

Abstract
Inflammation is strongly associated with pancreatic ductal adenocarcinoma (PDAC), a highly lethal malignancy. Dysregulated RNA splicing factors have been widely reported in tumorigenesis, but their involvement in pancreatitis and PDAC is not well understood. Here, we report that the splicing factor SRSF1 is highly expressed in pancreatitis, PDAC precursor lesions, and tumors. Increased SRSF1 is sufficient to induce pancreatitis and accelerate KRASG12D-mediated PDAC. Mechanistically, SRSF1 activates MAPK signaling—partly by upregulating interleukin 1 receptor type 1 (IL1R1) through alternative-splicing-regulated mRNA stability. Additionally, SRSF1 protein is destabilized through a negative feedback mechanism in phenotypically normal epithelial cells expressing KRASG12D in mouse pancreas, and in pancreas organoids acutely expressing KRASG12D, buffering MAPK signaling and maintaining pancreas-cell homeostasis. This negative-feedback regulation of SRSF1 is overcome by hyperactive MYC, facilitating PDAC tumorigenesis. Our findings implicate SRSF1 in the etiology of pancreatitis and PDAC, and point to SRSF1-misregulated alternative splicing as a potential therapeutic target.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) has an extremely poor prognosis, attributable to the presence of distant metastases at the time of diagnosis, which limits traditional cancer treatments like surgery or chemotherapy (1). Understanding the process of PDAC initiation is especially critical for PDAC prevention and effective treatment. Somatic activating mutations in KRAS—a member of the RAS superfamily of small GTPases with guanine-nucleotide-binding capability— are present in >90% of PDAC, dominated by substitution from glycine to aspartate (G12D) at the 12th residue, within the G domain that is critical for GTPase activity (2,3). The mutation impairs GTPase activity and stabilizes RAS-GTP, resulting in constitutive activation of KRAS signaling pathways (4). Studies involving the genetically engineered KC mouse model (LSL-KrasG12D/+; Pdx1-Cre) support the notion that mutant KRASG12D triggers the formation of pre-neoplastic pancreatic alterations, such as acinar-to-ductal metaplasia (ADM)—a process through which pancreatic acinar cells differentiate into ductal-like progenitor cells—and pancreatic intraepithelial neoplasia (PanIN)—a histologically well-defined precursor to invasive ductal adenocarcinoma of the pancreas—and results in a protracted onset of PDAC (3,5,6). However, the majority of pancreatic cells from KC mice remain morphologically normal for a long time, and very few are transformed into high-grade PanINs. This observation suggests that mutant KRASG12D expression is insufficient to induce pancreas-cell transformation, and that additional oncogenic alterations are required for further PDAC genesis and progression.
Epidemiological studies suggest a strong association between chronic pancreatitis and PDAC development (7,8). Although in most experimental murine models, pancreatitis alone cannot induce PDAC development, pancreatitis accelerates mutant-KRAS-mediated tumorigenesis (9). The IL1/IL1R1 pathway is strongly associated with an inflammatory environment for both cancer initiation and progression (10). IL1 secreted from PDAC cells can enforce malignancy through either an autocrine mechanism (11) or a paracrine mechanism that stimulates cancer-associated fibroblasts (12). In addition, a significantly elevated IL1 receptor IL1R1 is present in human PDAC tumors, compared with adjacent normal pancreatic tissue (13). Blocking IL1R1 with a specific antagonist, Anakinra, decreases the level of pERK and inhibits PDAC tumor growth (11).
RNA splicing is an enzymatic process that removes the introns from precursor mRNA and joins the exons to generate mature RNA in the nucleus, contributing to proteome diversity and gene-expression regulation (14). Dysregulated expression or mutations of oncogenic splicing factors can contribute to tumor development by altering pre-mRNA alternative splicing events that impinge on multiple signaling pathways (14–16). The serine/arginine-rich (SR) protein family is phylogenetically conserved and involved in constitutive and alternative splicing. Multiple SR proteins have oncogenic properties across different cancer types, notably SRSF1, which is overexpressed in many cancers and can promote transformation (17–23). However, its involvement in pancreatitis and PDAC tumorigenesis was unknown. Here we report that SRSF1 is associated with and promotes pancreatitis in mouse through increased MAPK signaling effected by alternative splicing of Il1r1. Interestingly, SRSF1 protein is initially destabilized as a negative feedback mechanism in response to KRASG12D mutation, maintaining pancreatic cell homeostasis. This negative feedback regulation of SRSF1 is transcriptionally bypassed by MYC, accelerating KRASG12D-mediated PDAC initiation and progression.
RESULTS
SRSF1 Promotes Pancreatitis in Mice
Given that precise regulation of SRSF1 (formerly SF2/ASF)—which is 100% conserved at the protein level and 95% conserved within the mRNA 3’ untranslated region (UTR) sequence between human and mouse (17)—is essential for cell homeostasis and tissue renewal (24–27), we examined the expression of SRSF1 in a mouse pancreatitis model induced by cerulein—a decapeptide analog of cholecystokinin that stimulates the secretion of pancreatic enzymes, provoking pancreatic injury, pancreatitis, and ADM formation (6,28). Reanalyzing the public gene-expression datasets GSE65146, GSE41418, and GSE109277, we found strong and consistent increases in Srsf1 mRNA expression in cerulein-induced mouse pancreatitis (Fig. 1A). To confirm this effect at the SRSF1 protein level, we generated an acute pancreatitis model by injecting mice with cerulein hourly for 8 h on 2 consecutive days. The pancreata were harvested 48 h after the last injection (Fig. 1B). Immunohistochemistry (IHC) staining showed increased SRSF1 protein in the ADM lesions associated with pancreatitis (Fig. 1B). Additionally, the pancreas regions with severe inflammation exhibited higher SRSF1 protein intensity than those with a mild phenotype (Fig. 1B). These observations suggest that elevated SRSF1 expression is associated with pancreatitis.
Figure 1.
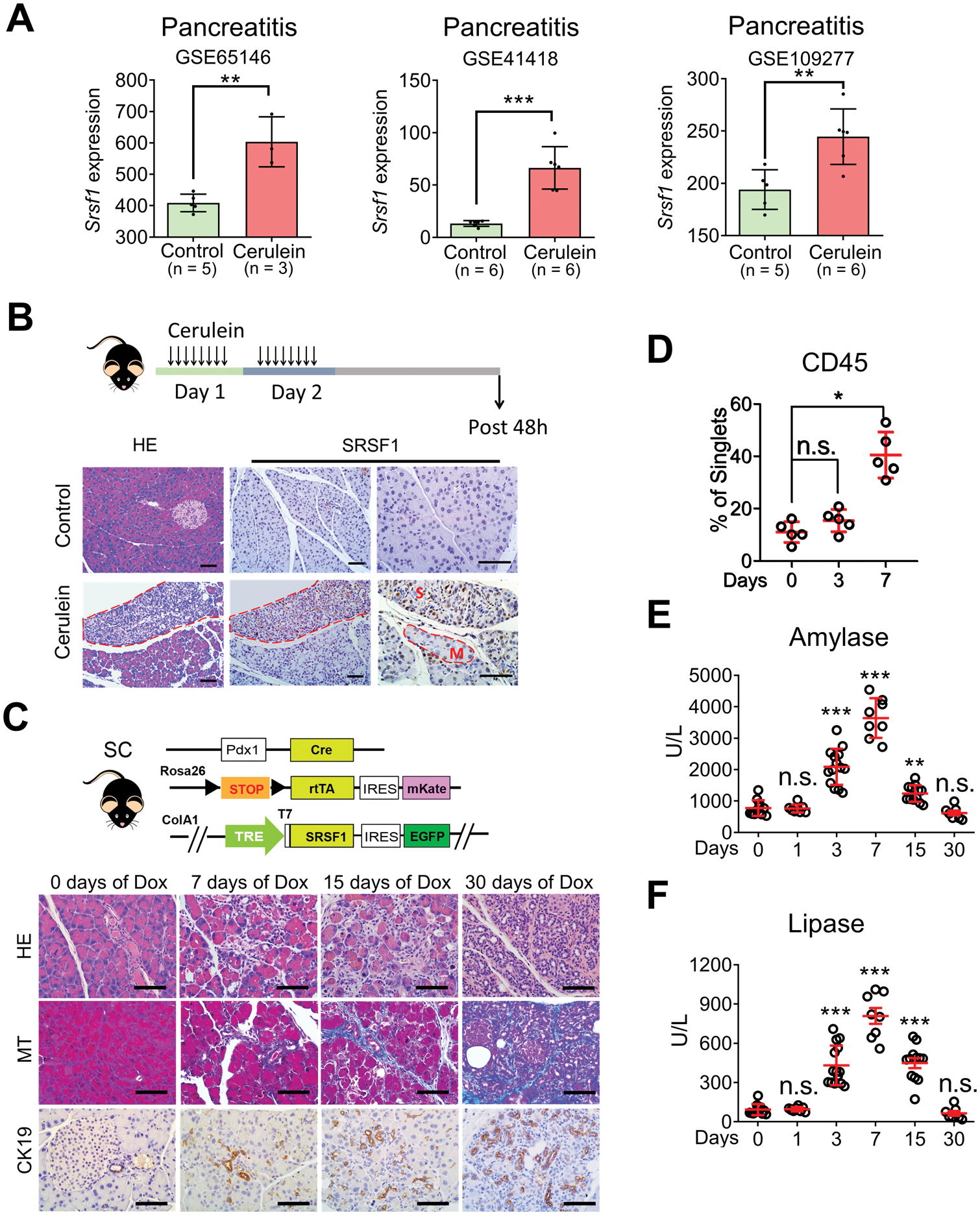
SRSF1 is associated with and promotes pancreatitis. A, Srsf1 expression in control and cerulein-treated mouse pancreas from reanalysis of GSE65146, GSE41418, and GSE10927 datasets. B, Scheme for cerulein-induced pancreatitis model (top); HE and SRSF1 IHC staining (bottom) of pancreata from control (saline) and cerulein-treated mice (M, mild; S, severe pancreatitis regions). Scale bars, 50 μm. n = 5 per group. C, Scheme of SC mice and their histological evaluation by HE staining, Masson’s trichrome staining (MT) (blue indicates collagen deposition), and IHC staining of cytokeratin CK19 to identify metaplastic ductal lesions, after treatment with Dox. Scale bars, 50 μm. n = 5 per group. D, Immune-cell infiltration evaluated by flow cytometry in SC mice treated with Dox. n = 5 per group. E-F, Circulating levels (U/L) of the pancreatic enzymes amylase (E) and lipase (F) in SC mice after treatment with Dox (days). Each data point represents a measurement from an individual mouse. A, Linear models and empirical bayes methods. (D-F) Kruskal-Wallis test followed by pairwise comparisons using Wilcoxon rank-sum test. Family-wise error rate was adjusted using Bonferroni-Holm method. *p <0.05, **p < 0.01, ***p <0.001. Error bars represent mean ± SD.
To determine whether SRSF1 can induce pancreatitis as a causative factor, or its accumulation is a consequence of the cellular-identity transition during pancreatitis, we generated a mouse model dubbed SC (tetO-Srsf1; LSL-rtTA; Pdx1-Cre), with doxycycline (Dox)-inducible expression of SRSF1 in the pancreas (Fig. 1C). Dox-treatment-induced SRSF1 expression in pancreata from SC mice was comparable to SRSF1 expression in cerulein-induced pancreatitis (Fig. 1A and B; Supplementary Fig. S1A and S1B). Additionally, Dox-treated SC mice exhibited histologic signs of pancreatitis, including interstitial edema, lymphocyte infiltration, collagen deposition, and accumulation of metaplastic ductal lesions (Fig. 1C). Expression of the proliferation marker Ki67 was elevated in SC mice after Dox treatment for 3 days, whereas the apoptotic marker cleaved caspase 3 was not detected (Supplementary Fig. S1B), suggesting that SRSF1 promotes pancreatic cell proliferation at the initial stage of pancreatitis. Additionally, flow cytometry revealed an influx of immune cells into the pancreas, especially macrophages (Fig. 1D; Supplementary Fig. S1C), which we confirmed by immunofluorescence staining (Supplementary Fig. S1D). Moreover, we detected elevated serum levels of the pancreatic enzymes amylase and lipase upon short-term Dox treatment, and these enzymes returned to their normal levels after 30 days of SRSF1 induction, consistent with a transition from acute to chronic pancreatitis (Fig. 1E and F) (28). SRSF1-induced pancreatitis was reversible after a recovery period following Dox withdrawal (Supplementary Fig. S1E and S1F), suggesting that SRSF1 contributes to the duration of inflammation. Of note, we did not detect PanIN lesions in SC mice, even with continuous Dox treatment for two months. These data, along with the observed increase in SRSF1 in the cerulein-induced pancreatitis model, demonstrate that elevated SRSF1 is associated with pancreas-cell plasticity and de-differentiation, and results in pancreatitis and ADM.
Elevated SRSF1 is Associated with PDAC Tumorigenesis and Progression
We previously reported that slight upregulation of SRSF1 promotes cell transformation and tumorigenesis in multiple cell contexts, including fibroblasts and mammary epithelium (25,26). To investigate if SRSF1 expression is also elevated in PDAC tumors, we examined its expression in human PDAC and tumors from KPC mice (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre). Both SRSF1 mRNA and SRSF1 protein are upregulated in PDAC tumors (Fig. 2A; Supplementary Fig. S2A–S2D), and both are associated with poor prognosis in PDAC patients (Fig. 2B; Supplementary Fig. S2E). In addition, we found that SRSF1 is upregulated in ADM and PanIN lesions, compared to morphologically normal pancreas acinar and ductal cells (Fig. 2C; Supplementary Fig. S2F), suggesting its involvement in PDAC initiation.
