

Abstract
Background:
Young women treated for breast cancer with cytotoxic therapies are at risk for clonal hematopoiesis of indeterminate potential (CHIP), a condition in which blood cells carrying a somatic mutation associated with hematologic malignancy comprise at least 4% of the total blood system. CHIP has primarily been studied in older patient cohorts with limited clinical phenotyping.
Materials and Methods:
We performed targeted sequencing on longitudinal blood samples to characterize the clonal hematopoietic landscape of 878 women treated for breast cancer enrolled in the prospective Young Women’s Breast Cancer Study.
Results:
We identified somatic driver mutations in 252 study subjects (28.7%), but only 24 (2.7%) had clones large enough to meet criteria for CHIP. The most commonly mutated genes were DNMT3A and TET2, similar to mutations observed in non-cancer cohorts. At nine years median follow up, we found no association between the presence of a somatic blood mutation (regardless of clone size) and adverse breast cancer (distant relapse-free survival) or non-breast cancer-related outcomes in this cohort. A subset of paired blood samples obtained over four years showed no evidence of mutant clonal expansion, regardless of genotype. Finally, we identified a subset of patients with likely germline mutations in genes known to contribute to inherited cancer risk, such as TP53 and ATM.
Conclusions:
Our data show that for young women with early-stage breast cancer, CHIP is uncommon after cytotoxic exposure, is unlikely to contribute to adverse outcomes over the decade-long follow up and may not require additional monitoring if discovered incidentally.
Keywords: Clonal hematopoiesis, CHIP, breast cancer, young women
INTRODUCTION
Despite significant advances in breast cancer therapy, adjuvant radiation and chemotherapy remain mainstays of treatment for a large proportion of women with localized breast cancer. Though effective at reducing the risk of breast cancer relapse, these treatments carry risks of short- and long-term complications, including increased rates of cardiovascular disease, development of premature menopause, and evolution of therapy-related myelodysplastic syndrome (t-MDS) and acute myeloid leukemia (t-AML).(1–4) These risks are of heightened importance in young patients who, when cured of their breast cancer, have the potential to live for many decades.
Clonal hematopoiesis (CH) is the expansion of clonally related blood cells due to the acquisition of somatic mutations that accumulate during the normal aging of hematopoietic stem cells.(5) Clonal Hematopoiesis of Indeterminate Potential (CHIP), a subset of CH in which the somatic mutation in the blood clone is present at variant allele fraction (VAF) of at least 0.02 in an individual with no overt hematologic malignancy, is common and age-associated.(6) The mutations found in CHIP are largely restricted to a select number of genes, the most common mutated gene in an aging population being DNTM3A, TET2, and ASXL1.(7–9) Individuals with CHIP are not only at significantly elevated risk of developing a myeloid malignancy, but also have increased rates of numerous additional ill-health effects including ischemic cardiovascular disease, chronic obstructive pulmonary disease, osteoporosis, and premature menopause.(10–16)
In patients receiving cytotoxic cancer therapies, CHIP is both more common and more likely to harbor mutations in genes that regulate the cellular DNA damage response such as TP53 and PPM1D, the former being associated with a particularly elevated risk of developing MDS and AML.(17–19) To date, the majority of studies of CHIP in cancer populations have focused on either non-selected populations or specific cohorts, many of which are older or have been heavily pretreated with multiple rounds and types of chemotherapy.(17–21) In many of these populations, including patients with solid tumors, lymphoma, and multiple myeloma, CHIP is associated with increased risk of therapy-related MDS and AML and inferior overall survival.(18,19,22,23) The prevalence and consequences of CHIP in younger adult cancer patients, however, have not been assessed. In this study, we investigated the prevalence, mutational landscape, and clinical significance of CHIP in a prospectively collected cohort of young women treated for breast cancer.
MATERIALS AND METHODS
Young Women’s Breast Cancer Study Cohort and Biospecimens
The Young Women’s Breast Cancer Study (YWS) is a prospective cohort of women with breast cancer aged 40 years old or younger at diagnosis between 2006 and 2016.(24) Enrollment occurred at academic and community centers in the United States and Canada. The study was performed in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board at Dana-Farber/Harvard Cancer Center and other participating sites. Participants signed informed consent prior to enrollment. When possible, peripheral blood samples were obtained from patients near the time of diagnosis (“baseline”), approximately one year after diagnosis (“one-year”), and approximately four years after diagnosis (“four-year”).
YWS participants completed a baseline survey and were then surveyed twice a year for the first three years following diagnosis and annually thereafter. Cancer stage at diagnosis was ascertained from pathology reports and medical record review. Primary breast cancer treatment, including surgery, chemotherapy, radiotherapy and/or endocrine therapy (within the first year after diagnosis), were obtained through a combination of survey data and medical record review. Similarly, events during follow-up including comorbidities (premature menopause, ischemic cardiovascular disease, AML/MDS), distant breast cancer recurrence and survival events were self-reported during follow-up and/or extracted from medical records.
