

Abstract
Although the United States and Europe have shifted to the prescription use of diclofenac due to several serious incidences of cardiotoxicity, it is one of the most commonly used over-the-counter (OTC) medicine in major parts of the world. We elucidated the quantitative and tissue-specific contribution of uridine diphosphate-glucuronosyltransferases 17 (UGT2B17) in diclofenac metabolism and pharmacokinetics. UGT2B17 is one of most deleted genes in humans with the gene deletion frequency ranging from ~20% in Caucasians to 90% in Japanese. The human intestinal and liver microsomes isolated from the high-UGT2B17 expressing individuals showed 21- and 4-fold greater rate of diclofenac glucuronide (DG) formation than in the null-UGT2B17 carriers, respectively. The greater contribution of intestinal UGT2B17 was confirmed by a strong correlation (R= 0.78, p<0.001) between UGT2B17 abundance and DG formation in individual intestinal microsomes (n=14). However, because UGT2B17 is a minor UGT isoform in the liver, DG formation rate correlated better with the expression of UGT2B7. The proteomics-informed physiologically-based pharmacokinetic model explains the reported higher exposure of diclofenac in females consistent with ~3-fold lower expression of UGT2B17. Similarly, our predictions also corroborate with the reported higher exposure and lower standard clinical dose of diclofenac in Japanese population. Therefore, UGT2B17 mediated metabolism of oral diclofenac is a concern, especially in the developing countries where it is still used as an OTC drug. The ontogeny data of UGTs in human hepatocytes can be utilized in developing PBPK models for predicting pharmacokinetics in pediatric population.
Introduction
Uridine 5’-diphospho-glucuronosyltransferase 2B17 (UGT2B17) is an important enzyme that metabolizes drugs and steroids and exhibits high inter-individual variability in its expression and activity. Hepatic UGT2B17 expression varies by more than 3000-fold, and the copy number variation (CNV), single nucleotide polymorphisms (SNPs), age, and sex are associated with its activity as well as with mRNA and protein expression.1 UGT2B17 is sparsely expressed in children below 9 yr, and males express 3-fold higher UGT2B17 protein than females.1 Interestingly, the frequency of UGT2B17 gene deletion varies greatly across populations, with 25% to 90% frequency in Caucasians versus Japanese, respectively.2 Although UGT2B17 variability contributes to inconsistent pharmacokinetics (PK) or efficacy of clinically important newer drugs such as verinurad, belzutifan, and vorinostat,3–5 the role of UGT2B17 is not well studied for drugs developed before 2010. One of the possible reasons for the lack of investigations on UGT2B17 is its low expression in the liver relative to other UGT isoforms (e.g., UGT2B7, UGT2B4, and UGT2B15) in abundance. Recent studies including proteomics data from our laboratory revealed that UGT2B17 expression is abundant in the intestine contributing to more than 50% of the total intestinal UGTs in the expressors of UGT2B17.6 The high intestinal expression of UGT2B17 has been linked to the extensive and variable first-pass metabolism of its substrates. For instance, MK-7246, an investigational drug for asthma, showed 25-fold higher plasma exposure (AUC) in individuals with UGT2B17 gene deletion (*2/*2) as compared to those with the reference allele (*1/*1), however, the plasma half-life showed no change confirming an extensive first-pass metabolism.7 These data suggest that the quantitative role of UGT2B17 variability on the PK of its substrates should be investigated, especially if they are orally administered.
Here, we estimated the quantitative contribution of UGT2B17 variability on diclofenac metabolism and PK. Diclofenac is one of the top 100 prescribed medicines of non-steroidal anti-inflammatory drug (NSAID) category, with over 10 million prescriptions annually in the United States for rheumatoid arthritis, osteoarthritis, ankylosing spondylitis, mild to moderate pain, inflammation, swelling, and acute gouty arthritis.8 Diclofenac is used either orally as tablets or capsules (38%) or topically (48%).9 Although diclofenac is effective in managing pain and inflammation, its safety and tolerability are the key concerns due to the side effects such as ulceration, bleeding, and upper perforation of gastrointestinal (GI), cardiovascular (CV), and nephrotoxicity.10 In particular, serious incidences of cardiotoxicity of diclofenac were observed in multiple clinical investigations between 2005-2013 which resulted in a major shift in its use from over-the-counter (OTC) to prescription.11, 12 Interestingly, the cardiotoxicity risk of diclofenac is similar to another NSAID, rofecoxib13 which was withdrawn from the market after the toxicity reports.14 The regulatory agencies also released an advisory for the use of diclofenac and contraindicated in patients with various heart problems such as congestive heart failure (CHF), ischemic heart disease (IHD), and cerebrovascular disease (CVD).10 Recently, an increased risk of acute kidney injury with rhabdomyolysis has also been reported in patients receiving lipid-lowering agents along with diclofenac.15 Unfortunately, diclofenac is still used OTC in other countries such as India, China, Russia, Australia, Italy, and African countries.16–19 Diclofenac glucuronide (DG) is the major circulating metabolite of the drug,20 it is known that UGT2B17 can metabolize diclofenac along with UGT2B7, UGT2B4, UGT1A3, and UGT1A9.21, 22 However, the quantitative contribution of UGT2B17 in the intestinal and hepatic metabolism of diclofenac has not been characterized. We hypothesized that the higher intestinal UGT2B17 abundance can lead to a greater contribution of UGT2B17 variability to diclofenac bioavailability and PK. We first confirmed the UGT isoforms involved in DG formation. Second, we determined the effect of UGT2B17 variability on diclofenac glucuronidation in well-characterized human liver and intestinal microsomes (HLM and HIM, respectively), followed by the estimation of the tissue-specific fraction of diclofenac glucuronidation (fgluc) by UGT2B17. To confirm the role of UGT2B17 in intestinal metabolism, the correlation of diclofenac metabolizing UGTs with DG formation was tested in individual HIM. The effect of age on DG formation and UGTs involved in diclofenac metabolism was also investigated in pediatric hepatocytes using quantitative proteomics and in vitro activity assay. Finally, proteomics-informed physiologically-based pharmacokinetic (PBPK) modeling was employed to characterize the effect of UGT2B17 gene deletion, sex, and age on diclofenac PK. The mechanistic in vitro data and proteomics-informed PBPK modeling presented here elucidated the potential role of UGT2B17 variability in diclofenac oral PK and highlighted a greater risk of cardiotoxicity in UGT2B17 poor metabolizers.
Material and Methods
Chemicals and reagents
Diclofenac, alamethicin, UDP-glucuronic acid (UDPGA), magnesium chloride (MgCl2), and mono- and di-basic potassium phosphate were purchased from Sigma-Aldrich (St. Louis, MO). Ammonium bicarbonate (ABC) (98% pure), bovine serum albumin (BSA), dithiothreitol (DTT), iodoacetamide (IAA), and trypsin were obtained from Thermo Fisher Scientific (Rockford, IL). The deuterated testosterone glucuronide (TG-d3) standard was obtained from Cerilliant Corporation (Round Rock, TX). Recombinant human UGT (rUGT) enzymes were purchased from Corning Life Sciences (Riverfront, NY). Individually genotyped HLM samples were from our previous study,6 procured from the University of Washington School of Pharmacy Liver Bank (Seattle, WA). Individual HIM samples and human pediatric cryopreserved hepatocytes (n=50), hepatocyte thawing (HT) media, and hepatocyte incubation (HI) media were provided by BioIVT (Westbury, NY) (Table S1). Synthetic light peptides with amino acid analysis and their corresponding stable-isotope labeled (SIL) forms were purchased from New England Peptides (Boston, MA) and Thermo Fisher Scientific (Rockford, IL), respectively.
In vitro characterization of diclofenac glucuronidation
Diclofenac glucuronidation assay in recombinant human UGT enzymes
Eleven recombinant human UGT enzymes (rUGT1A1, rUGT1A3, rUGT1A4, rUGT1A6, rUGT1A9, rUGT1A10, rUGT2B4, rUGT2B7, rUGT2B10, rUGT2B15, and rUGT2B17) were tested for their potential to metabolize diclofenac to its glucuronide. Briefly, 10 μg of rUGT protein was added to a potassium phosphate buffer (100 mM, pH 7.4) containing 5 mM MgCl2, 0.01% BSA, and 0.1 mg/ml of alamethicin. After 15 min of pretreatment on ice, the reaction was initiated by adding diclofenac (10 μM, final concentration) and 2.5 mM UDPGA. The incubations were performed for 45 min (37 °C) at 300 rpm in triplicates. The reaction was quenched with two times the volume of ice-cold acetonitrile containing TG-d3 as the internal standard. The sample was centrifuged at 10,000 x g for 5 min (4 °C) and the supernatant was transferred to an LC vial.