Figure 2.
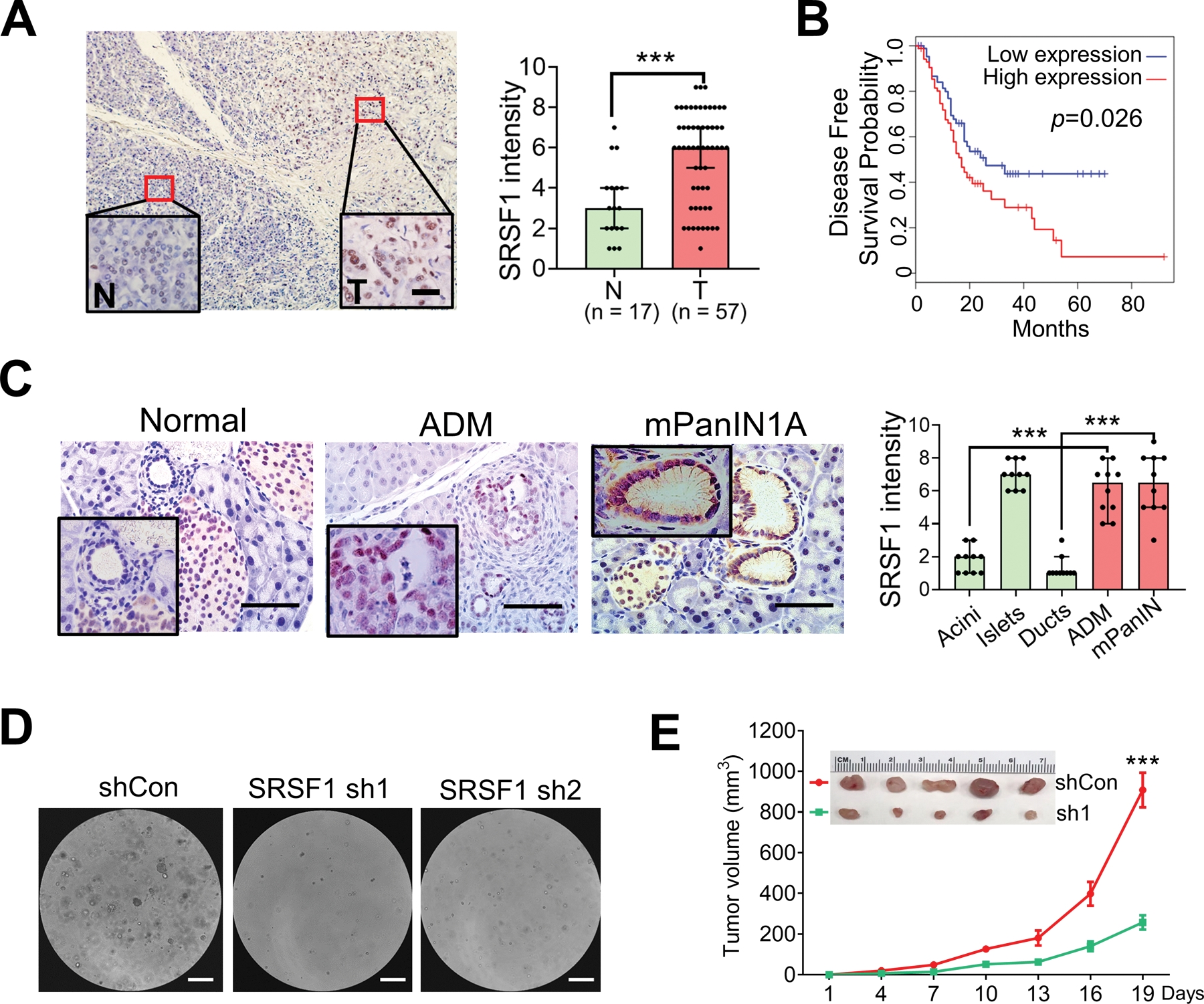
Elevated SRSF1 in PDAC and precursor lesions is associated with tumor progression. A, IHC staining (left) and intensity score (right) of SRSF1 in human PDAC tumor versus normal tissue. Insets show higher magnification. Scale bars, 20 μm. Unpaired, two-tailed t test. ***p < 0.001. Error bars represent mean ± SD. B, Kaplan-Meier survival analysis of patients with PAAD from TCGA. Group cut-offs were set at the SRSF1 median mRNA expression (n = 89 per group). Log-rank Mantel-Cox test was performed. C, IHC staining (left) and intensity score (right) of SRSF1 in pancreas cells from KC mice. Insets show higher magnification of normal duct, ADM, and murine PanIN1A. Scale bars, 50 μm. For quantification of intensity, n = 10 per group. One-way ANOVA with Tukey’s multiple comparison test. ***p < 0.001. D, Human tumor hT60 organoids following knockdown with control (shCon) or SRSF1sh hairpins (sh1 & sh2). Scale bar = 200 μm. n = 3 replicates per condition. E, Tumor growth curve of subcutaneous tumors formed with SUIT-2 cells, following knockdown with control (shCon) or SRSF1sh hairpins (sh1), n = 5 per group. Upper panel represents xenograft tumors. Linear mixed-effects model with the experimental group, time and group by time interaction as fixed effects, and tumor-specific random intercept was used to fit the model. ***P <0.001.
Moreover, we found that knocking down SRSF1 with short hairpin RNAs (Supplementary Fig. S2G) suppressed human PDAC organoid growth (Fig. 2D) and SUIT-2 cell line soft-agar colony formation and migration (Supplementary Fig. S2H and S2I). Likewise, SUIT-2 subcutaneous xenografts exhibited a significant decrease in tumor growth and expression of the proliferative marker Ki67 upon SRSF1 knockdown (Fig. 2E; Supplementary Fig. S2J). These results suggest that SRSF1 contributes to PDAC progression.
To dissect the role of the functional domains of SRSF1 in a PDAC context, we knocked down SRSF1 in hT60 human tumor organoids and expressed previously described truncated, or RRM1-inactivated (FF-DD, Phe56 and Phe58 substituted with Asp residues) SRSF1 proteins (Supplementary Fig. S2K) (25,29,30). We found that all the domain-deletion proteins had reduced rescue activity in cell proliferation (Supplementary Fig. S2L), with RRM1-deleted protein having the lowest rescue efficiency. Consistently, RRM1-inactivated SRSF1 could not rescue organoid proliferation. These results suggest that RRM1 is critical for SRSF1’s function in PDAC maintenance. We also used an NRS construct—a C-terminal fusion of SRSF1 to the nuclear-retention signal from SRSF2—to dissect the nuclear versus cytoplasmic functions of SRSF1 (25). NRS rescued organoid proliferation to a greater extent than the other constructs, with ~75% the activity of wild-type SRSF1. These results suggest that SRSF1’s nuclear functions account for the oncogenic role of SRSF1 in the PDAC context.
SRSF1 is Required for and Accelerates KRASG12D-Mediated PDAC Tumorigenesis and Progression
Considering that increasing SRSF1 induces pancreatitis, and the known strong association between pancreatitis and PDAC progression (7,8), we sought to investigate whether SRSF1 can cooperate with KRASG12D to promote tumorigenesis. We generated the KSC mouse strain by intercrossing SC and LSL-KrasG12D/+ mice (Fig. 3A). Compared to the pancreata of two-month-old KC mice, which were mostly morphologically normal, rarely exhibiting ADM or low-grade PanINs, the vast majority of the pancreas cells in KSC mice were transformed and exhibited neoplasia, interstitial edema, and collagen deposition (Fig. 3A and B; Supplementary Fig. S3A). In addition, KSC mice exhibited extensive, high-grade PanIN lesions, characterized by loss of cell polarity, significant nuclear atypia, and budding of cell clusters into the ductal lumen (Fig. 3A and C). The positive GFP staining of the transformed cells confirmed that they were derived from SRSF1-overexpressing cells (Supplementary Fig. S3A).
Figure 3.
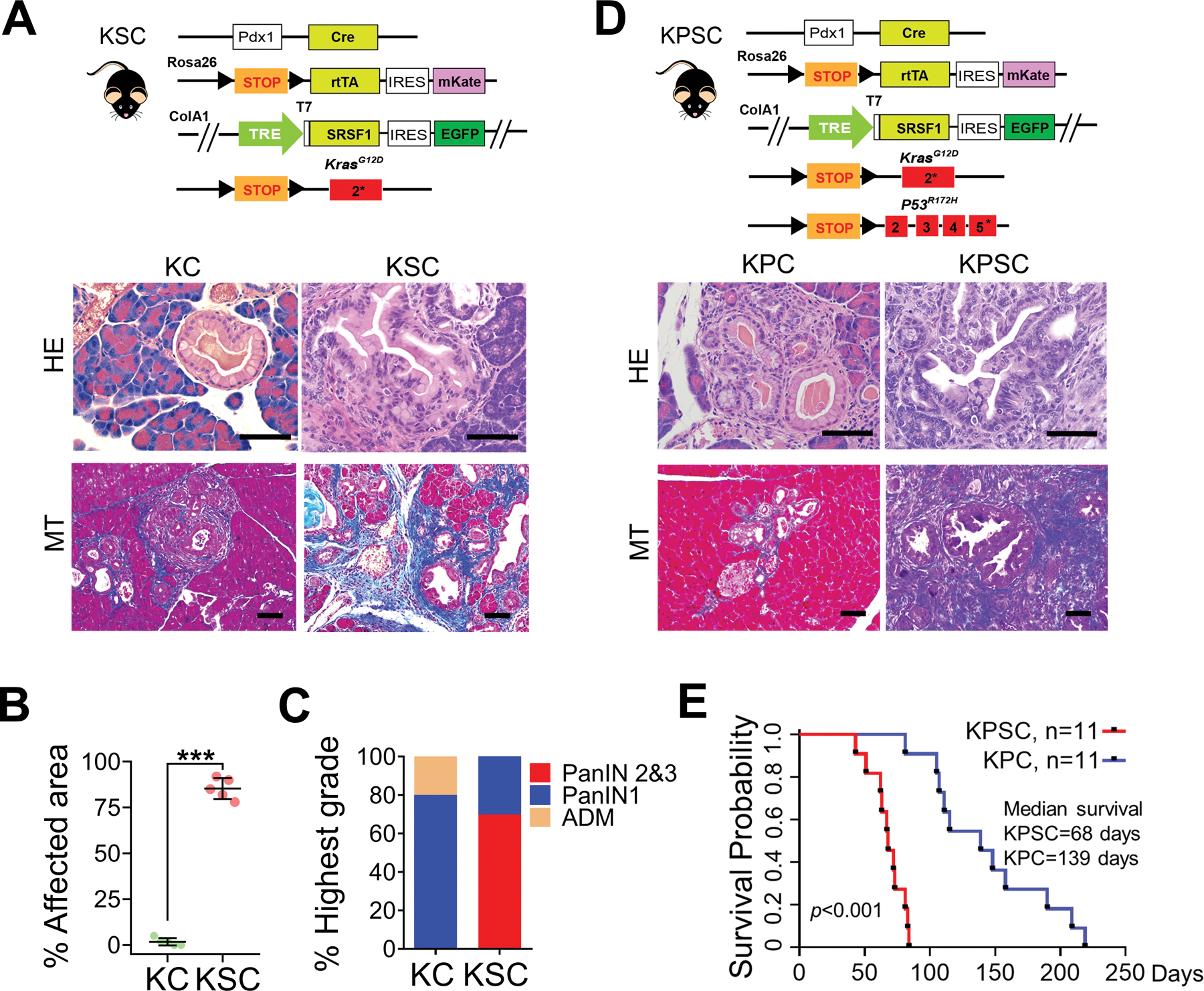
SRSF1 accelerates KRASG12D-mediated tumorigenesis. A, Scheme of KSC mouse strain (top), HE and MT staining of pancreata from two-month-old KC and KSC mice (n = 5 per group). Scale bars, 50 μm. B, Quantification of the percentage of pancreatic area exhibiting histological signs of ADM and neoplasia from two-month-old KC and KSC mice (n = 5 per group). Unpaired two-tailed t test, ***p <0.001. Error bars represent mean ± SD. C, Classification of highest-grade PanIN lesions present in two-month-old KC and KSC mice (n = 5 per group). D, Scheme of KPSC mouse strain, HE and MT staining of pancreata from one-month-old KPC and KPSC mice (n = 5 per group). Scale bars, 50 μm. E, Kaplan-Meier survival analysis of KPC and KPSC mice (n = 11 per group). The p value was determined by a log-rank Mantel-Cox test.
We reported that SRSF1 can stabilize p53 and trigger oncogene-induced senescence (OIS) in primary fibroblast cells (31). To examine whether elevated SRSF1 expression can induce OIS in KSC pancreata, we performed IHC staining of p53 and senescence-associated beta-galactosidase (SA-β-gal) staining. We did not detect a significant increase in p53 in KSC compared with KC pancreas sections, nor in SA-β-gal staining (Supplementary Fig. S3B and S3C). Additionally, Ki67 was significantly increased in the high-grade PanINs in KSC mice, compared to the low-grade PanINs in KC mice (Supplementary Fig. S3B). Thus, the modest SRSF1 overexpression levels achieved in our models appear to be insufficient to stabilize p53, allowing SRSF1 to promote proliferation in PanIN lesions, as opposed to senescence or apoptosis.