Targeted Sequencing of Peripheral Blood Samples
Targeted sequencing to identify CH was performed on all samples. DNA was extracted from peripheral blood samples (Qiagen, Hilden, Germany) and subject to error-corrected next generation sequencing. Hybrid capture using RNA probes from Twist Biosciences (California, USA) was performed for all samples with subsequent sequencing on the Illumina platform (Illumina, California, USA) as previously described.(20) The genes sequenced for the one-year samples are shown in Supplemental Table S1 and for the baseline and four-year samples in Supplemental Table S2. For the one-year samples, mutations with three or more alternative reads were considered in analysis, and curated based on likely pathogenicity and frequency in the population as previously described.(18) Mutations with a variant allele fraction (VAF) greater than 0.35 were excluded to avoid inclusion of germline variants. The mutations identified in the one-year samples were then assessed in the paired baseline and 4-year samples without specific alternative read cutoffs.
Statistical Analysis
The primary endpoints were overall survival (OS) among women diagnosed with stage I-IV cancer and distant recurrence free survival (DRFS) among women diagnosed with stage I-III cancer as evaluated through Cox proportional hazards regression. The co-primary endpoints uniformly share the type I error of 0.10 used for determining significance. OS was defined as time from diagnosis to death; DRFS was time first distant recurrence and excluded women with Stage IV disease at diagnosis. Patients without events were censored at last follow-up.
Secondary endpoints were ischemic cardiovascular disease, t-MDS/AML, and premature menopause. Tertiary analyses included the association between baseline patient characteristics and CHIP at one-year and adjuvant treatment for breast cancer and CHIP at one-year. All secondary and tertiary analyses were conducted with a type I error of 0.05. Analyses of outcome associations with CH at VAF 0.02 were considered exploratory. Continuous variables were evaluated using t-tests or Wilcoxon Rank-Sum tests where appropriate, and categorical variables were evaluated using Fisher’s exact test. Patients with stage 0 were excluded from the survival analyses, and stages III and IV were combined given the rarity of stage IV disease in this cohort.
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.
RESULTS
Somatic Clonal Hematopoietic Landscape in Young Women with Breast Cancer
To determine the frequency and distribution of clonal hematopoietic mutations in young women treated for breast cancer, we performed deep, error-corrected sequencing on peripheral blood DNA obtained from one-year samples from 878 individuals (Fig. 1A). The clinical characteristics of the cohort are shown in Table 1. All study participants were women and the median age at time of one-year blood draw was 38 years old. At the time of diagnosis, 830 (95%) had localized breast cancer (stage 0-III) while the remainder had metastatic disease. Within the first year of treatment, 366 women (42%) received radiation and chemotherapy, 308 (35%) received chemotherapy only, 61 (7%) received radiation only, and 493 (56%) were started on hormone therapy (tamoxifen or aromatase inhibitor). At a median follow-up of 9 years, 764 (87%) of patients were alive and 728 (83%) of patients were breast-cancer free (data not shown).
Fig. 1: Clonal Hematopoiesis of Indeterminate Potential (CHIP) in the YWS.
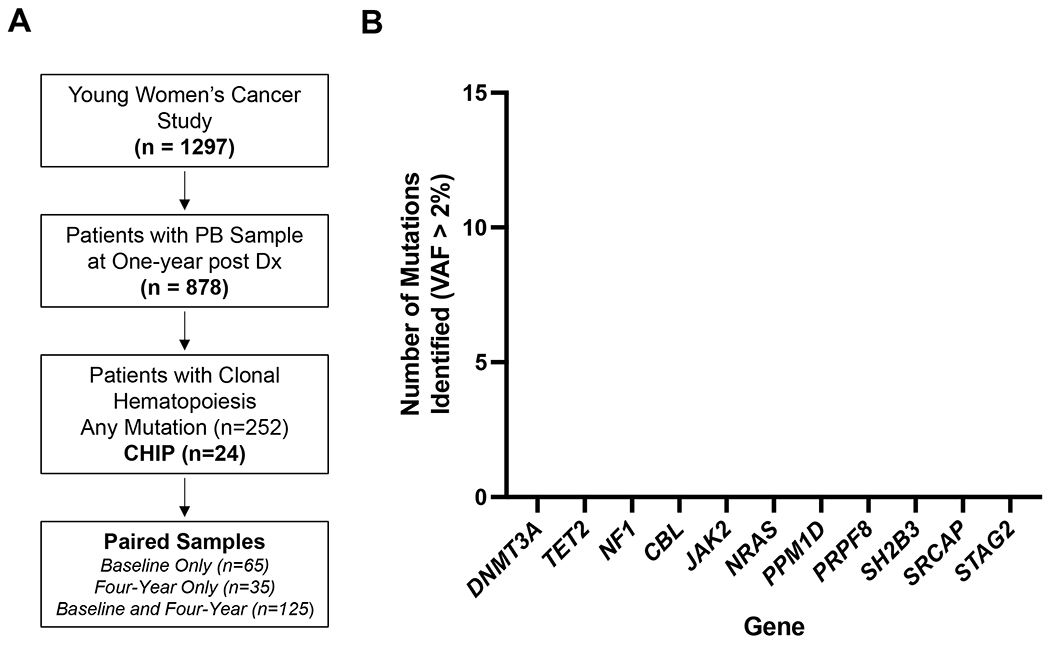
(A) Schematic of cohort and sequencing. Of the 1297 patients in YWS, 878 had a one-year peripheral blood (PB) sample available, from which DNA was isolated and sequenced for clonal hematopoiesis. Of the 252 patients in which a clonal hematopoietic mutation was identified at one year, we obtained, when available, paired baseline and four-year samples to assess for longitudinal clonal changes.
