

Abstract
BACKGROUND:
Despite successful preclinical treatment studies to improve neurocognition in the Ts65Dn mouse model of Down syndrome, translation to humans has failed. This raises questions about the appropriateness of the Ts65Dn mouse as the gold standard. We used the novel Ts66Yah mouse that carries an extra chromosome and the identical segmental Mmu16 trisomy as Ts65Dn without the Mmu17 non-Hsa21 orthologous region.
METHODS:
Forebrains from embryonic day 18.5 Ts66Yah and Ts65Dn mice, along with euploid littermate controls, were used for gene expression and pathway analyses. Behavioral experiments were performed in neonatal and adult mice. Because male Ts66Yah mice are fertile, parent-of-origin transmission of the extra chromosome was studied.
RESULTS:
Forty-five protein-coding genes mapped to the Ts65Dn Mmu17 non-Hsa21 orthologous region; 71%–82% are expressed during forebrain development. Several of these genes are uniquely overexpressed in Ts65Dn embryonic forebrain, producing major differences in dysregulated genes and pathways. Despite these differences, the primary Mmu16 trisomic effects were highly conserved in both models, resulting in commonly dysregulated disomic genes and pathways. Delays in motor development, communication, and olfactory spatial memory were present in Ts66Yah but more pronounced in Ts65Dn neonates. Adult Ts66Yah mice showed milder working memory deficits and sex-specific effects in exploratory behavior and spatial hippocampal memory, while long-term memory was preserved.
CONCLUSIONS:
Our findings suggest that triplication of the non-Hsa21 orthologous Mmu17 genes significantly contributes to the phenotype of the Ts65Dn mouse and may explain why preclinical trials that used this model have unsuccessfully translated to human therapies.
Ninety-five percent of cases of Down syndrome (DS) are caused by the presence of a freely segregating third copy of human chromosome 21 (Hsa21), which carries 235 protein-coding genes, 411 noncoding genes, and 188 pseudogenes (https://useast.ensembl.org). The multigenic nature of DS complicates the understanding of its etiology and the development of mouse models that recapitulate the human karyotype, genotype, and phenotype. Hsa21 orthologous genes map to 3 syntenic regions on mouse chromosomes (Mmu) 10 (from Pdxk to Prmt2, 2.1 Mb, 39 genes), 16 (from Lipi to Zbtb21, 22.5 Mb, 119 genes), and 17 (from Umodl1 to Rrp1b, 1.1 Mb, 19 genes). Because Mmu16 carries the largest number of Hsa21 orthologous genes, it has been used to generate several partial trisomy models, including the Ts(1716)65Dn/J (Ts65Dn) mouse (1–5).
For the past 25 years, the Ts65Dn mouse has been the gold standard model in which most preclinical treatment studies have been conducted, many of which have shown promising positive effects on brain and behavior phenotypes. Translation of these treatments to human trials, however, has not been successful (6,7). As in humans, the Ts65Dn mouse carries a freely segregating extra chromosome that was generated by a translocation of the distal region of Mmu16 onto the centromeric region of Mmu17. This results in triplication of Hsa21 orthologous genes and ~50–60 Mmu17 non-Hsa21 orthologous protein-coding genes (8,9).
The contribution of the triplicated non-Hsa21 orthologous Mmu17 genes to the Ts65Dn phenotype has not been specifically elucidated. Here, we used the new Ts66Yah model, in which CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 was used to eliminate non-orthologous Mmu17 genes from the Ts65Dn extra chromosome (10), to investigate the effects of the Mmu17 non-Hsa21 orthologous genes. We hypothesized that these genes may trigger transcriptional and pathway dysregulation that is unrelated to DS and may be one reason why therapies that looked promising in the Ts65Dn mouse failed to translate in humans with DS.
METHODS AND MATERIALS
Animal Rederivation and Breeding
All experiments were approved by the National Human Genome Research Institute Institutional Animal Care and Use Committee (Protocol G-17-1). Mice were housed in standard cages with food and water ad libitum in a controlled environment (temperature: 20 °C; humidity: 60%; 12 hour light/dark cycle with lights on at 7:00 am). The Ts65Dn and Ts66Yah mouse strains used in these studies were all maintained on the same B6C3H mixed genetic background (Jackson Laboratory stock No. 003647).
We obtained 3 Ts66Yah founder female mice from the Herault Laboratory on a B6C3B genetic background (with the C3B line as a C3H/HeH congenic line for the BALB/c allele at the Pde6b locus) (11). The founder Ts66Yah females were mated with B6EiC3Sn.BLiAF1/J (F1 hybrid) males. The sperm of 4 different Ts66Yah males were then used via in vitro fertilization to expand the colony.
Ts(1716)65Dn/DnJ (Ts65Dn; stock No. 005252) mice were obtained from the Jackson Laboratory. Because Ts65Dn male mice are infertile (12,13), Ts65Dn female mice were mated with B6EiC3Sn.BLiAF1/J (F1 hybrid) males. Thus, the extra chromosome could only be transmitted through the female parent.
To investigate the effects of parent-of-origin transmission of the extra chromosome, 2 different breeding schemes were used for the Ts66Yah mice: 1) cohort 1 was generated by mating Ts66Yah females with F1 males to mimic breeding in Ts65Dn mice and 2) cohort 2 was generated by mating Ts66Yah males with F1 euploid (Eup) females. The F1 Eup dams more accurately represent human pregnancies in which the mother is euploid.
Timed matings were set up as described previously (14,15). On embryonic day 18.5 (E18.5), pregnant females were euthanized, and embryonic forebrains were dissected and snap frozen in liquid nitrogen for gene expression studies (Ts66Yah = 45, EupTs66Yah = 59, Ts65Dn = 49, and EupTs65Dn = 44).
Genotyping and Gene Expression Studies
To determine genotype and sex, 50 ng of purified DNA from tail snips or ear punches was analyzed using multiplex polymerase chain reaction as described previously (15). Primers specific to Ts66Yah, Ts65Dn, and SRY were used (10,14).
RNA was isolated from E18.5 forebrains in both strains and hybridized on Clariom S HT arrays (Thermo Fisher Scientific). Gene expression analysis was performed on the normalized data as described previously (14,15). A Benjamini-Hochberg false discovery rate of 10% was used for multiple comparison correction of differentially expressed (DEX) genes. The marginally expressed genes (expression ratios < 0.8 and > 1.2 and raw p values < .01) were used for pathway analyses as described previously (14,15). Pathway analysis was performed using Ingenuity Pathway Analysis (IPA) and the Database for Annotation, Visualization, and Integrated Discovery (DAVID).
Behavioral Studies
Detailed descriptions of behavioral tests performed are in the Supplement. These were conducted in the light phase in neonates and adults (14,15). For neonates (between postnatal days 2 and 12), the open field was used to investigate motor development, ultrasonic vocalization (USV) to investigate communication development, and the homing test to examine olfactory spatial memory. Animal numbers for neonatal studies were Ts66Yah (11 females and 19 males for cohort 1 and 21 females and 40 males for cohort 2); Ts65Dn (17 females and 16 males); and Eup littermates (26 females and 19 males for EupTs66Yah cohort 1; 50 females and 37 males for EupTs66Yah cohort 2; and 35 females and 22 males for EupTs65Dn).
For adult mice, exploratory behavior (open field), motor coordination (rotarod), working memory (Y-maze), long-term memory (novel object recognition [NOR]), contextual hippocampal memory (fear conditioning), and hippocampal-dependent spatial (Morris water maze [MWM]) tests were examined in Ts66Yah and Eup littermates starting at 3–4 months of age. Because our group and others previously reported behavioral deficits in Ts65Dn adult mice using these tests (14,16,17), we did not repeat them. All behavioral tests using Ts66Yah mice were performed in cohorts 1 and 2, except for the MWM, which was only performed in cohort 2. For all experiments, the investigator was blinded to the genotype. Cohort 1 included 11 females and 17 males for Ts66Yah and 24 females and 17 males for Eup. Cohort 2 included 13 females and 36 males for Ts66Yah and 13 females and 34 males for Eup.
Statistical Analysis
In all analyses, trisomic mice were compared with their Eup littermates and matched by sex. Data analysis was performed using GraphPad Prism software (https://www.graphpad.com/).
Differences between genotypes or sexes were calculated using the t test when the data were normally distributed and the nonparametric Mann-Whitney test when the normality assumption was not satisfied. Differences between 3 or more groups were tested using one-way analysis of variance (ANOVA) when the data were normally distributed. The nonparametric Kruskal-Wallis test was used when the normality assumption was not satisfied. Repeated-measures ANOVA and mixed-effects model were used for time course analyses. Statistically significant results were further analyzed using the post hoc Tukey-Kramer or Conover test, as indicated. A p value < .05 was considered statistically significant.
RESULTS
Expression of Mmu16 Trisomic Genes in Embryonic Forebrain
The Ts66Yah and Ts65Dn models are both trisomic for the distal Mmu16 region encompassing Mrp139 to Zbtb21 (104 genes). The forebrains from both strains showed upregulation of most genes in this region (Figure 1A; Table S1). The average expression ratios for the triplicated genes on Mmu16 in the brains of Ts65Dn and Ts66Yah embryos were 1.21 and 1.14, respectively (range 1.15–1.5). In Ts65Dn and Ts66Yah E18.5 forebrains, 95% and 91% of the respective overexpressed Mmu16 genes overlapped (Figure 2A).
Figure 1.