Diclofenac glucuronidation activity assay in HIM and HLM
The diclofenac glucuronidation kinetic assay was carried out in individual HIM and HLM with known UGT2B17 abundance in triplicates.23 Briefly, 10 μg of microsomal protein was added to a 100 mM potassium phosphate buffer (pH 7.4) containing 5 mM MgCl2, 0.01% BSA, and 0.1 mg/ml of alamethicin. After 15 min of pretreatment on ice, the reaction was initiated by adding diclofenac (1.5 to 1000 μM, final concentration range) and 2.5 mM UDPGA followed by incubation for 30 min at 37 °C with gentle shaking at 300 rpm. The reaction was quenched with two times the volume of ice-cold acetonitrile containing TG-d3. The samples were centrifuged at 8000 x g for 5 min (4 °C) and the supernatant was transferred to an LC vial for analysis by LC-MS/MS. Similarly, the diclofenac glucuronidation activity assay was also tested in 14 individual adult HIM samples using a single concentration of diclofenac (100 μM). Other assay conditions were similar as described for the enzyme kinetic assay.
Diclofenac glucuronidation activity assay in pediatric hepatocytes
DG formation activity was also assessed in cryopreserved pediatric hepatocytes. Briefly, hepatocytes were thawed and suspended according to a previously defined protocol.6 The hepatocyte vial was completely immersed into a 37 °C water bath for 90-120 s and poured gently into the pre-warmed HT medium in a 10 ml conical tube, mixed gently by inverting the tube, and centrifuged at 50 x g for 5 min at room temperature. The supernatant was decanted and the cell pellet was resuspended in 3 ml of HT medium. Hepatocytes were counted using the trypan-blue exclusion method using Auto T4 cellometer (Nexcelom Bioscience, Lawrence, MA) and a suspension of 1 million cells per ml was made in HI media. The reaction was initiated by adding 5 μl diclofenac (12 mM) in 300 μl of hepatocyte suspension in 12-well plates. The reactions were performed in triplicates, and the DG formation was monitored after quenching the reaction with two times the volume of ice-cold acetonitrile containing TG-d3 as the internal standard by LC-MS.
LC-MS/MS analysis of diclofenac glucuronide (DG)
DG was analyzed on Xevo-TQ-XS MS (Waters, Milford, MA), coupled with standard electrospray ionization (ESI) source, microflow LC and Acquity UPLC HSS T3 column (100Å, 1.8 μm, 1 mm x 100 mm). The LC parameters included 50 μL/min flow rate and 1 μL injection volume. Mobile phase A (water with 0.1% formic acid) and B (acetonitrile with 0.1% formic acid) were used and the following gradient program was applied: 0.0-0.5 min (10% B), 0.5-3.0 min (10% to 30% B), 3.0-4.5 min (30% to 95% B), and 4.5-8.0 min (95% B), followed by 2.0-min equilibration with 10% B. The mass spectrometer was set to the positive ion (ESI+) mode with cone voltage (CV) of 30 V. The MRM transitions for DG and TG-d3 were m/z 472.05→ 296.0, 250.01 (collision energy (CE), 25 eV) and m/z 468.2→ 97.1, 109.1 (CE, 40 eV), respectively.
The enzyme kinetic parameters, i.e., Michaelis-Menten constant (Km) and the maximum glucuronidation rate (Vmax) were calculated using Eq. 1 in GraphPad Prism v. 8.4.3.
| Eq. 1 |
Where, Y is the rate of DG formation (peak area or response/min/mg protein) and X is diclofenac concentration. The Akaike information criterion (AIC) was used to check the goodness of model fit for UGT2B17 kinetics data, which was also evaluated by visual inspection, 95% confidence intervals (CIs) of the parameter estimates, and residual plots with baseline.
UGT protein quantification in HIM, HLM, rUGTs, and pediatric hepatocytes
We quantified UGTs involved in DG formation (UGT1A3, UGT1A9, UGT2B4, UGT2B7, and UGT2B17) in HLM, HIM, rUGT enzymes, and hepatocyte homogenate using an optimized targeted LC-MS/MS approach.24, 25 Briefly, 80 μL protein sample containing either 80 μg total protein of HLM, HIM, and rUGTs or 1 million pediatric hepatocytes was mixed with 30 μL ammonium bicarbonate (100 mM) and 20 μL BSA (0.02 mg/ml). The samples were denatured at 95 °C for 10 min with 10 μL of 250 mM DTT with gentle shaking at 300 rpm. After cooling for 10 min at room temperature, samples were alkylated in the dark for 30 min with 10 μL of 100 mM IAA. The samples were precipitated with ice-cold acetone (1 mL) and incubated at −80 °C for 1 hr before centrifugation at 16,000 x g (4 °C) for 10 min. Five hundred μL of ice-cold methanol was added to the samples to remove residual acetone and centrifuged at 16,000 × g (4 °C) for 10 min. The pellet was dried at room temperature for 30 min and resuspended in 60 μL ammonium bicarbonate buffer (50 mM, pH 7.8). Twenty μL of trypsin (protein: trypsin ratio, approximately 50:1) was added to the reconstituted protein samples and digested for 16 hr at 37 °C. The reaction was stopped by adding 5 μL of 0.5 percent formic acid in water, and the sample was vortex mixed and centrifuged at 8000 × g for 5 min. The supernatant was mixed with a cocktail of SIL peptide internal standard (IS) (Table S2) and transferred to an LC vial. The data was collected on an M-class Waters UPLC system with a Waters Xevo® TQ-XS microflow LC-MS/MS instrument connected to a standard ESI source, with optimized parameters shown in Table S3. The proteolytic peptides were separated on a standard HSS T3 C18 column (1.8 μm, 1.0 × 100 mm). A list of proteotypic peptides and their corresponding SIL peptides is provided in Table S2.
Calculation of the fractional metabolism of diclofenac by UGT enzymes (fgluc)
The fgluc of UGT2B17 for DG formation in the high-, average-, and null-UGT2B17 expressors was estimated using Eqs. 2–4. Where Vmax, expressor, Vmax, average, and Vmax, deletion indicate maximum diclofenac formation rates in the high-, average-, and null-UGT2B17 expressors, respectively. The protein abundance of UGT2B17 in the high and average expressors was obtained from our previous study.1
| Eq. 2 |
| Eq. 3 |
| Eq. 4 |
Protein-activity correlation in individual HIM and pediatric hepatocytes
The correlation between DG formation rate (response/min/mg protein) versus UGT2B7 and UGT2B17 abundance (pmol/mg protein) was evaluated using Pearson correlation in HLM. Similarly, in pediatric hepatocytes, the correlation of DG formation rate (per min/mg protein) versus UGT1A3, UGT1A9, UGT2B4, UGT2B7, and UGT2B17 abundance (pmol/mg protein) was tested using Pearson correlation and visualized using hierarchical clustering and heatmap.