(B) Frequency of mutations in each gene meeting criteria for CHIP with variant allele fraction (VAF) > 2% was identified in the cohort. Note that the number of patients with these mutations total 24 (2.7%).
Table 1.
Cohort Characteristics of Individuals Studied from the YWS
| Characteristic | Full Cohort (n= 878) |
|---|---|
| Age at 1-year blood draw (median, range) | 38 [22, 42] |
| Smoking Status | |
| Never | 513 (58%) |
| Current/Former | 258 (29%) |
| Unknown | 107 (12%) |
| Cancer Stage | |
| 0 | 55 (6%) |
| I | 269 (31%) |
| II | 361 (41%) |
| III | 145 (17%) |
| IV | 48 (5%) |
| Surgery | |
| Yes | 763 (87%) |
| Unknown | 115 (13%) |
| Radiation | |
| Yes | 427 (49%) |
| No | 451 (51%) |
| Chemotherapy | |
| Yes | 674 (77%) |
| No | 204 (23%) |
| Hormone Therapy (within 1 year of diagnosis) |
|
| Yes | 493 (56%) |
| No | 235 (27%) |
| Unknown | 150 (17%) |
We identified CHIP, defined by the presence of a somatic mutation with a VAF greater than 0.02, in 24 patients (2.7% of cohort) (Fig. 1B, Supplemental Table S3). The most common CHIP mutations were in DNMT3A and TET2, consistent with what has been observed in most studies of patients without a prior exposure to cytotoxic therapy.(8,9) In this study, there was one patient with PPM1D-mutant CHIP and no patients with TP53-mutant CHIP. In exploratory analyses of patients with CH below the VAF threshold for CHIP of 0.02, we identified 252 patients (28.7% of cohort) with pathogenic somatic mutations (minimum VAF 0.0002) (Supplemental Table S4, Supplemental Fig. S1). Among these 252 patients, DNMT3A mutations were most common (187 mutations in 152 patients), followed by TET2 (23 mutations in 22 patients), PPM1D (22 mutations in 17 patients), ASXL1 (14 mutations in 13 patients), SRCAP (13 mutations in 13 patients), and TP53 (9 mutations in 9 patients) (Supplemental Table S4).(25)
Association of Clonal Hematopoiesis with Clinical Characteristics and Outcomes
With a median follow up of 9 years, we did not detect any significant differences in either clinical characteristics (patient and tumor characteristics, including pathologic subtype) or survival outcomes between individuals with and without CHIP (HR 0.85 (95% CI 0.27-2.69) and 1.06 (95% CI 0.33-3.39) for OS and DRFS, respectively), though given the limited number of CHIP cases observed in this cohort, we were not powered to identify small differences (Fig. 2A–B, Table 2). During the nine-year follow-up period, the rarity of t-MDS/AML (one case overall in the no CHIP group) and ischemic cardiovascular disease (one case overall, occurred in the CHIP group) precluded formal risk analyses, but did suggest that CHIP was not strongly associated with these outcomes in this cohort. Neither the presence of CHIP nor any CH mutation was associated with self-reported cessation of menopause at the one-year time point after controlling for age and receipt of chemotherapy.(8,11,13) In an exploratory analysis of patients with other VAF cutoffs including 1% and any detectable CH clone, we again did not find any significant associations (Supplemental Fig.S2, Supplemental Table S5).
Fig. 2: Presence of Clonal Hematopoiesis of Indeterminate Potential (CHIP) and Survival in the YWS.
