

Abstract
Tissue-resident memory T cells (Trms) are an important subset of lymphocytes that are lodged within non-lymphoid tissues and carry out diverse functions to control local pathogen replication. CD103 has been used to broadly define subsets of Trms within the intestine, with CD103+ and CD103− subsets having unique transcriptional profiles and effector functions. Here we identify signal transducer and activator of transcription 4 (STAT4) as an important regulator of CD103− Trm differentiation. STAT4-deficient cells trafficked to the intestine and localized to areas of infection but displayed impaired Trm differentiation with fewer CD103− Trms. Single-cell RNA-sequencing demonstrated that STAT4-deficiency led to a reduction in CD103− Trm subsets and expansion of a single population of CD103+ cells. Alterations in Trm populations were due, in part, to STAT4-mediated inhibition of transforming growth factor (TGF)-β-driven expression of Trm signature genes. STAT4-dependent Trm populations expressed genes associated with cytokine production and cell migration, and STAT4-deficient Trm cells had altered localization within the tissue and reduced effector function after reactivation in vivo. Overall, our data indicate that STAT4 leads to increased differentiation of CD103− Trms, in part by modulating the expression of TGF–β-regulated genes, and results in increased Trm heterogeneity and function within the intestinal tissue.
INTRODUCTION
Memory CD8+ T cells are generated in response to infection and provide long-term protective immunity to subsequent infections with the same pathogen. Tissue-resident memory T cells (Trms) are a critical component of T cell memory, as they are maintained in the peripheral tissues, do not require replenishment by circulating cells, and remain anatomically poised for tissue surveillance and rapid recall responses1. Upon reactivation, Trms produce cytokines that induce changes in the local tissue microenvironment that enhance general pathogen resistance and the activation of other resident immune cell populations2,3. Interferon (IFN)-γ produced by Trms can also induce local chemokine secretion to amplify the recruitment of innate and adaptive immune cells and circulating antibodies to the site of infection4–7. In comparison to circulating memory T cells, CD8+ Trms provide superior protection and control pathogen replication at the site of the infection8,9. Trms can also exit certain tissues and contribute to immune responses in local lymph nodes10–14, highlighting the importance of these cells for both local and systemic immune function.
During primary infection, the adaptation of infiltrating effector CD8+ T cells to their tissue environment is required for Trm differentiation and robust tissue immunity. CD8+ T cells undergo rapid changes in their gene expression profile that enable their long-term maintenance as Trm cells, and a broad program of transcription factors, including Hobit, Blimp1, Runx3, Nur77, Notch, and Bhlhe40, have been identified as central regulators of tissue residency15–19. As they differentiate, Trm cells downregulate tissue egress markers and upregulate markers that support tissue retention, including CD69, CD49a, and CD1031,20. Although CD103 plays a role in the maintenance of Trms in the brain21, skin22, and intestinal epithelium23,24, it is not expressed on all Trms, and substantial numbers of CD103− Trms are found in the small intestine lamina propria (LP), colon, liver, and other tissues25–28. Although many transcription factors that regulate Trm populations as a whole or are specifically required for CD103+ Trm cells, the factors underlying the differentiation of the CD103− Trm population remain poorly understood.
In tissues that support the maintenance of both the CD103− and CD103+ Trm populations, these subsets have distinct gene expression signatures, with CD103− Trms displaying enhanced functional capabilities14,28–31. Even within subsets with uniform CD69 and CD103 expression, single-cell transcriptional profiling has identified subsets having heightened secondary memory potential and effector function32,33. Together, this suggests that CD8+ Trms with a spectrum of functions and memory potentials develop in response to the conditions within the local environment, with CD103 Trms being specialized for secondary expansion30,31. Understanding the signals that drive heterogeneity within the Trm compartment will allow us to manipulate the quality of the Trm response and improve the secondary responses within the tissue. These studies have identified signal transducer and activator of transcription 4 (STAT4) as a critical regulator of the differentiation of CD103− Trms in the intestine, with STAT4-dependent Trm populations displaying unique gene expression signatures indicative of enhanced migration, effector function, and improved responses to reactivation within the tissue.
RESULTS
STAT4 regulates the magnitude of the intestinal CD103− CD8+ T-cell response during infection
We previously identified interleukin (IL)-12 as a regulator of CD103− Trm differentiation34, implicating STAT4 in this process. To examine this directly, naïve wild-type (WT) and Stat4−/− OT-I T cells were transferred into congenically distinct hosts before infection with Yersinia pseudotuberculosis expressing ovalbumin (Yptb-OVA). At 9 days post infection (p.i.), we examined the expansion and intestinal trafficking of OT-Is. There was a modest increase in the expansion of WT compared with Stat4−/− OT-Is in the mesenteric lymph node (MLN); however, the ratio of WT to Stat−/− T cells was similar across all tissues (Figs. 1A and 1B), indicating that STAT4 does not regulate T-cell trafficking. There was a significant reduction in the CD69+CD103− population of intraepithelial (IE) and LP Stat4−/− OT-Is compared with WT cells, with an increase in the Stat4−/− CD69+CD103+ subset (Figs. 1C and 1D). Distinct localization of Trm subsets has been observed in response to Yptb infection, with T cells that cluster around areas of bacterial colonization preferentially differentiating into CD103− Trms26. We investigated whether impaired localization of Stat4−/− OT-Is to these proinflammatory microenvironments might account for their altered differentiation. Immunofluorescence microscopy demonstrated the localization of both WT and Stat4−/− OT-Is to lymphocyte clusters (Fig. 1E), and the ratio of WT:Stat4−/− OT-Is was similar in T-cell clusters and the total ileum (Fig. 1F). Our results indicate that Stat4−/− CD8+ T cells localize to the lymphocyte clusters at the sites of infection, but their early expression of CD103 is enhanced relative to WT cells.
Fig. 1.
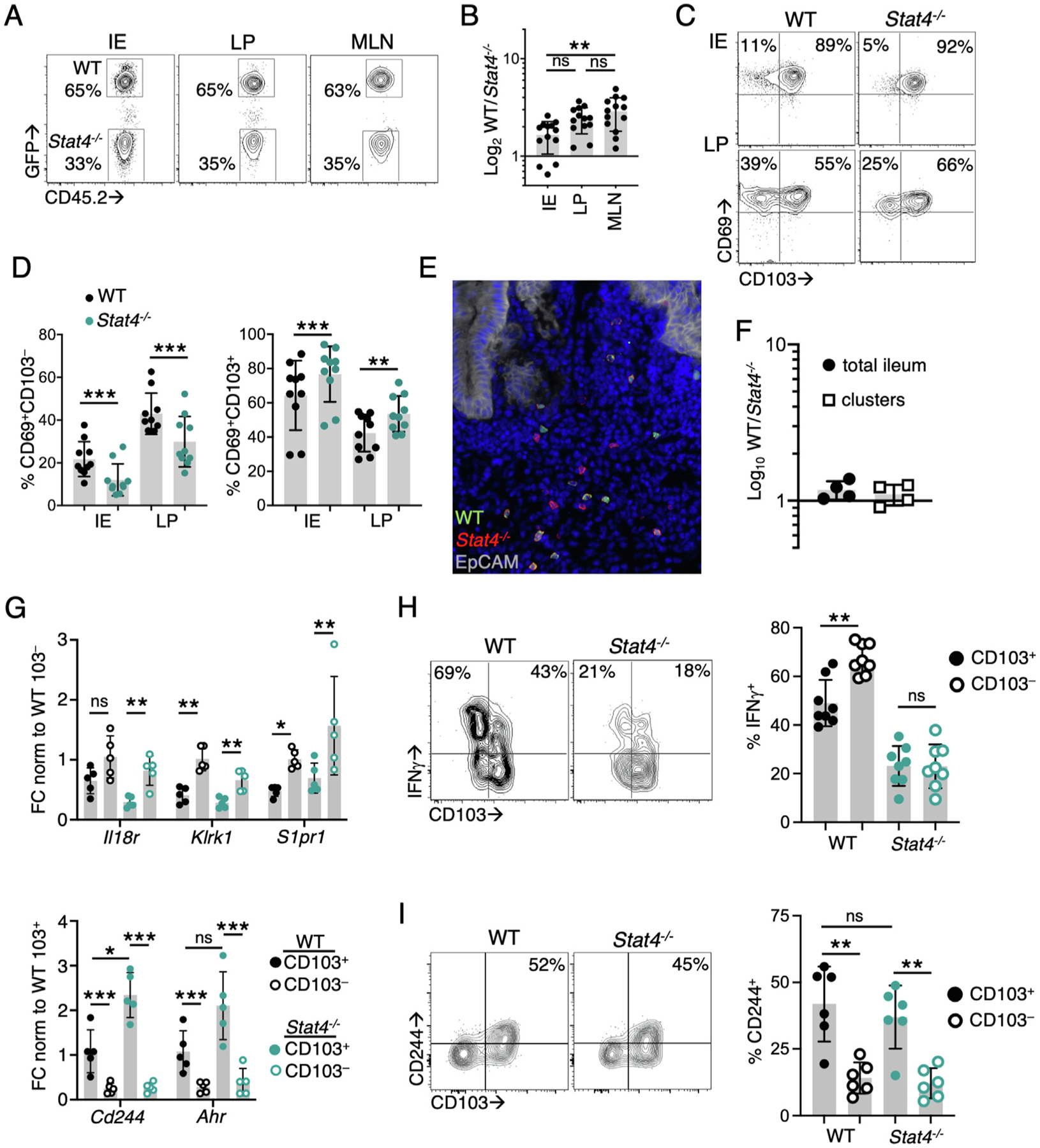
STAT4 regulates the magnitude of the intestinal CD103− CD8+ T-cell response during infection. CD45.1+ mice received an equal number of GFP+CD45.2+ WT and CD45.2+ Stat4−/− OT-I T cells (A–F, H–I) or WT and Stat4−/− YopE T cells (G) before oral gavage with Yptb-OVA. Tissues were harvested at 9–10 days p.i. (A) flow cytometry of populations of WT and Stat4−/− OT-I cells in the IE, LP and MLN. Numbers represent percent OT-Is of total transferred cells; (B) ratio of WT to Stat4−/− OT-1s in the indicated tissues (n = 13); (C–D): CD103 and CD69 expression by WT and Stat4−/− OT-Is. Numbers in quadrants indicate percent of total OT-Is (n = 10); (E–F) immunohistochemistry analyzing the distribution of OT-I T cells in the ileum of host mice. Tissue sections were stained with anti-GFP (green), anti-CD45.2 (red), anti-EpCam (white), and DAPI (blue). The ratio of WT to Stat4−/− OT-Is was determined in the total ileum or lymphocyte clusters (n = 4); (G) gene expression in sorted CD69+CD103+ and CD69+CD103− populations was determined by quantitative reverse transcription-polymerase chain reaction and normalized to Gapdh. FC relative to WT CD69+CD103+ (top panel) or WT CD69+CD103− (bottom panel) cells (n = 5); (H) lymphocytes were stimulated with SIINFEKL and representative flow cytometry plots of CD103 and IFN-γ production in CD69+ OT-Is are shown (n = 8); (I) representative flow cytometry plots of CD103 and CD244 expression in CD69+ OT-Is. Numbers are the percentage of positive cells within the CD103+ subset, with quantification in right panel (n = 6). Symbols represent individual mice and mean and standard deviations are shown. Data points are pooled from two to four independent experiments with two to four mice/group. *p < 0.05; **p < 0.005; **p < 0.0005 using one-way analysis of variance with Tukey test (B), paired Student’s t test (D, H, I), or two-way analysis of variance (G). FC = fold change; GFP = green fluorescent protein; IE = intraepithelial; IFN = interferon; LP = lamina propria; MLN = mesenteric lymph node; ns = not significant; p.i. = post infection; WT = wild-type; DAPI = 4′,6-diamidino-2-phenylindole.
