

INTRODUCTION
1.1. The genetic basis of human evolutionary traits
Elucidating the genetic changes underlying the evolution of human traits remains an unfinished puzzle. Genetic analyses have historically relied on single-nucleotide variants (SNVs; see Table 1 for a complete list of acronyms) for the identification of species differences and selection signatures. Although complex genomic variation has long been recognized as a force underlying phenotypic diversity—e.g., transposable elements in maize (McClintock, 1931), and chromosomal inversions (Sturtevant, 1913) and duplications (Bridges, 1936) in Drosophila—as well as a key driver of primate evolution (Bailey & Eichler, 2006), methodological difficulties have limited the understanding of their functional and evolutionary impact. Scientists are now poised to explore this question at unprecedented resolution with the large-scale adoption of high-throughput sequencing technologies (Goodwin, McPherson, & McCombie, 2016). Together, the widespread availability of high-quality reference genomes and population-level whole-genome sequencing datasets have reignited interest in studying the role of complex genomic variation in human/primate traits.
Table 1.
Acronyms used in this review
| Abbreviation | Definition |
|---|---|
| 1KGP | 1000 Genomes Project |
| AFR | African ancestry |
| AMR | American ancestry |
| BNG | Bionanogenomics |
| bp | basepair |
| CNV | copy-number variant |
| DAVID | Database for Annotation, Visualization and Integrated Discovery |
| EAS | East asian ancestry |
| EUR | European ancestry |
| eQTL | expression quantitative trait locus |
| FST | fixation index |
| gnomAD | Genome Aggregation Database |
| GoNL | Genome of the Netherlands |
| GTEx | Genotype-Tissue Expression |
| hESC | human embryonic stem cells |
| HGSVC | Human Genome Structural Variation Consortium |
| Hi-C or HIC | high-throughout chromatin conformation capture |
| HiFi | high fidelity |
| HPRC | Human Pangenome Reference Consortium |
| HSD | human-specific segmental duplication |
| HSE | human-specific expansion |
| i2QTL | Integrated iPSC QTL |
| IGC | interlocus gene conversion |
| IL | Illumina |
| iPSC | induced Pluripotent Stem Cell |
| kbp | kilobasepairs |
| kya | thousand years ago |
| LD | linkage disequilibrium |
| LRS | long-read sequencing |
| Mbp | megabasepairs |
| mCNV | multiple (multiallelic) copy-number variant |
| MESA | Multi-Ethnic Study of Atherosclerosis |
| mya | million years ago |
| NAHR | non-allelic homologous recombination |
| NHEJ | non-homologous end-joining |
| ONT | Oxford Nanopore Technologies |
| PacBio or PB | Pacific Biosciences |
| PSVs | paralog-specific variants |
| SAS | South Asian ancestry |
| SD | segmental duplication |
| SINE | short interspersed nuclear element |
| SNV | single-nucleotide variant |
| SRS | short-read sequencing |
| SV | structural variant |
| SVA | SINE-R-VNTR-Alu |
| T2T | Telomore-to-Telomere |
| TAD | topologically-associated domain |
1.2. Genomic structural variation
Broadly speaking, structural variants (SVs) are defined as complex genomic differences larger than 50 bp (Reviewed by Alkan, Coe, & Eichler (2011)) (Figure 1). These include copy number variants (CNVs) that can change the dosage of a gene or genomic region, such as deletions and duplications. Larger (>1 kbp) duplications with high sequence identity (>90%) are termed segmental duplications (SDs) or low-copy repeats (Bailey et al., 2002; Bailey, Yavor, Massa, Trask, & Eichler, 2001). Other types of SVs include insertions, which can comprise novel sequence and mobile elements such as retrotransposons (for a comprehensive review see Kazazian & Moran (2017)), translocations, and inversions.
Figure 1. Examples of genomic structural variation.
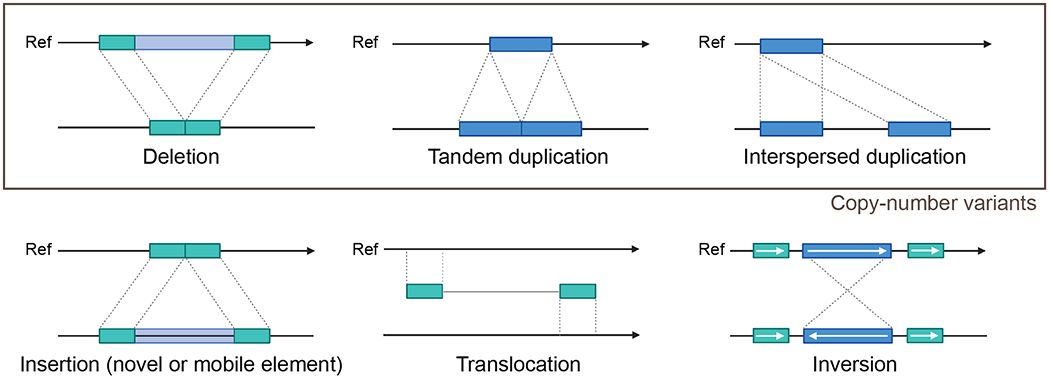
SVs exist as deletions and duplications (with the largest, most similar duplications termed segmental duplications, or SDs) that change the copy of a genomic segment (i.e., CNVs). Other types of SVs include insertions, translocations, inversions, as well as more complex events not pictured. Figure is adapted from (Alkan, Coe, et al., 2011) via “Genome Structural Variations” by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.
1.3. Goals and outline of this review
In general, both the larger size and low-complexity content that often accompany SVs inhibit their reliable discovery using genomic approaches, making this class of genetic variant historically understudied. However, rapid advancements in both sequencing technologies and computational tools are dramatically improving the study of SVs. The goal of this review is to introduce readers to genomic SVs, describe their known importance in human genome structure and evolution, outline ongoing developments in SV discovery and characterization, and provide resources to incorporate SVs in their own research. This will be achieved via the following topics:
The impact of structural variation in shaping human and related primate genomes
How SVs are used in genetic studies today with examples of variants contributing to human traits and evolution as well as gene function and regulation
Detail the roles that human-specific duplicated genes have played in brain development and evolution
Provide tips and describe the limitations and artifacts that arise from the study of SVs
Lay out ongoing genomic advancements that promise to transform our knowledge of the existing SV landscape, and propose how readers can prepare to use upcoming datasets/tools in their own research
2. HOW STRUCTURAL VARIATION HAS SHAPED HUMAN GENOMES
2.1. Hominid SDs and the “core” duplicons
Comparative genomic analyses—propelled recently by the availability of high-quality primate genomes (Chimpanzee Sequencing and Analysis Consortium, 2005; Gordon et al., 2016; Kronenberg et al., 2018; Lander et al., 2001; Locke et al., 2011; Mao et al., 2021; Nurk et al., 2022; Prüfer et al., 2012; Scally et al., 2012; Warren et al., 2020)—can provide insight into genetic instances explaining distinct phenotypic characteristics (Rogers & Gibbs, 2014). Genomic surveys have revealed thousands of genetic variants between species potentially leading to phenotypic innovations across primates (Kronenberg et al., 2018; Prado-Martinez et al., 2013; Yousaf, Liu, Ye, & Chen, 2021). One striking result of this comparative approach is the observed increased rate of accumulation of SDs in great apes compared to other primates (Marques-Bonet, Girirajan, & Eichler, 2009; Marques-Bonet, Kidd, et al., 2009a; Sudmant et al., 2013) given the known potential of duplications to serve as a source for phenotypic innovation (Ohno, 1970). In particular, the branch leading to African great apes (Figure 2) has experienced a three-fold increase in duplication activity 8–12 million years ago (mya) together with a clocklike rate of deletions and a decreased rate of SNVs, chromosomal rearrangements, and retrotransposition activity (Marques-Bonet, Kidd, et al., 2009b; Sudmant et al., 2013). As a result, SDs account for 7% (218 Mbp) of the human genome, according to the sequence of the first complete telomere-to-telomere (T2T-CHM13) reference genome (Vollger et al., 2021).
Figure 2. Cladogram of Hominidae family.
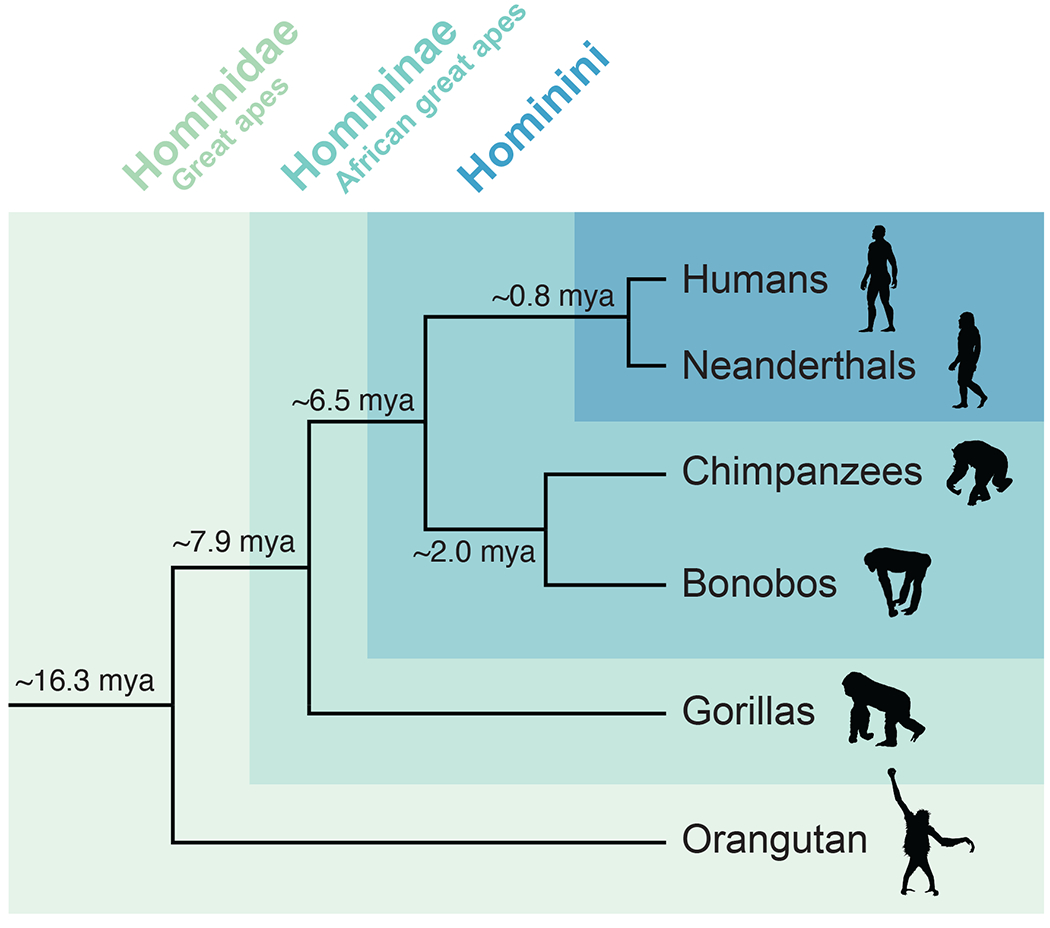
Divergence time estimates were obtained from Sudmant et al. (2013).
Hominid SDs are non-randomly distributed across the genome and organized in large blocks (>250 kbp) that display a complex structure of duplications-within-duplications arranged around sequence elements known as ‘core’ or ‘seed’ duplicons (Dennis & Eichler, 2016; Jiang et al., 2007; Marques-Bonet & Eichler, 2009). This differs from most other sequenced mammals—like mice, dogs, and cows—where SDs are primarily organized in tandem (Liu et al., 2009; Nicholas et al., 2009; She, Cheng, Zöllner, Church, & Eichler, 2008). These regions represent the focal point from which duplications accrue, with younger events located farther away from the core. In the human genome, hierarchical clustering of 437 duplicated blocks identified 24 core duplicons of ~15 kbp in size, of which fourteen were confined to one chromosome and ten were mixed across non-homologous chromosomes, mostly within subtelomeric and pericentromeric regions (Jiang et al., 2007). Evidence suggests that core duplicons have been reused independently and recurrently in different primate lineages (Cantsilieris et al., 2020; Johnson et al., 2006) and also exist at the breakpoints of large scale chromosomal translocation and inversions representing cytogenetic differences across hominids (Gagneux & Varki, 2001; Marques-Bonet, Girirajan, et al., 2009).
The core duplicons themselves are enriched for transcribed genes. Human core duplicon gene families exhibit signatures of positive selection (NBPF, RGPD, PMS2P, SPATA31, TRIM51, GOLGA8, NPIP [i.e., Morpheus], TBC1D3) (Johnson et al., 2001; Lorente-Galdos et al., 2013) and are among the most copy-number polymorphic gene families in the human genome (e.g., SPATA31, NPIP, and LRRC37A) (Redon et al., 2006; Sharp et al., 2005). Since their original discovery, only three of these gene families have been functionally characterized (TBC1D3 [detailed in Section 4.3], NBPF, and SPATA31), leaving the function of most core duplicon genes unknown (Bekpen & Tautz, 2019).
2.2. Molecular mechanisms contributing to structural variation
Different molecular mechanisms have been proposed to explain the origin of primate SDs. The enrichment of Alu short interspersed nuclear elements (SINE)—the most abundant interspersed repeats in the human genome—at the boundaries of interstitial (euchromatic) and pericentromeric SDs suggests Alu-mediated origins (Bailey & Eichler, 2006; Bailey, Liu, & Eichler, 2003). As such, it has been proposed that the primate-specific ‘burst’ of Alu retrotransposition activity that occurred 35–40 mya sensitized the ancestral primate genome to Alu-mediated recombination events, that later propelled duplication events via non-allelic homologous recombination (NAHR) and homology-directed repair. However, association with Alu elements significantly decreases for younger SDs and CNVs, suggesting newly-emerging molecular mechanisms underlying structural variation formation, with newer events driven by other repeat classes or different molecular mechanisms such as non-homologous end-joining (NHEJ) (Balachandran et al., 2022; Kim et al., 2008). In the case of the LCR16a core duplicon, which contains the rapidly-evolving primate-specific gene family Morpheus (NPIP), their interchromosomal and intrachromosomal expansions have been linked to the hominid-specific retrotransposon SINE-R-VNTR-Alu (SVA) (Damert, 2022).
As large stretches of homologous sequences provide a substrate for recombination, SDs sensitize the genome to NAHR, resulting in genomic rearrangements such as unequal crossing-over and interlocus gene conversion (IGC), where a donor sequence overwrites an acceptor sequence (Chen, Cooper, Chuzhanova, Férec, & Patrinos, 2007). Analysis of the 1000 Genomes Project (1KGP) dataset estimates that at least 2.7% of SNVs within SDs can be explained by IGC (Dumont, 2015; Genomes Project, Consortium et al., 2010), while a recent survey of variants identified from comparisons of whole human genome assemblies suggest IGC accounts for >7 Mbp of SD sequence per human haplotype (Vollger, DeWitt, et al., 2022). Duplicated non-functional pseudogenes can lead to disease by exchanging deleterious variants with functional paralogs via IGC. This is the case of SMN2, a nonfunctional HSD paralog of SMN1, which encodes the survival motor neuron protein involved in the maintenance of motor neurons (Rochette, Gilbert, & Simard, 2001). Unidirectional variant exchange via IGC causes SMN2 to “overwrite” functional SMN1 leading to the most common form of spinal muscular atrophy (Larson et al., 2015). Conversely, IGC events can also “rescue” non-functional duplicate gene paralogs from pseudogenization, as observed for NOTCH2NL, a gene implicated in human brain evolution (discussed in more detail in Section 4.3) (Fiddes et al., 2018; Suzuki et al., 2018). SDs preferentially exist at regions of genome instability, or ‘hotspots’, prone to recurrent genomic rearrangements (Stankiewicz & Lupski, 2002, 2010). Core duplicons seem to be preferential sites for rearrangement hotspots (Dennis & Eichler, 2016). A subgroup of CNVs, termed microdeletions and microduplications, have been implicated in certain conditions—such as autism, schizophrenia, and epilepsy—at certain genomic hotspots (Carvalho & Lupski, 2016; Coe et al., 2014; Inoue & Lupski, 2002; Mefford & Eichler, 2009; Perry et al., 2006; Sharp et al., 2005; Stankiewicz & Lupski, 2002, 2010; Watson, Marques-Bonet, Sharp, & Mefford, 2014; Zhang, Carvalho, & Lupski, 2009), including chromosomes 7q11.23 deletion (Williams-Beuren syndrome) and duplication (autism), 15q11–q13 deletion (Prader-Willi and Angelman syndromes), and 1q21.1 microdeletion (intellectual disability, schizophrenia) and duplication (autism). Overall, these examples suggest that gene duplication—a well-established driver of gene innovation—has conferred advantages to human evolution, while also increasing genome instability and disease risk.
3. CONTRIBUTIONS OF STRUCTURAL VARIATION TO HUMAN EVOLUTION, ADAPTATION, AND TRAITS
3.1. Variation landscape across modern humans
The availability of high-coverage population-level short-read sequencing (SRS) data provided by large-scale consortia projects have shown that SVs are an important source of genomic diversity across great apes (Prado-Martinez et al., 2013; Sudmant et al., 2013) and within human populations (see Section 5.2 for a comprehensive list of available human datasets and studies). Based on our current knowledge of SVs, which we recognize as incomplete due to difficulties in their discovery with SRS (detailed in Section 5.3) (De Coster, Weissensteiner, & Sedlazeck, 2021), lower-bound estimates suggest that around 9% of the human genome is affected by insertions, deletions, and inversions alone (~279 Mbp) (Ebert et al., 2021), while at least 7% of the human genome (Sudmant, Mallick, et al., 2015) and ~16% of hominid genomes (Sudmant et al., 2013) are variable because of CNVs. Individually, each diploid genome harbors at least 18.4 Mbp (0.6%) of SVs, accounting for more than five times as many affected base pairs as SNVs (~0.1%) (Sudmant, Rausch, et al., 2015). Focusing specifically on inversions, a more recent study estimates that an individual’s genome can harbor, on average, ~12 Mbp (~0.4%) of inverted sequence per haploid genome, affecting twice as many nucleotides as deletions and insertions and fourfold as many nucleotides as SNVs (1KGP Consortium et al., 2015; Ebert et al., 2021; Porubsky, Höps, et al., 2022). Further, NAHR between flanking SDs increases the likelihood of recurrent inversions, a process described as “inversion toggling” (Zody et al., 2008), with a majority of inversions displaying evidence of recurrence (Porubsky, Höps, et al., 2022). The existence of 27 inversions shared among different apes suggests inversion toggling also exists across species and/or as a result of incomplete lineage sorting (Porubsky et al., 2020).