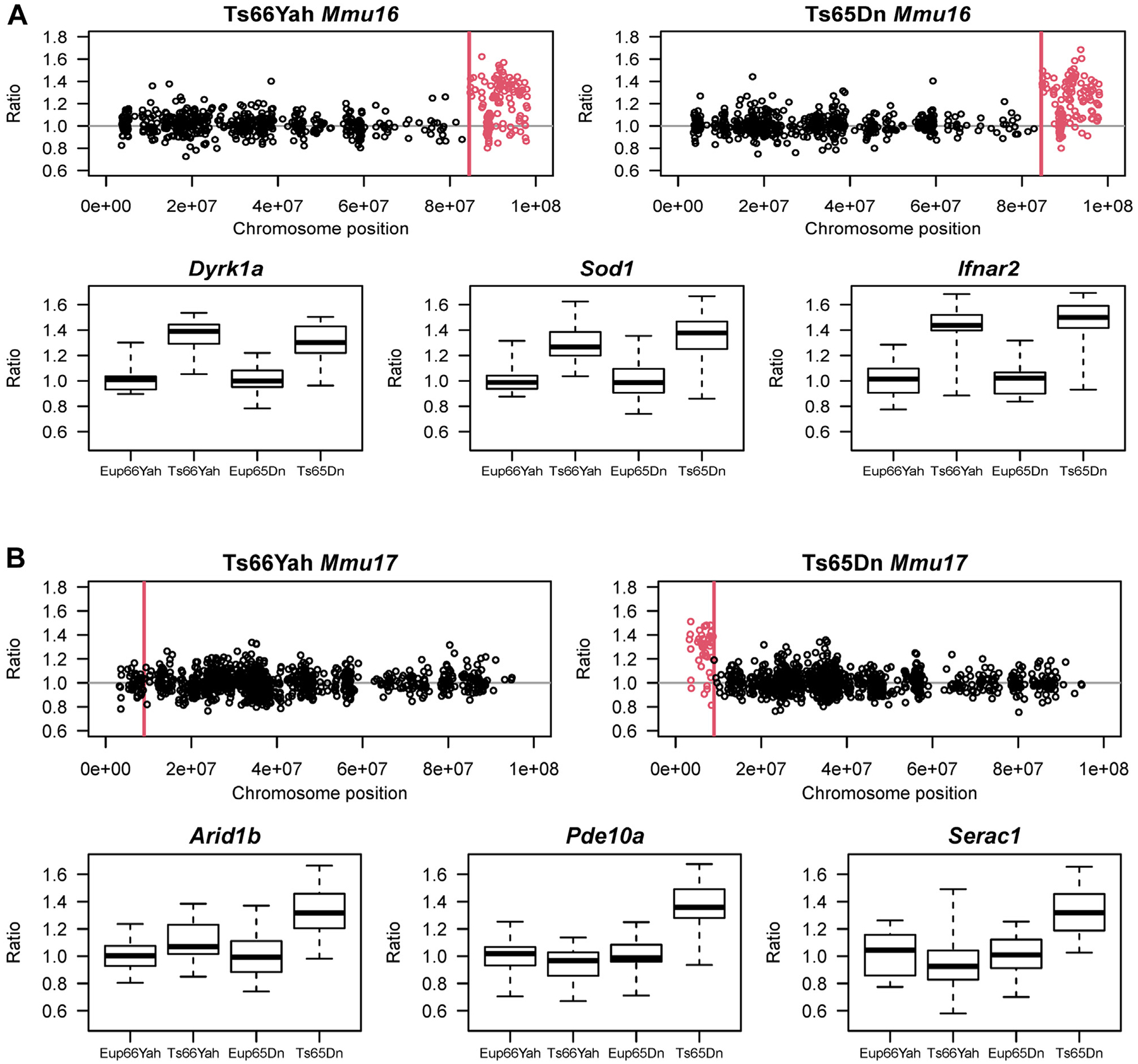
Expression of Mmu16 orthologous and Mmu17 non-Hsa21 orthologous genes in the Ts66Yah and Ts65Dn E18.5 embryonic forebrain. (A) (Top) Chromosomal map showing the overexpression of Mmu16 trisomic genes (red open circles) in the Ts66Yah and Ts65Dn embryonic forebrain. Chromosomal position is represented on the x-axis as the distance (in base pair) from the start of Mmu16. For both Ts65Dn and Ts66Yah, the Mmu16 trisomic region starts at Mrpl39 located 84,514,464 bp (84.5 Mb) distally from the start of chromosome 16. (Bottom) Examples of Mmu16 orthologous genes that are overexpressed in both mouse models, including Dyrk1a, Sod1, and Ifnar2. (B) (Top) Chromosomal map showing the expression of the Mmu17 non-Hsa21 orthologous genes trisomic only in the Ts65Dn mouse model. As expected, these genes were only overexpressed in the Ts65Dn embryonic forebrain (open red circles) but not in the Ts66Yah embryonic forebrain. (Bottom) Expression of some key Mmu17 non-Hsa21 orthologous genes (Arid1b, Pde10a, and Serac1) in the Ts66Yah and Ts65Dn embryonic forebrain.
Figure 2.
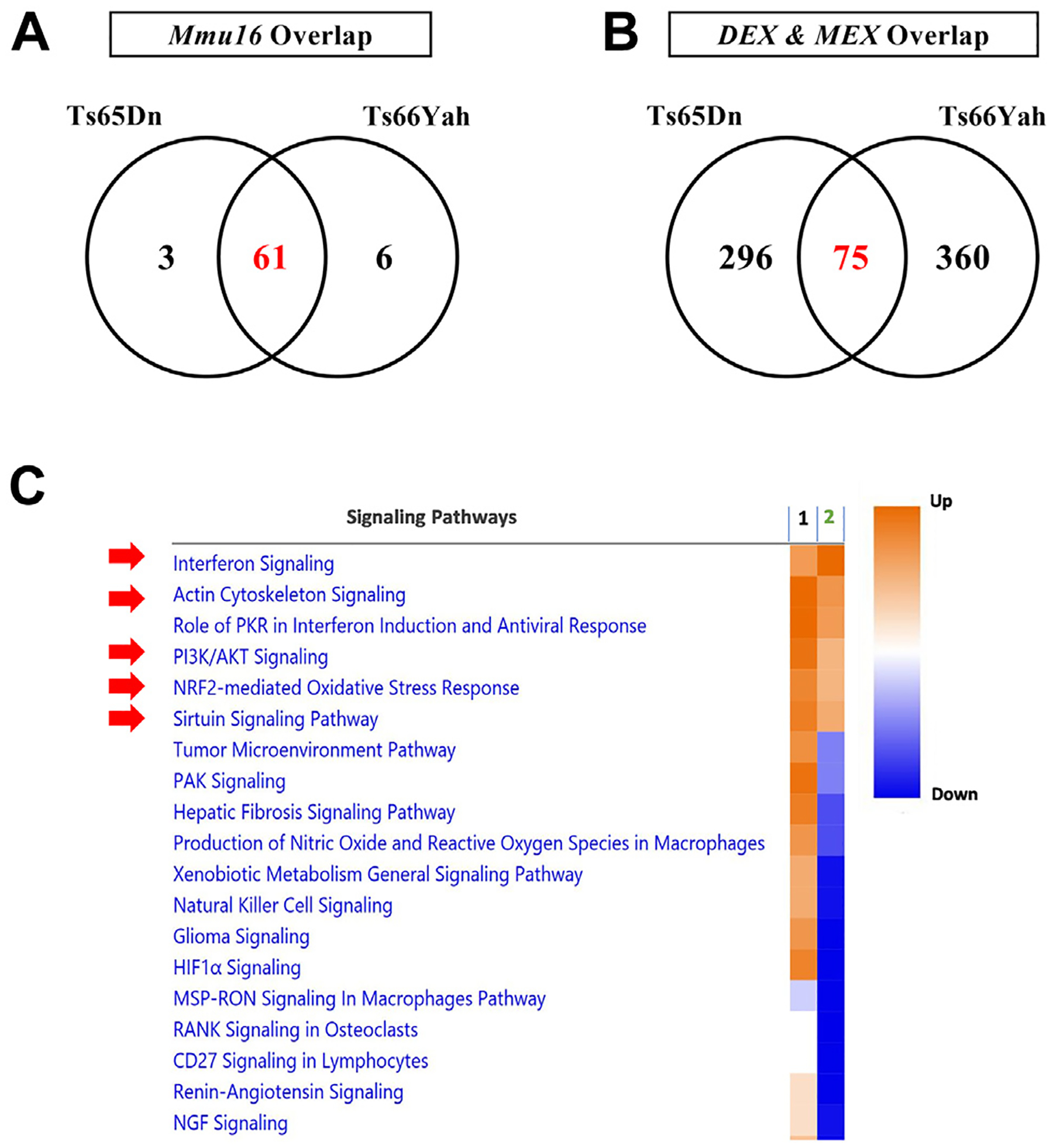
Overlap in dysregulated genes and pathways in the Ts65Dn and Ts66Yah E18.5 embryonic forebrains. (A) Overexpression of Mmu16 orthologous genes is conserved between the Ts65Dn and Ts66Yah mouse models. (B) Little overlap in the DEX and MEX genes is present between the Ts65Dn and Ts66Yah models, despite the conserved overexpression of Mmu16 trisomic genes. (C) Dysregulated pathways in the Ts65Dn and Ts66Yah embryonic forebrain. 1 = Ts66Yah and 2 = Ts65Dn. As a result of the distinct secondary genome-wide differences in dysregulated genes, Ts65Dn and Ts66Yah mice share very few dysregulated signaling pathways. DEX, differentially expressed; MEX, marginally expressed.