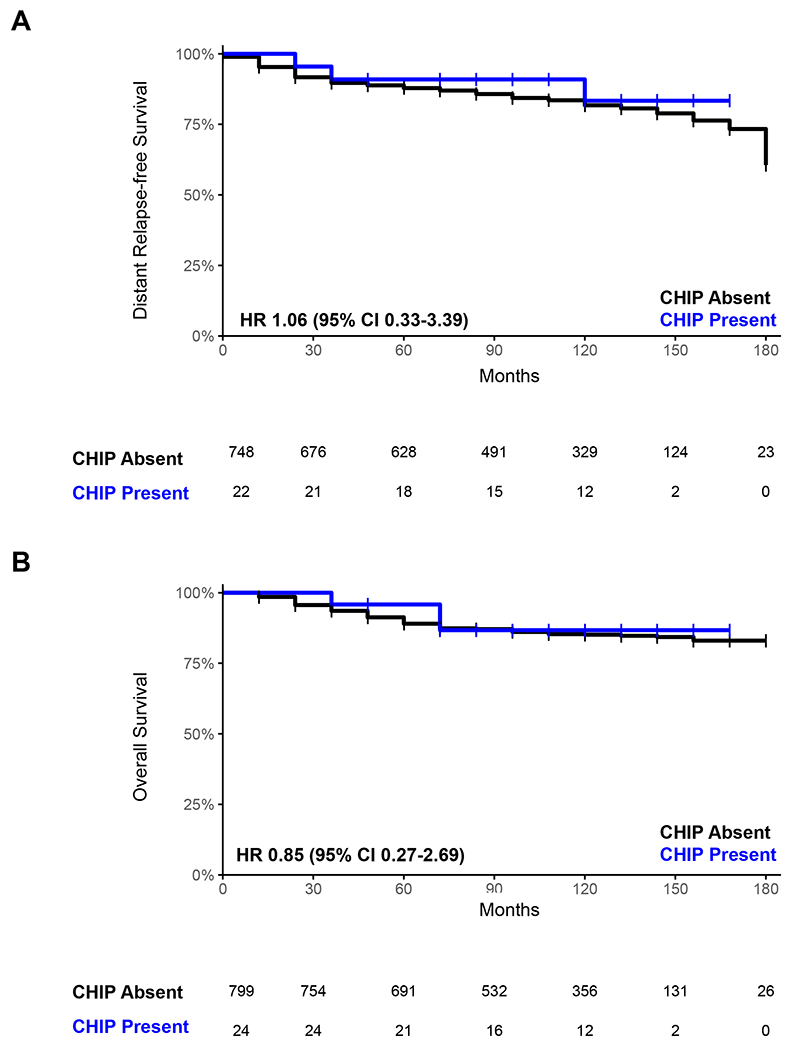
Comparison of distant relapse free (A) and overall (B) survival between individuals with CHIP (blue) and those without CHIP (black).
Table 2.
Cohort Characteristics of Study Population Stratified by Presence of Clonal Hematopoiesis of Indeterminate Potential (CHIP)
| Characteristic | CHIP+ | CHIP− | p-value |
|---|---|---|---|
| (n=24) | (n=854) | ||
| Age at blood draw (median, range) | 38 [25, 42] | 39 [22,42] | 0.133 |
| Smoking Status (baseline) | 0.337* | ||
| Never | 11 (46%) | 502 (59%) | |
| Current/Former | 9 (38%) | 249 (29%) | |
| Unknown | 4 (17%) | 103 (12%) | |
| Cancer Stage | 0.257 | ||
| 0 | 0 (0%) | 55 (6%) | |
| I | 10 (42%) | 259 (30%) | |
| II | 11 (46%) | 350 (41%) | |
| III | 1 (4%) | 144 (17%) | |
| IV | 2 (8%) | 46 (5%) | |
| Surgery | 0.759 | ||
| Yes | 22 (92%) | 741 (87%) | |
| Unknown | 2 (8%) | 114 (13%) | |
| Radiation | 0.838 | ||
| Yes | 11 (46%) | 416 (49%) | |
| No | 13 (54%) | 438 (51%) | |
| Chemotherapy | >0.999 | ||
| Yes | 19 (79%) | 655 (77%) | |
| No | 5 (21%) | 199 (23%) | |
| Hormone Therapy (within 1 year of diagnosis) | 0.802* | ||
| Yes | 13 (54%) | 480 (56%) | |
| No | 5 (21%) | 230 (27%) | |
| Unknown | 6 (25%) | 144 (17%) |
p-value estimated excluding the unknown group
Age was tested using a Wilcoxon Rank-Sum test, all other variables were tested with a Fisher’s Exact test.
Some prior studies of CH in cancer survivors who received cytotoxic therapy reported an increased rate of CHIP and a mutation distribution enriched for genes involved in the DNA damage response, such as PPM1D and TP53.(17,18,21) We did not observe a similar distribution (Fig.1B and Supplemental Fig.S1), nor were there significant associations between treatments administered for breast cancer (chemotherapy, radiation, and endocrine therapy) and the presence of CHIP or the frequency of CHIP mutations within a given gene. Comparing those who received either chemotherapy or radiation (n=758) with those who did not (n=119), we found no significant difference in the frequency of CHIP (2.6% vs. 3.4%, p=0.55) or any CH mutation (29% vs. 26%, p=0.59). While there was a higher rate of chemotherapy or radiation receipt among CH patients with mutations of any VAF in TP53 or PPM1D (n=24) compared to those without such a mutation (n=228), the results did not reach statistical significance (92% vs 74%, p=0.078 for chemotherapy; 67% vs 46%, p=0.085 for radiation; 96% vs. 87%, p=0.33 for chemotherapy or radiation) (Supplemental Table S6).