Per generation, at least 4.1 kbp are associated with de novo SV events, a 90-fold increase with respect to de novo SNVs (Kloosterman et al., 2015). Further, preliminary comparisons of genome assemblies from the Human Pangenome Reference Consortium (HPRC) suggests that SNV mutation rates are elevated by ~60% across SDs compared to non-duplicated genomic regions, likely driven by IGC (Vollger, DeWitt, et al., 2022; Wang et al., 2022). Albeit different in genome distribution and affected sequence, polymorphism of SVs and SNVs share population genetic properties and global distribution patterns. Frequency-wise, most variants are rare and those with higher allele frequencies are shared among the five human continental superpopulations. All SV classes can broadly recapitulate SNV-derived ancestries (Sudmant, Rausch, et al., 2015), including CNVs (Jakobsson et al., 2008). In concordance with SNVs, individuals of African ancestry exhibit more heterozygous SVs than other populations (Sudmant, Rausch, et al., 2015).
SVs can also exist in linkage disequilibrium (LD) with neighboring SNVs. Alleles in LD are observed together at higher than expected frequencies, which can be exploited to understand evolutionary history and fine-mapping of gene associations with diseases and traits. We note that the use of LD in human evolutionary and clinical genomics has been extensively reviewed by others (Slatkin, 2008). When SVs and SNVs consistently exist on the same haplotype (or a collection of alleles on the same chromosome), we say that the SVs are in LD with (or “tagged” by) SNVs; therefore, if a tagging SNV exhibits association with a trait, the SV in LD can also be considered a possible causal variant without having to be directly genotyped for the trait. Likewise, tagging SNVs displaying signatures of selection provide information about the evolutionary relevance of associated SVs. Numerous SNV genotyping- (Hinds, Kloek, Jen, Chen, & Frazer, 2006; Locke et al., 2006; McCarroll et al., 2006) and sequencing-based (Beyter et al., 2021; Conrad et al., 2010; Hehir-Kwa et al., 2016; Saitou, Masuda, & Gokcumen, 2021; Yan et al., 2021) studies have identified SVs in LD with surrounding SNVs. SVs within duplication-rich regions, however, show a weaker correlation with surrounding SNVs than those situated in less complex regions (Locke et al., 2006; Sudmant, Mallick, et al., 2015), likely due to methodological difficulties in SNV detection within large duplications as well as SV recurrence (e.g., the same inversion occurring separately in two individuals carrying different SNV haplotypes) and IGC (Saitou & Gokcumen, 2019a) (Figure 3). As a result, NAHR-derived inversions also commonly lack LD with surrounding SNVs (Giner-Delgado et al., 2019).
Figure 3. Difficulties appraising haplotypes between SVs and neighboring SNVs.
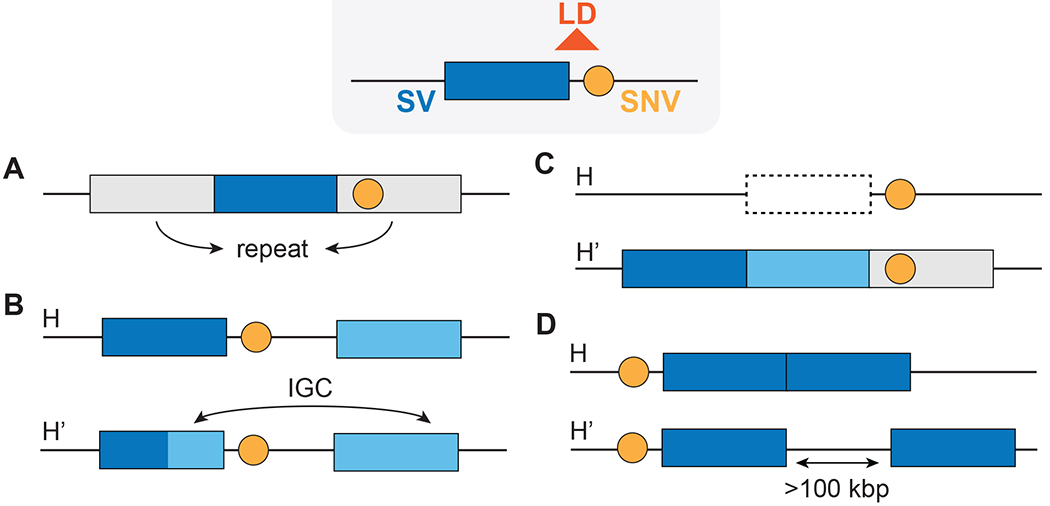
(A) Neighboring SNVs (orange circles) are difficult to detect when an SV (colored rectangle) is embedded in repeat-rich regions. (B, C) Haplotypes can be disrupted by (B) IGC and (C) recurrent deletions (H) and duplication (H’). (D) mCNVs can be in the same locus (H) or several kilobases apart (H’).
Multicopy CNVs (mCNVs), also known as multiallelic CNVs, are particularly challenging for LD-based analyses, as the duplicated paralog might not exist at the same locus of origin. Microarray-based approaches have estimated that 40% of common mCNVs are in LD with nearby SNVs (Campbell et al., 2011), while recent studies employing whole-genome sequencing (see Section 5.1 for more details) estimate that 73% of CNVs (>1% allele frequency) are in medium to strong LD (r2 > 0.6) with nearby SNVs (Sudmant, Mallick, et al., 2015). Considering the importance of the underlying LD architecture for genome-wide association studies and population genetics analyses, the lack of linkage information has hindered genotype–phenotype studies and selection scans of SV-associated regions (Saitou & Gokcumen, 2019a).
Despite methodological difficulties, the function and disease implication of some SVs have been inferred based on strong LD with surrounding associated SNVs or performing association tests with phenotype cohorts (Aguirre, Rivas, & Priest, 2019; Beyter et al., 2021). Linkage information, in particular, shows that SVs are 1.5 times more likely to be in strong LD with genome-wide association study hits than SNVs (Sudmant, Rausch, et al., 2015).
3.2. Natural selection and human structural variation
Over evolutionary timescales, SVs can be subjects of strong selective pressures. As such, a majority of SV hotspots develop in gene-poor regions, evolving under relaxed negative selection or neutrality (Lin & Gokcumen, 2019). For example, it has been proposed that relaxation of negative selection allowed for extensive copy-number variation of olfactory receptor genes in the primate lineage, with a relatively lower proportion of protein-encoding genes in humans and other primates versus dogs or rodents (Young et al., 2008). Conversely, functionally relevant sites—including coding regions and regulatory elements (e.g., enhancers and promoters)—are both depleted in SVs and enriched in rare SVs (Beyter et al., 2021), a signature consistent with purifying selection. As one might expect, selection against protein-truncating SVs has been shown to be stronger than in noncoding elements (Collins et al., 2020). Per genome, SVs are predicted to account for 17.2% of strongly deleterious variants, with rare SVs being 841 times more likely to be deleterious than rare SNVs (Abel et al., 2020). Among CNVs, deletions show stronger signatures of purifying selection than duplications, as they can severely impact gene functions by fully or partially ablating transcripts, regulatory elements, and chromatin organized units of the genomes (i.e., topologically-associated domains or TADs). Consequently, deletions are significantly depleted within functional elements in humans (Locke et al., 2006; Mills et al., 2011) and certain great apes (Fudenberg & Pollard, 2019; Soto et al., 2020).
Nevertheless, several examples of adaptive SVs exhibiting signatures of positive or balancing selection have been described in the literature, mostly implicated in local adaptation to dietary changes, environmental changes (e.g., pigmentation, thermoregulation, xenobiotic), and resistance to infectious diseases (Table 2). Here, we highlight some interesting examples, while also pointing the reader to recent reviews on this topic (Dennis & Eichler, 2016; Hollox, Zuccherato, & Tucci, 2022; Saitou & Gokcumen, 2019a). Positive selection of mCNVs has been associated with gene dosage effect (Handsaker et al., 2015). This is the case of the β-defensin genes, where copy-number gains lead to greater protein expression on the mucosal surface and higher antimicrobial activity (Hollox, Armour, & Barber, 2003). Other immune-related loci rich in common CNVs, such as the major histocompatibility complex, are thought to maintain their genetic diversity through the action of balancing and diversifying selection (Lin & Gokcumen, 2019).
Table 2.
Examples of large-scale SVs and whole-gene CNVs exhibiting signatures of natural selection in human populations.
| Gene/locus | Region | Variant | Selection type | Category | Putative trait | References |
|---|---|---|---|---|---|---|
| GSTM1 | 1p13.3 | Deletion | Positive (East Asian) | Metabolism | Xenobiotic metabolism | (Saitou, Satta, & Gokcumen, 2018) |
| Amylase (AMY1 / AMY2) | 1p21.1 | mCNV | Positive | Diet | Adaptation to high-starch diet | (Pajic et al., 2019) |
| LCEB, LCEC | 1q21.3 | Deletion | Balancing | Immune response / Pigmentation | Psoriasis / Natural vaccination | (Pajic et al., 2016) |
| UGT2B17 | 4q13.2 | Deletion | Balancing (European); Positive (East Asian) | Metabolism | Xenobiotic metabolism | (Xue et al., 2008) |
| Glycophorin (GYPA / GYPB / GYPE) | 4q31.2 | Complex duplication (GYPB-GYPA gene fusion) | Positive (East African) | Immune response | Resistance to malaria infection | (Leffler et al., 2017) |
| TCAF1 / TCAF2 | 7q35 | Non-duplicated haplogroup | Positive (Archaics) | Diet / Thermoregulation | Unknown | (Hsieh et al., 2021) |
| ORM1 | 9q32 | “Runaway” duplication | Positive (European) | Immune response | Unknown | (Handsaker et al., 2015) |
| HERC2 | 15q13.1 | Duplication | Negative (European) | Pigmentation | Unknown | (Saitou & Gokcumen, 2019b) |
| BOLA2 | 16p11.2 | mCNV | Positive | Diet | Protection against iron deficiency | (Giannuzzi et al., 2019) |
| α-Globin (HBA1/HBA2) | 16p13.3 | Deletion | Balancing (East African) | Immune response | Resistance to malaria infection | (Williams et al., 2005) |
| HPR | 16q22.2 | “Runaway” duplication | Positive (African) | Immune response | Resistance to trypanosomiasis infection | (Handsaker et al., 2015; Hardwick et al., 2014) |
| KANSL1 | 17q21.31 | Inversion, duplication | Positive (European) | Fecundity | Increased fertility | (Stefansson et al., 2005) |
| SIGLEC14 / SIGLEC5 | 19q13.41 | Deletion (gene fusion) | Positive | Immune response | Reduced risk of chronic obstructive pulmonary disease | (Angata et al., 2013; Yamanaka, Kato, Angata, & Narimatsu, 2009) |
| GSTT1 / GSTT1P1 | 22q11.23 | Deletion (gene fusion) | Balancing (African) | Diet | Xenobiotic metabolism | (Lin, Pavlidis, Karakoc, Ajay, & Gokcumen, 2015) |
| APOBEC3B | 22q13.1 | Deletion | Positive | Immune response | Unknown | (Kidd, Newman, Tuzun, Kaul, & Eichler, 2007) |
Some deletions have been maintained within populations by the action of balancing selection for thousands of years (Aqil, Speidel, Pavlidis, & Gokcumen, 2022), even before the divergence of modern humans and Neanderthals, estimated to have occurred ~800 kya (Gómez-Robles, 2019). A well-known example of this phenomenon is a common 32-kbp deletion impacting genes LCE3B and LCE3C associated with psoriasis. This deletion emerged in a common ancestor with Neanderthals and was maintained through balancing selection, likely due to increased effectiveness of the acquired immune system, albeit higher susceptibility to autoimmune disorders (Pajic, Lin, Xu, & Gokcumen, 2016). Interestingly, CNVs impacting GSTM1 and UGT2B17 are polymorphic in humans and chimpanzees, suggesting inter-species balancing selection. However, further analyses revealed that deletions of GSTM1 arose separately in both lineages (Saitou, Satta, & Gokcumen, 2018; Saitou, Satta, Gokcumen, & Ishida, 2018), while the evolutionary history of UGT2B17 remains unknown.
Inversions have also played significant roles in the evolution of great apes, representing a common large-scale rearrangement differentiating species (Catacchio et al., 2018; Locke et al., 2003; Nickerson & Nelson, 1998; Yunis, Sawyer, & Dunham, 1980; Yunis & Prakash, 1982), including nine pericentric inversions that distinguish humans and chimpanzees (Gross et al., 2006). Generally, inversions are strong candidates for speciation and selection because suppressed recombination allows for the accumulation of mutations between the derived and ancestral state (Fuller, Koury, Phadnis, & Schaeffer, 2019; Kirkpatrick & Barton, 2006; Noor, Grams, Bertucci, & Reiland, 2001). Further, they have the capacity to disrupt three-dimensional genome architecture, alter gene expression, and, in humans, exhibit a close relationship with disease-associated genomic hotspots (Koolen et al., 2006; Lakich, Kazazian, Antonarakis, & Gitschier, 1993; Lupiáñez et al., 2015; Osborne et al., 2001; Puig, Casillas, Villatoro, & Cáceres, 2015; Sturtevant, 1917). The chromosome 17q21.31 900-kbp inversion polymorphism represents an example of an SV exhibiting positive selection. The locus harbors two main distinct haplogroups, H1 (direct) and H2 (inverted), with little evidence of recombination for the last ~3 million years (Stefansson et al., 2005). The H2 haplogroup is rare in Africans and Asians while prevalent among Europeans (~20%) indicative of positive selection possibly due to its association with increased fertility in females (Stefansson et al., 2005). Both H1 and H2 haplogroups have evolved independently and experienced complex rearrangements, with recurrent partial duplications of KANSL1 (Boettger, Handsaker, Zody, & McCarroll, 2012; Steinberg et al., 2012), a gene for which haploinsufficiency causes the chromosome 17q21.31 microdeletion syndrome (also known as Koolen-De Vries syndrome) (Moreno-Igoa et al., 2015).
SVs involved in local adaptation—the genetic changes experienced by a population to adapt to local environmental conditions (Rees, Castellano, & Andrés, 2020)—are prime targets of positive selection. The identification of many adaptive SVs has relied on genome-wide scans of population stratification (Conrad et al., 2010; Redon et al., 2006; Saitou et al., 2021; Sudmant et al., 2010; Yan et al., 2021), as allele frequency differences between populations are robust to haplotype-disruptive recurrence and IGC. Population-stratified SNVs are frequently identified using the fixation index (FST). For mCNVs, the statistic VST (Redon et al., 2006) has been adapted from FST to account for multiple copy numbers. One of the most well-studied adaptive CNVs in humans impact the amylase genes, involved in starch digestion in mammals. The copy number of the salivary amylase gene, AMY1, has been found to be positively correlated with dietary starch consumption in humans (Perry et al., 2007) and several starch-consuming mammals such as dogs (Pajic et al., 2019), evidencing positive selection. Although AMY1 copy number has a dosage effect on salivary amylase production, it accounts for a small portion of the variability observed among individuals (Carpenter, Mitchell, & Armour, 2017). Interestingly, population-scale Vst analyses have led to the discovery of adaptive SVs in out-of-Africa populations that have introgressed from archaic genomes (Hsieh et al., 2019; Yan et al., 2021). Among Melanesians, for example, 19 positively-selected CNVs at chromosomes 16p11.2 and 8p21.3 likely introgressed from Denisovans and Neanderthals, respectively (Hsieh et al., 2019).
Some adaptive CNVs display a unique expansion pattern, where unusually high copy numbers are seen in one population, remaining low in the rest, a pattern termed ‘runaway duplications’ (Almarri et al., 2020; Handsaker et al., 2015). This is the case of HPR, encoding the haptoglobin-related protein which confers defense against trypanosome infection, that shows a copy-number increase in African populations consistent with the geographic distribution of the infection (Almarri et al., 2020; Handsaker et al., 2015; Hardwick et al., 2014). Other identified runaway duplications include the expansion of ORM1 in Europeans (Handsaker et al., 2015), a private expansion downstream of TNFRSF1B in the Biaka group, an expansion upstream of the olfactory receptor OR7D2 in individuals with East Asian ancestry, and expansions in medically-relevant genes HCAR2 in the Kalash group and SULT1A1 in Oceanians (Almarri et al., 2020).
3.3. Gene regulation
SVs impact not only genes but also their regulatory elements. The vast majority (>98%) of the human genome is noncoding, with changes to regulatory regions thought to be better tolerated than changes to protein-coding sequences. Enhancers possess cell-type specificity, so regulatory mutations tend to be modular, impacting the quantity, location, or developmental time of gene expression while leaving the genes themselves intact (Arnone & Davidson, 1997). Accordingly, gene regulation is a major contributor to variation within and between species (Fay & Wittkopp, 2008; Fraser, 2013; Wray et al., 2003). This was suspected even before the genomic era (Ohno, 1972), as comparison of human and chimpanzee sequences suggested that coding differences were insufficient to explain the phenotypic divergence between the species, and that most changes were likely regulatory (King & Wilson, 1975). Given that SVs constitute a major component of intra- and interspecific variation, they may underlie much of this regulatory divergence, and indeed contribute to regulatory differences within humans and between primate species (Iskow et al., 2012; McLean et al., 2011; Stranger et al., 2007). At the same time, proper development relies on finely tuned spatiotemporal expression patterns, and many disease etiologies result from aberrant cis-regulatory activity of promoters and enhancers. Strikingly, many are also caused by structural rearrangements (Kleinjan & Coutinho, 2009).