As the second most common genetic alteration following mutations in KRAS in PDAC, the TP53 tumor suppressor is mutated in nearly 70% of PDAC, mostly co-occurring with KRAS mutations (32). We further examined the effect of SRSF1 in PDAC development by introducing a conditional LSL-Trp53R172H allele, generating the KPSC strain (Fig. 3D). Even though KPC mice exhibit advanced PDAC with markedly shortened median survival, compared to KC mice (5), we detected only rare low-grade PanINs in one-month-old KPC mice (Fig. 3D; Supplementary Fig. S3D). In contrast, KPSC mice rapidly succumbed to PDAC tumors, with even shorter median survival (68 days) than KPC mice (139 days) (Fig. 3D and E; Supplementary Fig. S3D). We conclude that elevated SRSF1 accelerates KRASG12D-mediated cell transformation, PDAC initiation and progression.
To further examine the dependence of PDAC initiation on SRSF1, we also generated KSKOC (LSL-KrasG12D/+; Srsf1fl/fl; Pdx-1-Cre) mice with conditional knockout of SRSF1 in the pancreas (Supplementary Fig. S4A). We found markedly fewer and lower-grade neoplastic lesions with less Ki67 staining in 6-month-old KSKOC mice than in KC mice (Supplementary Fig. S4B–S4E), demonstrating that SRSF1 deficiency perturbs KRASG12D-mediated PDAC initiation.
Given that elevated SRSF1 can induce pancreatitis, and that pancreatitis promotes KRASG12D-mediated PDAC initiation, we assessed the role of SRSF1 in pancreatitis-accelerated PDAC initiation. We performed cerulein-induced pancreatitis experiments in 2-month-old KC and KSKOC mice. KSKOC mice exhibited fewer and lower-grade neoplasias with less Ki67 staining (Supplementary Fig. S4F–S4I). These results demonstrate that SRSF1 is necessary for KRASG12D-mediated PDAC initiation, both autonomously or in a pancreatitis-accelerated manner.
SRSF1 activates MAPK signaling, a prerequisite for KRASG12D-mediated PDAC initiation
To address the underlying mechanisms by which elevated SRSF1 induces pancreatitis and accelerates KRASG12D-mediated disease progression, we generated organoids from SC and KSC mice. Deep sequencing of RNA harvested from the organoid lines indicated comparable induction of SRSF1 after Dox treatment (Supplementary Fig. S5A). Interestingly, pathway-enrichment analysis showed opposite patterns resulting from KRASG12D mutation versus SRSF1 upregulation (Fig. 4A; Supplementary Fig. S5B and S5C). Among the affected pathways, MAPK signaling was negatively enriched in the presence of KRASG12D (Fig. 4A; Supplementary Fig. S5B), which is consistent with multiple reported negative-feedback loops in response to RAS activation that dampen the RAS signaling output (33–35).
Figure 4.
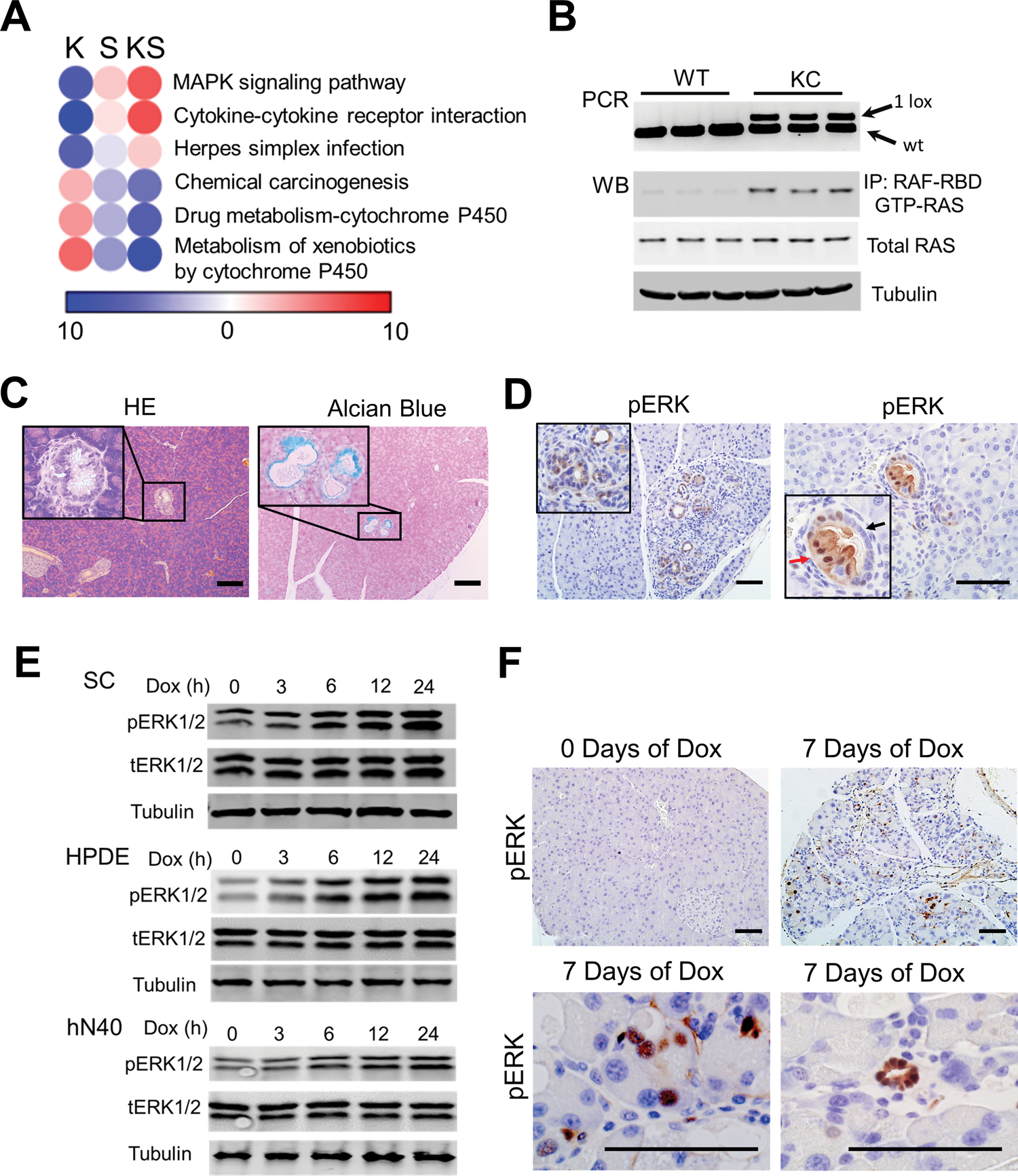
SRSF1 activates MAPK signaling, a prerequisite for KRASG12D-mediated PDAC initiation. A, Pathway-enrichment analysis of genes with increased (red) or decreased (blue) expression, in KSC organoids compared to SC organoids, both without Dox treatment (K), in Dox-treated SC organoids compared to untreated SC organoids (S), and in Dox-treated KSC organoids compared to untreated KSC organoids (KS). n = 3 replicates per group. Color bar represents −log10 (p value). B, PCR of pancreatic DNA (top), and western blot of Ras-GTP and total RAS levels (bottom) from 2-month-old wild-type (WT) and KC mice (n = 3 per genotype). C, Hematoxylin and eosin (HE), and Alcian blue staining of mucin of pancreata from two-month-old KC mice (n = 5). Insets show higher magnification of murine PanIN1A lesion. Scale bars, 100 μm. D, IHC staining of pERK in 2-month-old KC pancreas tissue (n = 5). Red arrow indicates ductal cells with early-stage neoplasia; black arrow indicates normal ductal cells. Scale bars, 50 μm. E, Western blotting of phosphorylated ERK1/2 (pERK1/2), total ERK1/2 (tERK1/2), and Tubulin in SC organoids, Dox-inducible T7-SRSF1 expressing HPDE cells, and hN40 organoids, with Dox treatment. n = 3 replicates per condition. F, pERK IHC staining of SC mice after Dox treatment (n = 5 per group). Scale bars, 50 μm.
KC mice express Cre recombinase under the control of the pancreatic-specific promoter Pdx1 (Pdx1-Cre) (3). Cre recombinase excises the “lox-stop-lox” (LSL) cassette—comprising an SV40 polyadenylation signal and a PGK-puromycin selection cassette—in an LSL-KrasG12D/+ allele in the pancreatic epithelium (Supplementary Fig. S6A). After recombination of the LSL cassette, a single LoxP site (34 bp) is present in the first intron of the Kras gene, which can be amplified by specific primers (Supplementary Fig. S6A). Genomic PCR showed a prevalent excised LSL cassette in pancreatic cells from two-month-old KC mice, indicative of an activated KrasG12D allele (Fig. 4B). Accordingly, western blotting of total protein extracted from pancreata of two-month-old KC mice revealed increased activated (GTP-bound) RAS (Fig. 4B), indicating that the conditional LSL-KrasG12D/+ alleles were efficiently recombined and activated in the pancreata from KC mice. However, consistent with a previous report, the majority of pancreatic cells from these early-stage KC mice remain morphologically normal, with only rarely detected PanIN1A lesions with abundant mucin content (Fig. 4C) (3). To precisely examine the status of the LSL-KrasG12D/+ allele in the pancreatic cells from KC mice in situ, we designed two sets of RNA fluorescent in situ hybridization (FISH) probes complementary to the sense and antisense transcripts expressed from the LSL cassette, respectively (Supplementary Fig. S6A). Notably, the majority of pancreas cells had a recombined LSL cassette, indicated by loss of the FISH signal, and corresponding to an active KrasG12D allele (Supplementary Fig. S6B and S6C). These observations imply the existence of a compensatory feedback mechanism against mutant KRASG12D, which preserves intracellular signaling homeostasis and pancreatic cell identity.
Consistent with previous reports (36,37), only the subset of pancreas cells with hyperactive MAPK signaling exhibited pancreatic metaplasia and neoplasia, even within individual ducts from two-month-old KC mice (Fig. 4D). These results point to the pivotal role of MAPK signaling in cell homeostasis, and the need for this pathway to be activated in the context of cell transformation. Remarkably, strengthened MAPK signaling was detected in the SC mouse organoids, as well as in immortalized human normal pancreatic duct epithelial (HPDE) cells and a human normal organoid line (hN40) with Dox-inducible SRSF1 expression (Fig. 4E). Accordingly, induction of SRSF1 increased pERK intensity in the pancreata from SC mice (Fig. 4F), as well as from KSC and KPSC mice (Supplementary Fig. S7A). We conclude that SRSF1 promotes MAPK signaling activation, which is a prerequisite for KRASG12D-mediated pancreas-cell transformation and PDAC initiation.
We then sought to examine the dependence on MAPK-signaling activation of the SRSF1-induced pancreatitis phenotype. We treated SC mice with Dox, with or without trametinib, a MEK inhibitor drug. On day 15, mice were euthanized, and the pancreas tissues were collected for pathological analysis. Trametinib treatment significantly suppressed SRSF1-induced pancreatitis (Supplementary Fig. S7B and S7C), indicating that MAPK signaling contributes substantially to SRSF1-induced pancreatitis.
Acute KRASG12D Expression Decreases SRSF1 by Promoting Its Ubiquitination
Given that the KRASG12D mutation is impeded by negative feedback mechanisms that attenuate MAPK signaling, whereas SRSF1 elevation can enhance MAPK signaling associated with cellular plasticity, we next measured SRSF1 expression in morphologically normal pancreas cells expressing KRASG12D in KC mice. We harvested the pancreata from two-month-old KC mice and Pdx1-Cre control littermates. In contrast to its elevated expression in focal ADM, PanIN, and PDAC lesions, SRSF1 protein was decreased in protein extract from entire pancreata of two-month-old KC mice (Fig. 5A). IHC staining indicated that SRSF1 is highly expressed in the rare ADM regions, compared to adjacent untransformed cells in two-month-old KC mice and to pancreas from WT mice. In particular, normal cells in WT mice exhibited higher SRSF1 expression than untransformed cells in KC mice (Supplementary Fig. S8A).
Figure 5.