Longitudinal Tracking of Clonal Hematopoietic Mutations
As the YWS includes prospective collection of biospecimens at multiple timepoints, we obtained all available peripheral blood samples collected at baseline and at four years after diagnosis for patients in whom any CH mutation was identified in the one-year sample. Among all the patients for whom a CH mutation was identified at one-year post treatment, we sequenced paired baseline, one-year, and four-year samples for 125 patients, paired baseline and one-year samples for 65 patients, and paired one-year and four-year samples for 35 patients (Fig. 3A). Mutations identified in the one-year samples were assessed by targeted sequencing, and VAFs of detectable mutations were compared across timepoints, as described in Methods.
Fig. 3: Longitudinal Mutation Analysis in the YWS.
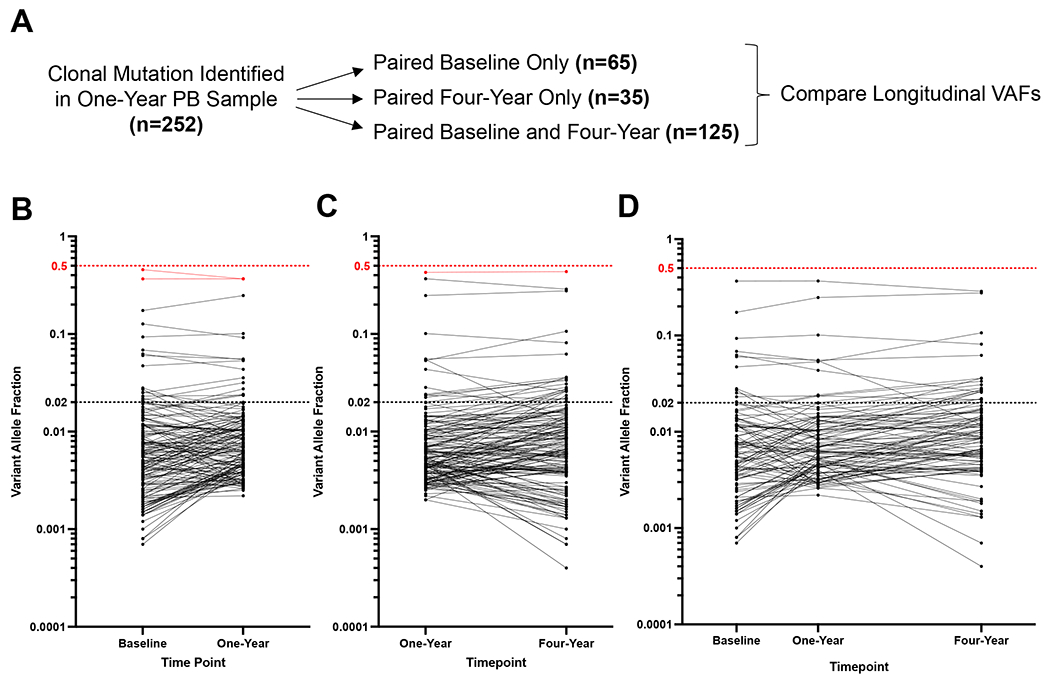
(A) Schematic of longitudinal sample acquisition and sequencing.
(B-D) Change in variant allele fraction (VAF) for mutations identified in paired samples. The VAF for all mutations identified in the baseline and one-year samples (B), one-year and four-year samples (C), or baseline, one-year, and four-year samples (D) are plotted. The horizontal dashed line at VAF 0.02 reflects the cutoff for CHIP.
Note that mutations presented are only in genes assessed on both of the sequencing panels used at the different time points (see Supplemental Tables 1 and 2).
For each mutation, the VAFs were highly concordant across time points, with no evidence of significant clonal expansion or contraction over the time interval within which the specimens were collected. This was true regardless of the mutated gene or the VAF of the mutation identified at the one-year timepoint (Fig.3B–D). Specifically, we did not observe significant changes in clone size over time when stratified by specific gene mutation (Supplemental Fig.S3A–D). Additionally, when comparing the frequency of mutations between time points, we did not observe significant differences in the prevalence of mutations in specific genes, suggesting that patient exposures in the intervals between sample acquisition did not drive selection for mutations in particular genes (Supplemental Fig.S3E). Taken together, these data suggest that over the longitudinal sample acquisition, mutations in this cohort were stable without evidence of significant clonal expansion or variation.
Identification of Germline Pathogenic Variants in Cohort
The distribution of VAFs for all mutations identified revealed a set of mutations with VAFs greater than 0.35 that were suspicious for being germline heterozygous variants, particularly those with VAFs of approximately 0.5 (Fig. 4A). Among these, we identified 15 mutations that were likely to impair protein function (nonsense, splicing, and frameshift), most commonly in the genes ATM and TP53 (Supplemental Table S7), both of which have been implicated as germline-risk alleles.(25,26) We also identified non-synonymous variants in ATM (n=37) and TP53 (n=6), many of which localized to functional domains of the proteins, suggesting that they may in fact impair protein function (Fig. 4B).
Fig. 4: Identification of Germline Variants in the YWS.