Proteomics-informed PBPK model development of diclofenac in the adult population
A PBPK model of diclofenac was developed using Simcyp software (version 21, Certara, NY) (Figure S1). The system-dependent parameters (e.g., organ weight, body composition, and blood flow rates) were inbuilt into the software, whereas drug-dependent parameters were added from the literature (Table S4). The intravenous diclofenac full PBPK disposition model was developed and validated using published diclofenac clinical studies.26, 27 The tissue plasma partition coefficient was predicted using Poulin and Theil method. The steady-state volume of distribution (Vss) was predicted using logP and Kp scalar values (ratio of the drug concentration in tissue and plasma), which were adjusted to fit the observed in vivo clinical data in adults reported by Willis et al. Diclofenac is an extensively metabolized drug by CYP and UGT enzymes, hence only these metabolic pathways of diclofenac clearance were considered. The intrinsic clearance (Clint) was retrospectively calculated from the reported clinical data (Table S4). In particular, the fractional contributions of CYP versus UGT-mediated metabolism of diclofenac was first estimated in average population from the reported metabolite levels excreted in the urine. The CYP-mediated metabolism was assigned to be CYP2C9 mediated, whereas DG formation was assumed to be mainly mediated by UGT2B7 and UGT2B17. fm values for UGT2B7 and UGT2B17 were estimated based on in vitro kinetics and protein abundance data (Eqs. 2–4). The first-order oral absorption model was developed using absorption parameters such as fraction dose absorbed (fa), absorption rate constant (ka), and lag time. Simulations were conducted at two diclofenac dose strengths (50-75 mg) to match the available clinical data. To ensure that the reported clinical values for diclofenac fell within the 5th and 95th percentiles of the acceptance range, the simulated mean plasma concentration-time profile was compared to the reported clinical data.26–30 The predicted/observed ratio was calculated for the PK endpoints, i.e., AUC and Cmax, with the acceptance criteria of 0.5-2-fold.
We also developed the UGT2B17 ontogeny-informed pediatric PBPK model using Simcyp to predict the effect of age-dependent UGT2B17 variability on diclofenac PK. The UGT2B17 mediated Clint value was extrapolated from adults to different pediatric age groups (neonates-adolescents) based on the UGT2B17 protein abundance from our previous study. 1 The pediatric simulated data were compared to the reported in vivo clinical data, and visual inspections were performed to ensure that the reported clinical data were within the predicted 5th and 95th confidence intervals for diclofenac plasma concentrations (Figure S2).31, 32 The mean PK parameter predicted/observed ratio was calculated with the 0.5-2-fold acceptance threshold.
Diclofenac clinical PK data were curated from the literature 24–30 and digitized using the GetData Graph Digitizer (http://www.getdata-graph-digitizer.com/index.php).
The potential effect of copy number variation (CNV), sex, and age on diclofenac bioavailability
The validated intravenous and oral adult PBPK models were used to predict the effect of CNV and sex, whereas the oral pediatric PBPK model was used to predict the impact of age on diclofenac PK. In this exercise, only the UGT2B17 Clint was changed based on UGT2B17 abundance in the high- and null-expressors, men versus women, and various pediatric age ranges.1 The predicted AUC and plasma clearance of diclofenac was estimated in adults and pediatric populations after oral administration.
Statistical analysis
The DG formation and UGT abundance differences across age groups were evaluated using the Kruskal-Wallis followed by the Dunn multiple-comparison tests.
Results
UGT isoforms involved in diclofenac glucuronidation
The UGT abundance normalized rate of DG formation in the recombinant system revealed a predominant role of UGT2B17 in diclofenac metabolism (Figure 1A). The rate of DG formation in rUGT2B4, rUGT1A3, rUGT2B7, and rUGT1A9 was ~ 21-, 10-, 4-, and 1.5- fold lower than in rUGT2B17 incubation. DG formation was not detected in the in vitro incubations containing rUGT1A1, rUGT1A4, rUGT1A6, rUGT1A10, rUGT2B10, and rUGT2B15.
Figure 1. Diclofenac glucuronide (DG) formation in recombinant human UGT enzymes (rUGTs), human liver microsomes (HLM), and human intestinal microsomes (HIM).
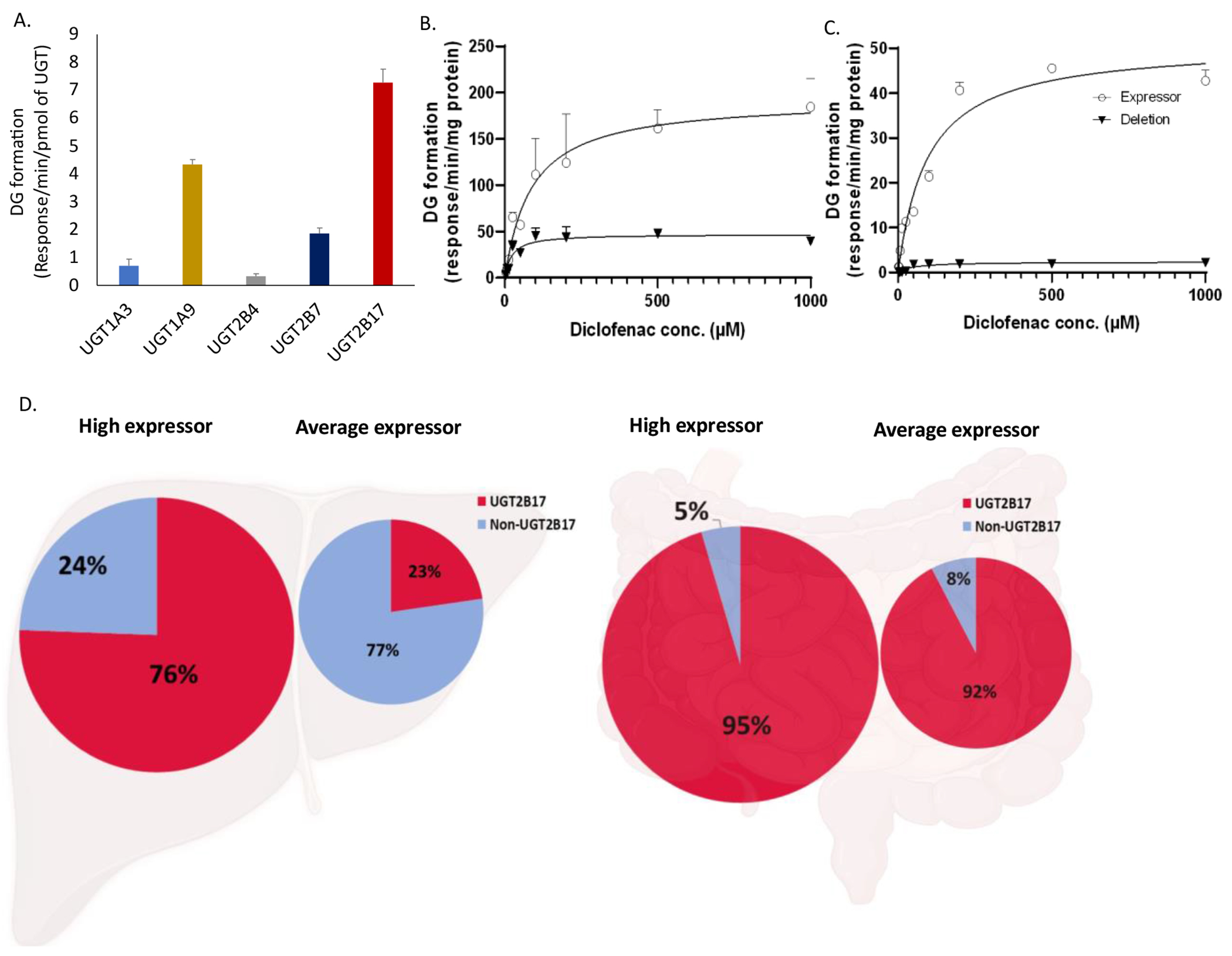
DG formation by individual rUGTs (A). Effect of UGT2B17 deletion on DG formation kinetics in the high (open circle) and null (solid triangle) UGT2B17-expressing HLM (B) and HIM (C), respectively. Data represent the mean ± SD of triplicate experiments. The estimated tissue-specific fraction glucuronidated (fgluc) by UGT2B17 versus other UGTs in human HLM and HIM (D) representing the high- and average- UGT2B17 expressors.
Enzyme kinetic parameters of diclofenac in the high and null-UGT2B17 expressing HLM and HIM
The protein abundance of UGT2B17 was 8.5 and 21.6 pmol/mg protein in the high-expressor HLM and HIM, respectively, whereas the levels of other UGTs in both the high- and null-UGT2B17 expressors were comparable (Figure S3). These data suggest that in the UGT2B17 high expressors, the protein abundance normalized activity of UGT2B17 towards DG formation in the intestine is ~ 10-fold higher as compared to the UGT2B7 activity. Consistent with the protein abundance data, the Vmax values of DG formation in the high- versus null-UGT2B17-expressing HLM and HIM were 4 and 21-fold, respectively (Figures 1B and 1C). The Km values of DG formation were higher by up to 4-fold in the high UGT2B17 expressor versus null-expressor due to the involvement of other UGTs in the null-expressors (Table 1, Figures 1B and 1C). Although only apparent Km values are reported, the higher affinity of other UGTs suggests that these enzymes could be saturated in the intestine, leading to potential greater contribution of UGT2B17.