Expression of Mmu17 Non-Hsa21 Orthologous Genes in Embryonic Forebrain
The Mmu17 non-Hsa21 orthologous (hereafter referred to as Mmu17 non-orthologous) region in the Ts65Dn mouse encompasses 45 protein-coding and 81 non–protein-coding genes (University of California Santa Cruz and Ensembl Genome Browsers, GRCm39 build). Using probe signal intensity from our microarray dataset and publicly available data from the Mouse Genome Informatics database, we found that most of the protein-coding genes in this region (82% in our microarray data and 71% in the Mouse Genome Informatics data) are expressed in the developing forebrain (Table S2).
In the Ts65Dn embryonic forebrain, the average expression ratios of these Mmu17 non-orthologous genes was 1.28 compared with Eup (Figure 1B; Table S1). When compared with our previously published gene expression data in the Ts65Dn embryonic forebrain at E15.5 (14), we identified 14 Mmu17 non-orthologous genes that were consistently overexpressed at both E15.5 and E18.5 (Table 1; Table S1). In contrast, the average expression ratio of genes that map to the Mmu17 Scaf8 to Pde10a region was 0.99 in the Ts66Yah embryonic brain versus Eup.
Table 1.
List of Mmu17 Non-Hsa21 Orthologous Genes Overexpressed in Ts65Dn Embryonic Forebrain at Mid and Late Gestation
| Gene Symbol | Ts65Dn E15.5 Ratio | Ts65Dn E18.5 Ratio | Ts66Yah E18.5 Ratio | Gene Function During Embryonic Development |
|---|---|---|---|---|
| Arid1b | 1.330 | 1.342 | 1.104 | Belongs to the neural progenitor-specific chromatin remodeling complex (np-BAF) and the neuron-specific chromatin remodeling complex (nBAF). During brain development, Arid1b regulates the switch from neural progenitors to postmitotic neurons. Haploinsufficiency of ARID1B in humans is associated with Coffin-Siris syndrome, autism spectrum disorder, intellectual disability, and corpus callosum agenesis. |
| Pde10a | 1.322 | 1.387 | 0.934 | Regulates signal transduction by regulating the intracellular concentration of cyclic nucleotides. PDE10A protein plays a crucial role in striatal cAMP signaling in the regulation of basal ganglia circuitry and movement. Homozygous mice on the C57BL/6N genetic background (PDE10AC57) exhibit multiple behavioral abnormalities, decreased locomotor activity, and delayed learning. In humans, mutations in PDE10A cause hyperkinetic movement disorders. |
| Serac1 | 1.340 | 1.331 | 0.931 | Plays an essential role in mitochondrial function and intracellular cholesterol trafficking through regulation of phosphatidylglycerol remodeling. In humans, mutations in SERAC1 are associated with deafness, encephalopathy, and Leigh-like syndrome. Individuals with SERAC1 mutations have psychomotor retardation, dystonia, and brain lesions on MRI. |
| T (Tbxt) | 1.204 | 1.214 | 1.028 | Involved in transcriptional regulation of genes involved in mesoderm formation and differentiation during embryonic development. Allelic variants of TBXT are associated with higher risk of neural tube defects and lumbosacral myelomeningocele. |
| Snx9 | 1.195 | 1.335 | 1.073 | Plays an important role in cell cycle progression through mitosis and cytokinesis by regulating endocytosis. SNX9 protein also promotes degradation of EGFR after EGF signaling and interacts with dynamin-1 and N-WASP to coordinate synaptic vesicle endocytosis. In humans, mutations in SNX9 were associated with higher risk of schizophrenia. |
| Ezr | 1.171 | 1.472 | 1.076 | Strongly expressed in placental syncytiotrophoblast and is involved in several cellular functions, including cell adhesion, migration, and the organization of cell surface structures. Although rare, mutations in EZR are associated with intellectual and speech delay, hypotonia, and severely delayed psychomotor development. Imaging studies in some individuals carrying EZR mutations reported brain atrophy, enlarged ventricles, and hypoplasia of the corpus callosum. |
| Rps6ka2 | 1.368 | 1.481 | 1.083 | Protein kinase that acts downstream of ERK signaling and plays an important role in stress-induced response and regulation of translation, cell proliferation, and differentiation. During embryonic development, Rps6ka2 is highly expressed in the neural and sensory tissues. The role of RSP6KA2 protein during human and mouse brain development is unknown. |
| Gtf2h5 | 1.329 | 1.398 | 0.991 | GTF2H5 protein is a component of the transcription and DNA repair factor IIH core (TFIIH) complex which is involved in general and transcription-coupled NER of damaged DNA. The role of GTF2H5 protein during brain development is unknown. |
| Scaf8 | 1.278 | 1.362 | 0.968 | Plays an important role in transcription regulation by preventing early mRNA termination. |
| Tfb1m | 1.355 | 1.512 | 1.084 | Methyltransferase required for basal transcription of mitochondrial DNA through interaction with POLRMT and TFAM. In humans, TFB1M point mutation A1555G in mitochondrial DNA causes maternally inherited deafness. The role of TFB1M protein in brain development is unknown. |
| Rsph3a | 1.215 | 1.392 | 1.046 | Acts as a protein kinase A-anchoring protein that scaffolds the cAMP-dependent protein kinase holoenzyme. Interacts with LRRC23 and RSPH3B to elicit the correct organization of radial spokes during spermatogenesis. The role of RSPH3A protein during brain development is unknown. |
| Tmem242 | 1.362 | 1.367 | 0.977 | Scaffold protein that participates in the c-ring assembly of mitochondrial ATP synthase. In humans, copy number variants of a 16p11.2 region encompassing TMEM242 were associated with neurodevelopmental delay. |
| Tulp4 | 1.296 | 1.366 | 0.987 | Member of the tubby-like family of proteins, but its function is poorly studied. Vieira et al. (37) found a strong association between the rs651333 TULP4 variant and cleft palate. |
| Zdhhc14 | 1.307 | 1.440 | 1.046 | Palmitoyltransferase that plays a role in cell differentiation and apoptosis. In mice, ZDHHC14 protein is strongly expressed in the developing hippocampus and interacts with PSD93 and Kv1 channels to regulate action potential in hippocampal neurons. |
cAMP, cyclic adenosine monophosphate; MRI, magnetic resonance imaging; mRNA, messenger RNA; NER, nucleotide excision repair.
Differentially and Marginally Expressed Genes
Ts66Yah E18.5 forebrains had 97 differentially expressed (DEX) genes (91 up- and 6 downregulated); 54 (55.7%) mapped to the Mmu16 trisomic region, while the 43 remaining DEX genes were disomic (Table S3). Ts65Dn E18.5 forebrains had 91 DEX genes (87 up- and 4 downregulated); 54 (59.3%) mapped to the Mmu16 trisomic region. Twenty-five of the DEX genes in the Ts65Dn E18.5 forebrain mapped to the Mmu17 non-orthologous region while only 12 DEXs were disomic (Table S3). Although Ts66Yah and Ts65Dn mice shared 90% of Mmu16 DEX genes, only 5 disomic protein-coding DEX genes overlapped between these 2 models, including Fam173a, Ifitm2, Rbm3, Pik3r3, and Cdkl3 (Table S3).
Comparison of marginally expressed genes in both strains showed overlap of only 63 Mmu16 trisomic and 12 disomic genes, including the 5 common disomic genes cited above (Figure 2B).
Dysregulated Signaling Pathways
Ts65Dn embryonic forebrains had more dysregulated pathways and cellular processes than Ts66Yah using IPA and DAVID (Table S4). Ts66Yah analyses demonstrated upregulation of several inflammation-associated pathways, including T-cell exhaustion, TH1, interferon signaling, and STAT3 signaling. Additionally, upregulation of the NRF2-mediated oxidative stress response, FGF signaling, and HIF1A signaling was observed. In contrast, downregulated pathways included translation initiation, G-protein signaling, and aryl hydrocarbon receptor signaling (Figure 2C).
Cellular processes and signaling pathways that were commonly dysregulated in both models included interferon signaling, sirtuin signaling, PI3K/AKT signaling, NRF2-mediated oxidative stress response, cytoskeleton organization, and zinc ion binding (Figure 2C; Table S4).
Neonatal Behavioral Testing
Neonatal behavioral data details are presented in Table S5.
Motor Development (Open Field).
During the first week of life, the movement of pups is limited and restricted to body rotation. As they acquire more physical strength, pups increasingly move and explore their environment. We measured movement and rotation as a proxy for motor development and maturation.
Mixed-effects analysis demonstrated that cohort 1 Ts66Yah males exhibited significant delays in motor development compared with Eup as evidenced by a lower number of body rotations and reduced total distance traveled (Figure 3A, B). Ts66Yah females, however, showed normal motor development profiles (Figure 3C, D). For cohort 2, both male and female pups showed delayed motor development with significant reduction in the number of body rotations (Figure 3).
Figure 3.
