

Abstract
DNA editing enzymes perform chemical reactions on DNA nucleobases. These reactions can change the genetic identity of the modified base or result in gene expression modulation. Interest in DNA editing enzymes has burgeoned in recent years due to the advent of CRISPR-Cas systems, which can be used to direct their DNA editing activity to specific genomic loci of interest. In this review, we showcase DNA editing enzymes that have been repurposed or redesigned and developed into programmable base editors. This includes cytidine and adenosine deaminases, glycosylases, methyltransferases, and demethylases. We highlight the astounding degree to which these enzymes have been redesigned, evolved, and refined, and present these collective engineering efforts as a paragon for future efforts to repurpose and engineer other families of enzymes. Collectively, base editors derived from these DNA editing enzymes facilitate programmable point mutation introduction and gene expression modulation by targeted chemical modification of nucleobases.
Keywords: base editors, deaminases, methyltransferases, demethylases, glycosylases, protein engineering
1. INTRODUCTION
DNA is conventionally considered a hard-coded and read-only repository of information, written in a language consisting of four canonical nucleotides. However, this simplified descriptor downplays the importance of DNA nucleobase modifications and the enzymes that introduce them (Figure 1a). These noncanonical bases have important roles in rewriting and modulating the genetic code.
Figure 1.
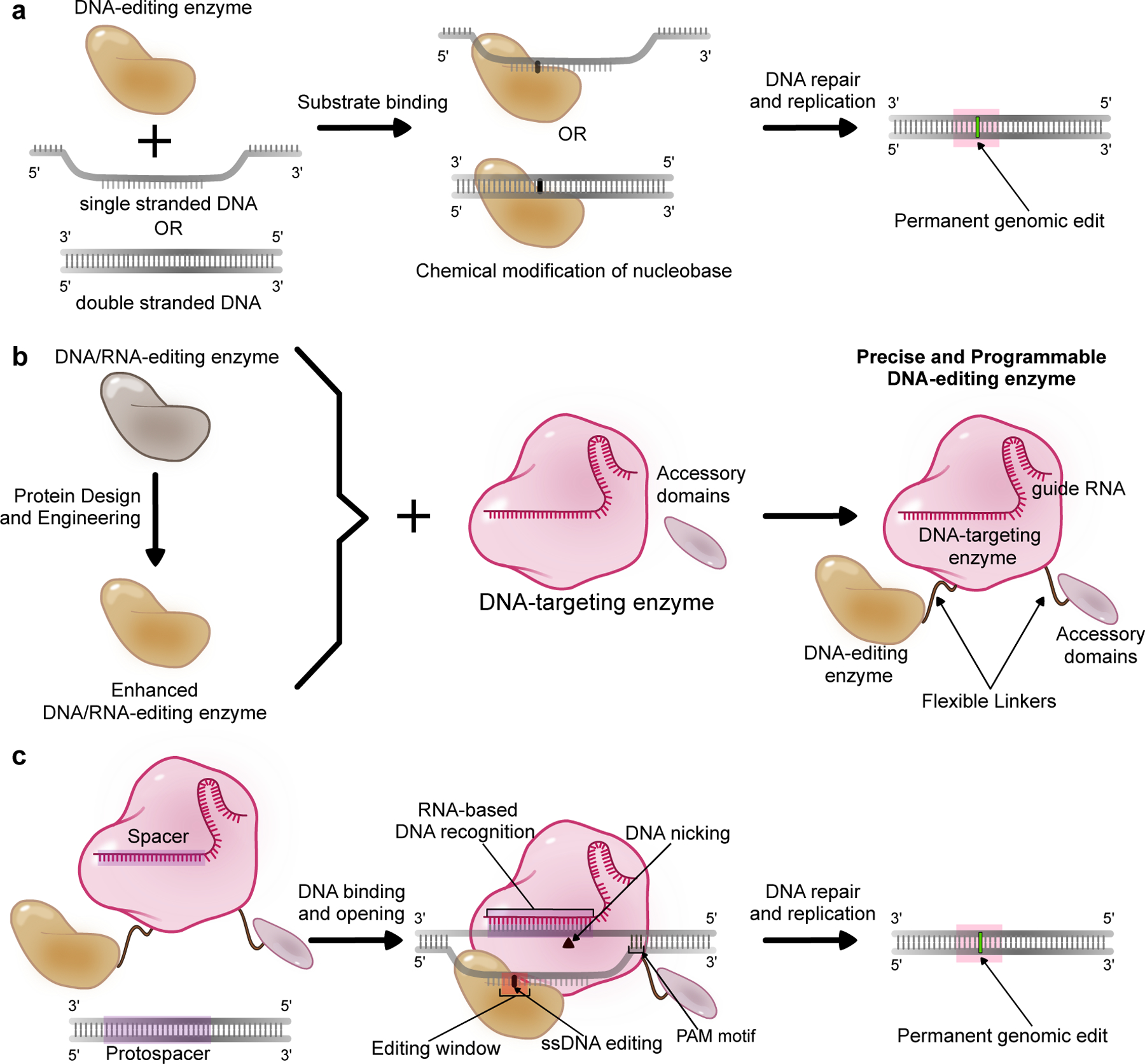
A schematic representation of DNA editing enzymes and their design and application as base editors. (a) “DNA editing enzymes” refer to enzymes that bind to DNA and chemically modify a target nucleobase. The resulting modified nucleobase is processed or interpreted by the cellular repair, replication, or transcriptional machinery as an alternate base, resulting in coding or epigenetic changes in the genome. (b) Naturally-occurring DNA editing enzymes as well as enhanced or engineered variants can be combined with precise and programmable DNA-targeting enzymes such as CRISPR-Cas proteins, along with optional accessory domains (which sometimes are DNA editing enzymes themselves) to produce modular enzyme complexes called base editors. (c) The DNA-targeting module of a base editor (usually CRISPR-Cas9) recognizes and binds to the target genomic locus via base-pairing between a guide RNA (gRNA) molecule and the genomic DNA (the “protospacer”). The protospacer must also be directly next to a protospacer adjacent motif (PAM) to facilitate Cas enzyme binding (for the most commonly used Cas9 system, this PAM sequence is NGG). The Cas protein unwinds the DNA double helix and exposes a small region of single-stranded DNA. If the DNA editing enzyme’s substrate is ssDNA, its DNA editing activity is focused on this ssDNA ’editing window’, and will chemically modify target nucleobases within this window. If, however, the DNA editing enzyme targets double-stranded DNA, it can modify target nucleobases within the general vicinity of the protospacer. Once processed by the cell’s DNA repair and replication machinery, these DNA edits become permanently incorporated into the genome.
We define “DNA editing” here as a chemical reaction that modifies a specific nucleobase in genomic DNA. This can include reactions that change the genetic identity of the base of interest (such as cytosine deamination to uracil) or those that install epigenetic changes (such as cytosine methylation to 5-methylcytosine). Given the importance of the fidelity of genomic DNA, DNA editing reactions and the enzymes that catalyze them have been extensively studied for their diverse roles in DNA repair, carcinogenesis, adaptive immunity, and epigenetic regulation. Despite our extensive knowledge of DNA editing enzymes and their roles in physiologically relevant contexts, interest in these enzymes was mainly confined to further understanding their canonical roles in cellular pathways until very recently.
The advent of programmable clustered regularly interspaced short palindromic repeat-associated (CRISPR-Cas) enzymes unwittingly ushered DNA editing enzymes into the era of genome and epigenome editing, where they have been repurposed into research tools capable of perturbing genomic DNA at will. Specifically, the ease and simplicity with which DNA targeting enzymes such as Cas9 can be re-programmed has been leveraged to transform DNA editing enzymes into precise, efficient, and programmable base editors. (Figure 1b, 1c). Moreover, the editing efficiencies and specificities of these DNA editing enzymes have been enhanced through extensive protein engineering and redesign (Figure 1b). This renewed interest in naturally occurring DNA editing enzymes combined with the push to expand the repertoire of genome and epigenome editors has also led to the development of artificial DNA editing enzymes through protein evolution and redesign. In this review, we highlight DNA editing enzymes that have been repurposed and applied to the fields of genome and epigenome editing. We discuss their structure, function, native biological roles, and reuse for genome editing.
2. DEAMINASES
Deamination of nucleobases in DNA as well as RNA is catalyzed by enzymes in the Cytidine Deaminase (CDA) superfamily. These enzymes consist of a zinc-dependent active site with a highly conserved H[A/V]E-x24−36-PCxxC motif that is supported by a five-stranded β-sheet core wrapped inside varying number of α-helices (Figure 2, 3)(1, 2). Despite their overall structural similarity, members of this superfamily can target purine or pyrimidine nucleobases in ssDNA, ssRNA, dsRNA, nucleotides, nucleosides, or free nucleobases. Given this extensive chemical diversity, it is not surprising that these enzymes are known to play critical roles in various physiological settings, ranging from conferring immunity through antibody diversification to causing certain forms of cancers via C·G→T·A hypermutation(3–5).
Figure 2.
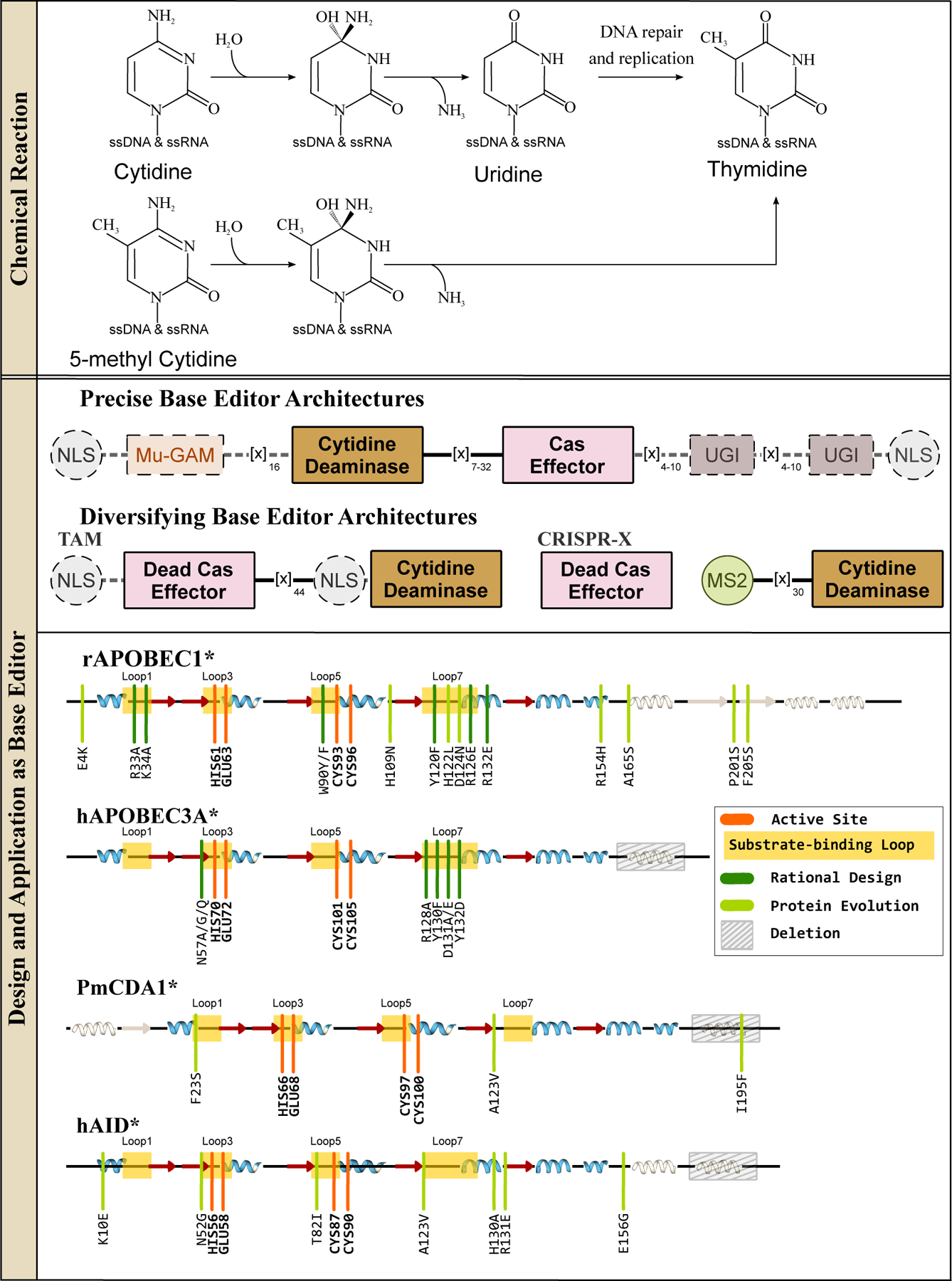
ssDNA cytidine deaminases and their design and applications in genomic DNA base editing. (Top) These enzymes hydrolytically deaminate cytosines or 5-methylcytosines in ssDNA and RNA to yield a uridine or thymine base, respectively. Overall, these reactions give rise to C·G→T·A base pair conversions. (Middle) Representative CBE architectures are shown, with essential and non-essential components indicated with solid and dashed outlines, respectively. (Bottom) Secondary structure alignments of APOBEC and AID deaminases are shown, with an emphasis on the similarity of their core CDA fold. Locations of the substrate-binding loops and active site residues are indicated, and key mutations discovered using either rational design or directed evolution approaches to enhance certain properties of the corresponding CBE are shown in dark and light green, respectively.
Figure 3.
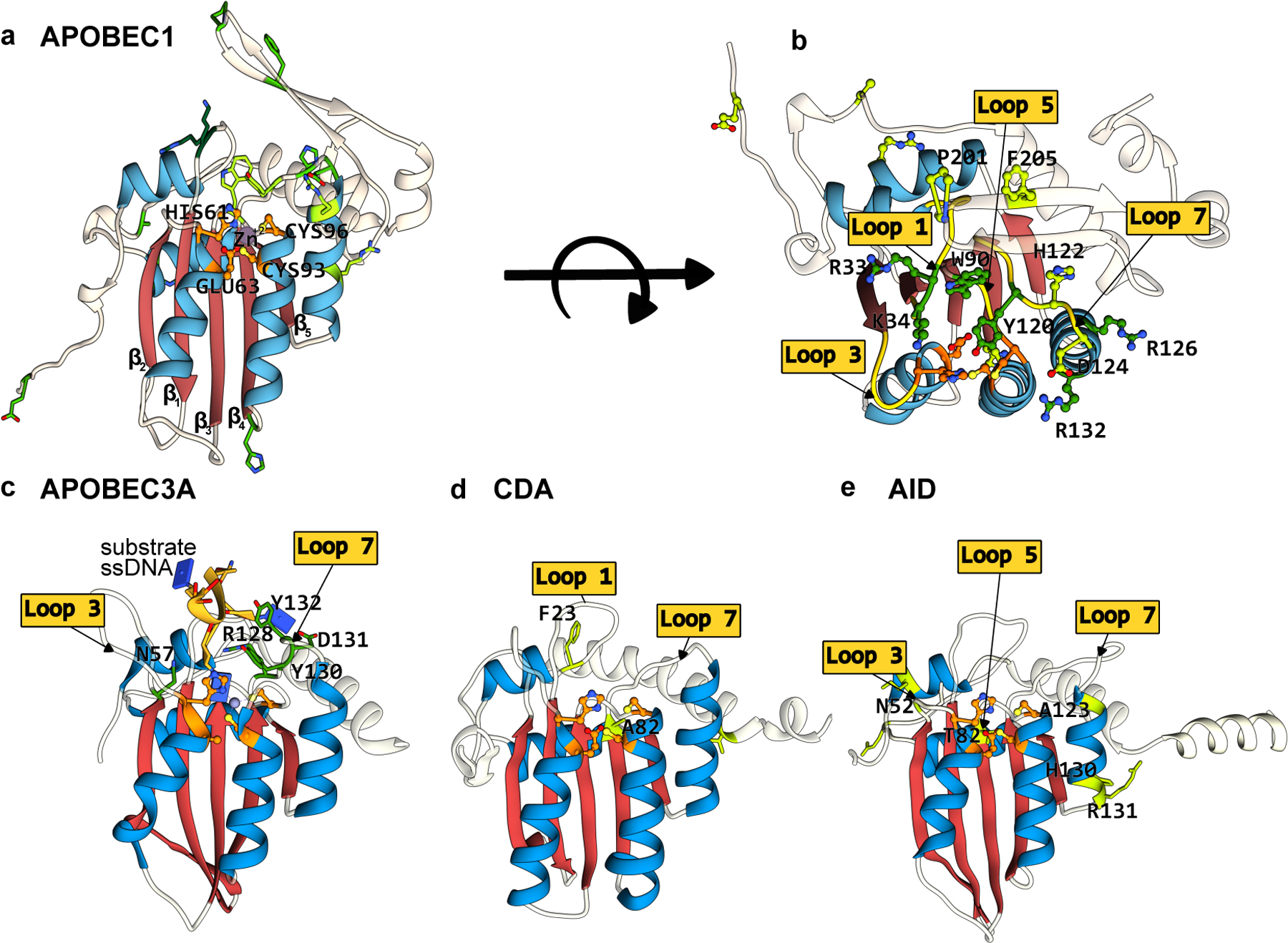
Structures of APOBEC/AID deaminases, which form the basis of the most extensively used CBEs. The conserved CDA fold is color-coded as follows: the β-sheet core is in red and the peripheral α-helices are in blue. The active site residues are shown in orange, with mutations that have enhanced certain properties of these deaminases (when used as a base editor) shown in green. (a) Side-view of the APOBEC1 structure (generated using Alphafold2(37)) depicting the CDA-family fold and the active site residues. (b) Top-view of the APOBEC1 structure, highlighting key amino acid mutations shown to enhance its base editing activity and the critical active site loops where these residues are located. (c-e) Side-view of APOBEC3A-ssDNA(PDB ID:5KEG(60)), CDA, and AID (generated using Alphafold2(37)), respectively. Active site loops and key amino acids are highlighted. Labels for active site residues are omitted for clarity. (cf. Figure 2)
2.1. Cytidine deaminases
Cytidine deaminase enzymes catalyze the deamination of cytidine to uridine in ssDNA and/or ssRNA (Figure 2). As uridine has the base-pairing properties of thymidine, cytidine deamination in DNA causes C·G→T·A point mutations, and cytidine deamination in RNA will directly recode the transcriptome. Enzymes in this family include activation-induced cytidine deaminase (AID), which is involved in antibody diversification, cytidine deaminase (CDA), which is involved in the pyrimidine salvage pathway, and various apolipoprotein B mRNA-editing enzyme, catalytic polypeptides (APOBECs), which generate protein diversity via editing of various mRNAs within the cell(Figure 3). Expression level changes and mutations in these genes are associated with cancer due to increased levels of genome-wide C·G→T·A mutagenesis. These enzymes’ potential to be repurposed into targeted C·G→T·A point mutation introduction tools was identified early on. However, prior to the mechanistic elucidation of CRISPR-Cas systems, there were no compatible DNA-targeting modules that could be used with cytidine deaminases, due to their substrate requirement of single-stranded nucleic acid(6).