Table 1:
Diclofenac glucuronide formation kinetic parameters in HLM and HIM with known UGT2B17 protein abundance
| UGT2B17 abundance (pmol/mg protein) | Km (μM) | Vmax (response/min/mg protein) | |
|---|---|---|---|
| HLM | 8.5 | 86.85 (51.75-145.4) | 193.2 (166.6-225.6) |
| 0 | 22.35 (13.7-35.63) | 47.18 (41.89-52.86) | |
| HIM | 21.6 | 96.10 (72.09-127.4) | 51.04 (46.74-55.75) |
| 0 | 48.79(31.51-75.38) | 2.33 (2.05-2.64) |
Tissue-specific fractional glucuronidation (fgluc) of diclofenac by UGT2B17 in the liver and intestine
Diclofenac tissue-specific fgluc by UGT2B17 was estimated by comparing the activity data between the high- and null-UGT2B17 expressors. UGT2B17 fgluc was 95% and 76% in the high-expressing HIM and HLM, respectively, whereas in the average expressors, the fgluc values were 92% and 24%, respectively (Figure 1D). The greater contribution of UGT2B17 in DG formation in the intestine versus liver suggest that UGT2B17 genetic variability mainly affect the oral bioavailability of diclofenac.
Correlation of DG formation with UGT abundance in individual HIM and hepatocytes
The rate of DG formation was compared with the protein abundance of UGTs in HIM and pediatric hepatocytes. In HIM, the rate of DG formation showed a stronger correlation with UGT2B17 (R=0.78, p<0.001) as compared to UGT2B7 (R=0.56, p<0.05) (Figure 2A). Whereas the hierarchical cluster heatmap of the pediatric hepatocyte data showed a strong correlation of DG formation activity with the abundance of UGT2B7 (R=0.65, p<0.0001) followed by UGT2B4 (R=0.59, p<0.001). Although we could only detect UGT2B17 levels in three pediatric hepatocytes, the DG formation showed a positive trend of correlation (R=0.27, p>0.05). UGT1A3 and UGT1A9 abundance showed no correlation with DG formation (Figure 2B).
Figure 2.
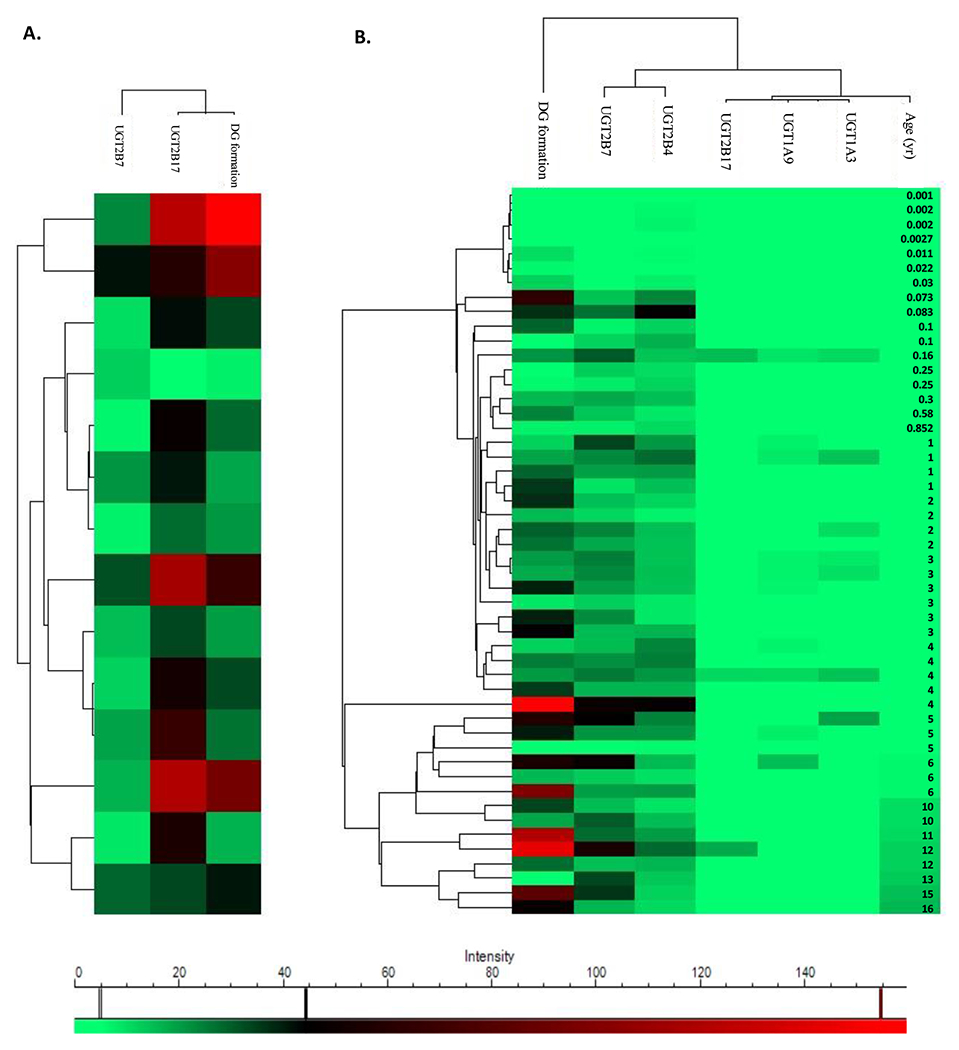
Hierarchical cluster heatmap representing correlations of diclofenac glucuronide (DG) formation, protein abundance of UGT isoforms involved in DG formation (2B7, 2B4, 2B17, 1A3, and 1A9), and age in individual human intestinal microsomes (HIM, N=14; A) and pediatric hepatocytes (N=50; B). The correlations were evaluated by Pearson correlation analysis.
Effect of age on DG formation rate and UGT abundance in hepatocytes
In pediatric hepatocytes, the DG formation rate was increased from neonates to infants, early-childhood, middle-childhood, and adolescents by 1.5, 3, 6, and 4-fold, respectively (Figure 3A). Similarly, as compared to neonates, the abundance of UGT2B7 was increased by 10, 13, 15, and 16-fold from infants to early-childhood, middle-childhood, and adolescents, respectively (Figures 3B and 3C). Age-dependent changes in UGT2B4 levels were moderate (within 2.5-fold). Consistent with our previous data,1 only three out of fifty pediatric hepatocytes showed UGT2B17 expression (Figure 3D).
Figure 3.