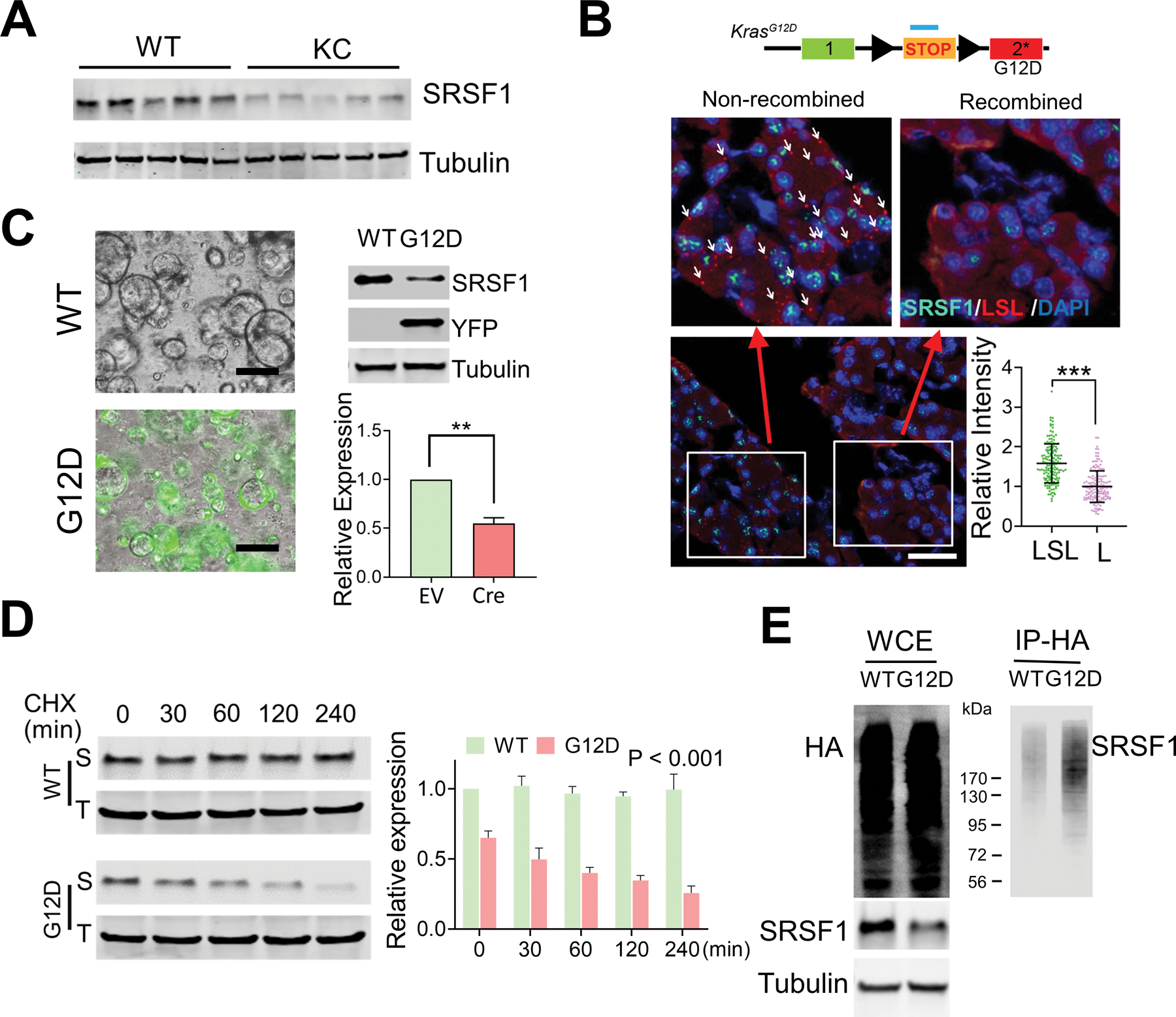
Reduced expression of SRSF1 in morphologically normal KRASG12D-expressing pancreatic cells. A, Western blotting of SRSF1 protein in pancreatic protein lysates from WT and KC mice (n = 5 per group). B, Multiplexed RNA-FISH/IF staining of STOP-cassette and SRSF1 protein in pancreata isolated from KC mice. RNA FISH probes were designed to target the STOP cassette. Reflecting the mosaic expression of Cre recombinase in KC mice, pancreatic cells with the STOP cassette recombined and non-recombined can be distinguished by RNA-FISH probes. Upper left: non-recombined region, with white arrows indicating some of the positive FISH signals; upper right: recombined region, where most cells have undergone recombination and hence the FISH signal is lost; Bottom right, quantification of SRSF1 intensity in LSL-positive or -negative pancreas cells. Scale bars, 50 μm. Unpaired, two-tailed t test. ***p < 0.001. Error bars represent mean ± SD. C, YFP fluorescence (left), and western blotting and quantification of SRSF1 (right) of LSLKrasG12D/+; R26-LSl-YFP ductal organoids infected with adeno-empty (WT) or adeno-Cre (G12D) (n = 3 biological replicates). Unpaired, two-tailed t test. **p < 0.01. Error bars represent mean ± SD. D, Western blotting of SRSF1 (S) and Tubulin (T), and quantification in LSLKrasG12D/+; R26-LSl-YFP ductal organoids infected with adeno empty (WT) or adeno-Cre (G12D), respectively, followed by exposure to cycloheximide (10 mg/ml) for the indicated times. Linear mixed effects model with the experimental group, time and group by time interaction as fixed effects, and sample-specific random intercept was used to estimate the relative expression. E, In vivo ubiquitination assay of HA-ubiquitin stable-expressing LSL-KRASG12D/+; R26LSL-YFP organoids infected with adeno empty (WT) and adeno-Cre (G12D), respectively. Following immunoprecipitation of cell lysates, immunoprecipitates (IP) and whole-cell extracts (WCE) were analyzed by immunoblotting with the indicated antibodies.
Due to the mosaic expression of Cre recombinase in KC mice, a small proportion of pancreas cells retain an intact LSL cassette and a dormant KrasG12D allele (Supplementary Fig. S6C) (3). Therefore, we combined FISH and immunofluorescence staining to examine the expression pattern of SRSF1 in KC mice in situ. Remarkably, SRSF1 expression was reduced in the population of morphologically normal pancreas cells with an active KrasG12D allele, compared with those harboring a dormant KrasG12D allele (Fig. 5B). Furthermore, we generated normal ductal organoids from LSL-KrasG12D/+; R26-LSL-YFP mice, and activated the repressed KRASG12D and YFP expression using adenovirus-Cre-mediated LSL recombination (Supplementary Fig. S8B). We confirmed the recombination of the LSL cassette by YFP expression and genomic PCR of the LSL-KrasG12D allele (Fig. 5C; Supplementary Fig. S8C). Consistently with our in vivo data, we observed a significant decrease in SRSF1 expression upon acute activation of KrasG12D (Fig. 5C). Remarkably, SRSF1 knockdown suppressed MAPK signaling in mouse PDAC organoids (Supplementary Fig. S8D and S8E). Considering that its elevated expression in PDAC precursor lesions and tumors is associated with tumorigenesis, our observation that SRSF1 decreases in morphologically normal cells with KRASG12D suggests that SRSF1 downregulation is part of the compensatory feedback to KRASG12D.
We next sought to understand the mechanism of SRSF1 downregulation upon acute KRASG12D expression. We did not detect significant changes in Srsf1 mRNA in LSL-KrasG12D/+; R26-LSL-YFP organoids infected with adenovirus-Cre (Supplementary Fig. S8F), suggesting that SRSF1 downregulation does not involve transcriptional or mRNA-decay mechanisms. The 3’ UTR of SRSF1 is ultraconserved and is necessary and sufficient for SRSF1 autoregulation by alternative splicing in HeLa cells (17). We detected the same six Srsf1 isoforms generated by alternative splicing of the 3’ UTR in murine Srsf1 mRNA from LSL-KrasG12D/+; R26-LSL-YFP mouse pancreas organoids (Supplementary Fig. S8G). Unlike isoform I, isoforms II-VI are either retained in the nucleus or degraded by nonsense-mediated mRNA decay (17). Interestingly, we found that isoform I—the canonical Srsf1 transcript—was upregulated upon KRASG12D expression, suggesting that the splicing alterations in the 3’ UTR occur to compensate for the decrease in SRSF1, rather than being the cause of the decrease (Supplementary Fig. S8H).
During T-cell activation, there is a reduction in SRSF1 protein, which is associated with increased SRSF1 ubiquitination and degradation, even though there is an increase in SRSF1 mRNA (22). We therefore measured SRSF1 protein stability in mouse pancreas organoids by cycloheximide-chase analysis (38). KRASG12D expression strongly promoted SRSF1 protein degradation (t1/2 ~1h) (Fig. 5D). To determine the SRSF1 ubiquitination state, we conducted immunoprecipitations from LSL-KrasG12D/+; R26-LSL-YFP organoids expressing HA-Ubiquitin, and detected increased SRSF1 ubiquitination upon KRASG12D expression (Fig. 5E). These results indicate that the compensatory feedback to KRASG12D decreases SRSF1 by promoting its ubiquitination and degradation.
Hyperactive MYC Upregulates SRSF1 in PDAC
Given the high level of SRSF1 in PDAC precursors and tumors, we sought to understand the mechanism by which SRSF1 becomes upregulated, escaping the feedback regulation by KRASG12D. Our previous work (with lung-cancer cell lines) demonstrated that SRSF1 is a critical transcriptional target of MYC (18,39), which is often overexpressed in PDAC (40,41). We found a significant positive correlation between MYC and SRSF1 expression in PDAC tumors (Supplementary Fig. S9A and S9B). We obtained further evidence of SRSF1 as a transcriptional target of MYC in the pancreas, by reanalyzing RNA-seq data from a mouse model with acute activation of a quasi-physiologic level of MYC in this organ (42). This analysis showed increased SRSF1 mRNA upon MYC activation (Supplementary Fig. S9C). Intriguingly, Gene Ontology analysis revealed that acute MYC activation results in negative enrichment of genes related to protein ubiquitination (Supplementary Fig. S9D), implying that MYC potentially further contributes to SRSF1 protein elevation by attenuating KRASG12D-induced SRSF1 degradation. Accordingly, ectopic expression of MYC increased SRSF1 in HPDE cells, and in human and mouse normal organoids (Supplementary Fig. S9E). These data suggest that hyperactive MYC in PDAC upregulates SRSF1 during tumorigenesis.
SRSF1-Regulated IL1R1 Alternative Splicing Contributes to Pancreatitis and Transformation
Considering that the SRSF1 expression level is strongly associated with the cellular homeostatic response to KRASG12D and with PDAC initiation, we next sought to identify direct splicing targets of SRSF1 associated with MAPK signaling, and involved in the compensatory feedback to KRASG12D mutation. We found that multiple MAPK-signaling-pathway genes, such as Il1r1, Axl, Sgk1, and Ets1, were both upregulated upon increased SRSF1 expression and downregulated in response to KRASG12D (Fig. 6A and B; Supplementary Fig. S5C). Interestingly, interleukin 1 receptor type 1 (Il1r1)—which encodes a cytokine receptor for IL1α, IL1β, and IL1RA—was identified as the top SRSF1-regulated splicing target associated with the MAPK signaling pathway (Fig. 6C). SRSF1 consistently promoted inclusion of exon 3 in the 5’UTR of Il1r1 mRNA in SC and KSC organoids (Fig. 6D), as well as in the conserved human region in HPDE cells and hN40 organoids with Dox-inducible SRSF1 (Fig. 6E; Supplementary Fig. S10A). Furthermore, an Il1r1 minigene co-transfection assay showed enhanced inclusion of exon 3 upon overexpression of SRSF1 (Fig. 6F and G). To test the dependence of exon 3 inclusion on SRSF1 binding to Il1r1 pre-mRNA, we mutated two SRSF1 motifs in exon 3 (Fig. 6F). Mutating the motifs individually or simultaneously suppressed SRSF1-induced exon 3 inclusion (Fig. 6G), consistent with SRSF1 directly binding to exonic splicing enhancers in Il1r1 pre-mRNA to promote exon 3 inclusion.
Figure 6.
