang K., Dong G.Q., Li Y., He S.P., Wu Y., Wu S.C., et al. Discovery of novel indoleamine 2,3-dioxygenase 1 (IDO1) and histone deacetylase (HDAC) dual inhibitors. ACS Med Chem Lett. 2018;9:312–317. doi: 10.1021/acsmedchemlett.7b00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Mehndiratta S., Qian B., Chuang J.Y., Liou J.P., Shih J.C. N-methylpropargylamine-conjugated hydroxamic acids as dual inhibitors of monoamine oxidase a and histone deacetylase for glioma treatment. J Med Chem. 2022;65:2208–2224. doi: 10.1021/acs.jmedchem.1c01726. [DOI] [PubMed] [Google Scholar]
- 253.Wang Y.J., Yang L.M., Hou J.Y., Zou Q., Gao Q., Yao W.H., et al. Hierarchical virtual screening of the dual MMP-2/HDAC-6 inhibitors from natural products based on pharmacophore models and molecular docking. J Biomol Struct Dyn. 2019;37:649–670. doi: 10.1080/07391102.2018.1434833. [DOI] [PubMed] [Google Scholar]
- 254.Chen W., Dong G.Q., Wu Y., Zhang W.N., Miao C.Y., Sheng C.Q. Dual NAMPT/HDAC inhibitors as a new strategy for multitargeting antitumor drug discovery. ACS Med Chem Lett. 2018;9:34–38. doi: 10.1021/acsmedchemlett.7b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Yuan Z.G., Chen S.P., Sun Q.S., Wang N., Li D., Miao S.H., et al. Olaparib hydroxamic acid derivatives as dual PARP and HDAC inhibitors for cancer therapy. Bioorg Med Chem. 2017;25:4100–4109. doi: 10.1016/j.bmc.2017.05.058. [DOI] [PubMed] [Google Scholar]
- 256.Rabal O., Sanchez-Arias J.A., Cuadrado-Tejedor M., de Miguel I., Perez-Gonzalez M., Garcia-Barroso C., et al. Discovery of in vivo chemical probes for treating alzheimer's disease: dual phosphodiesterase 5 (PDE5) and class I histone deacetylase selective inhibitors. ACS Chem Neurosci. 2019;10:1765–1782. doi: 10.1021/acschemneuro.8b00648. [DOI] [PubMed] [Google Scholar]
- 257.Rabal O., Sanchez-Arias J.A., Cuadrado-Tejedor M., de Miguel I., Perez-Gonzalez M., Garcia-Barroso C., et al. Design, synthesis, biological evaluation and in vivo testing of dual phosphodiesterase 5 (PDE5) and histone deacetylase 6 (HDAC6)-selective inhibitors for the treatment of alzheimer's disease. Eur J Med Chem. 2018;150:506–524. doi: 10.1016/j.ejmech.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 258.Zhou Y., Liu X.T., Xue J.X., Liu L.L., Liang T., Li W., et al. Discovery of peptide boronate derivatives as histone deacetylase and proteasome dual inhibitors for overcoming bortezomib resistance of multiple myeloma. J Med Chem. 2020;63:4701–4715. doi: 10.1021/acs.jmedchem.9b02161. [DOI] [PubMed] [Google Scholar]
- 259.Liu M., Gao S., Liang T., Qiu X.T., Yang X.Y., Fang H., et al. Discovery of novel src homology-2 domain-containing phosphatase 2 and histone deacetylase dual inhibitors with potent antitumor efficacy and enhanced antitumor immunity. J Med Chem. 2022;65:12200–12218. doi: 10.1021/acs.jmedchem.2c00866. [DOI] [PubMed] [Google Scholar]
- 260.Cincinelli R., Musso L., Artali R., Guglielmi M.B., La Porta I., Melito C., et al. Hybrid topoisomerase I and HDAC inhibitors as dual action anticancer agents. PLoS One. 2018;13 doi: 10.1371/journal.pone.0205018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Zhang X., Bao B., Yu X.H., Tong L.J., Luo Y., Huang Q.Q., et al. The discovery and optimization of novel dual inhibitors of topoisomerase II and histone deacetylase. Bioorg Med Chem. 2013;21:6981–6995. doi: 10.1016/j.bmc.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 262.Chen J.W., Li D., Li W.L., Yin J.X., Zhang Y.Y., Yuan Z.G., et al. Design, synthesis and anticancer evaluation of acridine hydroxamic acid derivatives as dual topo and HDAC inhibitors. Bioorg Med Chem. 2018;26:3958–3966. doi: 10.1016/j.bmc.2018.06.016. [DOI] [PubMed] [Google Scholar]