A subset of CD103− Trm signature genes is modulated by STAT4
Many of the genes that define the CD103− Trm subset are directly regulated by STAT4 (Klrk1, Il18r1, S1pr1, Ifng)35,36, and we examined the degree to which STAT4-deficiency influences expression of these genes during differentiation. CD103+ and CD103− T cells were sorted from the LP at day 9 p.i. for gene expression analysis. S1pr1 and Klrk1 expression was significantly increased in WT and Stat4−/− CD103− T cells compared with CD103+ T cells; however, no difference in expression of these genes was observed between in WT and Stat4−/− CD103− T cells (Fig. 1G). IFN-γ production was also examined after ex vivo peptide stimulation, and WT CD103− OT-Is produced significantly more IFN-γ than CD103+ cells. Stat4−/− LP OT-Is had reduced IFN-γ production compared with WT OT-Is and the difference was between LP T cell subsets was abrogated (Fig. 1H). We also examined the expression of CD103+ Trm signature genes (Cd244, Ahr), and their upregulation in CD103+ LP T cells was not impacted by STAT4-deficiency (Figs. 1G and 1I). Overall, these data indicate that Stat4−/− CD103− T cells are reduced in number but retain many of the features of WT cells, with STAT4 regulating IFN-γ production but having little effect on other CD103− Trm signature genes.
STAT4 is required for the differentiation of CD103– Trm cells
To determine whether this early reduction in intestinal Stat4−/− CD103− T cells persisted into the memory phase, intestinal Trm populations were analyzed at 30 days p.i. The ratio of WT to Stat4−/− cells within the CD103− Trm subsets demonstrated skewing toward the WT population, whereas similar numbers of WT and Stat4−/− cells were observed in the epithelial and LP CD103+ Trm populations and the MLN (Figs. 2A and 2B). We determined the number of OT-I T cells in each subset during and after infection and found that WT CD103+ and CD103− LP Trms both notably contract after the resolution of infection; WT CD103+ LP Trm cells were reduced by 10.1-fold and CD103− cells by 4.4-fold (Fig. 2C). Although there was a significantly lower number of Stat4−/− CD103− OT-I cells than WT CD103− cells, both WT and Stat4−/− populations contracted similarly (Fig. 2C). This suggests STAT4 is acting soon after T cells have entered the tissue to program Trm differentiation.
Fig. 2.
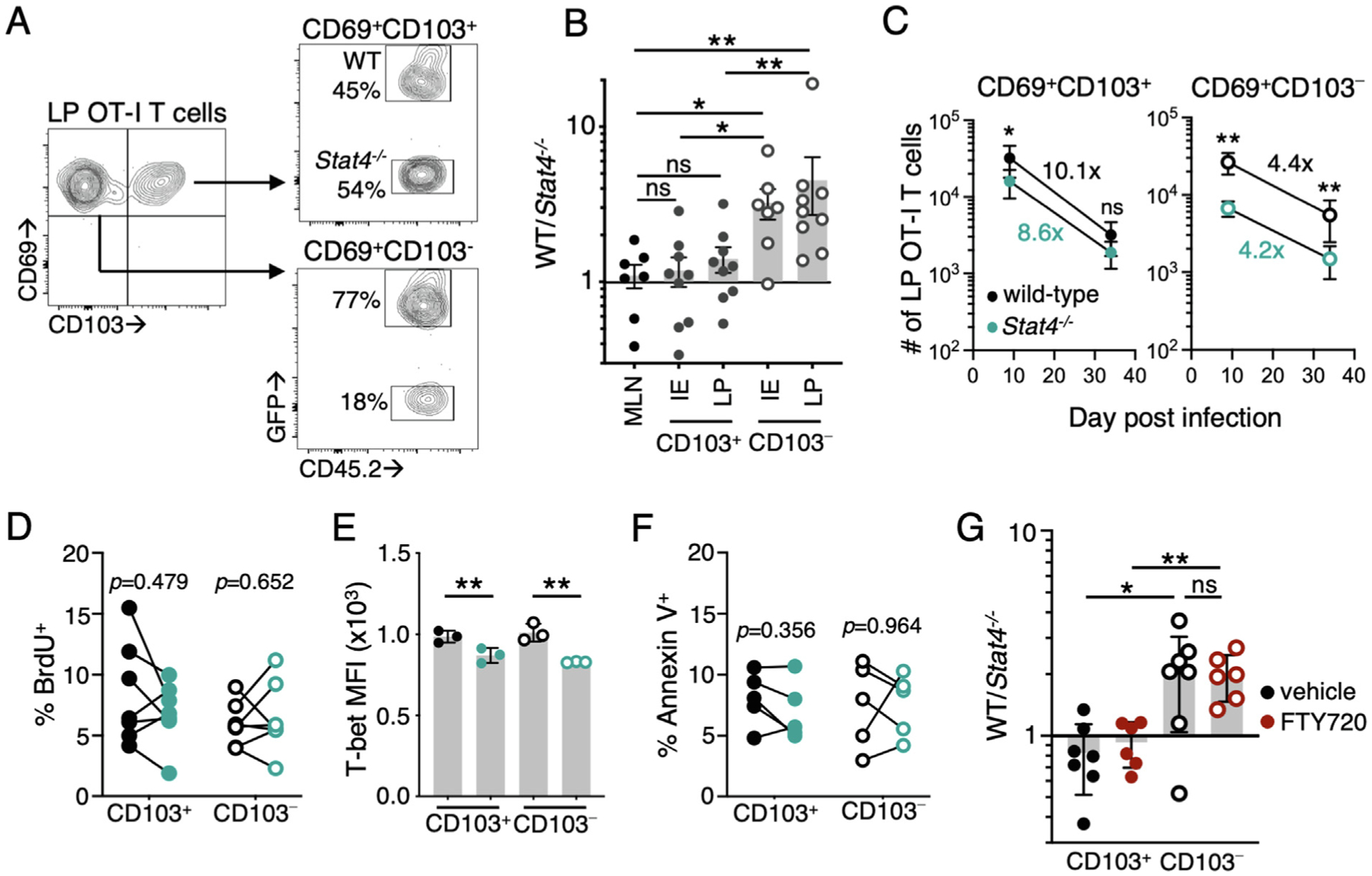
STAT4 is required for the differentiation of CD103− Trm cells. (A–F) CD45.1+ mice received an equal number of GFP+CD45.2+ WT and CD45.2+ Stat4−/− OT-Is before oral gavage with Yptb-OVA. Tissues were harvested at 30–40 days p.i. (A) flow cytometry plots WT and Stat4−/− OT-Is within the CD69+CD103− and CD69+CD103+ subset in the LP; and (B) ratio of WT to Stat4−/− OT-Is within intestinal Trm subsets or total OT-Is from the MLN (n = 7–9); (c) quantification of the number of WT and Stat4−/− CD103− and CD103+ cells in the LP at the indicated days p.i. Numbers indicate the fold reduction in each population over time (n = 9); (D) mice were given BrdU on day 7 p.i. and sacrificed 1 hour later. Percentage of cells that have incorporated BrdU within the CD69+CD103+ and CD69+CD103− LP Trm subsets (n = 6); (E) expression of T-bet at day 9 p.i. in CD69+CD103+ and CD69+CD103− LP Trm subsets, mean fluorescence intensity for each genotype; (F) Percentage of cells that are Annexin V+ within the CD69+CD103+ and CD69+CD103− LP Trm subsets at day 9 p.i (n = 5). Symbols represent individual mice and lines connect data points from the same mouse in (D, F); (G) CD45.1+ mice received an equal number of CD45.1/2+ WT and CD45.2+ Stat4−/− YopE T cells before oral gavage with ΔyopM Yptb. Mice were administered FTY720 or vehicle control intraperitoneally every other day from day 7 p.i. to harvest on day 20. The ratio of WT to Stat4−/− YopE T cells within intestinal Trm subsets (n = 6–7/group). Mean and standard deviation (E,G) or standard error of mean (B, C) are shown. Data points are pooled from at least two independent experiments with three to four mice/group. IE samples with no Stat4−/− CD69+CD103− Trms were excluded from the analysis in (B). *p < 0.05; **p < 0.005 calculated using Mann-Whitney test (B, G), paired Student’s t test (D–F), and Wilcoxon test (C). BrdU = bromodeoxyuridine; GFP = green fluorescent protein; IE = intraepithelial; LP = lamina propria; MLN = mesenteric lymph node; ns = not significant; p.i. = post infection; Trm = tissue-resident memory T cell; WT = wild-type.