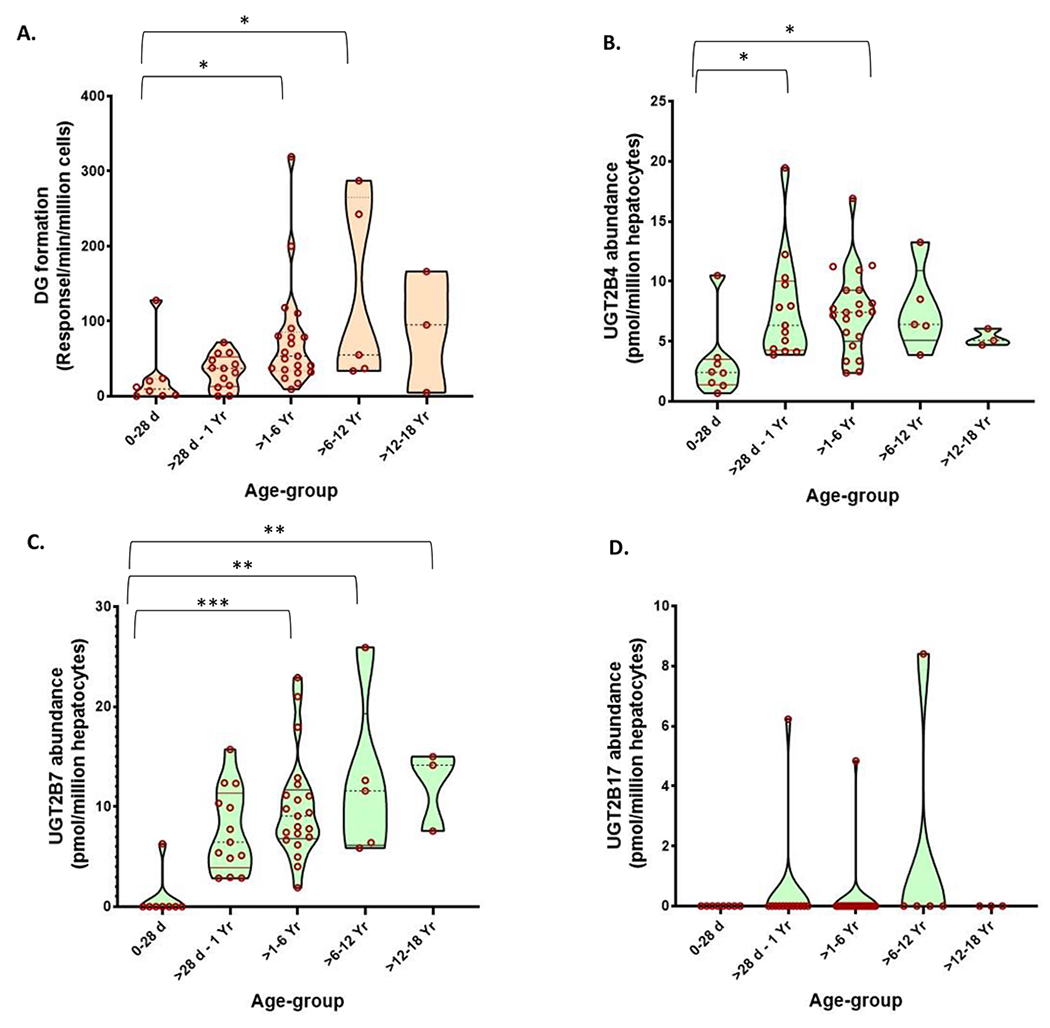
Ontogeny of diclofenac glucuronide (DG) formation (A) and the protein abundance of UGT2B7 (B), UGT2B4 (C), and UGT2B17 (D) in pediatric cryopreserved human hepatocytes (N= 50). The violin (truncated) plots indicate the range, median, and 25th to 75th percentiles of protein abundance values. The data were compared across different age groups by Kruskal Wallis test. *, **, and *** indicate p values of <0.05, <0.001, and < 0.0001, respectively.
Proteomics-informed diclofenac PBPK model predictions
The predicted plasma concentrations of diclofenac after intravenous and oral dosing (50-75 mg) were comparable to the observed data in adults and pediatric populations (Figures 4A and 4B). The observed diclofenac clinical data fell within 90% confidence intervals of the predicted plasma concentrations and the mean predicted/observed PK parameters (AUC, Cmax, and CL) were within 2-fold (Table 2).
Figure 4.
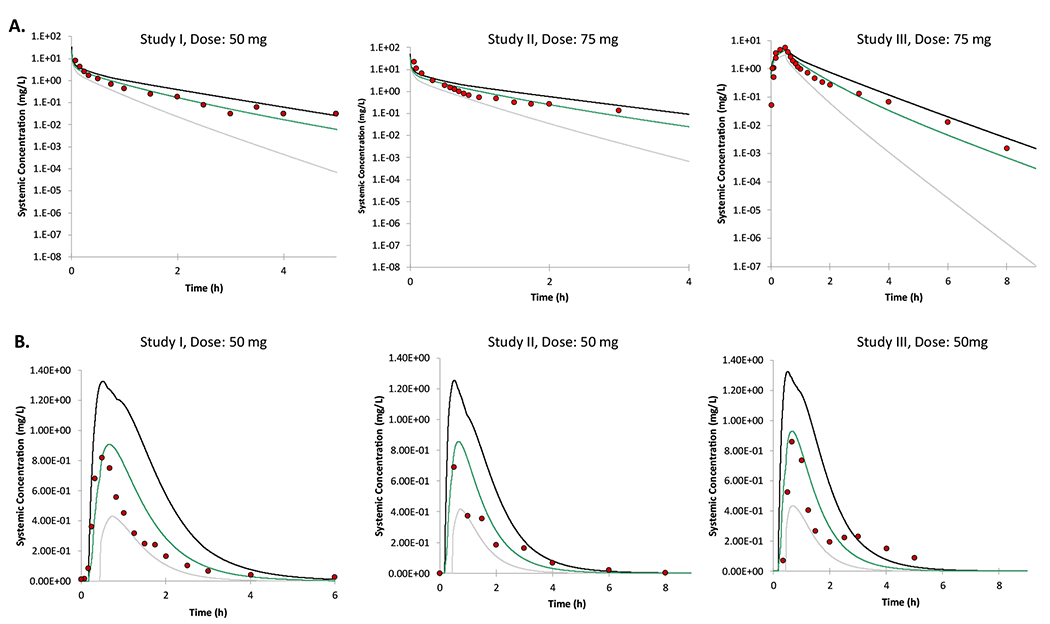
PBPK model predicted versus clinically observed plasma concentration-time profiles of diclofenac after iv doses (A) and PO doses in adults (B.). The PBPK model predicted mean plasma concentrations from individual trials are shown by colored lines with 5% and 95% confidence intervals (grey and black outer lines). The observed data are shown by filled circles.22–26
Table 2:
Predicted versus observed24–28 mean AUC, Cmax and clearancevalues of diclofenac in adults after iv and oral doses
| Drug | Age (yr) | Sex | Dosing regimen | Ratio | |
|---|---|---|---|---|---|
|
| |||||
| AUC (mg/L*h) | CL (L/h) | ||||
|
|
|||||
| 20-22 | Female | Single iv dose, 50 mg | 0.88 | 1.16 | |
| Diclofenac | ≥18 | Mixed | 30-minute iv infusion, 75 mg | 1.08 | 0.97 |
| Single iv dose, 75 mg | 1.05 | 0.99 | |||
|
|
|||||
| AUC (mg/L*h) | Cmax (mg/L) | ||||
|
|
|||||
| 20-31 | Male | Single oral dose, 50 mg | 1.40 | 1.23 | |
| 20-44 | Mixed | 1.19 | 0.83 | ||
| 18.45 | Mixed | 1.23 | 0.81 | ||
Consistent with the tissue-specific UGT2B17 fgluc values (Figure 1D), the poor intestinal glucuronidation of diclofenac in the null-expressors was associated with 8-fold higher predicted bioavailability of diclofenac as compared to the high-expressors, whereas the effect of UGT2B17 deletion on the plasma concentration of diclofenac after intravenous administered was only ~2-fold (Figure 5A). Similarly, oral pharmacokinetics of diclofenac was affected by sex-dependent differences in UGT2B17 abundance, where the predicted diclofenac AUC was ~66% higher in females than males after oral administration (Figure 5B). Based on UGT2B17 ontogeny data, we predicted 43-fold higher diclofenac AUC in neonates compared to adults (Figure 5C). Similarly, the estimated fraction gut-escaped (fg) is higher in the null (fg=0.96) versus high (fg=0.37) UGT2B17 expressors (Figure 5D).
Figure 5.