The key to unlocking cytidine deaminases’ potential as programmable base editors was the formation of an R-loop by Cas9 when bound to DNA (Figure 1c). This R-loop generates a localized section of accessible ssDNA, the native substrate of these enzymes. Komor et al.(7) recognized this, and evaluated several APOBEC/AID deaminases as potential DNA editing enzymes when directly fused to Cas9. Befittingly, APOBEC1 (Figure 3a, 3b), the titular founding member of this family, formed the basis for the first cytosine base editor (CBE), named BE1(2, 7). BE1 consists of rat APOBEC1 (rAPOBEC1) fused to a catalytically inactivated Cas9 (dCas9) enzyme via a flexible linker. In in vitro experiments, BE1 facilitated efficient, targeted cytosine deamination within a ∼5 nucleotide window in the Cas9 protospacer. While this prototypical base editor architecture established precedence that the functionalities of an ssDNA-specific deaminase and Cas9 can be combined successfully to achieve targeted C→U editing, the BE1 prototype did not function efficiently beyond the test tube. This is because mammalian cells contain another DNA editing enzyme called uracil N-glycosylase (UNG, see Section 3 for more details). UNG identifies aberrant uracils in the genome and cleaves the glycosidic bond between the uracil nucleobase and the deoxyribose sugar to initiate base excision repair (BER), in effect undoing the edits made by the BE1 enzyme. To circumvent this issue, Komor et al.(7) appended the short peptide uracil DNA glycosylase inhibitor (UGI, which binds to and inhibits the UNG enzyme) to the C-terminal end of BE1, resulting in BE2. With this new accessory domain, BE2 outperformed the BE1 prototype, but its editing activity was still modest. To further improve the efficiency of the C·G→T·A outcome, Komor et al.(7) swapped dCas9 with Cas9n. This small change results in the Cas9n enzyme nicking the DNA backbone of the strand opposite the introduced uracil, in effect “tricking” the cell’s DNA repair enzymes to replace the broken strand and in the process use the uracil-containing strand as a template. This final design, rAPOBEC1-Cas9n-UGI (Figure 2), is called BE3 and its basic architecture has served as a template for the development of subsequent base editors. BE3 facilitates C·G→T·A point mutation introduction with low concurrent indel formation, as it avoids the use of DSBs.
Due to the remarkable modularity of the BE3 system, the number of CBE enzymes has increased dramatically since the development of BE3(7) through the addition, replacement, and/or improvement of its various modules. For example, the rAPOBEC1 deaminase used in the original BE3 construct has been replaced by many other cytidine deaminase enzymes, which will be discussed in the upcoming sections. The basic BE3 architecture can also be “accessorized” with additional protein modules (Figure 2) to further increase editing efficiency or precision. For example, the fourth generation BE4 architecture contains longer linkers and two UGI subunits instead of one (8), resulting in increased C·G→T·A editing efficiency(9). Addition of Mu-Gam, a DSB end-binding protein, to the BE4 architecture reduced indel formation, therefore improving base editing precision (9). Fusion of optimized nuclear localization signal (NLS) peptides to the BE4 architecture has led to the development of BE4max and “FNLS” CBE variants, which boast enhanced nuclear uptake of the CBE and therefore higher editing efficiencies (Figure 2)(8, 10).
The Streptococcus pyogenes (Sp) Cas9 homolog was used in the initial CBE constructs as the DNA-targeting module. However, it can be replaced by other DNA-targeting modules, including other Cas9 homologs(11), Cas12 enzymes(12, 13), and engineered Cas variants(11, 13–32). However, as the focus of this review is on DNA editing enzymes, we refer readers interested in the design specifics and functional characteristics of the various accessory domains and Cas variants integrated into the CBE architecture to relevant publications cited above and extensively reviewed elsewhere (33–35). Here, we predominantly focus on DNA editing domains and the extensive redesign and engineering efforts made when applying them as base editors.
2.1.1. APOBECs.
Although there is no structure currently available for rAPOBEC1, the structure of its human analog (hA1)(36), which shares 69% homology with the deaminase domain of rAPOBEC1, as well as its Alphafold2 predicted structure(37) suggest that rAPOBEC1 adopts the canonical CDA superfamily fold (Figure 3a, 3b). APOBEC1 enzymes’ most well-known target is cytidine 6666 (present in a 5′-AC−3′ motif) in the Apolipoprotein B (APOB) mRNA(5), but they additionally target hundreds of other mRNA transcripts. While their primary targets are mRNA, APOBEC1 enzymes were found to also have activity on ssDNA, and prefer 5′-TC−3′ motifs(38, 39).
When incorporated into the CBE architecture, wild-type(wt)-rAPOBEC1 retains its high preference for 5′-TC−3′ in ssDNA, with an aversion against 5′-GC−3′, which is a major limiting factor for wt-rAPOBEC1-based CBEs(7). The editing window for these editors was found to depend on the sequence motif of the target C (the window for 5′-GC−3′’s is narrower than for 5′-TC−3′’s), but generally is nucleotides 4 through 8 within the protospacer (Figure 1c). If multiple target Cs are present within this window, they are often all deaminated (known as ”bystander editing”) as the rAPOBEC1 enzyme is quite processive. Lastly, these early CBEs have been shown to deaminate 5′-AC−3′’s transcriptome-wide (known as off-target RNA editing), as rAPOBEC1 inherently acts on both ssDNA and ssRNA substrates(40–43).
Attempts to address these limitations of wt-rAPOBEC1-based CBEs have led to extensive engineering and redesign of the deaminase domain. Many of the mutations that have been discovered in these studies are concentrated in the substrate-binding active site loops of rAPOBEC1 (Figure 2 and 3a, 3b). Previous work had established the importance of a conserved Trp residue in the SWS-containing active site loop 5 of the APOBECs(44) for substrate binding and formation of a hydrophobic catalytic cavity(45). Kim et al. (11) mutated this residue (W90 in rAPOBEC1) to Tyr in BE3 to slightly reduce the hydrophobicity of the active site and therefore potentially lessen the processivity of the enzyme (Figure 2 and 3b). This resulted in a narrower editing window, albeit with a slight reduction in the overall editing activity. In the same study, two Arg residues in another active site loop (R126 and R132 on loop 7, (Figure 2 and 3b) known to make contacts with the phosphate backbone of the target ssDNA were also mutated. These mutations, R126E and R132E, also narrowed the editing window, potentially as a result of a reduction in ssDNA binding due to electrostatic repulsion (Figure 2 and 3b). These critical substrate binding mutations were combined to yield four new CBE variants with narrower editing windows, and therefore greatly reduced bystander editing, than the canonical wt-rAPOBEC1 based BE3: YE1 (W90Y+R126E), YE2 (W90Y+R132E), EE (R126E+R132E), and YEE (W90Y+R126E+R132E).
These active site loops have also been implicated in imparting sequence preferences to the APOBEC enzymes(42, 44). However, altering the target preference of rAPOBEC1 from 5′-TC−3′ has required more extensive protein engineering methods than traditional structure-guided redesign. Using a phage-assisted continuous evolution approach called BE-PACE(46) to select for mutants with increased activity and reduced sequence specificity, several mutations in rAPOBEC1 were discovered that eliminated any bias for the base 5′ to the target C. Many of the mutations identified through BE-PACE lie far away from the substrate-binding pocket, and are therefore hypothesized to enhance editing activity by improving expression levels of the protein. However, two of the most crucial mutations discovered in this directed evolution effort, H122L and D124N, are located in active site loop 7, which is likely to interact with the nucleotide 5′ to the target C based on homology among the APOBECs(42, 44, 46) (Figure 2 and 3b). Additionally, two mutations (P201S and F205S) were identified in the hydrophobic C-terminal β-strands (Figure 2 and 3b). This C-terminal region is thought to serve as a flap that shields the hydrophobic active site of APOBEC1 from water molecules(36). Mutations in this active site flap, especially polar mutations such as these, may open up the active site in the evoAPOBEC-CBE variant and allow the enzyme to better access ssDNA substrates, resulting in improved activity and a reduced sequence bias(46). This C-terminal flap domain was completely removed in the “FERNY-CBE” variant, which was designed via an ancestral reconstruction (ASR) of 78 APOBEC1 homologs. The deaminase domain of FERNY-CBE is therefore 30% smaller than rAPOBEC1, and this base editor exhibits an increased editing efficiency, particularly on non-5′-TC−3′ motifs(46). BE-PACE of FERNY-CBE resulted in evoFERNY-CBE, which also contains mutations in the active site loop 7 (H102P and D104N, (Figure 2), corresponding to residues H122 and D124N evoAPOBEC1 variant(46).
ASR has now become a popular protein engineering method, especially for enzymes with a large number of extant homolog sequences(47). ASR on APOBEC1 has resulted in the Anc689-BE variant in addition to FERNY-CBE, which differs from rAPOBEC1 by 45 amino acids but demonstrates an improved editing profile(8).
It was recently discovered that the canonical BE3 enzyme induces transcriptome-wide RNA off-target edits, which can be detrimental for therapeutic applications(40, 41). To combat this, Grünewald et al. (40) used structure-guided design to introduce two neutralizing amino acid mutations in the active site loop 1 of rAPOBEC1, R33A and K34A (Figure 2 and 3b). This produced the “SECURE-CBE” variant, which has reduced off-target RNA editing compared to BE3. Interestingly, these residues constitute a cluster of positively charged surface residues on loop 1 (Figure 2 and 3b), which is speculated to serves as a binding interface with the apobec-1 complementation factor (ACF) cofactor, which facilitates docking of APOBEC1 to its target mRNA. The R33A and K34A mutations are therefore speculated to inhibit the interaction between SECURE-CBE and ACF to reduce off-target RNA-binding affinity(48), but there is some conflicting evidence against this theory(36). While off-target RNA editing is an issue with the wt-rAPOBEC1-based CBEs, many evolved/mutated variants, particularly the rationally designed YE1 variant, show greatly reduced RNA editing activity while retaining high on-target DNA editing activity(49).
Many other wt-cytidine deaminase enzymes have been used in place of rAPOBEC1 in the basic BE3 and BE4 architectures, including APOBEC1 homologs(50, 51), APOBEC2(50), and APOBEC3s (A3A:(19, 51–53), A3B:(51–54), A3C: (50, 51, 54), A3D:(50, 51, 54), A3F:(50, 51, 54), A3G:(8, 50, 51, 54, 55), and A3H:(51, 54)) Further, similar development and design trajectories have been followed for these APOBEC homologs(8, 9, 19, 50–52, 54–56), with the most extensively used being the human APOBEC3A (hA3A) deaminase enzyme(19, 41, 50, 51).
APOBEC3A(A3A), like APOBEC1, also edits both ssDNA and RNA substrates, however, prefers to edit cytidines in a 5′-TC-3′ motif (Figure 2 and 3c). Wang et al.(51) demonstrated that the rAPOBEC1-based BE3 does not efficiently edit methylated cytosines. Given the high occurrence of 5-methylcytosines (5mC) in CpG repeats at promoter regions and their significance in epigenetic regulation (See Section 4), CBEs capable of editing methylated cytosines would be highly useful tools. It also should be noted that due to the extra methyl group on 5mC, deamination of this modified base results in the canonical DNA base T (Figure 2). The authors, therefore, compared editing efficiencies at methylated CpG sites of various APOBEC and AID cytidine deaminases in the BE3 architecture. They established that hA3A-CBE was the most efficient wt cytidine deaminase enzyme out of those tested for targeted deamination of methylated cytosines(51). This can be attributed to A3A’s relatively short loop 1 (Figure 3c), which has previously been shown to be critical for its unique 5mC→T deamination activity(57–59). The hA3A-CBE variant has also been subjected to structure-based redesign to reduce bystander editing by imparting the deaminase with an extreme bias towards the 5′-TC−3′ motif(19). These engineered variants harbor the N57G (which was named “eA3A-CBE”), N57A, or N57Q and Y130F mutations within hA3A, which all occur in active site loops 3 or 7 and therefore flank the target C base on either side (Figure 2 and 3c)(60). A similar strategy was pursued with a CBE derived from the human APOBEC3G (hA3G) deaminase(61). While previous work with the full-length hA3G deaminase demonstrated that hA3G-CBEs typically had greatly reduced editing efficiency compared to rAPOBEC1-CBEs(9, 51), Liu et al. (61) used only the C-terminal catalytic domain of hA3G. This “eA3G-CBE” edits preferentially at 5′-CC−3′ motifs.
As the canonical target of wt-hA3A is DNA and not RNA, the A3A-CBE was evaluated for RNA off-target editing and found to have reduced, but still substantial, off-target RNA editing activity compared to the wt-rAPOBEC1-CBE(41, 70). This off-target activity is substantially suppressed by incorporating the R128A or Y130F mutations into the deaminase, which are in loop 7 and predicted to interact directly with the nucleic acid substrate (Figure 2 and 3c)(41). The eA3A-CBE variant (which harbors the N57G mutation in loop 3) was also found to have significantly reduced off-target RNA editing activity, which collectively suggests that single mutations in the active site loops can reduce the RNA editing activity of the deaminase while maintaining ssDNA editing activity(70). Interestingly, when the Y130F mutation is combined with Y132D (both residues are present in loop 7, (Figure 2 and 3c), the editing window of hA3A-CBE is narrowed(51). Furthermore, the C-terminal α helix of hA3A is deemed to be highly flexible, and truncations to this helix (to produce “A3A∆-CBEs”) have been shown to modulate the editing window as well(21).
2.1.2. CDAs.
On the heels of the original rAPOBEC1-CBE work, Nishida et al. (62) engineered their own CBE using the ancestral APOBEC protein from sea lamprey, PmCDA1. Despite having less than 20% sequence similarity to APOBEC1, PmCDA1 adopts the same conserved fold (Figure 2 and 3d). This work additionally unwittingly showcased the modularity of the base editor architecture. In this base editor, called “Target-AID”, PmCDA1 is tethered to the C-terminal end of Cas9n via a 100-aa long linker. Due to the inherently higher deamination activity of the PmCDA1 enzyme, Target-AID displays no bias towards the base 5′ upstream of target C. When pmCDA1 was incorporated into the BE4 architecture, it was found to have a much wider (10 nucleotides) editing window than its rAPOBEC1 counterpart (5 nucleotides)(46). This CDA1-CBE (in the BE4 architecture) was subjected to BE-PACE under selective pressure for enhanced editing activity. Three mutations across the protein (F23S, A123V, and I195F) were identified, which collectively expanded the editing window of evoCDA1-CBE to 13 nucleotides and caused increased off-target RNA editing(46). It is interesting to note that despite the evoCDA1 mutations being identified via directed protein evolution (in which unbiased mutagenesis is utilized), F23S and A123V lie on substrate-binding loops 1 and 7, respectively (Figure 2 and 3d). The I195F mutation of evoCDA1 resides on the C-terminal α-helix (Figure 2), which is speculated to be a nuclear-export signal and therefor may be specific to the evolution conditions (which occur in bacteria). In fact, truncating this C-terminal helix results in “CDA1∆-CBEs” with narrower editing windows and reduced transcriptome-wide off-target compared to the CDA1-CBE(21).
2.1.3. AIDs.
Unlike their homologs in the APOBEC family, the human AID (hAID) enzyme can edit only ssDNA targets, although it can bind to both RNA and DNA. Furthermore, it demonstrates a strong preference for two bases upstream of the target cytosine, preferring 5′-WRC-3′ motifs(W = dA or dT and R = dA or dG)(5). This lack of specificity for the bases upstream of the target C, despite having a structure that is similar to the APOBECs, might be attributed to the existence of multiple positively charged residues on the Loop 7 of AID (Figure 2 and 3e). The native biological role of hAID is to increase antibody diversification in host cells by somatic deamination of immunoglobulin genes. Given this unique gene diversification capability, AID enzymes have typically been applied toward targeted random mutagenesis. Two distinct, AID-based diversifying base editors have been reported thus far: Targeted AID-mediated Mutagenesis (TAM)(56) and CRISPR-X(63) (Figure 2). Both systems rely on dCas9 as the DNA-targeting module, which recruits hAID to mutagenize cytosines and guanines in a broad (>20 base pairs) region both within and around the target protospacer. The TAM system fuses hAID to the C-terminal end of dCas9 (similarly to Target-AID)(56, 62). Truncations of the C-terminal α-helix, which serves as the nuclear export signal, have produced “TAM-AIDx” variants (P182X or R190X, where X=deletion), which further increases the activity of AID(56). The CRISPR-X system does not covalently tether dCas9 and hAID, but instead relies on an RNA aptamer-coat protein non-covalent interaction (specifically, the MS2 coat protein-MS2 hairpin loop interaction) to recruit hAID to the target locus(63) (Figure 2). CRISPR-X boasts a editing window of over 50 base pairs surrounding the target protospacer. This window can be further broadened to 100 base pairs by using a C-terminally truncated AID variant (AID∆), similarly to TAM(56, 63). Protein evolution of AID has identified the substrate binding loop mutations N52G, T82I, and A123V (Figure 2 and 3e) which enhance the deamination activity of AID to produce the hyperactive variant “eAID”(64, 65). Combining these mutations with the C-terminal truncations improves the activity of both TAM and CRISPR-X.
2.2. Adenosine Deaminases
The Tad/ADAT2, ADAR, and ADAT1 enzymes are distant relatives of the APOBEC/AID family, and catalyze the deamination of adenine to inosine (Figure 4). As inosine has the base-pairing properties of guanine, targeted adenine deamination was quickly identified as a viable strategy to programmably introduce A·T→G·C point mutations. These enzymes also conform to the CDA superfamily fold and contain an identical zinc-based active site(5, 44). However, unlike the APOBEC/AID deaminases, none of these extant adenine deaminases accept ssDNA as their native substrate. Instead, they are only known to act on either ssRNA (mainly loop regions within transfer RNA [tRNA]) or double-stranded RNA(66). Thus, the strategy of merely inserting these naturally-occurring adenine deaminases(E. coli TadA [ecTadA], human ADAR2, mouse ADA, or human ADAT2) into the canonical CBE architecture led to no measurable A·T→G·C activity(67). Therefore, Gaudelli et al. (67) resorted to directed evolution to create a ssDNA-specific adenine deaminase enzyme. Starting from ecTadA, a tRNA adenosine deaminase that edits the wobble base A34 of tRNAArg2(68) (Figure 5a, 5b), seven rounds of directed evolution identified 14 amino acid mutations that transformed this tRNA editing enzyme into a highly efficient ssDNA editing adenine base editor (ABE, (Figure 4 and 5c). The wt-ecTadA has a strong sequence preference for 5′-UACG-3′ sites in tRNA (Figure 5a, 5b), which was inherited in early generation mutant TadAs. Therefore, a subset of these 14 mutations was identified specifically to broaden the substrate scope of the enzyme.