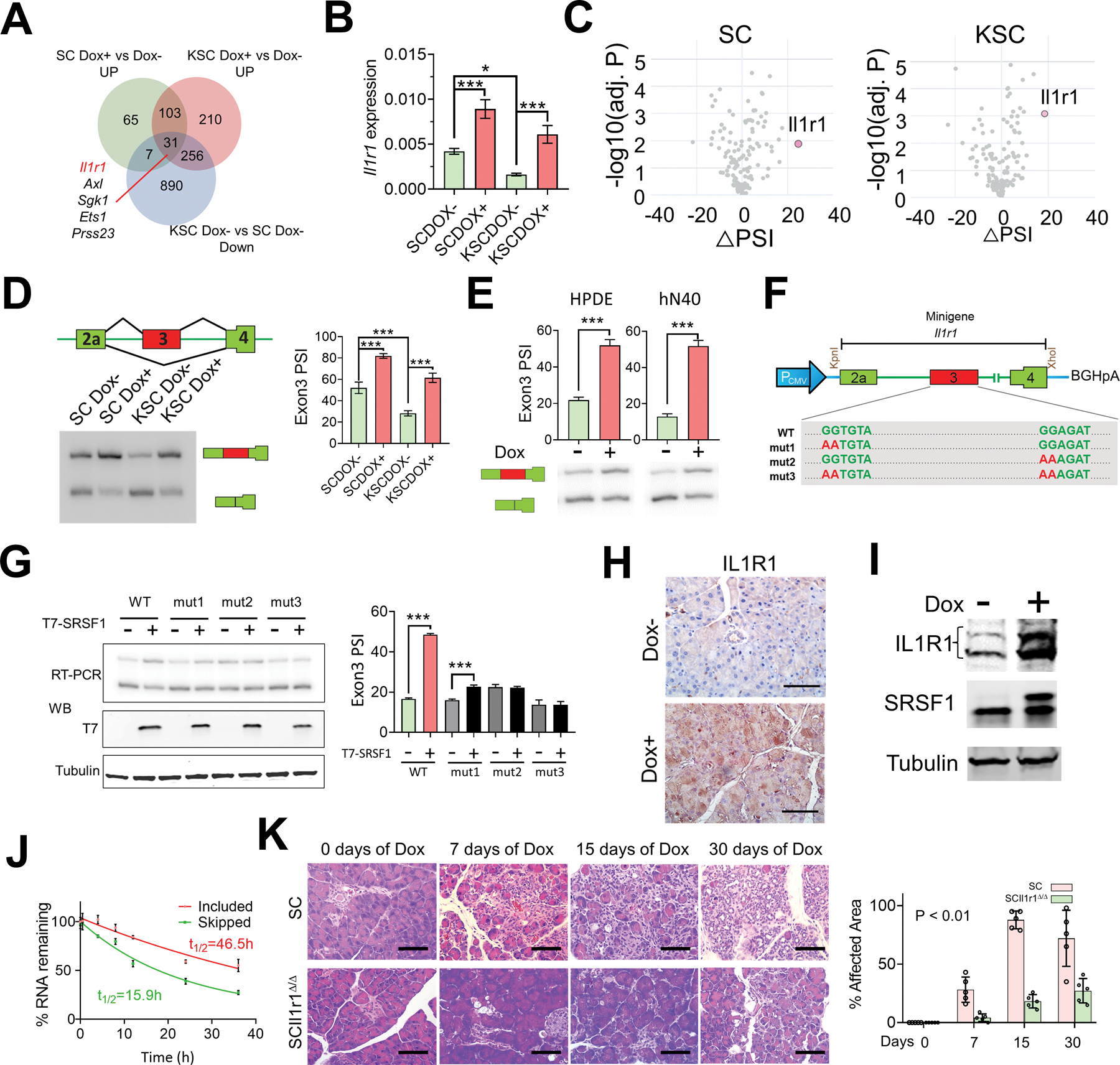
SRSF1 increases IL1R1 expression through alternative splicing, contributing to pancreatitis and transformation. A, Venn diagram of genes upregulated in SC and KSC organoids treated with Dox versus untreated; and genes downregulated in KSC organoids compared to SC organoids, both without Dox treatment. B, Real-time PCR validation of Il1r1 expression changes in SC and KSC organoids treated with Dox (n = 3 biological replicates). One-way ANOVA with Tukey’s multiple comparison test. *p <0.05, ***p <0.001. Error bars represent mean ± SD. C, Volcano plots of splicing changes in MAPK signaling pathway-associated genes from SC and KSC organoids treated with Dox. D, Scheme and radioactive RT–PCR of SRSF1-regulated splicing event in Il1r1 pre-mRNA from SC and KSC organoids treated with Dox. The percent spliced in (PSI) was quantified for each condition (right, n = 3 biological replicates). One-way ANOVA with Tukey’s multiple comparison test. ***p <0.001. Error bars represent mean ± SD. E, Radioactive RT–PCR of SRSF1-regulated splicing event in Il1r1 pre-mRNA from Dox-inducible T7-SRSF1-expressing HPDE cells and human normal pancreatic hN40 organoids treated with Dox. The percent spliced in (PSI) was quantified for each condition (top panel, n = 3 biological replicates). Unpaired two-tailed t test. ***p <0.001. Error bars represent mean ± SD. F, Diagram of Il1r1 minigene. Mutations were introduced to disrupt SRSF1 binding motif(s). G, Radioactive RT-PCR results of Il1r1 minigene assays (top left), and Western blotting of T7-tag (bottom left) in 293T cells overexpressing T7-SRSF1. The percentage of exon 3 inclusion was quantified for each condition (right, n = 3 biological replicates). One-way ANOVA with Tukey’s multiple comparison test. ***p <0.001. Error bars represent mean ± SD. H, IHC staining of IL1R1 in pancreas tissues from SC mice with Dox treatment for 3 days (n = 5 per group). Scale bars, 50 μm. I, Western blotting of IL1R1, endogenous and T7-tagged SRSF1 in SC organoids treated with Dox. J, mRNA-decay assay of the Il1r1 isoforms in SC organoids harvested at the indicated times after actinomycin D treatment. mRNAs were quantified by real-time PCR, normalized to Gapdh levels, and expressed as a percentage of the levels at time 0 h (n = 3 biological replicates). K, HE staining of pancreata of SC and SCIl1r1Δ/Δ mice with Dox treatment (n = 5 per group). Scale bars, 50 μm. The affected area was quantified on the right. Statistical analysis was performed by a linear mixed-effects model with the experimental group, time, and group by time interaction as fixed effects, and the sample-specific random intercept was used to estimate the % affected area.
Activation of IL1R1 leads to the production of inflammatory mediators and the regulation of biological responses that include tissue vascularity, adipogenesis, lipid metabolism, and inflammation (12,43–45). Conversely, suppression of IL1R1 with Anakinra, a recombinant form of the endogenous antagonist IL1RA, attenuates the level of pERK and suppresses PDAC tumor growth (11). We found elevated IL1R1 within pancreatitis lesions, as well as in the PanIN lesions in KC mice (Supplementary Fig. S10B), consistent with the reported association of IL1R1 with PDAC progression (46). IL1R1 protein also increased in response to SRSF1 induction in mouse pancreas tissue and organoids (Fig. 6H and I; Supplementary Fig. S10B), as well as in the human HPDE and hN40 lines (Supplementary Fig. S10C). Moreover, we analyzed a clinical data repository—PanCuRx (47)—and found that PDAC tumors with higher SRSF1 expression exhibited higher IL1R1 mRNA levels (Supplementary Fig. S10D).
The conserved regulation of IL1R1 pre-mRNA splicing and expression by SRSF1 in human and mouse prompted us to further investigate the relationship between alternative splicing and expression of IL1R1. We measured the decay rate of the two Il1r1 mRNA isoforms in SC organoids, following transcription inhibition by actinomycin D treatment, and found that the mRNA isoform that includes exon 3 was more stable (Fig. 6J), indicating that SRSF1 promotes IL1R1 expression through alternative splicing in the 5’UTR that enhances mRNA stability.
Finally, given the importance of IL1R1 in inflammation, we assessed the contribution of IL1R1 to SRSF1-mediated pancreatitis and transformation. We found that Il1r1 knockout in SC mice suppressed SRSF1-induced pancreatitis and ADM (Fig. 6K; Supplementary Fig. S11A). Additionally, Il1r1 deficiency reduced immune infiltration to the pancreas (Supplementary Fig. S11B and S11C). These results demonstrate that the upregulation of IL1R1 by SRSF1 greatly contributes to SRSF1-induced pancreatitis and transformation.
DISCUSSION
Patients with pancreatitis have a higher risk of developing PDAC (8,28). A substantial body of evidence obtained with multiple experimental models supports the idea that inflammation in pancreatic tissues hastens KRASG12D-mediated formation of high-grade PanINs and progression to PDAC (48–50). Precise regulation of pre-mRNA alternative splicing is critical for maintaining cellular identity, and for tissue and organ development; furthermore, this process is often altered in ways that can contribute to tumorigenesis (24,51–55). However, the contribution of aberrant RNA splicing to pancreatitis and to PDAC had not been extensively studied. We report that expression of the highly-conserved proto-oncogenic splicing factor SRSF1 is increased in pancreatitis. Moderate SRSF1 overexpression in our mouse model—which is constrained by conserved auto-regulation via multiple mechanisms (17,18,21)—mimicking the pathological levels seen in pancreatitis and PDAC tumors, stimulated inflammatory-cell infiltration through multiple signaling pathways to promote pancreatitis and ADM.
Related to inflammatory signaling, we showed that SRSF1 regulates alternative splicing in the 5’UTR of Il1r1 pre-mRNA to generate a more stable mRNA isoform, thus resulting in accumulated IL1R1 protein. IL1R1 is a key receptor for IL1α/β, and is competitively blocked by the endogenous antagonist IL1RA. Accumulated IL1R1 therefore may facilitate the binding of IL1α/β secreted by epithelial cells and tumor stromal cells, which can further confer positive feedback activation of IL1 signaling (12,56). In addition to the prominent role of IL1 signaling in tumor stromal-driven inflammation, our results confirm and extend the importance of IL1R1 in the PDAC precursor and tumor cells during inflammation and progression (13,57). Of note, the recombinant IL1R1 antagonist Anakinra is being evaluated in PDAC clinical trials, in combination with standard chemotherapy (NCT02021422, NCT02550327, NCT04926467). Our observations suggest that SRSF1-regulated splicing changes, including IL1R1, could serve as potential markers and/or therapeutic targets for pancreatitis and PDAC.
Accumulated somatic mutations have been identified in normal tissues; e.g., up to 19% of adult bladder epithelial cells represent driver mutations (58–61). Though some of these mutations affect well-known oncogenes, they do not lead to cell transformation or malignancy, or do so with long latency. KRAS mutation is an oncogenic driver in nearly all PDAC tumors, yet the majority of pancreas cells expressing KRASG12D in two-month old KC mice remain morphologically normal (3), suggesting that normal pancreas cells are refractory to KRASG12D-initiated cell transformation, and that additional oncogenic alterations are required for further PDAC initiation.
Pancreatic acinar cells exhibit high plasticity and can undergo ADM—a transition from differentiated acinar cells to proliferative cells with ductal cell traits, which is an important feature that facilitates pancreas regeneration after injury. Multiple factors have been implicated in mediating ADM, including KRAS hyperactivation and inflammatory signaling (6). It is well established that KRASG12D promotes cellular transformation by activating downstream signaling pathways, including RAF/MEK/ERK and PI3K/AKT. Although both Raf/MEK/ERK and PI3K/AKT pathways are hyperactive in PDAC and mediate tumor progression, only the former is required for ADM (62). MAPK signaling regulates a wide variety of cellular processes—including proliferation, differentiation, and apoptosis—and hyperactivation of this signaling pathway is a crucial prerequisite for KRASG12D-mediated cell transformation and tumorigenesis (36,37). Conversely, multiple negative feedback loops constrain MAPK signaling to maintain cell homeostasis (33–35).
We found that SRSF1 alone is sufficient to increase IL1R1 expression and activate MAPK signaling in ductal organoids, consistent with an intrinsic process. Furthermore, both SRSF1-induced MAPK signaling activation and pancreatitis were relieved by Il1r1 depletion. And treatment with the MEK inhibitor trametinib suppressed SRSF1-induced pancreatitis. These results indicate that SRSF1 causes pancreatitis specifically depending on MAPK signaling, although we cannot rule out a possible non-specific contribution from acinar-cell damage.
Our data demonstrate that SRSF1 is a key regulator in the cellular-homeostasis response to KRASG12D, and in KRASG12D-mediated PDAC tumorigenesis. SRSF1 was decreased in normal pancreas cells expressing KRASG12D in vivo, and in an acute KRASG12D-expressing organoid model, whereas elevated SRSF1 disrupted cellular homeostasis to accelerate PDAC initiation and progression. Transcriptomic analysis of organoids with KRASG12D mutation, or elevated SRSF1 expression, or both, demonstrated that SRSF1 protein levels decrease as part of the KRASG12D-driven cellular homeostatic response to buffer MAPK signaling, and conversely, that increasing SRSF1 promotes MAPK signaling and accelerates PDAC initiation and progression.
Different susceptibilities to KRASG12D-initiated tumorigenesis have been reported for different epithelia (63). It will be of interest to determine if there is a similar involvement of SRSF1 in the homeostatic response to KRASG12D across other normal tissues, besides the pancreas, considering that SRSF1 has also been reported to be oncogenic in other contexts, such as sarcoma (26), breast cancer (64), and glioma (65).
Besides MAPK signaling, pathway-enrichment analysis identified additional pathways with opposite regulation in response to KRASG12D versus SRSF1. One example is cytokine-cytokine receptor interaction, which implies a regulatory function of the microenvironment in cell plasticity, homeostasis, and PDAC initiation. Intriguingly, several pathways, including PI3K-AKT signaling, were negatively enriched in the presence of the KRASG12D mutation, and positively enriched upon induced SRSF1 only when combined with KRASG12D, but not in the wild-type KRAS context. These data point to the critical role of SRSF1 in the cellular homeostatic response to KRASG12D through more complex signaling networks, and indicate that elevated SRSF1 and KRASG12D have synergistic effects in tumorigenesis, which warrant further study.
The nuclear and cytoplasmic functions of SRSF1 have been studied in multiple models and contexts: i) In contrast to the early embryonic lethality of Srsf1−/− knockout mice (66), Srsf1NRS/NRS embryos expressing nuclear-retained SRSF1 instead of shuttling SRSF1 protein are viable and survive postnatally, but exhibit ciliary defects due to defective translation of a subset of mRNAs (67); ii) In the context of breast cancer, we reported that SRSF1’s nuclear functions, particularly splicing, are sufficient for mammary epithelial cell transformation (25); iii) Abrogating the nucleo-cytoplasmic shuttling of SRSF1 prevents its oncogenic potential in immortal liver-progenitor cell transformation (68). Our data here indicate that nuclear-retained SRSF1 retained its major oncogenic properties in PDAC, whereas RRM1-deleted or RRM1-inactivated mutants failed to rescue PDAC proliferation, suggesting that SRSF1’s nuclear functions account for the oncogenic role of SRSF1 in the PDAC context, which requires the RNA binding function of RRM1. These dissimilar observations in various systems are indicative of context-specific oncogenic roles of SRSF1.
Previous studies, including our own, indicated that SRSF1 expression is regulated via complex transcriptional, post-transcriptional, translational, and protein-degradation mechanisms (17–22). Here, we showed that acute expression of KRASG12D does not affect Srsf1 mRNA expression (transcriptional or mRNA-stability mechanisms). Our data showed the conservation of complex alternative splicing patterns and regulation in the SRSF1/Srsf1 3’UTR between mouse and human, consistent with the presence of an ultraconserved region (17). We found that Srsf1 alternative splicing favors the expression of the canonical protein-coding mRNA isoform upon KRASG12D expression, which we attribute to a feedback loop in response to the decrease in SRSF1 protein elicited by KRASG12D expression. Several lines of evidence have implicated post-translational regulation of splicing factor proteins contributing to their protein levels, decoupled from their transcriptional regulation (22,69). For example, T-cell activation is accompanied by a rapid increase in SRSF1 mRNA; conversely, the SRSF1 protein level decreases, mainly as a result of enhanced ubiquitin-proteasome degradation (22). Similarly, we found here that acute KRASG12D activation in LSL-KrasG12D/+; R26-LSL-YFP organoids promotes SRSF1 degradation via increased ubiquitination, which may result from mutation-induced changes in KRAS protein function or in its proximal interactome (4,70,71).