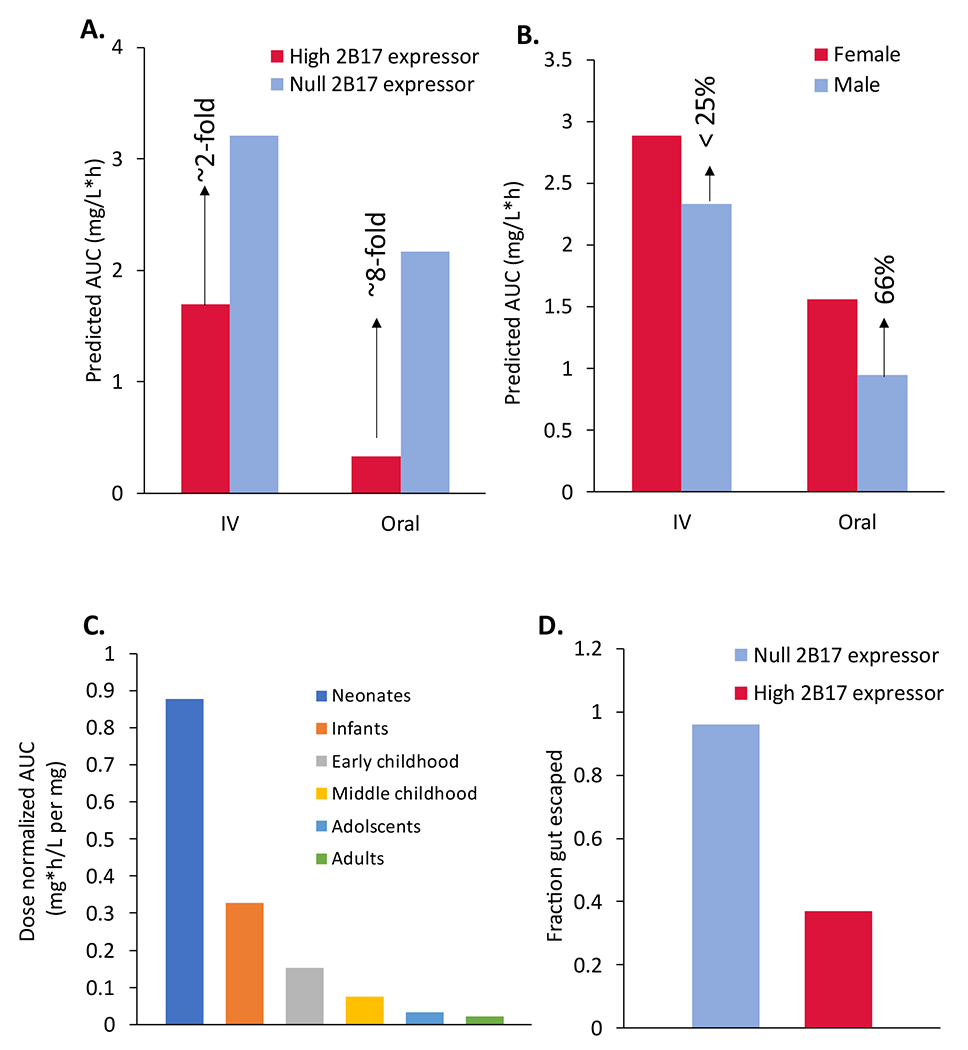
The potential effect of UGT2B17 copy number variation (A), sex (B), and age (C) on diclofenac pharmacokinetics. The estimated fraction of drug-eliminating gut metabolism (fg; D) shows significant intestinal first-pass metabolism in the high UGT2B17 expressors.
Discussion
UGT2B17 is one of most polymorphic genes in humans with the frequency of gene deletion carriers ranging from ~20% in Caucasians to 90% in Japanese populations.2 Further, the expression of UGT2B17 is ~3-fold lower in females than males and this enzyme is scarcely expressed in children below 9 years of age.1 Although the FDA recommends screening new chemical entities (NCE) for their potential metabolism by UGTs, UGT2B17 remained an underappreciated enzyme.6 Notably, the role of UGT2B17 in drug metabolism is not well characterized for drugs developed before 2010. Recently, we have characterized UGT2B17 abundance in the human intestine and found that its abundance is ~5-fold higher in the intestinal segments as compared to the liver, which indicates its role in first-pass metabolism.6 This suggests that UGT2B17 substrates are more prone to variable and unpredictable clinical PK as evident by the data from Phase I trials of MK-72467 and PT2385 (a novel hypoxia-inducible factor 2α inhibitor).33 UGT2B17 is also associated with the metabolism, PK, efficacy, or toxicity of clinically important drugs such as vorinostat, belzutifan, asciminib, and exemestane.4, 5, 34–36 Taken together, UGT2B17 may have been associated with clinical trial failure, variable PK, or toxicity of its substrates but remained unnoticed. To avoid variable clinical PK, drug developers should ideally rule out the liability of UGT2B17 mediated glucuronidation for their compounds in the early stages of drug discovery. This can be accomplished first by conducting a reaction phenotyping study using recombinant human UGT2B17 enzyme. Second, an in vitro assay involving the intestinal and hepatic microsomes with known UGT2B17 genotypes (deletion versus expressors) will provide an accurate estimate of UGT2B17 fm as illustrated for diclofenac in this study. If both Phase I and Phase II enzymes are involved in drug metabolism, such studies can be performed in hepatocytes and intestinal cell models with known UGT2B17 genotypes. If an investigational UGT2B17 substrate drug is moved into clinical trial phases, a PBPK model can be used to assess the impact of UGT2B17 population variability1 and if necessary, a precision clinical trial approach37 can ensure the predictable PK and safety. Because UGT1B17 expression is also regulated by non-genetic factors1, 38, 39, a phenotypic biomarker method can be used for stratifying subjects during clinical trials. We have previously40 established such clinical UGT2B17 biomarker, i.e., the urinary ratio of testosterone glucuronide to androsterone glucuronide (TG/AG). Further, UGT2B17 variability associated with the race (e.g., Asian versus Caucasian) and sex1 should be considered if an investigational UGT2B17 substrate drug is moved into clinical stages. We also recommend that high UGT2B17 may be noted in the regulatory guidelines to ensure safe and effective clinical trials of UGT2B17 substrate drugs.
Diclofenac is one of most commonly used NSAIDs, which is still used OTC in many countries, particularly in Asian population where UGT2B17 gene deletion frequency is 60-90%. Our in vitro data and in vitro to in vivo translation experiments indicate that UGT2B17 is the major contributor in the intestinal metabolism of diclofenac (>80% contribution). In fact, the variable bioavailability (60-90%),41 greater AUC28, 42 and, higher risk of cardiotoxicity in women12, 43 is explained by our data. Similarly, our data explain ~3-fold higher AUC of diclofenac and the requirement of 3-fold reduced dose in Japanese population.28–30, 44 We explored for the first time the potential role of UGT2B17 in the first-pass metabolism of diclofenac. Our protein abundance normalized in vitro UGT screening data confirmed that UGT2B17 is most efficient in DG formation followed by UGT1A9> UGT2B7> UGT1A3, and UGT2B4 (Figure 1A). We also assessed the DG formation rates in HLM and HIM with known UGT2B17 levels, confirmed the significant role of UGT2B17 (Figures 1B and 1C). Consistent with the higher expression of UGT2B17 in the intestine, we found that UGT2B17 efficiently metabolize diclofenac in the intestine. Whereas, despite only 2-3-fold lower expression than UGT2B17 (Figure S3), UGT2B7 activity towards DG formation in the intestine was 21-fold lower (Figure 1). Thus, UGT2B17 plays a major role in the intestinal first-pass metabolism of diclofenac and could be one of the factors of variable diclofenac bioavailability.26,45Although diclofenac PBPK model shown in this study is a preliminary attempt to demonstrate the extreme scenario, the model predicted up to 8-fold higher AUC in the UGT2B17 null- versus high-expressors after oral dose. Because the lack of clinical validation, our PBPK model should not be used for prospective PK predictions. Nevertheless, the model findings are hypothesis-generating for developing a systematic prospective clinical study to investigate the effect of UGT2B17 gene deletion on diclofenac PK. The higher and variable expression of UGT2B17 in the intestinal segments could also lead to variable intestinal tissue concentration resulting in variable intestinal toxicity in the null- versus high-UGT2B17 expressors. Thus, UGT2B17 high expressors may experience lower enterocyte concentration due to higher UGT2B17 metabolism, compared to the null-expressors. Because UGT2B7 is less abundant in the small intestine, UGT2B17 mediated glucuronidation is the major pathway of diclofenac glucuronidation in the intestine.