- 263.Guerrant W., Patil V., Canzoneri J.C., Oyelere A.K. Dual targeting of histone deacetylase and topoisomerase II with novel bifunctional inhibitors. J Med Chem. 2012;55:1465–1477. doi: 10.1021/jm200799p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Xie R., Li Y., Tang P.W., Yuan Q.P. Rational design, synthesis and preliminary antitumor activity evaluation of a chlorambucil derivative with potent DNA/HDAC dual-targeting inhibitory activity. Bioorg Med Chem Lett. 2017;27:4415–4420. doi: 10.1016/j.bmcl.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 265.Xie R., Tang P.W., Yuan Q.P. Rational design and characterization of a DNA/HDAC dual-targeting inhibitor containing nitrogen mustard and 2-aminobenzamide moieties. MedChemComm. 2018;9:344–352. doi: 10.1039/c7md00476a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Griffith D., Morgan M.P., Marmion C.J. A novel anti-cancer bifunctional platinum drug candidate with dual DNA binding and histone deacetylase inhibitory activity. Chem. 2009;44:6735–6737. doi: 10.1039/b916715c. [DOI] [PubMed] [Google Scholar]
- 267.Chen C., Li X., Zhao H.J., Liu M., Du J.T., Zhang J., et al. Discovery of DNA-targeting HDAC inhibitors with potent antitumor efficacy in vivo that trigger antitumor immunity. J Med Chem. 2022;65:3667–3683. doi: 10.1021/acs.jmedchem.1c02225. [DOI] [PubMed] [Google Scholar]
- 268.Chen C., Chu H.R., Wang A.Y., Yin H.H., Gao Y.Q., Liu S.H., et al. Discovery of 2,5-diphenyl-1,3,4-thiadiazole derivatives as HDAC inhibitors with DNA binding affinity. Eur J Med Chem. 2022;241 doi: 10.1016/j.ejmech.2022.114634. [DOI] [PubMed] [Google Scholar]
- 269.Liang T., Zhou Y., Elhassan R.M., Hou X.B., Yang X.Y., Fang H. HDAC-Bax multiple ligands enhance Bax-dependent apoptosis in HeLa Cells. J Med Chem. 2020;63:12083–12099. doi: 10.1021/acs.jmedchem.0c01454. [DOI] [PubMed] [Google Scholar]
- 270.Zhou R.L., Fan S.Y., Zhang M.M., Zhang Q.S., Hu J., Wang M.P., et al. Design, synthesis, and bioactivity evaluation of novel Bcl-2/HDAC dual-target inhibitors for the treatment of multiple myeloma. Bioorg Med Chem Lett. 2019;29:349–352. doi: 10.1016/j.bmcl.2018.12.052. [DOI] [PubMed] [Google Scholar]
- 271.Wu Y.W., Chao M.W., Tu H.J., Chen L.C., Hsu K.C., Liou J.P., et al. A novel dual HDAC and HSP90 inhibitor, MPT0G449, downregulates oncogenic pathways in human acute leukemia in vitro and in vivo. Oncogenesis. 2021;10:39. doi: 10.1038/s41389-021-00331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Mehndiratta S., Lin M.H., Wu Y.W., Chen C.H., Wu T.Y., Chuang K.H., et al. N-alkyl-hydroxybenzoyl anilide hydroxamates as dual inhibitors of HDAC and HSP90, downregulating IFN-gamma induced PD-L1 expression. Eur J Med Chem. 2020;185 doi: 10.1016/j.ejmech.2019.111725. [DOI] [PubMed] [Google Scholar]
- 273.Zhang X., Kong Y.N., Zhang J., Su M.B., Zhou Y.B., Zang Y., et al. Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors. Eur J Med Chem. 2015;95:127–135. doi: 10.1016/j.ejmech.2015.03.035. [DOI] [PubMed] [Google Scholar]
- 274.Cao F.Y., Xiao Z.P., Chen S.W., Zhao C.L., Chen D., Haisma H.J., et al. HDAC/MIF dual inhibitor inhibits NSCLC cell survival and proliferation by blocking the AKT pathway. Bioorg Chem. 2021;117 doi: 10.1016/j.bioorg.2021.105396. [DOI] [PubMed] [Google Scholar]
- 275.Omidkhah N., Ghodsi R. NO-HDAC dual inhibitors. Eur J Med Chem. 2022;227 doi: 10.1016/j.ejmech.2021.113934. [DOI] [PubMed] [Google Scholar]
- 276.Luo G.S., Lin X., Ren S.N., Wu S.J., Wang X., Ma L.Y., et al. Development of novel tetrahydroisoquinoline-hydroxamate conjugates as potent dual SERDs/HDAC inhibitors for the treatment of breast cancer. Eur J Med Chem. 2021;226 doi: 10.1016/j.ejmech.2021.113870. [DOI] [PubMed] [Google Scholar]
- 277.Fischer F., Avelar L.A.A., Murray L., Kurz T. Designing HDAC-PROTACs: lessons learned so far. Future Med Chem. 2022;14:143–166. doi: 10.4155/fmc-2021-0206. [DOI] [PubMed] [Google Scholar]
- 278.Gao H.Y., Sun X.Y., Rao Y. PROTAC technology: opportunities and challenges. ACS Med Chem Lett. 2020;11:237–240. doi: 10.1021/acsmedchemlett.9b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Wang Y., Jiang X.Y., Feng F., Liu W.Y., Sun H.P. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B. 2020;10:207–238. doi: 10.1016/j.apsb.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Rodrigues D.A., Roe A., Griffith D., Chonghaile T.N. Advances in the design and development of PROTAC-mediated HDAC degradation. Curr Top Med Chem. 2022;22:408–424. doi: 10.2174/1568026621666211015092047. [DOI] [PubMed] [Google Scholar]