STAT4 can augment the proliferation of CD8 T cells in lymphoid tissues37 and could drive the expansion of CD103− Trms upon entry into lymphocyte clusters. To measure proliferation in situ, mice received bromodeoxyuridine (BrdU) on day 7 p.i. and tissues were harvested 1 hour later for analysis. We found no difference in BrdU incorporation between WT and Stat4−/− Trms in the intestine, regardless of Trm subset (Fig. 2D). STAT4 can drive T-bet expression, and we observed a modest reduction in T-bet expression in both CD103+ and CD103− Stat4−/− Trms (Fig. 2E). The loss of T-bet results in reduced IL-15R levels and impaired survival of skin Trms38; however, IL-15R expression was not impacted by STAT4-deficiency in this setting (Supplementary Fig. 1A) and we found no difference in apoptosis between Stat4−/− and WT Trms in either subset using Annexin V staining (Fig. 2F). Enhanced migration of CD103− Stat4−/− T cells out of the intestinal tissue could also explain their reduced numbers. At day 9 p.i., CD103− Stat4−/− Trm cells display a modest reduction in CD69 expression (Supplementary Fig. 1B), along with elevated S1pr1 (Fig. 1G), which facilitates tissue egress. To address this, mice were treated with FTY720 to inhibit S1PR1-mediated trafficking starting at day 7 p.i., after T cells were established in the intestinal tissue. We examined the Trm cells in the tissue at day 20 p.i. and found no difference in the ratio of WT to Stat4−/− cells in the either Trm subset when comparing control and FTY720 treated mice (Fig. 2G). Together, these data indicate that the reduction in number of Stat4−/− CD103− Trm cells was programmed early during infection and was not due to insufficient proliferation, impaired survival, or enhanced tissue egress.
Single-cell RNA-sequencing identifies transcriptionally distinct STAT4-dependent Trm subsets
To uncover the diversity within intestinal LP Trms and more clearly identify Trm populations that are regulated by STAT4, we performed single-cell RNA-sequencing (scRNA-seq) of WT and Stat4−/− LP OT-Is at day 35 p.i. Unsupervised clustering analysis of pooled cells was used to identify putative subsets of Trm cells and revealed eight distinct Trm clusters (P1–8; Fig. 3A). Several populations were altered in the absence of STAT4, with drastic reductions in P4 and P5 (57.3% and 61.1%, respectively) and less prominent reductions in P3 and P6 (Fig. 5A). Itgae (CD103 α chain) was expressed in a subset of cells in most clusters, except P4, which was primarily Itgae-negative (Fig. 3B). We examined the expression of genes that have been previously reported to be bound by STAT435,36 and found that individual clusters had unique patterns of STAT4-regulated gene expression, with the highest expression in P4 and P5 (Fig. 3C). STAT4-regulated gene expression was the lowest in P2 (Fig. 3C), and Stat4−/− Trm cells were overrepresented in this cluster, with almost 30% of the total Stat4−/− cells found in P2 compared with 6.3% of WT cells (Fig. 3A). These data indicate that STAT4-deficiency results in an enhanced differentiation of cells into the P2 population at the expense of other CD103− Trm subsets.
Fig. 3.
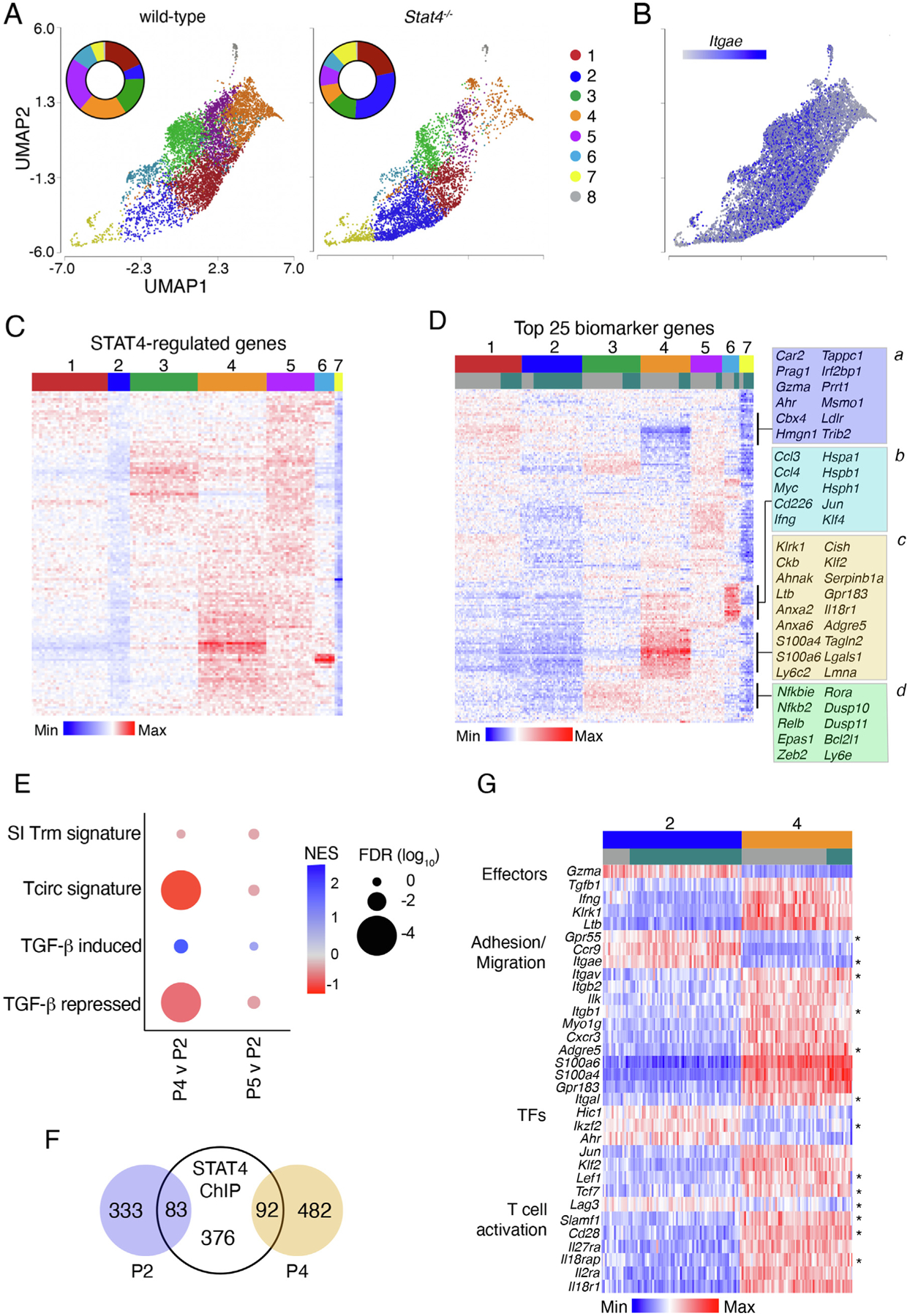
STAT4 regulates the abundance of multiple Trm subsets, increasing Trm heterogeneity. CD45.1+ mice received an equal number of GFP+CD45.2+ WT and CD45.2+ Stat4−/− OT-1 T cells before oral gavage with Yptb-OVA. Viable i.v.CD8a–CD8b+CD69+ OT-1 cells were sorted from the LP at 35 days post infection. (A) UMAP visualization LP Trms after non-linear dimensional reduction analysis and unsupervised clustering. Cells are colored by cluster and separated by genotype. Each point represents a single cell and data are pooled from two independent experiments (WT, 7074 cells; Stat4−/−, 5539 cells). Inset graph shows the proportion of cells in each cluster; (b) UMAP visualization of LP Trms showing expression of Itgae; (C–D) heatmap showing relative expression of genes identified by STAT4 ChIP in WT Trms (C); or expression of the top 25 biomarker genes for each cluster in both WT and Stat4−/− Trms (D); select modules are identified and genes are listed; (E) comparison of P2 and P4 or P5 by GSEA, NES and FDR are shown; (F) proportion of differentially expressed genes in P2 and P4 that were identified using STAT4 ChIPseq analysis; (G) heatmap showing relative expression of a subset differentially regulated genes in analysis of variance comparison of P2 and P4; FDR q < 0.05 and fold change > 1.5. Asterisks denote genes that are regulated by TGF-β in Trm cells. FDR = false discovery rate; LP = lamina propria; NES = normalized enrichment score; TGF = transforming growth factor; Trm = tissue-resident memory T cell; UMAP = uniform manifold approximation and projection; WT = wild-type.
Fig. 5.