Compared to SNVs, SVs are more likely to contribute to regulatory changes, since their large size allows them to alter the copy number and genomic context of genes and regulatory elements. CNVs of coding regions can directly impact gene dosage, while SVs of noncoding regions can cause indirect expression changes by deleting, duplicating, or rearranging regulatory elements rather than genes. Genome-wide, it is estimated that 3–7% of expression quantitative trait loci (eQTLs)—or variants associated with gene expression variation—are driven by SVs, with rare variants also associated with outlier expression levels (Chiang, 2019). Similarly, SVs are ~50 times more likely than SNVs to be the lead cause of eQTL signals, with large SVs having greater effect sizes. Further, estimates based on expression data representing diverse tissue types collected post mortem from 613 individuals from the Genotype-Tissue Expression (GTEx) project predict that common SVs are causal of 2.66% of eQTLs, which represents a 10.5-fold enrichment compared to SNVs, considering their relative abundance in the genome (Scott, Chiang, & Hall, 2021). Beyond simply altering mRNA levels, individual regulatory SVs can have marked phenotypic effects. For example, deletion or duplication of enhancers upstream of SOX9 causes XX and XY sex reversal, and a human-specific loss of a conserved GDF6 enhancer results in shortened hindlimb digits in mouse models (Croft et al., 2018; Indjeian et al., 2016).
The molecular mechanisms of SV-mediated non-coding changes have been well-studied in the context of promoter-enhancer “rewiring”, in which a variant alters endogenous regulatory contacts, leading to aberrant gene expression as enhancers interact with non-target genes. Functional dissection of the WNT6/IHH/EPHA4/PAX2 locus in humans and mice demonstrated that rearrangements relative to insulating elements allowed Epha4 enhancers to interact with other promoters in the locus, driving ectopic expression in the limb buds and causing digit malformation phenotypes (Lupiáñez et al., 2015). This mechanism has been implicated in other disease contexts, including “enhancer hijacking” in cancer (Franke et al., 2022; Northcott et al., 2014; Yang et al., 2020). It is likely that similar mechanisms are at work in typical human variation; for instance, different haplotypes of the aforementioned chromosome 17q21.31 inversion exhibiting signatures of positive selection in Europeans are associated with up- and down-regulation of multiple genes (de Jong et al., 2012; Stefansson et al., 2005).
More broadly speaking, comparison of great ape genome assemblies has identified hundreds of species-specific SVs putatively altering gene expression, though most of these have not been functionally investigated (Kronenberg et al., 2018). In particular, breakpoints of large inversions (>100 kbp) tend to colocalize at human TAD boundaries, with those disrupting boundaries associated with differential expression of genes across primates (Porubsky et al., 2020). Similar studies comparing inversions differentiating human with chimpanzee and rhesus also show significant depletions of inversion breakpoints at TAD boundaries, which could suggest selection against such events (Maggiolini et al., 2020; Soto et al., 2020). Examining diversity within species, an analysis of 42 common human polymorphic inversions identified 11 to be in strong LD with previously reported eQTLs from GTEx, impacting 62 genes (Giner-Delgado et al., 2019). One inversion in particular (HsInv0201) at chromosome 5q32 exhibited signatures of balancing selection and was associated with decreased expression of SPINK6, a gene known to play a role in immune response to Salmonella (Alasoo et al., 2018; Nédélec et al., 2016). In all, SVs are inextricably linked with the gene regulatory landscape.
4. FUNCTIONAL DUPLICATED GENES DRIVING HUMAN BRAIN EVOLUTION
4.1. Human brain evolution
Small canine teeth, reduced hair cover, elongated thumbs, language, bipedalism, and advanced tool usage represent example anatomical, social, physiological, and behavioral traits that distinguish humans from their closest primate relatives (Carroll, 2003; Pääbo, 2014; Varki & Altheide, 2005). Some of the most intriguing yet unanswered questions about distinct human characteristics involve the evolution of the human brain, given the enhanced cognitive capacity present in modern humans compared to other species (Defelipe, 2011; Pääbo, 2014). Since the divergence between humans and chimpanzees, estimated ~6 mya (Besenbacher, Hvilsom, Marques-Bonet, Mailund, & Schierup, 2019), a hallmark of the encephalization process in humans has been a rapid and continuous expansion [with the exception of a recent reduction in size observed in the last 3,000 years (DeSilva, Traniello, Claxton, & Fannin, 2021)]. As a result, modern human brains are almost three times the volume of modern chimpanzees (Defelipe, 2011; DeSilva et al., 2021; Molnár et al., 2019a). In particular, the neocortex represents a key driver of human brain evolution given its distinct anatomical and cellular characteristics (Geschwind & Rakic, 2013; Mora-Bermúdez et al., 2016; Rakic, 2009), including increases in neuronal density and connectivity (particularly in cortico-basal circuits), lengthening of prometaphase-metaphase stages in proliferating neuronal progenitors, and prolonged corticogenesis (Boyd et al., 2015; Enard et al., 2009; Herculano-Houzel, 2016; Liu, Hansen, & Kriegstein, 2011; Mora-Bermúdez et al., 2016). Collectively, these features are believed to play a key role in the development of higher cognitive abilities, such as language and perception (Molnár & Pollen, 2014). Importantly, these same characteristics are likely to also have contributed to a surge in neurodevelopmental conditions in modern humans, such as epilepsy (Tóth et al., 2018) and autism (Stoner et al., 2014).
4.2. Human-specific duplicated genes
Variants found exclusively in all humans are candidates for evolutionary-relevant changes underlying unique species traits (O’Bleness, Searles, Varki, Gagneux, & Sikela, 2012). Striking examples include frameshift mutations in MYH that led to its inactivity and a reduction in masticatory muscles (Stedman et al., 2004), regulatory changes in HACNS1 that produced a human-specific enhancer gain of function in limb development (Prabhakar et al., 2008), and the loss of penile spines due to a 60-kbp deletion near the androgen receptor (AR) gene (McLean et al., 2011). In the case of SVs, gene loss caused by fixation of lineage-specific deletions has been proposed as a common and rapid local adaptation mechanism, often associated with immune response and pathogen resistance (Olson, 1999). The Great Ape Genome Project identified 13.54 Mbp of human fixed deletions, containing 86 putative gene losses, 40 of which were human-specific, including known lost genes SIGLEC13 and CLECM4 (Sudmant et al., 2013). Conversely, human-specific SDs (HSDs)—large duplication events originating after the split from a common human-chimpanzees ancestor—and human-specific expansions (HSEs)—great ape gene duplications that reached higher copy numbers uniquely in humans—are also prime targets for the evolution of uniquely human traits (Dennis & Eichler, 2016).
Duplicated genes impact evolutionary history across the entire tree of life (Lallemand, Leduc, Landès, Rizzon, & Lerat, 2020; Taylor & Raes, 2004; Jianzhi Zhang, 2003), including notable examples of animal embryonic body patterning with the Hox genes (Wagner, Amemiya, & Ruddle, 2003), digestive abilities in leaf-eating monkeys with the ribonuclease genes (Zhang, Rosenberg, & Nei, 1998), resistance to plagues in soybean with the Rhg1 gene (Cook et al., 2012), and modifications to the visual system through the recruitment of crystallin genes (Piatigorsky, 2003). In this matter, humans are no exception to this trend. Through comparisons of genome assemblies and whole-genome sequences of hundreds of great apes, extensive progress has been made in identifying duplicated genes impacted uniquely in humans (Dennis et al., 2017; Fortna et al., 2004; Sudmant et al., 2013, 2010; Sudmant, Mallick, et al., 2015; Vollger, Guitart, et al., 2022). A recent study surveyed sequence data from modern humans (N=236) versus non-human primates (N=86) identifying 218 autosomal regions uniquely duplicated in humans but not other great apes (Dennis et al., 2017). Due to difficulties in assembling duplicated regions, the identified loci were enriched at gaps in the existing human reference genome. Targeted efforts to fix the sequence assembly of the largest HSD loci resulted in the identification of 33 gene families representing 80 gene paralogs, many of which clustered at the aforementioned NAHR hotspots implicated in neurodevelopmental conditions. Further, previous work by Sudmant et al., (2010) has shown enrichment of human-specific duplicated genes implicated in the neural functions, suggesting a functional connection may exist between human duplicated genes and brain evolution.
4.3. Examples of functional human duplicated genes important in neurodevelopment
A number of HSD and HSE genes have been associated with neurodevelopment. For example, GPRIN2 (Chen, Gilman, and Kozasa 1999) has been implicated in neurite outgrowth and branching. Further, human-specific HYDIN2—emerging from an incomplete duplication of ancestral HYDIN—likely adopted a new promoter increasing its expression in neural tissue (Dougherty et al. 2017) with CNVs affecting this gene associated with micro- and macrocephaly (Brunetti-Pierri et al. 2008). Collectively, in vivo knockout of ancestral orthologs and gain-of-function studies that introduce human paralogs of genes in mice and cortical organoids have been instrumental in delineating functions in neurodevelopment. Below, we highlight four additional HSD and HSE genes with compelling connections with human brain development (Figure 4).
Figure 4. Summary of human duplicated genes implicated in neurodevelopmental functions.
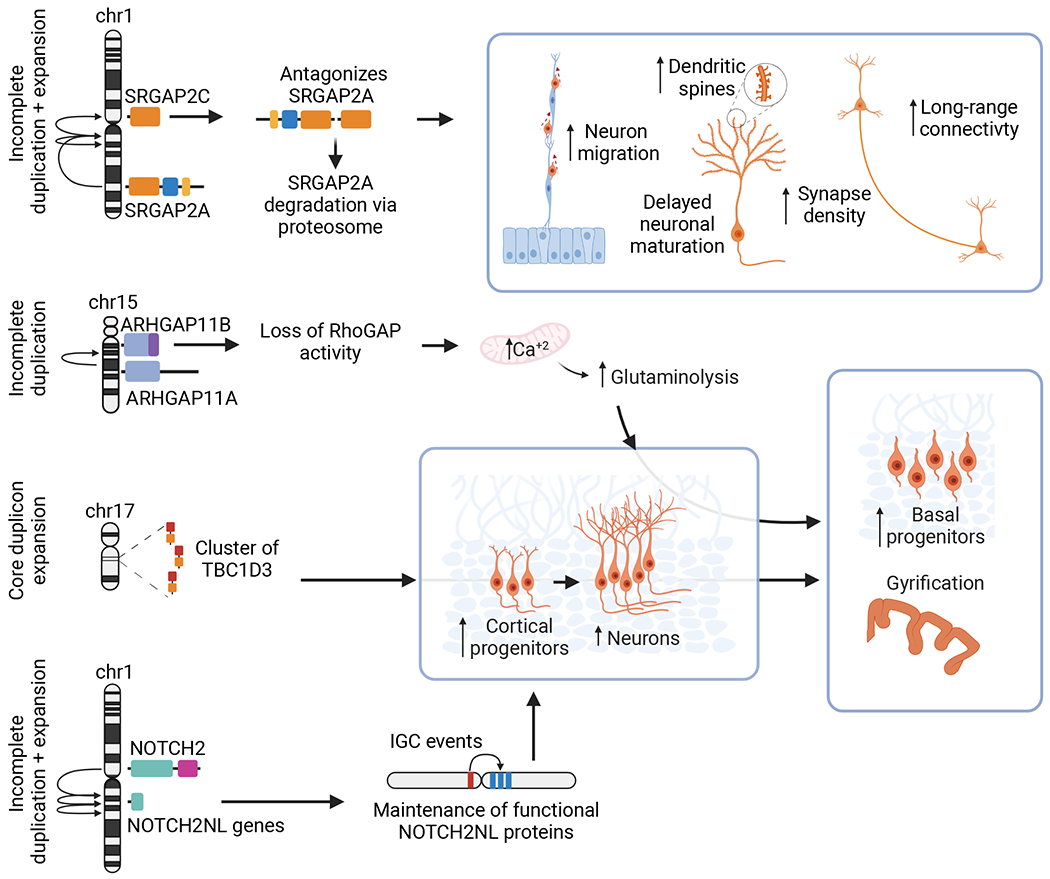
Partially duplicated SRGAP2C antagonizes ancestral SRGAP2A through dimerization of their F-BAR domains, causing its degradation and resulting in increases in the rate of neuronal migration, density of dendritic spines, long-range neuronal connections, and synapse density, as well as delayed neuronal maturation. Truncated ARHGAP11B carries 55 distinct terminal amino acids that result in a loss of its ancestral RhoGAP activity, increasing calcium levels in the mitochondria that result in increased glutaminolysis and a higher abundance of basal progenitors that lead to presence of gyrification. HSE of TBC1D3 located in a core duplicon has been expanded multiple times. Studies have revealed an increase in cortical progenitors and subsequently neurons in the presence of this gene, ultimately resulting in mice with gyrencephalic brains. Incomplete duplication of the N-terminal portion of NOTCH2 and subsequent expansion gave rise to several NOTCH2NL paralogs that remained functional likely due to IGC events and that have been found to directly increase the abundance of cortical progenitors and neurons. Figure created with BioRender.com.
SRGAP2C
In the search for functional human-specific duplicated genes impacting neurological traits, the SRGAP2 (SLIT-ROBO Rho-GTPase-activating protein 2) gene family has received considerable attention. SRGAP2 duplicated three independent times in the last ~3 million years along the human lineage resulting in the full-length ancestral SRGAP2 and truncated human-specific paralogs: SRGAP2B, SRGAP2C, and SRGAP2D (Dennis et al., 2012). SRGAP2, which encodes a homo-dimerizing protein comprising F-BAR, RhoGAP, and SH3 functional domains, is an important regulator of neuronal migration and outgrowth. Srgap2-knockdown mice generated using in utero electroporation of a short-hairpin RNA resulted in increased rate of neuronal migration and neurite outgrowth (Guerrier et al., 2009). These processes are largely driven by the homodimerization of Srgap2 F-BAR domains, which are widely known to participate in cytoskeleton remodeling (Liu, Xiong, Zhao, Yang, & Wang, 2015; Sporny et al., 2017). In utero electroporation of truncated SRGAP2C, encoding a truncated F-BAR domain while lacking RhoGAP and SH3, in mice phenocopied the Srgap2-knockdown with animals exhibiting neoteny during spine maturation and an increased density of dendritic spines (Charrier et al., 2012). Co-expression in COS7 cells revealed that SRGAP2C also interacts with SRGAP2A via their F-BAR domain resulting in degradation of the heterodimer produce in a proteasome-dependent manner (Schmidt, Kupferman, & Stackmann, 2019), explaining the mirroring effect between Srgap2-knockdown and SRGAP2C-injected mice. Moreover, SRGAP2A promotes maturation of excitatory and inhibitory synapses through interaction with key molecular players such as Homer, Gephyrin, and Rac1; functions that are inhibited in the presence of SRGAP2C, leading to delayed neuronal maturation and a higher density of synapses (both excitatory and inhibitory) (Fossati et al., 2016; Schmidt et al., 2019). Further, expression of SRGAP2C in mouse cortical pyramidal neurons resulted in increased long-range synaptic connectivity (Schmidt et al., 2021). SRGAP2C “humanized” mice also showed a higher and more selective response via whiskers stimulation, and evidenced a significantly increased cortical processing ability in a whisker-based texture-discrimination task. These results combined suggest a potential impact of SRGAP2C to functional features of human cortical connectivity that might have played a role in the emergence of unique cognitive capacities.
ARHGAP11B
Extensive studies have also focused on detailing the relevance of ARHGAP11B (Rho-type GTPase-activating protein 11), a gene implicated in human brain expansion. The duplicated paralog arose as a partial duplication of ARHGAP11A ~5.3 mya (Antonacci et al., 2014). Evaluations of multiple transcriptomic datasets of human fetal cell types revealed a high ARHGAP11B expression (more than 10-fold greater) in progenitor cells (basal glial) compared to differentiated neuronal cells (Florio et al., 2015, 2018) resulting in increased cortical gyrification in mouse (Florio et al., 2015), ferret, and marmoset models (Heide et al., 2020; Kalebic et al., 2018). A single substitution impacting an ARHGAP11B splice-donor site results in a truncated protein encoding a human-specific carboxy terminus with lost RhoGAP activity (Florio, Namba, Pääbo, Hiller, & Huttner, 2016) but new glutaminolysis mitochondrial functions by increasing Ca+2 concentrations (Namba et al., 2020). Mice expressing ARHGAP11B exhibit enhanced memory flexibility and reduced anxiety levels (Xing et al., 2021). Recent work using human- and chimpanzee-derived cortical organoids revealed the role of ARHGAP11B in basal progenitor amplification (Fischer et al., 2022). These combined findings provide an exciting case of a human-specific gene gaining a novel function that impacts the development of features highly relevant in the evolution of the human brain.
TBC1D3
Amplifications of the core duplicon containing TBC1D3 (TBC1 Domain Family Member 3) produced multiple paralogs in humans (TBC1D3A-K) whilst persisting as a reduced copy gene in chimpanzees (Perry et al., 2008). More recent comparisons across primate genomes revealed independent and recurrent expansions of TBC1D3 also in gorilla, orangutan, and macaque at different evolutionary times, but an evidently larger expansion in the human lineage ~2.3 mya (using the macaque sequence as outgroup) particularly in two genomic regions on chromosome 17 that are highly copy number polymorphic across humans, ranging between 2 and 14 copies (Vollger, Guitart, et al., 2022). Initial functional TBC1D3 studies reported a role in cell proliferation by modulating the signaling of growth factors (Hodzic et al., 2006; Wainszelbaum et al., 2008). More recently, expression of TBC1D3 paralogs in mice resulted in the expansion of self-renewing basal progenitors due to increased proliferation of outer radial glial cells, promoting folding of the neocortex (Ju et al., 2016), a hallmark feature of the human brain evolution (Geschwind & Rakic, 2013; Molnár et al., 2019b).
NOTCH2NL
Partial duplication, including the first four exons of the well-characterized NOTCH2 (Neurogenic locus notch homolog protein 2) signaling gene (Imayoshi, Sakamoto, Yamaguchi, Mori, & Kageyama, 2010; Irvin, Zurcher, Nguyen, Weinmaster, & Kornblum, 2001), and subsequent SD expansion resulted in three truncated paralogs (NOTCH2NLA, NOTCH2NLB, and NOTCH2NLC) on chromosome 1q21.1 and one paralog (NOTCH2NLR) on chromosome 1p11.2 (Fiddes et al., 2018). While gorillas and chimpanzees carry pseudogenized NOTCH2NL genes, human paralogs share >99.1% sequence similarity and encode functional proteins likely due to human-specific IGC events between NOTCH2 and the NOTCH2NL genes that acted to resurrect these previously non-functional genes. Recent studies overexpressing human-specific NOTCH2NLB in human embryonic stem cells and mouse cortical spheroids revealed clonal expansion of progenitors resulting in a higher neuronal count compared to controls (Fiddes et al., 2018; Suzuki et al., 2018). In utero electroporation of human-specific NOTCH2NL also increased the population of cycling basal progenitor cells in the embryonic mouse neocortical subventricular zone (Florio et al., 2018; Suzuki et al., 2018). These findings point to NOTCH2NLB as a potential key regulator in human brain expansion.