Interestingly, the standard dose of diclofenac tablet in Japan is 15- 25 mg, which is less than its dose (50-150 mg) in the US,46, 47 which correlates with 90% gene-deletion frequency in the Japanese population as compared to 20% frequency in Caucasians. Diclofenac is also administered as a suppository formulation, however, with a high abundance of UGT2B17 in the colon,6 diclofenac suppository would likely be susceptible to a greater effect of variable UGT2B17. We also quantified the ontogeny of diclofenac metabolizing UGTs using a large cohort (n= 50) of human hepatocytes for the first time. These data are highly applicable to developing pediatric PBPK models for prospective PK prediction in this vulnerable population, where clinical trials are often not conducted. Since UGT2B17 is sparsely expressed in children below 9 years, the pediatric population may experience higher plasma concentrations of diclofenac. Diclofenac use is associated with anaphylactic reactions in the pediatric population.48 Our pediatric hepatocytes activity assay showed lesser DG formation in kids below 6 yr compared to 12-18 yr (Figure 3A). Unlike reported microsomal data1, which are prone to variability due to differential enrichment of endoplasmic reticulum and contamination of mitochondrial and cytosolic proteins, the hepatocyte data were generated using direct cell homogenate in this study. Therefore, the ontogeny data presented here likely represent true age-dependent biological variability without any confounding effect of the sample preparation.
Diclofenac poses a serious risk of cardiotoxicity, which led to the contraindications and warnings for its recommended use by the US FDA and EMA. In addition, hepatotoxicity is also associated with diclofenac use, which is manifested with necrosis cholestatic hepatitis by oxidative stress and mitochondrial damage due to the covalent binding of DG to cellular proteins.49 Sex-dependent differences have been reported in cardiotoxicity and hepatotoxicity of diclofenac, with higher incidences of toxicity in women.12, 43, 50, 51 Banks et al. reported that out of 180 cases of diclofenac adverse events, 79% were female.52 Our previous study showed UGT2B17 abundance is ~3-fold lower in women than men,1 which correlated with ~66% higher predicted plasma concentration in women than men after oral administration of diclofenac, which in turn can explain the higher incidences of diclofenac toxicity in women. However, systematic clinical studies on diclofenac in male versus females are warranted to validate the preliminary PBPK model predictions.
We and others have previously shown that UGT2B17 can be inhibited by drugs and natural products. This suggests that oral administration of UGT2B17 inhibitors such as imatinib6 can influence the metabolism of diclofenac, potentially leading to its higher exposure and toxicity. The elevated risk of acute kidney injury with rhabdomyolysis has been recently reported in patients taking lipid-lowering agents with diclofenac.53 The mechanism of this DDI is unknown, but similar inhibition of CYP2C8 by gemfibrozil glucuronide leading DDI with statins,54 DG is also metabolized by CYP2C855 and has a potential to cause a similar DDI. Further, in vitro and in vivo studies are warranted to evaluate the DDI and toxicity of diclofenac with statins.
In conclusion, we observed that UGT2B17 is the primary enzyme responsible for diclofenac glucuronidation in the intestine, and variability in UGT2B17 abundance leads to differences in DG formation rate. We predicted a significant effect of UGT2B17 CNV, sex, and age on the intestinal metabolism of diclofenac. The findings from this study can be used to develop a mechanistic understanding of UGT2B17-mediated variability in the PK of its substrate drugs. In particular, considering its cardiotoxicity and high frequency of poor UGT2B17 metabolizers in Asian population, the OTC use of diclofenac should be evaluated for its safety. In this direction, the proteomics-informed PBPK model developed in this study can serve a hypothesis-generating purpose to develop prospective clinical trials of diclofenac to evaluate its oral PK and safety in poor UGT2B17 metabolizers. Overall, our study provides crucial direction for clinical researchers to perform prospective clinical studies to further establish links between UGT2B17 polymorphisms and diclofenac toxicity, efficacy, and pharmacokinetics.
Supplementary Material
Study Highlights.
What is the current knowledge on the topic?
UDP-glucuronosyltransferase 2B17 (UGT2B17) shows high interindividual variability in its activity and expression in humans due to factors such as copy number variation (CNV), single nucleotide polymorphisms (SNPs), age, and sex. UGT2B17 is sparsely expressed in children below 9 yr and males express 3-fold higher UGT2B17 protein than females.
What question did this study address?
We characterized the quantitative and tissue-specific contribution of the polymorphic UGT2B17 in the metabolism and pharmacokinetics of diclofenac after oral administration.
What does this study add to our knowledge?
The mechanistic in vitro data and proteomics-informed modeling elucidated the potential role of UGT2B17 population variability on diclofenac oral pharmacokinetics. The study highlights a greater risk of cardiotoxicity in UGT2B17 poor metabolizers.
How might this change clinical pharmacology or translational science?
The hypothesis-generating data from this study can be used to develop clinical studies of diclofenac to evaluate its oral pharmacokinetics and safety in poor UGT2B17 metabolizers, which when validated in clinic may provide directions for precision pharmacotherapy of diclofenac.
Funding
This work was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH) [Grant R01.HD081299] and the Department of Pharmaceutical Sciences, Washington State University, Spokane, WA [startup funds of BP].
Conflict of interest
BP is the co-founder of Precision Quantomics Inc. and recipient of research funding from Bristol Myers Squibb, Genentech, Gilead, Merck, Novartis, Takeda, and Generation Bio. All other authors declared no competing interests for this work.
Footnotes
SUPPORTING INFORMATION
Supplementary information accompanies this paper on the Clinical Pharmacology & Therapeutics website (www.cpt-journal.com).
References
- 1.Bhatt DK, et al. Hepatic Abundance and Activity of Androgen- and Drug-Metabolizing Enzyme UGT2B17 Are Associated with Genotype, Age, and Sex. Drug Metab Dispos. 46, 888–896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xue Y, et al. Adaptive evolution of UGT2B17 copy-number variation. Am J Hum Genet. 83, 337–346 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee CA, et al. Metabolism and Disposition of Verinurad, a Uric Acid Reabsorption Inhibitor, in Humans. Drug Metab Dispos. 46, 532–541 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Deeks ED Belzutifan: First Approval. Drugs. 81, 1921–1927 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Wong NS, et al. Impact of UDP-gluconoryltransferase 2B17 genotype on vorinostat metabolism and clinical outcomes in Asian women with breast cancer. Pharmacogenet Genomics. 21, 760–768 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, et al. Quantitative characterization of UDP-glucuronosyltransferase 2B17 in human liver and intestine and its role in testosterone first-pass metabolism. Biochem Pharmacol. 156, 32–42 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang YH, et al. UGT2B17 genetic polymorphisms dramatically affect the pharmacokinetics of MK-7246 in healthy subjects in a first-in-human study. Clin Pharmacol Ther. 92, 96–102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drug Usage Statistics, United States, 2013 - 2020. https://clincalc.com/DrugStats/Drugs/Diclofenac. Accessed 11/22/2022
- 9.Alfaro RA & Davis DD Diclofenac. In StatPearls (StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC., Treasure Island (FL), 2022). [Google Scholar]
- 10.Odom DM, et al. Relationship between diclofenac dose and risk of gastrointestinal and cardiovascular events: meta-regression based on two systematic literature reviews. Clin Ther. 36, 906–917 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Moore N, Salvo F, Duong M, Blin P & Pariente A Cardiovascular risks associated with low-dose ibuprofen and diclofenac as used OTC. Expert Opin Drug Saf. 13, 167–179 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Schmidt M, Sørensen HT & Pedersen L Diclofenac use and cardiovascular risks: series of nationwide cohort studies. Bmj. 362, k3426 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bresalier RS, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. New England Journal of Medicine. 352, 1092–1102 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Fernandez S The pain game. [Google Scholar]