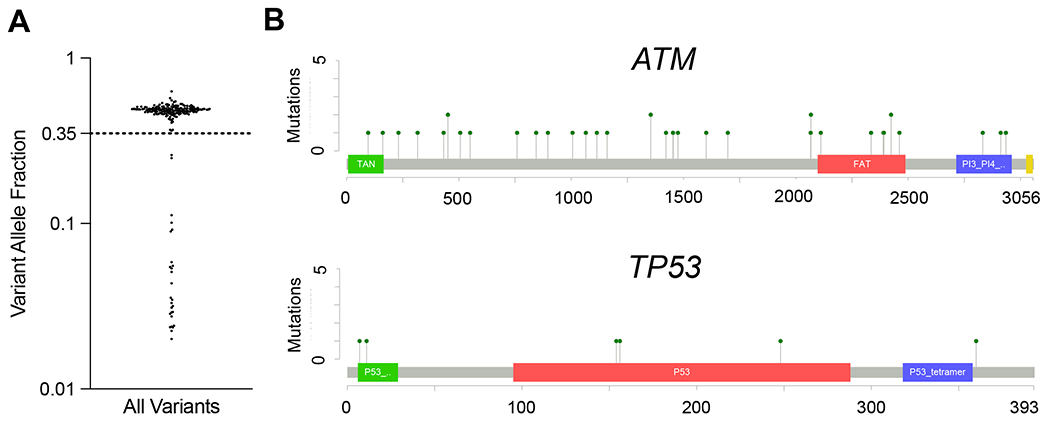
(A) Variant allele fraction (VAF) of all mutations identified with a VAF > 0.02. A cluster of mutations with VAF > 0.35 likely reflect germline variants.
(B) Lollipop plots of non-synonymous single nucleotide variants in ATM (top) and TP53 (bottom).
DISCUSSION
In this cohort of 878 young women with breast cancer, we identified CHIP in 2.7% of the patients from blood samples collected an average of one year after diagnosis. We did not observe any association between patient characteristics, tumor characteristics, or treatment with the prevalence or mutation pattern of CHIP, nor was there an association between CHIP and overall survival, distant recurrence-free survival, or the risk of adverse outcomes including t-MDS/t-AML, ischemic cardiovascular disease, or premature menopause. In contrast to other published series of CH in cancer patients, mutations in the DNA damage response genes TP53 and PPM1D were not enriched in this cohort.(17–19) Additionally, among the individuals with a CH mutation and longitudinal samples, we did not observe any significant change in the size of the mutant clone, suggesting that there is very limited clonal expansion or contraction over the four-year time-period studied. Finally, we identified a series of likely germline variants, including in the genes ATM and TP53, both of which have been shown to confer a risk of early-onset breast cancer.
The prevalence of CHIP in this cohort was lower than that reported in other studies of cancer patients treated with cytotoxic therapy, even when considering differences in technical factors such as sequencing depth and limits of detection for small clones.(17–19,21) We hypothesize that this is due to the comparatively young average age of our study subjects, consistent with the low rate of CHIP in healthy adults under age 40 in cross-sectional studies.(8–10) This may be related to the stochastic nature of mutation acquisition in hematopoietic stem cells (HSC), such that many adults do not have acquired CH-associated mutations when assessed at a young age. However, emerging evidence suggests that CH mutations can arise at any point in life, even during development in utero.(27) This suggests a model in which HSC clones with CH mutations are present in many young adults, but have not yet expanded enough for the mutations to be detectable by current sequencing methods.
Such a model may simply reflect a linear relationship to the time necessary for a sufficient number of cell divisions for a CH clone to reach a detectable size. However, the relative stability of many clones across years of observation, both in our study and others, suggests that other variables could also affect a CH clone’s expansion, such as the relative health or attrition of the non-mutant HSC pool.
The lack of clinical impact of CHIP in this study can be explained by several factors. First, since CHIP’s effect on clinical outcomes is driven primarily by large clones the rarity of such clones in this cohort limits our power to detect anything less than the largest effect sizes.(8,10,11) Second, available evidence suggests that CHIP’s impact on non-hematologic outcomes is best conceptualized as an amplifying influence, rather than as a root cause. For young patients in whom the baseline rate of cardiovascular and inflammatory disease is low, it may be unsurprising that CHIP does not measurably increase the incidence of these events. Finally, multiple studies have shown that single DNMT3A and TET2 mutations pose a low risk of evolution to hematologic malignancies.(28,29) The paucity of higher risk mutations, especially in TP53, is consistent with the low rate of therapy-related AML and MDS reported in previous studies of breast cancer patients.(1)
Most subjects in this cohort with CH had clones too small to be considered CHIP, which in turn had no significant impact on any clinical outcomes evaluated over the study period. However, the large CHIP clones associated with adverse clinical outcomes in other studies presumably began as small clones at some point in the past. While receipt of cytotoxic therapy did not lead to expansion of CH clones over the 4-year time interval captured in this study, we cannot rule out a model in which this exposure in young adulthood accelerates HSC attrition later in life, leading to rapid expansion of existing CH clones and uncertain impacts on CHIP-associated endpoints. This hypothesis could be addressed by a future study of CH in older populations of long-term cancer survivors.