4.4. The search for additional duplicated genes important in human brain evolution
Though the described examples showcase certain human duplicated genes and their roles in neurodevelopment, the functions of >100 discovered HSD genes remain to be characterized. Ongoing work optimizing protocols for cortical organoids promise to increase the scale and reproducibility necessary to globally test gene functions in neurodevelopment (Bray, 2019). In particular, great promise exists in understanding species differences directly by manipulating and directly comparing the development of chimpanzee and human induced Pluripotent Stem Cell (iPSC)-derived organoids (Pollen et al., 2019; Romero et al., 2015; Song et al., 2021). Ideally, expansion of iPSC resources for additional non-human primates would allow for more comprehensive comparisons (Fernandes, Klein, & Marchetto, 2021). Alternative to direct gene manipulations, functions of duplicated genes can also be delineated based on shared co-expression and protein interactions. To identify functional enrichments across a more comprehensive set of duplicated genes, we employ here an approach based on gene ontology. Using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Huang, Sherman, & Lempicki, 2009; Sherman et al., 2022) and the complete list of HSD genes identified by Dennis et al. (2017), we find significant enrichment for functions in nervous system development, actin cytoskeleton organization, and dendrite morphogenesis. Conversely, chimpanzee-specific duplicated genes identified by Sudmant et al (2013a) are enriched for functions in RNA secondary structure, positive regulation of interleukin production, and neurotransmitter secretion in muscles. The role of HSD genes in cytoskeleton organization is particularly interesting as actin plays a key role during brain development (Luo, 2002). Neuronal proliferation, migration, signaling, and differentiation all require considerable changes to cell morphology through coordinated actin cytoskeletal and membrane remodeling (Ayala, Shu, & Tsai, 2007; McKayed & Simpson, 2013). Indeed, duplicated genes SRGAP2 and ARHGAP11A both function in the Rho/Rac/Cdc42 pathways, key to actin cytoskeleton dynamics (Florio et al., 2015; Guerrier et al., 2009; Tapon & Hall, 1997). We propose other discovered HSD genes may play a role in neural functions via membrane dynamics that could be systematically tested using cell assays.
Though we expect to identify additional HSD paralogs that have retained their ancestral paralog function, undergone subfunctionalization, or acquired a new function (known as neofunctionalization), most duplicated genes, after a brief period of functional redundancy and relaxed selection, will accrue deleterious mutations and go the road of pseudogenization (Lynch & Conery, 2000). One way to assess this is through cell/tissue expression conservation between homologous genes. A comparison of cross-tissue expression data from 75 HSD gene paralogs between humans and chimpanzees suggests that human-specific paralogs broadly exhibit patterns consistent with both relaxed selection and neofunctionalization (Shew et al., 2021). Ancestral paralogs largely retain conserved expression patterns while duplicate paralogs either reduce expression in existing tissue and/or gain expression in novel tissues. This is consistent with long-read isoform sequencing of HSD genes in adult brain that found almost half of duplicate paralogs examined contained novel features, including exapted or truncated exons, new transcription start or end sites, or altered splicing (Dougherty et al., 2018).
Some of the studies highlighted above successfully identified functional duplicated genes important in brain development by their preferential expression in human fetal brain neural subtypes (Florio et al., 2015, 2018; Suzuki et al., 2018). For example, Florio et al. (2015 and 2018) narrowed in on both ARHGAP11B and NOTCH2NL paralogs, but also 13 additional human-specific genes with preferential expression in cortical progenitors, including the FAM72 gene family that exists directly adjacent to the SRGAP2 paralogs. Further, Suzuki et al. (2018) performed RNA-seq of human fetal cortical tissue extracted at different stages of development (7 to 21 gestational weeks) to identify 35 human- and hominid-specific genes displaying robust fetal brain expression profiles, including NOTCH2NL paralogs and several core duplicon-associated genes (e.g., GOLGA6/8, LRRC37A/B, NBPF, NPIP, and PMS2).
Employing a similar approach, for this review, we also assessed the propensity of a subset of HSD genes to function during critical stages of corticogenesis by re-quantifying expression across differentiated human embryonic stem cells (hESC)-derived cortical neurons from 0–77 days, as previous published analysis of these data did not include most duplicated genes (van de Leemput et al., 2014). To account for this, we used an approach demonstrated to accurately quantify gene paralog expression (Patro, Duggal, Love, Irizarry, & Kingsford, 2017; Shew et al., 2021) and detected expression of all tested HSD paralogs within one of the five stages of cortical neuron progression during development (Figure 5). We note high expression for ARHGAP11B during deep layer formation, coinciding with previous studies reporting its high expression in basal radial glial cells (Florio et al., 2015). NPY4RB also demonstrates a similarly high expression during deep layer formation and may be tested for impact on proliferation.
Figure 5. Transcriptional profiles of a subset HSD genes across corticogenesis using data from differentiated hESCs.
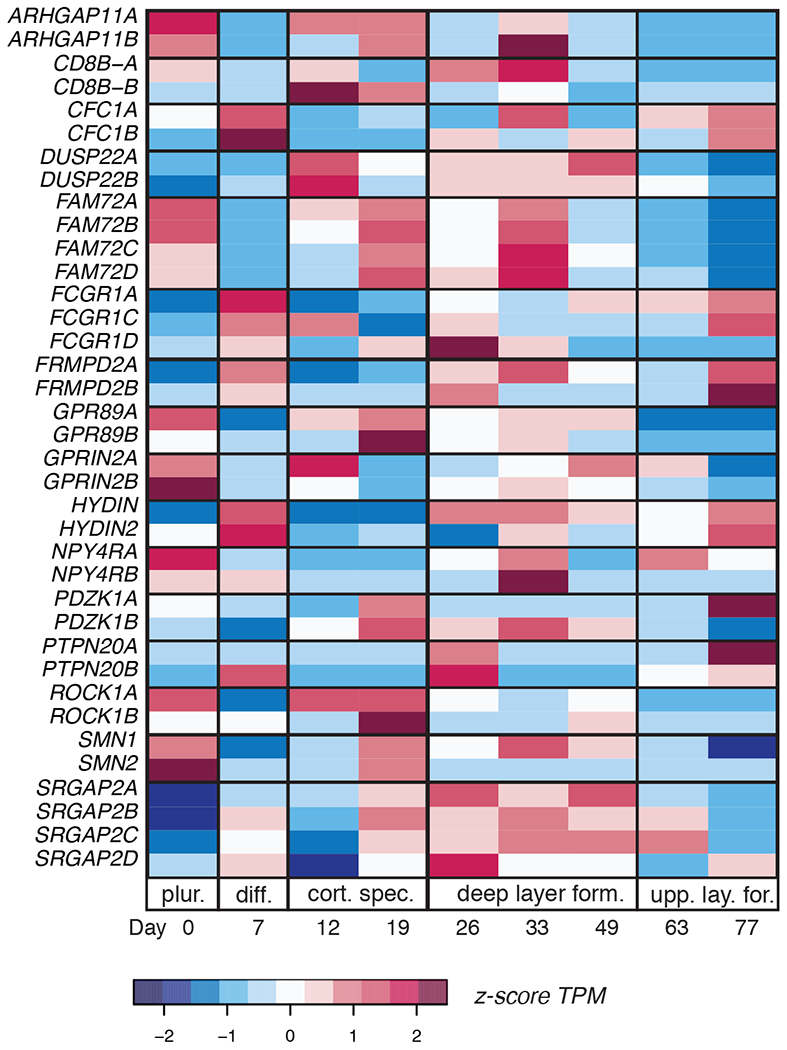
RNA-seq quantification across a time-course of 77 days to mimic developmental stages including: pluripotency (plur.), differentiation (diff.), cortical specification (cort. spec.), deep layer formation (form.) and upper layer formation (upp. lay. form.). Expression levels displayed as z-scores of HSD genes in relation to the complete transcriptome indicated as colors (red = high; blue = low expression).
With the increasing availability of transcriptomic datasets representing human fetal brain development, particularly as more studies employ longer sequence reads enabling more accurate assessment of highly-similar duplicate paralogs, we anticipate being able to identify more exciting functional candidates for future study.
5. INCORPORATING STRUCTURAL VARIATION IN YOUR OWN RESEARCH
5.1. Approaches to identify structural variation
SVs can be identified using a number of genomic techniques. Classical approaches, reviewed extensively by Alkan, et al. (2011) and others (Gresham, Dunham, & Botstein, 2008; Sharp, Cheng, & Eichler, 2006; Yang, 2020), use fluorescent probes targeting specific genomic loci coupled with DNA hybridization (e.g., fluorescent in situ hybridizations and SNP microarrays). More recently, with the advent of DNA sequencing and the exponential increase of available datasets (Section 5.2 below), current studies typically employ whole-genome sequencing (see “Beginner’s guide to next generation sequencing” for a recent, easy-to-follow review by Aigrain (2021)). The most commonly-used approach—Illumina or SRS—requires DNA fragments less than 1 kbp in size to generate up to billions of highly-accurate sequence reads as large as 300 bp, often representing “paired ends” of input DNA fragments. A majority of genomes are available as SRS based on its affordability (currently ~$700 for a genome at 30× coverage) and high accuracy. Less commonly used but increasingly adopted is long-read sequencing (LRS), via PacBio or Oxford Nanopore Technologies (ONT), with theoretically no DNA length limitation (e.g., over 1 Mbp for ONT) but at reduced fidelity (~90% accuracy) and increased costs. Recent improvements in both technologies, such as PacBio high-fidelity (HiFi) sequencing resulting in >99% accuracy for fragments ~20 kbp in length, has resulted in significantly improved accuracy of data (Vollger, Logsdon, et al., 2019; Wenger et al., 2019), while improvements in throughput are reducing costs (discussed in more detail in Section 5.4).
Typically, SRS is optimal for SNV detection, while LRS can span complex breakpoints enabling improved detections of SVs. Two recent reviews have nicely summarized existing bioinformatic approaches to identify SVs (Ho, Urban, & Mills, 2019; Mahmoud et al., 2019). Standard approaches rely on identifying optimal matches of sequence reads with a human reference genome (“mapping”) followed by discovery of differences in a sample versus the reference (Figure 6). SRS-based methods search for deviations from expectations of read depth as well as distance between read pairs to identify CNVs and SVs. In a simple scenario, a 10-kbp deletion residing on a single allele would result in half as many sequence reads spanning the reference region compared to the average sequence-read coverage of the rest of the genome (i.e., 15× across the deletion for a 30× coverage genome). That same deletion could also produce unexpected distances between read pairs with a majority of reads mapping ~500 bp apart (based on the fragment length of the sequence library), while paired reads spanning the deletion will map ~10 kbp apart (the length of the deletion). Further, both LRS- and SRS-based approaches can identify SVs when a single read maps to multiple locations in the reference (or “split read”). Considering our same deletion example, the entire 10 kbp will be missing from half of the reads and will result in no mapping to the deletion reference locus for these reads.
Figure 6. Short- and long-read SV discovery signals.
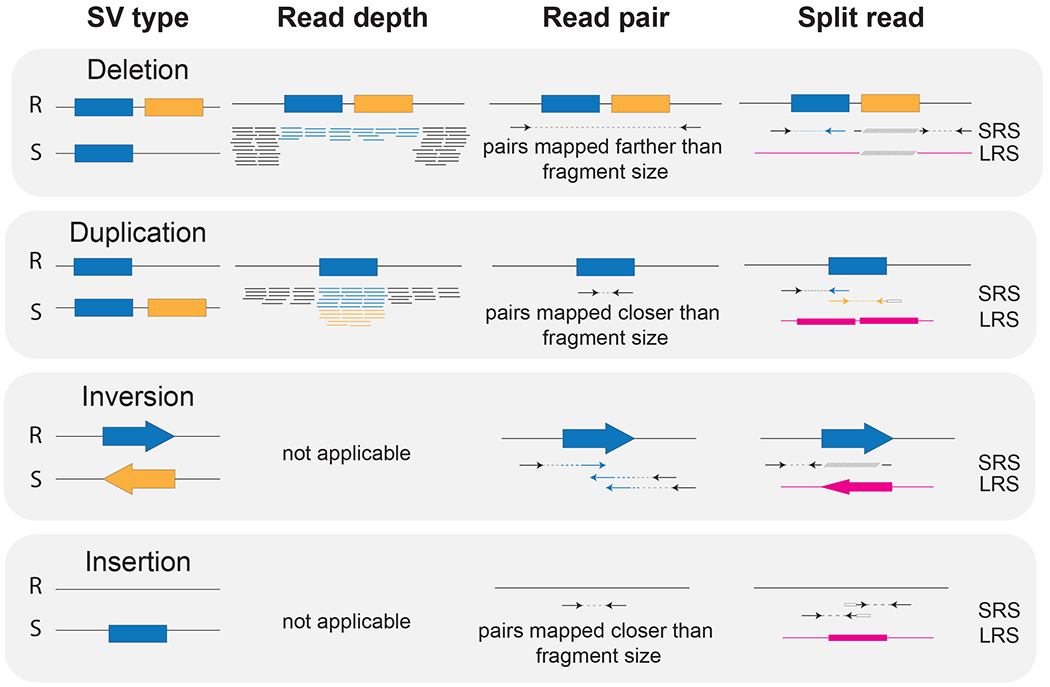
R: Reference. S: Sample. SRS: short-read sequencing. LRS: long-read sequencing. Dashed line connects two pairs from the same short-read sequencing DNA fragment. Pink shapes represent long reads.
Likewise, technologies also exist capable of maintaining long-range haplotype information, such as Strand-seq (Falconer et al., 2012), Hi-C (Belton et al., 2012), and optical mapping (Das et al., 2010), that have been used to detect SVs that are less dependent on nucleotide sequence quality and mappability of the target region. Strand-seq, which preserves directionality of reads by sequencing only replicating DNA strands within single cells, can identify balanced genomic rearrangements—inversion and translocation—as changes in orientation of mapped reads (Sanders, Falconer, Hills, Spierings, & Lansdorp, 2017; Sanders et al., 2016). Hi-C, or high-throughput chromatin conformation capture, detects both balanced and unbalanced large-scale genomic rearrangements as changes in the three-dimensional genome organization visualized in contact frequency heat maps (Harewood et al., 2017). Optical mapping fluorescently labels recognition sequences across single DNA molecules and identifies balanced and unbalanced SVs as changes in label spacing between the sample and a reference genome (Cao et al., 2014; Lam et al., 2012) (reviewed in Jeffet, Margalit, Michaeli, & Ebenstein (2021)).
5.2. Available human population datasets
A number of publicly-available sequencing datasets exist representing modern human populations (Table 3), enabling more comprehensive detection of SVs. Major human sequencing projects include the 1KGP (Byrska-Bishop et al., 2022; Sudmant, Rausch, et al., 2015), the Human Genome Diversity Project (HGDP) (Almarri et al., 2020; Bergström et al., 2020), the Genome Aggregation Database (gnomAD) (Collins et al., 2020), and the UK BioBank (Halldorsson et al., 2022), as well as a growing body of individuals from diverse backgrounds sequenced with long reads as part of the HPRC (Liao et al., 2022) and other projects (Aganezov et al., 2022; Audano et al., 2019; Ebert et al., 2021).
Table 3.
Population cohorts of human structural variation obtained from whole-genome sequencing data.
| Reference | Dataset | SV Discovery | SV Genotyping | ||||
|---|---|---|---|---|---|---|---|
| Cohort | Population(s) | Platform | Cohort | Population(s) | Platform | ||
| (Sudmant, Mallick, et al., 2015) | - | 236 | 125 populations | IL | - | - | - |
| (Sudmant, Rausch, et al., 2015) | 1KGP (low-cov) | 2,504 | AFR, EUR, EAS, SAS, AMR | IL | - | - | - |
| (Hehir-Kwa et al., 2016) | GoNL | 250 | Dutch | IL | - | - | - |
| (Chiang et al., 2017) | GTEx | 147 | AFR, EUR, American Indian, Asian | IL | - | - | - |
| (Chaisson et al., 2019) | HGSVC | 9 | AFR, EAS, AMR | IL, PB, ONT, BNG | - | - | - |
| (Audano et al., 2019) | - | 15 | AFR, EUR, EAS, SAS, AMR | PB | 440 | AFR, EUR, EAS, SAS, AMR | IL |
| (Jakubosky et al., 2020) | i2QTL | 719 | AFR, EUR, EAS, SAS, AMR | IL | - | - | - |
| (Almarri et al., 2020) | HGDP | 911 | 54 populations | IL | - | - | - |
| (Collins et al., 2020) | gnomAD | 14,891 | AFR, EUR, EAS, AMR | IL | - | - | - |
| (Abel et al., 2020) | CCDG | 17,795 | AFR, EUR, AMR | IL | - | - | - |
| (Quan et al., 2021) | - | 25 | EAS | ONT | - | - | - |
| (Ebert et al., 2021) | HGSVC | 32 | AFR, EUR, EAS, SAS, AMR | PB | 3,202 | AFR, EUR, EAS, SAS, AMR | IL |
| (Beyter et al., 2021) | - | 3,622 | Icelandics | ONT | - | - | - |
| (Yan et al., 2021) | - | - | - | - | 2,504 | AFR, EUR, EAS, SAS, AMR | IL |
| (Sirén et al., 2021) | - | - | - | - | 5,202 | AFR, EUR, EAS, SAS, AMR, MESA | IL |
| (Ebler et al., 2022) | HGSVC | 14 | AFR, EUR, EAS, AMR | PB | 300 | AFR, EUR, EAS, SAS, AMR | IL |
| (Aganezov et al., 2022) | - | 17 | AFR, EUR, EAS, SAS, AMR | PB, ONT | - | - | - |
| (Byrska-Bishop et al., 2022) | 1KGP (high-cov) | 3,202 | AFR, EUR, EAS, SAS, AMR | IL | - | - | - |
| (Halldorsson et al., 2022) | UK BioBank | 150,119 | British Irish, AFR, SAS | IL | - | - | - |
| (Jarvis et al., 2022; Liao et al., 2022) | HPRC HPRC+ |
29 18 |
AFR, EAS, AMR | PB, ONT, BNG, HIC | - | - | - |
1KGP: 1000 Genome Project. HGDP: Human Genome Diversity Project. HGSVC: Human Genome Structural Variation Consortium. gnomAD: Genome Aggregation Database. i2QTL: Integrated iPSC QTL. GoNL: Genome of the Netherlands Project. GTEx: Genotype-Tissue Expression Project. MESA: Multi-Ethnic Study of Atherosclerosis. HPRC: Human Pangenome Reference Consortium. AFR: African. EUR: European, EAS: East Asian. SAS: South Asian. AMR: American. IL: Illumina short reads. PB: PacBio long-reads. ONT: Oxford Nanopore Technologies long reads. BNG: Bionano Genomics. HIC: Hi-C chromatin conformation capture.