We found that the expression of SRSF1 eventually overcomes the negative regulation by mutant KRAS to promote premalignant changes and tumorigenesis. Aberrantly persistent MYC activation has been implicated in the majority of cancers, including PDAC (40,41). We showed that MYC activation can consistently increase SRSF1 expression, in accordance with SRSF1 being a direct transcriptional target of MYC (18,39). On the other hand, negative enrichment of protein ubiquitination pathways upon MYC activation also points to MYC potentially serving as a cooperating oncogenic partner of KRASG12D during PDAC tumorigenesis, through suppression of SRSF1 ubiquitination, in addition to transcriptionally activating it. Moreover, our finding that SRSF1 promotes ADM and pancreatitis, and its involvement in cytokine-cytokine receptor interaction, is consistent with the notion that MYC activation is associated with metaplasia, driving progression of KRASG12D-induced PanINs to PDAC by programming inflammation (42,72). MYC can be upregulated by multiple mechanisms, such as epigenetic modifications (73) or genetic amplification (41). Further investigation of the precise mechanisms underlying the SRSF1 expression changes involved in the homeostatic response to KRASG12D, including when and how MYC is hyperactivated to overcome this homeostatic response and to accelerate tumorigenesis, should facilitate PDAC prevention and therapeutic intervention.
In summary, SRSF1 expression is tightly regulated and is associated with pancreas-cell identity, plasticity, and inflammation. SRSF1 protein downregulation is involved in a negative-feedback cellular response to KRASG12D mutation, contributing to pancreas-cell homeostasis. Conversely, upregulated SRSF1 promotes pancreatitis and accelerates KRASG12D-mediated tumorigenesis through enhanced IL-1 and MAPK signaling (Fig. 7).
Figure 7.
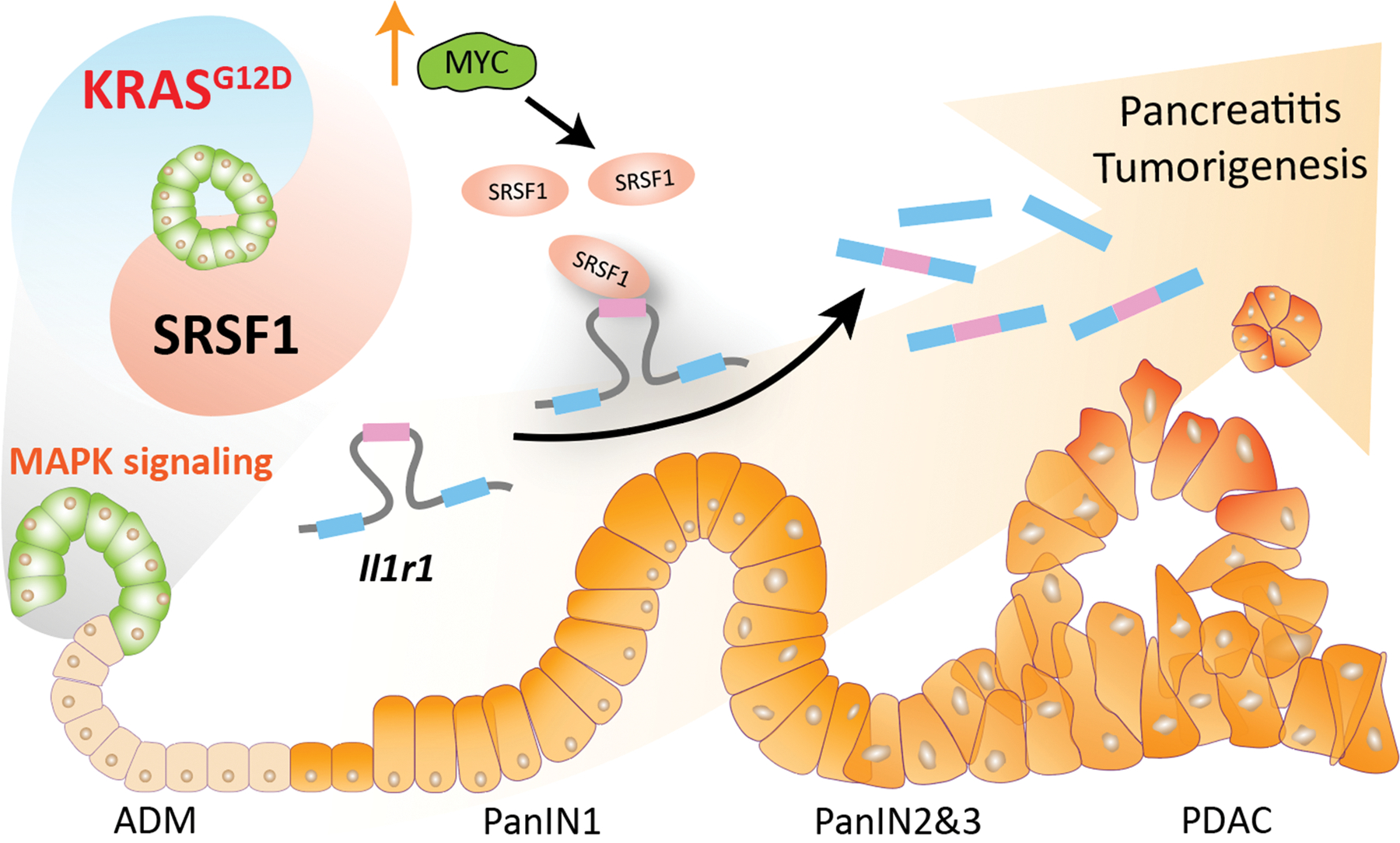
SRSF1 promotes pancreatitis and KRASG12D-mediated pancreatic cancer, and its downregulation is involved in negative feedback to KRASG12D to maintain cellular homeostasis. Splicing factor SRSF1 downregulation is a negative-feedback cellular response to KRASG12D mutation, which dampens MAPK signaling activity and contributes to pancreas-cell homeostasis. Conversely, increased SRSF1 enhances MAPK signaling through alternative splicing of Il1r1 pre-mRNA, promoting pancreatitis and ADM, thus accelerating KRASG12D-mediated tumorigenesis. One mechanism of SRSF1 upregulation is transcriptional activation by MYC, which can be amplified or epigenetically activated (↑).
METHODS
Analysis of Public Datasets
SRSF1 mRNA expression levels and relevant clinical information were retrieved from GEO DataSets, TCGA, and GTEx databases. The comparison between pancreatic tumor and normal tissue was performed with datasets GSE16515 (PAAD) and GSE28735 (PDAC) and GEPIA(74) based on TCGA and GTEx. The association of SRSF1 mRNA and protein levels with clinical prognosis was determined using GEPIA, and the http://survival.cshl.edu/ web portal(75), respectively. The correlation between SRSF1 and MYC in PDAC was analyzed using the PanCuRx dataset from the Ontario Institute for Cancer Research, funded by the Government of Ontario.
Statistics
Statistical tests were performed using GraphPad Prism software and R. Statistics are reported for each experiment in their corresponding figure legends. All graphs plot individual data points to indicate the n values for each treatment and genotype, with the horizontal lines representing the mean, and the error bars representing the standard deviation. p values are reported as * p<0.05, ** p<0.01, *** p<0.001.
Mice
All animal protocols were approved by the IACUC at Cold Spring Harbor Laboratory. KC (LSLKrasG12D/+; Pdx1-Cre) and KPC (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre) mice were described previously (3,5). The IL1R1 (028398) and SRSF1 (018020) conditional knockout strain was obtained from The Jackson Laboratory. A Dox-responsive element controlling SRSF1 expression was targeted downstream of the Cola1 locus by Flp/FRT recombinase-mediated site-specific integration in KH2 ES cells, as described (76). Injection of targeted ES cells into tetraploid blastocysts to produce fully ES-cell-derived transgenic mice was performed by the CSHL Gene Targeting Shared Resource. The offspring were further crossed with Rosa-LSL-rtTA3-IRES-mKATE and Pdx1-Cre strains to obtain SC mice (28,77). KSC and KPSC mice were generated by crossing SC mice with the LSL-KrasG12D/+ strain and the LSL-KrasG12D/+; LSL-Trp53R172H/+ strain, respectively. Mice at 6 weeks of age were enrolled in the studies. Dox chow was administered to deliver a daily dose of 2–3 mg of doxycycline, based on the consumption of 4–5 g/day (Harlan Teklad, TD.120769, Dox 625mg). For MEK inhibitor administration, Trametinib (S2673, Selleck Chemicals) was administered by oral gavage, at 2 mg/kg. For cerulein-induced pancreatitis, C57Bl/6J mice were injected with 100 μL cerulein (Sigma-Aldrich, St. Louis, MO, USA) at 50 μg/mL in PBS hourly for 8 h on 2 consecutive days; pancreata were harvested 48 h after the last injection.
Immunohistochemistry (IHC) and Immunofluorescence (IF)
Human Tissue microarrays were obtained from US Biomax (PA483e, PA1002b, BIC14011b). Mouse tissue samples were fixed in 4% PFA/PBS (EMS, Hatfield, PA, USA) and embedded in paraffin. Six-μm sections were deparaffinized and rehydrated. Endogenous peroxidase was blocked in H2O2/methanol for 15 min. Slides were then boiled in 10 mM sodium citrate, 0.05% Tween 20 for 5 min under pressure (for protein antigens). For IF studies, OCT-embedded sections were air-dried and fixed for 30 min in 4% paraformaldehyde. Slides were blocked for 30 min with peptide blocking solution (Innovex Biosciences, Richmond, CA, USA) and incubated with the indicated primary antibodies overnight at 4 °C. For IHC analysis, the signal was visualized with horseradish peroxidase (HRP)-labeled anti-rabbit polymer and DAB (DAKO, Carpinteria, CA, USA). Slides were counterstained with hematoxylin (Sigma-Aldrich) and mounted with Limonene mounting medium (Abcam, Cambridge, UK). For IF analysis, primary antibodies were visualized using species-appropriate cross-adsorbed secondary antibody Alexa Fluor 488 or 555 conjugates (Thermo Fisher Scientific); slides were mounted in Prolong Gold Antifade Mountant (Thermo Fisher Scientific).
SA–β-gal Assay
10-μm frozen sections of pancreatic tissue were fixed with 2% formaldehyde/0.2% glutaraldehyde in PBS for 3 to 5 minutes, washed with PBS, and stained at 37 °C for 12 to 16 h in X-Gal solution (1 mg/ml X-Gal, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, and 1 mM MgCl2 in PBS at pH 6.0), as described in the manufacturer’s protocol (Senescence β-Galactosidase Staining Kit, Cell Signaling). Sections were counterstained with Nuclear Fast Red (Sigma), dehydrated, and mounted with xylene-based mounting solution.
RNA FISH
RNA FISH was performed in formalin-fixed tissue sections, according to the manufacturer’s protocols for manual RNAscope® Multiplex Fluorescent Reagent Kit v2- Mm (Advanced Cell Diagnostics Inc., 323136). 4% paraformaldehyde-fixed pancreatic-tissue frozen sections were incubated at 60 °C for 1 h, followed by dehydration in ethanol. Heat-induced epitope retrieval was performed for 5 min at 100 °C using RNAscope® Target retrieval (Advanced Cell Diagnostics Inc, Hayward, CA-322000). Samples were incubated with RNAscope® protease III (Advanced Cell Diagnostics Inc, Hayward, CA-322330) at 40 °C and washed with milliQ water. Samples were incubated with their respective probe for 2 h at 40 °C and washed for 2 min in RNAscope® wash buffer (Advanced Cell Diagnostics Inc, Hayward, CA-310099. After amplification, samples were incubated with HRP probe. Signal detection was performed by incubating samples with Opal™ 620 Reagent Pack (Akoya, FP1495001KT) for 30 min. For FISH/IF multiplexing, IF was performed as described above. Slides were mounted in Prolong Gold Antifade Mountant. RNAScope probes 837571 and 561731 were used for probing the LSL-KrasG12D sense and antisense STOP cassette, respectively.
Cell and Organoid Culture
The HPDE cell line was from Kerafast, MA, USA, and was cultured in Keratinocyte SFM, + EGF + bovine pituitary extract (Invitrogen, MA, USA) following the provided culturing protocol. The human pancreatic cancer cell line SUIT-2 obtained from ATCC was propagated in 10% FBS, P/S in RPMI (GIBCO, MA, USA). Cells were authenticated by short tandem repeat (STR) profiling by University of Arizona Genetics Core (UAGC). Human pancreatic organoid lines were generated in the Tuveson laboratory (78) and cultured with assistance from the Organoids Shared Resource at Cold Spring Harbor Laboratory. Authentication of human organoids was performed by DNA sequencing. Mice were genotyped by TransnetX before organoids were generated as described (79). Briefly, the pancreas was harvested, followed by enzymatic digestion with 0.012% collagenase XI (Sigma-Aldrich) and 0.12% dispase (GIBCO) in DMEM containing 1% FBS (GIBCO). Ducts were collected and embedded in 100% growth-factor-reduced (GFR) Matrigel (Corning, AZ, USA). Organoids were passaged less than 10 times. All organoid and cell lines were routinely tested for Mycoplasma with MycoAlert Mycoplasma Detection Kit (LT07–318, Lonza).
Whole-Mount IF of Organoid Cultures
Organoids were fixed with 4% paraformaldehyde at 4 °C for 2 h. After washing with IF buffer (0.1% BSA, 0.05% NaN3, 0.2% Triton-X, 0.05% Tween-20 in PBS, pH 7.4) three times for 5 min each, samples were incubated in blocking buffer (10% goat serum in IF buffer) at room temperature for 1 h. Samples were then incubated with primary antibodies at 4 °C overnight, followed by washing with IF buffer three times at room temperature for 5 min each. Samples were then incubated with secondary antibodies diluted in blocking buffer at room temperature for 1 h. Slides were counterstained with DAPI and mounted in Prolong Gold Antifade Mountant.