Figure 4.
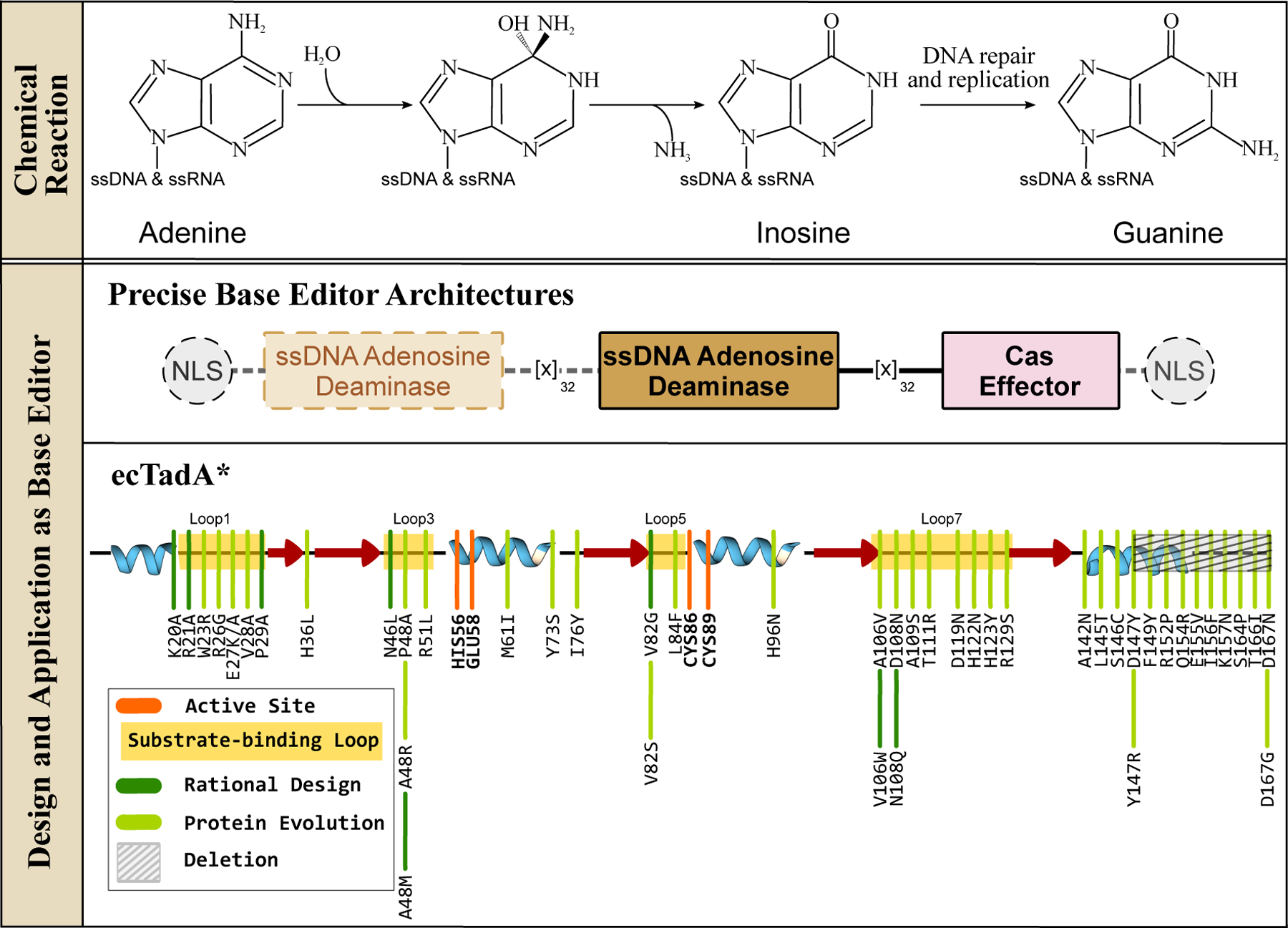
ssDNA adenosine deaminases and their design and application in genomic DNA base editing. (Top) These enzymes can hydrolytically deaminate adenines in ssDNA and ssRNA to yield an inosine, which is then processed into guanine via DNA replication and/or repair processes. Overall, this reaction gives rise to A·T→G·C base pair conversion. (Middle) Representative ABE architectures are shown, with the essential and non-essential components indicated with solid and dashed outlines, respectively. (Bottom) The secondary structure of the TadA enzyme is shown, with an emphasis on the core CDA fold. Locations of the substrate-binding loops and active site residues are indicated, and key mutations discovered using either rational design or directed evolution approaches to enhance certain properties of the corresponding ABE are shown in dark and light green, respectively.
Figure 5.
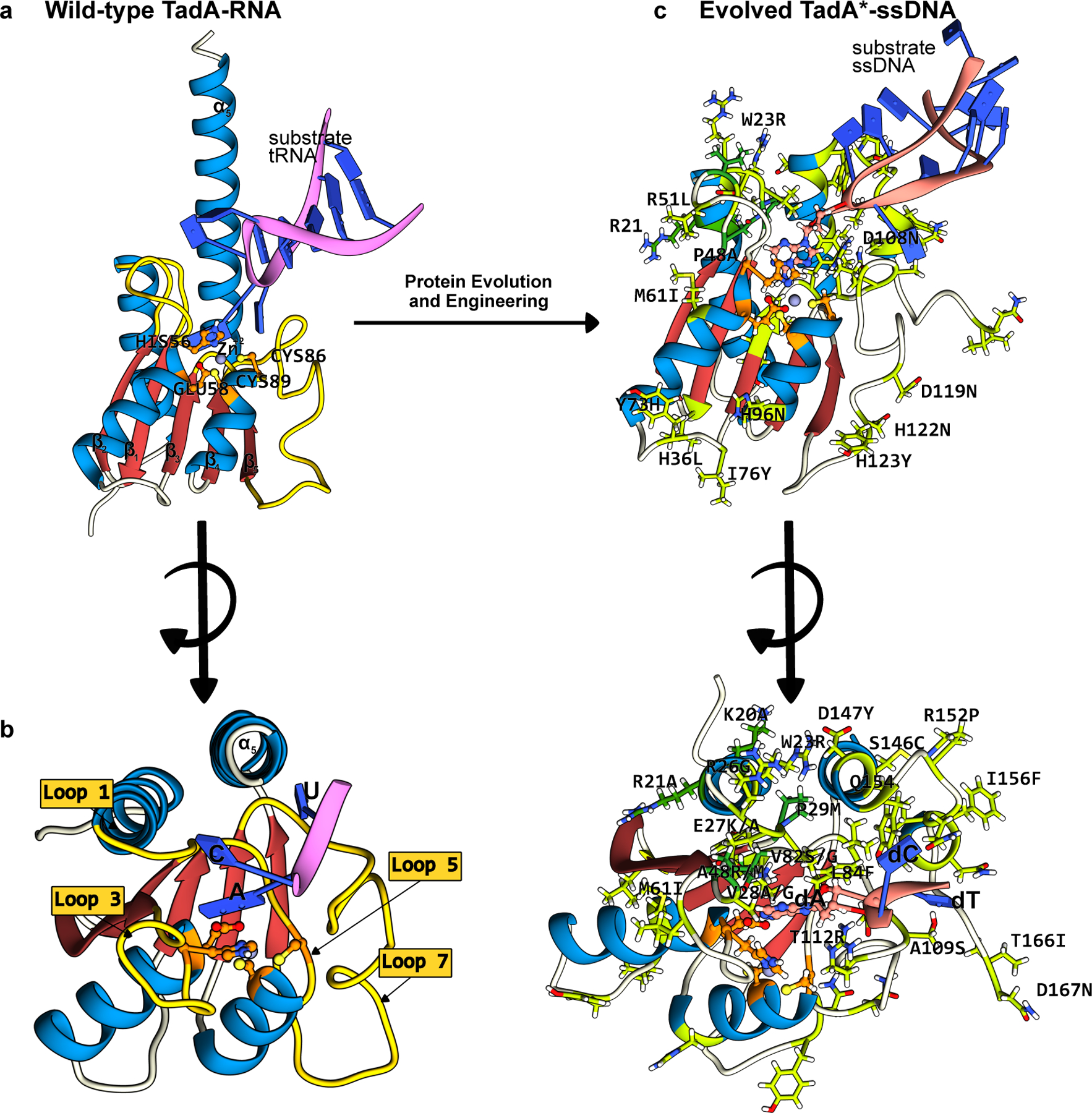
Structures of the wt-ecTadA enzyme bound to its substrate RNA, and the evolved TadA* bound to an ssDNA substrate. The conserved CDA fold is color-coded as follows: the β-sheet core is red and the peripheral α-helices are blue. The active site residues are shown in orange, with the mutations that have enhanced certain properties of the deaminase (when used as a base editor) shown in green. (a) Side-view of the wt-TadA-tRNA complex, generated using a combination of Alphafold2 predictions(37) and PDB ID:2B3J(82). (b) Top-view of the wt-TadA-tRNA complex. The RNA structure beyond the bases in the active site is omitted for clarity. (c) Side-view of the TadA*-ssDNA complex, generated from PDB ID:6VPC(75). (d) Top-view of the TadA*-ssDNA complex. The DNA structure beyond the bases in the active site is omitted for clarity. (cf. Figure 4)
The basic components of this prototypical ABE (referred to as ABE7.10) are very similar to the canonical CBE architecture. One important distinction, however, is that ABE7.10 lacks an equivalent UGI accessory domain (Figure 4). Inosine excision by endogenous glycosylase enzymes (see Section 3) such as Alkyl-Adenosine glycosylase (AAG) or endonuclease V (EndoV) is inefficient, most likely due to the inherently low spontaneous occurrence of inosines in genomic DNA(69). This is in direct contrast to the mutagenic uracil intermediate created by CBEs, which is actively and efficiently excised by UNG. Catalytically inactivated AAG and EndoV were tested as accessory domains in the ABE architecture (which would bind to the inosine intermediate, but not excise it, and in the process protect it from endogenous wt-AAG and wt-EndoV), but did not yield any noteworthy improvements in editing outcomes(67). An additional important difference between the CBE and ABE architectures is that ABE7.10 contains two copies of TadA fused to the Cas9n - one wt-ecTadA subunit and one evolved TadA (TadA*) subunit. The wt-TadA enzyme requires dimerization for activity on tRNA (one monomer aids in substrate binding, and the other performs the catalysis), and thus TadA was incorporated as a dimer in these early ABE constructs. It was found that using wt-TadA as the N-terminal monomer (which is the substrate binding monomer in ABE) improved editing yields compared to two TadA* subunits. The ABE7.10 final construct was therefore wt-TadA-TadA*-Cas9n. However, it was later found that the wt-TadA subunit is not necessary for efficient editing activity, as its removal has minimal impact on editing activity (70–73) (Figure 4). In fact, removal of the wt-TadA subunit is beneficial as it reduces the inherent residual RNA editing activity of ABE7.10, resulting in a better off-target profile(70, 74). However, it should be noted that removal of the covalently tethered wt-TadA subunit in the prototypical wt-TadA-TadA*-Cas9 architecture may not prevent these “miniABEs” from dimerizing in trans. Specifically, the recent cryo-EM structure of a later-generation ABE in complex with target DNA shows unambiguous dimerization between the TadA* subunits that belong to separate ABE molecules (PDB ID: 6VPC(75)).
The 14 mutations identified during the evolution of ABE7.10 are predominantly located in the substrate binding loops of the deaminase (loop 1: W23R, loop 3: P48A, R51L, loop 5: L84F, and loop 7: A106V, D108N, H123Y, and the C-terminal α-helix (S146C, D147Y, R152P, E155V, I156F, and K157N, (Figure 5c, 5d)). Out of these 14 mutations required for efficient ssDNA editing activity by TadA*, the D108N mutation in loop 7 has been shown to be absolutely critical by increasing the binding affinity between ssDNA and TadA*(71). Reversion of this single mutation back to the wt residue abrogates A·T→G·C activity, even when all 13 other mutations are present(71). The C-terminal helix mutations, especially R152P, kink the α-helix away from the active site (PDB ID: 6VPC) (Figure 5), making it more accessible to the ssDNA presented to the deaminase by the DNA-binding Cas effector(75), while simultaneously suppressing the RNA editing activity of TadA*(70, 74, 75). The significance of the C-terminal region of TadA* was highlighted by Gaudelli et al.(73) who in a series of sequential truncations showed that reducing the length of this α-helix too much can be detrimental to the ssDNA- as well as RNA editing activity of the deaminase. In fact, in addition to expanding the substrate scope of TadA to include ssDNA, these mutations may have additionally caused the enzyme to be more promiscuous, as Kim et al. (76) has reported low levels of C·G→T·A activity by ABE7.10, which would result from cytidine deamination by TadA*. This activity is suppressed by the introduction of the N108Q mutation(77). This substrate promiscuity of TadA* was exploited in three recent studies, in which the cytidine deamination activity of TadA* was enhanced through additional mutations in the active site loops. These mutations gave rise to a suite of TadA-derived CBEs(78–80).
Since the development of the prototypical ABE7.10, the TadA*7.10 deaminase has been subjected to further design and mutagenesis to improve its properties as an ABE. Directed evolution of TadA*7.10 using selective pressure for increased activity and reduced sequence specificity has resulted in ABE8e(72) and ABE8.20(73). Simultaneously, structure-based rational design has been employed to identify mutations that abolish the off-target RNA editing activity of TadA*, leading to the discovery of the K20A/R21A, V82G (referred to as “SECURE-ABEs”)(70), F148A(41), and V106W(81) mutations (Figure 4 and 5). More recently Chen et al. (77) employed a similar structure-based mutagenesis strategy in which they identified the L145T mutation, producing ABE9 which boasts a reduced editing window compared to the ABE8e variant. These higher-generation ABE8 and ABE9 mutations are localized primarily to the active site loops and the C-terminal α-helix, similar to the ABE7.10 mutations (Figure 4 and 5).
A noteworthy aspect of the design and development of ABEs is the heavy reliance on directed evolution of TadA*, which is in stark contrast with the design strategies used for developing CBEs. This is due to the lack of naturally occurring ssDNA editing adenosine deaminases compared to the plethora of ssDNA editing cytidine deaminases in the APOBEC/AID family. Hence, despite sharing many of the same design motivations (improving expression levels and nuclear import, improving on-target ssDNA editing, and suppressing off-target RNA editing), the development of ABEs has been unique and more challenging than that of CBEs.
2.3. Dual deaminase systems
As a testament to the modularity of the base editor architecture, five separate articles were published in 2020 that reported on various CBE/ABE “chimeras” that simultaneously introduce C·G→T·A and A·T→G·C point mutations(83–87). Briefly, these dual deaminase architectures combine TadA*7.10 on the N-terminal end of Cas9n and either PmCDA1 on the C-terminal end of Cas9n (SPACE(83), Target-ACEmax(84), and ACBE(87), Figure 6) or APOBEC3A/AID on the far N-terminal end of the TadA*7.10 (STEME(85) and A&C-BE(86), Figure 6). These systems additionally demonstrate the remarkable cooperativity of these two types of deaminases; none of the enzymes “out-compete” each other, as both C·G→T·A and A·T→G·C edits are observed in all five systems. In these systems, the mutations are confined within the canonical editing window in the protospacer of Cas9, but significant heterogeneity is observed in that C·G→T·A only, A·T→G·C only, and concurrent C·G→T·A and A·T→G·C editing products are observed.
Figure 6.
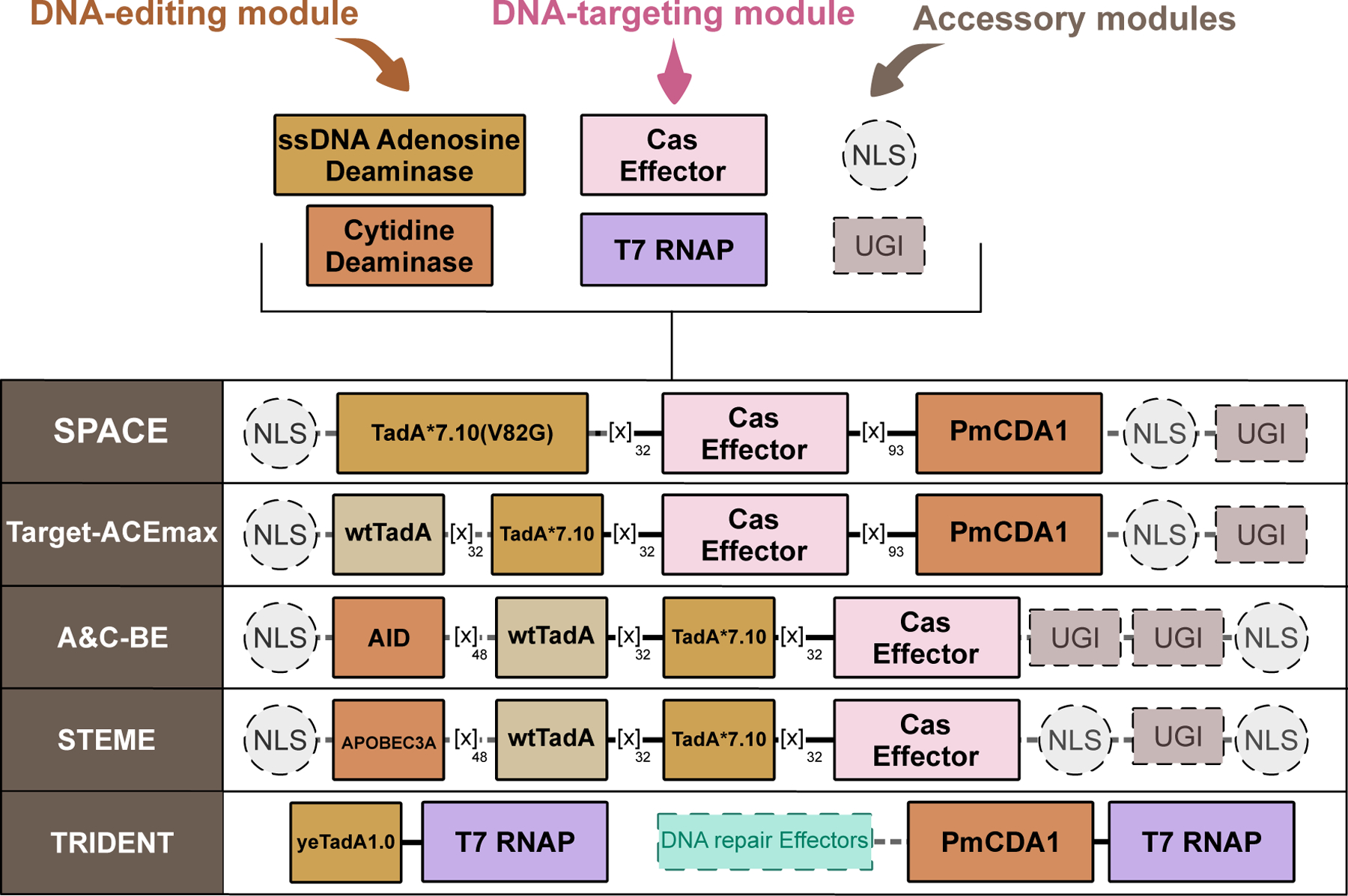
Design of cytosine and adenine deaminase dual base editors. Shown are representative dual base editor architectures, with essential and non-essential components indicated with solid and dashed outlines, respectively.