5.3. Challenges and opportunities across structurally complex genomic loci
Though we provide examples of the importance of structural variation in human evolution, traits, and diseases, the identification and analysis of these complex variants remain difficult. If you decide to ignore these loci, be aware of the technical artifacts that can arise due to SVs even in seemingly “simple” genomic regions. Please exercise caution and be aware of the limitations across complex regions in the various genomic technologies employed, which we will cover in this section.
Short-read sequencing technologies tend to underperform in complex genomic regions
The study of structural variation has faced several methodological challenges caused by the complex architecture of primate SDs. SVs, including duplicated regions, are historically difficult to assay using SRS technologies, the most widely available sequencing technology with thousands of whole-genome DNA samples sequenced in the public domain (Table 3). Short-read lengths (~50–300 bp) pose challenges for (i) the assembly of large repeats and SDs, (ii) reads mapping to repeat-rich regions, (iii) resolving SVs, and (iv) phasing haplotypes (Alkan, Sajjadian, & Eichler, 2011; Chaisson, Wilson, & Eichler, 2015). Since the emergence of SRS technologies, generating de novo assemblies results in gaps preferentially at nearly identical SDs, satellite DNA, and other repeat-rich regions (Chaisson, Wilson, et al., 2015; Treangen & Salzberg, 2011), in addition to AT- and GC-rich regions that suffer from low sequence coverage in DNA-amplification-dependent Illumina sequencing (Goodwin et al., 2016). In SRS assemblies, SDs tend to be either collapsed (missing copies) or misassembled (Eichler, 2001). This is an important limitation as errors in the representation of SDs in reference genomes give rise to false positive heterozygous calls that confound downstream genetic analyses and lead to departure from Hardy-Weinberg equilibrium (Aganezov et al., 2022) (Figure 7).
Figure 7. Genomic artifacts arising from errors in the human reference genome assembly.
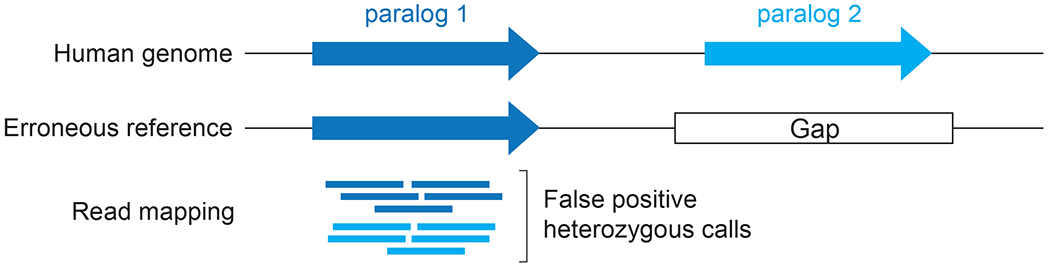
False positive heterozygous SNV calls originating from missing copies in the reference due to identification of paralog-sequence variants due to reads mapping from multiple paralogs.
However, when SDs are represented correctly, they are consistently tricky to assay using SRS technologies, as duplications are so similar that reads are unable to match to either paralog leading to ambiguous read mappings and inhibiting identification of true SNVs. These regions have been termed unmappable, inaccessible, “dark” or “camouflaged” (Ebbert et al., 2019) (Figure 8). HSD genes are particularly challenging as ancestral genes and their human-specific duplicate counterparts share on average ~99% sequence identity, with most also exhibiting varied copies in modern humans (Dennis et al., 2017; Vollger, DeWitt, et al., 2022). As a result, variation across HSDs are ignored in most genetic analyses (Hartasánchez, Brasó-Vives, Heredia-Genestar, Pybus, & Navarro, 2018; Havrilla, Pedersen, Layer, & Quinlan, 2019). To avoid false calls in duplicated regions when using SRS data, variants can be filtered according to accessibility masks, which delineate regions where variants can be confidently identified using base quality, mapping quality, and read-depth cutoffs. SRS accessibility-masks are available for several human reference genome versions, including GRCh38 (Zheng-Bradley et al., 2017) and T2T-CHM13 (Aganezov et al., 2022).
Figure 8. Differences in mappability between short and long reads in duplicated genes.
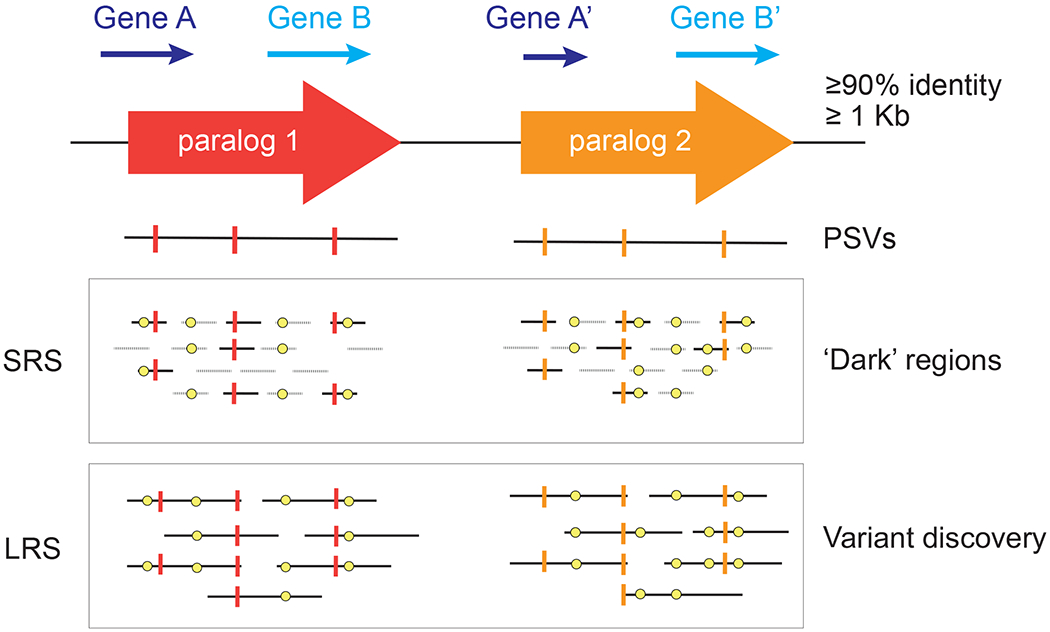
Paralog-specific variants (PSVs) (vertical lines) can distinguish paralogs enabling detection of polymorphic variation (yellow dots). Reads that do not carry PSVs (dashed lines) are unmappable in duplicated regions. SRS: short-read sequencing. LRS: long-read sequencing.
SRS technologies also show biases in their ability to detect different SV types. Deletions are often easier to discover, although not if they are embedded in SDs. Duplications and mCNVs can be detected using read-depth signatures (Alkan, Coe, et al., 2011), but often lack resolution of breakpoints, location of the insertion site of the duplicated sequence, and paralog specificity (Figure 6). Also, non-reference unique insertions larger than the average short-read length often go undetected (Almarri et al., 2020). Inversions are particularly challenging to identify with SRS because most are copy-number neutral, which is not suitable for read-depth approaches, and are enriched around repetitive DNA or flanked by highly-identical SDs (Porubsky, Harvey, et al., 2022; Porubsky, Höps, et al., 2022; Porubsky et al., 2020; Puig et al., 2020), hindering mappability and detection with discordant read-pairs (Chaisson et al., 2019; Lucas Lledó & Cáceres, 2013).
To leverage the hundreds of thousands of available SRS genomes while attempting to overcome limitations in the data, ‘ensemble’ algorithms employing a combination of tools (Ho et al., 2019) have been successfully used to discover SVs (Abel et al., 2020; Almarri et al., 2020; Byrska-Bishop et al., 2022; Collins et al., 2020).
Long-read and -range sequencing overcomes the limitations of short reads
In recent years, LRS technologies have overcome many of the limitations of SRS (Goodwin et al., 2016; Kovaka, Ou, Jenike, & Schatz, 2023; Mantere, Kersten, & Hoischen, 2019; Sedlazeck, Lee, Darby, & Schatz, 2018). PacBio and ONT can produce reads tens to hundreds of kilobases long. The first wave of LRS datasets enabled high-quality de novo assemblies of humans (Jain et al., 2018; Seo et al., 2016; Shafin et al., 2020; Shi et al., 2016; Wenger et al., 2019) and other non-human primates (Gordon et al., 2016; Kronenberg et al., 2018; Mao et al., 2021; Warren et al., 2020). Local assembly of the haploid human cell line CHM1 (Taillon-Miller et al., 1997) using bacterial artificial chromosome clones (CH17) allowed local reconstruction of misassembled regions of the human genome (Antonacci et al., 2014; Chaisson, Huddleston, et al., 2015; Dennis et al., 2017; Huddleston et al., 2014; O’Bleness et al., 2014; Steinberg et al., 2012; Vollger, Dishuck, et al., 2019; Vollger, Logsdon, et al., 2019).
A major achievement of LRS has been the completion of the first human genome sequence (T2T-CHM13), which was achieved by combining PacBio HiFi reads and ultra-long (≥100 kbp) ONT reads (Nurk et al., 2022). The new assembly filled in the missing 8% of the genome corresponding to repeat-rich regions including centromeres, telomeres, and the petite arms of the autosomal acrocentric chromosomes (13, 14, 15, 21, and 22). Additionally, T2T-CHM13 fixed euchromatic gaps and misassemblies, incorporating 51 Mbp of SDs (Vollger et al., 2021) and resolving ~8 Mbp of collapsed SDs compared to the previous reference genome (GRCh38), including previously missing HSD genes GPRIN2B and DUSP22B (Aganezov et al., 2022; Vollger, Dishuck, et al., 2019; Vollger, Guitart, et al., 2022). This complete reference genome significantly improved our ability to discover and interpret human genomic variation, including SNVs (Aganezov et al., 2022) and SVs (Aganezov et al., 2022; Porubsky, Harvey, et al., 2022). In particular, SV identification in 17 individuals sequenced with LRS showed a reduction in homozygous SVs observed in all the human samples assayed, indicating that T2T-CHM13 better represents the major structural allele. Additionally, T2T-CHM13 showed a more balanced ratio between deletions and insertions, fixing a bias towards insertions seen in previous incomplete assemblies (Aganezov et al., 2022). Similarly, T2T-CHM13 proved to increase inversion detection in 41 individuals sequenced with Strand-seq, enabling the discovery of 63 inversion polymorphisms, mostly overlapping novel or structurally-different loci between T2T-CHM13 and the previous version of the human reference assembly GRCh38, as well as correcting 26 misorientations (Porubsky, Harvey, et al., 2022).
LRS technologies have dramatically increased per-sample SV discovery (Figure 6). Employing a combination of long-read and -range sequencing technologies—including PacBio, ONT, Illumina, 10X Genomics linked reads, Bionano Genomics optical mapping, Strand-seq, and Hi-C—enabled identification of 27,622 SVs per human genome, representing a seven-fold increase in SV discovery with respect to SRS ensemble methods (Chaisson et al., 2019); a similar finding was obtained by ONT reads alone (22,636 SVs per human genome) (Beyter et al., 2021). For inversions, particularly larger ones (≥50 kbp) that are often flanked by highly-identical SDs, Strand-seq has shown to be the most sensitive platform because inversion detection does not rely on accurate mapping across repeat-rich regions (Chaisson et al., 2019), but it requires viable, dividing cells (Hanlon, Lansdorp, & Guryev, 2022).
Recently, population-scale LRS cohorts, ranging from dozens (PacBio) to thousands of individuals (ONT), are becoming available (reviewed in De Coster et al. (2021)). Direct mapping of these LRS datasets have identified >100,000 SVs in modern humans (Audano et al., 2019; Beyter et al., 2021; Ebert et al., 2021; Nurk et al., 2022). Importantly, initial discovery of these SVs via LRS has enabled subsequent genotyping across thousands of additional humans using existing SRS datasets to assess their functional and evolutionary impact (Yan et al., 2021).
An alternative approach to gather the full extent of genomic variation in human and non-human primate genomes is using LRS to generate fully-phased genome assemblies and SV detection via assembly-to-reference comparisons. Theoretically, fully-phased complete genomes have the ability to detect SVs of any kind and size (Alkan, Coe, et al., 2011; Mahmoud et al., 2019). The production of complete phased genomes is an area of active research. The current HPRC gold standard relies on PacBio HiFi reads with parental data to enable haplotype phasing (Jarvis et al., 2022); however, PacBio HiFi coupled with Strand-seq long-range information can achieve comparable results (Porubsky, Vollger, et al., 2022). Tools under development integrate both PacBio HiFi reads with ONT “ultralong” reads (>100 kbp) during the assembly process to resolve complex repeats without the need for manual curation (Cheng, Concepcion, Feng, Zhang, & Li, 2021; Rautiainen et al., 2022). These and other improvements will allow the HPRC to fulfill its promise of delivering 350 diploid high-quality fully phased human genomes in the next decade comprising the first human pangenome reference (Wang et al., 2022). Further, similar efforts in non-human primates will allow us to more comprehensively detect putative SV drivers of species’ differences.
Currently, nearly a hundred fully-phased LRS human genomes are available in the public domain, including 88 haplotypes generated by the Human Genome Structural Variation Consortium (HGSVC) (Ebert et al., 2021; Ebler et al., 2022) and 94 haplotypes generated by the HPRC (Jarvis et al., 2022; Liao et al., 2022). These genomes have detected thousands of SVs per haplotype, including 107,590 insertions/deletions (Wang et al., 2022) and 316 inversions identified in 64 haplotypes of unrelated individuals obtained by the HGSVC (Ebert et al., 2021), and >60,000 SVs (~17,000 per haplotype) obtained in HPRC genomes using a pangenome reference approach (Liao et al., 2022). Interestingly, less than 30% of the SVs discovered in phased genome assemblies have also been identified in SRS, highlighting the limitations of SRS in SV identification (Ebert et al., 2021).
Long reads have also shown improved mappability in “dark” regions of the human genome (Ebbert et al., 2019) (Figure 8). However, the original lower fidelity of LRS (~10–15%) hindered its implementation in SNV detection. Variant discovery using ONT reads in a human European individual (NA12878) yielded an overall accuracy of 91.40% (Jain et al., 2018). However, PacBio HiFi reads, averaging a base accuracy of 99.8% and variant-calling precision and recall over 99.4% (Wenger et al., 2019), now enables routine discovery of SNVs and small insertions/deletions (<50 bp) in duplicated regions.
5.4. Future directions for SV research
Despite the clear advantages in using LRS technologies to identify and characterize SVs across humans using both direct read mapping and phased assemblies, historical limitations in lower throughput and base-calling accuracy as well as higher costs versus SRS have limited the production of highly-accurate LRS datasets. Fortunately, biotechnological innovations expected in the coming year promise improvements in accuracy (“At NCM, Announcements Include Single-Read Accuracy of 99.1% on New Chemistry and Sequencing a Record 10 Tb in a Single PromethION Run,” 2020), as well as throughput and cost (“PacBio Announces Revio, a Revolutionary New Long Read Sequencing System Designed to Provide 15 Times More HiFi Data and Human Genomes at Scale for Under $1,000,” 2022); this suggests a near future where hundreds of thousands of LRS human genomes will be available for expanded population and disease studies. Increased ancestry representation and diversity of the samples sequenced will also aid in expanding the repertoire of SVs identified (Popejoy & Fullerton, 2016).
We note that, even with these impending technology improvements, LRS may not be feasible for certain anthropological questions. First, hundreds of thousands of human genomes and hundreds of primate genomes have already been sequenced with SRS (with many more to come) with short-read platforms likely to remain the most affordable whole-genome sequencing approach (Kovaka et al., 2023). Some of these genomes belong to communities that have given restricted consent for the use of their biospecimens and data. Other samples might come from endangered primate species where biospecimens are difficult to obtain. Additionally, anthropological specimens, such as from extinct hominids, are not suitable for LRS library preparations, which require large amounts of high-molecular weight and intact (non-degraded) DNA. As such, methods aimed at integrating SV discovery and genotyping using both SRS and LRS will remain relevant in the study of human and nonhuman primate evolution and demographic history.
Ongoing efforts using short reads for the study of SVs are focused on improvements of the computational algorithms to maximize SV discovery (Abel et al., 2020; Byrska-Bishop et al., 2022; Collins et al., 2020). LRS-based SV catalogs can aid this process by acting as truth-sets to fine tune algorithms. Alternatively, SVs initially discovered with LRS can be later genotyped in SRS cohorts, a strategy that has already efficiently discovered common SVs across human populations (Ebert et al., 2021; Ebler et al., 2022; Yan et al., 2021). Together, the incoming influx of human and non-human primate genomes sequenced with LRS in combination with large-scale SRS datasets are ushering in a new genomics era, promising to unveil the functional and evolutionary impact of complex variation in human traits and diseases.
ACKNOWLEDGEMENTS
This work was supported, in part, by the U.S. National Institutes of Health (NIH) grants from the Office of the Director and National Institute of Mental Health (DP2MH119424 to M.Y.D.) and National Human Genome Research Institute (F31HG011205 to C.J.S.), as well as the U.S. National Science Foundation CAREER Award (2145885 to M.Y.D.).