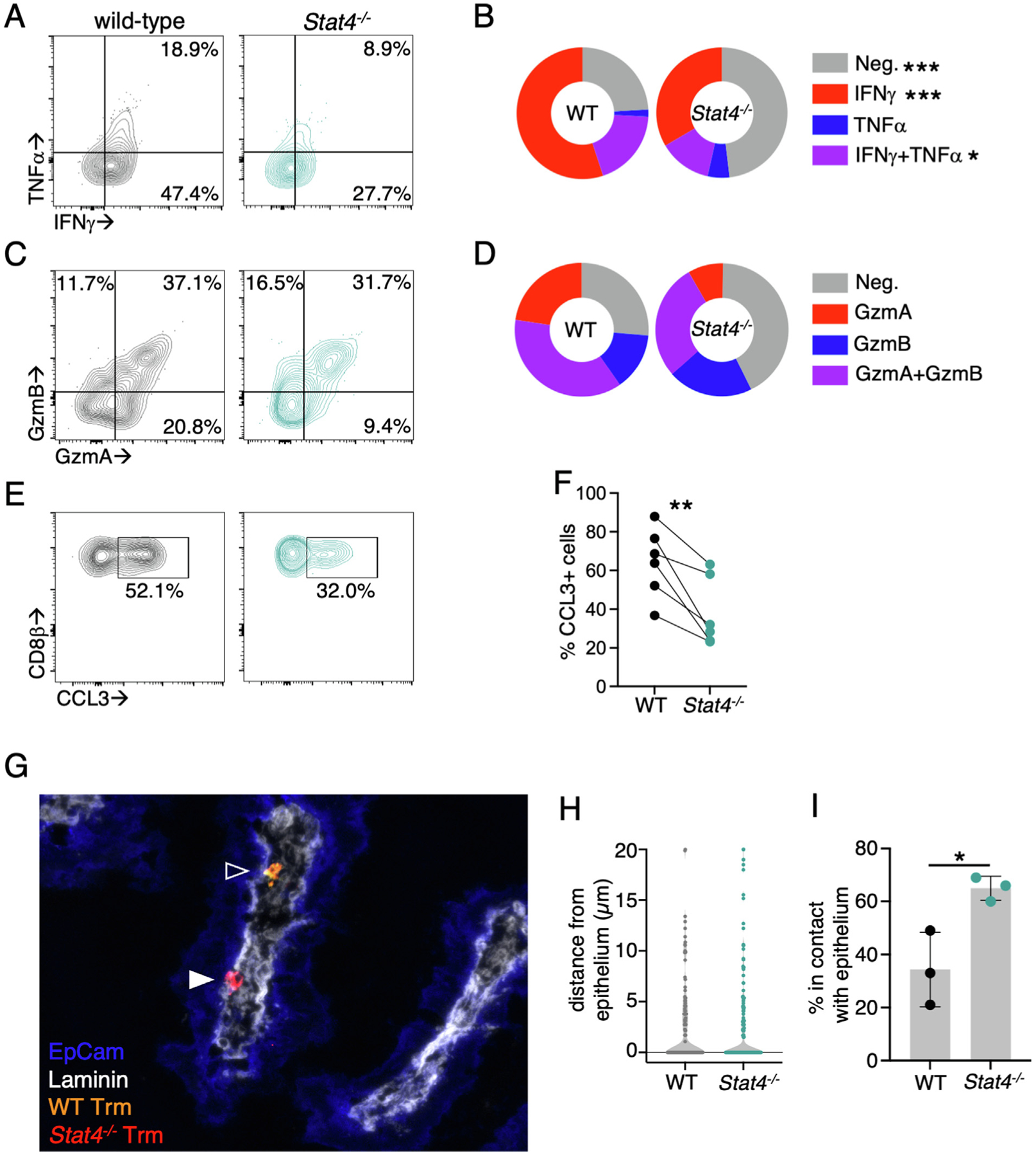
STAT4-deficient Trm cells have impaired effector function and altered localization within the LP. CD45.1+ mice received an equal number of CD90.1+CD45.2+ WT and CD45.2+ Stat4−/− YopE T cells before oral gavage with ΔyopM Yptb. (A–F) At 35 days p.i., mice were injected intraperitoneal with 10 μg YopE peptide and tissues were harvested 1 hour later (A) representative flow cytometry plots of TNF-α and IFN-γ production in total YopE T cells are shown; (B) percentage of cells from each genotype that are expressing cytokines; (C) representative flow cytometry plots of Gzm A and Gzm B expression in total LP YopE T cells; (D) percentage of cells from each genotype that are expressing granzymes. (E–F): To assess CCL3 production, brefeldin A was added to in vivo activated LP cells and they were incubated an additional 3 hours before analysis. Representative flow cytometry plots of CCL3 production by total LP T cells are shown (E) and quantified in (F). (G–I) At 35 days p.i., the ileum was isolated and the distribution of YopE T cells was examined by immunofluorescence microscopy. Tissue sections were stained for CD45.2 (red), CD90.1 (yellow), EpCam (blue), and laminin (white): (G) representative image with closed arrow indicating a Trm cell in contact with the epithelium and an open arrow indicated a Trm cell a short distance from the epithelium; (H) distance of individual Trm cells from the epithelium; and (I) the percentage of Trm cells in contact with the epithelium. Data are representative of two experiments with 3–4 mice/group. *p < 0.05; **p < 0.01 and ***p < 0.005. CCL = chemokine ligand; Gzm = granzyme; IFN = interferon; LP = lamina propria; Neg. = negative; ns = not significant; p.i. = post infection; TGF = transforming growth factor; TNF = tumor necrosis factor; Trm = tissue-resident memory T cell; WT = wild-type.
STAT4-dependent Trm populations display unique effector potential
The biomarker genes in each cluster were identified to determine how this shift from P4/P5 to P2 in a STAT4-deficient setting could influence Trm-mediated immunity (Supplementary Table 1). The top 25 biomarker genes were subjected to hierarchical clustering analysis, which revealed overlapping patterns of gene expression between the subsets (Fig. 3D). Multiple gene modules of interest were identified. Module a was present in all clusters, except P4 and P7, and contained elevated expression of genes associated with an intestinal Trm signature, including Ahr, Gzma, Prag1, and Car2. Module b genes were highly expressed in P6 but also present in P4 and P5 and contained heat shock proteins (Hspa1, Hspab1, Hsph1), the transcription factors Myc, Jun, and Klf4 and secreted effectors (Ccl3, Ccl4, and Ifng). Module c was elevated in P4, and representative genes include effector molecules (Klrk1, Ltb, Serpinb1a), genes involved in migration (S100a4, S100a6, Gpr183), and cytoskeletal components (Vim, Tagln2, Lmna). Module d was present in P3, P4, and P5 and had elevated expression of NF-κB genes (Nfkbie, Nfkb2, Relb). These data suggest that the loss of P4/P5 and expansion of P2 in the absence of STAT4 would lead to a population of Trm cells that retain cytotoxic potential but are impaired in a variety of other effector functions.
P2 and P4/P5 subsets were compared directly using gene set enrichment analysis (GSEA) to examine the expression of gut Trm and circulating T cell signatures. Although small intestinal Trm signature genes were not enriched in any populations, P4 had an elevated expression of circulating T cell signature genes compared with P2 (Fig. 3E). P2 and P4 were examined and ~750 genes were differentially regulated upon direct comparison of these populations (Supplementary Table 2), and only a subset of differentially expressed genes (~19%) are bound by STAT4 (Fig. 3F). P2 had elevated levels of transcription factors Ahr, Hic1, and Ikzf2 (Fig. 3G), which have been previously associated with intestinal T-cell function and maintenance33,39, whereas P4 maintained high levels of Lef1 and Tcf7, suggesting that this population might have an increased ability to expand during secondary infection40. There were also differences in genes associated with adhesion and migration. P2 expressed genes facilitating close association with the epithelium (Itgae and Ccr9) and Gpr55, which plays an inhibitory role in T-cell migration41. P4 expressed genes positively associated with migration (Myo1g, S100a4, S100a6, Gpr183) and adhesion molecules (Adgre5, Itgav, Itgab1, Itgab2) that could facilitate tissue retention or cell-cell interactions. P4 genes also indicated an increased potential for cytokine responsiveness and T-cell activation, including cytokine receptors (l18r1, Il18rap, Il27ra, Il2ra) and costimulatory molecules (Cd28, Slamf1). P4 also maintained greater expression of cytokines (Ifng, Ltb, Tgfb1) and other regulators of T-cell function (Tagln2, Ahnak, Lgals1, Vim) (Fig. 3D).
STAT4 modulates expression of CD103 and other TGF-β-regulated genes
TGF-β is an important signal that drives the differentiation of CD103+ Trm populations in several tissues22–24,42 and many of the differentially expressed genes between P2 and P4 were previously reported to be regulated by TGF-β in Trm cells14,43 (Fig. 3G, denoted by asterisks). GSEA revealed that TGF-β-induced genes were modestly enriched in the P2 subset, whereas TGF-β-repressed genes were enriched in P4 compared with P2 (Fig. 3E). In contrast, P5 did not display a significant enrichment of these gene sets compared with the P2 population. IL-12 can inhibit TGF-β-induced CD103 expression34, and we hypothesized that STAT4 could be more broadly modulating the responsiveness to TGF-β during Trm differentiation and inhibiting expression of CD103 and other CD103+ Trm signature genes.
We examined the additional TGF-β-regulated genes, and in general, TGF-β-induced genes were more highly expressed in population P2 and TGF-β-repressed genes in population P4 (Fig. 4A). However, there were some exceptions. Cish, an inhibitor of T cell receptor (TCR) signaling44, was reported to be induced in a TGF-β-dependent manner but was more highly expressed in P4 than P2. We used cytokine stimulation to model Trm gene expression patterns in vitro and understand the role of STAT4 in regulating TGF-β-mediated gene expression. Stimulation of activated T cells with IL-12 inhibited TGF-β-induced CD103 expression, as previously described34, and this reduction was dependent on STAT4 (Fig. 4B). CD103 regulation was at the transcriptional level because IL-12 completely blocked the expression of the CD103 α chain Itgae after TGF-β stimulation (Fig. 4C). To determine if IL-12 was able to regulate TGF-β signaling directly, we examined phosphorylation of SMAD2 in response to cytokine stimulation. TGF-β-induced Smad2 phosphorylation and pSMAD2 remained elevated in the presence of IL-12 (Figs. 4D and 4E), indicating that STAT4 did not impair TGF-β signaling. The expression of other TGF-β-regulated markers of CD103+ Trm cells were examined, and several were elevated after stimulation with TGF-β in vitro (Fig. 4F). The addition of IL-12 was able to prevent the expression of a subset of these genes, including Ikzf2, Litaf, and Chst12, whereas other genes were more moderately affected (Fig. 4F). The expression of these TGF-β-regulated genes was not altered by the addition of IL-12 in Stat4−/− cells (Fig. 4F). We also examined the expression of Cish and found that both IL-12 and TGF-β led to an increase in Cish levels (Fig. 4G), consistent with our finding that this gene does not follow the gene expression pattern of other TGF-β-induced genes in vivo (Fig. 4A). These data indicate that STAT4 modulates the expression of a subset of TGF-β-regulated genes that are a signature of Trm cells, and STAT4-deficiency results in a shift from CD103− to CD103+ Trm cells in vivo rather than the loss of the intestinal CD103– Trm population.