This same general strategy was utilized but modified for in vivo mutagenesis applications in yeast a year later when Cravens et al. (88) fused both TadA*7.10 and pmCDA1 to two separate T7 RNA Polymerase (T7RNAP) molecules, which they then co-expressed together. In this system, named “TRI-DENT”, genes downstream of a T7 promoter become mutagenized, as the DNA binding module (the T7RNAP) opens up the DNA and slides along the gene during transcription, while the deaminase enzymes modify cytosines and adenines across the gene (Figure 6). In this work, the authors additionally performed five rounds of directed evolution on TadA*7.10 to improve its activity and identified four mutations, three of which occur in the C-terminal α-helix (Q154R, S164P, and D167G, (Figure 4 and 5)), to yield “yeTadA1.0” (Figure 6)).
2.4. Deaminases for Mitochondrial DNA Editing
In addition to nuclear DNA, mammalian cells contain mitochondrial DNA (mtDNA), which is a multi-copy, circular, double-stranded DNA encoding for 13 essential proteins of the respiratory chain, as well as tRNAs and ribosomal RNAs (rRNAs) required to transcribe these essential proteins within the mitochondria. Anomalies in mtDNA, just as with nuclear DNA, are linked to many inheritable diseases. However, unlike nuclear DNA editing technologies, precise and efficient mtDNA editing has until very recently been elusive.
CRISPR-based genome editors have not been applied to mtDNA editing because reliable targeted delivery of nucleic acids (such as gRNAs) to mitochondria has not yet been achieved(101). As the delivery of proteins to mitochondria can be achieved using mitochondrial targeting signals (MTSs), mtDNA editing has been achieved using protein-only DNA-binding modalities such as transcription activator-like effector nucleases (TALENs) and zinc finger nucleases (ZFNs), the predecessors of Cas9. Introducing double-stranded breaks (DSBs) into the mtDNA at targeted loci using mitochondria-targeted TALENs and ZFNs (known as mitoTALENs and mitoZFNs) was therefore the only viable approach for mtDNA modification previously. Targeted introduction of DSBs in mtDNA results in large sequence deletions. Thus, the capabilities of these methods are limited. However, in 2020, Mok et al. (102) established a new milestone in the field of mtDNA editing by creating the first TALE-based CBE, called “DdCBE”.
Similar to the architecture of nuclear DNA editing CBEs, DdCBEs are also comprised of a DNA-targeting module fused to a DNA editing module, along with a UGI accessory domain and an MTS (instead of an NLS, Figure 7). However, the DNA-targeting module is now a pair of mitoTALEs that are designed to bind to neighboring half-sites. Furthermore, the DNA editing module is a dsDNA cytidine deaminase, since DNA binding by TALEs does not lead to the formation of exposed ssDNA (unlike Cas effectors). Mok et al. (102) therefore needed to utilize a non-APOBEC/AID deaminase as these enzymes are specific for ssDNA. Deaminase toxin A (DddAtox) is an elusive interbacterical toxin belonging to the type VI secretion system (T6SS) of Burkholderia cenocepacia(103, 104). Despite having a very different origin and native function from the traditional APOBEC/AID superfamily, DddAtox adopts a structure similar to the APOBECs/AID deaminases as it too exhibits a β-sheet core supporting a zinc-dependent active site (Figure 7 and 8). Given the potentially mutagenic and toxic nature of rampant dsDNA cytosine deamination facilitated by high intracellular expression levels of DddAtox, it was split into two non-toxic halves, which were each tethered to the pair of mitoTALEs (Figure 7 and 8)(102). Upon binding of the mitoTALEs to the two mtDNA target half-sites, the increased effective molar concentration of the N- and C-terminal halves of DddAtox facilitate its reassembly, allowing deamination of cytosines within the dsDNA between the two mitoTALEs half-sites (typically 14–18 base pairs). In this same work, the authors also generated an analogous Cas9 DddA-derived base editor for nuclear DNA editing (Figure 7). This strategy of splitting the DNA editing deaminase into separate fragments, which are then reassembled at the target site has also been applied with other DNA-deaminases(105–108) and RNA-deaminases(109) to afford temporal and/or spatial control of editing.
Figure 7.
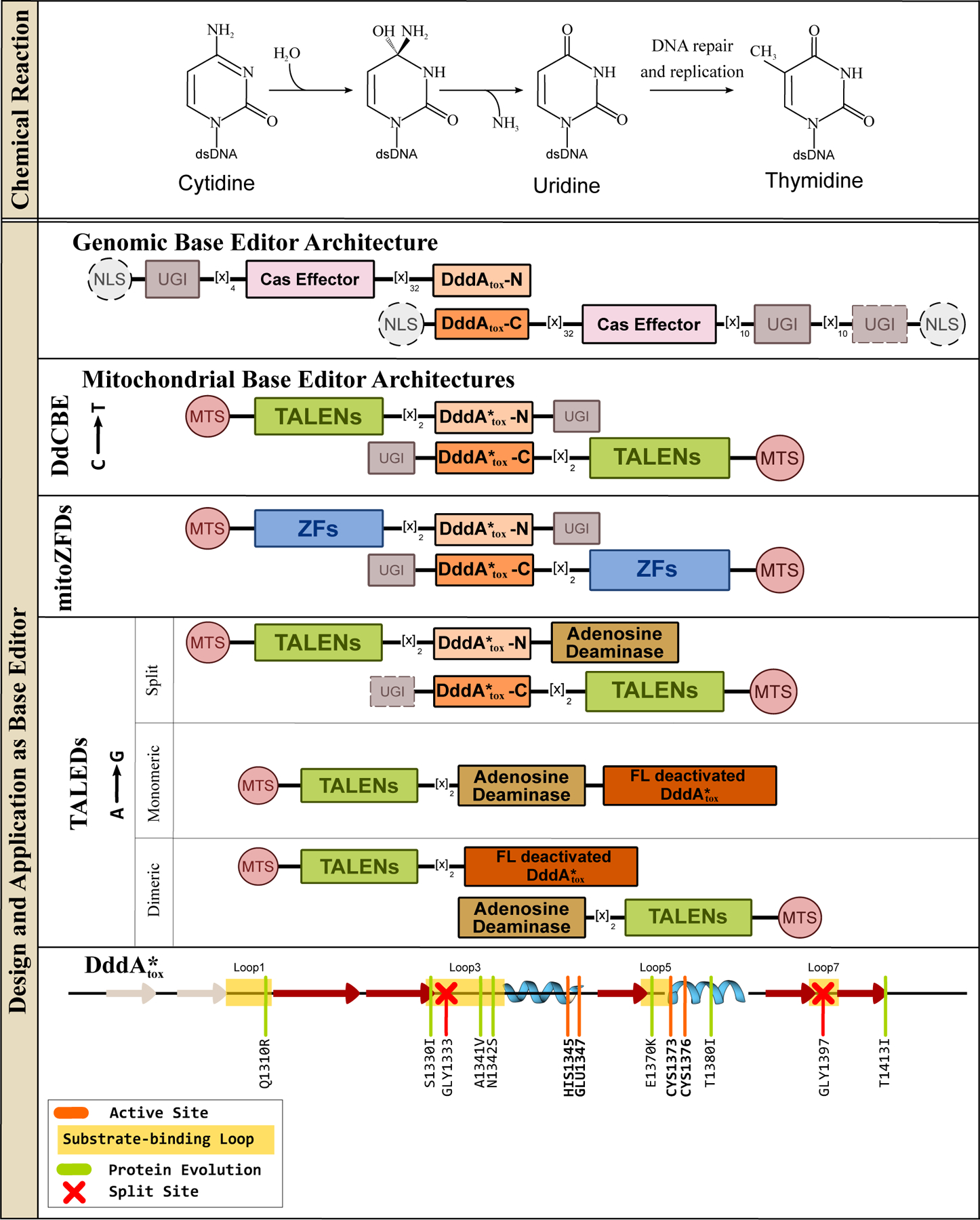
Deaminase toxin A (DddAtox), a dsDNA cytidine deaminase, and its design and application in genomic and mitochondrial base editing. (Top) DddAtox can hydrolytically deaminate cytosines in dsDNA to yield uridine, when is then processed into thymidine via DNA replication and/or repair processes. Overall, this reaction gives rise to a C·G→T·A base pair conversion. (Middle) Representative DdCBE, mitoZFDs, and DddAtox-based ABEs (TALEDs) architectures are shown, with essential and non-essential components indicated with solid and dashed outlines, respectively. (Bottom) Secondary structure alignment of the DddAtox enzyme is shown, with an emphasis on the core CDA fold. Locations of the substrate-binding loops and active site residues are indicated, and key mutations discovered using directed evolution to enhance certain properties of the corresponding DdCBE are shown in light green and the split sites are indicated with red crosses.
Figure 8.
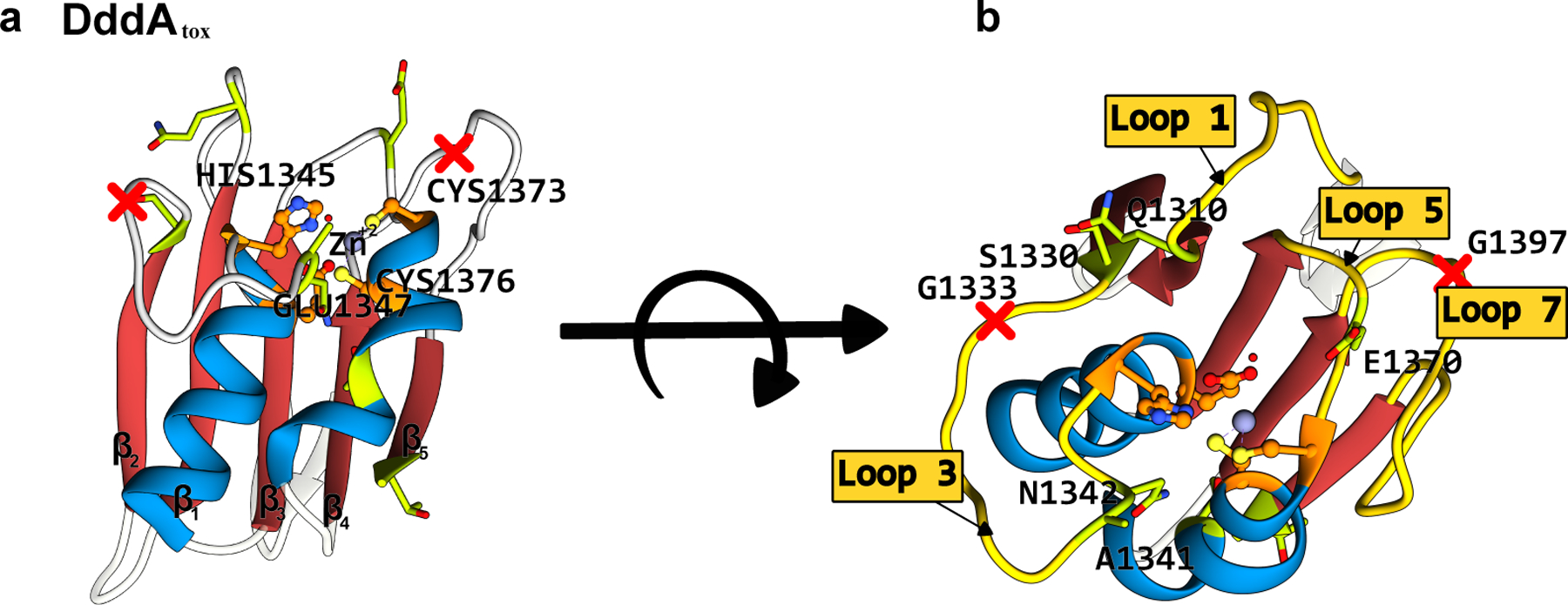
Structure of the B. cenocepacia DddAtox enzyme (PDB ID: 6U08)(102). (a) Side-view and (b) top-view. The conserved CDA fold is color-coded as follows: the β-sheet core is red and the peripheral α-helices are blue. The active site residues are shown in orange, with the mutations that have enhanced certain properties of the deaminase (when used as a base editor) shown in green. The split sites that were used to divide the enzyme into two inactive, non-toxic halves are shown as red crosses. (cf. Figure 7)
DddAtox-based DdCBEs displayed a preference for 5′-TC-3′ motifs, similar to the original nuclear DNA editing CBEs. In a follow up study, Mok et al.(110) evolved the DddAtox using BE-PACE to enhance its overall activity on both 5′-TC-3′ consensus targets as well as non-TC sites, giving rise to two DddAtox variants, DddA6 (Q1310R, S1330I, T1380I, and T1413I) and DddA11 (S1330I, A1341V, N1342S, E1370K, T1380I, and 1413I), respectively (Figure 7 and 8). Despite having a distinct origin and substrate from the APOBEC/AID deaminases, these evolved DddAtox mutations mimic the positioning of mutations identified in the evolved APOBECs and TadA*, as many of them reside in substrate-binding loops (Figure 7 and 8). However, some of the remote mutations (T1380I and T1413I specifically) may be responsible for enhancing activity by simply facilitating better reconstitution of the enzyme, as the evolution of DddAtox was conducted with the enzyme split in half (Figure 7 and 8).
mtDNA editing has also been extended to adenosine deamination(111). Simply fusing TadA*8e, the DNA editing domain from ABE8e (see section 2.2), to a mitoTALE did not facilitate significant A·T→G·C mitochondrial editing, again due to a lack of exposed ssDNA upon DNA binding by TALEs. Cho et al.(111) instead took the DdCBE architecture and replaced one of the UGI domains with TadA*8e to create a TALE-linked deaminase (“TALED”) construct (Figure 7). The resulting editor facilitates both C·G→T·A and A·T→G·C editing within the 16-bp region between the two TALE-binding sites. The act of dsDNA binding by the reconstituted DddA enzyme purportedly facilitates the ssDNA-specific TadA*8e enzyme to access the DNA for deamination. Targeted A·T→G·C editing with no C·G→T·A editing is achieved by simply omitting all UGI components from the TALED construct (Figure 7), or by catalytically inactivating the DddA enzyme (which will bind to DNA but not deaminate). Inactive DddA does not need to be split in half to avoid toxicity, so both dimeric and monomeric TALED constructs have been successfully used for A·T→G·C mtDNA editing (Figure 7). More recently, Lim et al. (112) substituted the TALEs in the DdCBE architecture with ZFs to yield mitoZFDs, once again demonstrating the modularity of the base editor architecture (Figure 7).
Lastly, while these TALE-derived base editors were developed to edit mtDNA in mammalian cells, a slightly modified version of DdCBE (with modified codon optimizations and localization sequences) has been shown to effectively edit mitochondrial and chloroplast DNA in plant cells(113). Further details about this novel plant organelle genome editing approach can be found in recent reviews by Barrera et al.(114) and Adashi et al. (115)
3. GLYCOSYLASES
The targeted chemical modification of a nucleobase by an editing enzyme is only the first step in base editing. The resulting modified nucleobase is typically recognized as DNA damage by the cell, and the endogenous DNA repair machinery will attempt to revert the base editing intermediate back to the original canonical base to restore the integrity of the genetic information. Therefore, the successful application of DNA editing enzymes as efficient base editors typically requires the incorporation of design elements that manipulate the endogenous DNA repair machinery. The most effective design element is the use of Cas9n to cleave the DNA strand opposite the modified base, but in certain cases this is not enough. The base editing intermediates U·G (for CBEs) and I·T (for ABEs) are canonical targets of the BER pathway, although the presence of the nick on the opposite strand may alter this. Nevertheless, the first step of BER is the recognition and excision of the damaged base by an enzyme from the DNA glycosylases superfamily (Figure 9)(116, 117). Although glycosylase enzymes are highly similar in their core catalytic domain and overall structure (Figure 10), individual members of this superfamily have evolved minute differences in their substrate recognition domains, allowing them to identify specific modified bases. Following damage recognition, glycosylases flip (or “evert”) the damaged base outside of the dsDNA helix, and excise it from the sugar-phosphate backbone by cleaving the N-glycosidic bond to generate an apurinic/apyrimidinic (AP) site (Figure 9). The resulting abasic site is then subsequently processed by AP endonucleases, DNA repair polymerases, and ligases to restore the original DNA sequence. Readers interested in further details of BER and other related DNA repair pathways (especially those that discuss these pathways’ involvement in base editing) are encouraged to consult recent reviews addressing this topic(118, 119). Here we will be focusing on DNA glycosylases, particularly UNG homologs, and how these DNA editing enzymes have been repurposed to yield novel “transversion” base editors.
Figure 9.