DATA SHARING
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, … McVean GA (2010). A map of human genome variation from population-scale sequencing. Nature, 467(7319), 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, … Abecasis GR (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel HJ, Larson DE, Regier AA, Chiang C, Das I, Kanchi KL, … Hall IM (2020). Mapping and characterization of structural variation in 17,795 human genomes. Nature, 583(7814), 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aganezov S, Yan SM, Soto DC, Kirsche M, Zarate S, Avdeyev P, … Schatz MC (2022). A complete reference genome improves analysis of human genetic variation. Science, 376(6588), eabl3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre M, Rivas MA, & Priest J (2019). Phenome-wide Burden of Copy-Number Variation in the UK Biobank. American Journal of Human Genetics, 105(2), 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigrain L (2021). Beginner’s guide to next-generation sequencing. The Biochemist, 43(6), 58–64. [Google Scholar]
- Alasoo K, Rodrigues J, Mukhopadhyay S, Knights AJ, Mann AL, Kundu K, … Gaffney. (2018). Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nature Genetics, 50(3), 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Coe BP, & Eichler EE (2011). Genome structural variation discovery and genotyping. Nature Reviews. Genetics, 12(5), 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Sajjadian S, & Eichler EE (2011). Limitations of next-generation genome sequence assembly. Nature Methods, 8(1), 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almarri MA, Bergström A, Prado-Martinez J, Yang F, Fu B, Dunham AS, … Xue Y (2020). Population Structure, Stratification, and Introgression of Human Structural Variation. Cell, 182(1), 189–199.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angata T, Ishii T, Motegi T, Oka R, Taylor RE, Soto PC, … Taniguchi N (2013). Loss of Siglec-14 reduces the risk of chronic obstructive pulmonary disease exacerbation. Cellular and Molecular Life Sciences: CMLS, 70(17), 3199–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonacci F, Dennis MY, Huddleston J, Sudmant PH, Steinberg KM, Rosenfeld JA, … Eichler EE (2014). Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nature Genetics, 46(12), 1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqil A, Speidel L, Pavlidis P, & Gokcumen O (2022). Balancing selection on genomic deletion polymorphisms in humans (p. 2022.04.28.489864). 10.1101/2022.04.28.489864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnone MI, & Davidson EH (1997). The hardwiring of development: organization and function of genomic regulatory systems. Development , 124(10), 1851–1864. [DOI] [PubMed] [Google Scholar]
- At NCM, announcements include single-read accuracy of 99.1% on new chemistry and sequencing a record 10 Tb in a single PromethION run. (2020, December 3). Retrieved January 16, 2023, from Oxford Nanopore Technologies website: https://nanoporetech.com/about-us/news/ncm-announcements-include-single-read-accuracy-991-new-chemistry-and-sequencing
- Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AE, … Eichler EE (2019). Characterizing the Major Structural Variant Alleles of the Human Genome. Cell, 0(0). 10.1016/j.cell.2018.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala R, Shu T, & Tsai L-H (2007). Trekking across the brain: the journey of neuronal migration. Cell, 128(1), 29–43. [DOI] [PubMed] [Google Scholar]
- Bailey JA, & Eichler EE (2006). Primate segmental duplications: crucibles of evolution, diversity and disease. Nature Reviews. Genetics, 7, 552. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, … Eichler EE (2002). Recent segmental duplications in the human genome. Science, 297(5583), 1003–1007. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Liu G, & Eichler EE (2003). An Alu transposition model for the origin and expansion of human segmental duplications. American Journal of Human Genetics, 73(4), 823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, & Eichler EE (2001). Segmental duplications: organization and impact within the current human genome project assembly. Genome Research, 11(6), 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran P, Walawalkar IA, Flores JI, Dayton JN, Audano PA, & Beck CR (2022). Transposable element-mediated rearrangements are prevalent in human genomes. Nature Communications, 13(1), 7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekpen C, & Tautz D (2019). Human core duplicon gene families: game changers or game players? Briefings in Functional Genomics, 18(6), 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton J-M, McCord RP, Gibcus JH, Naumova N, Zhan Y, & Dekker J (2012). Hi–C: A comprehensive technique to capture the conformation of genomes. Methods , 58(3), 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergström A, McCarthy SA, Hui R, Almarri MA, Ayub Q, Danecek P, … Tyler-Smith C (2020). Insights into human genetic variation and population history from 929 diverse genomes. Science, 367(6484). 10.1126/science.aay5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besenbacher S, Hvilsom C, Marques-Bonet T, Mailund T, & Schierup MH (2019). Direct estimation of mutations in great apes reconciles phylogenetic dating. Nature Ecology & Evolution, 3(2), 286–292. [DOI] [PubMed] [Google Scholar]
- Beyter D, Ingimundardottir H, Oddsson A, Eggertsson HP, Bjornsson E, Jonsson H, … Stefansson K (2021). Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nature Genetics, 53(6), 779–786. [DOI] [PubMed] [Google Scholar]
- Boettger LM, Handsaker RE, Zody MC, & McCarroll SA (2012). Structural haplotypes and recent evolution of the human 17q21.31 region. Nature Genetics, 44(8), 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JL, Skove SL, Rouanet JP, Pilaz L-J, Bepler T, Gordân R, … Silver DL (2015). Human-Chimpanzee Differences in a FZD8 Enhancer Alter Cell-Cycle Dynamics in the Developing Neocortex. Current Biology: CB, 25(6), 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N (2019). Inroads into cortical organoids. Nature Reviews. Neuroscience, 20(12), 717–717. [DOI] [PubMed] [Google Scholar]
- Bridges CB (1936). The bar “gene” a duplication. Science, 83(2148), 210–211. [DOI] [PubMed] [Google Scholar]
- Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, … Zody MC (2022). High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell, 185(18), 3426–3440.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CD, Sampas N, Tsalenko A, Sudmant PH, Kidd JM, Malig M, … Eichler EE (2011). Population-genetic properties of differentiated human copy-number polymorphisms. American Journal of Human Genetics, 88(3), 317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantsilieris S, Sunkin SM, Johnson ME, Anaclerio F, Huddleston J, Baker C, … Eichler EE (2020). An evolutionary driver of interspersed segmental duplications in primates. Genome Biology, 21(1), 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Hastie AR, Cao D, Lam ET, Sun Y, Huang H, … Xu X (2014). Rapid detection of structural variation in a human genome using nanochannel-based genome mapping technology. GigaScience, Vol. 3. 10.1186/2047-217x-3-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter D, Mitchell LM, & Armour JAL (2017). Copy number variation of human AMY1 is a minor contributor to variation in salivary amylase expression and activity. Human Genomics, 11(1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB (2003). Genetics and the making of Homo sapiens. Nature, 422(6934), 849–857. [DOI] [PubMed] [Google Scholar]
- Carvalho CMB, & Lupski JR (2016). Mechanisms underlying structural variant formation in genomic disorders. Nature Reviews. Genetics, 17(4), 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catacchio CR, Maggiolini FAM, D’Addabbo P, Bitonto M, Capozzi O, Lepore Signorile M, … Antonacci F (2018). Inversion variants in human and primate genomes. Genome Research, 28(6), 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, … Eichler EE (2015). Resolving the complexity of the human genome using single-molecule sequencing. Nature, 517(7536), 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, … Lee C (2019). Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nature Communications, 10(1), 1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Wilson RK, & Eichler EE (2015). Genetic variation and the de novo assembly of human genomes. Nature Reviews. Genetics, 16(11), 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrier C, Joshi K, Coutinho-Budd J, Kim J-E, Lambert N, de Marchena J, … Polleux F (2012). Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell, 149(4), 923–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Concepcion GT, Feng X, Zhang H, & Li H (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods, 18(2), 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Cooper DN, Chuzhanova N, Férec C, & Patrinos GP (2007). Gene conversion: mechanisms, evolution and human disease. Nature Reviews. Genetics, 8(10), 762–775. [DOI] [PubMed] [Google Scholar]
- Chiang C (2019). The Impact of Structural Variation on Human Gene Expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Scott AJ, Davis JR, Tsang EK, Li X, Kim Y, … Hall IM (2017). The impact of structural variation on human gene expression. Nature Genetics, 49(5), 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium. (2005). Initial sequence of the chimpanzee genome and comparison with the human genome. Nature, 437(7055), 69–87. [DOI] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, … Eichler EE (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nature Genetics, 46(10), 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, … Talkowski ME (2020). A structural variation reference for medical and population genetics. Nature, 581(7809), 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, … Hurles ME (2010). Origins and functional impact of copy number variation in the human genome. Nature, 464(7289), 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DE, Lee TG, Guo X, Melito S, Wang K, Bayless AM, … Bent AF (2012). Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science, 338(6111), 1206–1209. [DOI] [PubMed] [Google Scholar]
- Croft B, Ohnesorg T, Hewitt J, Bowles J, Quinn A, Tan J, … Sinclair A (2018). Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nature Communications, 9(1), 5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damert A (2022). SVA Retrotransposons and a Low Copy Repeat in Humans and Great Apes: A Mobile Connection. Molecular Biology and Evolution, 39(5). 10.1093/molbev/msac103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Austin MD, Akana MC, Deshpande P, Cao H, & Xiao M (2010). Single molecule linear analysis of DNA in nano-channel labeled with sequence specific fluorescent probes. Nucleic Acids Research, 38(18), e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster W, Weissensteiner MH, & Sedlazeck FJ (2021). Towards population-scale long-read sequencing. Nature Reviews. Genetics, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defelipe J (2011). The evolution of the brain, the human nature of cortical circuits, and intellectual creativity. Frontiers in Neuroanatomy, 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong S, Chepelev I, Janson E, Strengman E, van den Berg LH, Veldink JH, & Ophoff RA (2012). Common inversion polymorphism at 17q21.31 affects expression of multiple genes in tissue-specific manner. BMC Genomics, 13, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MY, & Eichler EE (2016). Human adaptation and evolution by segmental duplication. Current Opinion in Genetics & Development, 41, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MY, Harshman L, Nelson BJ, Penn O, Cantsilieris S, Huddleston J, … Eichler EE (2017). The evolution and population diversity of human-specific segmental duplications. Nature Ecology & Evolution, 1(3), 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MY, Nuttle X, Sudmant PH, Antonacci F, Graves TA, Nefedov M, … Eichler EE (2012). Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell, 149(4), 912–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSilva JM, Traniello JFA, Claxton AG, & Fannin LD (2021). When and Why Did Human Brains Decrease in Size? A New Change-Point Analysis and Insights From Brain Evolution in Ants. Frontiers in Ecology and Evolution, 0. 10.3389/fevo.2021.742639 [DOI] [Google Scholar]
- Dougherty ML, Underwood JG, Nelson BJ, Tseng E, Munson KM, Penn O, … Eichler EE (2018). Transcriptional fates of human-specific segmental duplications in brain. Genome Research, 28(10), 1566–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont BL (2015). Interlocus gene conversion explains at least 2.7% of single nucleotide variants in human segmental duplications. BMC Genomics, 16, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbert MTW, Jensen TD, Jansen-West K, Sens JP, Reddy JS, Ridge PG, … Fryer JD (2019). Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biology, 20(1), 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert P, Audano PA, Zhu Q, Rodriguez-Martin B, Porubsky D, Bonder MJ, … Eichler EE (2021). Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science, 372(6537). 10.1126/science.abf7117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebler J, Ebert P, Clarke WE, Rausch T, Audano PA, Houwaart T, … Marschall T (2022). Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes. Nature Genetics, 54(4), 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE (2001). Segmental duplications: what’s missing, misassigned, and misassembled--and should we care? Genome Research, 11(5), 653–656. [DOI] [PubMed] [Google Scholar]
- Enard W, Gehre S, Hammerschmidt K, Hölter SM, Blass T, Somel M, … Pääbo S (2009). A humanized version of Foxp2 affects cortico-basal ganglia circuits in mice. Cell, 137(5), 961–971. [DOI] [PubMed] [Google Scholar]
- Falconer E, Hills M, Naumann U, Poon SSS, Chavez EA, Sanders AD, … Lansdorp PM (2012). DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nature Methods, 9(11), 1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, & Wittkopp PJ (2008). Evaluating the role of natural selection in the evolution of gene regulation. Heredity, 100(2), 191–199. [DOI] [PubMed] [Google Scholar]
- Fernandes S, Klein D, & Marchetto MC (2021). Unraveling Human Brain Development and Evolution Using Organoid Models. Frontiers in Cell and Developmental Biology, 9. 10.3389/fcell.2021.737429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiddes IT, Lodewijk GA, Mooring M, Bosworth CM, Ewing AD, Mantalas GL, … Haussler D (2018). Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell, 173(6), 1356–1369.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J, Fernández Ortuño E, Marsoner F, Artioli A, Peters J, Namba T, … Heide M (2022). Human-specific ARHGAP11B ensures human-like basal progenitor levels in hominid cerebral organoids. EMBO Reports, 23(11), e54728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Albert M, Taverna E, Namba T, Brandl H, Lewitus E, … Huttner WB (2015). Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science, 347(6229), 1465–1470. [DOI] [PubMed] [Google Scholar]
- Florio M, Heide M, Pinson A, Brandl H, Albert M, Winkler S, … Hiller M (2018). Evolution and cell-type specificity of human-specific genes preferentially expressed in progenitors of fetal neocortex. eLife, 7. 10.7554/eLife.32332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Namba T, Pääbo S, Hiller M, & Huttner WB (2016). A single splice site mutation in human-specific ARHGAP11B causes basal progenitor amplification. Science Advances, 2(12), e1601941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortna A, Kim Y, MacLaren E, Marshall K, Hahn G, Meltesen L, … Sikela JM (2004). Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biology, 2(7), E207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossati M, Pizzarelli R, Schmidt ER, Kupferman JV, Stroebel D, Polleux F, & Charrier C (2016). SRGAP2 and Its Human-Specific Paralog Co-Regulate the Development of Excitatory and Inhibitory Synapses. Neuron, 91(2), 356–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke M, Daly AF, Palmeira L, Tirosh A, Stigliano A, Trifan E, … Trivellin G (2022). Duplications disrupt chromatin architecture and rewire GPR101-enhancer communication in X-linked acrogigantism. American Journal of Human Genetics, 109(4), 553–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB (2013). Gene expression drives local adaptation in humans. Genome Research, 23(7), 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, & Pollard KS (2019). Chromatin features constrain structural variation across evolutionary timescales. Proceedings of the National Academy of Sciences of the United States of America, 116(6), 2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller ZL, Koury SA, Phadnis N, & Schaeffer SW (2019). How chromosomal rearrangements shape adaptation and speciation: Case studies in Drosophila pseudoobscura and its sibling species Drosophila persimilis. Molecular Ecology, 28(6), 1283–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux P, & Varki A (2001). Genetic differences between humans and great apes. Molecular Phylogenetics and Evolution, 18(1), 2–13. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, & Rakic P (2013). Cortical evolution: judge the brain by its cover. Neuron, 80(3), 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannuzzi G, Schmidt PJ, Porcu E, Willemin G, Munson KM, Nuttle X, … Reymond A (2019). The Human-Specific BOLA2 Duplication Modifies Iron Homeostasis and Anemia Predisposition in Chromosome 16p11.2 Autism Individuals. American Journal of Human Genetics, 105(5), 947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giner-Delgado C, Villatoro S, Lerga-Jaso J, Gayà-Vidal M, Oliva M, Castellano D, … Cáceres M (2019). Evolutionary and functional impact of common polymorphic inversions in the human genome. Nature Communications, 10(1), 4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Robles A (2019). Dental evolutionary rates and its implications for the Neanderthal–modern human divergence. Science Advances, 5(5), eaaw1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin S, McPherson JD, & McCombie WR (2016). Coming of age: ten years of next-generation sequencing technologies. Nature Reviews. Genetics, 17(6), 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Huddleston J, Chaisson MJP, Hill CM, Kronenberg ZN, Munson KM, … Eichler EE (2016). Long-read sequence assembly of the gorilla genome. Science, 352(6281), aae0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, Dunham MJ, & Botstein D (2008). Comparing whole genomes using DNA microarrays. Nature Reviews. Genetics, 9(4), 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross M, Starke H, Trifonov V, Claussen U, Liehr T, & Weise A (2006). A molecular cytogenetic study of chromosome evolution in chimpanzee. Cytogenetic and Genome Research, 112(1–2), 67–75. [DOI] [PubMed] [Google Scholar]
- Guerrier S, Coutinho-Budd J, Sassa T, Gresset A, Jordan NV, Chen K, … Polleux F (2009). The F-BAR domain of srGAP2 induces membrane protrusions required for neuronal migration and morphogenesis. Cell, 138(5), 990–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldorsson BV, Eggertsson HP, Moore KHS, Hauswedell H, Eiriksson O, Ulfarsson MO, … Stefansson K (2022). The sequences of 150,119 genomes in the UK Biobank. Nature, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, & McCarroll SA (2015). Large multiallelic copy number variations in humans. Nature Genetics, 47(3), 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon VCT, Lansdorp PM, & Guryev V (2022). A survey of current methods to detect and genotype inversions. Human Mutation, 43(11), 1576–1589. [DOI] [PubMed] [Google Scholar]
- Hardwick RJ, Ménard A, Sironi M, Milet J, Garcia A, Sese C, … Hollox EJ (2014). Haptoglobin (HP) and Haptoglobin-related protein (HPR) copy number variation, natural selection, and trypanosomiasis. Human Genetics, 133(1), 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harewood L, Kishore K, Eldridge MD, Wingett S, Pearson D, Schoenfelder S, … Fraser P (2017). Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biology, 18(1), 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartasánchez DA, Brasó-Vives M, Heredia-Genestar JM, Pybus M, & Navarro A (2018). Effect of Collapsed Duplications on Diversity Estimates: What to Expect. Genome Biology and Evolution, 10(11), 2899–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havrilla JM, Pedersen BS, Layer RM, & Quinlan AR (2019). A map of constrained coding regions in the human genome. Nature Genetics, 51(1), 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehir-Kwa JY, Marschall T, Kloosterman WP, Francioli LC, Baaijens JA, Dijkstra LJ, … Guryev V (2016). A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nature Communications, 7, 12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heide M, Haffner C, Murayama A, Kurotaki Y, Shinohara H, Okano H, … Huttner WB (2020). Human-specific increases size and folding of primate neocortex in the fetal marmoset. Science, 369(6503), 546–550. [DOI] [PubMed] [Google Scholar]