Fig. 4.
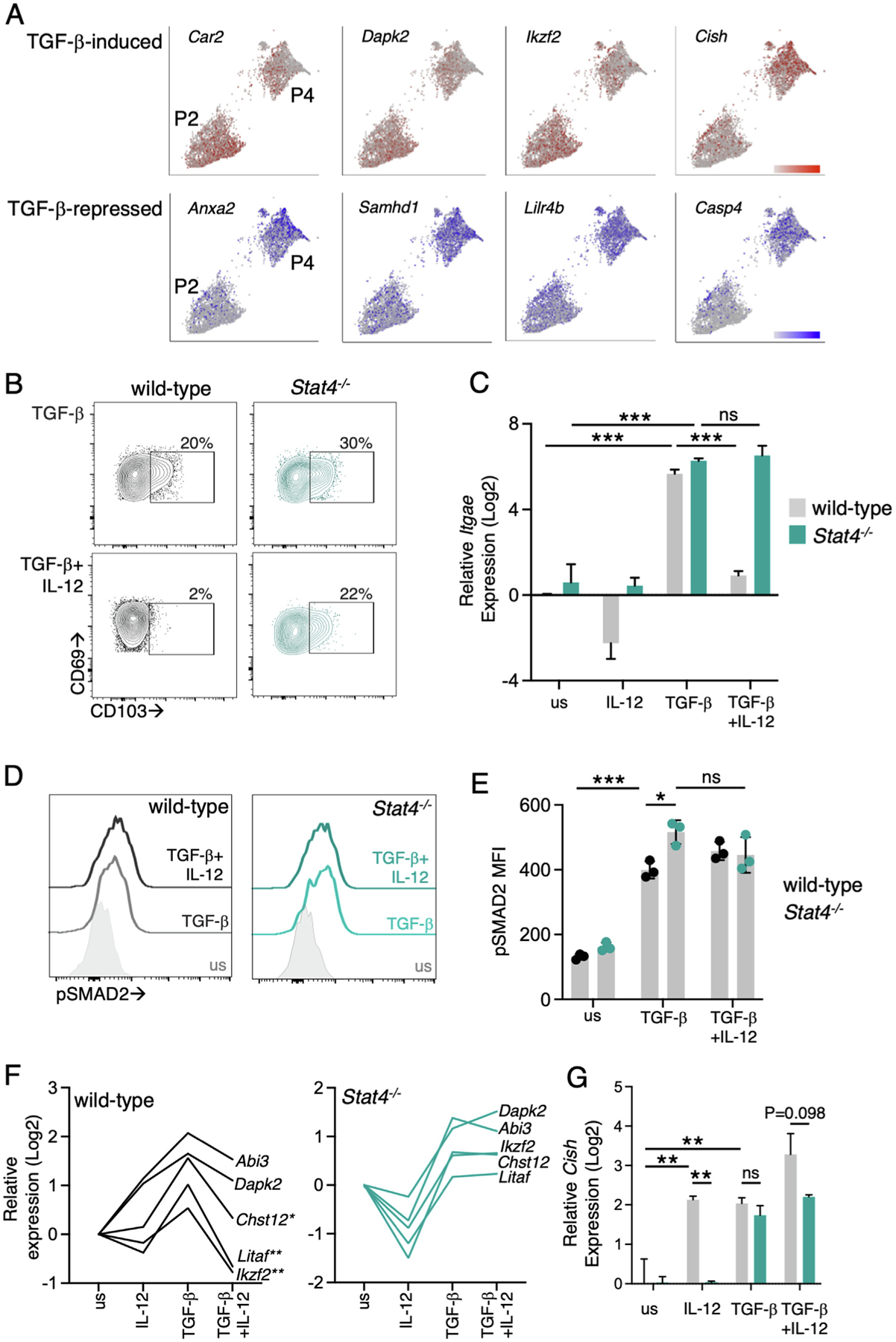
STAT4 modulates TGF-β-driven gene expression. Expression of TGF-β-induced (red) and -repressed (blue) genes in P2 and P4 using single-cell RNA-sequencing (A). WT and Stat4−/− YopE T cells were activated with 1 uM YopE69–77 peptide and IL-2 for 3 days and then stimulated with IL-12 and TGF-β for 3 hours (D, E) or 40 hours (B, C, F, G): representative flow cytometry plots of CD103 (B); and Itgae expression (C); after stimulation of WT and Stat−−/− YopE T cells with the indicated cytokines; representative flow plots of Smad2 phosphorylation (D) with data quantified in (E); expression of TGF-β-regulated genes in WT (F); and Stat4−/− (G) YopE T cells. Data points are biological replicates (E) and representative of two experiments or pooled data from two experiments (C, F, G). Mean and standard deviation are shown. *p < 0.05, **p < 0.005, ***p < 0.0001 using two-way analysis of variance with Tukey test for multiple comparisons. IL = interleukin; ns = not significant; TGF = transforming growth factor; WT = wild-type.
Reactivation of Trm populations reveal altered functionality of Stat4−/− Trm cells
We observed a reduction in the number of Stat4−/− Trm cells that develop in the LP, and scRNA-seq analysis indicates that on a per cell basis, Stat4−/− Trms would have distinct effector potential. To test this directly, mice received WT and Stat4−/− LP YopE-specific T cells, followed by infection with ΔyopM Yptb. At day 35 p.i., immune mice were administered YopE peptide to reactivate Trm cells and cytokine, chemokine, and granzyme production were assessed. There was a reduced cytokine production by Stat4−/− Trm cells during reactivation, with a significant increase in the percentage of cells that produced no cytokines and reduction in the IFN-γ+ and IFN-γ+ tumor necrosis factor (TNF)-α+ populations (Figs. 5A and 5B). We also examined the granzyme A and granzyme B levels, and there was no significant difference in the percentage of granzyme-producing populations (Figs. 5C and 5D). ScRNA-seq analysis also showed increased chemokine production by STAT4-dependent Trm populations, and we examined CCL3 production by WT and Stat4−/− LP Trm populations and found that Stat4−/− cells produced significantly less CCL3 (Figs. 5E and 5F).
The localization of WT and Stat4−/− Trm cells in the LP was also examined, as elevated expression of Ccr9, Gpr55, and Itgae/CD103 on Stat4−/− Trm cells could increase their proximity to the epithelium. Mice received WT and Stat4−/− LP YopE T cells, followed by infection with ΔyopM Yptb. The ileum was isolated from the mice at 35 days p.i., and tissue was stained with antilaminin and anti-Epcam antibodies to define the LP and intestinal epithelial layer. The distance between individual WT and Stat4−/− LP Trm cells and the intestinal epithelium was quantified, and there was no significant difference observed between WT and Stat4−/− Trm cells (Figs. 5G and 5H). However, LP Trm cells were frequently in contact with the epithelium at this time point, and significantly more Stat4−/− Trms were in direct contact with the epithelium than WT cells (65.0% vs. 34.3%). These data indicate that along with the reduction in cell number, STAT4-deficient cells have altered localization within the LP, which could impact access to antigen presenting cells and signals from other LP immune cells, and once reactivated, the STAT4-deficient Trm cells have reduced chemokine and cytokine production.
Human intestinal Trm cells have a STAT4-dependent Trm signature
We utilized data from a recently published scRNA-seq study examining human ileal lymphocytes was used to determine the prevalence of the identified STAT4-dependent Trm populations in humans45. Unsupervised clustering analysis of pooled ileal cells revealed nine lymphocyte clusters, and the clusters dominated by CD8β+ T cells were selected for further analysis. CD8β+ T cells were reclustered, and five distinct populations were identified (Fig. 6A). These populations had unique STAT4-regulated gene expression profiles, with human P4 having the highest level of STAT4-dependent gene expression (Fig. 6B). The top 100 signature genes from murine CD103− Trm P4 were examined in human ileal T cell subsets, and these genes were also upregulated in human P4 (Fig. 6C). These analyses suggest the ability of STAT4-regulated genes to define Trm populations is not specific to mice, and a subset of human CD8β+ LP Trms also expressed a STAT4-dependent gene expression signature and display similarities to murine CD103− Trm cells.
Fig. 6.