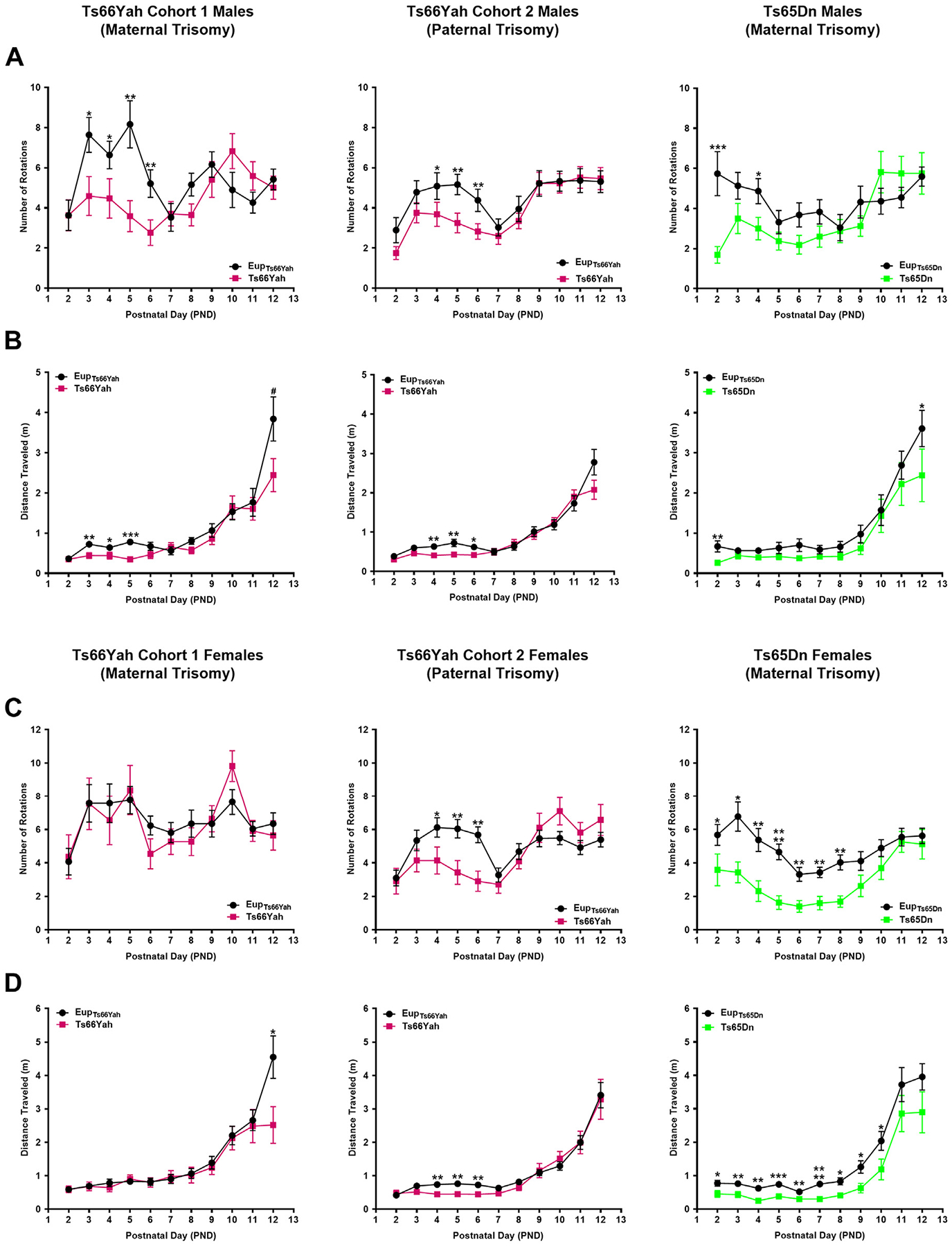
Neonatal motor development in the Ts66Yah and Ts65Dn mouse models. Comparison of the number of body rotations (A, C) and total distance traveled (B, D) in cohort 1 (from trisomic mothers) Ts66Yah, cohort 2 (from trisomic fathers) Ts66Yah, and Ts65Dn female and male pups between postnatal days 2 and 12. Significant differences are indicated as *p < .05, **p < .01, ***p < .001, and ****p < .0001.
In Ts65Dn mice, although females were more severely affected, neonates from both sexes displayed motor development delays versus their Eup littermates with a significant reduction in the number of body rotations and total distance traveled (Figure 3).
Communication Development (Ultrasonic Vocalization).
During the first postnatal week, the number of ultrasonic vocalizations (USVs) uttered by pups as a means of communication with their mother increases. It then steadily decreases during the second week to reach a minimum around the time the pups’ eyes open. Over these 2 weeks, pups develop a repertoire of USVs that can be classified into categories based on their duration and complexity.
In Ts66Yah cohort 1, both trisomic male and female neonates uttered significantly more USVs than Eup starting at the end of the first postnatal week (Figures 4, 5A). Similarly, cohort 2 Ts66Yah female pups exhibited increased USVs versus Eup, while Ts66Yah and Eup male neonates had similar numbers of USVs throughout the neonatal period (Figures 4, 5A).
Figure 4.
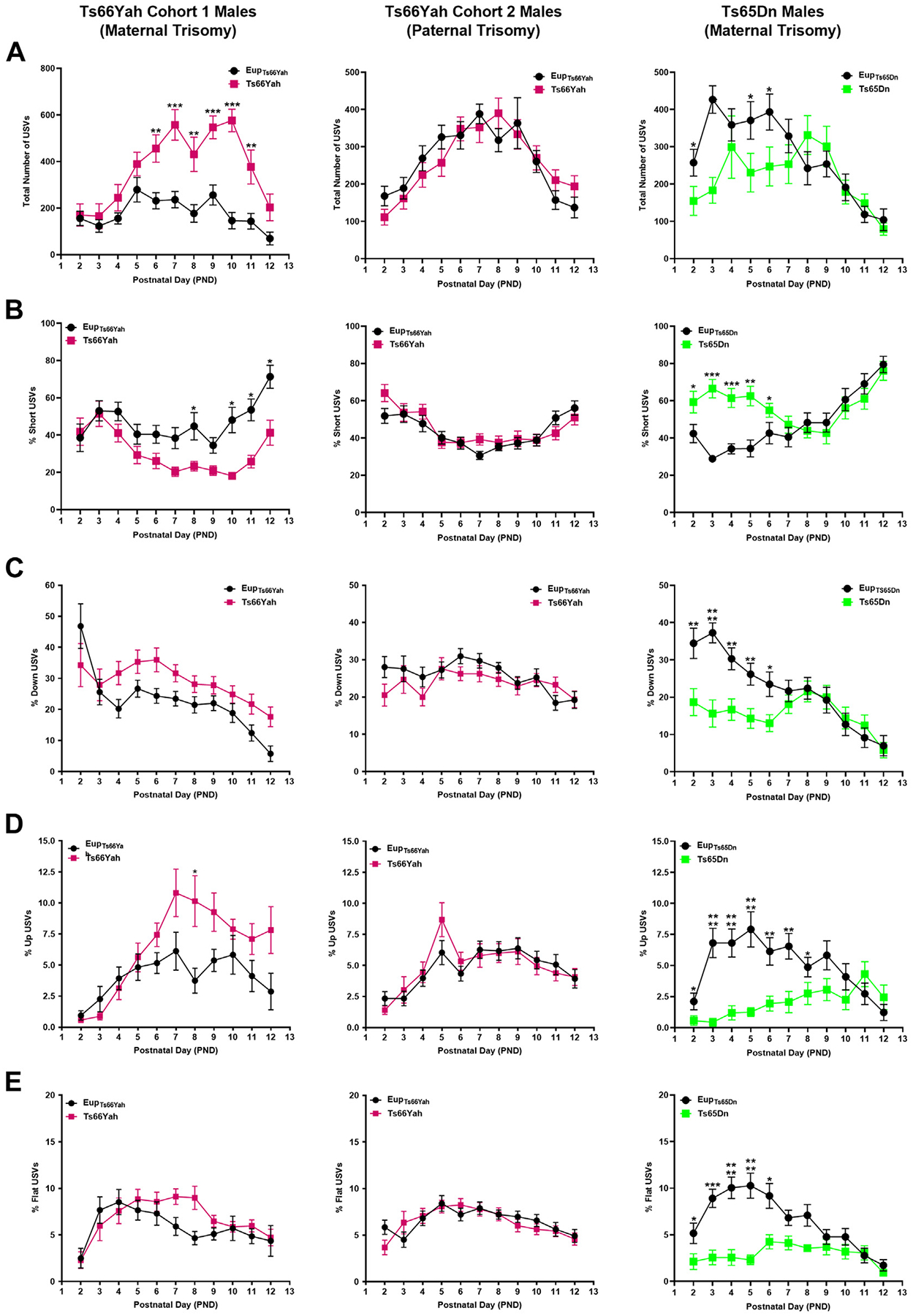
Communication in Ts66Yah and Ts65Dn male neonates. Comparison of the total number of USVs (A) and the percent of short (B), down (C), up (D), and flat (E) USVs as a proxy for neonatal communication in cohort 1 (from trisomic mothers) Ts66Yah, cohort 2 (from trisomic fathers) Ts66Yah, and Ts65Dn male neonatal mice between postnatal days 2 and 12. Significant differences are indicated as *p < .05, **p < .01, ***p < .001, and ****p < .0001. USV, ultrasonic vocalization.
Figure 5.