- Herculano-Houzel S (2016). The Human Advantage: A New Understanding of How Our Brain Became Remarkable. MIT Press. [Google Scholar]
- Hinds DA, Kloek AP, Jen M, Chen X, & Frazer KA (2006). Common deletions and SNPs are in linkage disequilibrium in the human genome. Nature Genetics, 38(1), 82–85. [DOI] [PubMed] [Google Scholar]
- Hodzic D, Kong C, Wainszelbaum MJ, Charron AJ, Su X, & Stahl PD (2006). TBC1D3, a hominoid oncoprotein, is encoded by a cluster of paralogues located on chromosome 17q12. Genomics, 88(6), 731–736. [DOI] [PubMed] [Google Scholar]
- Hollox EJ, Armour JAL, & Barber JCK (2003). Extensive normal copy number variation of a beta-defensin antimicrobial-gene cluster. American Journal of Human Genetics, 73(3), 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollox EJ, Zuccherato LW, & Tucci S (2022). Genome structural variation in human evolution. Trends in Genetics, Vol. 38, pp. 45–58. 10.1016/j.tig.2021.06.015 [DOI] [PubMed] [Google Scholar]
- Ho SS, Urban AE, & Mills RE (2019). Structural variation in the sequencing era. Nature Reviews. Genetics. 10.1038/s41576-019-0180-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh P, Dang V, Vollger MR, Mao Y, Huang T-H, Dishuck PC, … Eichler EE (2021). Evidence for opposing selective forces operating on human-specific duplicated TCAF genes in Neanderthals and humans. Nature Communications, 12(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh P, Vollger MR, Dang V, Porubsky D, Baker C, Cantsilieris S, … Eichler EE (2019). Adaptive archaic introgression of copy number variants and the discovery of previously unknown human genes. Science, 366(6463). 10.1126/science.aax2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, & Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 4(1), 44–57. [DOI] [PubMed] [Google Scholar]
- Huddleston J, Ranade S, Malig M, Antonacci F, Chaisson M, Hon L, … Eichler EE (2014). Reconstructing complex regions of genomes using long-read sequencing technology. Genome Research, 24(4), 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Yamaguchi M, Mori K, & Kageyama R (2010). Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 30(9), 3489–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indjeian VB, Kingman GA, Jones FC, Guenther CA, Grimwood J, Schmutz J, … Kingsley DM (2016). Evolving New Skeletal Traits by cis-Regulatory Changes in Bone Morphogenetic Proteins. Cell, 164(1–2), 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, & Lupski JR (2002). Molecular mechanisms for genomic disorders. Annual Review of Genomics and Human Genetics, 3, 199–242. [DOI] [PubMed] [Google Scholar]
- Irvin DK, Zurcher SD, Nguyen T, Weinmaster G, & Kornblum HI (2001). Expression patterns of Notch1, Notch2, and Notch3 suggest multiple functional roles for the Notch-DSL signaling system during brain development. The Journal of Comparative Neurology, 436(2), 167–181. [PubMed] [Google Scholar]
- Iskow RC, Gokcumen O, Abyzov A, Malukiewicz J, Zhu Q, Sukumar AT, … Lee C (2012). Regulatory element copy number differences shape primate expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 109(31), 12656–12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, … Loose M (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology, 36(4), 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, Fung H-C, … Singleton AB (2008). Genotype, haplotype and copy-number variation in worldwide human populations. Nature, 451(7181), 998–1003. [DOI] [PubMed] [Google Scholar]
- Jakubosky D, Smith EN, D’Antonio M, Jan Bonder M, Young Greenwald WW, D’Antonio-Chronowska A, … i2QTL Consortium. (2020). Discovery and quality analysis of a comprehensive set of structural variants and short tandem repeats. Nature Communications, 11(1), 2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis ED, Formenti G, Rhie A, Guarracino A, Yang C, Wood J, … Human Pangenome Reference Consortium. (2022). Automated assembly of high-quality diploid human reference genomes (p. 2022.03.06.483034). 10.1101/2022.03.06.483034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffet J, Margalit S, Michaeli Y, & Ebenstein Y (2021). Single-molecule optical genome mapping in nanochannels: multidisciplinarity at the nanoscale. Essays in Biochemistry, 65(1), 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, … Eichler EE (2007). Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nature Genetics, 39(11), 1361–1368. [DOI] [PubMed] [Google Scholar]
- Johnson ME, National Institute of Health Intramural Sequencing Center Comparative Sequencing Program, Cheng Z, Morrison VA, Scherer S, Ventura M, … Eichler EE (2006). Recurrent duplication-driven transposition of DNA during hominoid evolution. Proceedings of the National Academy of Sciences of the United States of America, 103(47), 17626–17631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ME, Viggiano L, Bailey JA, Abdul-Rauf M, Goodwin G, Rocchi M, & Eichler EE (2001). Positive selection of a gene family during the emergence of humans and African apes. Nature, 413(6855), 514–519. [DOI] [PubMed] [Google Scholar]
- Ju X-C, Hou Q-Q, Sheng A-L, Wu K-Y, Zhou Y, Jin Y, … Luo Z-G (2016). The hominoid-specific gene TBC1D3 promotes generation of basal neural progenitors and induces cortical folding in mice. eLife, 5. 10.7554/eLife.18197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalebic N, Gilardi C, Albert M, Namba T, Long KR, Kostic M, … Huttner WB (2018). Human-specific induces hallmarks of neocortical expansion in developing ferret neocortex. eLife, 7. 10.7554/eLife.41241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH, & Moran JV (2017). Mobile DNA in Health and Disease. The New England Journal of Medicine, 377(4). 10.1056/NEJMra1510092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Newman TL, Tuzun E, Kaul R, & Eichler EE (2007). Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genetics, 3(4), e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PM, Lam HYK, Urban AE, Korbel JO, Affourtit J, Grubert F, … Gerstein MB (2008). Analysis of copy number variants and segmental duplications in the human genome: Evidence for a change in the process of formation in recent evolutionary history. Genome Research, 18(12), 1865–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, & Wilson AC (1975). Evolution at two levels in humans and chimpanzees. Science, 188(4184), 107–116. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick M, & Barton N (2006). Chromosome inversions, local adaptation and speciation. Genetics, 173(1), 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan D-J, & Coutinho P (2009). Cis-ruption mechanisms: disruption of cis-regulatory control as a cause of human genetic disease. Briefings in Functional Genomics & Proteomics, 8(4), 317–332. [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Francioli LC, Hormozdiari F, Marschall T, Hehir-Kwa JY, Abdellaoui A, … Guryev V (2015). Characteristics of de novo structural changes in the human genome. Genome Research, 25(6), 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen DA, Vissers LEL, Pfundt R, de Leeuw N, Knight SJL, Regan R, … de Vries BBA (2006). A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nature Genetics, Vol. 38, pp. 999–1001. 10.1038/ng1853 [DOI] [PubMed] [Google Scholar]
- Kovaka S, Ou S, Jenike KM, & Schatz MC (2023). Approaching complete genomes, transcriptomes and epi-omes with accurate long-read sequencing. Nature Methods, 20(1), 12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg ZN, Fiddes IT, Gordon D, Murali S, Cantsilieris S, Meyerson OS, … Eichler EE (2018). High-resolution comparative analysis of great ape genomes. Science, 360(6393). 10.1126/science.aar6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakich D, Kazazian HH Jr, Antonarakis SE, & Gitschier J (1993). Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nature Genetics, 5(3), 236–241. [DOI] [PubMed] [Google Scholar]
- Lallemand T, Leduc M, Landès C, Rizzon C, & Lerat E (2020). An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice. Genes, 11(9). 10.3390/genes11091046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, … Kwok P-Y (2012). Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nature Biotechnology, 30(8), 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, … International Human Genome Sequencing Consortium. (2001). Initial sequencing and analysis of the human genome. Nature, 409(6822), 860–921. [DOI] [PubMed] [Google Scholar]
- Larson JL, Silver AJ, Chan D, Borroto C, Spurrier B, & Silver LM (2015). Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project. BMC Medical Genetics, 16, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler EM, Band G, Busby GBJ, Kivinen K, Le QS, Clarke GM, … Malaria Genomic Epidemiology Network. (2017). Resistance to malaria through structural variation of red blood cell invasion receptors. Science, 356(6343). 10.1126/science.aam6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W-W, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, … Paten B (2022). A Draft Human Pangenome Reference (p. 2022.07.09.499321). 10.1101/2022.07.09.499321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-L, & Gokcumen O (2019). Fine-Scale Characterization of Genomic Structural Variation in the Human Genome Reveals Adaptive and Biomedically Relevant Hotspots. Genome Biology and Evolution, 11(4), 1136–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-L, Pavlidis P, Karakoc E, Ajay J, & Gokcumen O (2015). The evolution and functional impact of human deletion variants shared with archaic hominin genomes. Molecular Biology and Evolution, 32(4), 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GE, Ventura M, Cellamare A, Chen L, Cheng Z, Zhu B, … Eichler EE (2009). Analysis of recent segmental duplications in the bovine genome. BMC Genomics, 10, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JH, Hansen DV, & Kriegstein AR (2011). Development and Evolution of the Human Neocortex. Cell, 146(1), 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Xiong X, Zhao X, Yang X, & Wang H (2015). F-BAR family proteins, emerging regulators for cell membrane dynamic changes-from structure to human diseases. Journal of Hematology & Oncology, 8, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke DP, Archidiacono N, Misceo D, Cardone MF, Deschamps S, Roe B, … Eichler EE (2003). Refinement of a chimpanzee pericentric inversion breakpoint to a segmental duplication cluster. Genome Biology, 4(8), R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke DP, Hillier LW, Warren WC, Worley KC, Nazareth LV, Muzny DM, … Wilson RK (2011). Comparative and demographic analysis of orang-utan genomes. Nature, 469(7331), 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke DP, Sharp AJ, McCarroll SA, McGrath SD, Newman TL, Cheng Z, … Eichler EE (2006). Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. American Journal of Human Genetics, 79(2), 275–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente-Galdos B, Bleyhl J, Santpere G, Vives L, Ramírez O, Hernandez J, … Marques-Bonet T (2013). Accelerated exon evolution within primate segmental duplications. Genome Biology, 14(1), R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas Lledó JI, & Cáceres M (2013). On the power and the systematic biases of the detection of chromosomal inversions by paired-end genome sequencing. PloS One, 8(4), e61292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L (2002). Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annual Review of Cell and Developmental Biology, 18, 601–635. [DOI] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, … Mundlos S (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell, 161(5), 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, & Conery JS (2000). [Review of The evolutionary fate and consequences of duplicate genes]. Science, 290(5494), 1151–1155. [DOI] [PubMed] [Google Scholar]
- Maggiolini FAM, Sanders AD, Shew CJ, Sulovari A, Mao Y, Puig M, … Antonacci F (2020). Single-cell strand sequencing of a macaque genome reveals multiple nested inversions and breakpoint reuse during primate evolution. Genome Research, 30(11), 1680–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud M, Gobet N, Cruz-Dávalos DI, Mounier N, Dessimoz C, & Sedlazeck FJ (2019). Structural variant calling: the long and the short of it. Genome Biology, 20(1), 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantere T, Kersten S, & Hoischen A (2019). Long-Read Sequencing Emerging in Medical Genetics. Frontiers in Genetics, 10, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Catacchio CR, Hillier LW, Porubsky D, Li R, Sulovari A, … Eichler EE (2021). A high-quality bonobo genome refines the analysis of hominid evolution. Nature. 10.1038/s41586-021-03519-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, & Eichler EE (2009). The evolution of human segmental duplications and the core duplicon hypothesis. Cold Spring Harbor Symposia on Quantitative Biology, 74, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Girirajan S, & Eichler EE (2009). The origins and impact of primate segmental duplications. Trends in Genetics: TIG, 25(10), 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Kidd JM, Ventura M, Graves TA, Cheng Z, Hillier LW, … Eichler EE (2009a). A burst of segmental duplications in the genome of the African great ape ancestor. Nature, 457(7231), 877–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Kidd JM, Ventura M, Graves TA, Cheng Z, Hillier LW, … Eichler EE (2009b). A burst of segmental duplications in the genome of the African great ape ancestor. Nature, 457(7231), 877–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, … International HapMap Consortium. (2006). Common deletion polymorphisms in the human genome. Nature Genetics, 38(1), 86–92. [DOI] [PubMed] [Google Scholar]
- McClintock B (1931). Cytological observations of deficiencies involving known genes, translocations and an inversion in Zea mays. Retrieved July 24, 2022, from https://mospace.umsystem.edu/xmlui/bitstream/handle/10355/52974/age000163.pdf?sequence=1
- McKayed KK, & Simpson JC (2013). Actin in action: imaging approaches to study cytoskeleton structure and function. Cells , 2(4), 715–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, … Kingsley DM (2011). Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature, 471(7337), 216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, & Eichler EE (2009). Duplication hotspots, rare genomic disorders, and common disease. Current Opinion in Genetics & Development, 19(3). 10.1016/j.gde.2009.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, … 1000 Genomes Project. (2011). Mapping copy number variation by population-scale genome sequencing. Nature, 470(7332), 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, Clowry GJ, Šestan N, Alzu’bi A, Bakken T, Hevner RF, … Kriegstein A (2019a). New insights into the development of the human cerebral cortex. Journal of Anatomy, 235(3), 432–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, Clowry GJ, Šestan N, Alzu’bi A, Bakken T, Hevner RF, … Kriegstein A (2019b). New insights into the development of the human cerebral cortex. Journal of Anatomy, 235(3), 432–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, & Pollen A (2014). How unique is the human neocortex? Development , 141(1), 11–16. [DOI] [PubMed] [Google Scholar]
- Mora-Bermúdez F, Badsha F, Kanton S, Camp JG, Vernot B, Köhler K, … Huttner WB (2016). Differences and similarities between human and chimpanzee neural progenitors during cerebral cortex development. eLife, 5. 10.7554/eLife.18683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Igoa M, Hernández-Charro B, Bengoa-Alonso A, Pérez-Juana-del-Casal A, Romero-Ibarra C, Nieva-Echebarria B, & Ramos-Arroyo MA (2015). KANSL1 gene disruption associated with the full clinical spectrum of 17q21.31 microdeletion syndrome. BMC Medical Genetics, 16, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba T, Dóczi J, Pinson A, Xing L, Kalebic N, Wilsch-Bräuninger M, … Huttner WB (2020). Human-Specific ARHGAP11B Acts in Mitochondria to Expand Neocortical Progenitors by Glutaminolysis. Neuron, 105(5), 867–881.e9. [DOI] [PubMed] [Google Scholar]
- Nédélec Y, Sanz J, Baharian G, Szpiech ZA, Pacis A, Dumaine A, … Barreiro LB (2016). Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell, 167(3), 657–669.e21. [DOI] [PubMed] [Google Scholar]
- Nicholas TJ, Cheng Z, Ventura M, Mealey K, Eichler EE, & Akey JM (2009). The genomic architecture of segmental duplications and associated copy number variants in dogs. Genome Research, 19(3), 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson E, & Nelson DL (1998). Molecular definition of pericentric inversion breakpoints occurring during the evolution of humans and chimpanzees. Genomics, 50(3), 368–372. [DOI] [PubMed] [Google Scholar]
- Noor MA, Grams KL, Bertucci LA, & Reiland J (2001). Chromosomal inversions and the reproductive isolation of species. Proceedings of the National Academy of Sciences of the United States of America, 98(21), 12084–12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, … Pfister SM (2014). Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature, 511(7510), 428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, … Phillippy AM (2022). The complete sequence of a human genome. Science, 376(6588), 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Bleness M, Searles VB, Dickens CM, Astling D, Albracht D, Mak AC, … Sikela JM (2014). Finished sequence and assembly of the DUF1220-rich 1q21 region using a haploid human genome. BMC Genomics, 15, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Bleness M, Searles VB, Varki A, Gagneux P, & Sikela JM (2012). Evolution of genetic and genomic features unique to the human lineage. Nature Reviews. Genetics, 13(12), 853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S (1970). Evolution by Gene Duplication. [Google Scholar]
- Ohno S (1972). An argument for the genetic simplicity of man and other mammals. Journal of Human Evolution, 1(6), 651–662. [Google Scholar]
- Olson MV (1999). When less is more: gene loss as an engine of evolutionary change. American Journal of Human Genetics, 64(1), 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, … Scherer SW (2001). A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nature Genetics, 29(3), 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pääbo S (2014). The Human Condition—A Molecular Approach. Cell, Vol. 157, pp. 216–226. 10.1016/j.cell.2013.12.036 [DOI] [PubMed] [Google Scholar]
- PacBio Announces Revio, a Revolutionary New Long Read Sequencing System Designed to Provide 15 Times More HiFi Data and Human Genomes at Scale for Under $1,000. (2022, October 26). Retrieved January 15, 2023, from PacBio website: https://www.pacb.com/press_releases/pacbio-announces-revio-a-revolutionary-new-long-read-sequencing-system-designed-to-provide-15-times-more-hifi-data-and-human-genomes-at-scale-for-under-1000/
- Pajic P, Lin Y-L, Xu D, & Gokcumen O (2016). The psoriasis-associated deletion of late cornified envelope genes LCE3B and LCE3C has been maintained under balancing selection since Human Denisovan divergence. BMC Evolutionary Biology, 16(1), 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajic P, Pavlidis P, Dean K, Neznanova L, Romano R-A, Garneau D, … Gokcumen O (2019). Independent amylase gene copy number bursts correlate with dietary preferences in mammals. eLife, 8. 10.7554/eLife.44628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, & Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods, 14(4), 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, … Stone AC (2007). Diet and the evolution of human amylase gene copy number variation. Nature Genetics, 39(10), 1256–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Tchinda J, McGrath SD, Zhang J, Picker SR, Cáceres AM, … Lee C (2006). Hotspots for copy number variation in chimpanzees and humans. Proceedings of the National Academy of Sciences of the United States of America, 103(21), 8006–8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Yang F, Marques-Bonet T, Murphy C, Fitzgerald T, Lee AS, … Redon R (2008). Copy number variation and evolution in humans and chimpanzees. Genome Research, 18(11), 1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatigorsky J (2003). Crystallin genes: specialization by changes in gene regulation may precede gene duplication. Journal of Structural and Functional Genomics, 3(1–4), 131–137. [PubMed] [Google Scholar]