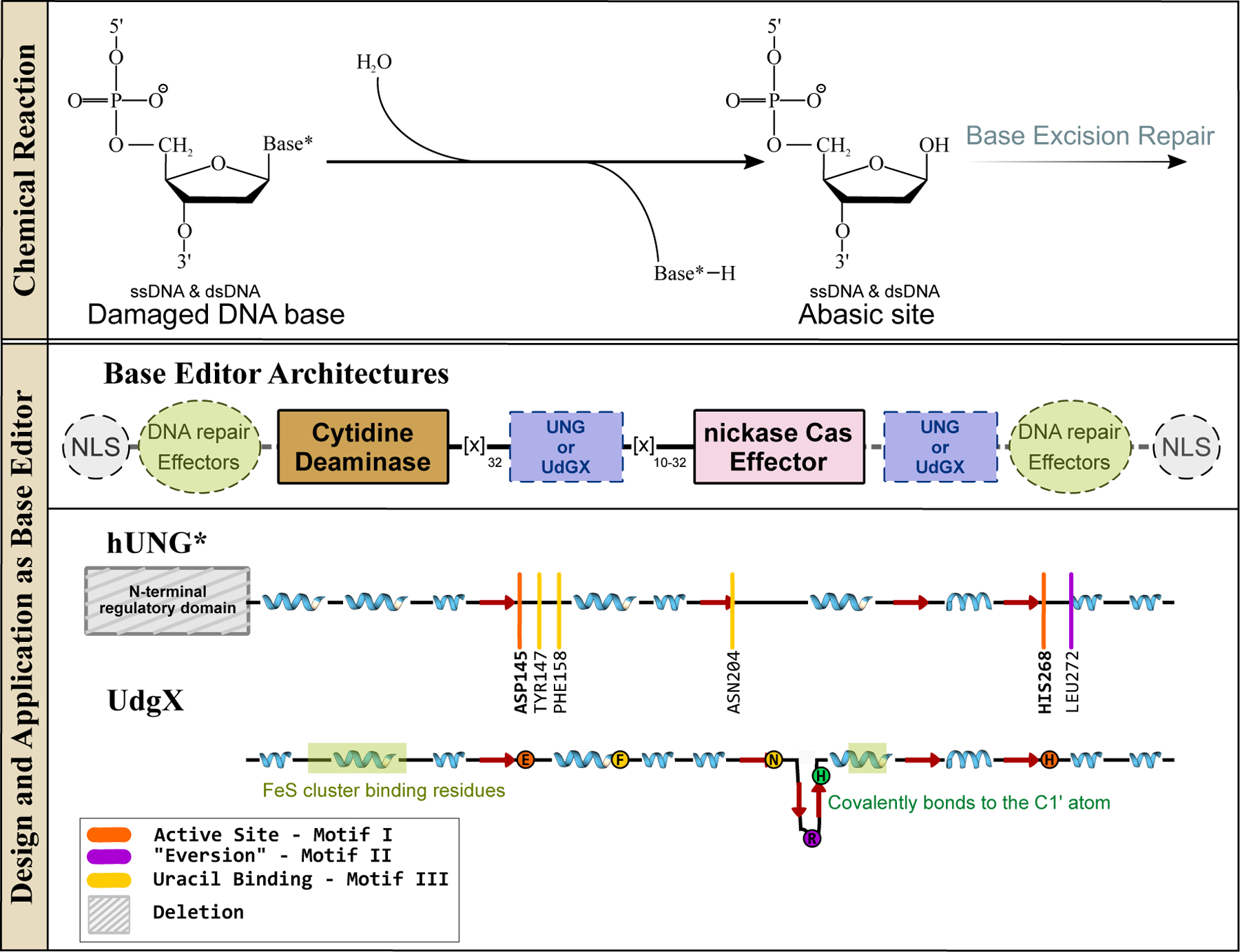
Glycosylases and their design and applications in genomic DNA base editing. (Top) Glycosylases initiate the BER pathway by excising modified nucleobases in dsDNA or ssDNA. (Bottom) Representative base editor architecture is shown, with essential and non-essential components indicated with solid and dashed outlines, respectively. The conserved β-sheet core of uracil glycosylases is shown, with an emphasis on the locations of the active site residues and critical DNA-binding sites, as well as other signature motifs.
Figure 10.
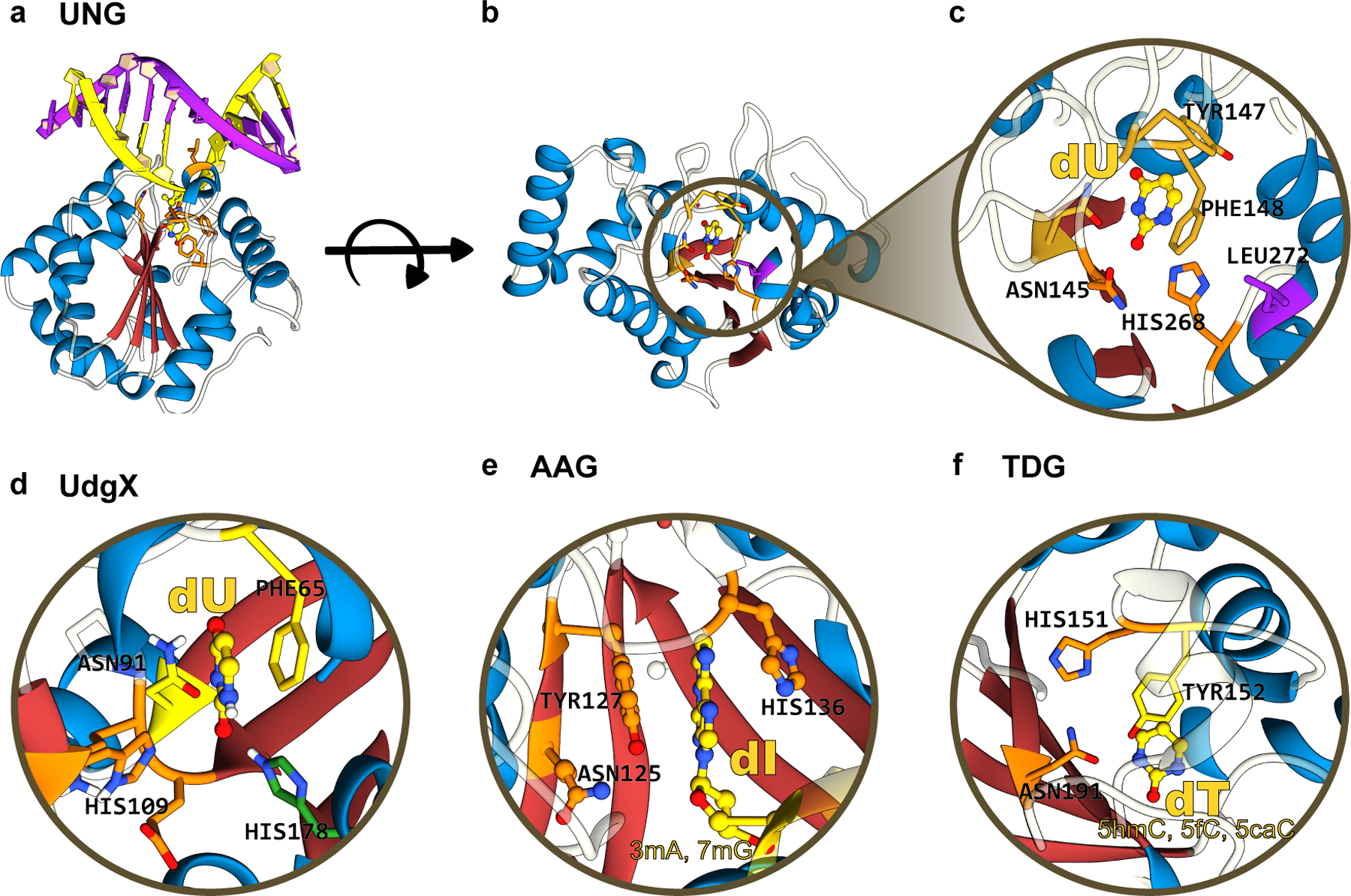
(a) Side-view, (b) top-view, and (c) zoomed-in structures of human UNG, which form the basis of current “transversion” base editors. The α/β fold of these glycosylases is color-coded as follows: the β-sheet core is in red and peripheral α-helices are in blue. (d), (e), and (f) correspond to the zoomed-in structure of UdgX, AAG, and TDG, respectively, highlighting the conserved β-stranded core. The aromatic residues(Phe/Tyr) responsible for stabilizing the “everted” nucleotide as well as the His and Asn residues critical for the excision reaction are annotated. UDG, UdgX, AAG, and TDG structures correspond to PDB ID: 1SSP(124), 6AIL(125), 1EWN(126), and 3UOB(177) respectively. (cf. Figure 9)
UNG enzymes, sometimes also referred to as Uracil DNA Glycosylase (UDG) enzymes, have a remarkably simple α/β-fold and carry out their excision reaction in a co-factor-independent manner. Various α-helices wrap around a deep but narrow active site groove supported by a highly conserved four-stranded parallel β-sheet architecture, into which fits a single nucleobase (Figure 10a–c). The core β-sheet consists of three conserved motifs: Motif I (situated on the β1 strand, which contains a highly conserved Asp residue responsible for activating the catalytic water molecule), Motif II (situated on the β4 strand, which harbors a highly conserved His residue responsible for stabilizing the target nucleobase, and a highly conserved Leu residue on the DNA-intercalation loop which fills the vacancy created in the minor groove of DNA by the “everted” target base), and Motif III (situated on the β2 strand, which contains an Asn residue that recognizes the target uracil base, and an aromatic residue, typically a Phe, which π-stacks against the target base) (Figure 9 and 10a–c). This basic architecture (a core β-sheet with three conserved motifs, surrounded by α-helices) is actually shared across many DNA glycosylases (Figure 10). While the sequences comprising each individual motif vary across different DNA glycosylases, the overall functionality of these motifs remains highly conserved. Moreover, the base “eversion” mechanism adopted by UNG enzymes is used by many other DNA editing and repair enzymes, which use it to probe the dsDNA and identify their target nucleobases (see Section 4).
With respect to CRISPR-based genome editors, the activity of DNA glycosylases was initially perceived as antagonistic to the intended base editing outcomes. Hence, the addition of UGI as an accessory domain to the CBE architecture was considered essential (Figure 2). Without this component, not only was the yield of C·G→U·G→T·A editing reduced but also varying levels of C·G→G·C and C·G→A·T editing was observed. UNG knock-out studies confirmed the role of UNG in this phenomenon(9).
While working against the BER pathway is necessary for precise C·G→T·A “transition mutation” introduction, working with BER to favor the creation of abasic sites by UNG has unlocked the possibility of targeted C·G→G·C (also known as “CGBEs”)(120–123) and C·G→A·T (also known as “CABEs”, which currently function only in bacteria)(122) “transversion” base editors. The overall architectures of these CGBEs and CABEs are derivatives of CBE architectures, as they all contain a cytidine deaminase DNA editing domain fused to a Cas9n DNA-targeting module (Figure 9). The cytidine deaminase introduces a programmable uracil DNA lesion as the critical first step of transversion base editing. The uracil intermediate is then excised by UNG to create an abasic site and initiate BER. These editors produce a mixture of outcomes, with the most common outcome being C·G→G·C. The cytidine deaminase variant used varies in each CGBE (the rAPOBEC1 R33A mutant in (120), hAID in (122), rAPOBEC1 in (121)). Additionally, a variety of methods to initiate BER were used, which included the direct fusion of UNG to Cas9n(120, 122, 123), the use of secondary DNA repair effectors (such as XRCC1 or EXO1) in the architecture(8, 121), the reliance on endogenous UNG to excise the uracil(120)), or the direct fusion of UNG homologs (E. coli UNG(120) and UdgX (Figure 10d), a UNG homolog with extremely tight binding to uracil(8)) to Cas9n. This once again demonstrates the modularity of the original base editor architecture. It should be noted that given the high reliance of CGBEs and CABEs on cellular BER pathways, their editing outcomes and efficiencies are highly sequence context-dependent, and are likely to be variable across different cell types.
Although only uracil glycosylases (UNG and UdgX) have been successfully utilized for programmable base editing, other enzymes from this superfamily (such as AAG, which was tested in the original ABE7.10 architecture, (Figure 10e)(67)) may also be explored for programmable transversion base editing. Additionally, incorporation of these glycosylases into mitochondrial base editor architectures (Figure 7) could in principle lead to mitochondrial transversion base editors. Overall, the development of CGBEs and CABEs not only expanded the base editor toolbox but also demonstrated the possibilities of combining the activities of multiple DNA editing enzymes to produce novel base editors.
4. METHYLASES AND DEMETHYLASES
Epigenetic regulation offers a secondary layer of control over the underlying genomic information by repressing or enhancing gene expression levels without changing the primary sequence of the genetic code. A variety of CRISPR-based targeted epigenetic modulators predate base editors, including gene repression using CRISPR interference (CRISPRi, which utilizes dCas9 fused to transcriptional repressor domains such as KRAB) and transcriptional activation using CRISPR activation (CRISPRa, which utilizes dCas9 fused to transcriptional activator domains such as VP16 and VP64). These methods facilitate expression level modulation via the secondary recruitment of repressive chromatin-modifying complexes (CRISPRi) or transcriptional machinery (CRISPRa)(128) rather than directly modifying the DNA and thus these will not be discussed here. Covalent modification of DNA nucleobases via methylation or demethylation facilitates stable and heritable “epigenetic editing” and is a form of direct DNA editing. One of the most extensively studied and well-understood forms of epigenetic editing is the deposition and removal of methyl groups at the 5-C position of cytosines (5mC) that occur in CpG islands of gene promoters (Figure 11). Methylation of C to 5mC induces transcriptional repression, while the 5mC→C demethylation reaction results in transcriptional activation. Given its impact on epigenetic regulation, it is not surprising that C↔5mC is implicated in numerous physiological processes in mammals including embryogenesis, X chromosome inactivation, cellular differentiation, genomic imprinting, chromatin structure modulation, aging, and even certain forms of cancer(129, 140). It should be noted that methylation, demethylation, phosphorylation, acetylation (and many other modifications) of histone tails also play a crucial role in epigenomic modulation, and often work synergistically with DNA nucleobase methylation and demethylation, but as we are focusing here exclusively on DNA editing enzymes, we will be restricting ourselves to nucleobase methylation and demethylation reactions and enzymes which catalyze them.
Figure 11.
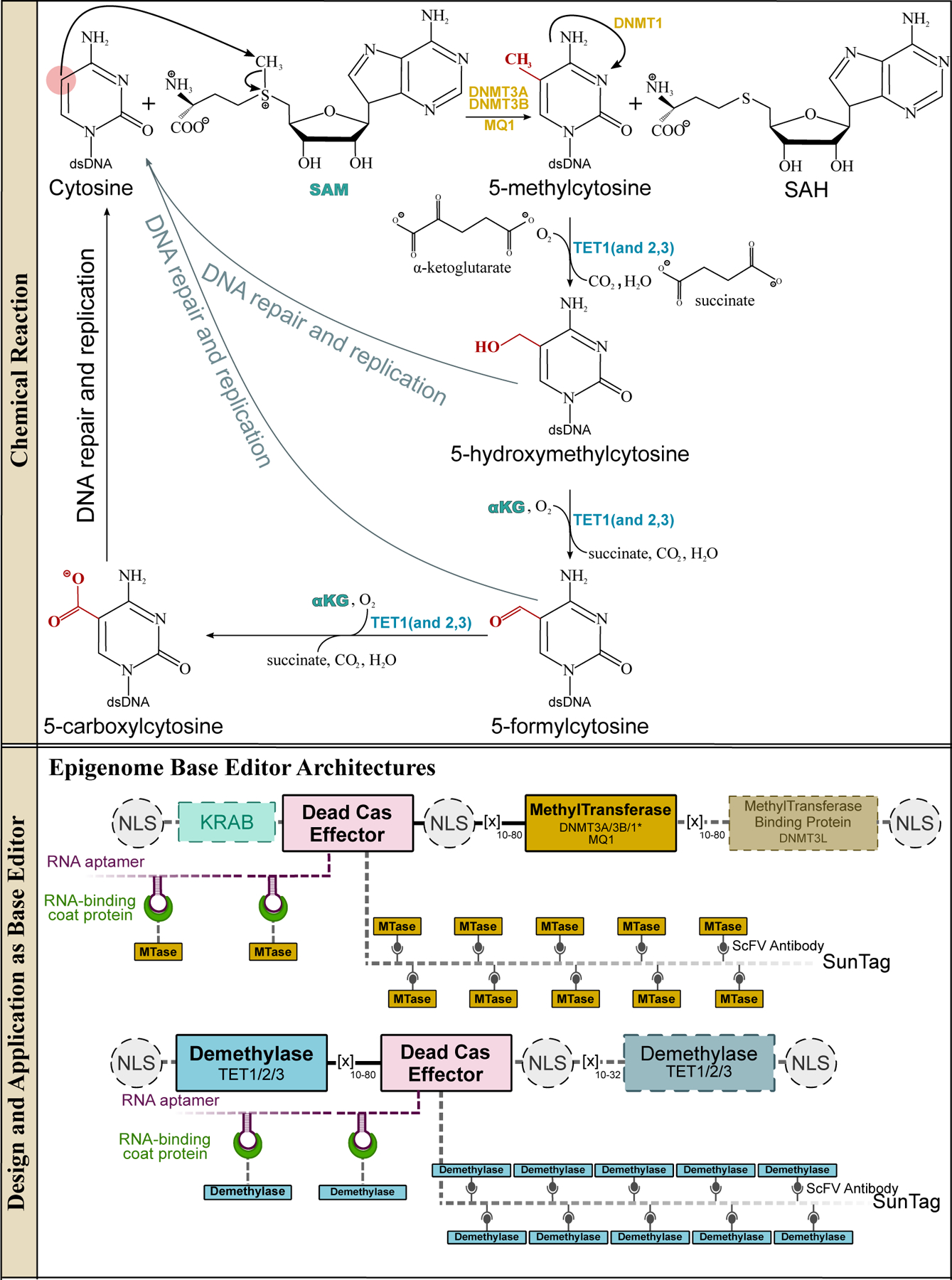
Methyltransferases and methylcytosine dioxygenases and their design and applications in genomic DNA base editing. The methyltransferase enzymes can methylate cytosines in 5′-CpG-3′ islands in dsDNA via a SAM-dependent mechanism to yield 5-methylcytosines (5mC), which act as gene expression suppressors. The demethylation of 5mC is triggered by the TET enzymes, which are iteratively oxygenated to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and finally 5-carboxycytosine (5caC). These oxygenated bases can all be recognized by TDG, which will excise the modified cytosine base. The BER pathway then installs a canonical cytosine base, resulting in demethylation of the 5mC. Overall, these reactions facilitate the full cyclic pathway of C·G→5mC·G and 5mC·G→C·G, which is critical for epigenetic regulation. (Bottom) Representative epigenome base editor architectures are shown, with essential and non-essential components indicated with solid and dashed outlines, respectively.
4.1. Methyltransferases
Methylation of cytidine nucleobases in DNA is catalyzed by the SAM (S-adenosyl L-methionine, also known as AdoMet)-dependent methyltransferase superfamily. These enzymes consist of a variable and flexible N-terminal domain that interacts with and recruits other epigenetic regulators, and a highly conserved SAM-dependent C-terminal catalytic domain that is supported by a core Rossman fold (alternating α/β folds, Figure 12). This C-terminal domain is comprised of seven key signature methyltransferase motifs (in sequential order): Motif I (a Gly-rich sequence on the β1-αA loop, which is responsible for SAM cofactor-binding), Motif IV (a PC[Q/N] motif on the β4-αD loop, which is responsible for initiating the nucleophilic attack on the C6 atom of the target dC), Motif VI (a ENV motif on the β4 strand, which is responsible for protonation of the N3 of the target dC), Motif VIII (a x3-A-x7−8-RxRxF motif on the β5-β6 loop, which is responsible for DNA binding), a variable target recognition domain (TRD), Motif IX (an Arg rich sequence, which is important for organizing the TRD), and Motif X (which is also responsible for SAM cofactor-binding) (Figure 12a)(131). The variability in the TRD domain and the C-terminal extensions diversify the substrate scope of this otherwise highly conserved methyltransferase catalytic core, and allow the enzymes from this superfamily to act on not just cytidines in dsDNA (C→5mC), but also adenosines in DNA (to facilitate A→[4mA/6mA], which occurs mostly in bacterial genomes), RNA, proteins, and even small biomolecules(132).
Figure 12.