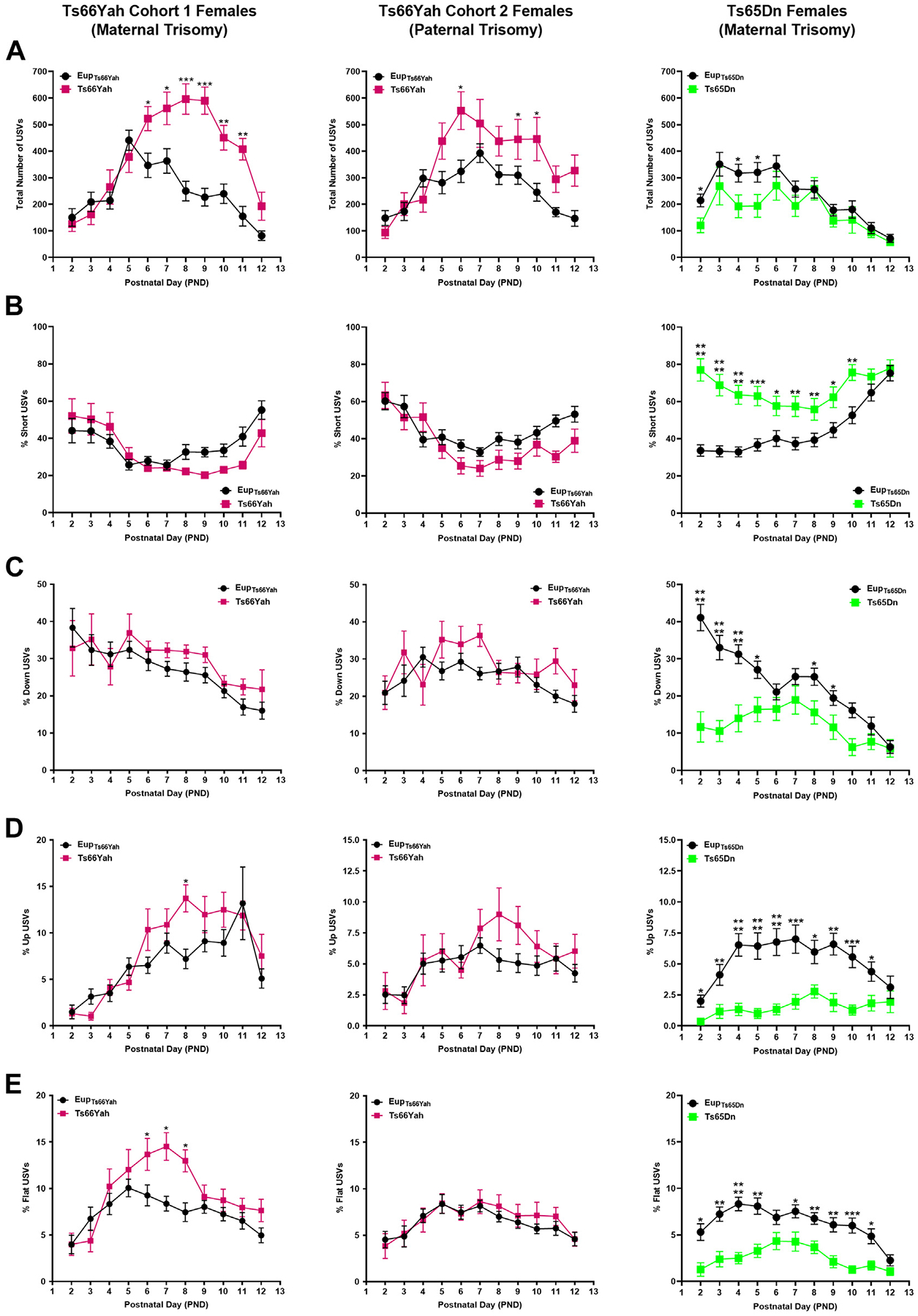
Communication in Ts66Yah and Ts65Dn female neonates. Comparison of the total number of USVs (A) and the percent of short (B), down (C), up (D), and flat (E) USVs as a proxy for neonatal communication in cohort 1 (from trisomic mothers) Ts66Yah, cohort 2 (from trisomic fathers) Ts66Yah, and Ts65Dn female neonatal mice between postnatal days 2 and 12. Significant differences are indicated as *p < .05, **p < .01, ***p < .001, and ****p < .0001. USV, ultrasonic vocalization.
When different USV categories were examined, cohort 1 male and female Ts66Yah pups uttered fewer short calls and more down, up, and flat USVs than Eup. These deficits were more pronounced in males (Figures 4, 5B). Similar trends were also observed in cohort 2 Ts66Yah female, but not in male, pups (Figures 4, 5).
By contrast, in Ts65Dn mice compared with Eup, the total number of USVs was significantly lower in both sexes during the first postnatal week (Figures 4, 5A). Analysis of the different USV categories demonstrated that the Ts65Dn male and female pups uttered a significantly higher proportion of short USVs but significantly less down, up, flat, step down, and step up when compared with Eup (Figures 4, 5B–E).
Spatial Olfactory Learning and Memory (Homing Test).
To investigate spatial olfactory memory, we used a modified version of the homing test in which pups were placed in the center of an open field arena with home bedding (home zone) on one side and clean bedding (clean zone) on the opposite side. Because the pups’ eyelids are still closed, they rely on their spatial olfactory navigation to reach the home zone instead of moving toward the clean zone.
Euploid neonates from both strains reached the home zone faster and spent significantly more time there than the clean zone, suggesting robust learning (Figure S1).
Ts66Yah pups of both sexes and in both cohorts exhibited increased latency to reach the home zone and spent less time there than Eup, but this did not reach statistical significance (Figure S1).
By comparison, both male and female Ts65Dn pups showed severe deficits in spatial olfactory memory as demonstrated by the increased latency to reach the home zone and the total time spent there versus Eup (Figure S1).
Adult Behavioral Testing
Adult behavioral data details are presented in Table S5.
Exploratory Behavior (Open Field).
For cohort 1, Ts66Yah males exhibited mildly hyperactive behavior in the open field test, as demonstrated by an increased distance traveled and average speed compared with Eup (Figure 6A). Male mice from cohort 1 and their Eup littermates explored the periphery of the arena more than its center, however, Ts66Yah mice traveled on average more in both zones (Figure 6B). Ts66Yah females exhibited normal exploratory behavior (Figure 6A, B).
Figure 6.
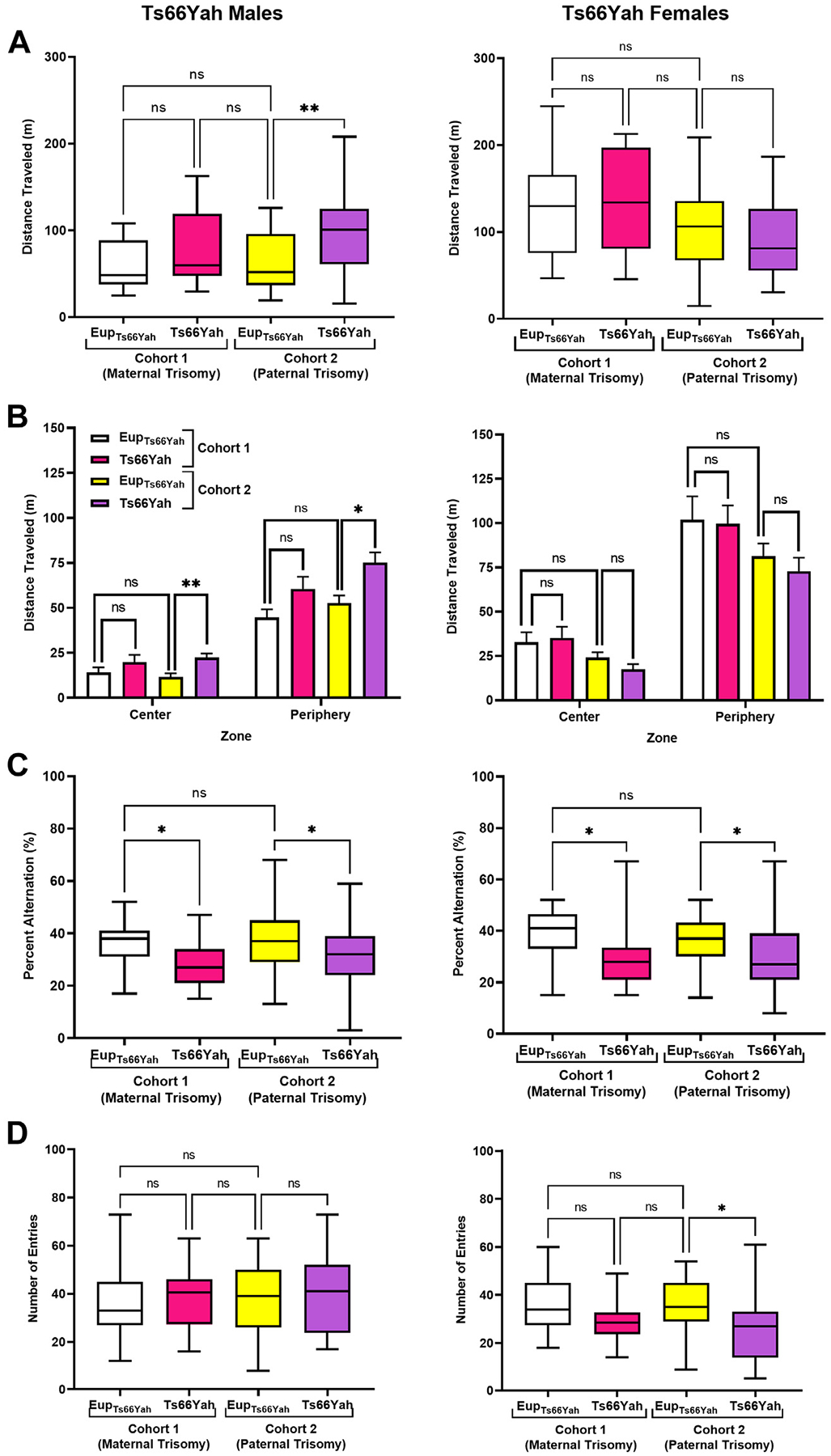
Exploratory behavior and working memory in Ts66Yah and Ts65Dn adult mice. (A, B) Exploratory behavior measured as total distance traveled in the open field as well as the distance traveled in the center vs. periphery in adult cohort 1 and cohort 2 Ts66Yah female and male mice. (C, D) Percent of alternation and total number of arm entries in the Y-maze in adult cohort 1 Ts66Yah and cohort 2 Ts66Yah female and male mice. Significant differences are indicated as *p < .05 and **p < .01. ns, not significant.