The strengths of our approach include the focus on a prospectively studied young cohort of patients with robust clinical phenotyping, use of a uniform sequencing platform in the one-year time point, and availability of serial samples for longitudinal analyses. Though our study of CHIP was the largest to date in young women with breast cancer, we recognize that future studies with even larger sample sizes may be able to identify clinical associations that we were not powered to assess. Additionally, though our cohort was relatively homogenous in terms of age, there was a range of treatment approaches. Further efforts to study patients exposed to a defined therapeutic regimen, such as in the context of a randomized clinical trial, may provide insights regarding specific treatment exposures, CHIP, and clinical outcomes.
The routine incorporation of next-generation sequencing into the evaluation of cancer patients will inevitably lead to the incidental discovery of CH in a subset of patients, and some centers have started to establish “CH clinics” that evaluate and monitor these patients for hematologic deterioration and evidence of leukemic transformation.(17–19,30–32) However, there are currently no approved interventions for CH, and its discovery can lead to significant patient anxiety and increased medical costs.(33) Taken together, our data show that CHIP is infrequent in young women treated for breast cancer and do not support routine testing for CHIP in this population. Furthermore, in the absence of any hematologic abnormalities, our data show that young breast cancer patients who are incidentally found to have CHIP via germline or somatic testing can be reassured and may not require long-term monitoring.
Supplementary Material
Statement of Translational Relevance.
Clonal Hematopoiesis of Indeterminate Potential (CHIP) is incidentally identified in cancer patients with increasing frequency, and there is a need for an evidentiary basis to guide counseling and clinical decisions for cancer patients who are found to have CHIP. Here, we assessed the genomic characteristics and clinical impact of CHIP in a large cohort of women under age 40 who were treated for breast cancer. In contrast to other studies of CHIP in cancer patients, we found that CHIP was infrequent and similar in mutation distribution compared to the non-cancer population. Importantly, we found that CHIP was not associated with impacts on relapse, non-relapse mortality, or overall survival over nine years of follow-up. These results should be reassuring to young women with breast cancer who are incidentally found to have CHIP, and to oncologists who use genomic assessments of peripheral blood in the care of their breast cancer patients.
Acknowledgments:
This work was supported by grants from the V Foundation, the National Institutes of Health (NIH) (K08-CA263181 [P.G.M.], K08-CA252174 [A.S.S.]), the Office of the Director (DP5-OD02958 [A.G.B.]), the Edward P. Evans Foundation (P.G.M..), the Burroughs Wellcome Foundation (A.G.B.), and the Pew Charitable Trusts and the Alexander and Margaret Stewart Trust (A.G.B.).
Disclosures:
P.G.M. reports consulting fees from Foundation Medicine and Roche; A.S.S. reports consulting fees from Adaptive Technologies and Roche. A.G.B. is a paid advisor and holds equity in TenSixteen Bio. D.D. is on the Academic Advisory Board for Oncology Analytics, Inc and receives research funding from Canon, Inc. A.H.P. receives royalties from UpToDate. S.M.R. receives grant funding from Pfizer/Conquer Cancer. D.N. received funding from Pharmacyclics, unrelated to this project, and has stock ownership in Madrigal Pharmaceuticals, outside of this project. All other authors declare no conflicts of interest.
REFERENCES
- 1.Wolff AC, Blackford AL, Visvanathan K, Rugo HS, Moy B, Goldstein LJ, et al. Risk of marrow neoplasms after adjuvant breast cancer therapy: the national comprehensive cancer network experience. J Clin Oncol 2015;33(4):340–8 doi 10.1200/JCO.2013.54.6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harbeck N, Ewer MS, De Laurentiis M, Suter TM, Ewer SM. Cardiovascular complications of conventional and targeted adjuvant breast cancer therapy. Ann Oncol 2011;22(6):1250–8 doi 10.1093/annonc/mdq543. [DOI] [PubMed] [Google Scholar]
- 3.Zagar TM, Cardinale DM, Marks LB. Breast cancer therapy-associated cardiovascular disease. Nat Rev Clin Oncol 2016;13(3):172–84 doi 10.1038/nrclinonc.2015.171. [DOI] [PubMed] [Google Scholar]