- Pollen AA, Bhaduri A, Andrews MG, Nowakowski TJ, Meyerson OS, Mostajo-Radji MA, … Kriegstein AR (2019). Establishing Cerebral Organoids as Models of Human-Specific Brain Evolution. Cell, 176(4), 743–756.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popejoy AB, & Fullerton SM (2016). [Review of Genomics is failing on diversity]. Nature, 538(7624), 161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Harvey WT, Rozanski AN, Ebler J, Höps W, Ashraf H, … Eichler EE (2022). Inversion polymorphism in a complete human genome assembly (p. 2022.10.06.511148). 10.1101/2022.10.06.511148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Höps W, Ashraf H, Hsieh P, Rodriguez-Martin B, Yilmaz F, … Korbel JO (2022). Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell, 185(11), 1986–2005.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Sanders AD, Höps W, Hsieh P, Sulovari A, Li R, … Eichler EE (2020). Recurrent inversion toggling and great ape genome evolution. Nature Genetics. 10.1038/s41588-020-0646-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Vollger MR, Harvey WT, Rozanski AN, Ebert P, Hickey G, … Eichler EE (2022). Gaps and complex structurally variant loci in phased genome assemblies (p. 2022.07.06.498874). 10.1101/2022.07.06.498874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, … Noonan JP (2008). Human-specific gain of function in a developmental enhancer. Science, 321(5894), 1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, … Marques-Bonet T (2013). Great ape genetic diversity and population history. Nature, 499(7459), 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, Munch K, Hellmann I, Akagi K, Miller JR, Walenz B, … Pääbo S (2012). The bonobo genome compared with the chimpanzee and human genomes. Nature, 486(7404), 527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig M, Casillas S, Villatoro S, & Cáceres M (2015). Human inversions and their functional consequences. Briefings in Functional Genomics, 14(5), 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig M, Lerga-Jaso J, Giner-Delgado C, Pacheco S, Izquierdo D, Delprat A, … Cáceres M (2020). Determining the impact of uncharacterized inversions in the human genome by droplet digital PCR. Genome Research, 30(5), 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan C, Li Y, Liu X, Wang Y, Ping J, Lu Y, & Zhou G (2021). Characterization of structural variation in Tibetans reveals new evidence of high-altitude adaptation and introgression. Genome Biology, 22(1), 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P (2009). Evolution of the neocortex: a perspective from developmental biology. Nature Reviews. Neuroscience, 10(10), 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautiainen M, Nurk S, Walenz BP, Logsdon GA, Porubsky D, Rhie A, … Koren S (2022). Verkko: telomere-to-telomere assembly of diploid chromosomes (p. 2022.06.24.497523). 10.1101/2022.06.24.497523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, … Hurles ME (2006). Global variation in copy number in the human genome. Nature, 444(7118), 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees JS, Castellano S, & Andrés AM (2020). The Genomics of Human Local Adaptation. Trends in Genetics: TIG, 36(6), 415–428. [DOI] [PubMed] [Google Scholar]
- Rochette CF, Gilbert N, & Simard LR (2001). SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Human Genetics, 108(3), 255–266. [DOI] [PubMed] [Google Scholar]
- Rogers J, & Gibbs RA (2014). Comparative primate genomics: emerging patterns of genome content and dynamics. Nature Reviews. Genetics, 15(5), 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Pavlovic BJ, Hernando-Herraez I, Zhou X, Ward MC, Banovich NE, … Gilad Y (2015). A panel of induced pluripotent stem cells from chimpanzees: a resource for comparative functional genomics. 10.7554/eLife.07103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, & Gokcumen O (2019a). An Evolutionary Perspective on the Impact of Genomic Copy Number Variation on Human Health. Journal of Molecular Evolution. 10.1007/s00239-019-09911-6 [DOI] [PubMed] [Google Scholar]
- Saitou M, & Gokcumen O (2019b). Resolving the Insertion Sites of Polymorphic Duplications Reveals a HERC2 Haplotype under Selection. Genome Biology and Evolution, 11(6), 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, Masuda N, & Gokcumen O (2021). Similarity-based analysis of allele frequency distribution among multiple populations identifies adaptive genomic structural variants. Molecular Biology and Evolution. 10.1093/molbev/msab313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, Satta Y, & Gokcumen O (2018). Complex Haplotypes of GSTM1 Gene Deletions Harbor Signatures of a Selective Sweep in East Asian Populations. G3 , 8(9), 2953–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, Satta Y, Gokcumen O, & Ishida T (2018). Complex evolution of the GSTM gene family involves sharing of GSTM1 deletion polymorphism in humans and chimpanzees. BMC Genomics, 19(1), 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AD, Falconer E, Hills M, Spierings DCJ, & Lansdorp PM (2017). Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nature Protocols, 12(6), 1151–1176. [DOI] [PubMed] [Google Scholar]
- Sanders AD, Hills M, Porubský D, Guryev V, Falconer E, & Lansdorp PM (2016). Characterizing polymorphic inversions in human genomes by single-cell sequencing. Genome Research, 26(11), 1575–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, … Durbin R (2012). Insights into hominid evolution from the gorilla genome sequence. Nature, 483(7388), 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ERE, Kupferman JV, & Stackmann M (2019). The human-specific paralogs SRGAP2B and SRGAP2C differentially modulate SRGAP2A-dependent synaptic development. Scientific Reports, 9. 10.1101/596940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ERE, Zhao HT, Park JM, Dipoppa M, Monsalve-Mercado MM, Dahan JB, … Polleux F (2021). A human-specific modifier of cortical connectivity and circuit function. Nature, 599(7886), 640–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AJ, Chiang C, & Hall IM (2021). Structural variants are a major source of gene expression differences in humans and often affect multiple nearby genes (p. 2021.03.06.434233). 10.1101/2021.03.06.434233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Lee H, Darby CA, & Schatz MC (2018). Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nature Reviews. Genetics, 19(6), 329–346. [DOI] [PubMed] [Google Scholar]
- Seo J-S, Rhie A, Kim J, Lee S, Sohn M-H, Kim C-U, … Kim C (2016). De novo assembly and phasing of a Korean human genome. Nature, 538(7624), 243–247. [DOI] [PubMed] [Google Scholar]
- Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, … Paten B (2020). Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nature Biotechnology. 10.1038/s41587-020-0503-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Cheng Z, & Eichler EE (2006). Structural variation of the human genome. Annual Review of Genomics and Human Genetics, 7, 407–442. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, … Eichler EE (2005). Segmental duplications and copy-number variation in the human genome. American Journal of Human Genetics, 77(1), 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, … Chang W (2022). DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Research. 10.1093/nar/gkac194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shew CJ, Carmona-Mora P, Soto DC, Mastoras M, Roberts E, Rosas J, … Dennis MY (2021). Diverse molecular mechanisms contribute to differential expression of human duplicated genes. Molecular Biology and Evolution. 10.1093/molbev/msab131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- She X, Cheng Z, Zöllner S, Church DM, & Eichler EE (2008). Mouse segmental duplication and copy number variation. Nature Genetics, 40(7), 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Guo Y, Dong C, Huddleston J, Yang H, Han X, … Wang K (2016). Long-read sequencing and de novo assembly of a Chinese genome. Nature Communications, 7, 12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirén J, Monlong J, Chang X, Novak AM, Eizenga JM, Markello C, … Paten B (2021). Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science, 374(6574), abg8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M (2008). Linkage disequilibrium — understanding the evolutionary past and mapping the medical future. Nature Reviews Genetics, Vol. 9, pp. 477–485. 10.1038/nrg2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JHT, Grant RL, Behrens VC, Kučka M, Roberts Kingman GA, Soltys V, … Kingsley DM (2021). Genetic studies of human–chimpanzee divergence using stem cell fusions. Proceedings of the National Academy of Sciences, Vol. 118. 10.1073/pnas.2117557118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto DC, Shew C, Mastoras M, Schmidt JM, Sahasrabudhe R, Kaya G, … Dennis MY (2020). Identification of Structural Variation in Chimpanzees Using Optical Mapping and Nanopore Sequencing. Genes, 11(3), 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporny M, Guez-Haddad J, Kreusch A, Shakartzi S, Neznansky A, Cross A, … Opatowsky Y (2017). Structural History of Human SRGAP2 Proteins. Molecular Biology and Evolution, 34(6), 1463–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, & Lupski JR (2002). Genome architecture, rearrangements and genomic disorders. Trends in Genetics: TIG, 18(2), 74–82. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, & Lupski JR (2010). Structural variation in the human genome and its role in disease. Annual Review of Medicine, 61, 437–455. [DOI] [PubMed] [Google Scholar]
- Stedman HH, Kozyak BW, Nelson A, Thesier DM, Su LT, Low DW, … Mitchell MA (2004). Myosin gene mutation correlates with anatomical changes in the human lineage. Nature, 428(6981), 415–418. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, … Stefansson K (2005). A common inversion under selection in Europeans. Nature Genetics, 37(2), 129–137. [DOI] [PubMed] [Google Scholar]
- Steinberg KM, Antonacci F, Sudmant PH, Kidd JM, Campbell CD, Vives L, … Eichler EE (2012). Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nature Genetics, 44(8), 872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, … Courchesne E (2014). Patches of disorganization in the neocortex of children with autism. The New England Journal of Medicine, 370(13), 1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, … Dermitzakis ET (2007). Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science, 315(5813), 848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtevant AH (1913). The linear arrangement of six sex-linked factors in Drosophila, as shown by their mode of association. Journal of Experimental Zoology, Vol. 14, pp. 43–59. 10.1002/jez.1400140104 [DOI] [Google Scholar]
- Sturtevant AH (1917). Genetic Factors Affecting the Strength of Linkage in Drosophila. Proceedings of the National Academy of Sciences of the United States of America, 3(9), 555–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Huddleston J, Catacchio CR, Malig M, Hillier LW, Baker C, … Eichler EE (2013). Evolution and diversity of copy number variation in the great ape lineage. Genome Research, 23(9), 1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, … Eichler EE (2010). Diversity of Human Copy Number Variation and Multicopy Genes. Science, 330(6004), 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, … Eichler EE (2015). Global diversity, population stratification, and selection of human copy-number variation. Science, 349(6253), aab3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, … Korbel JO (2015). An integrated map of structural variation in 2,504 human genomes. Nature, 526(7571), 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki IK, Gacquer D, Van Heurck R, Kumar D, Wojno M, Bilheu A, … Vanderhaeghen P (2018). Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell, 173(6), 1370–1384.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taillon-Miller P, Bauer-Sardiña I, Zakeri H, Hillier L, Mutch DG, & Kwok PY (1997). The homozygous complete hydatidiform mole: a unique resource for genome studies. Genomics, 46(2), 307–310. [DOI] [PubMed] [Google Scholar]
- Tapon N, & Hall A (1997). Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Current Opinion in Cell Biology, 9(1), 86–92. [DOI] [PubMed] [Google Scholar]
- Taylor JS, & Raes J (2004). Duplication and divergence: the evolution of new genes and old ideas. Annual Review of Genetics, 38, 615–643. [DOI] [PubMed] [Google Scholar]
- Tóth K, Hofer KT, Kandrács Á, Entz L, Bagó A, Erőss L, … Wittner L (2018). Hyperexcitability of the network contributes to synchronization processes in the human epileptic neocortex. The Journal of Physiology, Vol. 596, pp. 317–342. 10.1113/jp275413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, & Salzberg SL (2011). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nature Reviews. Genetics, 13(1), 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Leemput J, Boles NC, Kiehl TR, Corneo B, Lederman P, Menon V, … Fasano CA (2014). CORTECON: a temporal transcriptome analysis of in vitro human cerebral cortex development from human embryonic stem cells. Neuron, 83(1), 51–68. [DOI] [PubMed] [Google Scholar]
- Varki A, & Altheide TK (2005). Comparing the human and chimpanzee genomes: Searching for needles in a haystack. Genome Research, Vol. 15, pp. 1746–1758. 10.1101/gr.3737405 [DOI] [PubMed] [Google Scholar]
- Vollger MR, DeWitt WS, Dishuck PC, Harvey WT, Guitart X, Goldberg ME, … Eichler EE (2022). Increased mutation rate and interlocus gene conversion within human segmental duplications (p. 2022.07.06.498021). 10.1101/2022.07.06.498021 [DOI] [Google Scholar]
- Vollger MR, Dishuck PC, Sorensen M, Welch AE, Dang V, Dougherty ML, … Eichler EE (2019). Long-read sequence and assembly of segmental duplications. Nature Methods, 16(1), 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollger MR, Guitart X, Dishuck PC, Mercuri L, Harvey WT, Gershman A, … Eichler EE (2021). Segmental duplications and their variation in a complete human genome (p. 2021.05.26.445678). 10.1101/2021.05.26.445678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollger MR, Guitart X, Dishuck PC, Mercuri L, Harvey WT, Gershman A, … Eichler EE (2022). Segmental duplications and their variation in a complete human genome. Science, 376(6588), eabj6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollger MR, Logsdon GA, Audano PA, Sulovari A, Porubsky D, Peluso P, … Eichler EE (2019). Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Annals of Human Genetics. 10.1111/ahg.12364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GP, Amemiya C, & Ruddle F (2003). Hox cluster duplications and the opportunity for evolutionary novelties. Proceedings of the National Academy of Sciences of the United States of America, 100(25), 14603–14606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainszelbaum MJ, Charron AJ, Kong C, Kirkpatrick DS, Srikanth P, Barbieri MA, … Stahl PD (2008). The hominoid-specific oncogene TBC1D3 activates Ras and modulates epidermal growth factor receptor signaling and trafficking. The Journal of Biological Chemistry, 283(19), 13233–13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Antonacci-Fulton L, Howe K, Lawson HA, Lucas JK, Phillippy AM, … Human Pangenome Reference Consortium. (2022). The Human Pangenome Project: a global resource to map genomic diversity. Nature, 604(7906), 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren WC, Harris RA, Haukness M, Fiddes IT, Murali SC, Fernandes J, … Eichler EE (2020). Sequence diversity analyses of an improved rhesus macaque genome enhance its biomedical utility. Science, 370(6523). 10.1126/science.abc6617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CT, Marques-Bonet T, Sharp AJ, & Mefford HC (2014). The genetics of microdeletion and microduplication syndromes: an update. Annual Review of Genomics and Human Genetics, 15, 215–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, … Hunkapiller MW (2019). Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature Biotechnology. 10.1038/s41587-019-0217-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TN, Wambua S, Uyoga S, Macharia A, Mwacharo JK, Newton CRJC, & Maitland K (2005). Both heterozygous and homozygous alpha+ thalassemias protect against severe and fatal Plasmodium falciparum malaria on the coast of Kenya. Blood, 106(1), 368–371. [DOI] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, & Romano LA (2003). The Evolution of Transcriptional Regulation in Eukaryotes. Molecular Biology and Evolution, 20(9), 1377–1419. [DOI] [PubMed] [Google Scholar]
- Xing L, Kubik-Zahorodna A, Namba T, Pinson A, Florio M, Prochazka J, … Huttnet WB (2021). Expression of human-specific ARHGAP11B in mice leads to neocortex expansion and increased memory flexibility. The EMBO Journal, 40. 10.15252/embj.2020107093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Sun D, Daly A, Yang F, Zhou X, Zhao M, … Tyler-Smith C (2008). Adaptive evolution of UGT2B17 copy-number variation. American Journal of Human Genetics, 83(3), 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka M, Kato Y, Angata T, & Narimatsu H (2009). Deletion polymorphism of SIGLEC14 and its functional implications. Glycobiology, 19(8), 841–846. [DOI] [PubMed] [Google Scholar]
- Yang L (2020). A practical guide for structural variation detection in human genome. Current Protocols in Human Genetics / Editorial Board, Jonathan L. Haines … [et Al.], 107(1), e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Safavi S, Woodward EL, Duployez N, Olsson-Arvidsson L, Ungerbäck J, … Paulsson K (2020). 13q12.2 deletions in acute lymphoblastic leukemia lead to upregulation of FLT3 through enhancer hijacking. Blood, 136(8), 946–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SM, Sherman RM, Taylor DJ, Nair DR, Bortvin AN, Schatz MC, & McCoy RC (2021). Local adaptation and archaic introgression shape global diversity at human structural variant loci. eLife, 10. 10.7554/eLife.67615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JM, Endicott RM, Parghi SS, Walker M, Kidd JM, & Trask BJ (2008). Extensive copy-number variation of the human olfactory receptor gene family. American Journal of Human Genetics, 83(2), 228–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousaf A, Liu J, Ye S, & Chen H (2021). Current Progress in Evolutionary Comparative Genomics of Great Apes. Frontiers in Genetics, 12, 657468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunis JJ, & Prakash O (1982). The Origin of Man: A Chromosomal Pictorial Legacy. Science, Vol. 215, pp. 1525–1530. 10.1126/science.7063861 [DOI] [PubMed] [Google Scholar]
- Yunis JJ, Sawyer JR, & Dunham K (1980). The striking resemblance of high-resolution G-banded chromosomes of man and chimpanzee. Science, 208(4448), 1145–1148. [DOI] [PubMed] [Google Scholar]
- Zhang F, Carvalho CMB, & Lupski JR (2009). Complex human chromosomal and genomic rearrangements. Trends in Genetics: TIG, 25(7), 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J (2003). Evolution by gene duplication: an update. Trends in Ecology & Evolution, 18(6), 292–298. [Google Scholar]
- Zhang J, Rosenberg HF, & Nei M (1998). Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proceedings of the National Academy of Sciences of the United States of America, 95(7), 3708–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng-Bradley X, Streeter I, Fairley S, Richardson D, Clarke L, Flicek P, & 1000 Genomes Project Consortium. (2017). Alignment of 1000 Genomes Project reads to reference assembly GRCh38. GigaScience, 6(7), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zody MC, Jiang Z, Fung H-C, Antonacci F, Hillier LW, Cardone MF, … Eichler EE (2008). Evolutionary toggling of the MAPT 17q21.31 inversion region. Nature Genetics, Vol. 40, pp. 1076–1083. 10.1038/ng.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.