Western Blotting
Organoids were harvested with ice-cold Cell Recovery Solution (Corning) containing EDTA-free protease inhibitor cocktail (Sigma-Aldrich) and PhosSTOP (Sigma-Aldrich), followed with 10-min incubation on ice. Following centrifugation, the supernatant was discarded and the pellet was washed once with Cell Recovery Solution. For protein stability and half-life analysis, organoids were treated with 10 μg/ml cycloheximide (Sigma-Aldrich) for the indicated time points The pellet was lysed with RIPA buffer supplemented with protease and phosphatase inhibitor cocktail on ice for 30 min, clarified at 15,000 RCF for 15 min at 4 °C, and stored at −20 °C. Protein lysates were separated on an 8–16% precast SDS polyacrylamide gel (Bio-Rad, CA, USA), transferred onto a nitrocellulose membrane (Millipore, MA, USA), and blocked in 5% (w/v) BSA in Tween 20-TBST. Blots were incubated with primary antibodies overnight at 4 °C, followed by incubation with IR-Dye secondary antibodies (Licor, AL, USA). Bands were detected and quantified with an Odyssey Imaging System (Licor).
Detection of Active RAS Small-GTPases
Pancreas tissues were lysed and processed using the immunoprecipitation kits for detection of active RAS (Cell signaling) following the manufacturer’s instructions. Levels of active RAS small-GTPases were determined by western blotting after IP.
Flow Cytometry
Pancreata were harvested and separated from attached lymph nodes. Following mechanical dissociation, pancreas tissues were digested with 0.4 mg/mL liberase, 0.05 mg/mL collagenase D, and 0.1 mg/mL DNase I (Sigma-Aldrich) in DMEM (GIBCO) supplemented with 10% FBS at 37 °C for 15 min, and suspensions were passed through a nylon cell strainer. Cells were stained with antibodies in FACS buffer (Thermo Fisher Scientific) by incubating on a plate shaker for 15 min at 4 °C. Multicolor flow-cytometric analyses were performed using an LSR Fortessa cytometer (BD Biosciences, USA). Data files were analyzed using FlowJo v10 software (FlowJo, LLC, OR, USA).
Lentiviral Infection in Organoids
Lentivirus was produced in 293T cells by transfecting plasmids and packaging plasmids (pMD2 and psPAX2) with Lipofectamine™ 3000 transfection reagent. 293T cells were plated a day before transfection with 70%–80% confluency in 10-cm tissue-culture plates. The medium was changed with 9.6 ml of antibiotic-free DMEM supplemented with 10% FBS. 10 μg of plasmid DNA and Lipofectamine 2000 reagent (Thermo Fisher Scientific) were mixed in 400 μl of DMEM, incubated for 15 min at room temperature, and added to the cells. After 24 hr, the medium was replaced with fresh DMEM supplemented with 10% FBS. Retrovirus-containing supernatant was harvested at 48 hr and 72 hr after transfection and filtered through a 0.45 μm filter. Lentivirus was concentrated with PEG-it™ Virus Precipitation Solution (SBI, CA, USA), and the virus pellet was resuspended with organoid culture medium supplemented with Y-27632 (10 mM, Sigma-Aldrich). Organoid infections were performed as described (79). In brief, 5×104 single cells were resuspended with concentrated lentivirus and spinoculated at 600 RCF for 1 h at room temperature. Two days after infection, cells were treated with puromycin (2 μg/mL, Sigma-Aldrich) for selection.
Proliferation Assay
Organoids were dissociated into single cells by first triturating them in culture medium through a fire-polished glass pipette, and then by enzymatic dissociation with 2 mg/mL dispase dissolved in TrypLE (Life Technologies), until the organoids appeared as single cells under the microscope. Cells were counted, and diluted to 10 cells/mL in a mixture of complete medium, Rho kinase inhibitor Y-27632 (10.5 mM final concentration, Sigma), and Growth factor-reduced Matrigel (GFR-Matrigel, 10% final concentration). 100 mL of this mixture (1000 cells per well) was plated in 96-well plates (Nunc), whose wells had been previously coated with a bed of GFR-Matrigel to prevent attachment of the cells to the bottom of the plate. Cell viability was measured every 24 h using the CellTiter-Glo assay (Promega) and a SpectraMax I3 microplate reader (Molecular Devices).
Adenoviral Infection
Ductal organoids were prepared from LSL-KRASG12D/+; R26LSL-YFP mice and propagated in GFR-Matrigel (Corning) for at least 3 passages before infection with 500 pfu/cell of AdCMV-Cre or AdCMV-EV (University of Iowa Gene Transfer Vector Core Facility). Spinoculation was performed as described above.
Antisense Oligonucleotide Transfections
ASOs (uniformly modified with MOE sugars, phosphorothioate backbone, and 5-methyl cytosine) were purchased from Integrated DNA Technologies (IDT, IA, USA). Organoids were harvested and resuspended in DMEM containing ASO transfection mixture prepared with Lipofectamine 2000 (Thermo Fisher Scientific). Organoids were embedded in Matrigel after 4 h incubation at 37 °C. Organoids were harvested after 72 h for RT-PCR and western blotting analysis.
In Vivo Ubiquitination Assay
Stable HA-ubiquitin expressing LSL-KRASG12D/+; R26LSL-YFP organoids were infected with AdCMV-Cre or AdCMV-EV, respectively, as described above. Organoids were harvested and lysed in IP lysis buffer (25 mM Tris-HCl pH 7.5, 200 mM NaCl, 0.5% NP-40, 2 mM MgCl2, 2 mM EDTA, 100 mM NaF, and one tablet of EDTA-free protease inhibitor cocktail (Sigma-Aldrich) per 50 mL) supplemented with 25 U/mL Benzonase (Sigma-Aldrich) for 10 min on ice. Lysates were cleared by centrifugation, quantified and normalized with the Bradford assay, and incubated (3 h, 4 °C) with anti-HA coupled beads (ThermoFisher Scientific).
Subcutaneous Transplantation Model
For subcutaneous injections, 106 cells were suspended in 100 μl PBS and injected into each flank of BALB/c nu/nu nude mice (male, 6 weeks of age). Tumor growth was measured at 3-day intervals after injection using a caliper, and tumor volume was calculated according to the following formula: length × width2 × 0.5. Harvested tumor tissues were fixed in 10% buffered formalin, embedded in paraffin, sectioned, and stained.
Soft Agar Colony Formation
SUIT-2 cells in suspension (103 cells/well) were incubated in an upper layer of 0.3% agarose (ThermoFisher Scientific) in DMEM with 10% FBS. The plate was placed at room temperature until the top agarose solidified. The plate was then incubated at 37 °C/5% CO2 for at least 3 weeks, before staining with crystal violet. Visible colonies were then counted.
Serologic Measurements
Mouse whole blood was harvested by cardiac puncture and collected in SST-MINI Gold Top Tubes. The tubes were gently inverted for 1 min and kept for 15 min at room temperature to let the samples clot. The samples were then centrifuged at 2500 rpm for 15 min, and the serum was collected and stored at −20 °C. Amylase and lipase levels in serum were measured by IDEXX Pathology Services (ME, USA).
mRNA Decay Rate Assay
Mouse organoids were suspended in Matrigel and cultured as described (80). After 12 h, actinomycin D was added at a final concentration of 5 μg/ml. Cells were harvested at different time points, as described (81). The expression of isoforms was measured with specific primers by real-time PCR.
Primary Antibodies
CD45 (103138, clone 30-F11), CD3 (100206, clone 17A2), CD4 (100411, clone GK1.5), CD8 (100714, clone 53.6.7), CD19 (115520, clone 6D5), NKp46 (137610, clone 29A1.4), CD11b (101212, clone M1/70), Ly6G (127624, clone 1A8), CD11c (117318, clone N418), F4/80 (123128, clone BM8) were purchased from Biolegend for flow cytometry. SRSF1 (ab129108; Abcam), GFP (2956T; Cell Signaling Technology), CK19 (TROMA III, DSHB), phospho-ERK1/2 (4370, Cell Signaling), ERK1/2 (4695, Cell Signaling), GFP (2956, Cell Signaling), mIL1R1 (AF771-SP, Novus Biologicals), hIL1R1 (ab106278; Abcam), CD45 (70257, Cell Signaling), Cleaved Caspase-3 (9661, Cell Signaling), F4/80 (70076, Cell Signaling), P53 (NCL-L-p53-CM5p, Leica Biosystems) were used as primary antibodies for western blotting, immunohistochemistry, and immunofluorescence.
RT-PCR
Total RNAs were isolated using Trizol (Invitrogen). Then RT-PCR was performed using PrimeScript™ RT Reagent Kit (Takara, CA, USA) according to the manufacturer’s manual. Radioactive PCR was conducted using 32P-α-dCTP, 1.25 U of AmpliTaq (Invitrogen), and 26 cycles. Products were separated by 5% PAGE, and the bands were detected using a Typhoon FLA 7000 (GE Healthcare, IL, USA). Quantitative RT-PCR was performed on a QuantStudio 6-flex Real-time PCR instrument (Applied Biosystems, MA, USA). PCR analysis was performed with gene-specific primers (Supplementary Table S1). Expression levels were normalized to Gapdh.
Plasmids
The minigene for SRSF1-regulated splicing events in Il1r1 was constructed by insertion of intact exons 2a, 3, 4, intron 2, and truncated intron 3 (150 bp from each end) of murine Il1r1 in pcDNA 3.1(+) after KpnI and XhoI digestion. Mutations were introduced to disrupt SRSF1 binding motif(s). The percent spliced in (PSI) was quantified by radioactive RT-PCR using minigene-specific primers, T7 Universal primer, and bGH_rev primer (Table S1). Dox-inducible SRSF1 expressing plasmid was constructed by introducing T7-tag fused SRSF1 cDNA into pCW57.1 vector. pCW57.1 (41393), HA-Ubiquitin (74218), and pCDH-puro-MYC (46970) plasmids were purchased from Addgene.
Transwell Migration Assays
Cell migration was measured on Transwell chamber plates (24-well format; 8-μm pore size; Corning Costar, New York, USA) as described(82). Images of cells on the lower face of the filter were captured in fields at 10× magnification. To quantify the cells that had migrated across the filters, we used ImageJ (National Institutes of Health, USA) to measure the cell area on the lower face of the filter.
Gene Expression Analysis
RNA was extracted from organoid cultures using Trizol as described above. RNA libraries were generated using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB), following the manufacturer’s instructions. The libraries were sequenced on an Illumina NextSeq 2000 (150 bp, paired-end). Raw Illumina sequencing reads were aligned to genome assembly GRCm38 using STAR 2.5.3a. Transcript per million (TPM) values were quantified by using RSEM v1.3.0 with parameters: ‘--bowtie2 --calc-pme --calc-ci --ci-memory 4000 --seed 123 --no-bam-output --paired-end’ (83). Gene annotations of mouse (Mus_musculus.GRCm38.100.gtf) and human SRSF1 (hSRSF1) were concatenated to build a RSEM index file so that hSRSF1 expression could be quantified. TPM values smaller than 1 were normalized to 1. Fold-change values were calculated based on the difference of log2 (TPM+1) values in a pair-wise comparison. Two-sample T-test was used to calculate nominal p-values, and the q-value package of R was used to estimate false discovery rate (84). Differentially expressed genes whose |log2 (TPM+1)| > 0.585 and FDR < 0.05 were considered significant. The Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 was used to identify enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for up- and down-regulated genes, respectively (85). KEGG pathways with EASE score < 0.1 and FDR < 0.05 were considered significant.
For analyzing RNA-seq reads from transgenic mouse strain with acute MYC activation in the pancreas (42), raw counts for each annotated gene were counted by featureCounts (86), and differential expression was evaluated with edgeR version 3.12. TMM (Trimmed mean of M-values (87)) was applied for normalizing fold change. Down-regulated genes were selected based on log2 (fold change) < 0 and FDR < 0.05. Gene ontology analysis was done by using DAVID. GO terms with EASE score < 0.1 and FDR < 0.05 were considered significant.
RNA Splicing Analysis
ΔPSI and FDR values of splicing changes were produced by using PSI-Sigma v1.9j with parameters: ‘ --gtf Mus_musculus.GRCm38.100.stringtie.sorted.gtf --nread 20 --type 1 --fmode 3 --irrange 5’ (88). StringTie v2.1.4 was used to generate the ‘Mus_musculus.GRCm38.100.stringtie.sorted.gtf’ file for building the splicing database of PSI-Sigma (89).
Supplementary Material
SIGNIFICANCE.
We describe the regulation of splicing factor SRSF1 expression in the context of pancreas-cell identity, plasticity, and inflammation. SRSF1 protein downregulation is involved in a negative-feedback cellular response to KRASG12D expression, contributing to pancreas-cell homeostasis. Conversely, upregulated SRSF1 promotes pancreatitis and accelerates KRASG12D-mediated tumorigenesis through enhanced IL-1 and MAPK signaling.