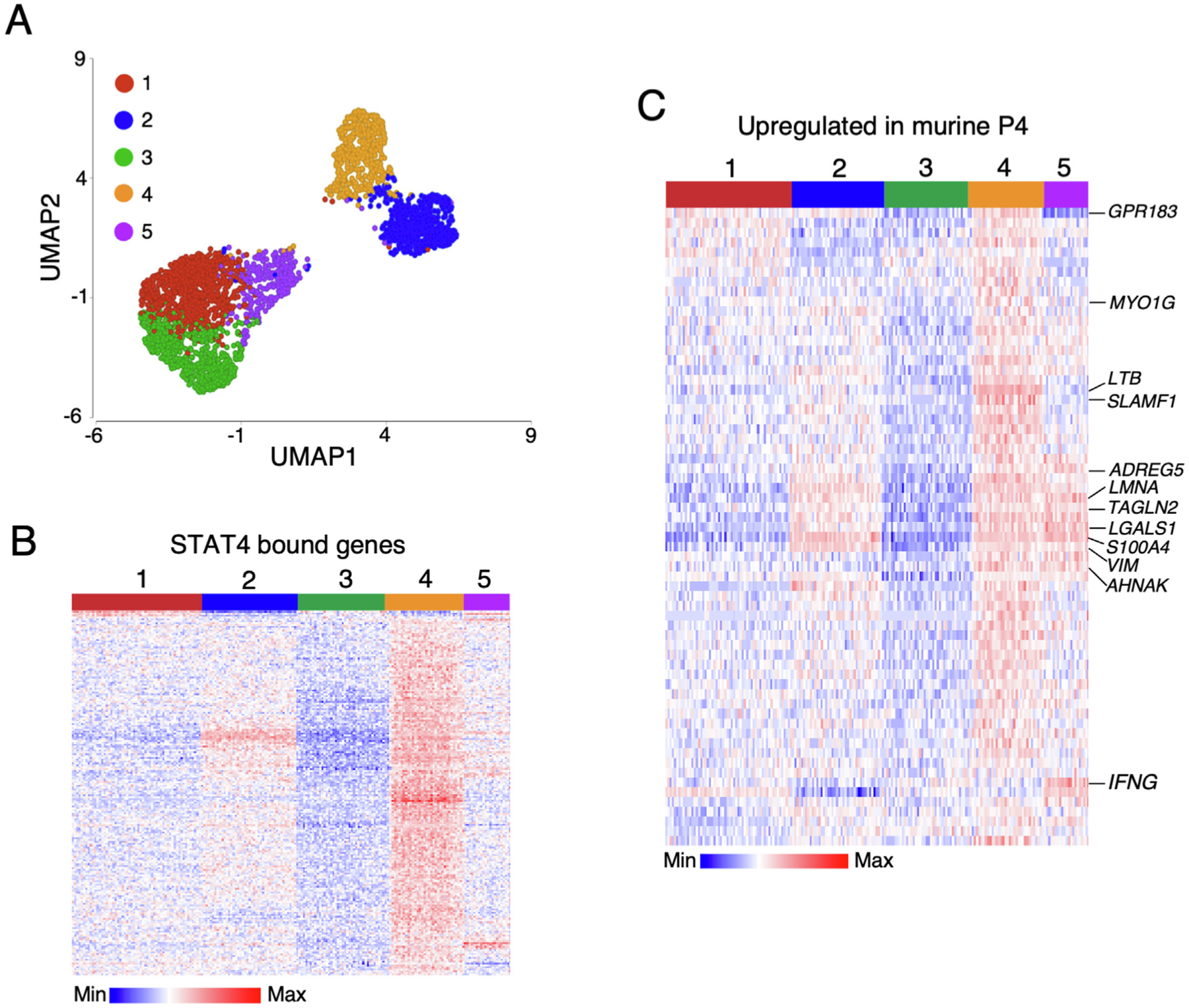
Human ileal T cells have a STAT4-dependent Trm signature. Analysis of lamina propria CD8β+ subsets from the ileum of healthy humans. (A) UMAP visualization of CD8β + cells after non-linear dimensional reduction analysis and unsupervised clustering. Cells are colored by cluster and each point represents a single cell (3090 total cells); (B) heatmap showing relative expression of genes identified by STAT4 ChIP in clusters identified in (A); (C) heatmap showing relative expression of top 100 genes identified in murine lamina propria Trm cluster 4. Trm = tissue-resident memory T cell; UMAP = uniform manifold approximation and projection.
DISCUSSION
These studies identify STAT4 as a critical regulator of the differentiation of the CD103− Trm subset in the intestinal tissue, and this is the first identification of a transcription factor that regulates the development of this Trm population exclusively. STAT4 is important early in Trm programming, and scRNA-seq at later time points revealed the maintenance of STAT4-regulated gene expression in a subset of LP Trm cells, along with expression of effector cytokines, genes involved in cell migration and adhesion, and cytoskeletal components. STAT4-deficiency lead to the loss of this population and expansion of a CD103+ population, in part due to the ability of STAT4 to modulate expression of TGF-β-regulated genes. There are functional consequences to this shift in Trm populations, and STAT4 deficiency leads to reduced cytokine and chemokine production and altered localization of Trm cells in the intestinal tissue. Our findings on the role of STAT4 in Trms are distinct from what is observed in circulating T-cell populations. In the lymphoid tissue, STAT4-activating cytokines support IFN-γ production and expansion of CD8+ T cells during infection37,46,47. IL-12 and STAT4 also lead to cell cycle-dependent downregulation of Tcf148, elevated expression of T-bet, and an increase in the proportion of cells that have a KLRG1+ effector memory phenotype49,50. However, Stat4−/− Trms were not impaired in their expansion within the tissue, Trms rapidly downregulate KLRG1 after their entry into the tissue23,34, and STAT4-dependent Trm populations expressed elevated levels of Tcf7 and Lef1, suggesting a less terminally differentiated phenotype. Our data indicate that STAT4 has unique roles based on tissue context; however, the activation of STAT4 ultimately results in a more heterogenous population of memory T cells in all tissues.
Although TGF-β drives CD103 expression and supports the maintenance of some Trm populations22–24,42, TGF-β-mediated changes in gene expression have also been implicated in the reduced function and plasticity of CD103+ Trms14. Recent findings indicate that CD103− Trms display increased proliferative potential compared with their CD103+ cells when isolated from the tissue and reintroduced into the circulation14; although, the proliferation of Trm cells of any subset is less robust than circulating memory populations14,51,52. CD103− Trms also proliferate more readily in situ than CD103+ Trms after infection in both the lung30 and the intestine31. CD103− Trms are likely exposed to TGF-β within the tissue; however, we have demonstrated that the presence of STAT4-activating cytokines can prevent the TGF-β-induced expression of CD10334 and other TGF-β-regulated genes and support the functional potential of CD103− Trms. Other transcription factors, including T-bet and Tcf1, can directly inhibit CD103 expression by Trms43,53, and Tcf7 expression is increased in STAT4-dependent Trm populations, which could also drive the unique gene expression signature in this subset. The counter regulatory functions of TGF-β and IL-12 in dictating the magnitude of CD8 T-cell responses and differentiation of Th1 cells is well described37,54. TGF-β can regulate IL-12 signaling by downregulating the expression of IL-12R subunits55,56 or direct inhibition of T-bet expression57. However, we demonstrate that after T cell activation in vitro, simultaneous exposure to IL-12 and TGF-β did not impact the signaling downstream of the opposing cytokine receptor. Importantly, the gene expression signature observed in vitro corresponded to what was observed in vivo and indicated that although CD103− LP Trm cells do not require TGF-β for their differentiation, this cytokine may be impacting their phenotype and downstream effector functions. Further studies are warranted to understand the interplay of STAT4 and other Trm-specific transcription factors during differentiation and maintenance of Trm cells.
Our scRNA-seq analysis revealed that although CD103 can broadly define Trm function in the intestine and other tissues, there is a significant heterogeneity within both the CD103+ and CD103− Trm subsets within the intestine. This is consistent with what has been observed in CD103+ intestinal intraepithelial Trms, and we observed a similar, although not identical, gene expression signatures among LP populations32,33. The dominant CD103+ subsets identified in WT Trm cells are P1 and P3. P3 expresses increased levels of cytokine receptors (Il12rb2, Il18rap) and NF-κB genes, along with Zeb2 and Ikzf2, which may indicate increased responses to bystander activation with a more terminally differentiated phenotype58,59. P1 expresses the Trm transcription factors Ahr and Runx3 and granzymes A and K and increased Bcl2 and Il7r, suggesting a long-lived subset with cytotoxic potential. Surprisingly, we did not detect the expression of Id3, a key regulator of intestinal epithelial Trm heterogeneity32, in any LP Trm subset. This indicates that there may be unique drivers of Trm diversity among LP CD103+ Trm populations. Our scRNA-seq analysis also identified a Trm subset that is primarily CD103− and expressed genes not identified in CD103+ LP and intraepithelial Trm subsets; these include S100a4, S100a6, Lgals1, Vim, Anxa2, and Nme1, among others. However, many of these genes are upregulated in Trm cells from the murine colon28 and human lung60, indicating that the CD103− Trm P4 we have identified may be a subset found in other mucosal tissues. Many of the CD103− Trm P4 genes are also expressed in circulating memory T cells and not Trm cells; however, we have previously demonstrated that the CD103− population maintains residency26. It is unclear how this population of cells remains in the tissue with increased expression of genes like S1pr1 or whether this allows the CD103− Trm subset to exit the tissue after reactivation. The extent to which circulating signature genes support the proliferation of CD103− Trms during secondary infection31 also remains an open question. Overall, our data have identified STAT4 as a critical regulator of CD103− Trm differentiation and highlight the importance of inflammation and STAT4 activation during primary infection in driving the differentiation of phenotypically and functionally diverse populations of Trm cells.
MATERIALS AND METHODS
Mice
C57BL/6, B6.SJL-Ptprca, C57BL/6J-Stat4em3Adiuj/J (Stat4−/−-), C57BL/6-Tg(UBC-GFP)30Scha/J, and C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-1) were purchased from Jackson Laboratories (Bar Harbor, ME) or bred in-house. YopE TCR transgenic mice were generated in-house with the assistance of the Rutgers Genome Editing Core31. All mice were housed and maintained under specific pathogen-free conditions at the Rutgers Cancer Institute of New Jersey animal facilities. Both male and female recipient mice at 7–14-weeks old were used for all experiments. All experiments were performed in accordance with the Institutional Animal Care and Use Committee of Rutgers New Jersey Medical School Comparative Medicine Resources.
Bacteria
YopM-deficient (ΔyopM Yptb), and ovalbumin expressing (Yptb-OVA)26 Yersinia pseudotuberculosis YPIII were used for these studies. Bacteria were routinely cultured on LB agar and grown shaking in Luria Broth (LB) at 26°C overnight before infection.