- 4.Calip GS, Malmgren JA, Lee WJ, Schwartz SM, Kaplan HG. Myelodysplastic syndrome and acute myeloid leukemia following adjuvant chemotherapy with and without granulocyte colony-stimulating factors for breast cancer. Breast Cancer Res Treat 2015;154(1):133–43 doi 10.1007/s10549-015-3590-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 2017;17(1):5–19 doi 10.1038/nrc.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126(1):9–16 doi 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20(12):1472–8 doi 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. The New England journal of medicine 2014;371(26):2488–98 doi 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England journal of medicine 2014;371(26):2477–87 doi 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2020;141(2):124–31 doi 10.1161/CIRCULATIONAHA.119.044362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. The New England journal of medicine 2017;377(2):111–21 doi 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller PG, Qiao D, Rojas-Quintero J, Honigberg MC, Sperling AS, Gibson CJ, et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood 2022;139(3):357–68 doi 10.1182/blood.2021013531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honigberg MC, Zekavat SM, Niroula A, Griffin GK, Bick AG, Pirruccello JP, et al. Premature Menopause, Clonal Hematopoiesis, and Coronary Artery Disease in Postmenopausal Women. Circulation 2021;143(5):410–23 doi 10.1161/CIRCULATIONAHA.120.051775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Honigberg MC, Patel AP, Lahm T, Wood MJ, Ho JE, Kohli P, et al. Association of premature menopause with incident pulmonary hypertension: A cohort study. PLoS One 2021;16(3):e0247398 doi 10.1371/journal.pone.0247398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Honigberg MC, Zekavat SM, Aragam K, Finneran P, Klarin D, Bhatt DL, et al. Association of Premature Natural and Surgical Menopause With Incident Cardiovascular Disease. JAMA 2019;322(24):2411–21 doi 10.1001/jama.2019.19191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim PG, Niroula A, Shkolnik V, McConkey M, Lin AE, Slabicki M, et al. Dnmt3a-mutated clonal hematopoiesis promotes osteoporosis. J Exp Med 2021;218(12) doi 10.1084/jem.20211872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet 2020;52(11):1219–26 doi 10.1038/s41588-020-00710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J Clin Oncol 2017;35(14):1598–605 doi 10.1200/JCO.2016.71.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017;21(3):374–82 e4 doi 10.1016/j.stem.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller PG, Gibson CJ, Mehta A, Sperling AS, Frederick DT, Manos MP, et al. Fitness Landscape of Clonal Hematopoiesis Under Selective Pressure of Immune Checkpoint Blockade. JCO Precis Oncol 2020;4 doi 10.1200/PO.20.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller PG, Sperling AS, Brea EJ, Leick MB, Fell GG, Jan M, et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Adv 2021;5(15):2982–6 doi 10.1182/bloodadvances.2021004554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mouhieddine TH, Sperling AS, Redd R, Park J, Leventhal M, Gibson CJ, et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat Commun 2020;11(1):2996 doi 10.1038/s41467-020-16805-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tahri S, Mouhieddine TH, Redd RA, Lampe LM, Nilsson KI, El-Khoury H, et al. Clonal hematopoiesis is associated with increased risk of progression of asymptomatic Waldenstrom macroglobulinemia. Blood Adv 2021. doi 10.1182/bloodadvances.2021004926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruddy KJ, Gelber SI, Tamimi RM, Ginsburg ES, Schapira L, Come SE, et al. Prospective study of fertility concerns and preservation strategies in young women with breast cancer. J Clin Oncol 2014;32(11):1151–6 doi 10.1200/JCO.2013.52.8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boettcher S, Miller PG, Sharma R, McConkey M, Leventhal M, Krivtsov AV, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019;365(6453):599–604 doi 10.1126/science.aax3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schon K, Tischkowitz M. Clinical implications of germline mutations in breast cancer: TP53. Breast Cancer Res Treat 2018;167(2):417–23 doi 10.1007/s10549-017-4531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams N, Lee J, Mitchell E, Moore L, Baxter EJ, Hewinson J, et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 2022;602(7895):162–8 doi 10.1038/s41586-021-04312-6. [DOI] [PubMed] [Google Scholar]
- 28.Gibson CJ, Kim HT, Zhao L, Murdock HM, Hambley B, Ogata A, et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. J Clin Oncol 2022;40(2):189–201 doi 10.1200/JCO.21.02286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malcovati L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017;129(25):3371–8 doi 10.1182/blood-2017-01-763425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Y, Ulrich BC, Supplee J, Kuang Y, Lizotte PH, Feeney NB, et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin Cancer Res 2018;24(18):4437–43 doi 10.1158/1078-0432.CCR-18-0143. [DOI] [PubMed] [Google Scholar]
- 31.Castillo D, Yuan TA, Nehoray B, Cervantes A, Tsang KK, Yang K, et al. Clonal Hematopoiesis and Mosaicism Revealed by a Multi-Tissue Analysis of Constitutional TP53 Status. Cancer Epidemiol Biomarkers Prev 2022;31(8):1621–9 doi 10.1158/1055-9965.EPI-21-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolton KL, Zehir A, Ptashkin RN, Patel M, Gupta D, Sidlow R, et al. The Clinical Management of Clonal Hematopoiesis: Creation of a Clonal Hematopoiesis Clinic. Hematol Oncol Clin North Am 2020;34(2):357–67 doi 10.1016/j.hoc.2019.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Sella T, Fell GG, Miller PG, Gibson CJ, Rosenberg SM, Snow C, et al. Patient perspectives on testing for clonal hematopoiesis of indeterminate potential. Blood Adv 2022. doi 10.1182/bloodadvances.2022008376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.