Acknowledgments
We gratefully acknowledge the assistance of the Cold Spring Harbor Laboratory (CSHL) Animal and Genetic Engineering, Organoids, Animal and Tissue Imaging, Microscopy, Flow Cytometry, Antibody, Next-Generation Sequencing, and Histology Shared Resources. We thank R. Jaenisch (Whitehead Institute) for KH2 ES cells; T. Ha (CSHL) for biostatistics consulting; and R. Karni (Hebrew University of Jerusalem), and H. H. Zhang and J. L. Tang (Zhejiang University) for helpful discussions. The TROMA III (CK19) antibody was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa. Access to the PanCuRx dataset was authorized by the Ontario Institute for Cancer Research, supported by the Government of Ontario. Funding: NCI Program Project Grant CA13106 Project 2 to A.R.K.; St. Giles Foundation support to A.R.K. CSHL Shared Resources used in this work were funded in part by NCI Cancer Center Support Grant 5P30CA045508; sequencing data analysis was performed with a high-performance computing cluster, supported by NIH grant S10OD028632-01; D.A.T. was supported by NIH grant R01 CA249002 and by the Lustgarten Foundation; Y.P. was supported by NCI grant R50 CA211506.
Disclosure of Potential Conflicts of Interest
ARK is a co-founder, Director, Chair of the SAB, and shareholder of Stoke Therapeutics. ARK is on the SABs and holds shares of Skyhawk Therapeutics, Envisagenics, Assembl.cns, and Autoimmunity Biologic Solutions, and is a consultant for Biogen. DAT is on the SAB and holds shares with Leap Therapeutics, Surface Oncology, Sonata, and Mestag Therapeutics. DAT is a scientific co-founder of Mestag Therapeutics. DAT receives research support from Mestag Therapeutics and ONO therapeutics.
Data Availability
RNA-seq data are available at the Sequence Read Archive of NCBI under the BioProject ID: PRJNA747810.
REFERENCES
- 1.Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. The Lancet 2020;395:2008–20. [DOI] [PubMed] [Google Scholar]
- 2.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012;72:2457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50. [DOI] [PubMed] [Google Scholar]
- 4.Cheng DK, Oni TE, Thalappillil JS, Park Y, Ting H-C, Alagesan B, et al. Oncogenic KRAS engages an RSK1/NF1 pathway to inhibit wild-type RAS signaling in pancreatic cancer. Proc Natl Acad Sci U S A 2021;118:e2016904118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–83. [DOI] [PubMed] [Google Scholar]
- 6.Storz P Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol 2017;14:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Practice & Research Clinical Gastroenterology 2010;24:349–58. [DOI] [PubMed] [Google Scholar]
- 8.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, et al. Pancreatitis and the risk of pancreatic cancer. N Engl J Med 1993;328:1433–7. [DOI] [PubMed] [Google Scholar]
- 9.Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007;11:291–302. [DOI] [PubMed] [Google Scholar]
- 10.Briukhovetska D, Dörr J, Endres S, Libby P, Dinarello CA, Kobold S. Interleukins in cancer: from biology to therapy. Nature Reviews Cancer 2021;21:481–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhuang Z, Ju HQ, Aguilar M, Gocho T, Li H, Iida T, et al. IL1 receptor antagonist inhibits pancreatic cancer growth by abrogating NF-κB activation. Clin Cancer Res 2016;22:1432–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 2019;9:282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi R, Macchini M, Sunagawa M, Jiang Z, Tanaka T, Valenti G, et al. Interleukin-1β-induced pancreatitis promotes pancreatic ductal adenocarcinoma via B lymphocyte–mediated immune suppression. Gut 2021;70:330–41. [DOI] [PubMed] [Google Scholar]
- 14.Rahman MA, Krainer AR, Abdel-Wahab O. SnapShot: Splicing alterations in cancer. Cell 2020;180:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshimi A, Lin KT, Wiseman DH, Rahman MA, Pastore A, Wang B, et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019;574:273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Escobar-Hoyos LF, Penson A, Kannan R, Cho H, Pan CH, Singh RK, et al. Altered RNA splicing by mutant p53 activates oncogenic RAS signaling in pancreatic cancer. Cancer Cell 2020;38:198–211.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun S, Zhang Z, Sinha R, Karni R, Krainer AR. SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat Struct Mol Biol 2010;17:306–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das S, Anczuków O, Akerman M, Krainer AR. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep 2012;1:110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verduci L, Simili M, Rizzo M, Mercatanti A, Evangelista M, Mariani L, et al. MicroRNA (miRNA)-mediated interaction between leukemia/lymphoma-related factor (LRF) and alternative splicing factor/splicing factor 2 (ASF/SF2) affects mouse embryonic fibroblast senescence and apoptosis. J Biol Chem 2010;285:39551–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meseguer S, Mudduluru G, Escamilla JM, Allgayer H, Barettino D. MicroRNAs-10a and -10b contribute to retinoic acid-induced differentiation of neuroblastoma cells and target the alternative splicing regulatory factor SFRS1 (SF2/ASF). J Biol Chem 2011;286:4150–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu H, Sun S, Tu K, Gao Y, Xie B, Krainer AR, et al. A splicing-independent function of SF2/ASF in microRNA processing. Mol Cell 2010;38:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moulton VR, Gillooly AR, Tsokos GC. Ubiquitination regulates expression of the serine/arginine-rich splicing factor 1 (SRSF1) in normal and systemic lupus erythematosus (SLE) T cells J Biol Chem 2014;289:4126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng X, Peng Q, Wang L, Zhang X, Huang L, Wang J, et al. Serine/arginine-rich splicing factors: the bridge linking alternative splicing and cancer. Int J Biol Sci 2020;16:2442–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu T, Cazares O, Tang AD, Kim H-Y, Wald T, Verma A, et al. SRSF1 governs progenitor-specific alternative splicing to maintain adult epithelial tissue homeostasis and renewal. Dev Cell 2022;57:624–37.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 2012;19:220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 2007;14:185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katsuyama T, Moulton VR. Splicing factor SRSF1 is indispensable for regulatory T cell homeostasis and function. Cell Rep 2021;36:109339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engle DD, Tiriac H, Rivera KD, Pommier A, Whalen S, Oni TE, et al. The glycan CA19–9 promotes pancreatitis and pancreatic cancer in mice. Science 2019;364:1156–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, et al. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell 2005;20:881–90. [DOI] [PubMed] [Google Scholar]
- 30.Cáceres JF, Krainer AR. Functional analysis of pre-mRNA splicing factor SF2/ASF structural domains. EMBO J 1993;12:4715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fregoso Oliver I, Das S, Akerman M, Krainer Adrian R. Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol Cell 2013;50:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Connor AA, Denroche RE, Jang GH, Lemire M, Zhang A, Chan-Seng-Yue M, et al. Integration of genomic and transcriptional features in pancreatic cancer reveals increased cell cycle progression in metastases. Cancer Cell 2019;35:267–82.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 2006;10:459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Z, Chen CC, Rillahan CD, Shen R, Kitzing T, McNerney ME, et al. Cooperative loss of RAS feedback regulation drives myeloid leukemogenesis. Nat Genet 2015;47:539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halbrook CJ, Wen HJ, Ruggeri JM, Takeuchi KK, Zhang Y, Pasca di Magliano M, et al. Mitogen-activated protein kinase kinase activity maintains acinar-to-ductal metaplasia and is required for organ regeneration in pancreatitis. Cell Mol Gastroenterol Hepatol 2017;3:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collins MA, Yan W, Sebolt–Leopold JS, Pasca di Magliano M. MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology 2014;146:822–34.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 2016;166:963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das S, Krainer AR. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol Cancer Res 2014;12:1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wirth M, Schneider G. MYC: a stratification marker for pancreatic cancer therapy. Trends in Cancer 2016;2:1–3. [DOI] [PubMed] [Google Scholar]
- 41.Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol 2002;15:462–9. [DOI] [PubMed] [Google Scholar]
- 42.Sodir NM, Kortlever RM, Barthet VJA, Campos T, Pellegrinet L, Kupczak S, et al. MYC instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov 2020;10:588–607. [DOI] [PubMed] [Google Scholar]
- 43.Gehrke N, Hövelmeyer N, Waisman A, Straub BK, Weinmann-Menke J, Wörns MA, et al. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J Hepatol 2018;68:986–95. [DOI] [PubMed] [Google Scholar]
- 44.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011;117:3720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 2018;281:8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sawai H, Funahashi H, Matsuo Y, Yamamoto M, Okada Y, Hayakawa T, et al. Expression and prognostic roles of integrins and interleukin-1 receptor type I in patients with ductal adenocarcinoma of the pancreas. Dig Dis Sci 2003;48:1241–50. [DOI] [PubMed] [Google Scholar]
- 47.Connor AA, Denroche RE, Jang GH, Timms L, Kalimuthu SN, Selander I, et al. Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncology 2017;3:774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Friedlander SYG, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009;16:379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guerra C, Collado M, Navas C, Schuhmacher Alberto J, Hernández-Porras I, Cañamero M, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011;19:728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carrière C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun 2009;382:561–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anczuków O, Krainer AR. Splicing-factor alterations in cancers. RNA 2016;22:1285–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paronetto MP, Passacantilli I, Sette C. Alternative splicing and cell survival: from tissue homeostasis to disease. Cell Death Differ 2016;23:1919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brumbaugh J, Di Stefano B, Hochedlinger K. Reprogramming: identifying the mechanisms that safeguard cell identity. Development 2019;146:dev182170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Agosto LM, Lynch KW. Alternative pre-mRNA splicing switch controls hESC pluripotency and differentiation. Genes Dev 2018;32:1103–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nature Reviews Molecular Cell Biology 2017;18:437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Science Signaling 2010;3:cm1. [DOI] [PubMed] [Google Scholar]
- 57.Garlanda C, Mantovani A. Interleukin-1 in tumor progression, therapy, and prevention. Cancer Cell 2021;39:1023–7. [DOI] [PubMed] [Google Scholar]
- 58.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015;348:880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wijewardhane N, Dressler L, Ciccarelli FD. Normal somatic mutations in cancer transformation. Cancer Cell 2021;39:125–9. [DOI] [PubMed] [Google Scholar]
- 60.Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019;565:312–7. [DOI] [PubMed] [Google Scholar]
- 61.Lawson ARJ, Abascal F, Coorens THH, Hooks Y, O’Neill L, Latimer C, et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 2020;370:75–82. [DOI] [PubMed] [Google Scholar]
- 62.Shi G, DiRenzo D, Qu C, Barney D, Miley D, Konieczny SF. Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 2013;32:1950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ray KC, Bell KM, Yan J, Gu G, Chung CH, Washington MK, et al. Epithelial tissues have varying degrees of susceptibility to KrasG12D-initiated tumorigenesis in a mouse model. PLoS One 2011;6:e16786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anczuków O, Akerman M, Cléry A, Wu J, Shen C, Shirole Nitin H, et al. SRSF1-regulated alternative splicing in breast cancer. Mol Cell 2015;60:105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou X, Wang R, Li X, Yu L, Hua D, Sun C, et al. Splicing factor SRSF1 promotes gliomagenesis via oncogenic splice-switching of MYO1B. The Journal of Clinical Investigation 2019;129:676–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu X, Yang D, Ding J-H, Wang W, Chu P-H, Dalton ND, et al. ASF/SF2-regulated CaMKIIδ alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 2005;120:59–72. [DOI] [PubMed] [Google Scholar]
- 67.Haward F, Maslon MM, Yeyati PL, Bellora N, Hansen JN, Aitken S, et al. Nucleo-cytoplasmic shuttling of splicing factor SRSF1 is required for development and cilia function. eLife 2021;10:e65104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimoni-Sebag A, Lebenthal-Loinger I, Zender L, Karni R. RRM1 domain of the splicing oncoprotein SRSF1 is required for MEK1-MAPK-ERK activation and cellular transformation. Carcinogenesis 2013;34:2498–504. [DOI] [PubMed] [Google Scholar]
- 69.Zhou Y, Han C, Wang E, Lorch AH, Serafin V, Cho B-K, et al. Posttranslational regulation of the exon Skipping machinery controls aberrant splicing in leukemia. Cancer Discov 2020;10:1388–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adhikari H, Counter CM. Interrogating the protein interactomes of RAS isoforms identifies PIP5K1A as a KRAS-specific vulnerability. Nature Communications 2018;9:3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kovalski JR, Bhaduri A, Zehnder AM, Neela PH, Che Y, Wozniak GG, et al. The functional proximal proteome of oncogenic Ras includes mTORC2. Mol Cell 2019;73:830–44.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 2017;171:1301–15.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011;478:524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 2017;45:W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith JC, Sheltzer JM. Systematic identification of mutations and copy number alterations associated with cancer patient prognosis. eLife 2018;7:e39217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jensen MA, Wilkinson JE, Krainer AR. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol 2014;21:189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dow LE, Nasr Z, Saborowski M, Ebbesen SH, Manchado E, Tasdemir N, et al. Conditional reverse Tet-transactivator mouse strains for the efficient induction of TRE-regulated transgenes in mice. PLoS One 2014;9:e95236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov 2018;8:1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boj Sylvia F, Hwang CI, Baker Lindsey A, Chio Iok In C, Engle Dannielle D, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rahman MA, Lin KT, Bradley RK, Abdel-Wahab O, Krainer AR. Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev 2020;34:413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nomakuchi TT, Rigo F, Aznarez I, Krainer AR. Antisense oligonucleotide–directed inhibition of nonsense-mediated mRNA decay. Nat Biotechnol 2016;34:164–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV. In vitro cell migration and invasion assays. JoVE 2014:e51046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Storey JD, Tibshirani R. Statistical significance for genomewide studies. PNAS 2003;100:9440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2008;37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013;30:923–30. [DOI] [PubMed] [Google Scholar]
- 87.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin KT, Krainer AR. PSI-Sigma: a comprehensive splicing-detection method for short-read and long-read RNA-seq analysis. Bioinformatics 2019;35:5048–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 2015;33:290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data are available at the Sequence Read Archive of NCBI under the BioProject ID: PRJNA747810.