T-cell transfers and infections
Naïve WT and Stat4−/− OT-I or YopE TCR transgenic cells were enriched from the spleen and lymph nodes using a CD8α+ T-cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, North Rhine-Westphalia, Germany) and depleted of memory T cells with the addition of biotinylated anti-CD44 (IM7; BD Biosciences, Franklin Lakes, NJ, USA) antibody. Mice received 104–105 congenically distinct transgenic T cells the day before oral gavage with 2 × 108 CFUs of Yptb-OVA or ΔyopM Yptb in 200 μl Phosphate Buffered Saline (PBS). To discriminate between intravascular circulating CD8+ T cells and tissue-resident CD8+ T cells, 3 μg of anti-CD8α-Allophycocyanin (APC)/R-phycoerythrin (PE) (53–6.7; Biolegend, San Diego, CA, USA) was administered intravenously 10 minutes before the mice were sacrificed. To examine the T-cell proliferation, 0.5 mg of BrdU (Thermo Fisher Scientific, Waltham, MA, USA) was administered intraperitoneally 1 hour before tissue harvest. To reactivate the Trm cells in vivo, the mice were given 10 μg of YopE peptide (SVIGFIQRM; Genemed Synthesis, San Antonio, TX, USA) intraperitoneally 1 hour before tissue harvest. FTY720 (Millipore Sigma, St. Louis, MO, USA) was given intraperitoneally at 1 mg/kg every other day from day 7 p.i. until tissue harvest on day 20 p.i..
Isolation of cells
For the isolation of intestinal epithelial and LP cells, Peyer patches were removed, the intestine was cut open longitudinally, and intestinal contents and mucus were removed by gentle scraping. The intestine was cut into 1 cm pieces and incubated in Hank balanced salt solution (HBSS) buffer containing 1 mM dithiothreitol (Thermo Fisher Scientific) and 10% FBS at 37°C with stirring for 20 minutes to isolate the intraepithelial lymphocytes. Intestinal tissue was transferred to HBSS containing 1.3 mM ethylenediaminetetraacetic acid and stirred at 37°C for 20 minutes to remove the epithelium. Intestinal pieces were then incubated in HBSS containing 5% FBS and 150 U/ml collagenase type 2 (Worthington Biochemical Corporation, Lakewood, NJ, USA) at 37°C with stirring for 45–60 minutes to isolate the LP cells. Intestinal lymphocytes were further purified by gradient centrifugation with 44% and 67% Percoll (Cytiva Life Sciences, Marlborough, MA, USA). The spleen and MLN cells were isolated by homogenizing tissue using a 100-μm screen and plunger and red blood cells were lysed to generate a single-cell suspension.
Flow cytometry
Before surface staining, single-cell suspensions were blocked with anti-mouse CD16/CD32 (24G2; BioXcell, Lebanon, NH, USA). Isolated cells were stained for surface markers with the following antibodies: anti-mouse CD8β (H35–17.2) BUV395/EF450, CD45.1 (A20) PeCy7/BUV395/EF450, CD45.2 (104) PerCpCy5.5, CD103 (M290) BUV395, CD103 (207) EF450, CD69 (H1.2F3) BV510/PeCy7, and CD244 (eBio244F4) APC, all from Invitrogen (Waltham, MA, USA). Cell viability was determined using a fixable LIVE/DEAD near-infrared cell-staining kit (Thermo Fisher Scientific). For intracellular staining, cells were permeabilized and stained using BD CytoFixPerm kit or eBioscience FoxP3/Transcription Factor Staining Buffer Set per manufacturer’s instructions. Intracellular staining was performed for anti-mouse IFN-γ (XMG1.2) PerCpCy5.5, TNF-α (MP6-XT22) FITC, Granzyme B (GRB05) APC, granzyme A (3G8.5; Biolegend) PE, CCL3 (DNT3CC) PE, and T-bet (4B10) PE, all from Invitrogen unless otherwise noted. To analyze BrdU incorporation, the cells were stained for surface markers and fixed in 2% paraformaldehyde before permeabilization and anti-BrdU APC staining, as detailed in the BrdU Flow Kit (Thermo Fisher Scientific). Annexin staining was performed using the Annexin V Apoptosis Detection kit per the manufacturer’s instructions (Thermo Fisher Scientific). The cells were analyzed on a LSRFortessa X-20 (BD Biosciences) or an Attune Nxt flow cytometer (Invitrogen) and analyzed with FlowJo software (BD Biosciences).
Immunofluorescence microscopy
The ileum was harvested at day 9 p.i., fixed in 2% paraformaldehyde, rehydrated in 20% sucrose in PBS, and flash frozen in optimal cutting temperature (OCT) media (Sakura Finetek, Torrance, CA, USA). The tissue sections were cut at a thickness of 7–8 μm and treated with ice cold acetone before storage at −80°C. Tissue sections were treated with avidin-biotin blocking reagents (Vector Laboratories, Newark, CA, USA) and stained with the following reagents: anti-CD45.2-biotin (104; Invitrogen), anti-CD90.1-APC (HIS51; Invitrogen), anti-laminin (L9393; Abcam, Cambridge, United Kingdom), anti-GFP-AF488 (Thermo Fisher Scientific), and anti-EpCam-AF647/BUV421 (G8.8; Biolegend), followed by Streptavidin-AF555 (Thermo Fisher Scientific), anti-rabbit AF488 (Thermo Fisher Scientific), and 1 μg/ml DAPI (Millipore Sigma). Stained slides were mounted with Prolong Gold antifade reagent, imaged using a Keyence fluorescence BZ-X series microscope (Osaka, Osaka, Japan), and analyzed using the Adobe Photoshop software. For the quantification of cells in lymphocyte clusters, the cells were counted in multiple sections for each mouse for > 700 total cells/mouse and > 200 cells in lymphocyte clusters/mouse. For examining the distance of LP Trm cells from the epithelium, the distance between the surface of each cell and the base of the nearest epithelial cell was determined for 150–360 cells/mouse.
Quantitative reverse transcription-polymerase chain reaction (RT-PCR) directly ex vivo
YopE T cells were sorted from the LP at 9 days after Yptb-OVA infection. To discriminate between intravascular CD8+ T cells and tissue-resident CD8+ T cells, 3 μg of anti-CD8α-APC was administered intravenously 10 minutes before the mice were sacrificed. Viable i.v.CD8a−CD8b+CD69+CD103+/− WT and Stat4−/− LP Trm cells were sorted using a FACSAriaII. The cells were resuspended in TRIzol (Millipore Sigma), and RNA was isolated using Arcturus PicoPure RNA kit. RT-PCR was performed using the One-Step TB Green PrimeScript RT-PCR Kit II (Takara Bio Inc., Kusatsu, Shiga, Japan) and gene expression was normalized to Gapdh and expressed as fold change relative to the WT Trm subset of interest. Primers used: Ahr F (5’-GCCCTTCCCGCAAGATGTTAT-3’) Ahr R (5’-TCAGCAGGGGTGGACTTTAAT-3’), Klrk1 (NKG2D) F (5’-CCTTGGCAATTCGATTCACCC-3’) Klrk1 (NKG2D) R (5’-CCTTGTTGCACAATACTGGCTG-3’), Cd244 (2B4) F (5’-CCACCTTCCAAAGCAAGTACA-3’) Cd244 (2B4) R (5’-AGAAACAACGATTGTGGGTGA-3’), Il18r1 F (5’-TGAGATCCAGGCAGGAGAACTC-3’) Il18r1 R (5’-AGAGCTCCACAGTTCCCAGAAC-3’), and S1pr120.
In vitro cytokine stimulation
T cells were isolated from the spleen of WT and Stat4−/− YopE TCR transgenic mice and activated in culture for 3 days with 1μM YopE69–77 peptide and 20 U/ml recombinant human IL-2 at 37°C and 7% CO2. Viable lymphocytes were purified using Histopaque 1083 (Millipore Sigma) gradient centrifugation before being stimulated with 20 ng/ml IL-12 and 1 ng/ml TGF-β. Phospho-Smad2-PE (E8F3R; Cell Signaling Technologies, Danvers, MA, USA) was measured 3 hours after culturing in the presence of cytokines. After 40 hours of cytokine stimulation, the cells were placed in TRIzol, and RNA was purified using the Arcturus PicoPure RNA Isolation kit (Thermo Fisher Scientific). RT was performed with the RNA to cDNA EcoDry Premix (doubleprimed) (Takara Bio Inc.) and quantitative PCR was done using TB Green Advantage qPCR Premix (Takara Bio Inc.) and the following primers: Itgae F(5’-CCTGTGCAGCATGTAAAAGAATG-3’) Itgae R(5’-CAAGGATCGGCAGTTCAGATAC-3’), Litaf F (5’-ATGTCGGCTCCAGGACCTTA-3’) Litaf R (5’-GGTAGTAACTGTTGACACCCACT-3’), Cish F (5’-ATGGTCCTTTGCGTACAGGG-3’), Cish R (5’-GGAATGCCCCAGTGGGTAAG-3’), Chst12 F (5’-TGGCTGGTACTAGGGTCGG-3’), Chst12 R (5’-GGAAGGGGTTCTAGGATGTGTG-3’), Abi3 F (5’-CTACTGCGAGGATAACTACTTGC-3’), Abi3 R (5’-CAGGTTACCCACTTGGTAGGC-3’), Ikzf2 F (5’-GAGCCGTGAGGATGAGATCAG −3’) Ikzf2 R (5’-CTCCCTCGCCTTGAAGGTC-3’), Dapk2 F (5’-CTCGATGAGGAGCCCAAATATG-3’) Dapk2 R (5’-CCCGGCACTTCTTCACGAT-3’). Fold gene expression change was calculated for individual genes using Gapdh and normalized to untreated samples.
scRNA-seq and analysis
OT-I T cells were sorted from the LP at greater at 35 days after Yptb-OVA infection, and samples were pooled from two to three mice to ensure a sufficient number of cells for sequencing. To discriminate between intravascular CD8+ T cells and tissue-resident CD8+ T cells, 3 μg of anti-CD8α-APC was administered intravenously 10 minutes before the mice were sacrificed. Viable i.v.CD8α−CD8β+CD69+ WT and Stat4−/− LP Trm cells were sorted using a FACSAriaII. A total of 5000–10,000 single cells at > 90% viability were resuspended in PBS with 0.05% bovine serum albumin, and gel beads in emulsion were generated using the Chromium Single Cell 3′ Reagent Kits v3 and 10X Chromium Controller (10X Genomics, Pleasanton, CA, USA), per the manufacturer’s instructions. Library quantity and quality control were performed using Qubit Fluorometer (Thermo Fisher Scientific) and TapeStation (Agilent Technologies, Santa Clara, CA, USA). The 3’ gene expression libraries were sequenced on an Illumina NovaSeq 6000 system (San Diego, CA, USA).