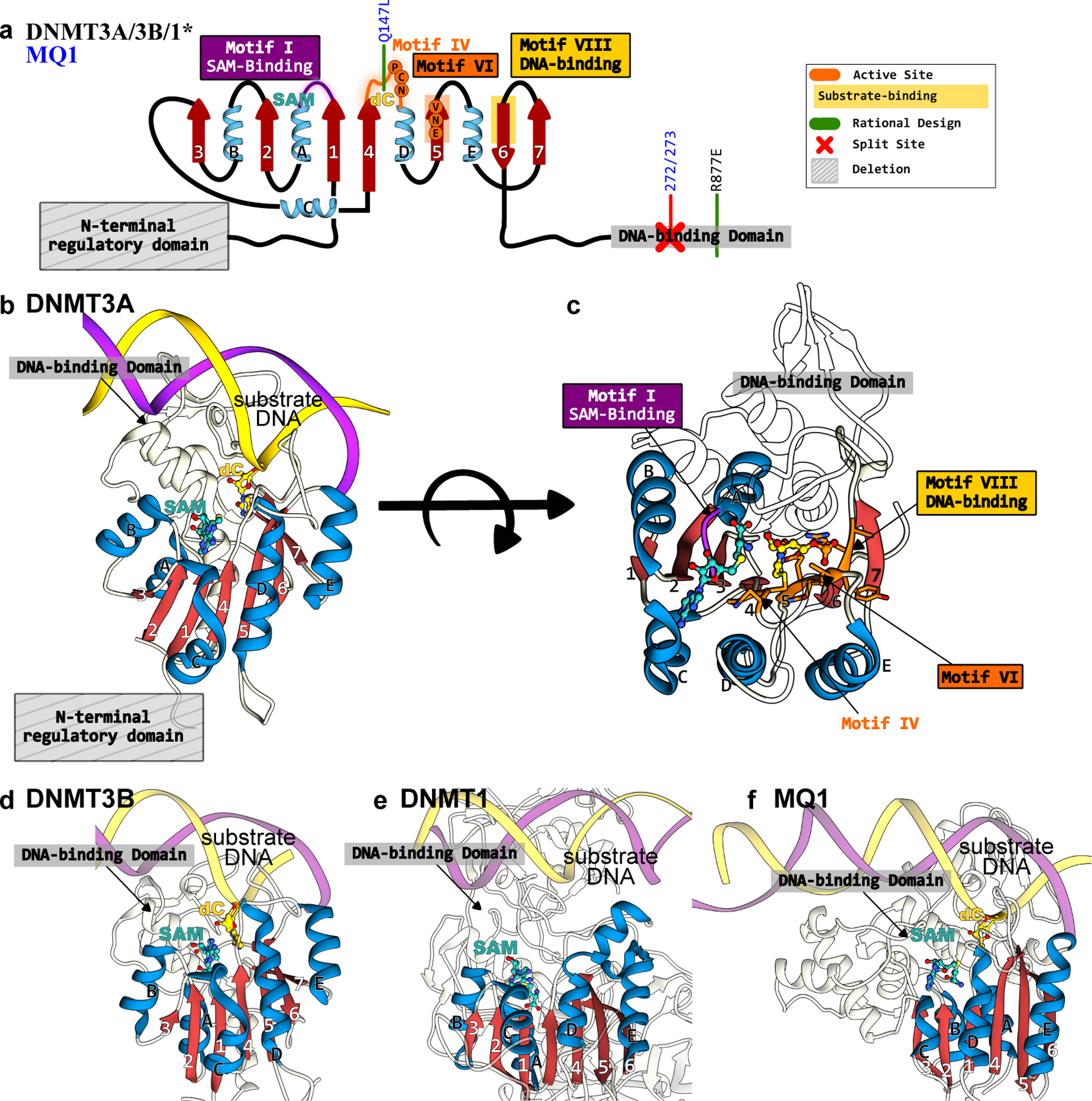
Structures of DNMTs and MQ1 methyltransferases that form the basis of epigenome base editors. (a) The conserved Rossman fold of the DNMT and MQ1 enzymes is color codes as follows: the β-sheet core is in red and peripheral α-helices are in blue. The active site residues, critical DNA- and cofactor-binding sites, and other signature motifs are annotated. Key mutations incorporated to increase activity or specificity are shown in green. (b) Side-view and (c) top-view of DNMT3A correspond to PDB ID: 6BRR(177). (d) Side-view of DNMT3B structure(6U8P(178)). (e) Side-view of DNMT1 structure (3PTA(179)). (f) Side-view of MQ1 structure, generated using Alphafold2(37) with the substrate dsDNA modeled in using its structural homolog(177).
Methylation of C to 5mC in mammalian genomes is catalyzed by a set of four DNA methyltransferase (DNMT) enzymes (Figure 12). These DNMTs all possess the conserved SAM-dependent methyltransferase fold (Figure 12a), but have different substrates and therefore serve slightly different physiological roles. De novo methylation of palindromic 5′-CpG-3′:3′-GpC-5′ sites is catalyzed by DNMT3A (Figure 12b, 12c) and DNMT3B (Figure 12d). The catalytic activities of these two enzymes are enhanced by spontaneous heterodimerization with their catalytically-impaired homolog, the DNMT3-like (DNMT3L) enzyme (which lacks critical residues within Motifs IV and VI) (Figure 12a). The resulting 5′-5mCpG-3′:3′-Gp5mC-5′ methylation pattern created by these two DNMT3 enzymes is maintained by the DNMT1 enzyme (Figure 12e), which preferentially methylates hemimethylated sites (5′-5mCpG-3′:3′-GpC-5′). DNMT1 is therefore considered a “maintenance methyltransferase”, and ensures that 5mC epigenetic modifications are propagated to daughter cells. While the mechanisms of CpG site recognition by DNMT enzymes are not precisely understood, their catalytic mechanisms have been well-characterized and are conserved across all three DNMT enzymes(133, 134). Once bound to the CpG island, the DNMT enzyme will “evert” the target cytosine base out from the dsDNA target. The “orphaned” guanine is stabilized by an Asn residue in the Motif IV loop. This is followed by nucleophilic attack of a conserved Cys residue in Motif IV on the target cytosine, resulting in the formation of a covalent protein-DNA intermediate. This intermediate is stabilized by residues in Motif VI, and subsequently abstracts the methyl group from SAM, eliminating the C5 proton and producing the 5mC product (Figure 11 and 12).
Because of their preference for dsDNA, targeted epigenetic base editing using DNMT enzymes predates CRISPR genome editing tools and was originally accomplished using TALEs and ZFs as the DNA-targeting module(135–142). These systems were superseded by CRISPR-based DNA-binding modules due to the enhanced simplicity and ease with which CRISPR systems are re-programmed compared to ZFs and TALEs. Specifically, the direct fusion of dCas9 to full-length DNMT3A(143), the C-terminal catalytic domain of DNMT3A(144–147), a combination of the catalytic domain of DNMT3A and its catalytically inactive binding partner DNMT3L(148–150), and full-length DNMT3B(151) have all been described (Figure 11). In addition, many studies have explored non-covalent fusion architectures such as the SunTag system (which uses a repurposed antibody-epitope system to recruit up to 24 copies of DNMT to each Cas9 protein)(152–154) and RNA aptamer systems (described previously)(155) (Figure 11).
Genome-wide off-target activity for the targeted dCas9-DNMT3A base editors has been reported to be equivalent to that of over-expressing DNMT3A. This is attributed to the ability of DNMT3A (even just the C-terminal catalytic domain) to bind to CpG sites in a Cas-independent manner and recruit the cell’s endogenous epigenetic machinery, resulting in ubiquitous methylation of the genomic DNA(147). To circumvent this issue, Hofacker et al.(156) introduced the R887E mutation into DNMT3A, which resides in the C-terminal DNA-binding extension (Figure 12a–c) and is involved in electrostatic interactions with the phosphate backbone of the strand opposite to the target C. This drastic charge substitution greatly reduces the non-specific DNA binding affinity and therefore improved the base editor’s specificity. Additionally, the human DNMTs can be replaced with their bacterial analogs to prevent the cell’s endogenous epigenetic machinery from being recruited to putative off-target sites. This route was adopted by Lei et al.(157) who replaced DNMT3A with MQ1, a well-characterized bacterial methyltransferase with the same conserved SAM-dependent C-terminal catalytic domain (Figure 11 and 12f). MQ1 is much smaller (approximately 40% smaller) in size compared to the human DNMTs and lacks their extensive recruitment domains. In this same study, the Q147L mutation in Motif IV of MQ1 (Figure 12a, 12f) was found to simultaneously increase editing efficiency and reduce off-target activity. Similarly, a “split MQ1” strategy was adopted by Xiong et al. (158) to reduce off-target editing (Figure 12a, 12f).
Epigenetic base editing using methyltransferases has also been seamlessly combined with other forms of transcriptional suppression. Specifically, O’Geen et al.(149) showed that co-expression of the dCas9-DNMT3a base editor with dCas9-histone modifying protein fusions (such as dCas9-Ezh2 or dCas9-KRAB, which cause methylation of histone lysines) that are targeted to the same genomic locus leads to prolonged gene repression (over at least 57 cell divisions). Synergistic gene repression by a combination of dCas9-targeted KRAB, DNMT3A, and DNMT3L has also been demonstrated (one of these systems was named “CRISPRoff”), and found to induce sustained and heritable gene silencing(145, 150).
4.2. Demethylases
Demethylation of 5mC back to the canonical C base, on the other hand, results in the upregulation of downstream genes. This essential epigenetic edit is carried out by the ten-eleven translocation (TET) enzymes belonging to the dioxygenases superfamily. These enzymes process 5mC into C via successive oxidation reactions (Figure 11). In each oxidation reaction, an oxygen atom from an O2 molecule is directly incorporated into the substrate, which is coupled with the decarboxylation of an α-ketoglutarate (α-KG) molecule to succinate, CO2, and H2O at an Fe(II) center in the active site. Overall, this process involves the iterative oxygenation reactions 5mC→5hmC→5fC→5caC (Figure 11). While each of these oxygenated intermediates is stable, they are less stable than the 5mC reactant, and are identified as DNA damage and thus acted upon by the cell’s DNA-repair machinery. In particular, thymine DNA glycosylase (TDG) (Figure 10f) can recognize any of the 5hmC/5fC/5caC:G intermediates and excise the modified cytidine. Therefore, overall, demethylation of 5mC by TET enzymes in fact involves iterative oxygenation of 5mC combined with excision by glycosylases. Intriguingly, the APOBEC/AID deaminases were initially speculated to be involved in this process by deaminating 5mC directly into T to generate T:G mismatches, the canonical substrate of TDG (159–161).
Of the three human TET enzymes (TET1, TET2, and TET3), the structures of full-length human TET1 and TET3 have not been resolved. However, their predicted structures(37), as well as the DNA-bound structure of human TET2(162), reveal that the C-terminal domain of these demethylases harbors the highly-conserved Fe(II)-containing active site. The Fe(II) ion is bound by a Hx[D/E]..H motif, which is supported by a doubled-stranded β-helix(DSβH) fold, also referred to as a “jelly-roll” fold (Figure 13a, 13b). This jelly-roll β-barrel core is a hallmark of the dioxygenases superfamily and is engulfed by various α-helices and loops that impart substrate diversity to these enzymes. An α-KG molecule acts as a bidentate ligand to the bound Fe(II), with the sixth coordination site of the Fe(II) center occupied by the O2 molecule(163) (Figure 13d). Furthermore, the catalytic core of all TET enzymes is preceded by a symmetric Cys-rich motif([Cx7C]x[Cx7C]) essential for DNA-binding (Figure 13a, 13c). Similar to the DNMT enzymes, upon DNA binding, the target nucleobase is everted out from the 5′-[5mC/5hmC/5fC]pG-3′ dsDNA into the active site of the TET enzymes.
Figure 13.
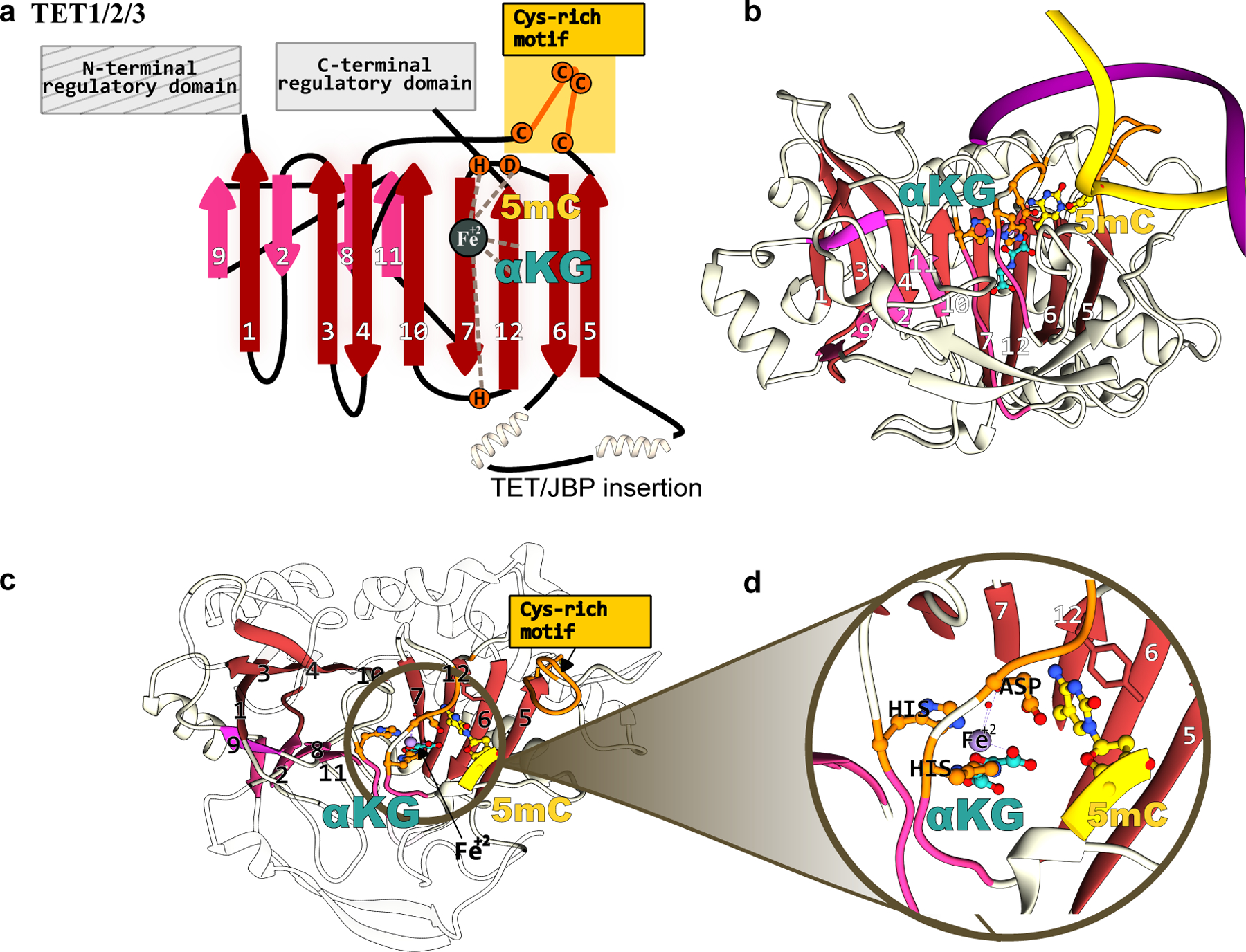
Structure of TET methylcytosine dioxygenases that form the basis of epigenome base editors. (a) The conserved jelly-roll fold of the TET1 enzyme consists of a β-sheet core, shown in red and pink. The active site residues, critical DNA- and cofactor-binding sites, and other signature motifs are annotated. (b) Side-view, (b) top-view, and (c) zoomed-in structural model of the TET1-DNA complex. The TET1 structure was predicted using Alphafold2(37) and the substrate dsDNA was modeled in using its structural homolog(180).
Just as with their methyltransferase counterparts, the ability of TET enzymes to edit dsDNA was initially exploited by fusing them to ZFs(164) and TALEs(165) to facilitate programmable DNA demethylation. However, with the advent of CRISPR-Cas9 for programmable DNA binding in mammalian cells, Both N-terminal(166) and C-terminal(143, 167–170) fusions of the catalytic domain of TET1 to dCas9 were quickly developed (Figure 11). Josipović et al. (171) demonstrated that N-terminal TET1-dCas9 fusions were generally more efficient than C-terminal fusions. They additionally demonstrated the modularity of epigenetic base editors, as they generated a TET1-dCas9 base editor with the S. aureus Cas9 homolog(171). Along the same lines, Xu et al. (172) substituted the TET1 catalytic domain with that of TET3 and demonstrated robust epigenome editing activity.
To increase the efficiency of 5mC demethylation by TET1-derived base editors, several groups have increased the number of TET1 enzymes recruited to each dCas9 protein. This was achieved via two non-covalent recruitment approaches that were both previously employed with DNMT base editors; the SunTag system(154, 173, 174) and RNA aptamer systems(175, 176) (Figure 11).
Lastly, again analogously to DNMT-derived base editors, TET-derived base editors have also been synergistically combined with other forms of transcriptional activation. Baumann et al. (170) directed both a dCas-TET1 base editor and a dCas9-VP64 fusion protein (which directly recruits transcriptional machinery) to the same genomic loci to achieve enhanced transcriptional activation of target genes compared to each individual construct. More recently, Nuñez et al. (150) combined an N-terminal TET1 catalytic domain-dCas9 fusion with simultaneous RNA aptamer-based recruitment of multiple transcriptional enhancers to create “CRISPRon”. “CRISPRon” is considered a synergistic combination of CRISPRa and TET1, and can reverse their “CRISPRoff” system (described previously), which itself is a synergistic combination of CRISPRi and DNMT enzymes.
While the on-target activities of the TET demethylation base editors have been explored extensively, their off-target activities have not been profiled thus far and remains an open question in the field of epigenetic base editing.
5. CONCLUSIONS AND FUTURE PERSPECTIVES
Base editors have become invaluable tools in the fields of biotechnology, basic science, and medicine. As they quickly move from the bench to the clinic, we have taken a step back here to discuss the fundamental biochemistry of the DNA editing enzymes that form the basis of these revolutionary technologies. Although each DNA editing enzyme discussed here followed a unique trajectory to become a programmable base editor, a few unifying themes have emerged. First, from an engineering and design perspective, base editors are quite modular in their composition. Under this design paradigm, the different modules are stitched together seamlessly to work in concert, and each component can be substituted for a similar effector. This has been demonstrated by varying the nature of the DNA editing module (deaminases, glycosylases, methyltransferases, and demethylases) and the DNA targeting module (different Cas effectors, TALEs, ZFs), or by adding “accessory” modules to the architecture to influence DNA repair outcomes (Figure 1b, 1c). Furthermore, the dual deaminase and glycosylase base editors demonstrated that two completely different DNA editing modules that facilitate different chemical reactions can be combined to achieve new editing outcomes. Second, the individual modules can be sourced and designed or redesigned independently to optimize overall functionality (Figure 1b). The motivations driving the engineering of the DNA editing modules of base editors are to enhance their on-target editing efficiency, reduce their off-target DNA and/or RNA editing activity, or modulate their substrate scope and specificity. While in this review we focused mostly on the DNA editing module, similar design motivations have also been extended to engineer the DNA targeting module. Third, regardless of the protein engineering approaches employed, the mutations that have been identified have largely resided on structural motifs that interact with the substrate (Figure 2 and4 and 7).