- 15.Russom M, Fitsum Y, Abraham A & Savage RL Diclofenac and the Risk of Rhabdomyolysis: Analysis of Publications and the WHO Global Pharmacovigilance Database. Drugs Real World Outcomes. 8, 263–275 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nie W, et al. Efficacy and safety of over-the-counter analgesics for primary dysmenorrhea: A network meta-analysis. Medicine (Baltimore). 99, e19881 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Non-steroidal anti-inflammatory drugs and diclofenac reviews. https://www.tga.gov.au/news/safety-alerts/non-steroidal-anti-inflammatory-drugs-and-diclofenac-reviews. Accessed 11/22/2022
- 18.China Diclofenac Investigation Market Report, 2019-2023. - ResearchAndMarkets.com. https://www.businesswire.com/news/home/20190822005517/en/China-Diclofenac-Investigation-Market-Report-2019-2023---ResearchAndMarkets.com. Accessed 11/22/2022
- 19.Diclofenac Sodium Market - Growth, Trends, COVID-19 Impact, and Forecasts (2022 - 2028). https://www.marketwatch.com/press-release/diclofenac-sodium-market---growth-trends-covid-19-impact-and-forecasts-2022---2028-2022-11-15. Accessed 11/22/2022
- 20.Zhang Y, et al. Diclofenac and Its Acyl Glucuronide: Determination of In Vivo Exposure in Human Subjects and Characterization as Human Drug Transporter Substrates In Vitro. Drug Metab Dispos. 44, 320–328 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Green MD, King CD, Mojarrabi B, Mackenzie PI & Tephly TR Glucuronidation of amines and other xenobiotics catalyzed by expressed human UDP-glucuronosyltransferase 1A3. Drug Metab Dispos. 26, 507–512 (1998). [PubMed] [Google Scholar]
- 22.King C, Tang W, Ngui J, Tephly T & Braun M Characterization of rat and human UDP-glucuronosyltransferases responsible for the in vitro glucuronidation of diclofenac. Toxicol Sci. 61, 49–53 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Basit A, et al. Characterization of Differential Tissue Abundance of Major Non-CYP Enzymes in Human. Mol Pharm. 17, 4114–4124 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Prasad B, et al. Toward a Consensus on Applying Quantitative Liquid Chromatography-Tandem Mass Spectrometry Proteomics in Translational Pharmacology Research: A White Paper. Clin Pharmacol Ther. 106, 525–543 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahire DS, Basit A, Karasu M & Prasad B Ultrasensitive Quantification of Drug-metabolizing Enzymes and Transporters in Small Sample Volume by Microflow LC-MS/MS. J Pharm Sci. 110, 2833–2840 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willis JV, Kendall MJ, Flinn RM, Thornhill DP & Welling PG The pharmacokinetics of diclofenac sodium following intravenous and oral administration. Eur J Clin Pharmacol. 16, 405–410 (1979). [DOI] [PubMed] [Google Scholar]
- 27.Mermelstein F, et al. Single-dose and multiple-dose pharmacokinetics and dose proportionality of intravenous and intramuscular HPβCD-diclofenac (Dyloject) compared with other diclofenac formulations. Pharmacotherapy. 33, 1012–1021 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Hynninen VV, et al. Effect of voriconazole on the pharmacokinetics of diclofenac. Fundam Clin Pharmacol. 21, 651–656 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Lissy M, Scallion R, Stiff DD & Moore K Pharmacokinetic comparison of an oral diclofenac potassium liquid-filled soft gelatin capsule with a diclofenac potassium tablet. Expert Opin Pharmacother. 11, 701–708 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Chen C, Bujanover S, Kareht S & Rapoport AM Differential pharmacokinetics of diclofenac potassium for oral solution vs immediate-release tablets from a randomized trial: effect of fed and fasting conditions. Headache. 55, 265–275 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Rømsing J, et al. Pharmacokinetics of oral diclofenac and acetaminophen in children after surgery. Paediatr Anaesth. 11, 205–213 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Haapasaari J, Wuolijoki E & Ylijoki H Treatment of juvenile rheumatoid arthritis with diclofenac sodium. Scand J Rheumatol. 12, 325–330 (1983). [DOI] [PubMed] [Google Scholar]
- 33.Xie C, et al. Metabolic Profiling of the Novel Hypoxia-Inducible Factor 2α Inhibitor PT2385 In Vivo and In Vitro. Drug Metab Dispos. 46, 336–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoch M, et al. Pharmacokinetics of Asciminib When Taken With Imatinib or With Food. Clin Pharmacol Drug Dev. 11, 207–219 (2022). [DOI] [PubMed] [Google Scholar]
- 35.Deeks ED Asciminib: First Approval. Drugs. 82, 219–226 (2022). [DOI] [PubMed] [Google Scholar]
- 36.Sun D, Chen G, Dellinger RW, Sharma AK & Lazarus P Characterization of 17-dihydroexemestane glucuronidation: potential role of the UGT2B17 deletion in exemestane pharmacogenetics. Pharmacogenet Genomics. 20, 575–585 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma S, et al. Dimethandrolone, a Potential Male Contraceptive Pill, is Primarily Metabolized by the Highly Polymorphic UDP-Glucuronosyltransferase 2B17 Enzyme in Human Intestine and Liver. Drug Metab Dispos. 50, 1493–1500 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pâquet S, et al. Differential expression of the androgen-conjugating UGT2B15 and UGT2B17 enzymes in prostate tumor cells during cancer progression. J Clin Endocrinol Metab. 97, E428–432 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Yang YA & Yu J Current perspectives on FOXA1 regulation of androgen receptor signaling and prostate cancer. Genes Dis. 2, 144–151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, et al. Normalized testosterone glucuronide as a potential urinary biomarker for highly variable UGT2B17 in children 7–18 years. Clinical Pharmacology & Therapeutics. 107, 1149–1158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davies NM & Anderson KE Clinical pharmacokinetics of diclofenac: therapeutic insights and pitfalls. Clinical pharmacokinetics. 33, 184–213 (1997). [DOI] [PubMed] [Google Scholar]
- 42.Crook PR, Willis JV, Kendall MJ, Jack DB & Fowler PD The pharmacokinetics of diclofenac sodium in patients with active rheumatoid disease. Eur J Clin Pharmacol. 21, 331–334 (1982). [DOI] [PubMed] [Google Scholar]
- 43.Farkouh A, et al. Sex-Related Differences in Drugs with Anti-Inflammatory Properties. J Clin Med. 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimamoto J, et al. Lack of differences in diclofenac (a substrate for CYP2C9) pharmacokinetics in healthy volunteers with respect to the single CYP2C9*3 allele. Eur J Clin Pharmacol. 56, 65–68 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Kendall MJ, Thornhill DP & Willis JV Factors affecting the pharmacokinetics of diclofenac sodium (Voltarol). Rheumatol Rehabil. Suppl 2, 38–46 (1979). [PubMed] [Google Scholar]
- 46.diclofenac sodium. https://www.pmda.go.jp/PmdaSearch/iyakuDetail/GeneralList/1147002F1. Accessed 11/22/2022
- 47.Voltaren - (diclofenac sodium enteric-coated tablets). https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/019201s038lbl.pdf. Accessed 11/22/2022
- 48.Huang WY, Chiu TM, Kuo SF, Chung WH & Tsai YG A Case Report of a 3-Year-Old Child With Anaphylactic Shock After a Diclofenac Suppository Confirmed by Serial Tryptase and a Basophil Activation Test. Front Pediatr. 9, 802715 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boelsterli UA Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol Appl Pharmacol. 192, 307–322 (2003). [DOI] [PubMed] [Google Scholar]
- 50.NIH. LiverTox: clinical and research information on drug-induced liver injury. https://www.ncbi.nlm.nih.gov/books/NBK547953/#Diclofenac.CASE_REPORTS. Accessed 12/18/2022
- 51.Diclofenac Adverse Events and Side Effects Reported to the FDA (AERS). Accessed 11/22/2022
- 52.Banks AT, Zimmerman HJ, Ishak KG & Harter JG Diclofenac-associated hepatotoxicity: analysis of 180 cases reported to the Food and Drug Administration as adverse reactions. Hepatology. 22, 820–827 (1995). [PubMed] [Google Scholar]
- 53.Gorriz J, et al. Rhabdomyolysis and acute renal failure associated with gemfibrozil therapy. Nephron. 74, 437–438 (1996). [DOI] [PubMed] [Google Scholar]
- 54.Shitara Y, Hirano M, Sato H & Sugiyama Y Gemfibrozil and its glucuronide inhibit the organic anion transporting polypeptide 2 (OATP2/OATP1B1:SLC21A6)-mediated hepatic uptake and CYP2C8-mediated metabolism of cerivastatin: analysis of the mechanism of the clinically relevant drug-drug interaction between cerivastatin and gemfibrozil. J Pharmacol Exp Ther. 311, 228–236 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Kumar S, et al. Extrapolation of diclofenac clearance from in vitro microsomal metabolism data: role of acyl glucuronidation and sequential oxidative metabolism of the acyl glucuronide. J Pharmacol Exp Ther. 303, 969–978 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.