The reads from scRNA-seq were aligned (mm 10) and collapsed into unique molecular identifier (UMI) counts using 10X Genomics Cell Ranger Software 3.1.0, and all samples had > 90% of reads that mapped to the genome. Raw reads were loaded into Partek Flow Genomic Analysis Software (Partek Inc., Chesterfield, MO, USA) and the dataset was filtered to exclude cells with < 500 genes and > 10% of UMIs mapping to mitochondria. The genes that were not present at > 1 UMI in more than 1% of cells were removed. The reads were normalized by count depth scaling, log2 transformed, and corrected for batch effects from separate sequencing runs using the Seurat3 Integration function. WT and Stat4−/− cells were combined, and principal component analysis- and graph-based clustering was performed using the top 20 principal components. Differentially expressed genes in individual clusters were calculated using the Partek Biomarker function, and the top 25 genes were included in subsequent analyses (p < 0.05 and fold change > 1.5). GSEA was performed using Trm and circulating signature genes17,61 and the TGF-β-induced and -repressed genes from skin Trm cells14. Direct comparison between clusters 2 and 4 was done using the analysis of variance (FDRq < 0.05 and FC > 1.5). The same strategy was used for filtering and normalization of human ileal Trm populations45. Total ileal cells were analyzed and clusters with a significant percentage of cells expressing CD8β (43.3%–67.0%), compared with approximately 5% in remaining clusters, were selected for further analysis. Filtered populations were again subjected to clustering and further analysis.
Statistical analysis
Statistical significance was determined with Prism software (GraphPad, Boston, MA, USA). Two-tailed, paired, or unpaired Student’s t test, or one- or two-way analysis of variance were used for comparisons and noted in each figure legend. A p value < 0.05 was considered significant.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Derek Sant’Angelo for reagents and assistance generating TCR transgenic mice. Products in support of the research were generated by Rutgers Cancer Institute of New Jersey (Genome Editing) Shared Resource P30CA072720-5922, with the assistance of Dr. Peter Romanienko. Flow cytometry and cell sorting was performed in the Rutgers New Jersey Medical School Flow Cytometry Core with the assistance of Dr. Sukhwinder Singh and Tammy Mui-Galenkamp.
FUNDING
This work was supported by the Feldstein Medical Foundation Grant, New Jersey Consortium on Cancer Research Bridge Grant, and the National Institutes of Health grants R01AI153096 and R21AI148900 (TB, HYF, MT).
Footnotes
DECLARATIONS OF COMPETING INTEREST
The authors have no completing interests to declare.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mucimm.2023.03.002.
DATA AVAILABILITY
Data availability Single-cell RNA-sequencing data is available through the GEO database under the accession code GSE185545.
REFERENCES
- 1.Masopust D & Soerens AG Tissue-resident T cells and other resident leukocytes. Annu. Rev. Immunol 37, 521–546 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schenkel JM et al. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346, 98–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ariotti S et al. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 346, 101–105 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Iijima N & Iwasaki A Access of protective antiviral antibody to neuronal tissues requires CD4 T-cell help. Nature 533, 552–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han SJ et al. White adipose tissue is a reservoir for memory T cells and promotes protective memory responses to infection. Immunity 47, 1154–1168.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schenkel JM, Fraser KA, Vezys V & Masopust D Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol 14, 509–513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schenkel JM, Fraser KA & Masopust D Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J. Immunol 192, 2961–2964 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iijima N & Iwasaki A T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346, 93–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang X et al. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 483, 227–231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beura LK et al. T cells in nonlymphoid tissues give rise to lymph-node-resident memory T cells. Immunity 48, 327–338.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wijeyesinghe S et al. Expansible residence decentralizes immune homeostasis. Nature 592, 457–462 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stolley JM et al. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J. Exp. Med 217:e20192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Behr FM et al. Circulating memory CD8+ T cells are limited in forming CD103 + tissue-resident memory T cells at mucosal sites after reinfection. Eur. J. Immunol 51, 151–166 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Christo SN et al. Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nat. Immunol 22, 1140–1151 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Li C et al. The transcription factor Bhlhe40 programs mitochondrial regulation of resident CD8+ T cell fitness and functionality. Immunity 51, 491–507.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackay LK et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Milner JJ et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hombrink P et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol 17, 1467–1478 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Boddupalli CS et al. ABC transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells. J. Clin. Invest 126, 3905–3916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skon CN et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol 14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wakim LM, Woodward-Davis A & Bevan MJ Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc. Natl Acad. Sci. U. S. A 107, 17872–17879 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackay LK et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol 14, 1294–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Sheridan BS et al. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 40, 747–757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang N & Bevan MJ Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandez-Ruiz D et al. Liver-resident memory CD8+ T cells form a front-line defense against malaria liver-stage infection. Immunity 45, 889–902 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Bergsbaken T & Bevan MJ Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat. Immunol 16, 406–414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casey KA et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol 188, 4866–4875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin YH et al. Small intestine and colon tissue-resident memory CD8+ T cells exhibit molecular heterogeneity and differential dependence on Eomes. Immunity 56, 207–223.e8 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wakim LM et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol 189, 3462–3471 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paik DH & Farber DL Influenza infection fortifies local lymph nodes to promote lung-resident heterosubtypic immunity. J. Exp. Med 218: e20200218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fung HY, Teryek M, Lemenze AD & Bergsbaken T CD103 fate mapping reveals that intestinal CD103− tissue-resident memory T cells are the primary responders to secondary infection. Sci. Immunol 7, eabl9925 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milner JJ et al. Heterogenous populations of tissue-resident CD8+ T cells are generated in response to infection and malignancy. Immunity 52, 808–824.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurd NS et al. Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol 5, eaaz6894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergsbaken T, Bevan MJ & Fink PJ Local inflammatory cues regulate differentiation and persistence of CD8+ tissue-resident memory T cells. Cell Rep. 19, 114–124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei L et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity 32, 840–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Good SR et al. Temporal induction pattern of STAT4 target genes defines potential for Th1 lineage-specific programming. J. Immunol 183, 3839–3847 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Starbeck-Miller GR, Xue HH & Harty JT IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J. Exp. Med 211, 105–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mackay LK et al. T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity 43, 1101–1111 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Burrows K et al. The transcriptional repressor HIC1 regulates intestinal immune homeostasis. Mucosal Immunol. 10, 1518–1528 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Zhao X, Shan Q & Xue H-H TCF1 in T cell immunity: a broadened frontier. Nat. Rev. Immunol 22, 147–157 (2022). [DOI] [PubMed] [Google Scholar]
- 41.Sumida H et al. GPR55 regulates intraepithelial lymphocyte migration dynamics and susceptibility to intestinal damage. Sci. Immunol 2, eaao1135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mani V et al. Migratory DCs activate TGF-β to precondition naïve CD8+ T cells for tissue-resident memory fate. Science 366, eaav5728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J et al. T cell Factor 1 suppresses CD103+ lung tissue-resident memory T cell development. Cell Rep. 31:107484. [DOI] [PubMed] [Google Scholar]
- 44.Palmer DC et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med 212, 2095–2113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaeger N et al. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat. Commun 12, 1921 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen KB et al. Critical role for STAT4 activation by Type 1 interferons in the interferon-γ response to viral infection. Science 297, 2063–2066 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Gil MP et al. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood 120, 3718–3728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Danilo M, Chennupati V, Silva JG, Siegert S & Held W Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T cell differentiation. Cell Rep. 22, 2107–2117 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Mollo SB, Ingram JT, Kress RL, Zajac AJ & Harrington LE Virus-specific CD4 and CD8 T cell responses in the absence of Th1-associated transcription factors. J. Leukoc. Biol 95, 705–713 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cui W, Joshi NS, Jiang A & Kaech SM Effects of Signal 3 during CD8 T cell priming: bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine 27, 2177–2187 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Behr FM et al. Tissue-resident memory CD8+ T cells shape local and systemic secondary T cell responses. Nat. Immunol 21, 1070–1081 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Fonseca R et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat. Immunol 21, 412–421 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laidlaw BJ et al. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ Tcells duringinfluenza viralinfection. Immunity 41, 633–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li MO, Wan YY, Sanjabi S, Robertson A-K-L & Flavell RA Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol 24, 99–146 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Bright JJ & Sriram S TGF-beta inhibits IL-12-induced activation of Jak-STAT pathway in T lymphocytes. J. Immunol 161, 1772–1777 (1998). [PubMed] [Google Scholar]
- 56.Gorham JD, Güler ML, Fenoglio D, Gubler U & Murphy KM Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J. Immunol 161, 1664–1670 (1998). [PubMed] [Google Scholar]
- 57.Gorelik L, Constant S & Flavell RA Mechanism of transforming growth factor β–induced Inhibition of T helper Type 1 Differentiation. J. Exp. Med 195, 1499–1505 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Naluyima P et al. Terminal effector CD8 T cells defined by an IKZF2+IL-7R−transcriptional signature express FcγRIIIA, expand in HIV infection, and mediate potent HIV-specific antibody-dependent cellular cytotoxicity. J. Immunol 203, 2210–2221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Omilusik KD et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med 212, 2027–2039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szabo PA et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun 10, 4706 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nath AP et al. Comparative analysis reveals a role for TGF-β in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS One 14:e0210495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data availability Single-cell RNA-sequencing data is available through the GEO database under the accession code GSE185545.