Overall, we have described here the many different extant DNA editing enzymes (cytidine deaminases, glycosylases, methyltransferases, and demethylases) that have been exploited and repurposed as base editors. What is particularly noteworthy is that even when an enzyme with the desired functionality did not exist, extensive protein engineering was conducted to either evolve the desired function from a suitable starting point (adenosine deaminase), or “stitch together” multiple existing DNA editing enzymes to produce the desired functionality (dual deaminases and glycosylase-containing base editors). Specifically, while inosine excision by AAG is not efficient enough to impact adenine base editing precision, we envision that directed evolution or engineering efforts on this enzyme could lead to the development of adenine transversion base editors. Further, there are a plethora of underutilized enzymes that post-transcriptionally modify RNA nucleobases which could be evolved into base editors that facilitate the introduction of additional types of point mutations(181). Leveraging the current structural and biochemical knowledge of these editing enzymes could the key to designing and developing them into novel base editors. Finally, we hope inspiration can be taken from the extensive design, engineering, and evolution efforts that have been performed on DNA editing enzymes and apply these principles to other systems, such as natural product enzyme assembly lines and post-translational modification enzymes.
Base Editors:
fusion proteins comprising a DNA-targeting module (such as Cas9) and DNA editing enzyme which can create targeted base edits in a programmable manner
APOBECs:
a family of C→U DNA and RNA editors that have a role in diverse biological functions.
AID:
a C→U DNA editing enzyme, which is essential for antibody affinity maturation and diversification.
dCas9:
dead/deactivated Cas9; a catalytically inactivate mutant of Cas9 that is not able to introduce DNA breaks
Cas9n:
Cas9 nickase; a catalytically impaired mutant of Cas9 that is able to introduce single-stranded DNA breaks
Editing Window:
The nucleotides within the exposed ssDNA portion of the R-loop that are accessible to the DNA editing module of deaminase-derived base editors.
Off-target Editing:
DNA editing by base editors at genomic loci other than the protospacer, or across the transcriptome.
Bystander Editing:
Undesirable DNA editing events that can occur at bases in close proximity to the target nucleotide within the ssDNA editing window.
Indels:
Insertion and/or deletion sequences introduced as a by-product of double-stranded break lesions due to Cas nucleases or as a by-product of base excision repair (BER) processing of base editor intermediates
Directed Evolution:
A protein engineering method that involves iterative cycles of mutagenesis and screening/selection under an artificially applied survival pressure to identify beneficial mutations that impart enhanced properties onto the enzyme
CpG islands:
5′-C-phosphate-G-3′ islands; C and G rich ( 55%) stretches of DNA comprised of various CpG dinucleotide motifs, typically 0.4–3 kb in length, that are located in promoter regions of 76% of human genes
KRAB:
The Krüppel associated box (KRAB) domain is a transcriptional repression domain that recruits a variety of protein partners via protein-protein interactions to mediate transcriptional control of downstream genes.
VP64:
VP64 is a transcriptional activator composed of four tandem copies of the Herpes Simplex Viral Protein 16 (VP16). VP64 functions by forming a complex with the transcription factor Oct-1 and the host cell factor (HCF) to induce transcription of downstream genes.
SunTag-antibodies:
An antibody-epitope system repurposed to recruit multiple copies of a protein of interest to a “carrier protein”.
RNA-aptamers:
RNA oligonucleotides that fold into specific 3-dimensional structures and bind with high affinity and specificity to a peptide target.
ZFNs and TALENs.
Zinc finger nucleases (ZFNs)(89–91) were among the first designer nucleases (proteins that have been engineered to recognize and cleave a predefined site in the genome). They are comprised of a zinc finger (ZF) DNA-binding domain fused to the non-specific DNA cleavage domain of the FokI nuclease(92). ZFs are comprised of repetitive ∼30 amino acid long DNA-binding modules. Each module contains a Cys2His2 motif that coordinates a zinc atom to form an overall ββα fold(93). Furthermore, each module recognizes a 3-bp DNA sequence via specific H-bonding interactions between four residues within the α-helix and the DNA. A typical ZF array is comprised of three or four modules fused in tandem that recognize a specific 9- or 12-bp DNA sequence. A “ZF recognition code” provides genome editors with instructions on which amino acids to incorporate at each of the four positions within the α-helix to recognize a predefined DNA sequence(94–97). ZFNs are designed in pairs, in which each ZFN recognizes a half-site, which are 5- to 7-bp away from each other. This allows the FokI nuclease to dimerize between the two half-sites, which is necessary for DSB introduction.
Transcription activator-like effector nucleases (TALENs) were the successor to ZFNs, which similarly consisted of a tandem array of transcription activator-like effectors (TALEs) fused to the FokI nuclease domain(98). TALEs are comprised of repetitive 33–34 amino acid long DNA binding modules. Each module recognizes a single DNA base via H-bonding between two TALE residues (called the repeat variable diresidue, or RVD) and the DNA. Again, a “TALE recognition code” provides genome editors with instructions of which RVD recognizes which DNA base(99, 100). TALENs are also designed in pairs to facilitate FokI dimerization, and each TALEN typically consists of ∼12 DNA binding modules.
Mechanism and significance of Cas effectors in base editors.
CRISPR-Cas systems are RNA-guided endonuclease complexes that impart innate immunity to bacteria and archaea against invading phages. Cas effector complexes recognize target nucleic acid sequences via simple base pairing rules between the Cas complex’s guide RNA (gRNA) and the target DNA or RNA (Figure 1c). The Cas protein first binds to a gRNA molecule, which is comprised of a “gRNA backbone” (the portion of the gRNA that the Cas protein recognizes and binds to) and a spacer sequence (typically 20–25 bases), which is complementary to the target genomic sequence (Figure 1b). The resulting ribonucleoprotein (RNP) complex then searches the genome for protospacer adjacent motifs (PAMs), which are short sequences (typically 2–7 bases) that the Cas protein recognizes and requires for DNA binding. Once the RNP complex finds a PAM that is directly adjacent to a match to the gRNA spacer sequence (the “protospacer”), it will unwind the DNA and base-pair the protospacer with the gRNA spacer (Figure 1c). Binding of the Cas:gRNA complex to the protospacer creates an R-loop, where the “non-target” DNA strand (the strand that is not base-paired with the gRNA) lacks a base-pairing partner. While most of this strand is enveloped within the Cas protein, a subset of this strand is exposed to the surrounding cellular environment. Following successful DNA binding, the endonuclease domains of the Cas enzyme will introduce a double-stranded DNA break (DSB) into the target locus. “Traditional” genome editing methods utilize wild-type Cas enzymes and perform genome editing using DSBs. Competing and stochastic cellular DSB repair pathways that process the DSB intermediate result in unwanted genome editing “byproducts”. However, “nontraditional” genome editing methods, such as base editing, utilize partially or fully catalytically inactivated Cas enzymes and rely on non-DSB DNA damage product as intermediates, thereby avoiding many of the issues faced by “traditional” Cas-based methods (Figure 1b, 1c).
Overall, in the context of base editing, Cas effectors have the following crucial functions:
Carrying the DNA editing enzyme to the target genomic locus and localizing the enzymatic activity of the DNA editing enzyme to a nucleobase of interest
Exposing a “bubble” of ssDNA substrate to the DNA editing enzyme via R-loop formation (this is important when the DNA editing enzyme’s substrate is ssDNA)
Nicking the backbone of the DNA strand opposite from the edited target nucleobase, to stimulate DNA repair
ACKNOWLEDGMENTS
A.C.K. and K.L.R. are partially funded by NIH grant 1R35GM138317.
Footnotes
DISCLOSURE STATEMENT
A.C.K. is a member of the SAB of Pairwise Plants, is an equity holder for Pairwise Plants and Beam Therapeutics, and receives royalties from Pairwise Plants, Beam Therapeutics, and Editas Medicine via patents licensed from Harvard University. A.C.K.’s interests have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. All other authors declare no competing financial interests. K.L.R. declares no competing interests.
LITERATURE CITED
- 1.Conticello S, Langlois M, Neuberger M. 2007. Insights into DNA deaminases. Nat. Struct. Mol. Biol 14:7–9 [DOI] [PubMed] [Google Scholar]
- 2.Conticello S, Thomas C, Petersen-Mahrt S, Neuberger M. 2005. Evolution of the AID/APOBEC family of polynucleotide (deoxy) cytidine deaminases. Mol. Biol. Evol 22:367–377 [DOI] [PubMed] [Google Scholar]
- 3.Di Noia J, Neuberger M. 2007. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem 76:1–22 [DOI] [PubMed] [Google Scholar]
- 4.Swanton C, McGranahan N, Starrett G, Harris R. 2015. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity APOBEC and Cancer Heterogeneity. Cancer Discov 5:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pecori R, Di Giorgio S, Paulo LJ, Papavasiliou FN. 2022. Functions and consequences of AID/APOBEC-mediated DNA and RNA deamination. Nat. Rev. Genet pp:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L, Briggs A, Chew W, Mali P, Guell M, et al. 2016. Engineering and optimising deaminase fusions for genome editing. Nat. Commun 7:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Komor A, Kim Y, Packer M, Zuris J, Liu D. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. 2016. Nature 533:420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koblan L, Doman J, Wilson C, Levy J, Tay T, et al. 2018. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol 36:843–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komor A, Zhao K, Packer M, Gaudelli N, Waterbury A, et al. 2017. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv 3:eaao4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zafra M, Schatoff E, Katti A, Foronda M, Breinig M, et al. 2018. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol 36:888–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y, Komor A, Levy J, Packer M, Zhao K, Liu D. 2017. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol 35:371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Wang Y, Liu Y, Yang B, Wang X, et al. 2018. Base editing with a Cpf1–cytidine deaminase fusion. Nat. Biotechnol 36:324–327 [DOI] [PubMed] [Google Scholar]
- 13.Kleinstiver B, Sousa A, Walton R, Tak Y, Hsu J, et al. 2019. Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol 37:276–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, et al. 2018. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361:1259–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller S, Wang T, Randolph P, Arbab M, Shen M, et al. 2020. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol 38:471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu J, Miller S, Geurts M, Tang W, Chen L, et al. 2018. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556:57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walton R, Christie K, Whittaker M, Kleinstiver B. 2020. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368:290–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J, Jeong E, Lee J, Jung M, Shin E, et al. 2018. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun 9:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gehrke J, Cervantes O, Clement M, Wu Y, Zeng J, et al. 2018. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol 36:977–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Y, Xu W, Wang F, Zhao S, Feng F, et al. 2019. Increasing cytosine base editing scope and efficiency with engineered Cas9-PmCDA1 fusions and the modified sgRNA in rice. Front. Genet 10:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan J, Zhang F, Karcher D, Bock R. 2020. Expanding the genome-targeting scope and the site selectivity of high-precision base editors. Nat. Commun 11:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong Z, Sretenovic S, Ren Q, Yang L, Bao Y, et al. 2019. Improving plant genome editing with high-fidelity xCas9 and non-canonical PAM-targeting Cas9-NG. Mol. Plant 12:1027–1036 [DOI] [PubMed] [Google Scholar]
- 23.Endo M, Mikami M, Endo A, Kaya H, Itoh T, et al. 2019. Genome editing in plants by engineered CRISPR–Cas9 recognizing NG PAM. Nat. Plants 5:14–17 [DOI] [PubMed] [Google Scholar]
- 24.Doman J, Raguram A, Newby G, Liu D. 2020. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol 38:620–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren B, Liu L, Li S, Kuang Y, Wang J, et al. 2019. Cas9-NG greatly expands the targeting scope of the genome-editing toolkit by recognizing NG and other atypical PAMs in rice. Mol. Plant 12:1015–1026 [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Shan H, Chen S, Chen M, Zhang Q, et al. 2019. Improved base editor for efficient editing in GC contexts in rabbits with an optimized AID-Cas9 fusion. FASEB J 33:9210–9219 [DOI] [PubMed] [Google Scholar]
- 27.Huang T, Zhao K, Miller S, Gaudelli N, Oakes B, et al. 2019. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol 37:626–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rees H, Komor A, Yeh W, Caetano-Lopes J, Warman M, et al. 2017. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun 8:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang P, Sun H, Sun Y, Zhang X, Xie X, et al. 2017. Effective gene editing by high-fidelity base editor 2 in mouse zygotes. Protein Cell 8:601–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu W, Song W, Yang Y, Wu Y, Lv X, Yuan S, et al. 2019. Multiplex nucleotide editing by high-fidelity Cas9 variants with improved efficiency in rice. BMC Plant Biol 19:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan J, Ma Y, Huang T, Chen Y, Peng Y, et al. 2019. Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol. Cell 72:380–394 [DOI] [PubMed] [Google Scholar]
- 32.Tóth E, Varga E, Kulcsár P, Kocsis-Jutka V, Krausz S, et al. 2020. Improved LbCas12a variants with altered PAM specificities further broaden the genome targeting range of Cas12a nucleases. Nucleic Acids Res 48:3722–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang T, Newby G, Liu D. 2021. Precision genome editing using cytosine and adenine base editors in mammalian cells. Nat. Protoc 16:1089–1128 [DOI] [PubMed] [Google Scholar]
- 34.Rees H, Liu D. 2018. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet 19:770–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porto E, Komor A, Slaymaker I, Yeo G. 2020. Base editing: advances and therapeutic opportunities. Nat. Rev. Drug Discov 19:839–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolfe A, Li S, Goedderz C, Chen X. 2020. The structure of APOBEC1 and insights into its RNA and DNA substrate selectivity. NAR Cancer 2:zcaa027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saraconi G, Severi F, Sala C, Mattiuz G, Conticello S. 2014. The RNA editing enzyme APOBEC1 induces somatic mutations and a compatible mutational signature is present in esophageal adenocarcinomas. Genome Biol 15:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris R, Petersen-Mahrt S, Neuberger M. 2002. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Molecular Cell 10:1247–1253 [DOI] [PubMed] [Google Scholar]
- 40.Grünewald J, Zhou R, Garcia S, Iyer S, Lareau C, Aryee M, Joung J. 2019. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569: 433–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou C, Sun Y, Yan R, Liu Y, Zu E, et al. 2019. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 571:275–278 [DOI] [PubMed] [Google Scholar]
- 42.Salter J, Bennett R, Smith H. 2016. The APOBEC protein family: united by structure, divergent in function. Trends Biochem. Sci 41:578–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenberg B, Hamilton C, Mwangi M, Dewell S, Papavasiliou FN. 2011. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA editing targets in transcript 3′ UTRs. Nat. Struct. Mol. Biol 18:230–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conticello S 2008. The AID/APOBEC family of nucleic acid mutators. Genome Biol 9:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen K, Harjes E, Gross P, Fahmy A, Lu Y, et al. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 452:116–119 [DOI] [PubMed] [Google Scholar]
- 46.Thuronyi B, Koblan L, Levy J, Yeh W, Zheng C, et al. 2019. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol 37:1070–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gumulya Y, Gillam E. 2017. Exploring the past and the future of protein evolution with ancestral sequence reconstruction: the ‘retro’approach to protein engineering. Biochem. J 474:1–19 [DOI] [PubMed] [Google Scholar]
- 48.Park S, Beal P. 2019. Off-target editing by CRISPR-guided DNA base editors. Biochemistry 58:3727–3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zuo E, Sun Y, Yuan T, He B, Zhou C, et al. 2019. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nat. Methods 17:600–604 [DOI] [PubMed] [Google Scholar]
- 50.Yu Y, Leete T, Born D, Young L, Barrera L, et al. 2020. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun 11:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Li J, Wang Y, Yang B, Wei J, et al. 2018. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat. Biotechnol 36:946–949 [DOI] [PubMed] [Google Scholar]
- 52.Coelho M, Li S, Pane L, Firth M, Ciotta G, et al. 2018. BE-FLARE: a fluorescent reporter of base editing activity reveals editing characteristics of APOBEC3A and APOBEC3B. BMC Biol 16:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin A St., Salamango D, Serebrenik A, Shaban N, Brown W, et al. 2018. A fluorescent reporter for quantification and enrichment of DNA editing by APOBEC–Cas9 or cleavage by Cas9 in living cells. Nucleic Acids Res 46:e84–e84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin A, Salamango D, Serebrenik A, Shaban N, Brown W, Harris R. 2019. A panel of eGFP reporters for single base editing by APOBEC-Cas9 editosome complexes. Sci. Rep 9:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee S, Ding N, Sun Y, Yuan T, Li J, et al. 2020. Single C-to-T substitution using engineered APOBEC3G-nCas9 base editors with minimum genome-and transcriptome-wide off-target effects. Sci. Adv 6:eaba1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma Y, Zhang J, Yin W, Zhang Z, Song Y, Chang X. 2016. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods 13:1029–1035 [DOI] [PubMed] [Google Scholar]
- 57.Ito F, Fu Y, Kao S, Yang H, Chen X. 2017. Family-wide comparative analysis of cytidine and methylcytidine deamination by eleven human APOBEC proteins. J. Mol. Biol 429:1787–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hou S, Lee J, Myint W, Matsuo H, Yilmaz N, Schiffer C. 2021. Structural basis of substrate specificity in human cytidine deaminase family APOBEC3s. J. Biol. Chem 297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maiti A, Hou S, Schiffer C, Matsuo H. 2021. Interactions of APOBEC3s with DNA and RNA. Curr. Opin. Struct. Biol 67:195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kouno T, Silvas T, Hilbert B, Shandilya S, Bohn M, et al. 2017. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun 8:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Z, Chen S, Shan H, Jia Y, Chen M, Song Y, et al. 2020. Precise base editing with CC context-specificity using engineered human APOBEC3G-nCas9 fusions. BMC Biology 18:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, et al. 2016. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353:aaf8729 [DOI] [PubMed] [Google Scholar]
- 63.Hess G, Frésard L, Han K, Lee C, Li A, et al. 2016. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13:1036–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang M, Yang Z, Rada C, Neuberger M. 2009. AID upmutants isolated using a high-throughput screen highlight the immunity/cancer balance limiting DNA deaminase activity 16:769–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qiao Q, Wang L, Meng F, Hwang J, Alt F, Wu H. 2017. AID recognizes structured DNA for class switch recombination. Mol. Cell 67:361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schaub M, Keller W. 2002. RNA editing by adenosine deaminases generates RNA and protein diversity. Biochimie 84:791–803 [DOI] [PubMed] [Google Scholar]