For cohort 2, Ts66Yah males displayed hyperactivity when compared with Eup (Figure 6A). When the distances traveled in the center and periphery of the arena were compared, Ts66Yah males traveled significantly more in both zones versus Eup (Figure 6B). As observed in cohort 1, females that were born to both Ts66Yah mothers and Ts66Yah fathers showed normal exploratory behavior (Figure 6A, B).
Working Memory (Y-Maze).
For both cohorts 1 and 2, Ts66Yah males exhibited significant working memory deficits as demonstrated by the significant decrease in the percent alternation (Figure 6C).
Like Ts66Yah males from both cohorts, Ts66Yah females also exhibited defective working memory in the Y-maze test with significant reduction of percent alternation when compared with Eup (Figure 6C).
Motor Coordination.
For cohort 1, Ts66Yah mice of both sexes had comparable performances in the fixed speed version of the rotarod test as compared with Eup (Figure S2A). Cohort 2 Ts66Yah males performed significantly better in the rotarod test versus Eup (Figure S2A). Cohort 2 females and Eup controls exhibited similar motor coordination phenotypes (Figure 2A).
In the accelerating speed version of the rotarod, cohort 1 Ts66Yah males and females fell from the rotarod faster than their controls, but this did not reach statistical significance (Figure S2B).
As in the fixed speed trial, cohort 2 Ts66Yah male mice remained significantly longer on the rotarod in the accelerating speed trial than Eup, whereas cohort 2 Ts66Yah female mice fell at a slightly faster rate versus Eup. This did not reach statistical significance (Figure S2A, B).
Hippocampal Contextual Memory (Fear Conditioning).
For both cohorts, Ts66Yah males and females exhibited normal hippocampal contextual memory. The average percent of freezing 24 hours after the electrical shock was not significantly different between Ts66Yah mice and Eup (Figure S2C, D).
Long-term Memory (Novel Object Recognition).
For both cohorts, male and female Ts66Yah mice showed comparable familiarity indices during day 1 (training) (Figure S3A) and similar long-term memory performance as their Eup littermates with no differences seen in the recognition indices after 24 hours of training (Figure S3B).
Spatial Hippocampal Memory (Morris Water Maze).
In the visible platform phase of the MWM, no visual learning deficits were observed in Ts66Yah mice of either sex. Repeated-measures ANOVA revealed that the performance of Ts66Yah mice improved at a similar rate as Eup with repeated testing sessions (Figure 7A).
Figure 7.
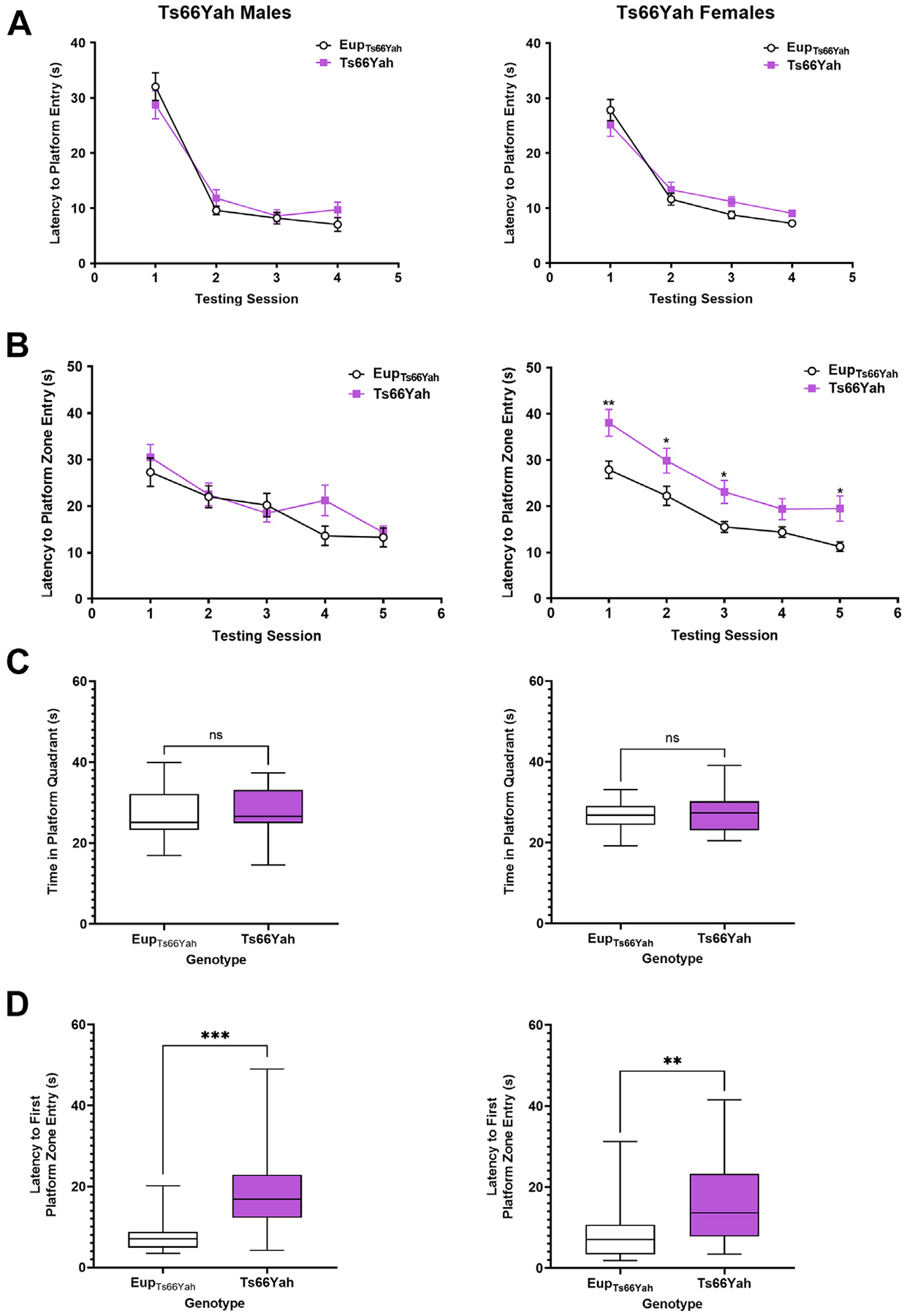
Hippocampal-dependent spatial memory in Ts66Yah adult mice. Hippocampal-dependent spatial learning/memory was measured in the Morris water maze test in adult Ts66Yah female and male mice as latency to reach the platform during the visible platform phase (A) and the hidden platform phase (B). During the probe trial, the total time spent in the platform quadrant (C) and the latency to first enter the platform zone was also analyzed (D). Significant differences are indicated as *p < .05, **p < .01, and ***p < .001. ns, not significant.
In the hidden phase, Ts66Yah females exhibited hippocampal-dependent spatial memory deficits as indicated by a significant increase in the latency to reach the platform in trisomic females compared with Eup. Repeated-measures ANOVA demonstrated a significant genotype effect in Ts66Yah females; however, no spatial memory deficits were observed in Ts66Yah males versus Eup (Figure 7B).
In the probe trial, although the total time spent in the platform quadrant was not significantly different between Ts66Yah and Eup in either sex, the latency to first entry to the platform zone was significantly higher in trisomic mice (Figure 7C, D).
DISCUSSION
Historically, most DS preclinical therapeutic studies have relied on the Ts65Dn mouse (18,19). This gold standard model, however, also carries a trisomy of the centromeric segment of Mmu17 that is not triplicated in humans with trisomy 21 (8,9,20).
Here, we studied the novel Ts66Yah mouse (10), comparing prenatal gene expression and postnatal behavioral phenotypes in Ts66Yah and Ts65Dn mice to gain a better understanding of the role of the Mmu17 non-orthologous genes. Our findings highlight widespread differences between these 2 models (Table 2).
Table 2.