- 281.Smalley J.P., Adams G.E., Millard C.J., Song Y., Norris J.K.S., Schwabe J.W.R., et al. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem Commun. 2020;56:4476–4479. doi: 10.1039/d0cc01485k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Smalley J.P., Baker I.M., Pytel W.A., Lin L.Y., Bowman K.J., Schwabe J.W.R., et al. Optimization of class I histone deacetylase PROTACs reveals that HDAC1/2 degradation is critical to induce apoptosis and cell arrest in cancer cells. J Med Chem. 2022;65:5642–5659. doi: 10.1021/acs.jmedchem.1c02179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Baker I.M., Smalley J.P., Sabat K.A., Hodgkinson J., Cowley S.M. Comprehensive transcriptomic analysis of novel class I HDAC proteolysis targeting chimeras (PROTACs) Biochemistry. 2023;62:645–656. doi: 10.1021/acs.biochem.2c00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Xiao Y.F., Wang J., Zhao L.Y., Chen X.Y., Zheng G.R., Zhang X., et al. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem Commun. 2020;56:9866–9869. doi: 10.1039/d0cc03243c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Yang K., Song Y., Xie H., Wu H., Wu Y.T., Leisten E.D., et al. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg Med Chem Lett. 2018;28:2493–2497. doi: 10.1016/j.bmcl.2018.05.057. [DOI] [PubMed] [Google Scholar]
- 286.Yang K., Zhao Y., Nie X., Wu H., Wang B., Almodovar-Rivera C.M., et al. A cell-based target engagement assay for the identification of cereblon E3 ubiquitin ligase ligands and their application in HDAC6 degraders. Cell Chem Biol. 2020;27:866–876. doi: 10.1016/j.chembiol.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Sinatra L., Yang J., Schliehe-Diecks J., Dienstbier N., Vogt M., Gebing P., et al. Solid-phase synthesis of cereblon-recruiting selective histone deacetylase 6 degraders (HDAC6 PROTACs) with antileukemic activity. J Med Chem. 2022;65:16860–16878. doi: 10.1021/acs.jmedchem.2c01659. [DOI] [PubMed] [Google Scholar]
- 288.An Z.X., Lv W.X., Su S., Wu W., Rao Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell. 2019;10:606–609. doi: 10.1007/s13238-018-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Yang H.Y., Lv W.X., He M., Deng H.T., Li H.T., Wu W., et al. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem Commun. 2019;55:14848–14851. doi: 10.1039/c9cc08509b. [DOI] [PubMed] [Google Scholar]
- 290.Wu H., Yang K., Zhang Z.R., Leisten E.D., Li Z.Y., Xie H.B., et al. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J Med Chem. 2019;62:7042–7057. doi: 10.1021/acs.jmedchem.9b00516. [DOI] [PubMed] [Google Scholar]
- 291.Yang K., Wu H., Zhang Z.R., Leisten E.D., Nie X.Q., Liu B.K., et al. Development of selective histone deacetylase 6 (HDAC6) degraders recruiting von hippel-lindau (VHL) E3 ubiquitin ligase. ACS Med Chem Lett. 2020;11:575–581. doi: 10.1021/acsmedchemlett.0c00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Chotitumnavee J., Yamashita Y., Takahashi Y., Takada Y., Iida T., Oba M., et al. Selective degradation of histone deacetylase 8 mediated by a proteolysis targeting chimera (PROTAC) Chem Commun. 2022;58:4635–4638. doi: 10.1039/d2cc00272h. [DOI] [PubMed] [Google Scholar]
- 293.Huang J.B., Zhang J., Xu W.C., Wu Q., Zeng R.S., Liu Z.C., et al. Structure-based discovery of selective histone deacetylase 8 degraders with potent anticancer activity. J Med Chem. 2022;66:1186–1209. doi: 10.1021/acs.jmedchem.2c00739. [DOI] [PubMed] [Google Scholar]
- 294.Xiong Y., Donovan K.A., Eleuteri N.A., Kirmani N., Yue H., Razov A., et al. Chemo-proteomics exploration of HDAC degradability by small molecule degraders. Cell Chem Biol. 2021;28:1514–1527. doi: 10.1016/j.chembiol.2021.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Petrylak D.P., Gao X., Vogelzang N.J., Garfield M.H., Taylor I., Moore M.D., et al. First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI) J Clin Oncol. 2020;38:3500. [Google Scholar]
- 296.Berdeja J., Ailawadhi S., Horwitz S.M., Matous J.V., Mehta-Shah N., Martin T., et al. A phase 1 study of CFT7455, a novel degrader of IKZF1/3, in multiple myeloma and non-hodgkin lymphoma. Blood. 2021;138:1675. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.