- 67.Gaudelli N, Komor A, Rees H, Packer M, Badran A, Bryson D, Liu D. 2017. Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature 551:464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim J, Malashkevich V, Roday S, Lisbin M, Schramm V, Almo S. 2006. Structural and kinetic characterization of Escherichia coli TadA, the wobble-specific tRNA deaminase. Biochemistry 45:6407–6416 [DOI] [PubMed] [Google Scholar]
- 69.Alseth I, Dalhus B, BjørM. 2014. Inosine in DNA and RNA. Current Opinion In Genetics, Development 26:116–123 [DOI] [PubMed] [Google Scholar]
- 70.Grünewald J, Zhou R, Iyer S, Lareau C, Garcia S, Aryee M, Joung J. 2019. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol 37:1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rallapalli K, Komor A, Paesani F. 2020. Computer simulations explain mutation-induced effects on the DNA editing by adenine base editors. Sci. Adv 6:eaaz2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Richter M, Zhao K, Eton E, Lapinaite A, Newby G, et al. 2020. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol 38:883–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gaudelli N, Lam D, Rees H, Solá-Esteves N, Barrera L, et al. 2020. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol 38:892–900 [DOI] [PubMed] [Google Scholar]
- 74.Rallapalli K, Ranzau B, Ganapathy K, Paesani F, Komor A. 2022. Combined Theoretical, Bioinformatic, and Biochemical Analyses of RNA Editing by Adenine Base Editors. CRISPR J 5:294–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lapinaite A, Knott G, Palumbo C, Lin-Shiao E, Richter M, et al. 2020. DNA capture by a CRISPR-Cas9–guided adenine base editor. Science 369:566–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim H, Jeong Y, Hur J, Kim J, Bae S. 2019. Adenine base editors catalyze cytosine conversions in human cells. Nat. Biotechnol 37:1145–1148 [DOI] [PubMed] [Google Scholar]
- 77.Chen L, Zhang S, Xue N, Hong M, Zhang X, et al. 2022. Engineering a precise adenine base editor with minimal bystander editing. Nat. Chem. Biol. [DOI] [PubMed] [Google Scholar]
- 78.Neugebauer M, Hsu A, Arbab M, Krasnow N, McElroy A, et al. 2022. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nat. Biotechnol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen L, Zhu B, Ru G, Meng H, Yan Y, et al. 2022. Re-engineering the adenine deaminase TadA-8e for efficient and specific CRISPR-based cytosine base editing. Nat. Biotechnol [DOI] [PubMed] [Google Scholar]
- 80.Gaudelli N, Yu Y, Slaymaker I, Gehrke J, Lee S, et al. 2022. Nucleobase editors having reduced off-target deamination and methods of using same to modify a nucleobase target sequence. US Patent App 17/427,422 [Google Scholar]
- 81.Rees H, Wilson C, Doman J, Liu D. 2019. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv 5:eaax5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Losey H, Ruthenburg A, Verdine G. 2006. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol 13:153–159 [DOI] [PubMed] [Google Scholar]
- 83.Grünewald J, Zhou R, Lareau C, Garcia S, Iyer S, et al. 2020. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol 38:861–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sakata R, Ishiguro S, Mori H, Tanaka M, Tatsuno K, et al. 2020. Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat. Biotechnol 38:865–869 [DOI] [PubMed] [Google Scholar]
- 85.Li C, Zhang R, Meng X, Chen S, Zong Y, et al. 2020. Targeted, random mutagenesis of plant genes with dual cytosine and adenine base editors. Nat. Biotechnol 38:875–882 [DOI] [PubMed] [Google Scholar]
- 86.Zhang X, Zhu B, Chen L, Xie L, Yu W, et al. 2020. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat. Biotechnol 38:856–860 [DOI] [PubMed] [Google Scholar]
- 87.Xie J, Huang X, Wang X, Gou S, Liang Y, et al. 2020. ACBE, a new base editor for simultaneous C-to-T and A-to-G substitutions in mammalian systems. BMC Biol 18:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cravens A, Jamil O, Kong D, Sockolosky J, Smolke C. 2021. Polymerase-guided base editing enables in vivo mutagenesis and rapid protein engineering. Nat. Commun 12:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim Y, Smith J, Durgesha M, Chandrasegaran S. 1998. Chimeric restriction enzyme: Gal4 fusion to Fokl cleavage domain [DOI] [PubMed] [Google Scholar]
- 90.Kim Y, Chandrasegaran S. 1994. Chimeric restriction endonuclease. Proc. Natl. Acad. Sci. U.S.A 91:883–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim Y, Cha J, Chandrasegaran S. 1996. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. U.S.A 93:1156–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li L, Wu L, Chandrasegaran S. 1992. Functional domains in Fok I restriction endonuclease. Proc. Natl. Acad. Sci. U.S.A 89:4275–4279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pavletich N, Pabo C. 1991. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 Å. Science 252:809–817 [DOI] [PubMed] [Google Scholar]
- 94.Isalan M, Klug A, Choo Y 1998. Comprehensive DNA recognition through concerted interactions from adjacent zinc fingers. Biochemistry 37:12026–12033 [DOI] [PubMed] [Google Scholar]
- 95.Wolfe S, Greisman H, Ramm E, Pabo C. 1999. Analysis of zinc fingers optimized via phage display: evaluating the utility of a recognition code. Journal Of Molecular Biology 285:1917–1934 [DOI] [PubMed] [Google Scholar]
- 96.Pabo C, Peisach E, Grant R. 2001. Design and selection of novel Cys2His2 zinc finger proteins. Annual Review Of Biochemistry 70:313–340 [DOI] [PubMed] [Google Scholar]
- 97.Sera T & Uranga C Rational design of artificial zinc-finger proteins using a nondegenerate recognition code table. Biochemistry 41, 7074–7081 (2002) [DOI] [PubMed] [Google Scholar]
- 98.Christian M, Cermak T, Doyle E, Schmidt C, Zhang F, et al. 2010. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186:757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moscou M, Bogdanove A. 2009. A simple cipher governs DNA recognition by TAL effectors. Science 326:1501–1501 [DOI] [PubMed] [Google Scholar]
- 100.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, et al. 2009. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512 [DOI] [PubMed] [Google Scholar]
- 101.Gammage P, Moraes C, Minczuk M. 2018. Mitochondrial genome engineering: the revolution may not be CRISPR-Ized. Trends Gene 34:101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mok B, Moraes M, Zeng J, Bosch D, Kotrys A, et al. 2020. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583:631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iyer L, Zhang D, Rogozin I, Aravind L. 2011. Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucleic Acids Res 39:9473–9497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moraes M, Hsu F, Huang D, Bosch D, Zeng J, et al. 2021. An interbacterial DNA deaminase toxin directly mutagenizes surviving target populations. Elife 10:e62967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Berríos K, Evitt N, DeWeerd R, Ren D, Luo M, et al. 2021. Controllable genome editing with split-engineered base editors. Nat. Chem. Biol 17:1262–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Long J, Liu N, Tang W, Xie L, Qin F, et al. 2021. A split cytosine deaminase architecture enables robust inducible base editing. FASEB J 35:e22045. [DOI] [PubMed] [Google Scholar]
- 107.Levy J, Yeh W, Pendse N, Davis J, Hennessey E, et al. 2020. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng 4:97–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ryu S, Koo T, Kim K, Lim K, Baek G, et al. 2018. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol 36:536–539 [DOI] [PubMed] [Google Scholar]
- 109.Katrekar D, Xiang Y, Palmer N, Saha A, Meluzzi D, Mali P. 2022. Comprehensive interrogation of the ADAR2 deaminase domain for engineering enhanced RNA editing activity and specificity. Elife 11:e75555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mok B, Kotrys A, Raguram A, Huang T, Mootha V, Liu D. 2022. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat. Biotechnol 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cho S, Lee S, Mok Y, Lim K, Lee J, et al. 2022. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell 185:1764–1776 [DOI] [PubMed] [Google Scholar]
- 112.Lim K, Cho S, Kim JS. 2022. Nuclear and mitochondrial DNA editing in human cells with zinc finger deaminases. Nat. Commun 13:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kang B, Bae S, Lee S, Lee J, Kim A, et al. 2021. Chloroplast and mitochondrial DNA editing in plants. Nat. Plants 7:899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barrera-Paez J, Moraes C. 2022. Mitochondrial genome engineering coming-of-age. Trends Gene [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Adashi E, Rubenstein D, Mossman J, Schon E, Cohen I. 2021. Mitochondrial disease: Replace or edit?. Science 373:1200–1201 [DOI] [PubMed] [Google Scholar]
- 116.Lindahl T 1979. DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog. Nucleic Acid Res. Mol. Bio 22:135–192 [DOI] [PubMed] [Google Scholar]
- 117.Krokan H, Bjoras M. 2013. Base excision repair. Cold Spring Harb. Perspect. Biol. 5:a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gu S, Bodai Z, Cowan Q, Komor A. 2021. Base editors: Expanding the types of DNA damage products harnessed for genome editing. Gene Genome Ed 1:100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nambiar T, Baudrier L, Billon P, Ciccia A. 2022. CRISPR-based genome editing through the lens of DNA repair. Mol. Cell 82:348–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kurt I, Zhou R, Iyer S, Garcia S, Miller B, et al. 2021. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol 39:41–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen L, Park J, Paa P, Rajakumar P, Prekop H, et al. 2021. Programmable C: G to G: C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat. Commun 12:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhao D, Li J, Li S, Xin X, Hu M, et al. 2021. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol 39:35–40 [DOI] [PubMed] [Google Scholar]
- 123.Koblan L, Arbab M, Shen M, Hussmann J, Anzalone A, et al. 2021. Efficient C• G-to-G• C base editors developed using CRISPRi screens, target-library analysis, and machine learning. Nat. Biotechnol 39:1414–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Parikh S, Mol C, Slupphaug G, Bharati S, Krokan H, Tainer J 1998. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J 17:5214–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ahn W, Aroli S, Kim J, Moon J, Lee G, et al. 2019. Covalent binding of uracil DNA glycosylase UdgX to abasic DNA upon uracil excision. Nat. Chem. Biol 15:607–614 [DOI] [PubMed] [Google Scholar]
- 126.Lau A, Wyatt M, Glassner B, Samson L, Ellenberger T 2000. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc. Natl. Acad. Sci. U.S.A 97:13573–13578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang L, Lu X, Lu J, Liang H, Dai Q, et al. 2012. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol 8:328–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hsu P, Lander E, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Moore L, Le T, Fan G. 2013. DNA methylation and its basic function. Neuropsychopharmacology 38:23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Holtzman L, Gersbach C. 2018. Editing the epigenome: reshaping the genomic landscape. Annu. Rev. Genomics Hum. Genet 19:43–71 [DOI] [PubMed] [Google Scholar]
- 131.Pósfai J, Bhagwat A, Pósfai G, Roberts R. 1989. Predictive motifs derived from cytosine methyltransferases. Nucleic Acids Res 17:2421–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Martin J, McMillan F. 2002. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol 12:783–793 [DOI] [PubMed] [Google Scholar]
- 133.Bestor T, Verdine G. 1994. DNA methyltransferases. Curr. Opin. Cell Biol 6:380–389 [DOI] [PubMed] [Google Scholar]
- 134.Cheng X 1995. Structure and function of DNA methyltransferases.. Annu. Rev. Biophys. Biomol. Struct 24:293–318 [DOI] [PubMed] [Google Scholar]
- 135.Carvin C, Parr R, Kladde M. 2003. Site-selective in vivo targeting of cytosine-5 DNA methylation by zinc-finger proteins. Nucleic Acids Res 31:6493–6501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mlambo T, Nitsch S, Hildenbeutel M, Romito M, Müller M, et al. 2018. Designer epigenome modifiers enable robust and sustained gene silencing in clinically relevant human cells. Nucleic Acids Res. 46:4456–4468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li F, Papworth M, Minczuk M, Rohde C, Zhang Y, et al. 2007. Chimeric DNA methyltransferases target DNA methylation to specific DNA sequences and repress expression of target genes. Nucleic Acids Res. 35:100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rivenbark A, Stolzenburg S, Beltran A, Yuan X, Rots M, et al. 2012. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 7:350–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kungulovski G, Jeltsch A. 2016. Epigenome editing: state of the art, concepts, and perspectives. Trends In Genetics 32:101–113 [DOI] [PubMed] [Google Scholar]
- 140.Holtzman L, Gersbach C. 2018. Editing the epigenome: reshaping the genomic landscape. Annu. Rev. Genomics Hum. Genet. 19:43–71 [DOI] [PubMed] [Google Scholar]
- 141.Groote M, Verschure P, Rots M. 2012. Epigenetic editing: targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic Acids Res 40:10596–10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thakore P, Black J, Hilton I, Gersbach C. 2016. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat. Methods 13:127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liu X, Wu H, Ji X, Stelzer Y, Wu X, et al. 2016. Editing DNA methylation in the mammalian genome. Cell 167:233–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vojta A, Dobrinić P, Tadić V, Bočkor L, Korać P, et al. 2016. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res 44:5615–5628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Amabile A, Migliara A, Capasso P, Biffi M, Cittaro D, Naldini L, Lombardo A. 2016. Inheritable silencing of endogenous genes by hit-and-run targeted epigenetic editing. Cell 167:219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McDonald J, Celik H, Rois L, Fishberger G, Fowler T, et al. 2016. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol. Open 5:866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Galonska C, Charlton J, Mattei A, Donaghey J, Clement K, et al. 2018. Genome-wide tracking of dCas9-methyltransferase footprints. Nat. Commun 9:1–9 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stepper P, Kungulovski G, Jurkowska R, Chandra T, Krueger F, et al. 2017. Efficient targeted DNA methylation with chimeric dCas9–Dnmt3a–Dnmt3L methyltransferase. Nucleic Acids Res 45:1703–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.
- 150.Nuñez J, Chen J, Pommier G, Cogan J, Replogle J, et al. 2021. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 184:2503–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lin L, Liu Y, Xu F, Huang J, Daugaard T, et al. 2018. Genome-wide determination of on-target and off-target characteristics for RNA-guided DNA methylation by dCas9 methyltransferases. Gigascience 7:giy011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pflueger C, Tan D, Swain T, Nguyen T, Pflueger J, et al. 2018. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res 28:1193–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Huang Y, Su J, Lei Y, Brunetti L, Gundry M, et al. 2017. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol 18:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Marx N, Grünwald-Gruber C, Bydlinski N, Dhiman H, Ngoc Nguyen L, et al. 2018. CRISPR-based targeted epigenetic editing enables gene expression modulation of the silenced beta-galactoside alpha-2, 6-sialyltransferase 1 in CHO cells. Biotechnol. J 13:1700217. [DOI] [PubMed] [Google Scholar]
- 155.Lu A, Wang J, Sun W, Huang W, Cai Z, et al. 2019. Reprogrammable CRISPR/dCas9-based recruitment of DNMT1 for site-specific DNA demethylation and gene regulation. Cell Discov 5:1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hofacker D, Broche J, Laistner L, Adam S, Bashtrykov P, Jeltsch A. 2020. Engineering of effector domains for targeted DNA methylation with reduced off-target effects. Int. J. Mol. Sci 21:502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lei Y, Zhang X, Su J, Jeong M, Gundry M, et al. 2017. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat. Commun 8:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Xiong T, Meister G, Workman R, Kato N, Spellberg M, et al. 2017. Targeted DNA methylation in human cells using engineered dCas9-methyltransferases. Sci. Rep. 7:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kohli R, Zhang Y. 2013. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502: 472–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wu H, Zhang Y.2014. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156:45–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ranzau B, Komor A. 2018. Genome, epigenome, and transcriptome editing via chemical modification of nucleobases in living cells. Biochemistry 58:330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hu L, Lu J, Cheng J, Rao Q, Li Z, et al. 2015. Structural insight into substrate preference for TET-mediated oxidation. Nature 527:118–122 [DOI] [PubMed] [Google Scholar]
- 163.Aik W, McDonough M, Thalhammer A, Chowdhury R, Schofield C. 2012. Role of the jelly-roll fold in substrate binding by 2-oxoglutarate oxygenases. Curr. Opin. Struct. Biol 22: 691–700 [DOI] [PubMed] [Google Scholar]
- 164.Chen H, Kazemier H, Groote M, Ruiters M, Xu G, Rots M. 2014. Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res 42:1563–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Maeder M, Angstman J, Richardson M, Linder S, Cascio V, et al. 2013. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 31:1137–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Choudhury S, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. 2016. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 7:46545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Okada M, Kanamori M, Someya K, Nakatsukasa H, Yoshimura A. 2017. Stabilization of Foxp3 expression by CRISPR-dCas9-based epigenome editing in mouse primary T cells. Epigenetics Chromatin 10:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liu X, Wu H, Krzisch M, Wu X, Graef J, et al. 2018. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172:979–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kang J, Park J, Ko J, Kim Y. 2019. Regulation of gene expression by altered promoter methylation using a CRISPR/Cas9-mediated epigenetic editing system. Sci. Rep. 9:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Baumann V, Wiesbeck M, Breunig C, Braun J, Köferle A, et al. 2019.Targeted removal of epigenetic barriers during transcriptional reprogramming. Nat. Commun 10:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Josipović G, Tadić V, Klasić M, Zanki V, Beuceheli I, et al. 2019. Antagonistic and synergistic epigenetic modulation using orthologous CRISPR/dCas9-based modular system. Nucleic Acids Res. 47:9637–9657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xu X, Tan X, Tampe B, Wilhelmi T, Hulshoff M, et al. 2018. High-fidelity CRISPR/Cas9-based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun 9:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hanzawa N, Hashimoto K, Yuan X, Kawahori K, Tsujimoto K, et al. 2020. Targeted DNA demethylation of the Fgf21 promoter by CRISPR/dCas9-mediated epigenome editing. Sci. Rep 10:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Morita S, Noguchi H, Horii T, Nakabayashi K, Kimura M, Okamura K, et al. 2016. Targeted DNA demethylation in vivo using dCas9–peptide repeat and scFv–TET1 catalytic domain fusions. Nat. Biotechnol 34:1060–1065 [DOI] [PubMed] [Google Scholar]
- 175.Xu X, Tao Y, Gao X, Zhang L, Li X, et al. 2016. A CRISPR-based approach for targeted DNA demethylation. Cell Discov 2:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Taghbalout A, Du M, Jillette N, Rosikiewicz W, Rath A, et al. 2019. Enhanced CRISPR-based DNA demethylation by Casilio-ME-mediated RNA-guided coupling of methylcytosine oxidation and DNA repair pathways. Nat. Commun 10:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang Z, Lu R, Wang P, Yu Y, Chen D, et al. 2018. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 554:387–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gao L, Emperle M, Guo Y, Grimm S, Ren W, et al. 2020. Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat. Commun 11:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Song J, Rechkoblit O, Bestor T, Patel D. 2011. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331:1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hashimoto H, Pais J, Zhang X, Saleh L, Fu Z, et al. 2014. Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature 506:391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sood A, Viner C, Hoffman M 2019. DNAmod: the DNA modification database. J. Cheminform 11:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]