Comparative Summary of Phenotypes in Ts65Dn and Ts66Yah Mice
| Parameters Measured | Ts65Dn (Trisomic Mothers) | Cohort 1 Ts66Yah (Trisomic Mothers) | Cohort 2 Ts66Yah (Trisomic Fathers) |
|---|---|---|---|
| Karyotype | Freely segregating marker chromosome | Freely segregating marker chromosome | |
| Mmu16 Orthologous Genes | Overexpressed | Overexpressed | |
| Mmu17 Non-Orthologous Genes | Overexpressed | Unchanged | |
| Motor Development (Neonates) | Severely delayed in females and males | Mildly delayed in females and males | |
| USV (Neonates) | Severe deficit in females and males (lower number of USVs, more short USVs, and less USVs of other categories) | Severe deficit in females and males (higher number of USVs, less short USVs, and more USVs of other categories) | Deficit only in females (higher number of USVs, less short USVs, and more USVs of other categories) |
| Neonatal Spatial Olfactory Memory (Neonates) | Severe deficits in females and males | Mild deficits in females and males | Mild deficits in females and males |
| Hyperactivity (Open) | Present in males only | Mild in males only | Present in males only |
| Motor Coordination | Normal | Normal | Normal |
| Working Memory | Severe deficits | Severe deficits | Severe deficits |
| Long-term Memory | Severe deficits | Normal | Normal |
| Contextual Hippocampal Memory | Mild in males | Normal | Mild in males |
| Spatial Hippocampal Memory | Severe deficits | Severe deficits in females Mild deficits in males | Not assessed |
USV, ultrasonic vocalization.
Overexpression of Mmu17 Non-Hsa21 Orthologous Genes Only in the Ts65Dn Forebrain
We found that over 90% of the Mmu16 overexpressed genes in the E18.5 forebrains of both Ts66Yah and Ts65Dn embryos overlapped, indicating that the primary transcriptomic effects of the segmental Mmu16 trisomy in these 2 models is highly conserved.
We have previously shown that ~30% of the Mmu17 non-orthologous genes are overexpressed in the Ts65Dn mouse during midgestation (E15.5) (14). Here we showed that, as embryonic brain development progresses, overexpression of more Mmu17 non-orthologous genes (60%) is observed later in gestation (E18.5) in Ts65Dn mice. In Ts66Yah embryonic forebrains, expression of Mmu17 non-orthologous genes was similar to Eup littermates.
Several of the Mmu17 non-orthologous genes overexpressed only in Ts65Dn are known to be essential during embryonic brain development, including Arid1b, Pde10a, Serac1, Rps6ka2, and Snx9. ARID1B protein belongs to the neural progenitor-specific and the neuron-specific chromatic remodeling complexes npBAF and nBAF and plays an important role in neuronal differentiation (21). ARID1B haploinsufficiency in humans is associated with autism spectrum disorder, corpus callosum agenesis, and growth delays (22,23). Pde10a encodes a cyclic nucleotide phosphodiesterase that plays a major role in regulating signal transduction through cAMP (cyclic adenosine monophosphate) and cGMP (cyclic guanosine monophosphate). PDE10A protein is highly expressed in the fetal brain, particularly in the medium spiny neurons of the striatum and controls striatocortical movement (24,25). In humans, PDE10A mutations are associated with infantile-onset limb and orofacial dyskinesia, striatal degeneration, and schizophrenia (24,26,27). A description of the function and phenotypes associated with the mutations (in humans)/knockout (in mice) of the remaining genes can be found in Table 1 and Table S1.
Distinct Genome-wide Transcriptional Dysregulation in Ts65Dn and Ts66Yah Models
While the primary trisomic effects were highly conserved in the Ts66Yah and Ts65Dn embryonic forebrains, the downstream secondary effects on the rest of the genome were distinct. These unexpected results suggest that overexpression of the Mmu17 non-orthologous genes in Ts65Dn triggers significant genome-wide dysregulation, which might ultimately lead to the major differences in their phenotypes.
Despite these differences, we identified 12 disomic genes that were consistently dysregulated in these 2 strains, suggesting that the expression of these genes is directly or indirectly regulated by Mmu16 trisomic genes (Table S3). As examples, Cdkl3 encodes a kinase required for neurogenesis and neurite outgrowth; mutations in this gene are associated with intellectual disability in humans (28,29). Rbm3 encodes a stress response protein that is abundant in the brain during embryonic and early postnatal development. Its suppression in neural stem cells significantly impairs neurogenesis, while its overexpression enhances cell proliferation and neuroprotection during hypoxic insults (30–32).
The limited overlap in dysregulated genes between Ts66Yah and Ts65Dn resulted in very few commonly dysregulated pathways, including neuroinflammation, interferon signaling, oxidative stress response, and the sirtuin pathway. Understanding how these dysregulated pathways affect brain development and cognition in DS is critical to developing effective treatment interventions.
Ts66Yah Mice Have Milder Behavioral Deficits With Sex-Specific Differences
Ts66Yah neonates exhibited motor deficits, abnormal USV profiles, and delayed spatial olfactory memory versus Eup. Motor deficits were milder than those observed in Ts65Dn neonates. Additionally, in both strains, USV profiles went in opposite directions for most categories, suggesting that the underlying molecular and cellular mechanisms leading to these deficits are quite different.
Ts66Yah adult mice exhibited hyperactivity (open field) in males, defective working memory (Y-maze) in both sexes and abnormal hippocampal-dependent spatial memory (MWM) that is more apparent in females. Ts66Yah mice did not show any deficits in motor coordination (rotarod), long-term memory (NOR), or contextual hippocampal memory (contextual fear conditioning).
Duchon et al. (10) compared behavior in adult Ts66Yah and Ts65Dn male progeny from trisomic mothers. They demonstrated that Ts65Dn males exhibit hyperactivity in the open field and circadian activity tests and severe deficits in the Y-maze, NOR, and MWM tests. By contrast, Ts66Yah showed slight increases in the light phase of the circadian activity test but not in the open field test, mild deficits in the MWM, and significant delays in the Y-maze and NOR. They examined sex-specific differences for Y-maze and NOR, but did not examine parent-of-origin differences, which we report here.
Overall, the findings from these 2 studies are consistent, except for the NOR results. In our study, Ts66Yah mice did not show deficits in the NOR test. Testing was performed in an open field arena, and animals were exposed to 2 trials with similar objects during day 1. This was associated with better discrimination when animals were exposed to the novel object 24 hours later but might have consolidated learning, resulting in the annihilation of long-term memory differences. Duchon et al. (10) used 2 protocols with either an open or a V-shaped arena with only one training session on day 1. This resulted in a lower discrimination index of the novel object on day 2. Further studies using different configurations of the NOR tests are needed to better characterize nonspatial long-term memory in the Ts66Yah mouse.
Our previous studies, and those of others, showed that Ts65Dn males exhibit hyperactivity, severe deficits in long-term memory and hippocampal-dependent memory, mild deficits in contextual hippocampal memory, and normal motor coordination (14,16,33–35). To our knowledge, only one study extensively examined behavioral deficits in both sexes using behavioral paradigms like ours (17). These investigators demonstrated that both female and male Ts65Dn mice had hyperactivity and severe deficits in working, long-term, contextual, and spatial memory.
Parental Origin of the Trisomy
In human pregnancies, a fetus with trisomy 21 develops in a normal intrauterine environment of a euploid mother. In the Ts65Dn mouse, trisomic males are sterile. Thus, transmission of the trisomic marker chromosome is only possible through an aneuploid dam. The impact of an abnormal intrauterine environment on the development of Ts65Dn embryos and their euploid littermates is unknown.
Here we were able to investigate the effects of maternal versus paternal trisomy in the Ts66Yah mouse by taking advantage of male fertility in this model. Although Ts66Yah offspring from trisomic mothers and fathers exhibited similar behavioral deficits in most paradigms, maternal trisomy induced more significant changes in the USV profiles of the Ts66Yah male and female pups than paternal trisomy. The reasons for these differences are still under investigation.
Conclusions
Comparative phenotyping of the Ts66Yah and Ts65Dn mouse models of DS uncovered a considerable influence of the trisomic Mmu17 non-Hsa21 orthologous genes on brain development and behavioral outcomes in Ts65Dn mice, with Ts66Yah showing fewer dysregulated pathways and milder behavioral deficits than Ts65Dn. These data provide quantitative evidence of the impact of the extraneous triplicated genes in Ts65Dn, which has important implications for human clinical trials that are based solely on preclinical studies in the Ts65Dn model (36). Our findings suggest that Ts66Yah may be a good alternative model for DS preclinical studies, because it more closely mimics the human DS karyotype and genotype.
Supplementary Material
ACKNOWLEDGMENTS AND DISCLOSURES
This research was supported, in part, by the intramural program of the National Human Genome Research Institute (NHGRI) at the National Institutes of Health, Grant # 1ZIA HG200399 (to DWB).
FG and DWB conceptualized the study; FG, JLAP, LAB, EK, YH, and DWB developed the methods; FG, JLAP, EK, and DWB validated the methods; FG and JLAP performed formal analyses; FG, JLAP, and DWB performed the investigation; FG and DWB drafted the manuscript; and FG, JLAP, LAB, EK, YH, and DWB reviewed and edited the manuscript.
We thank the NHGRI and Institut de Génétique et de Biologie Moléculaire et Cellulaire Technology transfer offices and the NHGRI Office of Laboratory Animal Medicine (Dr. Tannia Clark, Wendy Pridgen, and Irene Ginty) for facilitating the transfer of the Ts66Yah mouse model to our laboratory. We also acknowledge the NHGRI Embryonic Stem Cell and Transgenic Mouse Facility for performing the initial in vitro fertilization rederivation of the Ts66Yah mouse model. We would like to also thank the National Institute of Mental Health Rodent Behavioral Core staff, directed by Dr. Yogita Chudasama, for granting us access to the Rodent Behavioral Core behavioral equipment and analysis platforms. Lastly, we acknowledge Dr. Laura Baxter’s assistance with mapping the Mmu17 non-orthologous genes to various databases.
All gene expression data have been deposited into the Gene Expression Omnibus, identifier GSE222355.
The authors report no biomedical financial interests or potential conflicts of interest.
Footnotes
Supplementary material cited in this article is available online at https://doi.org/10.1016/j.biopsych.2023.02.012.
REFERENCES
- 1.Davisson MT, Schmidt C, Akeson EC (1990): Segmental trisomy of murine chromosome 16: A new model system for studying Down syndrome. Prog Clin Biol Res 360:263–280. [PubMed] [Google Scholar]
- 2.Davisson MT, Schmidt C, Reeves RH, Irving NG, Akeson EC, Harris BS, Bronson RT (1993): Segmental trisomy as a mouse model for Down syndrome. Prog Clin Biol Res 384:117–133. [PubMed] [Google Scholar]
- 3.Muñiz Moreno MDM, Brault V, Birling MC, Pavlovic G, Herault Y (2020): Modeling Down syndrome in animals from the early stage to the 4.0 models and next. Prog Brain Res 251:91–143. [DOI] [PubMed] [Google Scholar]
- 4.Tosh J, Tybulewicz V, Fisher EMC (2022): Mouse models of aneuploidy to understand chromosome disorders. Mamm Genome 33:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herault Y, Delabar JM, Fisher EMC, Tybulewicz VLJ, Yu E, Brault V (2017): Rodent models in Down syndrome research: Impact and future opportunities. Dis Model Mech 10:1165–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SE, Duran-Martinez M, Khantsis S, Bianchi DW, Guedj F (2020): Challenges and opportunities for translation of therapies to improve cognition in Down syndrome. Trends Mol Med 26:150–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rueda N, Flórez J, Dierssen M, Martínez-Cué C (2020): Translational validity and implications of pharmacotherapies in preclinical models of Down syndrome. Prog Brain Res 251:245–268. [DOI] [PubMed] [Google Scholar]
- 8.Reinholdt LG, Ding Y, Gilbert GJ, Czechanski A, Solzak JP, Roper RJ, et al. (2011): Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm Genome 22:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y (2011): Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: Relevance for modeling Down syndrome. Mamm Genome 22:674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duchon A, Del Mar Muñiz Moreno M, Chevalier C, Nalesso V, Andre P, Fructuoso-Castellar M, et al. (2022): Ts66Yah, a mouse model of Down syndrome with improved construct and face validity. Dis Model Mech 15:dmm049721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoelter SM, Dalke C, Kallnik M, Becker L, Horsch M, Schrewe A, et al. (2008): “Sighted C3H” mice—A tool for analysing the influence of vision on mouse behaviour? Front Biosci 13:5810–5823. [DOI] [PubMed] [Google Scholar]
- 12.Roper RJ, St John HK, Philip J, Lawler A, Reeves RH (2006): Perinatal loss of Ts65Dn Down syndrome mice. Genetics 172:437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villar AJ, Belichenko PV, Gillespie AM, Kozy HM, Mobley WC, Epstein CJ (2005): Identification and characterization of a new Down syndrome model, Ts[Rb(12.1716)]2Cje, resulting from a spontaneous Robertsonian fusion between t(171)65Dn and mouse chromosome 12. Mamm Genome 16:79–90. [DOI] [PubMed] [Google Scholar]
- 14.Aziz NM, Guedj F, Pennings JLA, Olmos-Serrano JL, Siegel A, Haydar TF, Bianchi DW (2018): Lifespan analysis of brain development, gene expression and behavioral phenotypes in the Ts1Cje, Ts65Dn and Dp(16)1/Yey mouse models of Down syndrome. Dis Model Mech 11:dmm031013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guedj F, Pennings JL, Ferres MA, Graham LC, Wick HC, Miczek KA, et al. (2015): The fetal brain transcriptome and neonatal behavioral phenotype in the Ts1Cje mouse model of Down syndrome. Am J Med Genet A 167A:1993–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olmos-Serrano JL, Tyler WA, Cabral HJ, Haydar TF (2016): Longitudinal measures of cognition in the Ts65Dn mouse: Refining windows and defining modalities for therapeutic intervention in Down syndrome. Exp Neurol 279:40–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faizi M, Bader PL, Tun C, Encarnacion A, Kleschevnikov A, Belichenko P, et al. (2011): Comprehensive behavioral phenotyping of Ts65Dn mouse model of Down syndrome: Activation of beta1-adrenergic receptor by xamoterol as a potential cognitive enhancer. Neurobiol Dis 43:397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta M, Dhanasekaran AR, Gardiner KJ (2016): Mouse models of Down syndrome: Gene content and consequences. Mamm Genome 27:538–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guedj F, Bianchi DW, Delabar JM (2014): Prenatal treatment of Down syndrome: A reality? Curr Opin Obstet Gynecol 26:92–103. [DOI] [PubMed] [Google Scholar]
- 20.Akeson EC, Lambert JP, Narayanswami S, Gardiner K, Bechtel LJ, Davisson MT (2001): Ts65Dn – Localization of the translocation breakpoint and trisomic gene content in a mouse model for Down syndrome. Cytogenet Cell Genet 93:270–276. [DOI] [PubMed] [Google Scholar]
- 21.Moffat JJ, Jung EM, Ka M, Smith AL, Jeon BT, Santen GWE, Kim WY (2019): The role of ARID1B, a BAF chromatin remodeling complex subunit, in neural development and behavior. Prog Neuropsychopharmacol Biol Psychiatry 89:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sim JC, White SM, Lockhart PJ (2015): ARID1B-mediated disorders: Mutations and possible mechanisms. Intractable Rare Dis Res 4: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moffat JJ, Smith AL, Jung EM, Ka M, Kim WY (2022): Neurobiology of ARID1B haploinsufficiency related to neurodevelopmental and psychiatric disorders. Mol Psychiatry 27:476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diggle CP, Sukoff Rizzo SJ, Popiolek M, Hinttala R, Schülke JP, Kurian MA, et al. (2016): Biallelic mutations in PDE10A lead to loss of striatal PDE10A and a hyperkinetic movement disorder with onset in infancy. Am J Hum Genet 98:735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujishige K, Kotera J, Yuasa K, Omori K (2000): The human phosphodiesterase PDE10A gene genomic organization and evolutionary relatedness with other PDEs containing GAF domains. Eur J Biochem 267:5943–5951. [DOI] [PubMed] [Google Scholar]
- 26.Mencacci NE, Kamsteeg EJ, Nakashima K, R’Bibo LDS, Lynch DS, Balint B, et al. (2016): De novo mutations in PDE10A cause childhood-onset chorea with bilateral striatal lesions. Am J Hum Genet 98:763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siuciak JA, McCarthy SA, Chapin DS, Martin AN, Harms JF, Schmidt CJ (2008): Behavioral characterization of mice deficient in the phosphodiesterase-10A (PDE10A) enzyme on a C57/Bl6N congenic background. Neuropharmacology 54:417–427. [DOI] [PubMed] [Google Scholar]
- 28.Liu Z, Xu D, Zhao Y, Zheng J (2010): Non-syndromic mild mental retardation candidate gene CDKL3 regulates neuronal morphogenesis. Neurobiol Dis 39:242–251. [DOI] [PubMed] [Google Scholar]
- 29.Liu Z, Tao D (2015): Inactivition of CDKL3 mildly inhibits proliferation of cells at VZ/SVZ in brain. Neurol Sci 36:297–302. [DOI] [PubMed] [Google Scholar]
- 30.Pilotte J, Cunningham BA, Edelman GM, Vanderklish PW (2009): Developmentally regulated expression of the cold-inducible RNA-binding motif protein 3 in euthermic rat brain. Brain Res 1258:12–24. [DOI] [PubMed] [Google Scholar]
- 31.Jackson TC, Kotermanski SE, Kochanek PM (2018): Infants uniquely express high levels of RBM3 and other cold-adaptive neuroprotectant proteins in the human brain. Dev Neurosci 40:325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan J, Goerne T, Zelmer A, Guzman R, Kapfhammer JP, Wellmann S, Zhu X (2019): The RNA-binding protein RBM3 promotes neural stem cell (NSC) proliferation under hypoxia. Front Cell Dev Biol 7:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sago H, Carlson EJ, Smith DJ, Rubin EM, Crnic LS, Huang TT, Epstein CJ (2000): Genetic dissection of region associated with behavioral abnormalities in mouse models for Down syndrome. Pediatr Res 48:606–613. [DOI] [PubMed] [Google Scholar]
- 34.Shaw PR, Klein JA, Aziz NM, Haydar TF (2020): Longitudinal neuro-anatomical and behavioral analyses show phenotypic drift and variability in the Ts65Dn mouse model of Down syndrome. Dis Model Mech 13:dmm046243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coussons-Read ME, Crnic LS (1996): Behavioral assessment of the Ts65Dn mouse, a model for Down syndrome: Altered behavior in the elevated plus maze and open field. Behav Genet 26:7–13. [DOI] [PubMed] [Google Scholar]
- 36.Manfredi-Lozano M, Leysen V, Adamo M, Paiva I, Rovera R, Pignat JM, et al. (2022): GnRH replacement rescues cognition in Down syndrome. Science 377:eabq4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vieira AR, de Carvalho FM, Johnson L, DeVos L, Swailes AL, Weber ML, et al. (2015): Fine mapping of 6q23.1 identifies TULP4 as contributing to clefts. Cleft Palate Craniofac. J 52:128–134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.